
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNovel latonduine derived proligands and their copper(II) complexes show cytotoxicity in the nanomolar range in human colon adenocarcinoma cells and in vitro cancer selectivity†
Felix
Bacher
*a,
Christopher
Wittmann
a,
Márta
Nové
b,
Gabriella
Spengler
 b,
Małgorzata A.
Marć
c,
Eva A.
Enyedy
c,
Denisa
Darvasiová
d,
Peter
Rapta
d,
Thomas
Reiner
efg and
Vladimir B.
Arion
*a
b,
Małgorzata A.
Marć
c,
Eva A.
Enyedy
c,
Denisa
Darvasiová
d,
Peter
Rapta
d,
Thomas
Reiner
efg and
Vladimir B.
Arion
*a
aInstitute of Inorganic Chemistry of the University of Vienna, Währinger Strasse 42, A-1090 Vienna, Austria. E-mail: felix.bacher@univie.ac.at; vladimir.arion@univie.ac.at
bDepartment of Medical Microbiology and Immunobiology, University of Szeged, Dóm tér 10, H-6720 Szeged, Hungary
cDepartment of Inorganic and Analytical Chemistry, Interdisciplinary Excellence Centre, University of Szeged, Dóm tér 7, H-6720 Szeged, Hungary
dInstitute of Physical Chemistry and Chemical Physics, Slovak University of Technology in Bratislava, Radlinského 9, 81237 Bratislava, Slovak Republic
eDepartment of Radiology, Weill Cornell Medical College, New York City, NY 10065, USA
fDepartment of Radiology, Memorial Sloan Kettering Cancer Center, New York City, NY 10065, USA
gChemical Biology Program, Memorial Sloan Kettering Cancer Center, New York City, NY 10065, USA
First published on 10th May 2019
Abstract
Four Schiff bases derived from 7-hydrazin-yl-5,8-dihydroindolo[2,3-d][2]benzazepin-(6H)-one and its bromo-substituted analogue (HL1–HL4) and four copper(II) complexes 1–4 have been synthesised and fully characterised by standard spectroscopic methods (1H and 13C NMR, UV-vis), ESI mass spectrometry, single crystal X-ray diffraction and spectroelectrochemistry. In addition, two previously reported complexes with paullone ligands 5 and 6 were prepared and studied for comparison reasons. The CuII ion in 1–4 is five-coordinate and adopts a square-pyramidal or slightly distorted square-pyramidal coordination geometry. The ligands HL1–4 act as tridentate, the other two coordination places are occupied by two chlorido co-ligands. The organic ligands in 2 and 3 are bound tighter to copper(II) when compared to related paullone ligands in 5 and 6. The new compounds show very strong cytotoxic activity against human colon adenocarcinoma doxorubicin-sensitive Colo 205 and multidrug resistant Colo 320 cancer cell lines with IC50 values in the low micromolar to nanomolar concentration range.
Introduction
Indolobenzazepines are a class of compounds with a broad spectrum of biological activities, amongst them analgesic, antidepressant, antimalarial, antidiabetic, anticancer and antiparasitic.1–7 The search for potent kinase inhibitors led to indolo[3,2-d]benzazepines, known as paullones.8 Cyclin dependent kinases (cdks) are attractive targets for anticancer drugs, since they control the cell cycle.9 Research on structure–activity relationships (SARs) showed that electron-withdrawing groups in position 9 of the paullone scaffold (Chart 1, left) enhance the cdk inhibition, while the lactam moiety is essential for the anticancer activity. This finding resulted in the discovery of the lead compounds kenpaullone and alsterpaullone (Chart 1).10 Despite their potency, paullones possess some adverse properties. In particular, the low aqueous solubility and bioavailability hampered their clinical use. Later it was shown that the bioavailability can be improved by the attachment of suitable metal binding sites onto the paullone backbone and the consequent metal complex formation with gallium(III), ruthenium(II), osmium(II) and copper(II) ions. This led to highly cytotoxic compounds with an enhanced aqueous solubility, with copper(II) complexes being the most active ones.11–16 However, cdks seem not to be the main target for these metal complexes.17 Alternative mechanisms of action have been suggested, amongst them DNA intercalation.14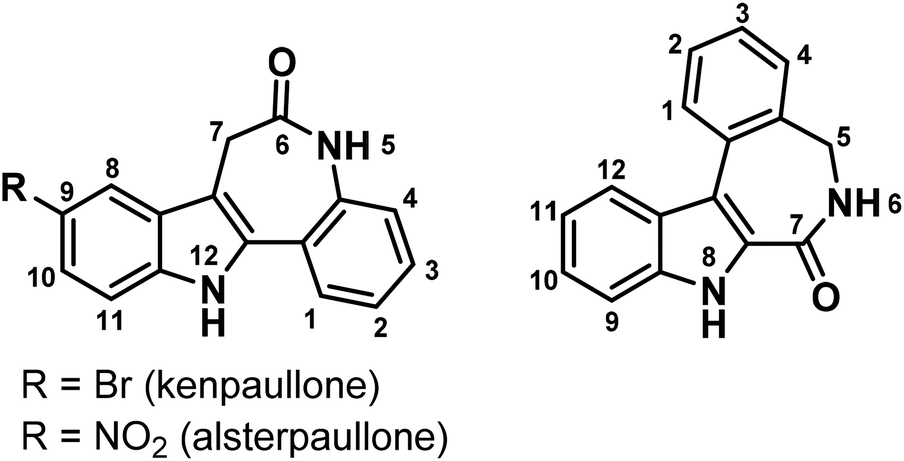 | ||
| Chart 1 The indolo[3,2-d]benzazepine (paullone) (left), vs. the indolo[2,3-d]benzazepine backbone (right) with numbering schemes. | ||
In our present work, we focused our attention on the isomeric indolo[2,3-d]benzazepine scaffold (Chart 1, right). It differs from the paullone core by the position of the lactam group in the azepine ring and features a flipped indole moiety with respect to the benzazepinone half of the molecule. These new compounds contain a typical latonduine structural motif. Latonduine-modified molecules usually exhibit prominent anticancer activity in vitro, along with the ability to inhibit tubulin polymerisation.18–21 Like paullones, these compounds do not possess any metal binding site. Therefore, it was of interest to us to chemically modify them and obtain proligands with potentially tridentate κN, κN′, κN′′ metal binding site, in particular, for copper(II). Copper is an essential trace metal and therefore considered to be safer than other metals used in chemotherapy, e.g. platinum.22,23 In addition, it is well-established that copper(II) is able to strongly enhance the cytotoxicity of biologically active ligands.24
Herein we report on the synthesis and characterisation of four novel latonduine core containing proligands and their copper(II) complexes. In order to obtain structure–activity relationships we modified the indolobenzazepine backbone by inserting a bromine substituent at position 11, the equivalent to position 9 in the paullone scaffold (compare kenpaullone). In addition, an aldehyde and a ketone containing a functional group in a position suitable for chelate formation were used for Schiff base condensation reactions, namely 2-formyl- and 2-acetylpyridine (Chart 2).
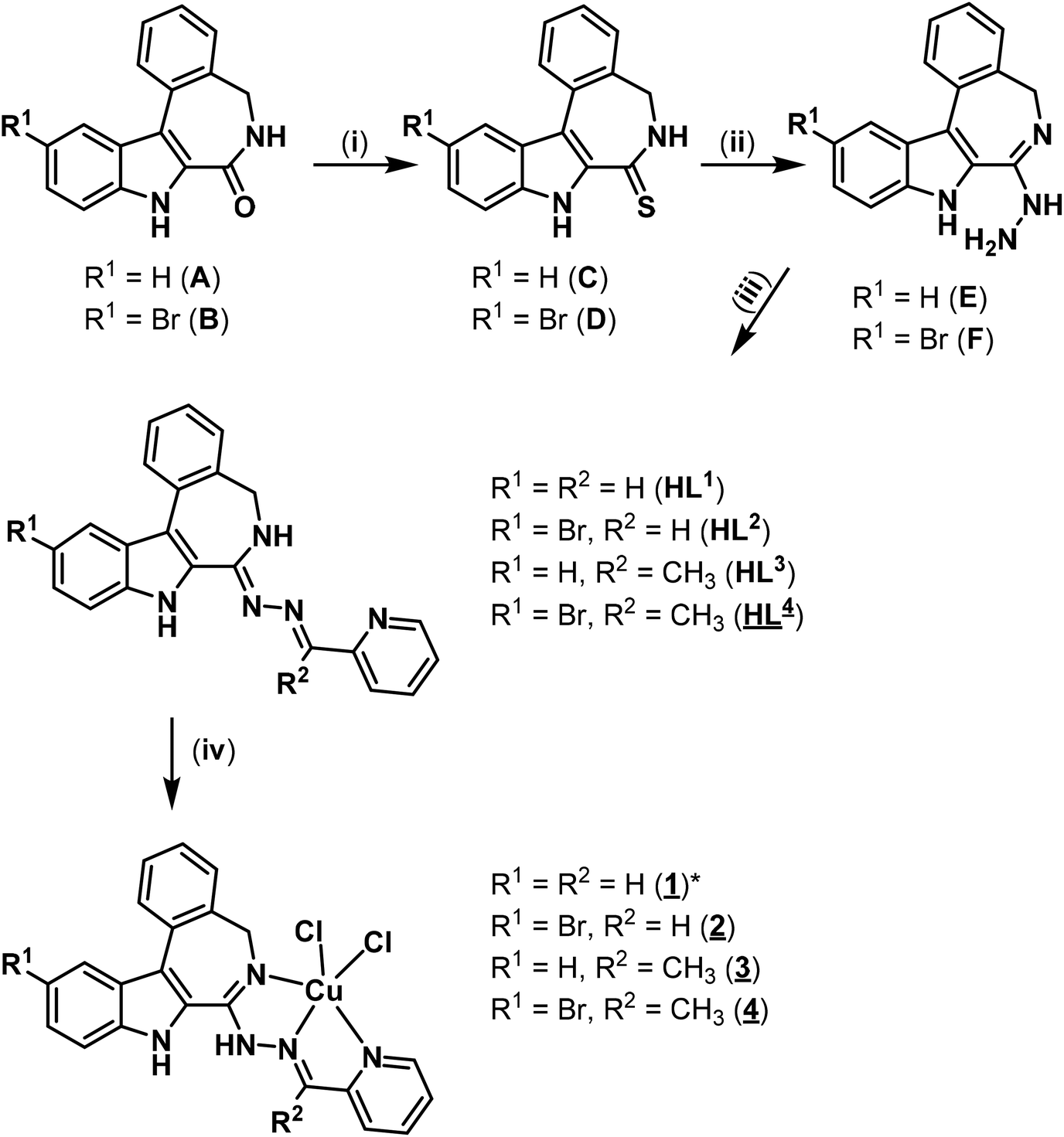 | ||
| Chart 2 New proligands and copper(II) complexes synthesised in this work.a Underlined numbers indicate compounds studied by X-ray diffraction. *1 crystallised from DMF as the trimer [Cu3Cl4(HL1)3]Cl2 (1trim). aReagents and conditions: (i) C: phosphorus pentasulfide/aluminium oxide, acetonitrile, 85 °C, overnight; D: phosphorus pentasulfide/aluminium oxide, tetrahydrofuran, 75 °C, overnight; (ii) hydrazine monohydrate, reflux, overnight; (iii) HL1, HL2: 2-formypyridine, ethanol, 85 °C, overnight; HL3, HL4: 2-acetylpyridine, ethanol, 85 °C, overnight; (iv) CuCl2·2H2O, isopropanol, reflux, 15 min. | ||
The new compounds have been characterised by 1H and 13C NMR spectroscopy (HL1–HL4), single crystal X-ray diffraction (HL4, 1–4) and ESI mass spectrometry; their purity was validated by elemental analysis. Solution equilibrium properties of HL3 and its copper(II) complex 3 were characterised in a DMSO–water solvent mixture by UV-vis titrations. The cytotoxicity of the proligands and the corresponding copper(II) complexes was tested in Colo 205 (chemosensitive) and Colo 320/MDR-LRP (multidrug resistant) human colon adenocarcinoma cell lines and one non-cancerous human embryonal lung fibroblast MRC-5 cell line and compared to those of the two previously reported paullone-derived copper(II) complexes 5 and 6 (Chart 3).15,25 The new compounds showed very high activity with IC50 values from the low micromolar to the nanomolar concentration range and were superior to 5 and 6 in cancer cell lines, while less cytotoxic in non-cancerous MRC-5 cells. Furthermore some selectivity for cancer cells over normal cells was observed in most cases, making this compound class pertinent for further development as anticancer drugs.
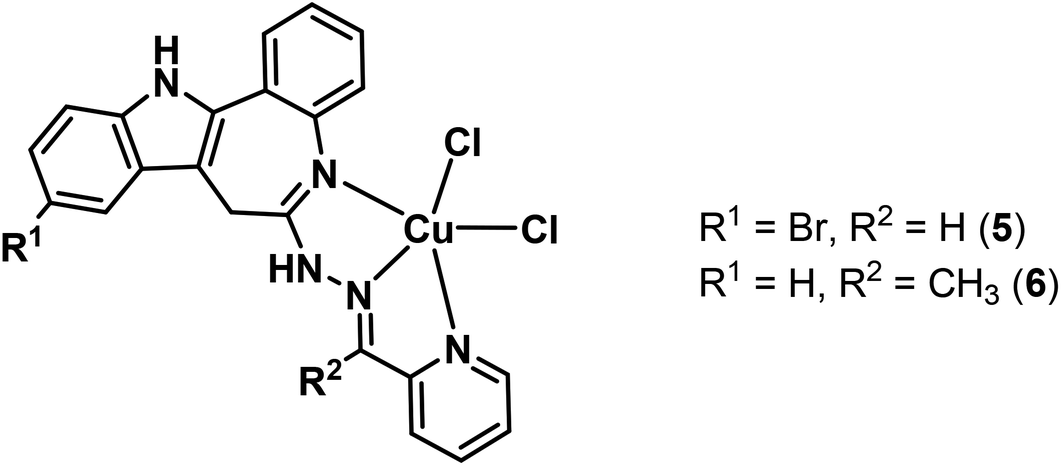 | ||
| Chart 3 Paullone-derived copper(II) complexes used in this study.15,25 | ||
Experimental
2-Iodobenzonitrile, ethyl-1H-indole-carboxylate and 5-bromo-ethyl-1H-indole-carboxylate were purchased from ABCR. Borane solution (1 M in THF), absolute DMF, dimethylaminopyridine, di-tert-butyl-dicarbonate, absolute acetonitrile, palladium(II) acetate, sodium bicarbonate, basic aluminium oxide, 2-acetylpyridine and 2-formylpyridine were bought from Fisher/Acros Organics. Ethoxy-methylchloride was obtained form TCI. Sodium hydride, phosphorus(V) sulfide, celite, hydrazine monohydrate and methyl iodide were purchased from Sigma Aldrich, while lithium hydroxide monohydrate and triphenylphosphine were from Alfa Aesar. 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide-hydrochloride was purchased from IRIS biotech, while silver(I) carbonate from Merck. 2-Iodobenzylamine was prepared by a known method.26 The unsubstituted indolo[2,3-d]benzazepine (A) was prepared by following published protocols.18–20 The 11-bromo-substituted species B was prepared using reported procedures,18–20 with some modifications. A detailed description of the synthesis of B is given in the ESI.†Synthesis of proligands
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 w/w)27 (1.60 g) was added and the reaction mixture was stirred at 85 °C overnight. The next day, the mixture was cooled to room temperature and filtered. The filtrate was concentrated in vacuo and taken up in water (50 mL). The pH was adjusted to 8 using saturated potassium carbonate solution. The solution was extracted with dichloromethane (DCM) (3 × 100 mL). The organic phases were combined and dried over magnesium sulfate. The dried organic phase was concentrated and the raw product was purified on silica using a mixture of DCM:methanol 99:1 as eluent. Yield: 841 mg, 82%. 1H NMR (500 MHz, DMSO-d6) δ 11.78 (s, 1H, NH), 10.57 (t, J = 5.5 Hz, 1H, NH), 7.98 (dd, J = 14.4, 7.9 Hz, 2H, H(Ar)), 7.64 (d, J = 8.3 Hz, 1H, H(Ar)), 7.53 (t, J = 7.5 Hz, 1H, H(Ar)), 7.48 (d, J = 6.9 Hz, 1H, H(Ar)), 7.41 (t, J = 7.3 Hz, 1H, H(Ar)), 7.36 (t, J = 7.4 Hz, 1H, H(Ar)), 7.20 (t, J = 7.5 Hz, 1H, H(Ar)), 4.41–4.28 (m, 1H, CH2), 4.04 (d, J = 12.2 Hz, 1H, CH2). ESI-MS (acetonitrile/methanol + 1% water), positive: m/z 265.08 [M + H]+.
:1 w/w)27 (2.20 g) were suspended in dry THF (50 mL) in an 100 mL Schlenk tube, under argon atmosphere. The reaction mixture was stirred overnight at 75 °C. The next day it was cooled to room temperature, the yellow precipitate was filtered off and washed with THF. The filtrate was evaporated and the residue was purified on silica by using DCM/methanol 99:1 as eluent. The product was obtained as yellow-orange powder after removal of the solvent. Yield: 635 mg, 60%. 1H NMR (500 MHz, DMSO-d6) δ 12.00 (s, 1H, NH), 10.67 (t, J = 5.6 Hz, 1H, NH), 8.10 (d, J = 1.7 Hz, 1H, H(Ar)), 7.93 (d, J = 7.2 Hz, 1H, H(Ar)), 7.60 (d, J = 8.7 Hz, 1H, H(Ar)), 7.55 (td, J = 7.5, 1.4 Hz, 1H, H(Ar)), 7.51–7.47 (m, 2H, H(Ar)), 7.43 (td, J = 7.4, 1.1 Hz, 1H, H(Ar)), 4.33 (s, 1H, CH2), 4.06 (d, J = 12.5 Hz, 1H, CH2). ESI-MS (acetonitrile/methanol + 1% water), positive, m/z 344.99 [M + H]+.
:1 and dried in vacuo. Yield: 266 mg, 99%. Anal. Calcd for C22H17N5·0.4C2H5OH (M 372.13 g mol−1): C, 73.71; H, 5.07, N, 19.09. Found: C, 73.91; H, 5.33; N, 18.82. 1H NMR (600 MHz, DMSO-d6) δ 11.93 (s, 1H, H12), 8.59 (d, J = 4.1 Hz, 1H, H18), 8.37 (d, J = 8.0 Hz, 2H, H15, H21), 8.31 (t, J = 5.4 Hz, 1H, H6), 7.98 (dd, J = 15.4, 7.6 Hz, 2H, H12, H3), 7.87 (td, J = 7.5, 1.2 Hz, 1H, H20), 7.60 (d, J = 8.2 Hz, 1H, H9), 7.53–7.49 (m, 1H, H2), 7.47 (d, J = 6.6 Hz, 1H, H4), 7.40–7.31 (m, 3H, H10, H1, H19), 7.20 (dd, J = 11.1, 4.0 Hz, 1H, H11), 4.36–4.02 (m, 2H, H5). 13C NMR (151 MHz, DMSO) δ 155.62 (Cq, C7), 154.52 (Cq, C16), 152.14 (CH, C15), 149.28 (CH, C18), 137.78 (Cq, C4a), 136.76 (Cq, C8a), 136.30 (CH, C20), 133.161 (Cq, C12c), 129.01(Cq, C7a), 128.09 (CH, C4), 128.06 (CH, C2), 127.41 (CH, C3), 126.33 (CH, C1), 124.94 (Cq, C12a), 124.10(CH, C10), 123.87 (CH, C19), 120.77 (CH, C21), 120.46 (CH, C11), 120.22(CH, C12), 117.12(Cq, C12b), 112.69(CH, C9), 46.00 (CH2, C5). For atom numbering scheme see ESI, Scheme S1.† ESI-MS (acetonitrile/methanol + 1% water), positive: m/z 352.26 [M + H]+.
:1 w/w)27 (1.60 g) was added and the reaction mixture was stirred at 85 °C overnight. The next day, the mixture was cooled to room temperature and filtered. The filtrate was concentrated in vacuo and taken up in water (50 mL). The pH was adjusted to 8 using saturated potassium carbonate solution. The solution was extracted with dichloromethane (DCM) (3 × 100 mL). The organic phases were combined and dried over magnesium sulfate. The dried organic phase was concentrated and the raw product was purified on silica using a mixture of DCM:methanol 99:1 as eluent. Yield: 841 mg, 82%. 1H NMR (500 MHz, DMSO-d6) δ 11.78 (s, 1H, NH), 10.57 (t, J = 5.5 Hz, 1H, NH), 7.98 (dd, J = 14.4, 7.9 Hz, 2H, H(Ar)), 7.64 (d, J = 8.3 Hz, 1H, H(Ar)), 7.53 (t, J = 7.5 Hz, 1H, H(Ar)), 7.48 (d, J = 6.9 Hz, 1H, H(Ar)), 7.41 (t, J = 7.3 Hz, 1H, H(Ar)), 7.36 (t, J = 7.4 Hz, 1H, H(Ar)), 7.20 (t, J = 7.5 Hz, 1H, H(Ar)), 4.41–4.28 (m, 1H, CH2), 4.04 (d, J = 12.2 Hz, 1H, CH2). ESI-MS (acetonitrile/methanol + 1% water), positive: m/z 265.08 [M + H]+.
:1 w/w)27 (2.20 g) were suspended in dry THF (50 mL) in an 100 mL Schlenk tube, under argon atmosphere. The reaction mixture was stirred overnight at 75 °C. The next day it was cooled to room temperature, the yellow precipitate was filtered off and washed with THF. The filtrate was evaporated and the residue was purified on silica by using DCM/methanol 99:1 as eluent. The product was obtained as yellow-orange powder after removal of the solvent. Yield: 635 mg, 60%. 1H NMR (500 MHz, DMSO-d6) δ 12.00 (s, 1H, NH), 10.67 (t, J = 5.6 Hz, 1H, NH), 8.10 (d, J = 1.7 Hz, 1H, H(Ar)), 7.93 (d, J = 7.2 Hz, 1H, H(Ar)), 7.60 (d, J = 8.7 Hz, 1H, H(Ar)), 7.55 (td, J = 7.5, 1.4 Hz, 1H, H(Ar)), 7.51–7.47 (m, 2H, H(Ar)), 7.43 (td, J = 7.4, 1.1 Hz, 1H, H(Ar)), 4.33 (s, 1H, CH2), 4.06 (d, J = 12.5 Hz, 1H, CH2). ESI-MS (acetonitrile/methanol + 1% water), positive, m/z 344.99 [M + H]+.
:1 and dried in vacuo. Yield: 266 mg, 99%. Anal. Calcd for C22H17N5·0.4C2H5OH (M 372.13 g mol−1): C, 73.71; H, 5.07, N, 19.09. Found: C, 73.91; H, 5.33; N, 18.82. 1H NMR (600 MHz, DMSO-d6) δ 11.93 (s, 1H, H12), 8.59 (d, J = 4.1 Hz, 1H, H18), 8.37 (d, J = 8.0 Hz, 2H, H15, H21), 8.31 (t, J = 5.4 Hz, 1H, H6), 7.98 (dd, J = 15.4, 7.6 Hz, 2H, H12, H3), 7.87 (td, J = 7.5, 1.2 Hz, 1H, H20), 7.60 (d, J = 8.2 Hz, 1H, H9), 7.53–7.49 (m, 1H, H2), 7.47 (d, J = 6.6 Hz, 1H, H4), 7.40–7.31 (m, 3H, H10, H1, H19), 7.20 (dd, J = 11.1, 4.0 Hz, 1H, H11), 4.36–4.02 (m, 2H, H5). 13C NMR (151 MHz, DMSO) δ 155.62 (Cq, C7), 154.52 (Cq, C16), 152.14 (CH, C15), 149.28 (CH, C18), 137.78 (Cq, C4a), 136.76 (Cq, C8a), 136.30 (CH, C20), 133.161 (Cq, C12c), 129.01(Cq, C7a), 128.09 (CH, C4), 128.06 (CH, C2), 127.41 (CH, C3), 126.33 (CH, C1), 124.94 (Cq, C12a), 124.10(CH, C10), 123.87 (CH, C19), 120.77 (CH, C21), 120.46 (CH, C11), 120.22(CH, C12), 117.12(Cq, C12b), 112.69(CH, C9), 46.00 (CH2, C5). For atom numbering scheme see ESI, Scheme S1.† ESI-MS (acetonitrile/methanol + 1% water), positive: m/z 352.26 [M + H]+.
Synthesis of copper(II) complexes
| Compound | HL4 | 1trim·2DMF·1.25H2O | 2·0.55MeOH | 3·1.2DMF·0.25H2O |
|---|---|---|---|---|
| a R 1 = ∑||Fo| − |Fc||/∑|Fo|. b wR2 = {∑[w(Fo2 − Fc2)2]/∑[w(Fo2)2]}1/2. c GOF = {∑[w(Fo2 − Fc2)2]/(n − p)}1/2, where n is the number of reflections and p is the total number of parameters refined. | ||||
| Empirical formula | C23H18BrN5 | C72H67.5Cl6Cu3N17O3.25 | C22.55H18.2BrCl2CuN5O0.55 | C26.6H27.9Cl2CuN6.2O1.45 |
| fw | 444.33 | 1626.25 | 582.37 | 592.09 |
| Space group | P21/c |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
C2/c |
P |
| a [Å] | 18.405(4) | 13.0443(3) | 37.232(30) | 9.7602(13) |
| b [Å] | 8.2958(16) | 14.9957(3) | 11.3330(11) | 13.4027(15) |
| c [Å] | 13.230(3) | 20.2999(4) | 11.6674(10) | 20.685(2) |
| α [°] | 92.0747(8) | 98.235(4) | ||
| β [°] | 103.962(8) | 103.4914(8) | 107.494(3) | 92.013(5) |
| γ [°] | 112.4054(8) | 93.367(4) | ||
| V [Å3] | 1960.4(7) | 3535.02(13) | 4695.3(7) | 2670.7(5) |
| Z | 4 | 2 | 8 | 4 |
| λ [Å] | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| ρ calcd [g cm−3] | 1.506 | 1.528 | 1.648 | 1.473 |
| Crystal size [mm] | 0.10 × 0.08 × 0.01 | 0.14 × 0.08 × 0.06 | 0.13 × 0.10 × 0.02 | 0.80 × 0.08 × 0.08 |
| T [K] | 100(2) | 100(2) | 100(2) | 296(2) |
| μ [mm−1] | 2.117 | 1.183 | 2.883 | 1.053 |
| R 1 | 0.0527 | 0.0387 | 0.0599 | 0.0725 |
| wR2b | 0.1203 | 0.0984 | 0.1650 | 0.1719 |
| GOFc | 1.041 | 1.033 | 1.149 | 0.943 |
| Compound | 3′·DMF | 4·2DMF |
|---|---|---|
| a R 1 = ∑||Fo| − |Fc||/∑|Fo|. b wR2 = {∑[w(Fo2 − Fc2)2]/∑[w(Fo2)2]}1/2. c GOF = {∑[w(Fo2 − Fc2)2]/(n − p)}1/2, where n is the number of reflections and p is the total number of parameters refined. | ||
| Empirical formula | C26H25ClCuN6O | C29H32BrCl2CuN7O2 |
| fw | 536.51 | 724.97 |
| Space group | P21/c |
P |
| a [Å] | 10.3347(4) | 12.0530(9) |
| b [Å] | 23.4454(7) | 16.7234(14) |
| c [Å] | 11.0757(4) | 17.8152(15) |
| α [°] | 105.698(3) | |
| β [°] | 117.731(1) | 105.962(3) |
| γ [°] | 106.665(3) | |
| V [Å3] | 2375.41(15) | 3060.2(4) |
| Z | 4 | 4 |
| λ [Å] | 0.71073 | 0.71073 |
| ρ calcd [g cm−3] | 1.500 | 1.574 |
| Crystal size [mm] | 0.10 × 0.08 × 0.05 | 0.19 × 0.08 × 0.03 |
| T [K] | 100(2) | 100(2) |
| μ [mm−1] | 1.065 | 2.234 |
| R 1 | 0.0566 | 0.0498 |
| wR2b | 0.1597 | 0.1177 |
| GOFc | 1.031 | 1.024 |
:water 30:70 (w/w) mixture as solvent at 25.0 ± 0.1 °C in the pH range from 2 to 11. An Orion 710A pH-meter equipped with a Metrohm combined electrode (type 6.0234.100) and a Metrohm 665 Dosimat burette were used for the pH measurements and titrations. The electrode system was calibrated to the pH = −log[H+] scale in the DMSO–water solvent mixture by means of blank titrations (HCl vs. KOH) similarly to the method suggested by Irving et al. in pure aqueous solutions.31 The average water ionisation constant (pKw) was 14.52 ± 0.05, which corresponds well to the literature data.32 Argon was passed over the solutions during the titrations. Proton dissociation constants (pKa) of the ligand, overall stability constants (logβ) of the copper(II) complexes and the individual spectra of the various species present in solution were calculated by the computer program PSEQUAD.33β (MpLqHr) is defined for the general equilibrium pM + qL + rH ⇌ MpLqHr as β (MpLqHr) = [MpLqHr]/[M]p[L]q[H]r where M denotes the copper(II) ion and L the completely deprotonated ligand. The calculations were always performed from the experimental titration data measured in the absence of any precipitate in the solution.
Attempts to determine distribution coefficients (D7.4) of HL1, HL3 and complexes 1, 3 were undertaken by the traditional shake-flask method in n-octanol/buffered aqueous solution at pH 7.40 (20 mM phosphate buffer, 0.10 M KCl) as described previously.34
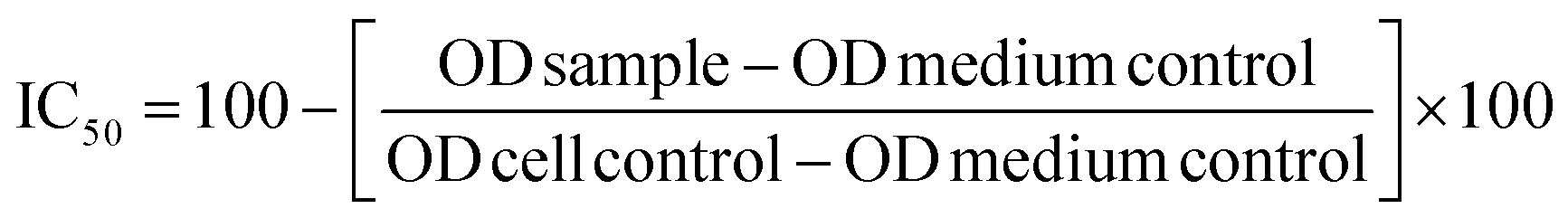
Results are expressed in terms of IC50, defined as the inhibitory dose that reduces the growth of the cells exposed to the tested compounds by 50%. The IC50 values were calculated using GraphPad Prism7 software.
Results and discussion
Synthesis
The synthesis of the main core structure A in Chart 2 was performed by following the published literature protocols. B was synthesised similarly.18–20 A detailed description for the synthesis of B is given in the ESI.† The lactam derivatives A and B were converted into thiolactams C and D by reaction with P2S5 on Al2O3 in dry boiling acetonitrile or tetrahydrofuran in 82 and 60% yield, respectively.27 Thionation with phosphorus pentasulfide bound to aluminium oxide offers the advantage that the reagent can be removed by filtration after the reaction. In addition, this method was chosen because the standard method for the thionation of paullones10 gave only small yields. Further reaction of C and D with hydrazine hydrate as reagent and solvent afforded hydrazines E and F in 86 and 92% yield, respectively. It should be noted that F exists in two tautomeric forms in DMSO solution, as evidenced by the appearance of two signal sets in the 1H NMR spectrum. The predominant species is the one shown in Chart 2 where N13 is protonated and the double bond is endocyclic. The NMR signal of N13H appears as a singlet at δ = 6.36 ppm. In the minor species, (ca. 15% in DMSO), N6 is protonated and the double bond is exocyclic. N6H appears as a triplet with the typical coupling constant of 5.3 Hz at δ = 8.49 ppm. NMR data is given only for the major species in the experimental part, because of the low signal intensity of the minor species and signal overlapping. For atom numbering scheme see Scheme S1, ESI.† Similar tautomerism has been observed in the case of paullones previously.14 The Schiff bases HL1–HL4 were prepared in 99, 31, 94 and 58% yield, respectively, from hydrazine species E and F and 2-formyl- and 2-acetylpyridine taken in 10% excess in boiling ethanol. 1H and 13C NMR spectra of HL1–HL4 were in agreement with the formulae proposed. Notably, HL4 showed a second set of NMR-signals with an intensity of about 10% of the main species in DMSO-d6. Tautomerism as in the case for F was excluded, because of the presence of the typical triplet signal for N6H in both species. It is likely that a mixture of E and Z isomers is present, similarly to previously reported paullone derived ruthenium(II) and osmium(II) complexes.17 However, low signal intensity and signal overlapping did not allow a more accurate analysis to be performed. ESI mass spectra measured in positive ion mode showed peaks with m/z 352.26, 432.06, 366.16 and 446.08 attributed to [M + H]+. The purity of HL1–HL4 (>95%) was confirmed by elemental analysis. The structure of HL4 was also established by single crystal X-ray diffraction (vide infra). Complexes 1–4 were prepared by reaction of HL1–HL4 in isopropanol by addition of a methanolic solution of CuCl2·2H2O in 1:1 mol ratio, respectively, in 77 to 99% yields. The positive ion ESI mass spectrum of 1 showed the presence of a peak with m/z 764.18 due to [CuII(L1)(HL1)]+. The mass spectra of 2–4 contain peaks at m/z 528.97 and 492.99, 463.04 and 427.07, 542.96 and 507.00 attributed to [CuIICl(HL2–4)]+ and [CuII(L2–4)]+, respectively. The elemental analysis was in agreement with the structures shown in Chart 2, providing the required purity of bulk samples for biological investigations. The complexes 1–4 as well as 3′ have been studied by X-ray diffraction.
X-ray diffraction
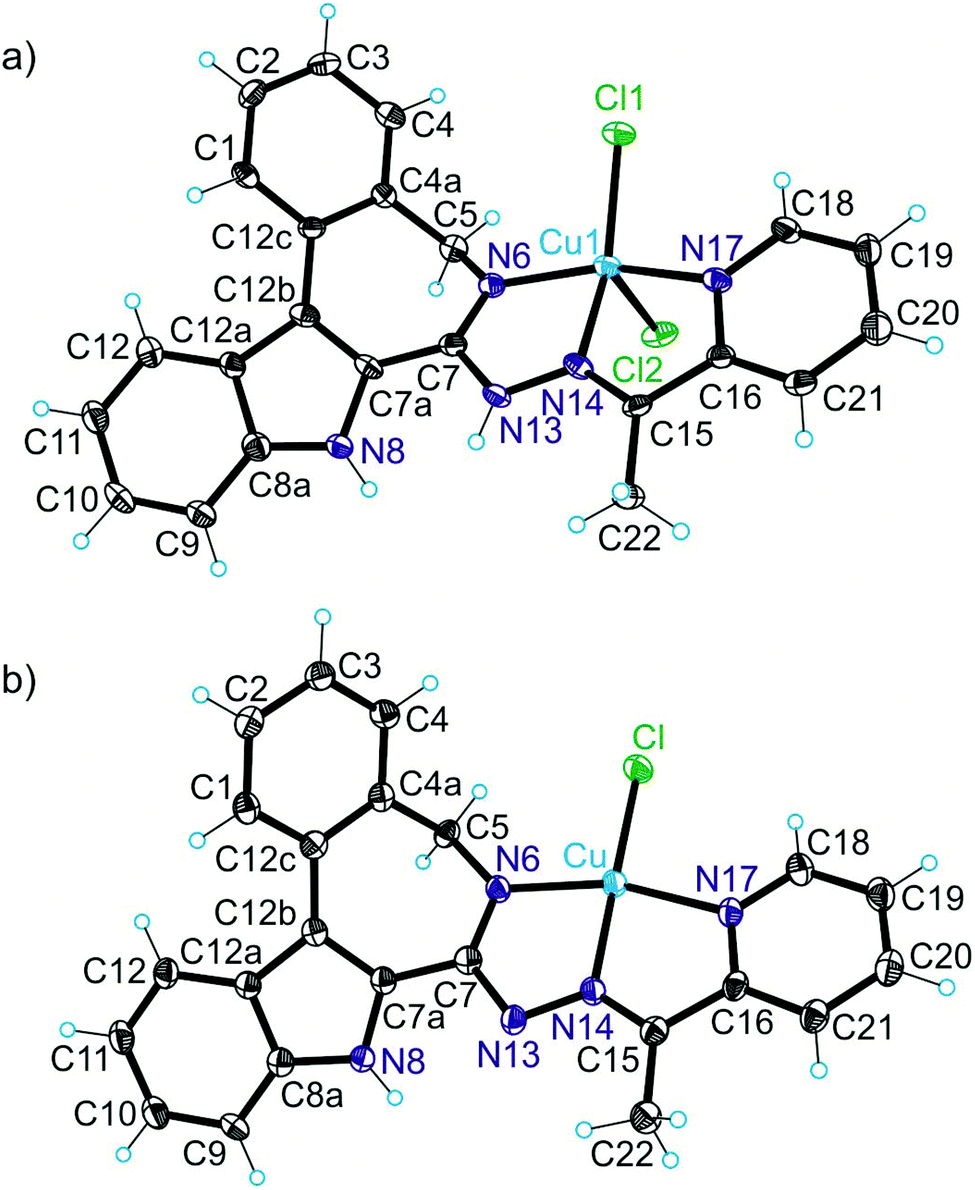
The results of X-ray diffraction studies for 1trim, 2, 3, 3′, 4 and HL4 are shown in Fig. 1–3, respectively, with selected bond lengths and bond angles quoted in the legends. Complex 1 crystallised as a trimer (1trim), while 2–4 and 3′ as monomeric entities. The corresponding ligand is protonated in 1trim, 2–4 and acts as a tridentate one being coordinated to copper(II) via azepine nitrogen, hydrazine nitrogen and pyridine nitrogen atoms. The ligands can be deprotonated in the presence of a base. In particular, deprotonation of HL3 in 3 in the presence of triethylamine was confirmed by X-ray diffraction study of 3′ (Fig. 2b). The coordination geometry of copper(II) ion in the trimer (1trim) and in mononuclear complexes 2–4 is square-pyramidal or distorted square-pyramidal (see legends to Fig. 1–3 quoting τ5-descriptor values35 for each copper(II) ion), while in 3′ it is slightly distorted square-planar. The coordination polyhedron in each case is completed by two or one chlorido co-ligands. Note that the same coordination geometry was previously established for complexes 5 and 6 with paullone derivatives (τ5 = 0.10 (5) and 0.05 (6)).15 The bond lengths Cu–N5 = 2.038(3) and 2.0220(15), Cu–N14 = 1.982(3) and 1.9787(15), Cu–N17 = 2.062(3) and 2.0466(15) in 5 and 6, respectively, are significantly longer than those in 2 and 3 (see legends to Fig. 1 and 2) by 0.01–0.05 and 0.005–0.04 Å, respectively, even though the metal binding moieties in the corresponding ligands are closely related. Overall a tighter binding of the ligand moiety in 2 and 3 is to be mentioned when compared to that in 5 and 6. The proligand HL4 and the ligand HL4 in 4 adopt different configurations and distinct proton tautomeric forms as shown in Fig. 3 and confirmed by the distribution of bond lengths (electron density) over the fragment C5–N6–C7–N13–N14–C15 quoted in the legend to Fig. 3.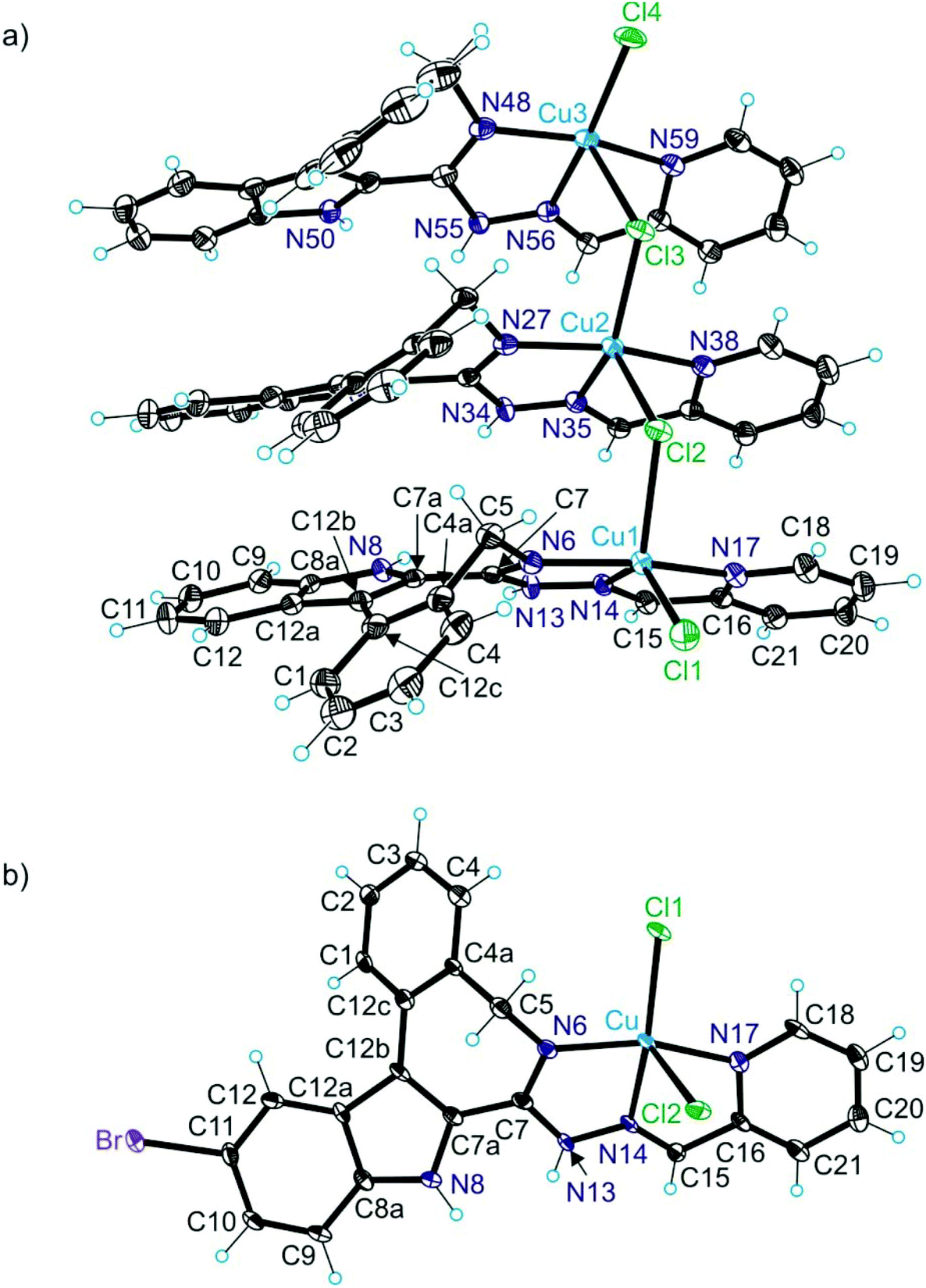 | ||
| Fig. 1 ORTEP views of (a) the complex cation [Cu3Cl4(HL1)3]2+ in 1trim and (b) of [CuCl2(HL2)] in 2. Selected bond distances (Å) and bond angles (deg) in 1trim: Cu1–N6 1.994(2), Cu1–N14 1.991(2), Cu1–N17 2.044(2), Cu1–Cl1 2.2685(8), Cu1–Cl2 2.4382(7), N6–Cu1–N14 78.75(10), N14–Cu1–N17 77.92(9), Cu2–N27 1.990(2), Cu2–N35 1.975(2), Cu2–N38 2.034(2), Cu2–Cl2 2.4492(8), Cu2–Cl3 2.2781(8), N27–Cu2–N35 79.39(10), N35–Cu2–N38 78.63(10), Cu3–N48 1.995(3), Cu3–N56 1.959(2), Cu3–N59 2.044(2), Cu3–Cl3 2.6404(8), Cu3–Cl4 2.2116(8), N48–Cu3–N56 79.51(10), N56–Cu3–N59 79.20(10). Details of coordination geometry in 1trim: τ5 (Cu1) = 0.35, τ5 (Cu2) = 0.25, τ5 (Cu3) = 0.17; in 2: Cu–N6 1.982(5), Cu–N14 1.971(5), Cu–N17 2.051(5), Cu–Cl1 2.2252(16), Cu–Cl2 2.5762(17), N6–Cu–N14 79.7(2), N14–Cu–N17 78.8(2). Details of coordination geometry in 2: τ5 (Cu) = 0.06. | ||
 | ||
| Fig. 2 ORTEP views of (a) one crystallographically independent complex [CuCl2(HL3)] in 3 (co-crystallised solvent is omitted) and (b) of [CuCl(L3)] in 3′. Selected bond distances (Å) and bond angles (deg) in 3: Cu1–N6 1.979(4), Cu1–N14 1.973(4), Cu1–N17 2.034(4), Cu1–Cl1 2.2460(12), Cu1–Cl2 2.4566(14), N6–Cu1–N14 78.72(17), N14–Cu1–N17 78.33(16), Cu2–N28 1.986(5), Cu2–N36 1.969(4), Cu2–N39 2.031(4), Cu2–Cl3 2.2309(14), Cu2–Cl4 2.5054(13), N28–Cu2–N36 79.09(17), N36–Cu2–N39 78.36(18). Details of coordination geometry in 2: τ5 (Cu1) = 0, τ5 (Cu2) = 0.06 (second crystallographically independent molecule); in 3′: Cu–N6 1.937(3), Cu–N14 1.953(3), Cu–N17 2.019(3), Cu–Cl 2.2058(9), N6–Cu–N14 79.52(12), N14–Cu–N17 80.27(12). | ||
 | ||
| Fig. 3 ORTEP views of (a) proligand HL4 and (b) of [CuCl2(HL4)] in 4 (co-crystallised solvent is omitted). Selected bond distances (Å) and bond angles (deg) in HL4: C5–N6 1.459(4), N6–C7 1.344(4), C7–N13 1.325(4), N13–N14 1.391(3), N14–C15 1.300(4); in 4: Cu1–N6 2.011(2), Cu1–N14 1.986(2), Cu1–N17 2.057(3), Cu1–Cl1 2.2612(8), Cu1–Cl2 2.4327(9), C5–N6 1.470(4), N6–C7 1.295(4), C7–N13 1.376(4), N13–N14 1.366(3), N14–C15 1.285(4), N6–Cu1–N14 78.83(10), N14–Cu1–N17 77.34(10), Cu2–N28 2.010(2), Cu2–N36 1.968(2), Cu2–N39 2.035(3), Cu2–Cl3 2.2204(8), Cu2–Cl4 2.5321(9), N28–Cu2–N36 79.40(10), N36–Cu2–N39 78.40(10). Details of coordination geometry in 4: τ5 (Cu1) = 0.28, τ5 (Cu2) = 0.28 (second crystallographically independent molecule). | ||
Of note is also that the two Cu–Cl bonds in 1–4 are markedly different. The bond length from copper(II) to basal chlorido co-ligand is by 0.17 to 0.43 Å shorter compared to that between copper(II) and the apical chlorido co-ligand. A similar situation is also typical for 5 and 6 reported previously.15,25 The corresponding bonds differ by 0.19 and 0.13 Å in 5 and 6, respectively.
Solution equilibrium studies
The proligand HL3 and complex 3 (Charts 1 and 2) were chosen for the detailed solution equilibrium studies. Two complexes with HL3 were obtained in the solid state, namely [CuCl2(HL3)] (3) and [CuCl(L3)] (3′) (Fig. 2), in which the ligand adopts different protonation states. We therefore aimed to study the transformation process of 3 into 3′ in solution. Both HL3 and complex 3 have fairly poor water solubility, thus UV-vis spectrophotometric titrations were performed in the presence of 30% (w/w) DMSO at low concentration (12.5 μM). The UV-vis spectra recorded for the proligand revealed small but characteristic changes upon increasing the pH (Fig. 4a) and two well separated proton dissociation processes could be observed. The first deprotonation process was accompanied by a blue shift (λmax: 351 nm → 340 nm) and an absorbance increase at ∼440 nm in the pH range between 2.5 and 3.25. The solution was yellow in this pH range. Upon increasing the pH the solution became colourless due to the decrease of absorbance in the visible wavelength range (415–490 nm) and the λmax is red shifted (340 nm → 360 nm). At pH >6.5 precipitation occurred in the solution most likely due to the formation of the neutral species, thus data collected below this pH were used for the calculations. The pKa values (Table 3) and the spectra of the individual ligand species (Fig. 5a) were calculated on the basis of deconvolution of recorded UV-vis spectra. The proligand HL3, which can be drawn in two tautomeric forms due to the rearrangement of the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–NH–N and NH–CN–N bonds, contains two additional protons in its fully protonated form (H2(HL3)2+, (Chart 4). The first deprotonation process (pKa1) can presumably be attributed to deprotonation of the pyridinium nitrogen, while the second step (pKa2) to the deprotonation of the benzazepinium nitrogen. The pKa values were predicted by using the software MarvinSketch as well36 (Table 3), showing the same order of the stepwise deprotonation processes as we suggest herein. However, these are somewhat different compared to the values obtained experimentally in the DMSO–water solvent mixture.
C–NH–N and NH–CN–N bonds, contains two additional protons in its fully protonated form (H2(HL3)2+, (Chart 4). The first deprotonation process (pKa1) can presumably be attributed to deprotonation of the pyridinium nitrogen, while the second step (pKa2) to the deprotonation of the benzazepinium nitrogen. The pKa values were predicted by using the software MarvinSketch as well36 (Table 3), showing the same order of the stepwise deprotonation processes as we suggest herein. However, these are somewhat different compared to the values obtained experimentally in the DMSO–water solvent mixture.
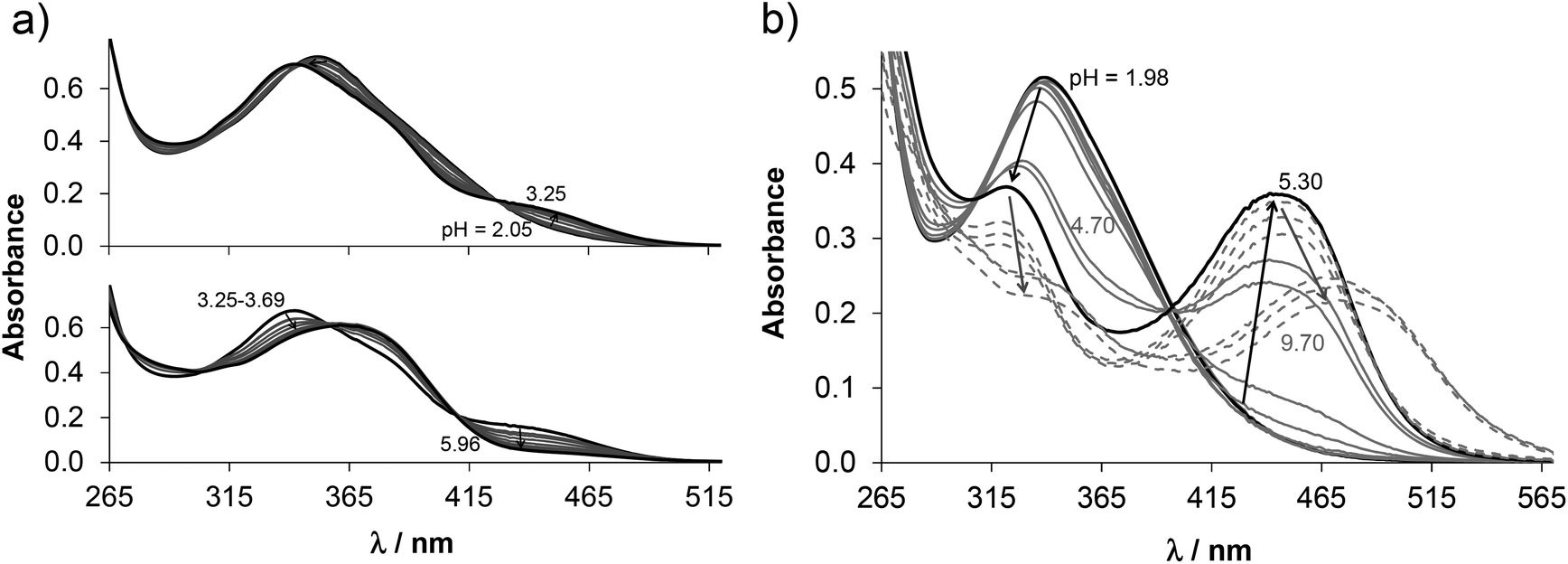 | ||
| Fig. 4 UV-vis spectra recorded for (a) HL3 and (b) complex 3 at various pH values in 30–70% (w/w) DMSO–water solvent mixture. {cligand = ccomplex = 12.5 μM; T = 298 K; I = 0.10 M (KCl); l = 2 cm}. | ||
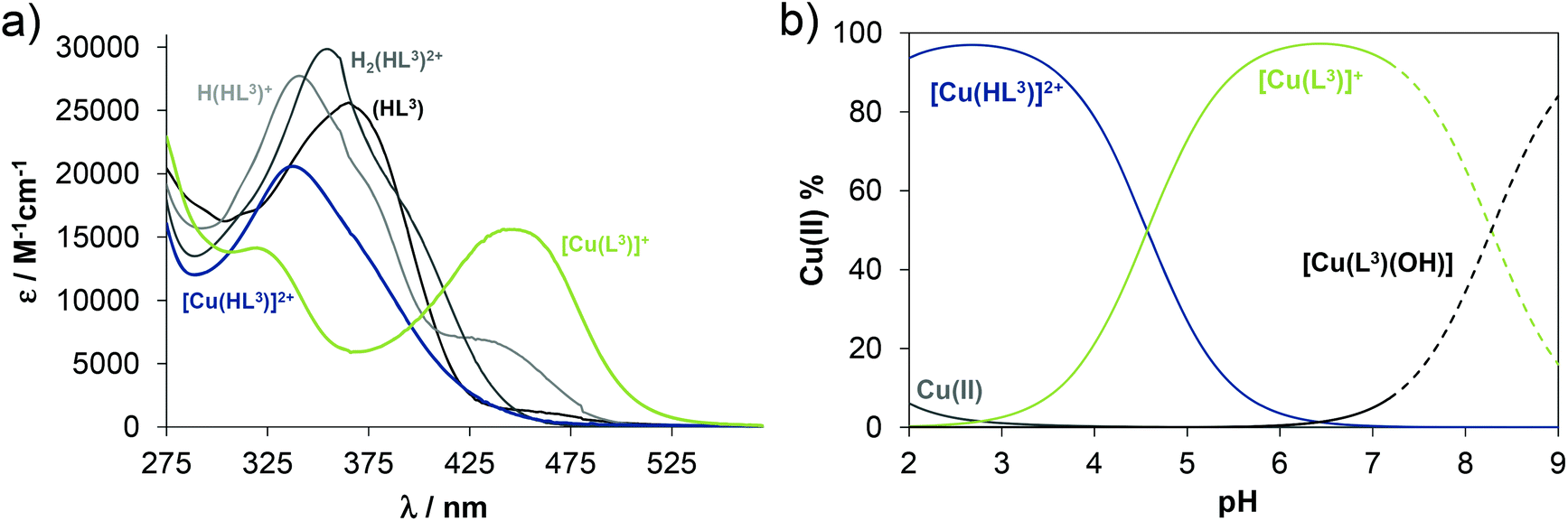 | ||
| Fig. 5 (a) Molar absorbance spectra computed for proligand HL3 and complex 3 in the various protonation states, and concentration distribution curves for 3 plotted against the pH. (Dashed lines show the region where precipitate appears.) {ccomplex = 12.5 μM; T = 298 K; I = 0.10 M (KCl); 30% (w/w) DMSO-H2O}. | ||
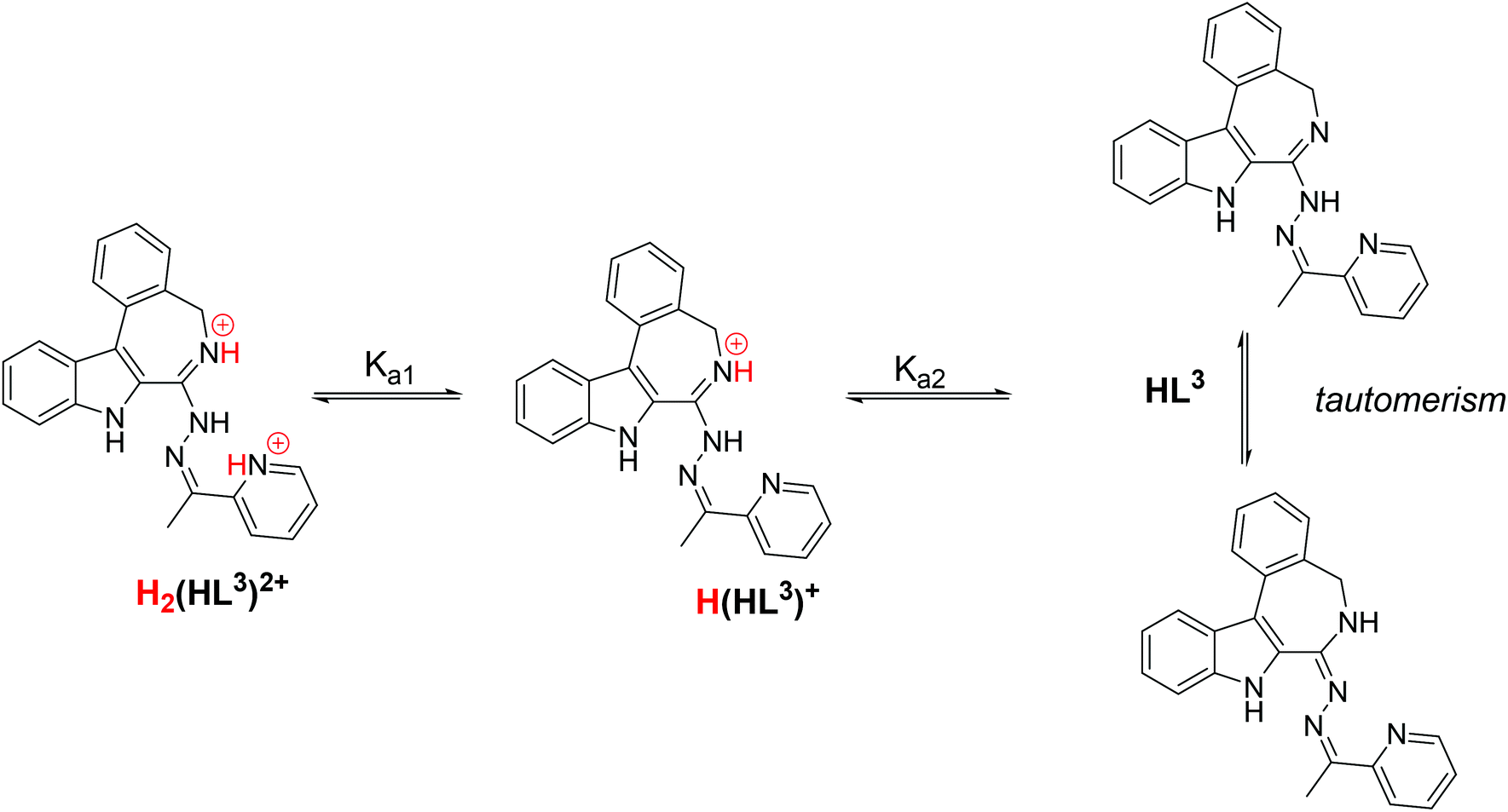 | ||
| Chart 4 Stepwise deprotonation processes of the fully protonated form of HL3 and its tautomeric forms. | ||
D7.4 values for HL3 and HL1 by the software MarvinSketch.36
| Constant | Predicted constant | ||
|---|---|---|---|
| pKa1 H2(HL3)2+ | 2.51 ± 0.01 | pKa1 H2(HL3)2+ | 1.82 |
| pKa2 H(HL3)+ | 5.03 ± 0.01 | pKa2 H(HL3)+ | 5.37 |
| logβ [Cu(HL3)]2+ |
10.96 ± 0.02 | logD7.4HL3 |
+4.03 |
| logβ [Cu(L3)]+ |
6.39 ± 0.02 | pKa1 H2(HL1)2+ | 2.00 |
| logβ [Cu(L3)(OH)] |
∼−1.9 | pKa2 H(HL1)+ | 5.39 |
| pKa [Cu(HL3)]2+ | 4.57 | logD7.4HL1 |
+4.75 |
| pKa [Cu(L3)]+ | ∼8.3 |
The UV-vis spectrum recorded for 3 at pH ∼2 (Fig. 4b) showed significant complex formation comparing to that of the free proligand. Thus the complex dissociates only partly under these conditions. Therefore, the tridentate coordination via the (N, N, N) donor set is assumed in [Cu(HL3)]2+ at such low pH. Upon increasing the pH the λmax is shifted to the lower wavelength (338 nm → 323 nm) concomitant with the development of a new band with λmax 444 nm implying a rearrangement in the copper(II) chromophore. Most probably the non-coordinating hydrazinic nitrogen is deprotonated, as is the case with 3′ (Fig. 2b). In addition a novel process starts at pH >7 as the λmax shows a further bathochromic shift (λmax ∼483 nm). Although it is accompanied by remarkable absorbance decrease in the whole wavelength range monitored due to the formation of precipitate of a neutral mixed hydroxido species [Cu(L3)(OH)]. On the basis of the recorded UV-vis spectra overall stability constants for the complexes [Cu(HL3)]2+ and [Cu(L3)]+ were computed (Table 3) in addition to their individual spectra (Fig. 5a), while logβ for [Cu(L3)(OH)] could be only estimated. Concentration distribution curves were computed using the determined stability constants (Fig. 5b) revealing the predominant formation of [Cu(L3)]+ at neutral pH.
The lipo-hydrophilic character is an important property of drugs as it strongly affects the passage via biological membranes. Therefore we attempted to determine the logD7.4 values for proligands HL1, HL3 and complexes 1, 3 using the traditional shake-flask method in n-octanol/buffered aqueous solution at pH 7.40. All these compounds were found to be so lipophilic that data could not be obtained experimentally since they remained in the octanol phase and only a minor fraction was found in the aqueous phase. Thus logD7.4 values were estimated for proligands HL1, HL3 by using the software MarvinSketch as well36 (Table 3). The predicted values indicate the strong lipophilic character of the compounds. The bromo substituents in HL2, HL4 are suggested to increase the lipophilicity even more.
Electrochemistry and spectroelectrochemistry
Cyclic voltammograms of 1–4 in DMSO/nBu4NPF6 at glassy carbon (GC) working electrode show two irreversible reduction peaks with cathodic peak potentials at E1pc = −0.75 V and E2pc = −1.13 V for 1, E1pc = −0.68 V and E2pc = −1.05 V for 2, E1pc = −0.74 V and E2pc = −1.12 V for 3 and E1pc = −0.76 V and E2pc = −1.11 V for 4vs. Fc+/Fc (see Fig. 6 and Fig. S1, ESI†). The first irreversible reduction step can be attributed to the Cu(II) → Cu(I) process. We suppose that unstable [Cu(I)L] complex decomposes with partial or full release of the proligand. The follow-up product is then irreversibly reduced in the next reduction step.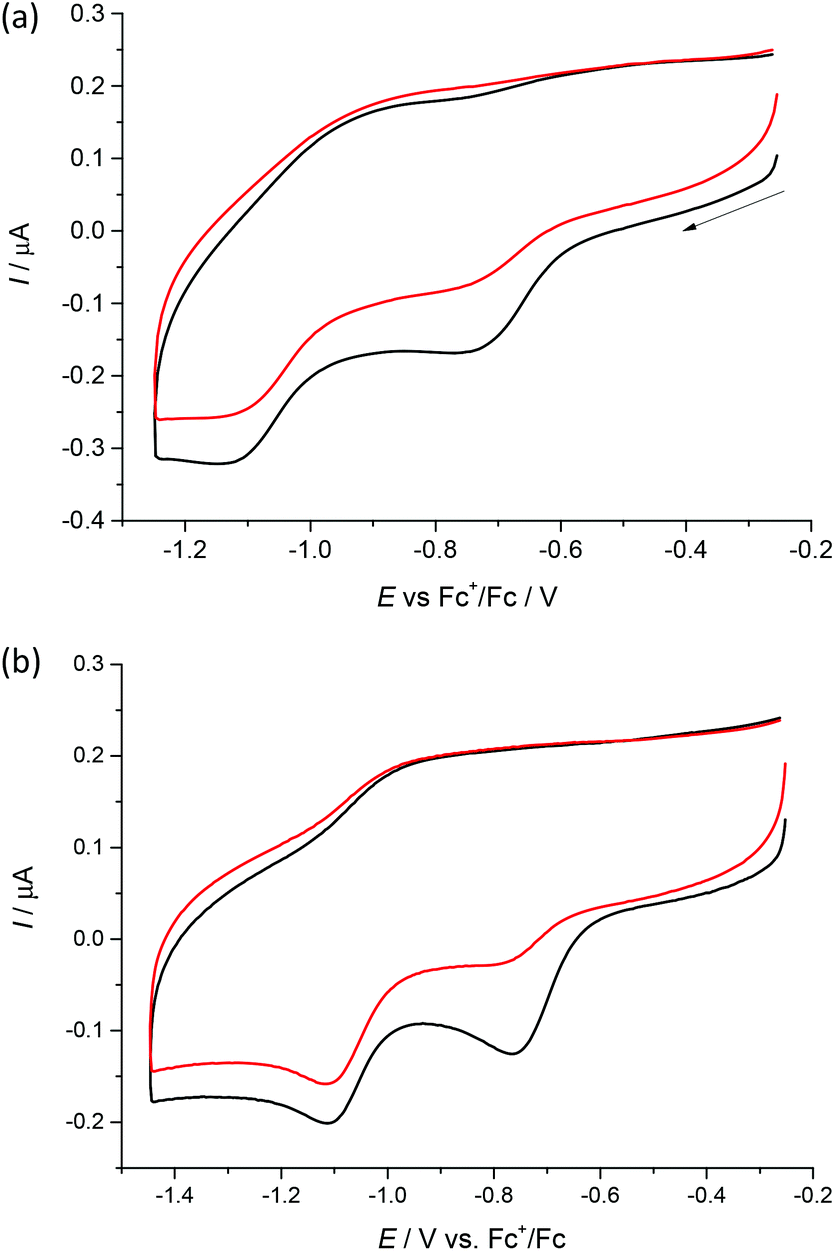 | ||
| Fig. 6 Cyclic voltammograms of (a) 1 and (b) 4 in DMSO/nBu4NPF6 at scan rate of 100 mV s−1 at GC working electrode (black traces represent the first scan, while red traces the second scan). | ||
To check the chemical reversibility of the first reduction step for 1–4, the in situ spectroelectrochemical UV-vis-NIR experiments were carried out under an argon atmosphere in a special thin layer spectroelectrochemical cell with a microstructured honeycomb working electrode. The UV-vis spectra measured upon cathodic reduction of 1 revealed in the region of the first reduction peak a new emerging optical band at 385 nm. In addition, a decrease of the intensity of the bands at 275 and 485 nm is observed (Fig. 7a). According to the findings of the UV-vis titrations (Fig. 5a), these spectral changes suggest the dissociation of the Cu(II) complex and the liberation of the proligand.
 | ||
| Fig. 7 In situ UV-vis-NIR spectroelectrochemistry for 1 in DMSO/nBu4NPF6 (scan rate of 10 mV s−1, Pt-microstructured honeycomb working electrode): (a) evolution of UV-vis spectra in 2D projection in forward scan in the region of the first reduction peak; (b) UV-vis spectra detected simultaneously upon the cyclic voltammetric scan in 3D projection (Inset: The corresponding cyclic voltammogram). | ||
Very similar spectroelectrochemical response was observed also for 2–4 in the region of the first reduction peak as illustrated for 3 in Fig. S2, ESI.† However, upon scan reversal the product that is formed upon reduction (Cu(II) to Cu(I)) is not reoxidised back to the initial state (Fig. 7b). This fact provides strong evidence that the Cu(I) oxidation state is unstable, resulting in the partial or complete decomposition of the complex. The presence of the Cu(II) ion in the cells may lead to the production of ROS in vivo because of Cu(II)/Cu(I) redox cycling due to Fenton like reactions.37,38
Cytotoxic activity assays
The latonduine derivatives HL1–HL4, their copper(II) complexes 1–4, and the paullone derived copper(II) complexes 5, 6 exhibited strong cytotoxic activity in cancer cells, exceeding tremendously that of cisplatin. The latonduine derivatives HL1–HL4 revealed strong cytotoxic activity in cancer cells, while they were less toxic in non-cancerous MRC-5 fibroblasts showing selectivity towards cancer cells (Table 4).| IC50 (μM), 72 h | SF(205)* | SF(320)* | |||
|---|---|---|---|---|---|
| Colo 205 | Colo 320 | MRC-5 | |||
| HL1 | 0.266 ± 0.012 | 0.294 ± 0.005 | 1.522 ± 0.547 | 6 | 5 |
| HL2 | 0.189 ± 0.040 | 0.051 ± 0.012 | 2.312 ± 0.032 | 12 | 45 |
| HL3 | 0.037 ± 0.004 | 0.042 ± 0.001 | 0.127 ± 0.004 | 3 | 3 |
| HL4 | 0.017 ± 0.002 | 0.018 ± 0.005 | 1.257 ± 0.463 | 74 | 70 |
| 1 | 0.346 ± 0.008 | 0.315 ± 0.023 | 0.461 ± 0.069 | — | — |
| 2 | 0.556 ± 0.028 | 0.104 ± 0.002 | 0.378 ± 0.004 | — | 4 |
| 3 | 0.017 ± 0.001 | 0.020 ± 0.001 | 0.162 ± 0.009 | 10 | 8 |
| 4 | 0.007 ± 0.001 | 0.015 ± 0.002 | 0.201 ± 0.001 | 29 | 13 |
| 5 | 1.704 ± 0.218 | 0.413 ± 0.044 | 0.152 ± 0.001 | — | — |
| 6 | 0.380 ± 0.032 | 0.120 ± 0.007 | 0.061 ± 0.003 | — | — |
| Cisplatin | 31.94 ± 2.17 | 4.81 ± 0.68 | 12.41 ± 0.367 | — | 3 |
Interesting structure-activity and selectivity relationships deserve to be mentioned. Acetylpyridine derived proligands HL3 and HL4 were by one order of magnitude more cytotoxic than formylpyridine derived HL1 and HL2. Bromo substituted HL2 and HL4 were about twice as cytotoxic compared to their unsubstituted counterparts HL1 and HL3. The bromo substituted proligands HL2 and HL4 were not only more active, but they also showed a more pronounced selectivity for cancer cells over normal cells, by a factor of 2 to 25 compared to unsubstituted analogues HL1 and HL3 (see Table 4). Interestingly HL2 is more active in the multidrug resistant Colo 320 cell line than in the chemosensitive Colo 205 cell line with selectivity factors of 45 vs. 12, respectively. The most active proligand, HL4, shows IC50 values in the nanomolar range and is around 70 times more toxic in the cancer cell lines (Colo 205, Colo 320) than in the normal fibroblast MRC-5 cell line.
Formylpyridine derived copper(II) complexes 1 and 2 showed activity comparable to the respective proligands HL1 and HL2, however, their selectivity for cancer cells over non-cancerous cell line is diminished. Copper(II) complex 2 was more active in the chemoresistant Colo 320 cancer cell line than in the chemosensitive Colo 205 cell line, similar to HL2, leading to the assumption that proligand HL2 and copper(II) complex 2 may interfere with the ABCB1 transporter (P-glycoprotein). This is the first characterised ATP-binding cassette (ABC) transporter, which is the most studied member of this protein superfamily. ABC transporters bind and hydrolyse ATP to provide energy needed to transport/extrude the substrates/drugs via the cell membranes. It has been shown that the overexpression of this protein is often associated with the development of multidrug resistant phenotype in cancer and disadvantageous clinical prognosis.39 An emerging research approach is the use of efflux pump inhibitors as adjuvant compounds (so-called “chemosensitisers”) to improve the efficacy of antitumour therapy, by co-administering them with chemotherapeutic agents. Computer-aided drug design techniques are used for the rapid assessment of chemical libraries in order to guide and speed up the early-stage development of new active compounds. In order to determine the ability of the compounds investigated in the present study to interact with P-glycoprotein molecular docking calculations based on crystallographic data reported by Ferreira et al.40 will be performed and the results will be reported in due course.
Acetylpyridine derived complexes 3 and 4 are by one to two orders of magnitude more active than their formylpyridine derived analogues 1 and 2 and about twice as cytotoxic as the corresponding proligands HL3 and HL4. The strong enhancement of cytotoxicity, when a methyl group is present at the imine carbon of the ligand, has been observed also in the case of paullone derived copper(II) complexes previously.15,25 Compound 4 is the most active drug in the whole series, with an astonishingly low IC50 value of 7 nM in the Colo 205 cancer cell line, being superior to the corresponding proligand HL4 with IC50 value of 17 nM in the same cell line. The presence of bromo-substituent in position 9 of the paullone41 or similar position in latonduine derivative and ketimine group instead of aldimine one increases the cytotoxic activity of both proligands and metal complexes, but we do not have any explanation of this observation at the molecular level now. Even though 4 is more cytotoxic to MRC-5 cells than HL4, it still shows marked selectivity for cancer cells over normal ones, with selectivity factors of 29 and 13 for the Colo 205 and Colo 320 cancer cell lines, respectively. Paullone derived complexes 5 and 6 are isomers of latonduine derived complexes 2 and 3. Intriguingly, 5 and 6 are less cytotoxic in the cancer cell lines (Colo 205, Colo 320) and more cytotoxic in MRC-5 cells, when compared to their counterparts 2 and 3. This means that the new latonduine derived copper(II) complexes presented in this work offer both enhanced activity and selectivity in comparison with previously reported paullone derivatives.
Conclusions
Four new proligands, which can be regarded as biologically active latonduine modified or isomeric to paullone derivatives and containing a potentially tridentate metal binding site, HL1–HL4 were prepared via a multistep synthesis. In addition, four copper(II) complexes 1–4 were synthesised by direct complex formation reactions of proligands with CuCl2·2H2O. For comparison two copper(II) complexes with paullone modified ligands reported previously, namely 5 and 6, were also used in this study. These two complexes form isomeric pairs with 2 and 3, respectively. The following features can be outlined from the performed investigations of the listed compounds. X-ray diffraction studies revealed, that like 5 and 6, complexes 1–4 are five-coordinate and adopt a square-pyramidal or slightly distorted square-pyramidal coordination geometry. Deprotonation of the ligand HL3 in 3 resulted in formation of 3′, in which a square-planar coordination geometry is adopted by copper(II) as confirmed by single crystal X-ray crystallography. The decrease of coordination number of copper(II) from five in 3 to four in 3′ led to shortening of Cu–N and Cu–Cl bond lengths due to less repulsion between the ligand/donor atoms (see legend to Fig. 2 for comparison of bond lengths in the two complexes). It should be also noted that the ligands HL2 and HL3 are bound stronger in terms of bond lengths to copper(II) when compared to those in isomeric paulone-based complexes 5 and 6, respectively. The Cu–N bond lengths were found to be by 0.01–0.05 and 0.005–0.04 Å shorter in 2 and 3 than in 5 and 6, respectively. All the studied compounds are significantly lipophilic, and the proligands are in their neutral forms (HL) in solution at neutral pH (in 30% DMSO/H2O), while complexes are found in their [Cu(L)]+ forms under these conditions.Cell tests showed that the new proligands and copper(II) complexes are highly active against Colo 205 and Colo 320 cancer lines and show selectivity for cancer cell lines over MRC-5, a non-cancerous fibroblast cell line. Structure-activity relationships were established, revealing that a methyl substituent at the imine carbon leads to a distinct enhancement of the cytotoxicity of the new proligands and copper(II) complexes and that a bromo substituent at position 11 of the indolo[2,3-d]benzazepine backbone enhances the activity as well as the selectivity. Intriguingly, complexes 2 and 3 were by factors 3 and 22 and by factors 4 and 6 more cytotoxic against Colo 205 and Colo 320 cells in terms of IC50 values than paullone-based complexes 5 and 6. In contrast, in non-cancerous cells 5 and 6 were more cytotoxic by factors 2.5 and 2.6 than 2 and 3, respectively. So, the new complexes 2 and 3 appear to be superior to isomeric copper(II) complexes with paullone-modified ligands (5 and 6) in terms of suitability for further preclinical development as anticancer drugs.
Conflicts of interest
The authors declare no competing interest.Acknowledgements
This work was supported by the Austrian Science Fund (FWF) via the Grant no. P31293-N37. EAE acknowledges the support by National Research, Development and Innovation Office-NKFIA through project FK 124240 and Ministry of Human Capacities, Hungary grant 20391-3/2018/FEKUSTRAT. The study was also supported by the project GINOP-2.3.2-15-2016-00038 of the University of Szeged. We thank Alexander Roller for the X-ray data collection. This work was also supported by the Science and Technology Assistance Agency under the contract no. APVV-15-0053 and SK-AT-2017-0017, and by the Slovak Grant Agency VEGA under contract no. 1/0416/17 and 1/0466/18. TR acknowledges the support of this work by National Institutes of Healthvia the grant P30 CA00874.References
- H. Blumberg, U.S. Pat., US3790675A, 1974 Search PubMed.
- L. Toscano, G. Grisanti, G. Fioriello, E. Seghetti, A. Bianchetti, G. Bossoni and M. Riva, J. Med. Chem., 1976, 19, 208–213 CrossRef CAS PubMed.
- H. L. Koh, M. L. Go, T. I. Ngiam and J. W. Mak, Eur. J. Med. Chem., 1994, 29, 107–113 CrossRef CAS.
- X. Xie, T. Lemcke, R. Gussio, D. W. Zaharevitz, M. Leost, L. Meijer and C. Kunick, Eur. J. Med. Chem., 2005, 40, 655–661 CrossRef CAS PubMed.
- D. H.-C. Chou, N. E. Bodycombe, H. A. Carrinski, T. A. Lewis, P. A. Clemons, S. L. Schreiber and B. K. Wagner, ACS Chem. Biol., 2010, 5, 729–734 CrossRef CAS PubMed.
- J. Ryczak, M. Papini, A. Lader, A. Nasereddin, D. Kopelyanskiy, L. Preu, C. L. Jaffe and C. Kunick, Eur. J. Med. Chem., 2013, 64, 396–400 CrossRef CAS PubMed.
- C. Reichwald, O. Shimony, U. Dunkel, N. Sacerdoti-Sierra, C. L. Jaffe and C. Kunick, J. Med. Chem., 2008, 51, 659–665 CrossRef CAS PubMed.
- E. A. Sausville, D. Zaharevitz, R. Gussio, L. Meijer, M. Louarn-Leost, C. Kunick, R. Schultz, T. Lahusen, D. Headlee and S. Stinson, Pharmacol. Ther., 1999, 82, 285–292 CrossRef CAS PubMed.
- A. Huwe, R. Mazitschek and A. Giannis, Angew. Chem., Int. Ed., 2003, 42, 2122–2138 CrossRef CAS PubMed.
- C. Schultz, A. Link, M. Leost, D. W. Zaharevitz, R. Gussio, E. A. Sausville, L. Meijer and C. Kunick, J. Med. Chem., 1999, 42, 2909–2919 CrossRef CAS PubMed.
- A. Dobrov, V. B. Arion, N. Kandler, W. Ginzinger, M. A. Jakupec, A. Rufińska, N. Graf von Keyserlingk, M. Galanski, C. Kowol and B. K. Keppler, Inorg. Chem., 2006, 45, 1945–1950 CrossRef CAS PubMed.
- G. Mühlgassner, C. Bartel, W. F. Schmid, M. A. Jakupec, V. B. Arion and B. K. Keppler, J. Inorg. Biochem., 2012, 116, 180–187 CrossRef PubMed.
- V. B. Arion, A. Dobrov, S. Göschl, M. A. Jakupec, B. K. Keppler and P. Rapta, Chem. Commun., 2012, 48, 8559–8561 RSC.
- W. F. Schmid, R. O. John, V. B. Arion, M. A. Jakupec and B. K. Keppler, Organometallics, 2007, 26, 6643–6652 CrossRef CAS.
- M. F. Primik, G. Mühlgassner, M. A. Jakupec, O. Zava, P. J. Dyson, V. B. Arion and B. K. Keppler, Inorg. Chem., 2010, 49, 302–311 CrossRef CAS PubMed.
- A. Dobrov, S. Göschl, M. A. Jakupec, A. Popović-Bijelić, A. Gräslund, P. Rapta and V. B. Arion, Chem. Commun., 2013, 49, 10007–10009 RSC.
- W. F. Schmid, R. O. John, G. Muehlgassner, P. Heffeter, M. A. Jakupec, M. Galanski, W. Berger, V. B. Arion and B. K. Keppler, J. Med. Chem., 2007, 50, 6343–6355 CrossRef CAS PubMed.
- L. Keller, S. Beaumont, J.-M. Liu, S. Thoret, J. S. Bignon, J. Wdzieczak-Bakala, P. Dauban and R. H. Dodd, J. Med. Chem., 2008, 51, 3414–3421 CrossRef CAS PubMed.
- A. Putey, F. Popowycz, Q.-T. Do, P. Bernard, S. K. Talapatra, F. Kozielski, C. M. Galmarini and B. Joseph, J. Med. Chem., 2009, 52, 5916–5925 CrossRef CAS PubMed.
- A. Putey, L. Joucla, L. Picot, T. Besson and B. Joseph, Tetrahedron, 2007, 63, 867–879 CrossRef CAS.
- V. Pons, S. Beaumont, M. E. Tran Huu Dau, B. I. Iorga and R. H. Dodd, ACS Med. Chem. Lett., 2011, 2, 565–570 CrossRef CAS PubMed.
- C. Marzano, M. Pellei, F. Tisato and C. Santini, Anticancer Agents Med. Chem., 2009, 9, 185–211 CrossRef CAS PubMed.
- F. Tisato, C. Marzano, M. Porchia, M. Pellei and C. Santini, Med. Res. Rev., 2010, 30, 708–749 CAS.
- F. Bacher, O. Dömötör, A. Chugunova, N. V. Nagy, L. Filipović, S. Radulović, É. A. Enyedy and V. B. Arion, Dalton Trans., 2015, 44, 9071–9090 RSC.
- M. F. Primik, S. Göschl, M. A. Jakupec, A. Roller, B. K. Keppler and V. B. Arion, Inorg. Chem., 2010, 49, 11084–11095 CrossRef CAS PubMed.
- L. wyffels, G. G. Muccioli, S. De Bruyne, L. Moerman, J. Sambre, D. M. Lambert and F. De Vos, J. Med. Chem., 2009, 52, 4613–4622 CrossRef CAS PubMed.
- V. Polshettiwar and M. P. Kaushik, Tetrahedron Lett., 2004, 45, 6255–6257 CrossRef CAS.
- SAINT-Plus, version 8.32B and APEX2, Bruker-Nonius AXS Inc., Madison, WI, 2016 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- M. N. Burnett and G. K. Johnson, ORTEPIII. Report ORNL-6895, OAK Ridge National Laboratory, Tennessee, 1996.
- H. M. N. H. Irving, M. G. Miles and L. D. Pettit, Anal. Chim. Acta, 1967, 38, 475–488 CrossRef CAS.
- É. A. Enyedy, N. V. Nagy, É. Zsigó, C. R. Kowol, V. B. Arion, B. K. Keppler and T. Kiss, Eur. J. Inorg. Chem., 2010, 1717–1728 CrossRef.
- L. Zékány and I. Nagypál, Computational Methods for the Determination of Stability Constants, Plenum Press, New York, 1985 Search PubMed.
- É. A. Enyedy, D. Hollender and T. Kiss, J. Pharm. Biomed. Anal., 2011, 54, 1073–1081 CrossRef PubMed.
- A. W. Addison, T. N. Rao, J. Reedijk, J. Van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- L. ChemAxon, Instant J Chem/MarvinSketch, ChemAxon Ltd., Budapest, Hungary, 2012 Search PubMed.
- M. Valko, C. J. Rhodes, J. Moncol, M. Izakovic and M. Mazur, Chem.-Biol. Interact., 2006, 160, 1–40 CrossRef CAS PubMed.
- M. Valko, H. Morris and M. T. D. Cronin, Curr. Med. Chem., 2005, 12, 1161–1208 CrossRef CAS PubMed.
- R. Evens, N. Beenaerts, T. Neyens, N. Witters, K. Smeets and T. Artois, Sci. Rep., 2018, 8, 3008 CrossRef PubMed.
- R. J. Ferreira, M.-J. U. Ferreira and D. J. V. A. dos Santos, J. Chem. Inf. Model., 2013, 53, 1747–1760 CrossRef CAS PubMed.
- M. F. Primik, L. F. Filak and V. B. Arion, in Organometallic Chemistry and Catalysis: The Silver/Gold Jubilee International Conference on Organometallic Chemistry Celebratory Book, ed. A. J. L. Pombeiro, John Wiley & Sons, 1st edn, 2014, pp. 605–617 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR numbering scheme for HL1–HL4 (Scheme S1), synthesis of B (Scheme S2), cyclic voltammograms of 2 and 3 (Fig. S1), UV-vis-NIR spectroelectrochemistry for 3 (Fig. S2), NMR spectra (Fig. S3–S20), ESI-MS spectra (Fig. S21–S32). CCDC 1903184–1903189. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9dt01238a |
| This journal is © The Royal Society of Chemistry 2019 |