 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Towards compartmentalized photocatalysis: multihaem proteins as transmembrane molecular electron conduits†
Anna
Stikane
 ab,
Ee Taek
Hwang
ab,
Emma V.
Ainsworth
c,
Samuel E. H.
Piper
c,
Kevin
Critchley
bd,
Julea N.
Butt
*c,
Erwin
Reisner
*e and
Lars J. C.
Jeuken
*ab
ab,
Ee Taek
Hwang
ab,
Emma V.
Ainsworth
c,
Samuel E. H.
Piper
c,
Kevin
Critchley
bd,
Julea N.
Butt
*c,
Erwin
Reisner
*e and
Lars J. C.
Jeuken
*ab
aSchool of Biomedical Sciences, University of Leeds, Leeds, LS2 9JT, UK. E-mail: L.J.C.Jeuken@leeds.ac.uk
bThe Astbury Centre for Structural Molecular Biology, University of Leeds, Leeds, LS2 9JT, UK
cCentre for Molecular and Structural Biochemistry, School of Chemistry and School of Biological Sciences, University of East Anglia, Norwich, NR4 7TJ, UK
dSchool of Physics and Astronomy, University of Leeds, Leeds, LS2 9JT, UK
eDepartment of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK
First published on 21st December 2018
Abstract
The high quantum efficiency of natural photosynthesis has inspired chemists for solar fuel synthesis. In photosynthesis, charge recombination in photosystems is minimized by efficient charge separation across the thylakoid membrane. Building on our previous bioelectrochemical studies of electron transfer between a light-harvesting nanoparticle (LHNP) and the decahaem subunit MtrC, we demonstrate photo-induced electron transfer through the full transmembrane MtrCAB complex in liposome membranes. Successful photoelectron transfer is demonstrated by the decomposition of a redox dye, Reactive Red 120 (RR120), encapsulated in MtrCAB proteoliposomes. The photoreduction rates are found to be dependent on the identity of the external LHNPs, specifically, dye-sensitized TiO2, amorphous carbon dots (a-CD) and graphitic carbon dots with core nitrogen doping (g-N-CDs). Agglomeration or aggregation of TiO2 NPs likely reduces the kinetics of RR120 reductive decomposition. In contrast, with the dispersed a-CD and g-N-CDs, the kinetics of the RR120 reductive decomposition are observed to be faster with the MtrCAB proteoliposomes and we propose that this is due to enhancement in the charge-separated state. Thus, we show a proof-of-concept for using MtrCAB as a lipid membrane-spanning building block for compartmentalised photocatalysis that mimics photosynthesis. Future work is focused on incorporation of fuel generating redox catalysts in the MtrCAB proteoliposome lumen.
Introduction
Global research efforts are continuously advancing strategies for harnessing solar energy into sustainable electricity, solar fuels and solar chemicals.1–3 The light harvesting stage, i.e., photo-induced charge separation and electron (or hole) transfer to the electrode or catalyst, remains the principal efficiency-limiting step in these strategies.3 In contrast, the stunning efficiency of biological light-harvesting systems results from a very precise and sophisticated arrangement of photosynthetic components: organic photosensitizers (e.g., P680, P700, etc.), electron relay (chlorophyll, pheophytin, quinones, tyrosine, etc.) and biocatalytic conversions (QB reduction/water splitting).4 These components are optimized in the dimensions of space (the relative location of the components), energy (excited-state and redox properties) and time (the rates of competing processes).5,6 The composition of biological photosynthetic assemblies allows efficient photon absorption at light harvesting antennae, after which energy is passed along a series of chromophores to the reaction centres (e.g., plant photosystems I and II), where it is used for excitation of the P680/P700 cofactors.4 Electrons ejected from P680/P700 are relayed along an electron transfer chain and the light energy is ultimately stored as a transmembrane proton gradient and reduced redox-active molecules such as NADPH7 (Fig. 1a).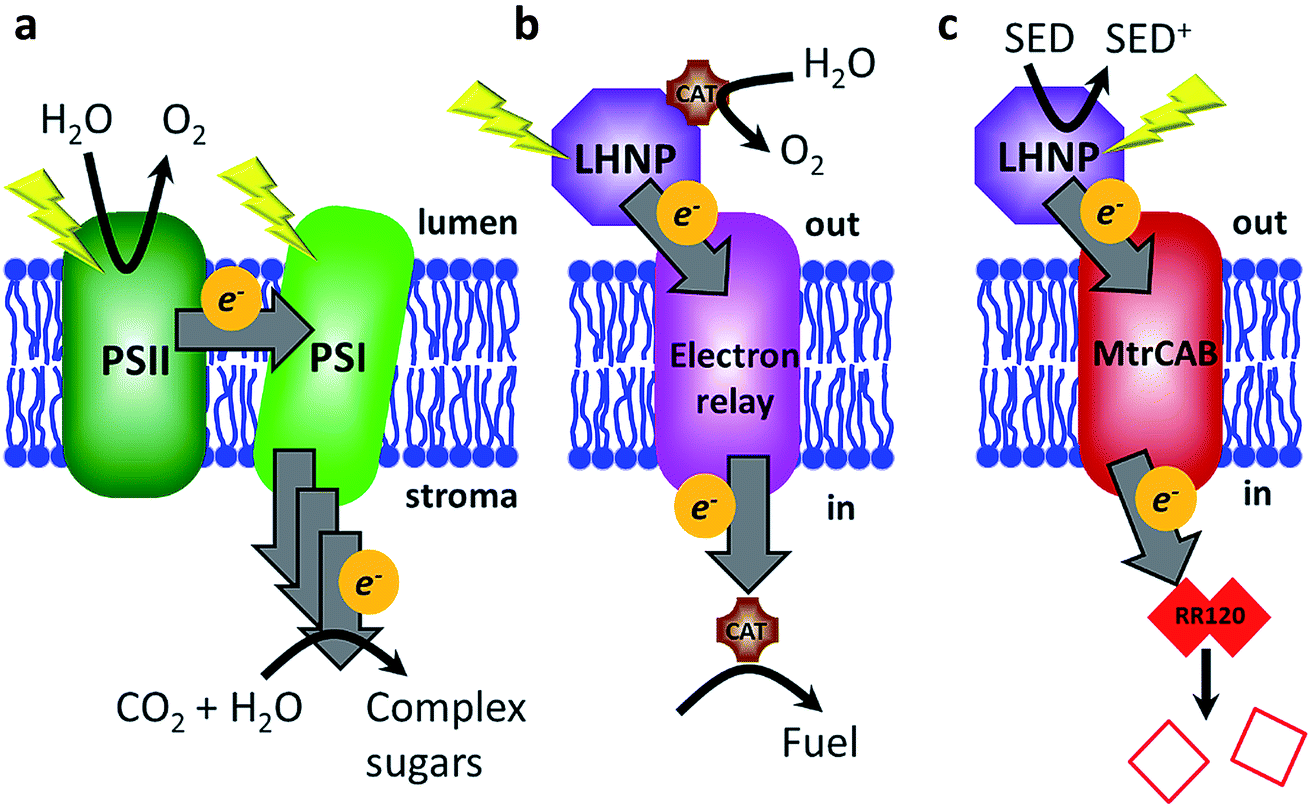 | ||
| Fig. 1 A schematic of light-driven electron transfer across the lipid membrane in nature (a), in the envisioned biomimicking system (b), and as presented in this study (c). (a) Photosystems I and II (PSI and PSII) are photo-excited and electrons are transferred via several electron acceptors across the membrane, where they are ultimately used for CO2 conversion into complex sugars. (b) External electrons are supplied photochemically from a light-harvesting nanoparticle (LHNP), which is regenerated by a water-oxidising catalyst (CAT). Electrons are relayed across the membrane to a catalyst leading to fuel generation within the compartment. (c) Electron transfer across the lipid bilayer is ensured via the transmembrane protein complex MtrCAB and monitored following reductive bleaching of an internalised red azo dye, Reactive Red 120 (RR120). SED – sacrificial electron donor. | ||
Features such as near-unity quantum yields and environmentally friendly operation put biological light-harvesting systems above any other known system with regard to the initial steps of light harvesting for production of solar electricity and/or chemical synthesis.7 Hence, there is a lot of interest in directly exploiting natural or genetically modified organisms3,8–11 or their components for energy harvesting in artificial bio-hybrid systems.12 Natural systems such as photosystem I, photosystem II and whole plant thylakoid membranes have been directly coupled to electrodes and inorganic catalysts in various photosynthetic devices to directly produce electricity, fuel (e.g., molecular hydrogen) or evolve oxygen.12–19 However, light-induced damage and degradation limits the use of pigment–protein complexes, especially photosystem II.12,20 Alternative approaches are being developed where synthetic light-harvesting analogues are interfaced to (bio)catalysts to biomimic the general principles of natural photosynthesis.6,7 Reported systems include examples in which photosensitizers (PSs) (e.g., porphyrins) and light-harvesting nanoparticles (LHNPs) (e.g., quantum dots) are interfaced with various conductive materials ranging from graphene to peptide nanotubes to semi-conductor nanoparticles, fuel producing enzymes and electron mediators to regenerate cofactors for redox enzymes.7,21 Efforts are also being made to explore the effects of photosynthetic component spatial organisation by mimicking natural systems such as stacked plant thylakoid membranes22 and chlorosomes of green sulfur bacteria.23
In this work, we aimed to mimic another aspect of plant photosynthesis, i.e., the use of a lipid membrane to arrange and spatially separate the photosynthetic components between the different environments of the thylakoid lumen and stroma (Fig. 1a).4 Specifically, the objective was to spatially separate the photo-oxidation and reduction reactions in the external and internal space of the liposome compartments, respectively (Fig. 1b). Thus, the envisioned system requires four components: (1) PSs or LHNPs to harvest light energy on the outside of liposomes, (2) transmembrane electron transfer, (3) a reduction catalyst within the liposome compartment (lumen) to chemically store the light energy and (4) an oxidation catalyst outside the liposome to regenerate the PS or LHNP.
We chose synthetic LHNPs over their natural equivalents (e.g., plant photosystems I and II) because they are simpler and cheaper to produce and because of their stability and chemical inertness.24–26 Three LHNPs were compared: dye-sensitised TiO2 nanoparticles and two types of carbon dots. Dye-sensitized TiO2 nanoparticles are well-studied and among the most active photocatalyst materials.24 We used TiO2 nanoparticles photosensitized with a Ru(II)(bipyridine)3 dye in which one of the bipyridines is phosphonated in the 4,4′-position to enable chemisorption to TiO2 (RuP-TiO2, see Hwang et al.27). Carbon dots form another group of emerging light-absorbing nanomaterials showing remarkable photo-stability, water solubility, low toxicity and a sustainable and cost-effective synthesis, avoiding the use of rare metals.25,26,28 Here we tested amorphous carbon dots (a-CD)25 and graphitic carbon dots with core nitrogen doping (g-N-CDs).28,29
To transfer electrons across the lipid membrane after light harvesting, an icosa-haem transmembrane protein, MtrCAB, was employed (Fig. 1c), which provides an electron-transfer relay through the otherwise insulating lipid membrane.30,31 MtrCAB is a heterotrimeric protein (MtrA, MtrB and MtrC) found in the bacterium Shewanella oneidensis MR1, where it forms a 20 haem long conductive molecular ‘wire’ across the bacterial outer membrane.30,31 This enables the bacterium to use insoluble minerals such as iron and manganese oxides as external electron acceptors for its anaerobic metabolism.32 When incorporated to span the lipid bilayer of a proteoliposome, MtrCAB exhibits fast transmembrane electron transfer, estimated to be 103–104 electrons per second by spectroscopic reduction of encapsulated methyl viologen.33 We have previously demonstrated efficient electron exchange between the soluble decahaem subunit MtrC and LHNPs.34–36
In the present study, electrons are transferred via MtrCAB to the liposome lumen, where we envision they could generate fuel (such as hydrogen) by a fuel-generating catalyst. In this proof-of-concept study, electron transfer is optically monitored (539 nm) by destructive reduction of an encapsulated azo dye, Reactive Red 120 (RR120, Fig. S1†).37–39 RR120 contains two azo bonds (R–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–R′), each of which requires a transfer of four electrons in order to be cleaved to a colourless (pale yellow) product (Fig. S1c†), i.e., 8 electrons per RR120.38 The optical signatures revealing haem redox status (Fig. S2†) are also monitored.
N–R′), each of which requires a transfer of four electrons in order to be cleaved to a colourless (pale yellow) product (Fig. S1c†), i.e., 8 electrons per RR120.38 The optical signatures revealing haem redox status (Fig. S2†) are also monitored.
Materials and methods
Unless stated otherwise, all of the chemical substances were obtained from commercial suppliers and used without further purification: 3-(N-morpholino)propansulfonic acid (MOPS, >99.5%), sodium sulphate (Na2SO4, analytical reagent grade), N,N-dimethyldodecylamine N-oxide (LDAO, BioXtra, >99%), sodium hydrosulfite (DT, >82%) and Reactive Red 120 azo dye (RR120) were purchased from Sigma-Aldrich. Ethylenediaminetetraacetic acid disodium salt dehydrate (EDTA, >99.5%) and n-octyl glucoside (OG, laboratory grade) were acquired from Melford and Triton X100 detergent (electrophoresis grade) was purchased from Fisher Chemicals. A Milli-Q system was used to generate ultrapure water (resistance 18.2 MΩ cm) which was used throughout. Ruthenium (Ru) dye sensitized TiO2 anatase nanoparticles (RuP-TiO2, diameter 6.8 ± 0.7 nm), g-N-CD (diameter 3.1 ± 1.1 nm) and a-CD (diameter 6.8 ± 2.3 nm) were synthesized an characterized as described previously.25,27,29,40,41Shewanella oneidensis MR1 protein MtrCAB was purified in Triton X-100 as described before.42 The detergent exchange into 5 mM LDAO and additional purity resolution were performed using a Superdex 200 Increase SEC column (GE Healthcare) eluted with 5 mM LDAO, 20 mM HEPES pH 7.8. The purity of the purified MtrCAB was confirmed by SDS-PAGE with the protein visualized by Coomassie and haem stain.43Escherichia coli polar lipid extracts were purchased from Avanti Polar Lipids and stored in 5 mg dry aliquots under a nitrogen atmosphere at −20 °C.Preparation of MtrCAB proteoliposomes
5 mg of E. coli polar lipid extract was dissolved by vigorous vortexing for up to 20 minutes in 294 μL of MOPS buffer (20 mM MOPS, 30 mM Na2SO4, pH 7.4) containing 6.6 mM RR120 and 85 mM OG. 50.5 μL of 10 μM MtrCAB (or 5 mM LDAO for control liposomes) was added to the lipid solution and kept on ice for a further 10 min. The sample was then rapidly diluted while mixing in 50 mL of ice-cold 20 mM RR120 in MOPS buffer. The sample was transferred to an ultracentrifuge tube (polycarbonate) and centrifuged for 100 min at 71![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g at 4 °C. The supernatant containing most of the non-encapsulated RR120 was discarded and the pellet was re-suspended in 500 μL of MOPS buffer. The resulting sample was then centrifuged at 5000g for about 5 min to pellet any aggregates. The remaining non-encapsulated RR120 was removed by two consecutive rounds of 60 min sample incubation with 0.6 g of Bio-Beads (Bio-Rad SM-2) per 1 mL of sample at 4 °C on a rolling shaker. The experiments were performed within 2 days of the liposome preparation.
000g at 4 °C. The supernatant containing most of the non-encapsulated RR120 was discarded and the pellet was re-suspended in 500 μL of MOPS buffer. The resulting sample was then centrifuged at 5000g for about 5 min to pellet any aggregates. The remaining non-encapsulated RR120 was removed by two consecutive rounds of 60 min sample incubation with 0.6 g of Bio-Beads (Bio-Rad SM-2) per 1 mL of sample at 4 °C on a rolling shaker. The experiments were performed within 2 days of the liposome preparation.
Liposome characterization
The concentration and size distribution of the liposomes was determined by nanoparticle tracking analysis (NTA) using Nanosight (NS300, Malvern Panalytical). The liposome size was also determined by dynamic light scattering (DLS) using a Zetasizer Nano Z (Malvern Panalytical). The size and volume of the liposomes were estimated by treating the liposomes as spherical particles, with the average diameter based on NTA data.The amount of reconstituted MtrCAB was determined using a BCA assay (ThermoFisher Scientific). As the absorbance of encapsulated RR120 overlaps with the BCA reagent absorbance, the liposomes were first lysed with 0.1% v/v Triton X100 and RR120 was removed by two consecutive desalting columns (0.5 ml Zeba™ Spin, ThermoFisher) according to the manufacturer’s protocol. The effectiveness of the desalting columns was confirmed using a control sample of RR120 loaded liposomes without MtrCAB.
Reduction of RR120 encapsulated in the MtrCAB proteoliposomes
Samples for the photo-reduction experiments were assembled under a nitrogen atmosphere (glovebox, O2 < 0.1 ppm) to ensure an anaerobic environment. MtrCAB proteoliposome samples were diluted 10-fold in MOPS buffer containing 50 mM sacrificial electron donor (EDTA). An appropriate amount of 10 mg ml−1 photosensitiser stock (27 μmol NP per L RuP-TiO2, 476 μM g-N-CD or 44 μM a-CD; mass of particles is estimated based on size determined by EM and density of material) was added to 1 μmol LHNP per L final concentration. The cuvette was then sealed airtight and removed from the glovebox for UV-vis absorbance spectroscopy (Cary 5000 UV-Vis-NIR, Agilent) fitted with an integrating sphere (Internal DRA-900, Agilent). UV-vis absorbance spectra were measured after 10 s, 50 s, 60 s, 120 s, or in some cases 300 s, of sample irradiation using a cold light source holding a 150 W (15 V) halogen lamp (OSRAM) with a fibre optic arm (Krüss KL5125). The sample was placed 10 cm from the light source and irradiated. The light intensity at the sample under these conditions was approximately 450 ± 40 mW cm−2 at 400 nm. Afterwards, the chemical reductant DT was added (final concentration of 27 mM) to monitor further possible reduction of the RR120. Finally, Triton X100 detergent was added (final concentration 0.045% v/v) to lyse the lipid vesicles and observe the reduction of any remaining RR120. Control experiments testing reduction by DT (i.e., without LHNP) were also performed. Photo-reduction control experiments with non-encapsulated RR120 were performed as above, but with 10 μM RR120, 50 mM EDTA and 1 μmol LHNP per L PS in MOPS buffer. The recovery yield of MtrCAB was observed to vary between the proteoliposome preparations (see Results). To account for this, comparisons of the encapsulated RR120 (photo)reduction by DT and the photosensitisers were made based on proteoliposomes from the same preparation.Treatment of UV-visible spectroscopy data
The spectroscopy data were corrected for sample dilution and for variation in the background signal (by setting the absorbance at 750 nm as the zero absorbance for each spectrum). Absorbance at 539 nm was selected to follow changes in the RR120 absorbance over time, because it is less influenced by the absorbance of reduced MtrCAB (α- and β- haem peaks at 552 and 522 nm). In order to correct for the contribution of liposome scattering, the optical density outside the RR120 absorbance peaks was measured at 440 and 610 nm, i.e., either side of the RR120 absorbance, and the average value was subtracted.Results
Characterization of MtrCAB liposomes
MtrCAB proteoliposomes loaded with the dye RR120 were prepared as described in the methods section. (Proteo)liposomes from each preparation were characterized to determine their size, concentration and the amount of reconstituted MtrCAB and encapsulated RR120, as described in the materials and methods. Although the size of the MtrCAB proteoliposomes showed some batch-to-batch variation, the proteoliposomes were consistently between 100 and 200 nm in diameter (Fig. S3†). The reconstitution protocol generated about 1013 liposomes per mL and thus an estimated total lumen volume in the order of 10–30 μL per mL of sample. Approximately 43 ± 13% of initial MtrCAB was present in the reconstituted proteoliposomes with an estimated ratio of 10–50 MtrCAB proteins per liposome (depending on the liposome size) assuming an even distribution across the liposomes.Estimation of the amount of RR120 encapsulated in the MtrCAB proteoliposomes was performed spectroscopically using optical absorbance at 534 nm (ε534 nm = 31.8 mM−1 cm−1 was determined here using titration). It was estimated that, on average, the RR120 concentration in the liposome lumen was ∼10 mM, i.e., the same order of magnitude as during the liposome formation.
MtrCAB provides electron transfer across the bilayer
The ability of MtrCAB to transfer electrons across the membrane and reductively degrade RR120 was confirmed using excess chemical reductant (DT; Fig. 2). DT (Em approximately −0.41 V vs. SHE at pH 7.4)44 reduced MtrCAB (haem potential window ranging from −0.45 to 0 V vs. SHE)45 within the time resolution of the experiment (<20 s), as indicated by a shift of the MtrCAB Soret peak due to haem absorbance (from 410 to 420 nm, Fig. 2a). This was followed by a slower (minutes) decrease in RR120 absorbance (450–570 nm, RR120 becomes reductively bleached at ≤−0.4 V vs. SHE46), confirming the destructive reduction of the encapsulated RR120 (Fig. 2a). Only ∼10% of RR120 was reduced in the control experiments using liposomes without MtrCAB, indicating that RR120 is protected from reductive bleaching when inside the liposomes and that reduction of encapsulated RR120 proceeds only if MtrCAB is present (Fig. 2b). As a positive control, detergent (Triton X100, TX) was added at the end of the experiment to lyse the liposomes. This was followed by the immediate reductive bleaching of any remaining and now released RR120 (Fig. 2a and b, green lines). The rates of reduction of encapsulated RR120 were observed to vary between the MtrCAB proteoliposome preparations, likely due to the fact that the MtrCAB recovery yields varied (see above). For this reason, (photo)reduction of encapsulated RR120 by different reductants (i.e., DT, LHNPs) was compared using proteoliposomes from the same preparation. In such studies the relative rates of RR120 reduction by the different LHNPs are as reported by the representative data shown below.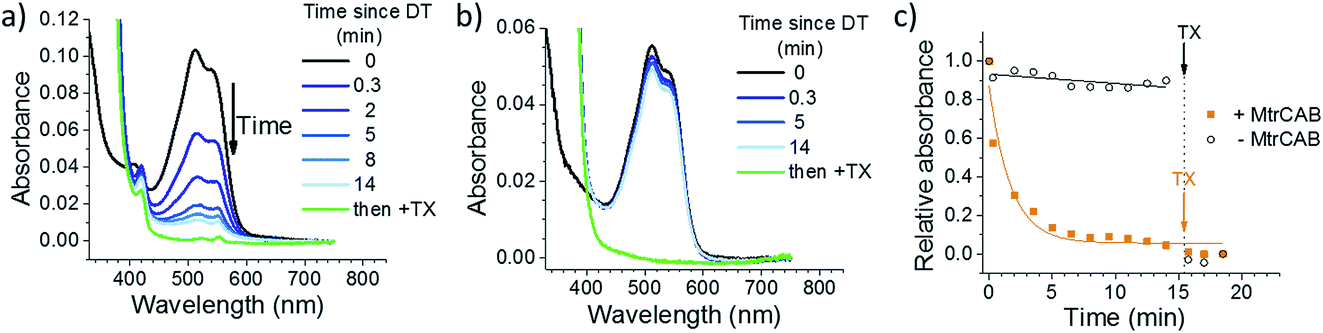 | ||
| Fig. 2 Chemical reduction of encapsulated RR120 by sodium dithionite (DT) with (a) and without (b) MtrCAB. Reduction is followed optically by monitoring the absorbance of the MtrCAB haems (oxidised peak at 410 nm, reduced peak at 420 nm) and RR120 (oxidised 450–570 nm region). Black – oxidized sample; blue – intact liposomes after the addition of sodium dithionite; green – sample after disruption of the proteoliposome bilayer by detergent (Triton X100, TX). Time points indicate the time passed since the addition of DT. (c) The decrease of RR120 absorption (λ = 539 nm) over time using liposomes with and without MtrCAB. The yellow and black lines show the exponential and linear fits to the data, respectively. | ||
Photoreduction across the membrane
Three different LHNPs, i.e., RuP dye-sensitized TiO2 nanoparticles (RuP-TiO2),27 amorphous carbon dots (a-CD)25 and graphitic carbon dots with core nitrogen doping (g-N-CDs),28,29 were tested for photoreduction of RR120 encapsulated in liposomes with and without MtrCAB (Fig. 3a and b). All of the LHNPs have been previously shown to have sufficiently low reducing potential (<−0.45 V vs. SHE)29,47 to be able to reduce methyl viologen, and thus MtrCAB and RR120. Consistent with the data above, in the absence of MtrCAB, the majority (>70%) of RR120 was protected from photoreduction inside the liposome compartments (Fig. 3a). However, subsequent addition of DT to all of the samples showed that slightly more RR120 was reduced in the samples exposed to g-N-CD and RuP-TiO2 compared to the ‘DT only’ control (compare the black open circles to the blue data points in Fig. 3a). This could suggest that small amounts of RR120 are released from the liposomes due to interactions between the RuP-TiO2/g-N-CD and the liposomes. To further quantify this, well-established vesicle leakage assays were performed using a self-quenching dye, carboxyfluorescein.48 No significant leakage was observed upon addition of any of the LHNPs, indicating that no, or very limited, damage is incurred to the vesicles by the LHNPs.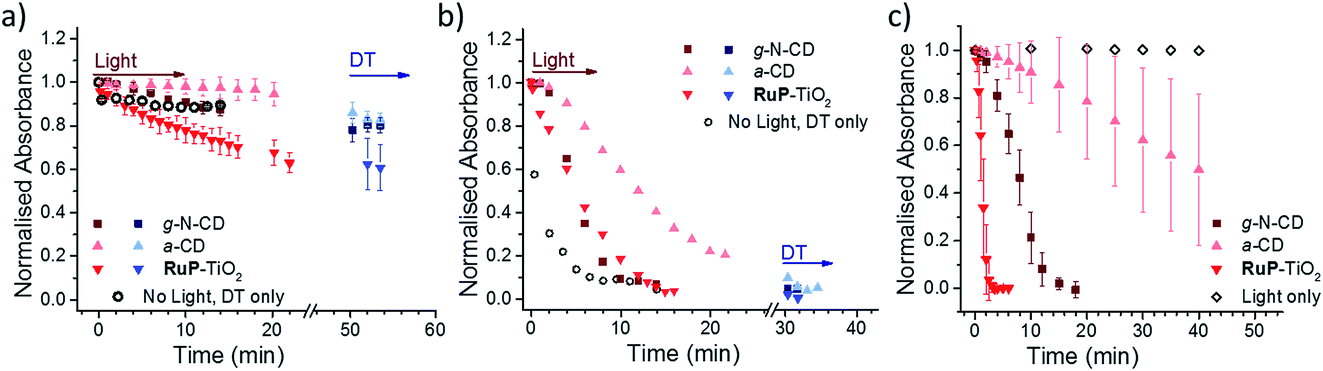 | ||
| Fig. 3 Photoreduction of RR120 encapsulated in liposomes without MtrCAB (a) and in MtrCAB proteoliposomes (b) followed by a decrease in the RR120 absorbance at 539 nm. Squares – g-N-CD; upward triangles – a-CD; downward triangles – RuP-TiO2; red – sample after irradiation; blue – sample after addition of DT; black circles – chemical reduction using DT added at t = 0 and without irradiation. Time points indicate the cumulative time of irradiation. In the case of DT, the time of DT addition is arbitrarily set to 50 and 30 min for (a) and (b), respectively, and the following time points indicate the time passed since the addition of DT. (c) Direct photo-reduction of 10 μM RR120 in solution by LHNPs. White rhombus – irradiation of RR120 without LHNPs. | ||
In the presence of MtrCAB, all three of the LHNPs photo-reduced the encapsulated RR120 (Fig. 3b). These experiments used 1 μM LHNPs, with an estimated ratio of 45 ± 2 LHNP per MtrCAB. RuP-TiO2 and g-N-CD showed the fastest photoreduction, but with a lower rate compared to that with DT. Both the g-N-CDs and a-CD showed a short 1–2 min delay from the start of irradiation until the onset of RR120 photoreduction. This delay is further referred to as the ‘lag phase’ throughout this paper. Quantification of MtrCAB haem photoreduction by all three of the LHNPs was also attempted. Unfortunately, haem difference spectra could not be used due to spectral overlap with changes in RR120 and DT absorbance. Instead the first derivatives of all of the spectra were used instead as this is less sensitive to the background absorbance (Fig. S2 and S4†). This approach suggested that most MtrCAB is photoreduced by RuP-TiO2 within the first minute of irradiation. In the case of the g-N-CDs and a-CD, it appeared that MtrCAB became reduced after several minutes, a time that coincides with the initial lag phase of RR120 reduction. After the lag phase, MtrCAB appeared to be fully reduced by the g-N-CDs, whereas only partial MtrCAB photo-reduction seems to be observed by the a-CDs. This suggest that with a-CD, photo-reduction of RR120 is in a large part rate limited by the photo-reduction of MtrCAB.
Finally, the photo-reduction of RR120 in the MtrCAB proteoliposomes was compared to the direct photo-reduction of non-encapsulated RR120 (Fig. 3c). RuP-TiO2 showed faster photoreduction compared to the MtrCAB proteoliposomes, clearing >90% in less than 2 min, in line with the conclusion that reduction in the proteoliposomes is rate limited by the interaction between RR120 and MtrCAB. In contrast, the g-N-CDs and a-CDs took significantly longer to directly photo-reduce RR120 compared to the MtrCAB proteoliposomes, i.e., about 20 min for the g-N-CDs and for the a-CDs it took more than 40 min to reduce even 50% of the RR120. Both of the LHNPs also showed longer and more variable kinetics, with lag phases of up to 5 min for the g-N-CDs and 10–20 min for the a-CDs. These variations in photo-reduction could reflect heterogeneity within the carbon dots, as observed before.49,50
Discussion
In plant photosynthesis, a lipid membrane is used as scaffolding to arrange and spatially separate photosynthetic components between the different environments of the thylakoid lumen and stroma.4 Here we mimic such physical separation and show biomimetic photo-reduction across an insulating lipid membrane, where energy generated by external LHNPs is transferred across the lipid membrane via MtrCAB conduits to reduce electron acceptors located in the lumen of liposomes.This system has several interfacial electron transfer steps: (1) LHNP to MtrCAB, (2) MtrCAB to RR120 and (3) SED to LHNP (Fig. 1c). All of the experiments used excess amounts of SED (50 mM EDTA) and we have previously shown that the SED is not rate limiting for the photo-reduction of MtrC by RuP-TiO2.27 As MtrCAB provides the electron relay across the membrane, the observed rate of RR120 reduction within the liposomes will be dependent on the amount and distribution of MtrCAB within the liposome population. Chemical reduction of MtrCAB with DT was instantaneous with respect to the time resolution of the experiments reported here. MtrCAB reduction by DT thus represents the fastest possible RR120 reduction within each liposome sample. The photoreduction by all three of the LHNPs was slower than the reduction by DT, confirming that the overall rate of RR120 reduction was at least partly limited by the electron supply from the LHNP to the MtrCAB. However, for the RuP-TiO2 and g-N-CD, MtrCAB was almost fully reduced during the photo-reduction experiments, suggesting that the reductive bleaching kinetics of RR120 were also rate limited by the reduction of RR120 by MtrCAB. MtrCAB orientation in liposomes is not known and likely random, possibly further complicating the observed kinetics.
TiO2 has high affinity for Glu/Asp protein residues,51–53 and RuP-TiO2 has been shown before to bind strongly to MtrC and MtrCAB.36 In addition, RuP-TiO2 showed the best direct photo-reduction of non-encapsulated RR120. Despite this, the photoreduction of RR120 in the MtrCAB liposomes with RuP-TiO2 was slower compared to the chemical reduction with DT. We attribute the slower photoreduction of RR120 in the MtrCAB proteoliposomes to the self-agglomeration or aggregation of the RuP-TiO2 particles, as observed with cryo-electron microscopy analysis (Fig. S5†). Hence, interaction between MtrCAB and RuP-TiO2 might have been impaired. In contrast, the interaction between MtrCAB and both the g-N-CDs and a-CDs is likely to be transient as no aggregation was detected upon mixing of the particles with the MtrCAB liposomes. Nevertheless, for both of the carbon dots, relaying the electrons via MtrCAB improved the bleaching rate of RR120 remarkably, which is up to four times faster in the MtrCAB proteoliposomes compared to the direct photoreduction of RR120. Encapsulation of RR120 at mM concentration in the small lumen of the liposomes (compared to 10 μM RR120 in the control experiments with direct photoreduction) will enhance the reduction kinetics by MtrCAB and, indeed, reduction of RR120 by MtrCAB was not observed to be rate limiting for a-CD. The enhanced photobleaching kinetics in the proteoliposomes are thus due to the faster reduction of MtrCAB by a-CDs compared to reduction of RR120 by a-CD (at concentrations ≪10 μM). We propose that this enhancement is due to the MtrCAB conduit, which can accumulate multiple electrons on its 20 haems, improving the rate of the multi-electron reduction required to bleach each RR120 molecule. In this respect, MtrCAB is able to stabilise the charge separated intermediate for the photo-reduction of RR120, mimicking the role of the chlorophyll/pheophytin/QA electron relay of the natural photosystems I and II.
These results provide an insight into how control over the nano-device organization and assembly can be used in artificial photosynthesis and solar-fuel catalyst design to enhance catalytic and quantum efficiencies. This work adds to the ongoing work in which the organisation of different photosynthetic components is exploited for (bio-)nanocatalysis.7 For example, stacked multilayers of lipid membranes containing PSII22 have been shown to increase production of ATP due to highly efficient exchange of substrates, while limiting the diffusion of photo- and catalytic centres. Besides lipid membranes, various other template materials such as viruses, graphene and peptide fibres have been used to gain control over precise physical distribution of porphyrin PSs and catalytic reaction centres (e.g., Pt, TiO2 and IrO2 clusters).54–59 A 10-times higher yield for selective CO2 conversion into methanol was reported using hollow graphene-doped nanofibers (G-fibers).59 In this case, multiple enzymes required for methanol generation were confined within the nanofibers, and the photo-excited electrons were transported through the graphene fibers from photosensitizers located on the outside.59 In a similar approach, photo-oxidation was separated from photo-reduction reactions by employing hierarchical cobalt oxide–silica core–shell nanotube arrays, where water oxidation and photo-reduction were confined to the inner and outer surface of the nanotubes, respectively.60 Many other ideas for building architectures with isolated environments for separated photo-oxidation and reduction can be drawn from the field of artificial nano-compartments, which has reported the use of various materials ranging from labile biological liposomes, protein cages and virus capsids to rigid synthetic polymersomes and hybrid vesicles.61–63
Conclusion and future perspective
Here, we show a proof-of-concept of using the transmembrane MtrCAB conduit for compartmentalized photo-reduction. Three LHNPs demonstrated efficient photo-reduction of a liposome-encapsulated dye using MtrCAB as an electron relay. The rate with which two different carbon dots photo-reduced the encapsulated dye was improved in the liposome system. This example demonstrated how incorporation of a scaffolding material to separate photo-oxidation and reduction reactions can be beneficial for the overall efficiency of solar energy harvest. In particular, we propose that MtrCAB can aid in the stabilisation of the charge separated state, improving the quantum yield. Such a component could be beneficial to further advance artificial photosynthesis strategies and other (bio-)nanocatalysis applications. To further explore the potential use of MtrCAB conduit and nano-compartments, MtrCAB compartments should be tested for photosynthetic production of solar fuels or solar chemicals. In this case, a catalyst can be encapsulated in the liposomes, which enables a PS/LHNP to function in a separate environment from the fuel-generating catalyst. Finally, the lipids and/or MtrCAB could be replaced by synthetic components to explore other compartmentalised and structured molecular nano-architectures.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was supported by the BBSRC (DTP studentship 1827308; grants BB/K009753/1, BB/K010220/1 and BB/K009885/1) and the Engineering and Physical Sciences Research Council (PhD studentship 1307196). We thank Dr Simone Payne for purifying MtrCAB, Dr Manuela A. Gross for synthesizing RuP, Dr Daisuke Hojo for the TiO2 particles and Dr Benjamin C. M. Martindale for providing the carbon dot samples.References
- M. A. Green, Nat. Energy, 2016, 1, 15015 CrossRef.
- S. Ardo, D. Fernandez Rivas, M. A. Modestino, V. Schulze Greiving, F. F. Abdi, E. Alarcon Llado, V. Artero, K. Ayers, C. Battaglia, J.-P. Becker, D. Bederak, A. Berger, F. Buda, E. Chinello, B. Dam, V. Di Palma, T. Edvinsson, K. Fujii, H. Gardeniers, H. Geerlings, S. M. H. Hashemi, S. Haussener, F. Houle, J. Huskens, B. D. James, K. Konrad, A. Kudo, P. P. Kunturu, D. Lohse, B. Mei, E. L. Miller, G. F. Moore, J. Muller, K. L. Orchard, T. E. Rosser, F. H. Saadi, J.-W. Schüttauf, B. Seger, S. W. Sheehan, W. A. Smith, J. Spurgeon, M. H. Tang, R. van de Krol, P. C. K. Vesborg and P. Westerik, Energy Environ. Sci., 2018, 11, 2768–2783 RSC.
- D. Lips, J. M. Schuurmans, F. Branco Dos Santos and K. J. Hellingwerf, Energy Environ. Sci., 2018, 11, 10–22 RSC.
- N. Nelson and A. Ben-Shem, Nat. Rev. Mol. Cell Biol., 2004, 5, 971–982 CrossRef CAS PubMed.
- M. Şener, J. Strümpfer, J. Hsin, D. Chandler, S. Scheuring, C. N. Hunter and K. Schulten, ChemPhysChem, 2011, 12, 518–531 CrossRef PubMed.
- V. Balzani, A. Credi and M. Venturi, ChemSusChem, 2008, 1, 26–58 CrossRef CAS PubMed.
- J. H. Kim, D. H. Nam and C. B. Park, Curr. Opin. Biotechnol., 2014, 28, 1–9 CrossRef CAS PubMed.
- W. Wei, P. Sun, Z. Li, K. Song, W. Su, B. Wang, Y. Liu and J. Zhao, Sci. Adv., 2018, 4, eaap9253 CrossRef PubMed.
- M. K. Sarma, S. Kaushik and P. Goswami, Biomass Bioenergy, 2016, 90, 187–201 CrossRef CAS.
- L. M. Lassen, A. Z. Nielsen, B. Ziersen, T. Gnanasekaran, B. L. Møller and P. E. Jensen, ACS Synth. Biol., 2014, 3, 1–12 CrossRef CAS PubMed.
- D. J. Vinyard, J. Gimpel, G. M. Ananyev, S. P. Mayfield and G. C. Dismukes, J. Am. Chem. Soc., 2014, 136, 4048–4055 CrossRef CAS PubMed.
- E. Musazade, R. Voloshin, N. Brady, J. Mondal, S. Atashova, S. K. Zharmukhamedov, I. Huseynova, S. Ramakrishna, M. M. Najafpour, J.-R. Shen, B. D. Bruce and S. I. Allakhverdiev, J. Photochem. Photobiol., C, 2018, 35, 134–156 CrossRef CAS.
- M. Miyachi, S. Ikehira, D. Nishiori, Y. Yamanoi, M. Yamada, M. Iwai, T. Tomo, S. I. Allakhverdiev and H. Nishihara, Langmuir, 2017, 33, 1351–1358 CrossRef CAS PubMed.
- F. Zhao, F. Conzuelo, V. Hartmann, H. Li, M. M. Nowaczyk, N. Plumeré, M. Rögner and W. Schuhmann, J. Phys. Chem. B, 2015, 119, 13726–13731 CrossRef CAS PubMed.
- M. Miyachi, K. Okuzono, D. Nishiori, Y. Yamanoi, T. Tomo, M. Iwai, S. I. Allakhverdiev and H. Nishihara, Chem. Lett., 2017, 46, 1479–1481 CrossRef CAS.
- L. M. Utschig, N. M. Dimitrijevic, O. G. Poluektov, S. D. Chemerisov, K. L. Mulfort and D. M. Tiede, J. Phys. Chem. Lett., 2011, 2, 236–241 CrossRef CAS.
- S. C. Silver, J. Niklas, P. Du, O. G. Poluektov, D. M. Tiede and L. M. Utschig, J. Am. Chem. Soc., 2013, 135, 13246–13249 CrossRef CAS PubMed.
- T. Noji, H. Suzuki, T. Gotoh, M. Iwai, M. Ikeuchi, T. Tomo and T. Noguchi, J. Phys. Chem. Lett., 2011, 2, 2448–2452 CrossRef CAS.
- K. P. Sokol, W. E. Robinson, J. Warnan, N. Kornienko, M. M. Nowaczyk, A. Ruff, J. Z. Zhang and E. Reisner, Nat. Energy, 2018, 57, 10595–10599 Search PubMed.
- F. Zhao, S. Hardt, V. Hartmann, H. Zhang, M. M. Nowaczyk, M. Rögner, N. Plumeré, W. Schuhmann and F. Conzuelo, Nat. Commun., 2018, 9, 1973 CrossRef PubMed.
- S. H. Lee, D. S. Choi, S. K. Kuk and C. B. Park, Angew. Chem., Int. Ed., 2018, 57, 7958–7985 CrossRef CAS PubMed.
- Y. Li, J. Fei, G. Li, H. Xie, Y. Yang, J. J. Li, Y. Xu, B. Sun, J. Xia, X. Fu and J. J. Li, ACS Nano, 2018, 12, 1455–1461 CrossRef CAS PubMed.
- M. Wang, J. Chen, T. Lian and W. Zhan, Langmuir, 2016, 32, 7326–7338 CrossRef CAS PubMed.
- S. H. Lee, J. H. Kim and C. B. Park, Chem.–Eur. J., 2013, 19, 4392–4406 CrossRef CAS PubMed.
- B. C. M. Martindale, G. A. M. Hutton, C. A. Caputo and E. Reisner, J. Am. Chem. Soc., 2015, 137, 6018–6025 CrossRef CAS PubMed.
- Y. Wang and A. Hu, J. Mater. Chem. C, 2014, 2, 6921–6939 RSC.
- E. T. Hwang, K. Sheikh, K. L. Orchard, D. Hojo, V. Radu, C.-Y. Lee, E. Ainsworth, C. Lockwood, M. A. Gross, T. Adschiri, E. Reisner, J. N. Butt and L. J. C. Jeuken, Adv. Funct. Mater., 2015, 25, 2308–2315 CrossRef CAS PubMed.
- G. A. M. Hutton, B. C. M. Martindale and E. Reisner, Chem. Soc. Rev., 2017, 46, 6111–6123 RSC.
- B. C. M. Martindale, G. A. M. Hutton, C. A. Caputo, S. Prantl, R. Godin, J. R. Durrant and E. Reisner, Angew. Chem., Int. Ed., 2017, 56, 6459–6463 CrossRef CAS PubMed.
- M. J. Edwards, G. F. White, C. W. Lockwood, M. C. Lawes, A. Martel, G. Harris, D. J. Scott, D. J. Richardson, J. N. Butt and T. A. Clarke, J. Biol. Chem., 2018, 293, 8103–8112 CrossRef CAS PubMed.
- M. Breuer, K. M. Rosso, J. Blumberger and J. N. Butt, J. R. Soc., Interface, 2015, 12, 20141117 CrossRef PubMed.
- J. K. Fredrickson, M. F. Romine, A. S. Beliaev, J. M. Auchtung, M. E. Driscoll, T. S. Gardner, K. H. Nealson, A. L. Osterman, G. Pinchuk, J. L. Reed, D. a Rodionov, J. L. M. Rodrigues, D. a Saffarini, M. H. Serres, A. M. Spormann, I. B. Zhulin and J. M. Tiedje, Nat. Rev. Microbiol., 2008, 6, 592–603 CrossRef CAS PubMed.
- G. F. White, Z. Shi, L. Shi, Z. Wang, A. C. Dohnalkova, M. J. Marshall, J. K. Fredrickson, J. M. Zachara, J. N. Butt, D. J. Richardson and T. A. Clarke, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 6346–6351 CrossRef CAS PubMed.
- C.-Y. Lee, B. Reuillard, K. Sokol, T. Laftsoglou, C. Lockwood, S. Rowe, E. T. Hwang, J.-C. Fontecilla-Camps, L. J. C. Jeuken, J. Butt and E. Reisner, Chem. Commun., 2016, 52, 7390–7393 RSC.
- B. Reuillard, K. H. Ly, P. Hildebrandt, L. J. C. Jeuken, J. N. Butt and E. Reisner, J. Am. Chem. Soc., 2017, 139, 3324–3327 CrossRef CAS PubMed.
- E. V. Ainsworth, C. W. J. Lockwood, G. F. White, E. T. Hwang, T. Sakai, M. A. Gross, D. J. Richardson, T. A. Clarke, L. J. C. Jeuken, E. Reisner and J. N. Butt, ChemBioChem, 2016, 17, 2324–2333 CrossRef CAS PubMed.
- F. Zhang, A. Yediler, X. Liang and A. Kettrup, J. Environ. Sci. Health, Part A: Toxic/Hazard. Subst. Environ. Eng., 2002, 37, 707–713 CrossRef.
- A. B. dos Santos, F. J. Cervantes and J. B. van Lier, Bioresour. Technol., 2007, 98, 2369–2385 CrossRef CAS PubMed.
- M. C. Costa, F. S. B. Mota, A. B. Dos Santos, G. L. F. Mendonça and R. F. do Nascimento, Quim. Nova, 2012, 35, 482–486 CrossRef CAS.
- S. A. Trammell, J. A. Moss, J. C. Yang, B. M. Nakhle, C. A. Slate, F. Odobel, M. Sykora, B. W. Erickson and T. J. Meyer, Inorg. Chem., 1999, 38, 3665–3669 CrossRef CAS PubMed.
- C. X. Guo, D. Zhao, Q. Zhao, P. Wang and X. Lu, Chem. Commun., 2014, 50, 7318 RSC.
- G. R. Heath, M. Li, I. L. Polignano, J. L. Richens, G. Catucci, P. O’Shea, S. J. Sadeghi, G. Gilardi, J. N. Butt and L. J. C. Jeuken, Biomacromolecules, 2016, 17, 324–335 CrossRef CAS PubMed.
- P. E. Thomas, D. Ryan and W. Levin, Anal. Biochem., 1976, 75, 168–176 CrossRef CAS PubMed.
- S. G. Mayhew, Eur. J. Biochem., 1978, 85, 535–547 CrossRef CAS PubMed.
- R. S. Hartshorne, C. L. Reardon, D. Ross, J. Nuester, T. a Clarke, A. J. Gates, P. C. Mills, J. K. Fredrickson, J. M. Zachara, L. Shi, A. S. Beliaev, M. J. Marshall, M. Tien, S. Brantley, J. N. Butt and D. J. Richardson, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 22169–22174 CrossRef CAS PubMed.
- C. Guarantini, A. G. Fogg and M. V. B. Zanoni, in Proceedings of the Symposium on Chemical and Biological Sensors and Analytical Electrochemical Methods, Pennington: Electrochemical Society Inc., 1997, vol. 97, ch. 19, pp. 467–476 Search PubMed.
- J. Willkomm, K. L. Orchard, A. Reynal, E. Pastor, J. R. Durrant and E. Reisner, Chem. Soc. Rev., 2016, 45, 9–23 RSC.
- J. N. Weinstein, R. Blumenthal and R. D. Klausner, in Methods Enzymology, Academic Press, 1986, vol. 128, pp. 657–668 Search PubMed.
- Y. Zhou, P. Y. Liyanage, D. L. Geleroff, Z. Peng, K. J. Mintz, S. D. Hettiarachchi, R. R. Pandey, C. C. Chusuei, P. L. Blackwelder and R. M. Leblanc, ChemPhysChem, 2018, 19, 2589–2597 CrossRef CAS PubMed.
- J. B. Essner, J. A. Kist, L. Polo-Parada and G. A. Baker, Chem. Mater., 2018, 30, 1878–1887 CrossRef CAS.
- E. Reisner, J. C. Fontecilla-Camps and F. a Armstrong, Chem. Commun., 2009, 550–552 RSC.
- E. Reisner, D. J. Powell, C. Cavazza, J. C. Fontecilla-Camps and F. A. Armstrong, J. Am. Chem. Soc., 2009, 131, 18457–18466 CrossRef CAS PubMed.
- Y. Kang, X. Li, Y. Tu, Q. Wang and H. Ågren, J. Phys. Chem. C, 2010, 114, 14496–14502 CrossRef CAS.
- Y. S. Nam, T. Shin, H. Park, A. P. Magyar, K. Choi, G. Fantner, K. A. Nelson and A. M. Belcher, J. Am. Chem. Soc., 2010, 132, 1462–1463 CrossRef CAS PubMed.
- Y. S. Nam, A. P. Magyar, D. Lee, J. Kim, D. S. Yun, H. Park, T. S. Pollom, D. A. Weitz and A. M. Belcher, Nat. Nanotechnol., 2010, 5, 340–344 CrossRef CAS PubMed.
- K. Liu, R. Xing, Y. Li, Q. Zou, H. Möhwald and X. Yan, Angew. Chem., Int. Ed., 2016, 55, 12503–12507 CrossRef CAS PubMed.
- K. Liu, M. Abass, Q. Zou and X. Yan, Green Energy Environ., 2017, 2, 58–63 CrossRef.
- J. H. Kim, M. Lee, J. S. Lee and C. B. Park, Angew. Chem., Int. Ed., 2012, 51, 517–520 CrossRef CAS PubMed.
- X. Ji, Y. Kang, Z. Su, P. Wang, G. Ma and S. Zhang, ACS Sustainable Chem. Eng., 2018, 6, 3060–3069 CrossRef CAS.
- W. Kim, B. A. McClure, E. Edri and H. Frei, Chem. Soc. Rev., 2016, 45, 3221–3243 RSC.
- S. Schmidt, K. Castiglione and R. Kourist, Chem.–Eur. J., 2018, 24, 1755–1768 CrossRef CAS PubMed.
- L. Klermund and K. Castiglione, Bioprocess Biosyst. Eng., 2018, 41, 1233–1246 CrossRef CAS PubMed.
- S. Khan, M. Li, S. P. Muench, L. J. C. Jeuken and P. A. Beales, Chem. Commun., 2016, 52, 11020–11023 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8fd00163d |
| This journal is © The Royal Society of Chemistry 2019 |