
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ambiphilicity of a mononuclear cobalt(III) superoxo complex†
Ting-Yi
Chen
ab,
Po-Hsun
Ho
a,
Can-Jerome
Spyra
b,
Franc
Meyer
 *b,
Eckhard
Bill
c,
Shengfa
Ye
*de and
Way-Zen
Lee
*af
*b,
Eckhard
Bill
c,
Shengfa
Ye
*de and
Way-Zen
Lee
*af
aDepartment of Chemistry, National Taiwan Normal University, Taipei 11677, Taiwan. E-mail: wzlee@ntnu.edu.tw
bUniversität Göttingen, Institut für Anorganische Chemie, D-37077 Göttingen, Germany. E-mail: franc.meyer@chemie.uni-goettingen.de
cMax-Planck-Institut für Chemische Energiekonversion, Mülheim an der Ruhr D-45470, Germany. E-mail: eckhard.bill@cec.mpg.de
dState Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China. E-mail: shengfa.ye@dicp.ac.cn
eMax-Planck-Institut für Kohlenforschung, Mülheim an der Ruhr, D-45470, Germany. E-mail: shengfa.ye@kofo.mpg.de
fDepartment of Medicinal and Applied Chemistry, Kaohsiung Medical University, Kaohsiung 807, Taiwan
First published on 15th October 2020
Abstract
Addition of HOTf to a mixture of CoIII(BDPP)(O2˙) (1, H2BDPP = 2,6-bis((2-(S)-diphenylhydroxylmethyl-1-pyrrolidinyl)methyl)pyridine) and Cp*2Fe produced H2O2 in high yield implying formation of CoIII(BDPP)(OOH) (3), and reaction of Sc(OTf)3 with the same mixture gave a peroxo-bridged CoIII/ScIII5. These findings demonstrate the ambiphilic property of CoIII-superoxo 1.
Metal-superoxo species are often believed to be the first intermediate following dioxygen (O2) association in the catalytic cycle of O2 activating metalloenzymes.1 Despite intensive work in the past, the chemistry of metal-superoxo complexes remains largely unexplored, and hence attracts significant attention from chemists and biochemists.2Inter alia, metal-superoxo intermediates can react with NO˙ or organic radicals to furnish metal-peroxynitrite3 and -alkylperoxo4 complexes via radical coupling. Furthermore, they exhibit considerable electrophilicity as indicated by their capability of performing hydrogen atom abstraction (HAA)5 from weak C–H and O–H bonds and oxygen atom transfer6 to triphenylphosphine or thiol anisoles. On the other hand, they can initiate deformylation processes when treated with 2-phenylpropionaldehyde, thereby revealing their nucleophilic character.7,8 Besides the aforementioned well know activities, in a given elementary transformation metal-superoxo intermediates may function not only as an electrophile but also as a nucleophile. In fact, ambiphilicity of metal-superoxo species has been postulated in a series of theoretical and experimentally investigations including O2 activation catalyzed by α-ketoglutarate dependent dioxygenases,9 and by Cu, Fe and Co model complexes.10 Only recently has such ambiphilic property been experimentally confirmed.8 In our continuing efforts devoted to investigating reactivity of metal-superoxo intermediates, some of us succeeded in preparing a range of homologous FeIII-, CoIII- and MnIII-superoxo species by reacting O2 with the corresponding divalent precursors.11 It has been shown that these trivalent metal-superoxo complexes can convert into the metal-hydroperoxo complexes via HAA. In particular, the reaction of MnIII(BDPBrP)(O2˙) (H2BDPBrP = 2,6-bis((2-(S)-di(4-bromo)-phenylhydroxylmethyl-1-pyrrolidinyl)methyl)pyridine) with trifluoroacetic acid (TFA) and Sc(OTf)3 yields rare examples of MnIV-hydroperoxo complexes, MnIV(BDPBrP)(OOH), and [MnIV(μ-OO)Sc(OTf)n](3−n)+ as evidenced by the combined spectroscopic and computational studies (Scheme 1).8 Obviously, these proton- and metal-coupled electron transfer processes provide the first experimental support for the proposed ambiphilicity of metal-superoxo species. In this regard, more examples are desired to fully understand how the ambiphilic property of metal-superoxo species affects their chemical reactivity. To this end, we examined the reaction of a CoIII-superoxo complex, CoIII(BDPP)(O2˙) (1, H2BDPP = 2,6-bis((2-(S)-diphenylhydroxylmethyl-1-pyrrolidinyl)methyl)pyridine) with TFA and Sc(OTf)3 together with external electron donors.
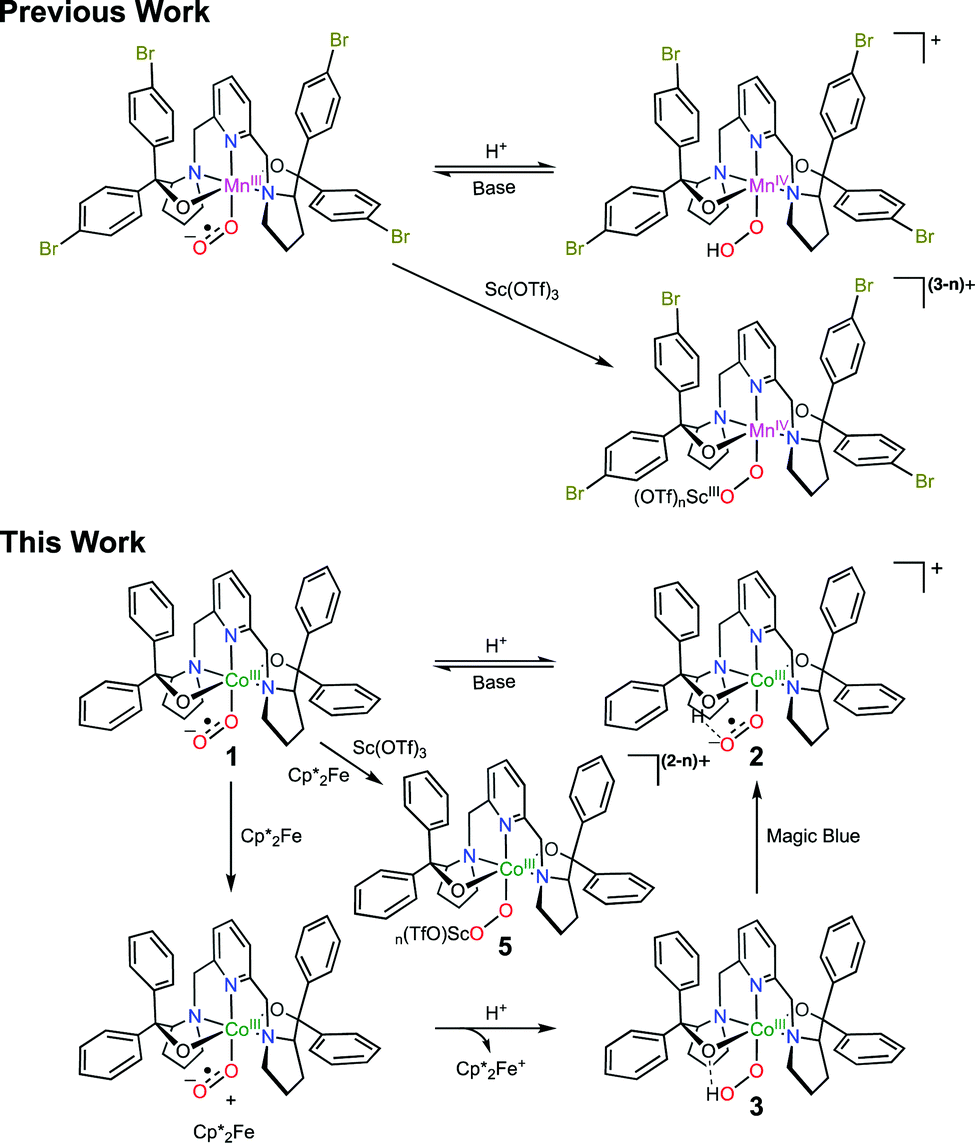 | ||
| Scheme 1 Ambiphilicity of metal-superoxo complexes. | ||
Treating 1 with HOTf in THF at −90 °C gave a gray-green solution attributed to intermediate 2 having two weak absorption bands at 470 and 640 nm, which reached maxima when 1 equiv. of HOTf was added (the inset of Fig. 1). The existence of an isosbestic point at 590 suggested that no intermediate was formed in the course of conversion of 1 to 2 (Fig. 1). Conversely, complex 1 can be retrieved from deprotonation of 2 by 1 equiv. of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) with a yield of 80% with respect to 1 (Fig. S1, ESI†). Moreover, complex 2 can be obtained from one-electron oxidation of the hydroperoxo complex CoIII(BDPP)(OOH) (3). Adding equimolar of tris(4-bromophenyl)ammoniumyl hexachloroantimonate, which is often referred to as magic blue, to a THF solution of 3 at −90 °C resulted in a gray-green solution, whose absorption spectrum displayed the same signature features as those found for 2 (Fig. S2, ESI†).
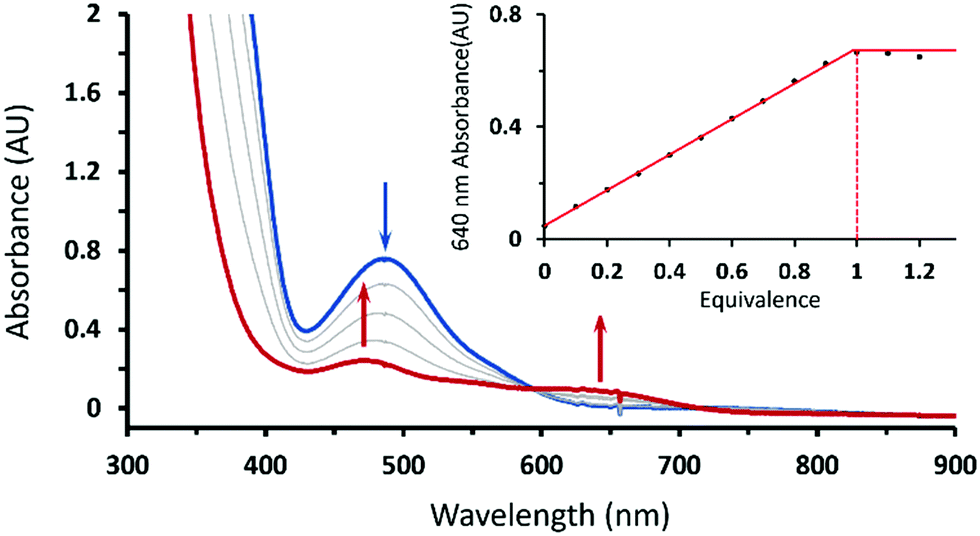 | ||
| Fig. 1 UV-vis spectral changes of the reaction of 1 (1.0 mM, blue trace) with 1 equiv. of HOTf in THF at −90 °C. Inset: Titration curve of 1 with HOTf. X-axis: the equivalents of HOTf, Y-axis: the conversion ratio of the absorption peak at 640 nm. | ||
To identify the exact nature of the resulting species 2, we have undertaken detailed spectroscopic characterization and DFT calculations. The EPR measurement of 2 exhibited a spectrum similar to that of 1 (ACo = 18 G) except for a slightly larger 59Co hyperfine coupling constant (ACo = 24 G) seen in Fig. 2, thus indicating that 2 still consists of a CoIII center coupled with a radical ligand yielding an overall doublet ground state. The radical ligand thus would be a hydroperoxyl radical or a superoxo having a strong hydrogen bonding interaction with the protonated BDPP2− ligand (Scheme 1) as suggested by the crystal structure of 3.11b However, the EPR spectrum of the product generated by reacting 1 with deuterated triflate acid (DOTf) is almost identical to that of 2 without discernable line broadening, which essentially rules out the possibility of the radical ligand being a hydroperoxyl radical (Fig. 2C). Repeated attempts to obtain the O–O vibrational frequencies of 2 from resonance Raman measurements did not accomplish, largely because 2 has only weak chromophores in the usual UV-vis region (Fig. 1). Consequently, the intensity of the O–O stretching signal is too low to be readily detected.
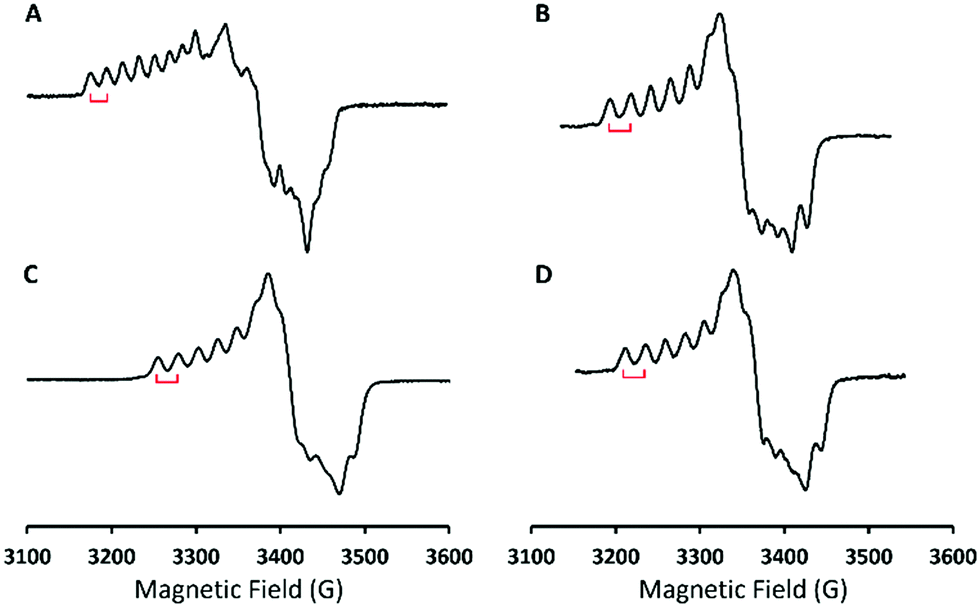 | ||
| Fig. 2 X-band EPR spectra of (A) 1 (g1 = 2.098, ACo = 18 G); (B) 1 with 1 equiv. of HOTf (g1 = 2.066, ACo = 24 G); (C) 1 with 1 equiv. of DOTf (g1 = 2.064, ACo = 24 G) and (D) 1 with 1 equiv. of Sc(OTf)3 (g1 = 2.066, ACo = 24 G). Measurement condition: T = 77 K, fmw = 9.6 GHz. | ||
DFT calculations also suggested the O donor of the BDPP2− ligand to be the favored protonation site of 1, consistent with experiment. Even when the starting geometry contained a OOH ligand in which the distal H atom forms a hydrogen bond with the BDPP2− ligand, the geometry optimizations invariably shifted the H atom back to the O atom of BDPP2− and eventually converged to A (Fig. 3). We also tested the initial geometry without the hydrogen bond by tilting the H atom upward. The computations indeed yielded a CoIII center bound to a hydroperoxyl radical ligand (C), but C lies 23.2 kcal mol−1 higher in energy above A (Fig. 3). Moreover, formation of a hydrogen bond between the superoxo motif and the proton of the OH group of the protonated BDPP2− ligand stabilized A by 10.4 kcal mol−1 relative to B (Fig. 3). Thus, A is best deemed as the most appropriate model for 2.
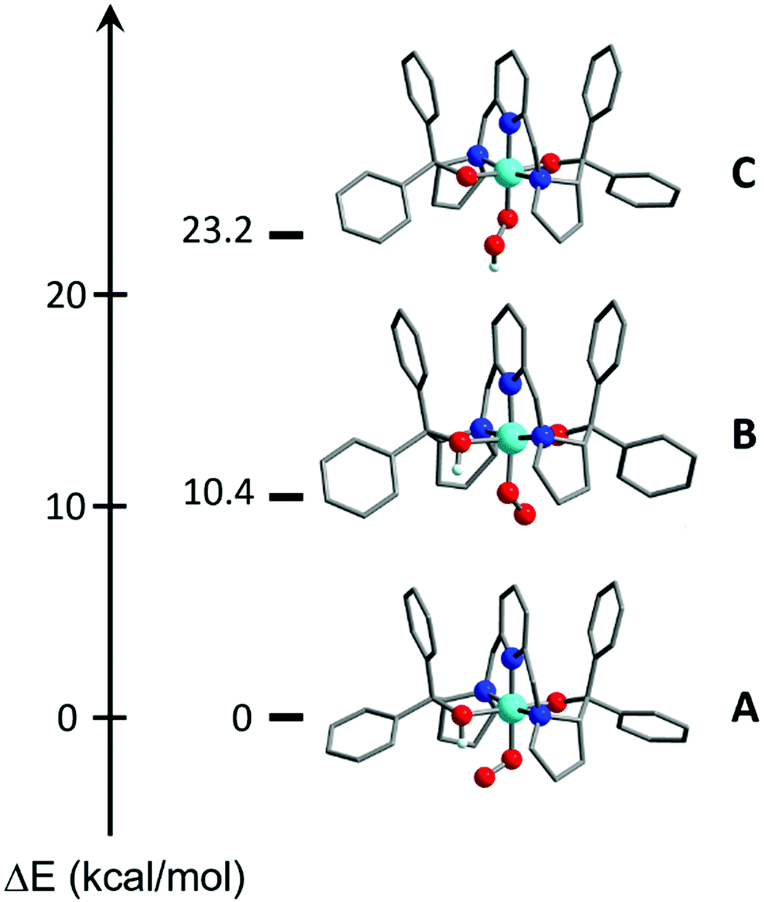 | ||
| Fig. 3 Computed models for 2. | ||
Alternatively, to transform 1 into the corresponding peroxo product, we then added 1 equiv. of decamethylferrocene (Cp*2Fe) or sodium naphthalenide (NaC10H8) to THF solutions of 1 at −90 °C, but UV-vis measurements suggested that no reactions occurred (Fig. S3 and S4, ESI†). Taken together, neither proton nor electron donors alone can realize the superoxo-to-peroxo conversion for 1.
Interestingly, upon treating a mixture containing equimolar 1 and Cp*2Fe with 1 equiv. of HOTf, the color of the reaction solution gradually changed from gray-green to dark green then orange; meanwhile, characteristic features of decamethylferrocenium (Cp*2Fe+) emerged suggesting that CoIII-superoxo 1 was reduced in the presence of both HOTf and Cp*2Fe (Fig. 4A). During this process, we did not observe the formation of CoIII-hydroperoxo 3. Instead, the reaction produced 19% of H2O2 with respect to 1, as determined by iodometric titration (Fig. S5, ESI†). When 2 equiv. of HOTf was added, 42% of H2O2 was furnished (Fig. S6, ESI†). Thus, we reasoned that the aforementioned reaction indeed generates 3; however, once formed, 3 further reacted with HOTf to produce H2O2. On the other hand, treating 2 with 1 equiv. of Cp*2Fe (Fig. 4B) also generated 23% of H2O2 (Fig. S7, ESI†). Therefore, all experimental findings revealed that transformation of 1 to 3 proceeds via concerted proton coupled electron transfer, which clearly demonstrated the ambiphilicity of 1.
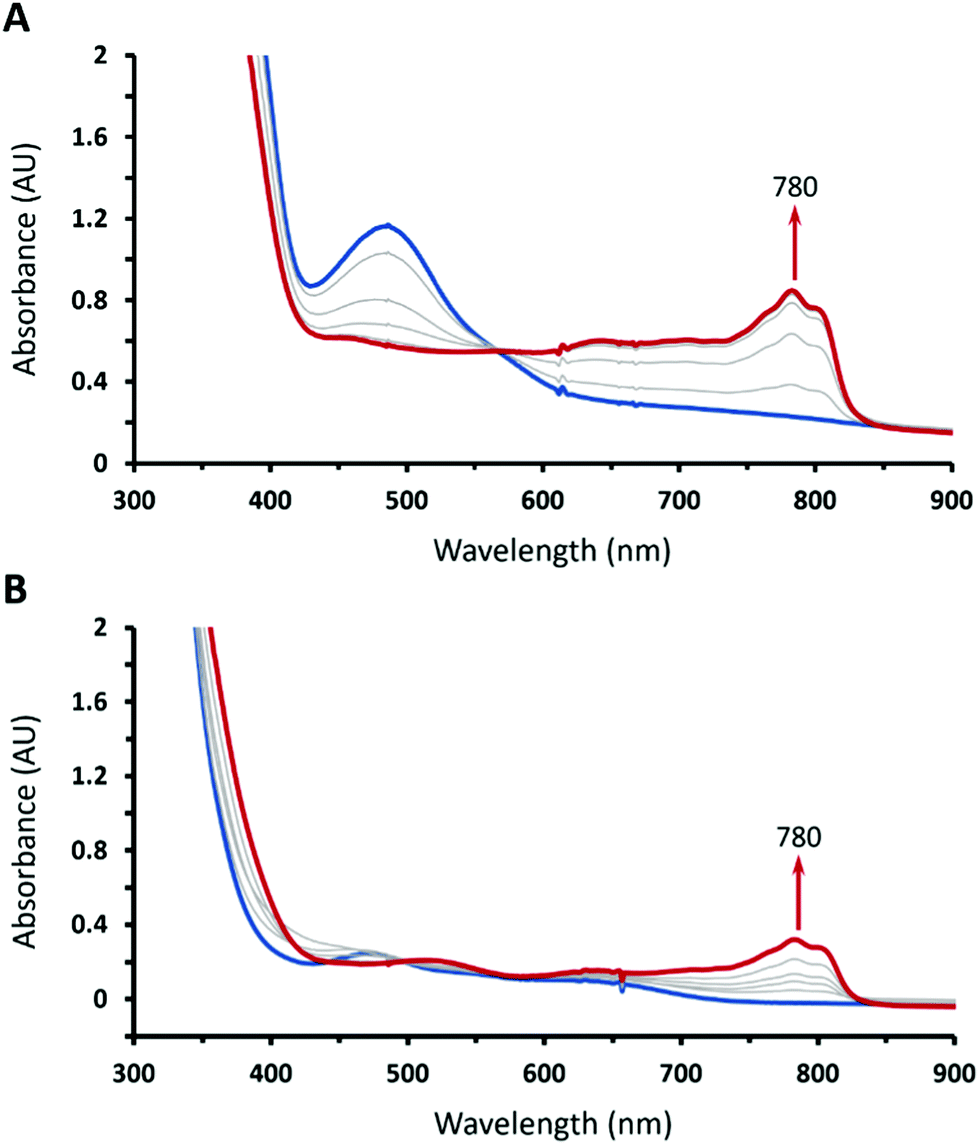 | ||
| Fig. 4 UV-vis spectral changes of addition of (A) 1 equiv. of HOTf into a solution of 1 (1.0 mM) in the presence of 1 equiv. of Cp*2Fe and (B) 1 equiv. of Cp*2Fe into a solution of 2 (1.0 mM) in THF at −90 °C. | ||
Reaction of 1 with Sc(OTf)3 in THF at −90 °C generated a product, which showed nearly identical absorption and EPR spectra to those of 2 (Fig. S8 (ESI†) and Fig. 2D). On the basis of these observations, we tentatively suggest that the reaction furnished a CoIII-superoxo⋯ScIII species, [Co(BDPP)(O2˙)⋯Sc(OTf)n](3−n)+ (4). Furthermore, addition of Sc(OTf)3 to the mixture of 1 and Cp*2Fe in THF at −90 °C afforded a CoIII-peroxo-ScIII complex, [Co(BDPP)(μ-OO)Sc(OTf)n](2−n)+ (5), as depicted in Scheme 1 (Fig. S9, ESI†). Addition of 2 equiv. of HOTf to the THF solution of 5 produced 76% H2O2 as quantified by iodometric titration experiments (Fig. S10, ESI†). Formation of H2O2 thus strongly supports the identity of peroxo-bridged binuclear 5. These findings further reinforce the ambiphilic property of 1.
In comparison with the similar reaction found for MnIII(BDPBrP)(O2˙) (Scheme 1), the difference can be readily attributed to the much higher oxidation potential of CoIII to CoIV than that of MnIII to MnIV, which can ultimately be rooted back to the distinct effective nuclear charge of low spin CoIII compared to high spin MnIII centers. Therefore, formation of an otherwise hydroperoxo O–H bond does not provide a sufficient driving force to trigger an electron transfer from the CoIII center to the superoxo ligand. Consequently, the superoxo motif is not electron rich enough to accommodate the incoming proton from HOTf and protonation of the supporting BDPP2− ligand is preferred. Therefore, to effect surperoxo-to-peroxo conversion for 1, an external electron source has to be provided in addition to Brønsted or Lewis acids.
In conclusion, treatment of CoIII-superoxo 1 with HOTf and Sc(OTf)3 afforded the ligand-protonated CoIII-superoxo 2 with a hydrogen bond formed between the O2˙− motif and the protonated BDPP2− ligand and a superoxo-bridged binuclear CoIII/ScIII4, and CoIII-superoxo 1 can be regenerated from deprotonation of 2 by DBU. However, addition of 2 equiv. of HOTf into the reaction mixture of 1 and Cp*2Fe produced 42% of H2O2 suggesting the formation of CoIII-hydroperoxo 3, and the reaction of Sc(OTf)3 with 1 in the presence of Cp*2Fe gave a peroxo-bridged binuclear CoIII/ScIII5. These findings provided strong experimental support for the ambiphilic property of CoIII-superoxo 1. Interestingly, the ligand-protonated CoIII-superoxo 2 can be prepared from one-electron oxidation of CoIII-hydroperoxo 3. The unveiled results underline the critical property of ambiphilicity for metal-superoxo species and direct us to design further investigation strategies towards better understanding O2 activation processes carried out by metalloenzymes and related catalysts.
We are grateful for the financial supports from the Ministry of Science and Technology of Taiwan (MOST 108-2113-M-003-009-MY3 to W.-Z. L.) and the Max-Planck Society. W.-Z. L. and S. Y. also acknowledge the MOST-DAAD Project-Based Personnel Exchange Program (MOST 107-2911-I-003-502 and DAAD 57320810). Open Access funding provided by the Max Planck Society.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) E. G. Kovaleva and J. D. Lipscomb, Nat. Chem. Biol., 2008, 4, 186–193 CrossRef CAS; (b) M. Costas, M. P. Mehn, M. P. Jensen and L. Que, Chem. Rev., 2004, 104, 939–986 CrossRef CAS; (c) A. J. Jasniewski and L. Que, Jr., Chem. Rev., 2018, 118, 2554–2592 CrossRef CAS; (d) C. E. Elwell, N. L. Gagnon, B. D. Neisen, D. Dhar, A. D. Spaeth, G. M. Yee and W. B. Tolman, Chem. Rev., 2017, 117, 2059–2107 CrossRef CAS; (e) K. Ray, F. F. Pfaff, B. Wang and W. Nam, J. Am. Chem. Soc., 2014, 136, 13942–13958 CrossRef CAS; (f) S. Sahu and D. P. Goldberg, J. Am. Chem. Soc., 2016, 138, 11410–11428 CrossRef CAS.
- (a) M. H. Dickman and M. T. Pope, Chem. Rev., 1994, 94, 569–584 CrossRef CAS; (b) S. Fukuzumi, Y. M. Lee and W. Nam, Dalton Trans., 2019, 48, 9469–9489 RSC; (c) H. Noh and J. Cho, Coord. Chem. Rev., 2019, 382, 126–144 CrossRef CAS; (d) X. Cai, S. Majumdar, G. C. Fortman, C. S. J. Cazin, A. M. Z. Slawin, C. Lhermitte, R. Prabhakar, M. E. Germain, T. Palluccio, S. P. Nolan, E. V. Rybak-Akimova, M. Temprado, B. Captain and C. D. Hoff, J. Am. Chem. Soc., 2011, 133, 1290–1293 CrossRef CAS; (e) R. Huacuja, D. J. Graham, C. M. Fafard and C.-H. Chen, J. Am. Chem. Soc., 2011, 133, 3820–3823 CrossRef CAS; (f) X. Zhang, G. R. Loppnow, R. McDonald and J. Takats, J. Am. Chem. Soc., 1995, 117, 7828–7829 CrossRef CAS.
- (a) S. Herold and W. H. Koppenol, Coord. Chem. Rev., 2005, 249, 499–506 CrossRef CAS; (b) R. D. Harcourt, Coord. Chem. Rev., 2018, 358, 178–180 CrossRef CAS; (c) P. R. Gardner, A. M. Gardner, L. A. Martin and A. L. Salzman, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 10378–10383 CrossRef CAS; (d) A. K. Das and M. Meuwly, Angew. Chem., Int. Ed., 2018, 57, 3509–3513 CrossRef CAS; (e) S. K. Sharma, A. W. Schaefer, H. Lim, H. Matsumura, P. Moënne-Loccoz, B. Hedman, K. O. Hodgson, E. I. Solomon and K. D. Karlin, J. Am. Chem. Soc., 2017, 139, 17421–17430 CrossRef CAS; (f) R. Cao, L. T. Elrod, R. L. Lehane, E. Kim and K. D. Karlin, J. Am. Chem. Soc., 2016, 138, 16148–16158 CrossRef CAS; (g) J. J. Liu, M. A. Siegler, K. D. Karlin and P. Moenne-Loccoz, Angew. Chem., Int. Ed., 2019, 58, 10936–10940 CrossRef CAS.
- (a) E. G. Kovaleva and J. D. Lipscomb, Science, 2007, 316, 453 CrossRef CAS; (b) P. Kumar, S. V. Lindeman and A. T. Fiedler, J. Am. Chem. Soc., 2019, 141, 10984–10987 CrossRef CAS.
- (a) M. N. Blakely, M. A. Dedushko, P. C. Yan Poon, G. Villar-Acevedo and J. A. Kovacs, J. Am. Chem. Soc., 2019, 141, 1867–1870 CrossRef CAS; (b) A. Kunishita, M. Kubo, H. Sugimoto, T. Ogura, K. Sato, T. Takui and S. Itoh, J. Am. Chem. Soc., 2009, 131, 2788–2789 CrossRef CAS; (c) R. L. Peterson, R. A. Himes, H. Kotani, T. Suenobu, L. Tian, M. A. Siegler, E. I. Solomon, S. Fukuzumi and K. D. Karlin, J. Am. Chem. Soc., 2011, 133, 1702–1705 CrossRef CAS; (d) J. Cho, J. Woo and W. Nam, J. Am. Chem. Soc., 2010, 132, 5958–5959 CrossRef CAS; (e) H. Kelm and H.-J. Krüger, Angew. Chem., Int. Ed., 2001, 40, 2344–2348 CrossRef CAS; (f) E. Tamanaha, B. Zhang, Y. Guo, W.-C. Chang, E. W. Barr, G. Xing, J. St Clair, S. Ye, F. Neese, J. M. Bollinger, Jr. and C. Krebs, J. Am. Chem. Soc., 2016, 138, 8862–8874 CrossRef CAS; (g) N. Kindermann, C.-J. Günes, S. Dechert and F. Meyer, J. Am. Chem. Soc., 2017, 139, 9831–9834 CrossRef CAS.
- (a) M. T. Kieber-Emmons, J. Annaraj, M. S. Seo, K. M. Van Heuvelen, T. Tosha, T. Kitagawa, T. C. Brunold, W. Nam and C. G. Riordan, J. Am. Chem. Soc., 2006, 128, 14230–14231 CrossRef CAS; (b) S. Yao, E. Bill, C. Milsmann, K. Wieghardt and M. Driess, Angew. Chem., Int. Ed., 2008, 47, 7110–7113 CrossRef CAS; (c) J. Cho, J. Woo and W. Nam, J. Am. Chem. Soc., 2012, 134, 11112–11115 CrossRef CAS; (d) K. Fujita, R. Schenker, W. Gu, T. C. Brunold, S. P. Cramer and C. G. Riordan, Inorg. Chem., 2004, 43, 3324–3326 CrossRef CAS; (e) T. Tano, Y. Okubo, A. Kunishita, M. Kubo, H. Sugimoto, N. Fujieda, T. Ogura and S. Itoh, Inorg. Chem., 2013, 52, 10431–10437 CrossRef CAS; (f) L.-L. Liu, H.-X. Li, L.-M. Wan, Z.-G. Ren, H.-F. Wang and J.-P. Lang, Chem. Commun., 2011, 47, 11146–11148 RSC.
- (a) P. Pirovano, A. M. Magherusan, C. McGlynn, A. Ure, A. Lynes and A. R. McDonald, Angew. Chem., Int. Ed., 2014, 126, 6056–6060 CrossRef; (b) W. D. Bailey, N. L. Gagnon, C. E. Elwell, A. C. Cramblitt, C. J. Bouchey and W. B. Tolman, Inorg. Chem., 2019, 58, 4706–4711 CrossRef CAS.
- Y.-H. Lin, Y. Kutin, M. van Gastel, E. Bill, A. Schnegg, S. Ye and W.-Z. Lee, J. Am. Chem. Soc., 2020, 142, 10255–10260 CrossRef CAS.
- (a) S. Ye, C. Riplinger, A. Hansen, C. Krebs, J. M. Bollinger, Jr. and F. Neese, Chem. – Eur. J., 2012, 18, 6555–6567 CrossRef CAS; (b) T. Borowski, A. Bassan and P. E. M. Siegbahn, Chem. – Eur. J., 2004, 10, 1031–1041 CrossRef CAS.
- (a) W. D. Bailey, D. Dhar, A. C. Cramblitt and W. B. Tolman, J. Am. Chem. Soc., 2019, 141, 5470–5480 CrossRef CAS; (b) S. Hong, K. D. Sutherlin, J. Park, E. Kwon, M. A. Siegler, E. I. Solomon and W. Nam, Nat. Commun., 2014, 5, 5440–5547 CrossRef CAS; (c) M. Sankaralingam, Y.-M. Lee, W. Nam and S. Fukuzumi, Coord. Chem. Rev., 2018, 365, 41–59 CrossRef CAS; (d) A. R. Corcos, O. Villanueva, R. C. Walroth, S. K. Sharma, J. Bacsa, K. M. Lancaster, C. E. MacBeth and J. F. Berry, J. Am. Chem. Soc., 2016, 138, 1796–1799 CrossRef CAS.
- (a) C. W. Chiang, S. T. Kleespies, H. D. Stout, K. K. Meier, P. Y. Li, E. L. Bominaar, L. Que, Jr., E. Munck and W. Z. Lee, J. Am. Chem. Soc., 2014, 136, 10846–10849 CrossRef CAS; (b) C. C. Wang, H. C. Chang, Y. C. Lai, H. Fang, C. C. Li, H. K. Hsu, Z. Y. Li, T. S. Lin, T. S. Kuo, F. Neese, S. Ye, Y. W. Chiang, M. L. Tsai, W. F. Liaw and W. Z. Lee, J. Am. Chem. Soc., 2016, 138, 14186–14189 CrossRef CAS; (c) Y. H. Lin, H. H. Cramer, M. van Gastel, Y. H. Tsai, C. Y. Chu, T. S. Kuo, I. R. Lee, S. Ye, E. Bill and W. Z. Lee, Inorg. Chem., 2019, 58, 9756–9765 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc05337f |
| This journal is © The Royal Society of Chemistry 2020 |