
Atomic spectrometry update: review of advances in elemental speciation
Robert
Clough
*a,
Chris F.
Harrington
b,
Steve J.
Hill
c,
Yolanda
Madrid
d and
Julian F.
Tyson
e
aBiogeochemistry and Environmental Analytical Chemistry Research Group, University of Plymouth, Plymouth, UK. E-mail: rclough@plymouth.ac.uk
bSupra-Regional Assay Service, Trace Element Laboratory, Surrey Research Park, 15 Frederick Sanger Road, Guildford, GU2 7YD, UK
cSpeciation and Environmental Analysis Research Group, University of Plymouth, Plymouth, UK
dDepartamento de Quimica Analitica, Facultad de Ciencias Quimicas, Universidad, Complutense de Madrid, Avda Complutense s/n, 28040 Madrid, Spain
eDepartment of Chemistry, University of Massachusetts Amherst, 710 North Pleasant, USA
First published on 23rd June 2017
Abstract
This is the ninth Atomic Spectrometry Update (ASU) to focus on advances in elemental speciation and covers a period of approximately 12 months from December 2015. This ASU review deals with all aspects of the analytical atomic spectrometry speciation methods developed for: the determination of oxidation states; organometallic compounds; coordination compounds; metal and heteroatom-containing biomolecules, including metalloproteins, proteins, peptides and amino acids; and the use of metal-tagging to facilitate detection via atomic spectrometry. The review does not cover fractionation, which is sometimes termed operationally defined speciation. As with all ASU reviews the focus of the research reviewed includes those methods that incorporate atomic spectrometry as the measurement technique. However, because speciation analysis is inherently focused on the relationship between the metal(loid) atom and the organic moiety it is bound to, or incorporated within, atomic spectrometry alone cannot be the sole analytical approach of interest. For this reason molecular detection techniques are also included where they have provided a complementary approach to speciation analysis. As in previous years, As and Se speciation continues to dominate the current literature, with reports of the speciation of Cr and Hg also increasing, and there are an ever rising number of publications concerning the analysis of ‘biomolecules’. Whilst most of this work is still the preserve of the research field some methods are now approaching the robustness and rapidity for use in the clinical setting.
1 Topical reviews
This latest update adds to that from last year1 and complements other reviews2–6 of analytical techniques in the series of atomic spectrometry updates. Two books which cover elemental speciation have recently been published. At nearly 500-pages a textbook on metallomics deals with analytical techniques and speciation methods.7 In addition to the editor, some 54 other scientists have contributed material to the text, much of which is devoted to elemental speciation analysis, both from the points of view of the analytical instrumentation (about one-fifth of the book) and of individual elements. The speciation of As and Se occupy a further fifth of the book, with Al, Cr, Hg and Sn about 12%. There are several sections devoted to speciation of compounds containing arsenic-sulfur bonds. Other elements that are mentioned include Ag, Fe, Mn, Pt and V. There are short chapters on (a) nanoparticles, and (b) novel techniques for bioimaging, and a long chapter on biomedical and pharmaceutical applications. A book dealing with the analysis of hair for drugs8 covers, according to the publishers description, “testing for metals, including inductively coupled plasma spectrometry (ICP-MS), hair multielemental analysis, and the new speciation analysis”. These latter two topics are further characterized by the author of the chapter in question as offering “an original challenge and interesting future applications”. The table of contents indicates that of all the elements considered, only for Hg is speciation analysis discussed.There has been, as has been the case for several of the past years, considerable activity relating to reviewing the elemental speciation literature. A number of these reviews are focused on the instrumental techniques involved. Delafiori et al.9 reviewed the clinical applications of HPLC-ICP-MS concentrating on As, Hg and Se. The text is a non-critical survey of 101 publications (whose titles are given) starting with an account of the work relating to each element. The first major table is a list of selected studies in reverse chronological order in which considerable detail regarding the HPLC conditions is provided, but there is no information about the operation of the mass spectrometer. A variety of sample preparation methods are considered in some detail, with particular focus on the hollow-fibre pre-concentration procedure. These methods are summarised in a table that also includes values for “percentage recoveries”. It is not clear what these are, as the only recoveries mentioned in the text are “column recoveries”. The values are probably not, therefore, recoveries of spikes. Despite stressing the importance of accuracy, the reviewers do not mention method validation, compound-dependent responses, or calibration. Milacic et al.10 have reviewed monolithic chromatography for the speciation analysis of metal-containing biomolecules. They point out that although monolithic columns (made of a single piece of rigid porous material sealed to the column wall) have been used in the industrial and preparative separation of biomolecules, they have not been widely adopted for elemental speciation analysis, mainly because the limited range of stationary phases available means that mobile phases contain high salt concentrations incompatible with both ICP-MS and ES-MS detection. However, the reviewers identify about relevant 20 articles (titles are not given) describing separation of compounds of Al, As, Cu, Fe, Ni, Pt, Ru, Se, and Zn. They point out the potential advantages, compared with conventional packed beds, of lower back-pressure (with consequent higher flow rates and shorter separation times), of the independence of resolution and capacity from flow rate, and of the lack of “void” volume. This latter claim really means “dead” volume, the point being that there is no diffusion into and out of stagnant volumes of mobile phase (so-called eddy diffusion) to contribute to peak broadening. One example of the separation of iAs, AB, DMAV and MMAV in urine is given, in which there is poor separation of AsIII and AB, though the separation time is just over 3 min. Barin et al.11 have reviewed the determination of elemental impurities in pharmaceutical products and related matrices by ICP-based methods. Although the bulk of the 144 cited articles (with titles) are concerned with the determination of total elements, some 28 articles are highlighted in terms of speciation analysis of As, Sb, Se (as impurities) and of Gd, I, and P as indicative of the active pharmaceutical ingredient. In this context of pharmaceutical product analysis, the reviewers characterise speciation analysis as a complex task with many drawbacks, requiring the multiple steps of extraction, derivatisation, pre-concentration, clean-up, separation, and detection. The writers conclude that speciation of impurities remains a difficult task requiring more development. They also point out, as a more general problem in this area, the lack of suitable CRMs.
Two reviews concerned with electrophoretic separations with mass spectrometric detection cover biological inorganic chemistry12 and applications to elemental speciation, metal–ligand interaction and indirect determinations.13 The first review is the latest in a continuing series and so covers approximately only research published in the last four to five years. The focus is on metallo-drugs containing Au, Bi, Gd, Eu, Pt and Sb, though studies of protein-bound metals are also mentioned, as are studies of metallo-drug–protein binding. About 20 of the 118 articles cited (no titles given) are concerned with the separation and detection of metallic NPs, mostly derived from Au, for which many of the separations have been performed by micellar electrokinetic chromatography. The focus of the review is on the applications of CE-ICP-MS, and so there is almost no discussion of developments in interface design, construction and performance. These topics are considered in some detail in the second review13 in which about 20 of the 172 articles cited (no titles) are concerned with tracking the developments in interface design and performance, with emphasis on the past 10 years or so. The review contains two extensive tables, the first of which is a list of applications, grouped by element, featuring Ag, Al, As, Br, Ca, Cr, Fe, Hg, I, Mg, Pb, Pt, Se, Sn, and Zn, together with NPs and lanthanides. The second table is a list of studies of metal–ligand binding and is organized by ligand featuring, ascorbate, carbonate, chloride, DNA, DTPA, fulvates, glutathione, glycinate, humates, HSA, hydroxide, sulfate, nitrate, oxalate, phytochelatins, and proteins. The reviewers conclude that CE is not as useful as HPLC because of the poorer sensitivity due to the small sample volume and subsequent dilution in the nebulizer and spray chamber interface as well as the relative fragility of the coupling. Nonetheless they are optimistic that future developments in sample stacking (and other pre-concentration procedures) and aerosol generation techniques will make CE emerge as a standard approach.
Although somewhat overshadowed by the popularity of HPLC (primarily with ICP-MS detection), the combination of GC separation with an element-specific detector has been quite widely applied in elemental speciation analysis. Li et al.14 have reviewed atomic spectrometric detectors for GC, covering some 142 articles (whose titles are provided) published since 2000, with an emphasis on plasma-based emission and mass spectrometric detectors, which have the capability of multi-element measurements. The review is organised by detector, and for each interface, design, analyte derivatisation and illustrative examples, which are summarized in a series of tables, are discussed. The techniques of capacitively coupled plasma and glow discharges with various types of hollow cathodes and barrier layers are also mentioned. The reviewers conclude that there is still plenty of room for improvement and highlight the possibilities for miniaturization and portability.
Although the current commercially available AFS instrumentation restricts application to a limited number of elements, there is considerable interested in the use of the technique. Butcher has reviewed15 advances in instrumentation and novel applications of AFS published in the primary literature in 2014 and 2015. The review starts with a discussion of 10 reviews, two of which are ASUs (atomic spectrometry, and biological materials and foods). There are 94 references (with titles), many of which are concerned with speciation analysis, for which emphasis was placed on As (some 20 citations) and Hg (some 17 citations). Other topics covered include diffusive gradients in thin films, photochemical vaporization, and novel atom cells. Applications to the analysis of food, biological materials, and environmental samples were discussed. There are no tables; summaries of the contents of each article mentioned are provided that include information on both analytical performance parameters (such as LOD) and the crucially important aspect of method validation. Almost all applications of elemental speciation by separation followed by AFS involve chemical vapour generation (CVG) reactions, many of which are based on reaction with tetrahydridoborate (commonly known as borohydride). D’Ulivo has reviewed16 the recent developments in the understanding of the mechanisms of chemical vapour generation by aqueous tetrahydridoborate (THB), with the goal of defining a more general reaction model that can be applied to CVG for analytical purposes (including the interferences and formation of by-products in arsane generation), nanoparticle synthesis and the catalytic hydrolysis of THB by metal nanoparticles. Of course, many of the 90 articles (whose titles are given) cited are to the prior work by D’Ulivo and his many collaborators, most of whom are primarily interested in the analytical applications of CVG. The topic of speciation is mentioned, but not dealt with directly in that readers cannot immediately identify reaction conditions that will allow the selective determination of, say, iAs in the presence of DMA. The experiments performed to elucidate reaction mechanisms, particularly those involving deuterated reagents, are discussed in some detail. The review concludes with a consideration of interference effects that includes (a) the nature of the volatile compounds detected when transition and noble metals react with THB, pointing out that (a) one possible mechanism is the extraction of metal free atoms and metal nanoparticles into small hydrogen bubbles, and (b) self-interference and cross-interference among hydride forming elements. In this latter section, the beneficial role of additives, such as cysteine and iodide, are discussed.
The role of nanomaterials in analytical reaction chemistry is the subject of two reviews. In the first,17 the applications of carbon-based materials are reviewed in an article that is part of a series of communications on the design and physicochemical studies of advanced materials at the Saint Petersburg University. The materials in question in this review are fullerenes, nanotubes, graphene, nanodiamond and nanodispersed active carbon, which have been used as high-performance sorbents for chromatographic separation and pre-concentration. Although the word speciation appears several times in the review, the authors do not seem to be using it is the sense of “determination of various chemical forms of an element,” and so the number of examples of the use of these materials for elemental speciation is rather limited. In the 364 articles cited (no titles) there is one citation to the determination of butyltin compounds (by GC separation on single-walled carbon nanotubes). In contrast, the second review18 specifically focuses on nanomaterials for elemental speciation, though the scope of the materials is broader, encompassing magnetic NPs, metal oxides, noble metal nanoparticles, and ion imprinted polymers as well as carbon nanostructures (23 examples are given in one of the tables). The elements that dominate the 164 references cited (no titles given) are, as might be expected, As, Cr, Hg and Se, with smaller contributions from Sb and Sn. A tutorial introduction to photocatalytic HG is given in an early section devoted to the use of CVG as the interface between LC separation and element-specific detection. A large section is devoted to non-chromatographic separations for which four strategies are identified, and examples are listed in which (a) “metal” oxide NPs (silicon is in the list), (b) magnetic NPs (c) noble metal NPs and (d) carbon NPs are used. There is a section on the applications of functionalized polymers. Finally the topic of noble metal NPs as optical nanosensors is described including methods based on localised surface plasmon resonance, surface enhanced Raman and fluorescence spectroscopies. The reviewers conclude that although speciation analysis is a well-established research field performing it is still an “analytical challenge”. They suggest that NPs have the potential to be deployed for high efficiency separations, though they concede that the achievable LOD values for routine sensor applications for most of the analyses described are much higher than relevant environmental concentrations. They also point out that a serious restriction on the practical employment of some nanocomposite materials is the complicated multi-step synthesis, which is often difficult to reproduce. Developments in nanotechnology and of new materials are on the list of factors identified in a review of recent advances in FIA that will ensure the continued use of the technique.19 The reviewers estimate that the FI literature is now in excess of 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 publications, so they concentrate on developments in the last 10 years and highlight developments in the last two to three years, citing a total of 458 sources (whose titles are given). In addition, details of the 14 books in English on various aspects of FIA are collected in a table. There is a table of review articles from the past 15 years divided into several sections. Of the 135 articles in this table, about 10 are concerned with methods in which an atomic spectrometric detector is used. These, together with another 33 articles, are discussed in a section devoted to flow injection atomic spectrometry. Speciation analysis is mentioned several times in the review, although not always in connection with elemental speciation by atomic spectrometry. The reviewers conclude by predicting that methods featuring atomic spectrometry will be increasingly used and that this development trend will also include non-chromatographic speciation analysis, employing various methods of on-line transformation.
000 publications, so they concentrate on developments in the last 10 years and highlight developments in the last two to three years, citing a total of 458 sources (whose titles are given). In addition, details of the 14 books in English on various aspects of FIA are collected in a table. There is a table of review articles from the past 15 years divided into several sections. Of the 135 articles in this table, about 10 are concerned with methods in which an atomic spectrometric detector is used. These, together with another 33 articles, are discussed in a section devoted to flow injection atomic spectrometry. Speciation analysis is mentioned several times in the review, although not always in connection with elemental speciation by atomic spectrometry. The reviewers conclude by predicting that methods featuring atomic spectrometry will be increasingly used and that this development trend will also include non-chromatographic speciation analysis, employing various methods of on-line transformation.
In a review of soft X-ray spectromicroscopy for speciation, quantification and nano-eco-toxicology of nanomaterials, Lawrence et al.20 point out that there is a critical need for methods that provide simultaneous detection, identification, quantification and visualization of nanomaterials at their interfaces with biological and environmental systems. The reviewers provide an overview of measurements made by ambient soft X-ray scanning transmission X-ray spectromicroscopy (STXM) on beamline 10ID1 at the Canadian Light Source (CLS, Saskatoon, SK, Canada). The spatial resolution (30 μm) is about one order of magnitude better than conventional optical microscopy but several orders of magnitude worse than transmission electron microscopy (TEM), and also somewhat worse than super-resolution optical microscopy. On the other hand, the chemical sensitivity is generally better than that of TEM. There are 104 references with titles. The work reviewed includes the interaction of nanomaterials with microbial cells, biofilms and extracellular polymers, for which the studies involved speciation and mapping of metal-containing nanomaterials (Cu, Ni, Ti) and carbon-based nanomaterials (multiwalled carbon nanotubes, C-60 fullerene). The benefits of X-ray fluorescence detection in soft X-ray STXM are illustrated by a study of low concentrations of Ni in a natural river biofilm. Evidence for the presence of both NiII and NiIII was obtained.
In a slightly broader overview of the same area, Li et al.21 review synchrotron radiation techniques for nanotoxicology. There are 104 references with titles. The review opens with a tutorial introduction to the various techniques, and then describes a number of examples, including the speciation of Au in rat liver and spleen (after injection of Au nanorods), the transformation of metal oxide (Ce, La, Zn) NPs by plants (soybeans and cucumbers), the speciation of Ag and Zn in sludge from a wastewater treatment plant, and the reaction of Ag NP with kaolin. The reviewers emphasize the benefits of the XAFS technique, but point out that raising awareness about the unique capacities of SR techniques among nanotoxicologists is still necessary, even though inadequate beam time is available because of the wide-ranging application of SR techniques in many other scientific fields.
The specific area of metal and metalloid speciation topic of plant biochemistry has been reviewed by Kroukamp et al.22 The reviewers point out that the ability of plants to accumulate metals and metalloids could pose a health risk to both humans and animals as plants are a substantial dietary component. This uptake has also been exploited for bioremediation and biomonitoring. There are 119 citations with titles. The first table indicates the scope of the species considered with a list of 24 citations that includes As, Cd, Mn, Ni, Sb, Se, and U, but not Hg (or Pb, which is mentioned—barely, just two citations—towards the end of the review). So-called “direct techniques” are briefly considered, and it was concluded that although XANES has been shown to be a valuable tool in speciation analysis, poor detection limits, maintenance of the species integrity, and limitations on the number and variety of species within a sample that are quantifiable make calibration a challenge and reduce the technique’s applicability. The bulk of the review deals with measurement of the various chemical forms that appear after extraction, so there is considerable discussion of sample preparation (extraction) procedures, separation procedures, (including various modes of LC and CZE), and of the various element-specific detection techniques. The reviewers rightly point out that quality control is critical, and they highlight the importance of using CRMs whenever possible. They consider that future directions in speciation studies are likely to focus on the development of analytical methods that ensure that the original species profile is not altered. Future studies are also likely to focus on additional elements of toxicological and environmental relevance and in the evaluation of food biofortified with desired chemical species.
Das has surveyed green methodologies for the multi-elemental speciation analysis of environmental samples.23 The review contains 41 citations (without titles), only one of which is dated later than 2014. A summary table containing 17 citations shows that fractionation procedures are also being considered. The definition of “green” in this context is not given, though readers are directed to a suitable source. The author considers that the most widely used green separation–detection combination is HPLC-ICP-MS coupled with extraction procedures that avoid organic solvents. Examples of procedures for a wide range of metals and metalloids are given, including some not normally on the list, such as Mg and Zn.
2 CRMs and metrology
Species specific ID for the accurate determination of AB and MeHg in prawn and cuttlefish tissues by LC-MS-MS and SPME GC-ICP-MS has been reported as part of a new CRM validation exercise.24 Quantitation of AB and MeHg was achieved using a 13C-enriched AB spike (NRCC CRM CBET-1) and an enriched spike of (MeHg)–Hg-198 (NRCC CRM EMMS-1), respectively. The analyte mass fractions in enriched spikes were determined by reverse ID using natural abundance AB and MeHg primary standards. Validation was achieved using several biological CRMs (DORM-4, TORT-3, DOLT-5, BCR-627 and BCR-463) with good results. The developed methods were then applied for the determination of AB and MeHg in two new CRMs, prawn (PRON-1) and cuttlefish (SQID-1) produced jointly by Thailand Institute of Scientific and Technological Research (TISTR) and National Research Council Canada (NRCC). Following additional measurements of AB using LC-ICP-MS, standard additions calibration, and the use of external calibration at NRCC and TISTR, certified values for AB as As of 1.206 ± 0.058 and 13.96 ± 0.54 mg kg−1 were obtained for the new CRMs PRON-1 and SQID-1, respectively. It was found that AB comprised 69.7% and 99.0% of total As in the prawn and cuttlefish, respectively, whereas MeHg comprised 94.5% of total Hg in cuttlefish. In the second paper two analytical procedures for total Hg (ID-ICP-MS) and MeHg (ssID-HPLC-ICP-MS) mass fractions in two candidate CRMs (oyster and clam) were evaluated. For the total Hg measurements aliquots of ERM-AE640, which is enriched in 202Hg, were added to 0.2 mg of dried biota samples, followed by 4 mL of HNO3 (65%), 1 mL of HCl (36%), 0.5 mL of HF (49%) and 1 mL of H2O2 (30%). A MAE, ramping from ambient to 180 °C in 10 minutes followed by 30 minutes at 180 °C at a controlled maximum pressure of 30 bar, was used to solubilise Hg present in the samples. After evaporation to dryness in a heating block the residues were dissolved in 10 mL of 2% HNO3 which were subsequently diluted for analysis by ICP-MS. The MeHg content in the oyster and clam samples was solubilised as follows. An aliquot of IES-MeHg, which is enriched in Me202Hg, was added to 0.2 mg of sample followed by 10 mL of 15% HCL and MAE (80 W for 30 minutes) and centrifugation. The Hg species in the supernatant were separated on an AEX column and the Hg isotopic ratios in the collected fractions measured using ICP-MS. The full method is described in a separate cited paper. The methods were validated using a scallop CRM, IAEA-452 and full uncertainty budgets for the procedures were estimated. Subsequently, the candidate CRMs were certified for total Hg and MeHg via an interlaboratory comparison. This has produced two new CRMs, IAEA-461 (Clam) and IAEA-470 (Oyster) with total Hg and MeHg mass fractions of 390 ± 40 mg kg−1 and 62.3 ± 6.9 μg g−1 and 21.1 ± 2.1 mg kg−1 and 5.22 ± 0.87 μg g−1 respectively.An overview, from a metrological standpoint, of the efforts during the last three years to achieve reliable quantification of metalloproteins has also been published this year.25 The analytes covered in the review, which include haemoglobin (HGB), transferrin (TRF), superoxide dismutase (SOD), selenoproteins and ceruloplasmin, were chosen due to their importance in clinical diagnostics. The importance of the traceability of the analytical results to the SI, via a pure substance reference material, and the challenges faced in a clinical setting to achieve this is discussed. The authors suggest that ssIDMS, where the metal incorporated into the protein is in an isotopically enriched form for use as a spike material, is the most reliable approach to obtain traceable results of high accuracy and minimal uncertainty. The results obtained by various workers in the field (61 references in total) using both elemental speciation and other techniques are discussed in detail and also summarised in a table. The authors conclude that many analytical methods are too complex and time consuming, in particular those involving several separation steps, to be used as routine methods. However, they allow insight into different aspects of protein behaviour in a research context and also have the ability to be used produce pure substance CRMs that can be used to validate methodologies currently in use in the clinical setting.
3 Elemental speciation analysis
3.1 Antimony
There have been fewer publications covering Sb speciation this year, but the reported applications have been diverse. The speciation of Sb in soils, sediments and volcanic ashes has been undertaken following the first recorded eruption of the Chaiten Volcano in southern Chile.26 The total Sb content was determined by ICP-MS and SbV and SbIII determination by HPLC-HG-AFS. Microwave digestion using a 6 mL HNO3 and 0.5 mL HBF4 acid digestion mixture achieved total solubilization of Sb in solid samples and 30 minutes at 200 °C with 100 mmol L−1 of oxalic acid and 20 mmol L−1 of ascorbic acid proved best for Sb solubilisation and SbIII stabilization. A total of 1.8 ± 0.1 μg g−1 of Sb was found in the volcanic ash from the eruption. The Sb concentrations in sediments were 3 times higher than the Sb concentrations in the sediments from a reference zone. In the extracts of the volcanic ash and sediment, SbIII was the most abundant species, accounting for 70 to 97% of the total Sb found in the extracts.A method to determine Sb speciation in plant biomass and nutrient media has been reported utilising a separation method using an anion exchange cartridge with ET-AAS detection.27 The selectivity of the AE cartridge towards both SbIII and SbV reversed in the presence of deionized water or 2 mM citric acid. While SbV was retained by the cartridge in deionised water, SbIII was retained in citric acid media. At pH 6, SbIII and SbV both formed citrate complexes, but the cartridge had higher affinity towards the SbIII–citrate complex. Separation of SbIII was tested at various concentrations in fresh and spent growth media and plant tissues, and the results compared to analysis using HPLC-ICP-MS. The cartridge retained SbIII and showed 92–104% SbV recovery from As hyperaccumulator Pteris vittata roots treated with SbIII and SbV. The potential uptake pathway for SbV through erythrocyte membranes has been reported by Barrera et al.28 Using AFS with Laurdan and diphenylhexatriene probes, it was found that there was no interaction between SbV and the membrane lipids. By comparing the SbV entry percentages through lipid vesicles and sealed erythrocyte membranes, it was found that SbV required protein channels to pass through the membrane. The competitive inhibition results using HCO3− and Cl− showed that the SbV uptake rate through the membrane fell by 50–70% until full inhibition was reached, possibly due to the inhibition of the anion exchanger channel. Finally, AFS measurements with a 5-iodoacetamidofluorescein probe showed that the SbV interacted with membrane protein SH groups.
The biogeochemical cycles of the metalloid elements Sb and As in the East China Sea have been studied.29 The As results are discussed in the appropriate section below. Dissolved SbV and SbIII were determined by HG-AFS and showed that the total dissolved concentrations of iSbV + iSbIII were high near the Changjiang Estuary and the coastal area of Hangzhou Bay, but decreased away from the coast. The total dissolved iSb displayed moderate conservative behaviour, confirmed by the correlation of salinity with dissolved Al. The SbIII concentrations were extremely low in the area, with slightly higher concentrations near the bottom layer which indicated the contribution from the sediment–water interface. A preliminary box model was established to estimate the water-mass balance and Sb budgets for the East China Sea. The concentrations of dissolved iSb in the East China Sea are similar to other areas in the world.
The speciation analysis of iSbIIIand SbVin polyurethane foam loaded with bromopyrogallol red (PUF-BPR) as a selective sorbent for SbIII has been described.30 The quantification of Sb in the solutions was performed by ET-AAS, and analytical variables that could affect the performance of the method evaluated. The best conditions for the sorption of SbIII were achieved by shaking 50 mg of PUF-BPR with the solutions containing SbIII at pH 4.0 for 90 min. The retention of SbV was not significant whilst over 95% of SbIII was removed from the solution. The desorption of the SbIII from the loaded PUF-BPR foam was achieved by shaking with 7.5 mL of a 2.5 M L−1 HNO3 solution for 30 min. No interference of metal cations was observed on the SPE of SbIII. The developed method was applied to the speciation of Sb in river water samples fortified with different concentrations of SbIII and SbV, and the determination of SbIII in a sample of a Sb-based drug utilized in the treatment of leishmaniasis. The results showed that SbIII can be separated from high concentrations of SbV, with recoveries in the range of 81–110%. The LOD and LOQ values were 0.6 and 2 μg L−1, respectively, for SbIII and 1 and 3 μg L−1 for SbV.
Table 1 shows other applications of Sb speciation presented in the literature during the time period covered by this ASU.
| Analyte species | Technique | Matrix | Sample treatment | Separation | LOD | Validation | Reference |
|---|---|---|---|---|---|---|---|
| SbIII, SbV | FAAS | Milk and beverages | UAE, 30 °C, 5 min, centrifuged | Selective CPE, borate complexes (from Britton Robinson buffer) + pyronin B + sodium dodecyl sulfate + triton X-114 | 2–4 μg L−1 | NIST SRM 1549 (nonfat milk powder), SRM 1643e (trace elements in water) and spike recovery | 31 |
| SbIII, SbV | FAAS | Milk and beverages | MAE with HNO3 and H2O2 | Selective CPE SbIII as complex with 4-(2-thiazolylazo)resorcinol and 2-(2-thiazolylazo)-p-cresol + sodium dodecyl sulfate pH 6.0 mediated by nonionic surfactant, t-octylphenoxypolyethoxyethanol | 0.1–0.3 μg L−1 | NIST SRM 1549, SRM 1643e and spike recovery | 32 |
| SbIII, iSb, SeIV, iSe | HG-AAS | Waters, beverages, foods | Washed, peeled, sliced, frozen, ground, freeze dried. UAE with HNO3 and H2O2, 80 °C, filtered | Selective CPE, charge transfer sensitized ternary complexes of Sb(III) and Se(IV) with neutral red in presence of pyrogallol and CTAB at pH 6.0 into PONPE 7.5. SeVI and SbV reduced with L-cysteine and tartaric acid | 2–5 ng L−1 | NIST SRM 1573a (tomato leaves) and, SRM 1547a (peach leaves) and spike recovery | 33 |
| AsIII, AsV, CrVI | HPLC-ICP-MS, O2 reactant gas 91AsO+ | Water | Diluted, filtered | AEC, Hamilton PRP X-100 isocratic 22 mmol L−1 (NH4)2HPO4, 25 or 65 mmol L−1 NH4NO3 pH 9.2 | 0.06–0.15 μg L−1 | NRCC SLRS-5 (river water) TM 27.3 National Water Research Institute Canada (trace element fortified calibration standard) and spike recovery | 34 |
| AsIII, AsV SbIII, SbV CrIII, CrVI | HPLC-ICP-MS O2 reactant gas 91AsO+ | Drinking water | Filtered (0.22 mm) | AEC, Hamilton PRP X-1003, gradient 22 mmol L−1 Na2EDTA + 36 mmol L−1 NH4NO3 (A) pH 4.6, (B) pH 9.0 | 0.04–0.09 μg L−1 | NRCC SLRS-5 (river water) TM 27.3 National Water Research Institute Canada (trace element fortified calibration standard) and spike recovery | 35 |
| SbIII, SbV | HPLC-ICP-MS | River water and sediment | Waters, preserved with Na2EDTA or HNO3 sediments frozen, thawed, air-dried, sieved, UAE various extractants | AEC, Hamilton PRP X-100 gradient (a) 20 mmol L−1 NH4NO3 pH 8.7 (b) or 60 mmol L−1 NH4NO3 pH 8.7 | 0.009–0.01 μg L−1 | China National Analysis Center for Iron and Steel DC 73309 (stream sediment) | 36 |
3.2 Arsenic
Following the pattern of recent years, As speciation remains an active area of research. Most authors report on applications based on established techniques reflecting the maturity of methods employing HPLC-ICP-MS for As speciation, although various modifications have been reported as summarised in Table 2.| Analyte species | Technique | Matrix | Sample treatment | Separation | LOD | Validation | Reference |
|---|---|---|---|---|---|---|---|
| AsIII, AsV | Batch-HG-AAS | Various water | Acidified to 0.1 mol L−1 HCl and stored at 4 °C | Selective HG from AsIII. Reduction of AsV by KI + ascorbic acid | 1 μg L−1 | SCP science EnviroMAT drinking water-low, EP-L-2 and spike recovery | 90 |
| AsIII, AsV, DMAV | FI-HG-AAS | River water, sediment and plants | Dried, sieved, ultrasonicated, UAE with 6 mol L−1 HCl, centrifuged, filtered | Selective HG, varying acid (HCl and HNO3) and borohydride concentrations | 3–6 μg L−1 | Spike recovery | 91 |
| iAs | Batch or continuous flow HG-AAS | Waters | Adjusted to pH 6 | Adsorption on nickel/nickel boride NP. Dissolved in 2 mol L−1 HCl | 0.02 μg L−1 | EnviroMAT drinking water-low, EP-L-2 and spike recovery | 92 |
| iAs | FI-HG-AAS | Fish | Lyophilized and ground (<250 μm), MAE with 1 mol L−1 HCl | Selective HG of iAs in 3 mol L−1 HCl | 5 μg kg−1 | NRCC DORM-3 (fish protein) and spike recovery of iAs, MMA, DMA and AB | 93 |
| AsIII, AsV | FI-HG-AAS | Various waters, serum, urine | Filtered, frozen, thawed, diluted | Selective SPE of AsV on carboxylated nanoporous graphene at pH 3.5 | 0.002 μg L−1 for the high volume version | NIST 2669 (arsenic species in human urine) and spike recovery | 94 |
| AsIII, AsV and total As | FI-HG-ETAAS | Various waters | Filtered and stored at 5 °C | Selective HG to distinguish AsIII and AsV. Online photoxidation + TiO2 for total As. Arsine trapped on minicolumn of carbon nanotubes, eluted with 0.5% HNO3 | 0.008 μg L−1 | Spike recovery | 95 |
| AsIII, iAs and total As | Electrolytic HG-AFS | Rice and water | Dried, ground (<250 μm), frozen (−18 °C), thawed, MAE (protease and amylase), centrifuged, filtered | Selective HG by chemical modification of the cathode (thiourea or L-cysteine) and control of current | 0.1–0.2 μg L−1 (5–10 μg kg−1 in solid) | NIST 1568a (rice) and spike recoveries (water) | 96 |
| AsIII, iAs | ET-AAS, Pd and Mg modifier | Smokeless tobacco (saliva extract) | Dried, ground (<125 μm) USE with artificial saliva, centrifuged | CPE of AsIII and SPE for iAs | 0.02–0.03 μg L−1 (1–2 μg kg−1 in solid) | Atomic Energy Agency ICHTJ-cta-VTL-2 (Virginia tobacco leaves) and spike recovery | 97 |
| AsV, iAs | ET-AAS, nickel modifier | Spa, tap and ground waters | Acidified on-site, filtered | Selective SPE of AsV on Amberlite IRA 900 resin (anion exchange) at pH 4.0. iAs retained at pH 8.0 | 0.006 μg L−1 | SRM High Purity Standards CWW-TM-D (waste water) and spike recovery | 98 |
| AsV, iAs | ET-AAS, palladium modifier | Waters (sea, river, ground) | Filtered, acidified on-site. For iAs oxidation with permanganate | On-site selective retention of AsV as molybdoarsenate on activated carbon, elution with NH4OH | 0.05 μg L−1 | Spike recovery | 99 |
| AsIII, AsV, MMAV, DMAV | HPLC-HG-AFS | Human urine | Filtered, frozen, thawed, diluted | Ion-pair and AEC (PRP X-100) phosphate buffer (preferred) | 40–100 ng L−1 | NIST 2669 (arsenic species in frozen human urine), comparison with reference (cryotrap) method | 100 |
| AsIII, iAs | ETV-ICP-MS | Waters | Filtered, refrigerated | Selective LLE of AsIII as APDC complex into floating drop of 1-dodecanol | 0.4–0.1 ng L−1 | GSB Z50004-88 (water) and spike recovery | 101 |
| AsIII, AsV, MMAV, DMAV | HPLC-ICP-MS | Rice | UAE with 5 mmol L−1 malonic acid at 80 °C for 2 h, centrifuged, filtered | AEC. Hamilton PRP X-100, isocratic 5 mmol L−1 malonic acid pH 5.6 | 0.02 μg kg−1 | NMIJ 7503a (white rice flour) | 102 |
| AsIII, AsV, MMAV, DMAV AB, AC, tetramethylarsonium | HPLC-ICP-MS, Ge internal standard | Human urine | Centrifuged, filtered, diluted | AEC, gradient (a) water, (b) 20 mmol L−1 HNO3 both with 2% methanol | 0.3–0.9 μg L−1 | NIST 2669 (arsenic species in frozen human urine) | 103 |
| AsIII, AsV, MMAV, DMAV | HPLC-ICP-MS, AsO+ | Seafood, rice and rice products | UAE 1 + 4 methanol/water for up to 6 h | AEC. Narrow-bore Nucleosil 100 SB, 12 mmol L−1 ammonium dihydrogen phosphate + 2% methanol at 0.3 mL min−1 | 0.3 μg L−1 | Instituto Superiore di Sanita MURST ISS-A2 (Antarctic krill), NIST 1568a (rice flour), NRCC TORT-2 (lobster hepatopancreas) | 104 |
| AsIII, AsV, MMA, DMA, AB | HPLC-ICP-MS | River water and sediment | Waters, preserved with Na2EDTA or HNO3 sediments frozen, thawed, air-dried, sieved, UAE various extractants | AEC Dionex Ion Pack AS7, isocratic 1 mmol L−1 phthalic acid + 10 mmol L−1 Na2EDTA | 0.08–0.2 μg L−1 | China National Analysis Center for Iron and Steel DC 73309 (stream sediment) | 36 |
| AsIII, AsV | XANES | Coal fly ash | Pressed pellets 13 mm diameter 20% wax binder | Spectral interpretation | Not given | SRM NIST 1633b coal fly ash | 105 |
The feasibility of microchip-based electrochromatography interfaced with ICP-MS for elemental speciation has been reported by Cheng et al.37 Building on previous work, the authors utilised a homemade nanoflow nebulizer and heated single pass spray chamber. A monolithic polymer (poly(butyl methacrylate)) (BMA) monolith was fabricated in a 50 mm microchannel by UV initiated polymerisation after vinylisation pretreatment of the microchannels using 3-(trimethoxysilyl)propyl methacrylate. Negatively charged moieties (2-acrylamido-2-methyl-1-propanesulfonic acid, AMPS) at 1% wt were added into the monomer to enhance electroosmotic flow. The monolith not only eliminated the laminar flow induced by the nebulizer suction but also ensured variable-volume electrokinetic injection. Factors affecting the electroosmotic flow rate such as the amount of AMPS added and electrochromatographic conditions were optimized. The μ-electrochromatography-ICP-MS system offered satisfactory resolutions and repeatabilities (<4.4%) by individually using multiple metallic cations and iodine anions as the models. The LOD values of between 0.15–0.22 μL−1 for AsIII, AsV, MMA and DMA, were achieved and good agreement with As species in a reference human urine (GBW 09115) obtained. Mercury speciation in a certified fish tissue was also determined. A specific tri-n-butylphosphine functionalized polymer microsphere has been designed and used as the core adsorbent to comprehensively enrich both inorganic and organic As species in environmental waters by on line SPE-HPLC-ICP-MS.38 Avoiding any redox reagent, four As species (AsIII, DMA, MMA and AsV) were quantitatively retained on an in house made n-butylphosphine functionalized polymermini-column. The As species were then rapidly eluted, with a mixed solution of ammonium nitrate and ammonium dihydrogen phosphate at pH 8.8 onto an Hamilton PRP-X100 column coupled to an ICP-MS instrument. Following optimization, the enrichment factor obtained for AsIII and DMA with 25 mL sample solution was 28, while for MMA and AsV it was 30. The LOD values of 1.2 ng L−1, 0.96 ng L−1, 0.82 ng L−1 and 0.91 ng L−1, with RSDs of 3.9%, 5.6%, 3.2% and 4.5%, were obtained for AsIII, DMA, MMA and AsV, respectively. The method was validated using a water CRM (GSBZ 07-3171-2014), prior to use on lake, river and seawater samples.
Publications on the speciation of As in air are often directed at the determination of As associated with PM. This area is the focus of a review with 140 references by Sanchez-Rodas et al.39 which looks at sampling, sample preparation and instrumental analytical techniques for both total As determination and As speciation analysis. The origin, concentration and legislation concerning As in ambient air are also considered. The review describes the procedures for sample collection of total suspended particles (TSP) or particles with a certain diameter expressed in microns (e.g. PM10 and PM2.5), but also considers the collection of the gaseous phase As species. Sample digestion of the collecting media for PM is described, including procedures that use acids or mixtures of acids aided with different heating procedures. Detection techniques are summarised and compared (AFS, ICP-MS, ICP-OES and ET-AAS), as well those techniques capable of direct analysis of the solid sample (PIXE, INAA and XRF). Studies involving As speciation on PM are also discussed and include both GC and HPLC separation approaches. Arsenic species in tailings and windblown dust from a gold mining area in Brazil has been examined.40 Although the levels of As in the tailings were up to 2666 mg As kg−1, the As bioaccessibility in the same area was low (<4.4%). Mine samples were collected from four areas and windblown dust from eight sites. SR-XANES, μ-XANES, and μ-XRF spectroscopy were applied to determine As solid-phase speciation. The study demonstrated that As occurred in the AsV oxidation state, mostly as poorly crystalline ferric AsV, but it was also present as arsenopyrite (FeAsS) and its weathering products reflecting the importance of Fe oxides in immobilizing As in the terrestrial environment. All air particulate samples exhibited As-rich particles (up to 313 mg As kg−1). These solid-phase As species were very similar to those found in the mine samples indicating that As in the windblown dust was not readily available.
Regional studies of As in drinking water continue to be published. Most reports on As in drinking water are based on globally recognised As hotspots, notably places such as Bangladesh, India, parts of SE Asia and China. However, a study of urinary As profiles in the UK has revealed exposure to iAs from private drinking water supplies in Cornwall South West England.41 The study was based on 207 volunteers, from 127 households who use private water supplies for drinking, who were asked to provided urine and drinking water samples. This followed on from previous work which showed that private water supplies in Cornwall exceeded the current WHO guidance value and UK prescribed concentration or value (PCV) for As of 10 μg L−1 in 5% of the 497 properties surveyed. Total As was determined by ICP-MS and urinary As speciation by HPLC-ICP-MS. The As concentrations were found to exceed 10 μg L−1 in the private drinking water of 10% of the volunteers. Unadjusted total urinary As concentrations were poorly correlated with private drinking water As largely due to the use of spot urine samples and the dominance of AB from seafood sources. However, the osmolality adjusted sum of urinary iAs species, AsIII and AsV, and their metabolites, MMA and DMA, was found to strongly correlate with private drinking water As, indicating private water supplies as the dominant source of iAs exposure in the study population of private water supply users. The concentration of total As, AsIII, AsV, MMA, DMA, and AsB in drinking water, surface water, wastewater, and snow collected from the provinces of Wielkopolska, Kujawy–Pomerania, and Lower Silesia in Poland have been reported.42 The total As was determined by ICP-MS, and As species by HPLC-ICP-MS. The highest total As concentration determined in drinking water samples was 1.01 μg L−1, although total As in surface water reached 3778 μg L−1 due to geogenic As contamination. Total As concentrations in wastewater samples were comparable to those determined in drinking water samples. Significantly higher As concentration, 83.1 ± 5.9 μg L−1, was found in snow samples and AsV was present in all of the investigated samples, and in most of them, it was the sole species observed. However, in the snow sample, 97% (81 ± 11 μg L−1) was in the form of AsIII. Two similar papers have been published by the same group on As speciation in Polish reservoirs. The first focused on the Rybnik Reservoir43 selected because it is subjected to thermal anthropopressure. AsIII, AsV, SbIII, and SbV ions were separated on Dionex IonPac AS7 column connected to an ICP-MS. The LOD values were 0.18, 0.22, 0.009, 0.012, μg L−1, respectively. Water and bottom sediment samples were collected monthly at three points between January and November. The AsIII and SbIII speciation forms dominated in the bottom water and the oxidized As and Sb forms dominated in the bottom sediments in the heated water discharge zone of the Rybnik Power Plant. The second paper44 again reports on different As and Sb species in the water column and bottom sediments in order to verify if the bottom sediments deposited in the discussed reservoirs could pose an environmental threat due to the release of toxic ionic forms into the pelagic zone. More than 0.3 million individuals are subject to chronic exposure to As via their drinking water in Inner Mongolia, China. To determine the As methylation capacity profiles for such individuals, concentrations of urinary As metabolites were measured in 548 subjects using HPLC-HG-ICP-MS.45 The mean urinary concentrations of DMA, MMA, iAs, and total As were 201, 46.7, 53.0, and 300 μg L−1, respectively. Mean urinary % iAs and % MMA were higher in males, while urinary % DMA was higher in females. There was a strong positive correlation between % iAs and % MMA, with negative correlations between % iAs and % DMA, and % iAs and % MMA. In addition, % iAs and % MMA were positively associated with total As in drinking water, while % DMA was negatively related with total As in drinking water. Regression analysis indicated that the primary methylation index and secondary methylation index generally decreased with increasing total As in drinking water. Females had a higher As methylation capacity compared to males. Younger subjects had lower primary As methylation capacity. However, the secondary As methylation capacity was hardly affected by age. Moreover, both primary and secondary As methylation capacities were negatively related to total As in drinking water.
Arsenic speciation in river, groundwater, wastewater, and porewater have also been reported. Water, suspended particulate matter (SPM), and biota samples have been collected from the Taehwa River Estuary in S. Korea to determine the distribution, partitioning, and bioaccumulation of As species in the river water samples.46 Six forms of As (AsIII, AsV, MMA, DMA, AB and AC) were quantitated by the use of HPLC-ICP-MS. Arsenic was found mainly near urban and industrial areas, and iAsV was the predominant As form in both water and suspended particulate matter (SPM). Particulate As was found at the greatest concentrations in coarse particles (>180 μm), followed by medium (30–180 μm) and fine (0.45–30 μm) particles, in freshwater. Arsenic concentrations were similar across the three particle fractions in saltwater. Field-based distribution coefficient values for As depended strongly on SPM, with a less robust dependence on salinity. Concentrations of As were greater in macroalgae than in marine animals, such as fishes, bivalves, crabs, shrimps, and gastropods. XANES, in conjunction with analysis using ICP-MS has been used to understand As transfer and transformation within the freshwater Montezuma Well (central Arizona, USA) food web.47 The Montezuma Well water contained 110 μg L−1 As (100% H3AsO4), known to originate from ore deposits approximately 30 km to the west, and transported underground to enter Montezuma Well through vents in the bottom of this collapsed travertine spring. The Montezuma Well food web contains three trophic levels with only five organisms in the top two levels, making it possible to account for the As in >90% of the biomass of the food web. Arsenic diminution generally occurs between trophic levels, with 702 mg kg−1 As in the primary producers, 3.4 mg kg−1 As in the second trophic level, and <1.3 mg kg−1 As for the top-tier of the littoral zone food web. A notable exception to the biodiminution trend is the very high total As content (2810 mg kg−1) of Motobdella montezuma, an endemic leech and top predator. The biotransformed (sulfur-coordinated) As in M. montezuma appears to be present almost entirely on the surface of the organism, possibly suggesting a detoxification mechanism. XANES sample spectra and a linear combination of model As compound spectra indicated that As enters the food web from the well water entirely as iAsV and is transformed within the food web into more reduced and organic As species, including sulfur-coordinated and methylated compounds. A study on the treatment of AsIII or AsV containing industrial wastewater using a bio-column reactor containing Bacillus arsenicus MTCC 4380 immobilized on a sawdust (Syzygium cimini)/MnFe2O4 composite bed has been reported.48 The influence of bed height (20–100 cm), flow rate (251–837 mL h−1), empty bed contact time (8–20 h), and initial As (either as AsIII or AsV) concentration (2000–5000 mg L−1) on the As removal was investigated. Total As was determined by ET-AAS. The breakthrough curve for the bed height specified that a longer bed column extended the life span of the column with a maximum capacity of 87.5 and 89.0 mg g−1 for the AsIII and AsV columns, respectively. The impact of co-existing ions on biosorption/bioaccumulation of As was also investigated in column mode. The column was regenerated by eluting AsIII or AsV using 0.05 M NaOH after the biosorption/bioaccumulation studies. A simple method to preserve As species in simulated pore water has been evaluated.49 Synthetic pore water containing high levels of Fe, Mn and S (as sulfide) were synthesized and spiked with different As species (AsIII, AsV, MMA, DMA, AsO3S3, and AsS43) in different combinations. Solutions were analysed at intervals of one day, week, month and 2 months after preparation by HPLC-ICP-MS using an ion exchange column and ammonium carbonate buffer. Samples containing Fe and Mn spiked with AsIII, AsV, DMA, MMA showed adequate species preservation for two months when EDTA was added. The total As in samples containing Fe, Mn and S2− was preserved over 60 days. However, speciation was not preserved. Samples spiked with synthesized mono- and tetra-thioarsenateV species showed immediate degradation these species into unidentified As–S species. The results indicate that while EDTA may be adequate to preserve As species containing high Fe and Mn concentrations, the combination of EDTA and temperature was not successful in preserving As speciation in samples containing sulfides.
The speciation of As in the marine environment continues to received attention, although the number of publications is down on recent years. The biogeochemical cycles of the metalloid elements As and Sb in the East China Sea have been studied.29 The Sb results are discussed in the Sb section of this Update. Dissolved iAsV and AsIII were determined by HG-AFS and showed that the total dissolved iAs was depleted in the surface water and enriched in the deep water. The AsIII showed different vertical profiles to that of the total dissolved As, with significant surface enrichment in the middle shelf region where the concentrations of phosphate were extremely low. The speciation of dissolved As was subtly controlled by the stoichiometric molar ratio of AsV to phosphate and the average AsV/P ratio during the spring months was 10.8 × 10−3, which is higher than previously recorded. Hydrothermal vents are surficial expressions of subsurface geological and hydrological processes and fluids emitting from active vents are chemically distinct from bottom seawater, being enriched in dissolved metals and metalloids, including As. Vent organisms accumulate As, but the As speciation in these non-photosynthetic organisms is largely unknown. Maher et al.50 have measured As concentrations and chemical species in three deep sea hydrothermal vent worms (Ridgeia piscesae, Paralvinella sulfincola and Paralvinella palmiformis) from the Juan de Fuca Ridge in the northwest Pacific. Total As was measured by ET-AAS, and As speciation by HPLC-ICP-MS. Separations were achieved using a Supelocosil LC-SCX cation exchange column and a Hamilton PRP-X200 anion exchange column. XANES was also conducted on P. sulfincola and P. palmiformis and the exopolysaccharide mucus as there was insufficient sample mass to analyse individual R. piscesae. R. piscesae had similar As concentrations (3.8–35 μg g−1) to shallow water polychaetes while P. sulfincola and P. palmiformis have significantly higher As concentrations (420–1417 and 125–321 μg g−1 respectively). R. piscesae contains appreciable quantities of iAs (36 ± 14%), MMA (2 ± 2%), DMA (34 ± 21%), an unknown methyl As compound (7 ± 16%), OSO3-arsenosugar (5 ± 9%), tetramethylarsonium ion (4 ± 5%), thioPO4/thiodimethylarsenic ethanol (1 ± 2%) and an unknown thio-arsenical (12 ± 14%). These results suggests that host and symbionts are either involved in the methylation of As, or are bathed in fluids enriched in methylated As as a result of free-living microbial activity. The host carrying out methylation, however, cannot be ruled out. In contrast, 96–97% of the As in P. sulfincola and P. palmiformis was iAs, likely the result of As precipitation within and upon the mucus they ingest while feeding. While all worms had oxo- and thio arsenosugars (2–30%), Paralvinella also have small amounts of AB (<0.001–0.21%). The presence of arsenosugars, AB and other minor As species in the absence of photosynthesising algae/bacteria indicates that they may be formed by vent animals in the absence of sunlight, but at this time their formation could not be explained. Photochemical VG sample introduction coupled to ICP-MS has been described for the determination of As in seawater.51 A Plackett–Burman design (PBD) and central composite design (CCD) were employed to evaluate the significance of experimental variables relevant to the optimization of photochemical VG-ICP-MS detection. The impact of the saline matrix on the suppression of analyte signal was eliminated by use of a mixture of 20% (v/v) formic and 20% acetic acid (v/v) as the photochemical reductants. Optimized conditions yielded equivalent photochemical VG generation efficiencies for AsIII, AsV, MMAs and DMAs, permitting direct and rapid determination of total As in seawater without any other sample pretreatment. Quantitation was accomplished using gravimetric standard addition along with a spike of 82Se internal standard to compensate for signal drift and fluctuation during analysis. The method provided a LOD of 3 pg g−1 (3σ) and a 15-fold improvement over that obtained using direct solution nebulization, i.e. comparable to that for conventional HG-ICP-MS. Accuracy was demonstrated by analysis of two CRMs (NASS-6 and CASS-5 seawater) and precisions of 3.5% and 3.2% RSD for CASS-5 and NASS-6, respectively were obtained.
The determination of As in foodstuffs continues to be an area of extensive activity. Rice continues to attract considerable attention reflecting its importance as a staple food crop. Various analytical strategies, including sample pretreatment and pre-concentration steps, have been reviewed by Costa et al.52 Although a relatively short review (74 references), the authors critically discuss both the advantages and weaknesses of approaches used in the recent literature. Bralatei et al.53 have reported on the use of a screening method based on the Gutzeit methodology to detect iAs in rice within 1 h. After optimization, 30 rice commodities from the UK market were tested with the field method and then compared to a reference method using HPLC-ICP-MS. Inorganic As species were determined in all but three rice samples. The results showed no bias for iAs using the field method with a LOQ of about 50 μg kg−1, and reproducibility of ±12%. A method using RP ICP-MS for the simultaneous determination of As and Hg speciation analysis has been described.54 The effect of the ion-pairing reagent and pH on the separation of AsIII, AsV, MMA and DMA and HgII, MeHg and EtHg was investigated. The separation and re-equilibration time was less than 20 min following 1% HNO3 microwave-assisted extraction. Calibration curves based on peak area measurements were linear with a correlation coefficient greater than 0.9958 for each species. The LOD values obtained were in the range of 0.84–2.41 μg L−1 for As and 0.01–0.04 μg L−1 for Hg. The proposed method was applied to the simultaneous determination of As and Hg species in rice flour standard material (NIST 1568a rice flour) and two rice samples from local markets. The simultaneous separation and determination of AsIII, AsV, MMA, DMA, AsB, AsC in rice samples have been carried out on a single anion-exchange column by HPLC-ICP-MS.55 The competing anion, ionic strength, pH of elution buffer, and flow rate of mobile phase were investigated using a univariate approach. Under optimum conditions, baseline separation of the six As species was achieved within 10 min using a gradient elution program employing 4 mM NH4HCO3 at pH 8.6 as mobile phase A, and 4 mM NH4HCO3, 40 mM NH4NO3 at pH 8.6 as mobile phase B. The method LOD values for AsIII, AsV MMA, DMA, AsB, and AsC were 0.4, 0.9, 0.2, 0.4, 0.5, and 0.3 μg kg−1 respectively. The proposed method was applied to separation and quantification of As species in rice samples with iAs accounting for over 80% of the total As. Statistical experimental designs have also been employed to optimize the extraction condition of As species (AsIII, AsV MMA, DMA) in paddy rice using water as an extraction reagent.56 A two-level Plackett–Burman factorial design followed by a five-level central composite design were employed to optimize the extraction variables (extraction temperature, shaking time, rotation speed and solvent volume). The separation and determination of As species was achieved using HPLC-ICP-MS. All the As species exhibited linearity with correlation coefficients greater than 0.9995 and LOD values of 0.4, 0.9, 0.6 and 0.8 μg kg−1 were obtained for AsIII, AsV, MMA and DMA, respectively. The optimized method was applied to 2 CRMs (GBW-10043 and GBW-10045) and five rice samples. AsIII, AsV and DMA were detected in all of the rice samples in the order AsIII > AsV > DMA. The iAs concentration ranged from 96.1 to 186 μg kg−1. A spatial distribution map of total As concentration in rice samples grown in the Hunan Province, China has been produced in a study of 43 locally grown samples collected across the region.57 Coupled HPLC and ICP-MS was used to determine AsIII, AsV, MMA and DMA in the samples. The mean concentration of total As in the rice samples was 129.4 ± 49.2 μg kg−1, which is lower than the Chinese maximum contaminant levels of iAs in rice (200 μg kg−1), although higher levels of total As in the rice grains were observed in regions associated with mining activities and urbanization. The dominant species detected in the rice samples was AsIII.
As in previous years, As residues from the use of organoarsenicals, such as 3-nitro-4-hydroxyphenylarsonic acid (roxarsone) to prevent disease and to promote growth in the poultry industry has received attention.58 Although some previous studies have determined As species in chicken litter after composting or after application to agricultural lands, it is not always clear what As species were excreted by the chickens prior to biotransformation of As species during composting. This study reports the identification and quantitation of As species in chicken litter collected on days 14, 24, 28, 30, and 35 of a roxarsone-feeding study involving 1600 chickens of two strains. Species separation by HPLC with simultaneous detection by both ICP-MS and ES-MS provided complementary information to support the identification and quantitation of the As species. A new metabolite, N-acetyl-4-hydroxy-m-arsanilic acid (N-AHAA), was identified accounting for 3–12% of total As. Speciation analyses of litter samples collected from roxarsone fed chickens on days 14, 24, 28, 30, and 35 showed, the presence of N-AHAA, 3-amino-4-hydroxyphenylarsonic acid (3-AHPAA), AsIII, AsV, MMAV, DMAV, and roxarsone. 3-AHPAA accounted for 3–19% of the total As and iAs comprised 2–6% (mean 3.5%) of total As. The authors suggest that the detection of iAs, methylarsenicals, 3-AHPAA, and N-AHAA in the chicken litter support recent findings that roxarsone is actually metabolized by the chicken or its gut microbiome. The presence of the toxic metabolites in chicken litter is environmentally relevant as chicken litter is commonly used as fertilizer.
Although the subject of less publications this year, As speciation in fish (both marine and freshwater) has received attention. A study to evaluate the As and Hg species in muscle and liver of 26 individual fishes of seven freshwater fish species caught in the River Elbe, Germany has been reported.59 Arsenic-containing fatty acids and hydrocarbons were determined and partially identified in methanolic extracts of the fish by HPLC-ICP-MS and ES-Q-TOF-MS. While AsB was the dominant As species in the fish, six arsenolipids were detected and identified in the extracts of liver tissue in common bream (Abramis brama), ide (Leuciscus idus), asp (Aspius aspius) and northern pike (Esox Lucius). Four As-containing fatty acids and two As-containing hydrocarbons were reported in freshwater fish for the first time. The median concentrations of As were 162 μg kg−1 in liver and 92 μg kg−1 in muscle (both wet weight). The same research group have also have reported the synthesis of two new arsenolipids and their identification in fish by HPLC-ICP-MS and HR-MS.60 The molecular structures were elucidated on the basis of their exact mass and their product ion mass spectra. One of the synthesized arsenolipids has been already identified in fish samples, although the other is reported for the first time in fish. A study describing the identification and quantification of five new arsenolipids present in raw marine fish oil extracted mainly from Peruvian anchoveta (Engraulis ringens) has been published.61 The arsenolipids accumulated on bentonite which is used to clean-up raw fish oils in an industrial process for producing commercial fish oil rich in omega-3 fatty acids. The bentonite, which adsorbed the arsenolipids efficiently from the raw fish oil was extracted with a range of different solvents and subsequently cleaned up using normal phase chromatography which fractionated all absorbed compounds according to polarity. The As containing fatty acids (AsFA) and As containing hydrocarbons (AsHC) were determined by RP-HPLC-ICP-MS for quantification and ES-MS for identification. A mixture of methanol/chloroform (1:2 v/v) was sufficient for the extraction of the majority of the adsorbed As species (129 μg g−1 As bentonite). Pre-concentration using the adsorbent with subsequent fractionation made it possible to identify minor arsenolipids especially in the polar methanol fraction. Two major As containing hydrocarbons (C17H38AsO and C19H42AsO), three new As containing medium chain fatty acids of molecular mass 250, 278, 292 and two new As containing long chain fatty acids of mass 306 and 320 could be identified, although their concentrations were low (0.004 μg g−1 As). The presence of medium length chain fatty acids is significant since these compounds do not usually occur as free fatty acids, but conjugated to glycerol forming triglycerides. Confirmation was provided using a methanol extract which was directly analyzed without any clean up procedure and only contained traces of As containing long chain fatty acids. The study highlights that an appropriate pre-concentration and clean up procedure is essential when determining traces of minor arsenolipids, but it will not guarantee the integrity of all arsenolipid species.
A comparison of bioavailability and biotransformation of As species in two marine fish, the herbivorous rabbitfish Siganus fuscescens and the carnivorous seabass Lateolabrax japonicus has been made.62 Little detail of the analytical methodology is provided in the paper which refers back to a previous publication using HPLC-UV-HG-AFS and ICP-MS, although a tuna fish SRM (BCR-627) was used. The rabbitfish were fed with an artificial diet of three macroalgae, whereas the seabass were fed with an artificial diet of polychaete or bivalves for 28 days. The diets contained different proportions of AsIII, AsV, MMA, DMA and AsB. It was found that the trophic transfer factors of As in both fish were negatively correlated with the concentrations of iAs in the diets, but there was no relationship between trophic transfer factors and the AsB concentrations in the diets. Positive correlation was observed between the accumulated As concentrations and the AsB concentrations in both fish, suggesting that AsB was trophically more available than iAs. The biotransformation in seabass was higher than that in rabbitfish, which resulted in higher As accumulation in the seabass. The study demonstrated that different prey with different ratios of As species can result in diverse bioaccumulation of total As in different marine fish. The accumulation of As species (AsIII, AsV, AsC, AsB, MMA, and DMA) in 36 fish species collected from three coastal cities in Korea has been reported.63 The As levels varied substantially, ranging from 0.02 μg As g−1 w/w (Israeli carp) to 9.65 μg As g−1 w/w (Skate ray) with a median of 0.40 μg As g−1 w/w. As with previous studies in this area, AsB (mean fraction: 90.6%) was dominant among the six As species. Fish species with high water contents showed elevated levels of AsIII, but there were no further significant correlations between arsenicals and water/lipid contents. The concentrations of AsV were significantly lower than those of AsIII, which the authors state is due to AsV being reduced during biomethylation of iAs. Consequently they suggest that the toxicity of As (mainly derived from AsIII) can be increased by the reduction of AsV, especially for the fish species with higher water contents.
Arsenic and As species in shellfish and finfish from the western Arabian Gulf have been investigated with respect to consumer health risk assessment.64 The study determined the levels of total As and As species in marine biota such as clams (Meretrix meretrix; N = 21) and pearl oyster (Pinctada radiata; N = 5) collected from nine costal sites, and cuttlefish (Sepia pharaonis; N = 8), shrimp (Penaeus semisulcatus; N = 1), and seven commercially important finfish species (N = 23) collected from seven offshore sites in the western Arabian Gulf. Total As and As species such as DMA, AB, TMAO, AC, TMA, arsenosugar-glycerol (As-Gly) and iAs were determined using ICP-MS and HPLC-ICP-MS. In bivalves, the total As concentrations ranged from 16 to 118 mg kg−1 dry mass with the toxic iAs fraction contributing, overall, less than 0.8% of the total As, while the nontoxic AB fraction formed around 58%. Total As concentrations for the remaining seafood (cuttlefish, shrimp and finfish) ranged from 11 to 134 mg kg−1 dry mass and the iAs and AB fractions contributed on average 0.03% and 81% respectively of the total As. There was no significant relationship between the tissue concentrations of total As and iAs in the samples. There was also no significant relationship between As levels in seafood and geographical location or salinity of the waters from which samples were collected. Based on the results, the authors recommend introducing a maximum permissible level of As in seafood from the Gulf based on iAs content rather than total As. The analyses of health risks identified the greatest risk for consumers of bivalves and high-end consumers of seafood, although many uncertainties remain. A study by Sadee et al.65 has evaluated extraction techniques for As in staple diets (fish and rice) utilising both classical and enzymatic extraction methods. The extraction efficiency using enzymatic extraction was first evaluated for total As by analyzing CRM materials DORM-3 fish protein, NIES 106 rice flour and GBW10015 spinach. These were compared with total As concentration determined using microwave-assisted acid digestion and ICP-MS. The total As concentrations in the CRM materials were in good agreement with the certified values. Enzymatic hydrolysis using trypsin was successfully employed to extract As species in DORM-3 and fish samples. Whilst this method of hydrolysing the proteins worked well for the fish samples, an alternative approach was required to facilitate the digestion of cellulose in plant materials. However, enzymatic extraction using cellulase was found to give unsatisfactory results for both the NIES and GBW10015 CRM materials. A mild acid extractant, 1% HNO3, was found to give a more efficient extraction for As species in the these two CRM materials and rice samples. The study was extended to evaluate a range of real samples. The total As concentrations in 13 different types of fish tissue was then determined following microwave-assisted acid digestion using HNO3/H2O2 and speciation analysis using HPLC-ICP-MS. The results obtained for the fish were in the range of 3.53–98.8 μg g−1 As (dry weight). Similarly, the results of 17 rice samples were in the range of 0.054–0.823 μg g−1. The study demonstrates the importance of selecting an appropriate extraction technique for the quantitative measurement of As species in food.
The extraction of As species from leafy vegetables has been investigated using a combination of different methods (shaking, sonication and microwave), and extractants (ultrapure water, ammonium phosphate buffer and diluted HNO3).66 The extracted As species were separated and determined by HPLC-ICP-MS. Of the methods tested, a microwave assisted method using 1% HNO3 as the extractant provided satisfactory extraction efficiency (>90%) at 90 °C for 1.5 h. This method was then applied to extract As species from 13 different leafy vegetables. The predominant species in all the investigated vegetable samples were AsIII and AsV, although the percentage of AsV reduced with total As concentration, probably due to the conversion and transformation of AsV to AsIII after uptake. A study focusing on locally-grown and frequently-consumed vegetables, such as carrots (Daucus carota), beets (Beta vulgaris) and quinoa (Chenopodium) from the As-polluted Chiu Chiu area in Northern Chile has been reported.67 The region is affected both by As discharge from Cu mining activities and natural As contamination, leading to a high As content in local food and water. The As speciation in the edible part of the vegetables was investigated using LC-ICP-MS. The As bioaccessibility was assessed using an in vitro gastrointestinal digestion and As dietary exposure estimates calculated to assess the risk posed by the vegetables. For carrots, only AsIII and AsV species were found, whilst in quinoa, around 85% of the element was present as AsV, although the extraction efficiency was poor (40%). For beets, iAsV and an unknown As species (probably arsenosugars) were found. No significant transformation of the original As species was observed during the in vitro gastrointestinal digestion. Arsenic dietary exposure values obtained for the three vegetables (0.017–0.021 μg As per person per day) were much lower than the Joint FAO/WHO Expert Committee on Food Additives safety limit of 50 μg As per person per day. Levels of As in mushrooms can vary depending on the concentration level of this metalloid in the soil/substrate. A study to evaluate the As content in Boletus badius fruiting bodies collected from polluted and non-polluted sites has been reported.68 It was found that mushrooms from the As-polluted sites contained mean concentrations ranging from 49 to 450 mg As per kg dry matter, with the greatest content found in specimens growing in close proximity of sludge deposits (490 ± 20 mg As per kg dry matter). The mean total As in mushrooms from clean sites ranged from 0.03 to 0.37 mg kg−1. It was found that B. badius could tolerate As in soil substrate at concentrations in excess of 2500 mg kg−1. Over the 4 years of investigation, there were differences in the As species identified, as well as a general increase in the accumulation of organic As content. The results indicated that B. badius should not be collected for culinary purposes from any sites that may be affected by pollution. Seaweeds produced for consumption have continued to attract attention. A survey of As concentrations and speciation was conducted on 10 species of seaweed from commercial harvesters and from collection at two sites in New England, USA.69 Samples were extracted using HNO3 and microwave digestion prior to determination of the As species by ICP-MS (total As) and HPLC-ICP-MS (As species). Concentrations of As ranged from 4 to 106 mg kg−1, mostly in the form of arsenosugars, with the distribution of arsenosugar analogs varying between taxa. In brown algae, As levels were correlated with phosphate concentrations, and arsenosugar speciation reflected differences in sulfur and phosphate concentrations between taxa. Several samples of the brown algae species Laminaria digitata contained significant levels of iAs (2.8–20 mg kg−1). A weak acid extraction with microwave heating was compared with a methanol: water extraction method, and found to give slightly higher extraction efficiency with comparable relative concentrations of iAs, supporting the use of this faster and simpler extraction method for monitoring. In a comprehensive study to evaluate the movement of As between environmental compartments, As speciation in soil, the broad bean plant (Vicia faba) and irrigation water collected from the same agricultural site in Dokan, in the Kurdistan region of Iraq has been reported by Sadee et al.70 Total As and As speciation were determined in all materials by ICP-MS and HPLC-ICP-MS, respectively. Available As (11%) was also determined within the soil, together with a range of other elements (Cd, Cr, Cu, Ni, Zn, Fe and Mn). The concentrations of total As were: soil (5.32 μg g−1), irrigation water (1.06 μg g−1), roots (2.065 μg g−1) and bean (0.133 μg g−1). Stems, leaves and pods were also measured. Inorganic AsV dominated soil (90%) and root (78%) samples. However, organo-As (MMA, 48% and DMA, 19%) was the more dominant species in the edible bean. The study provided an insight into the uptake, preferred disposal route, speciation changes and loss mechanism involved for As within this plant.
The speciation of As in plants and plant derived materials is still a popular area for research. The determination of As species in the medicinal plant Solanum lyratum Thunb using CE-ICP-MS employing a microflow polyfluoroalkoxy nebulizer has been recorded by Shuai et al.71 Six As species, AsIII, AsV, MMA, DMA, AB and AC were baseline-separated and determined in a single run within 11 min under the optimized separation conditions. The LOD values obtained were in the range of 0.02–0.06 ng mL−1 with RSDs between 2.5 and 4.3% for the six As compounds. Samples were prepared using closed-vessel microwave-assisted extraction. The results showed that AB was the main As species in the Solanum lyratum Thunb samples, with concentrations between 0.42–1.30 μg g−1. The fern Pityrogramma calomelanos is known to be able to hyperaccumulate As, although the mechanisms involved are largely unknown. This subject was the focus of a study by Campos et al.72 who exposed ferns with five to seven fronds to varying As concentrations prior to characterization using GC-MS. As the treatment progressed, the total concentration of proteins and soluble phenols, enhanced peroxidase activities, and promoted disturbances in nitrogen and carbon metabolism were monitored. The reduction of the glucose pool was one of the responses to As. Changes in amino acids levels were also observed in As-treated plants, including those related to biosynthesis of glutathione and phenols, osmoregulation and two photorespiratory intermediates. The study revealed that P. calomelanos tolerates high concentration of As due to its capacity to upregulate biosynthesis of amino acids and antioxidants, without greatly disturbing central carbon metabolism. At extremely high As concentrations, however, this protective mechanism fails to block reactive oxygen species production, leading to lipid peroxidation and leaf necrosis. Arsenic compounds occurring in ruderal plant communities growing in As contaminated soils near former gold or silver mines in the Czech Republic have been investigated for total and speciated As.73 The most abundant plant families were Fabaceae, Lamiaceae, Asteraceae, Poaceae and Plantaginaceae. Total As concentrations in above ground biomass of the plants growing at three sites was determined by an ICP-MS and ranged from 0.02 mg As kg−1 (Stellaria spp.) to 39.30 ± 6.32 mg As kg−1 (Daucus carota L). The concentrations were dependent on both plant species and physico-chemical soil properties. For the As speciation HPLC-ICP-MS was used indicating AsIII and AsV as the prevalent species, although MMA, DMA, AB, AC, TMA and TMAO were identified as minor species. AB was found at significant concentrations in Carex praecox Schreb (28% of the extractable As) and P. lanceolata L. (1.2% of the extractable As). The concentrations of six As species (AsIII, AsV, MMA, DMA, TMAO and TMA) determined in tobacco leaves, ash and smoke of 10 kinds of cigarettes made from tobacco sourced from different countries has been assessed.74 Total As in the tobacco leaves, determined by ICP-MS after microwave-assisted digestion, ranged from 0.20 to 0.63 mg kg−1. Speciation by HPLC-ICP-MS following acid extraction identified AsIII and AsV, together with trace amounts of MMA, DMA, TMAO and TMA and some unidentified As species. The sum of the As species in tobacco leaves, ash and smoke was in reasonable agreement with the result of total As determination in each sample, with recoveries of 100 ± 10%.
The application of different ionic liquids as modifiers for chromatographic separation and determination of AsIII, AsV, MMA and DMA in wine samples, by RP-HPLC-HG-AFS has been reported.75 Factors influencing the chromatographic separation of the As species, such as pH of the mobile phase, buffer solution concentration, buffer type, ionic liquid concentration and length of alkyl groups in ionic liquids were evaluated. The complete separation of As species was achieved using a C-18 column in isocratic mode with a mobile phase composed of 0.5% (v/v) 1-octyl-3-methylimidazolium chloride and 5% (v/v) methanol at pH 8.5. A multivariate methodology was also used to optimize the AFS detection of As species after they were separated. Separation of As species was achieved within 18 min with a resolution better than 0.83. The LODs for of AsIII, AsV, MMA and DMA were 0.81, 0.89, 0.62 and 1.00 μg As L−1 respectively. The method was applied to As speciation analysis in white and red wine samples originating from different grape varieties.
Few clinical studies on As speciation have been reported this year. An HPLC-ICP-MS method has been developed to determine 13 common As metabolites possibly present in humans with special attention directed at thiolated or thiol conjugated As compounds.76 The thirteen species included in the study were AsIII, arsino-glutathione, AsV, MMAIII, monomethylarsinoglutathione, MMAV, DMAIII, S-(dimethylarsinic)cysteine, dimethylarsino-glutathione, DMAV, dimethylmonothioarsinic acid, dimethyldithioarsinic acid, and dimethylarsinothioyl glutathione. Following optimization of various mobile phases, significant baseline disturbance was observed due to high levels of acetonitrile present in the gradient elution program, although adjustment of the nebulizer flow rate reduced the problem. Under optimum conditions, a LOD of 1.25 g L−1 was quoted as indicative of all As species (individual LODs are not presented) and similarly the LOQ is quoted as 2.5 g L−1 for all species. The method was found to be linear between the tested ranges of 1.25–2500 μg L−1. The developed method was applied for the analysis of cancer cells that were incubated with darinaparsin (dimethylarsino-glutathione), a novel chemotherapeutic agent for refractory malignancies. A major cause of toxicity of AsIII compounds is reported to be due to their interaction with the sulfhydryl groups in proteins, particularly metallothionein. A study to investigate the sequestration of As ions by metallothionein to provide protection against acute arsenic toxicity has been reported by Garla et al.77 A rat model study on the metal stoichiometric analysis of MT1 isoform isolated from the liver of As treated, untreated and Zn treated animals was carried out using the combination of PIXE and ES-MS. The results revealed the absence of bound As in the samples isolated from As treated animals, although, both Cu and Zn ions were present in MT1 samples isolated from all the treatment groups. These authors suggest that the role of metallothionein during acute As toxicity is different from its already established role in case of Cd toxicity.
The speciation of As in body fluids has been reported by a number of research groups. The total and speciated urinary As levels in the Spanish subjects (124 volunteers aged 20–76 years; 88 women and 36 men) has been studied.78 Demographic, lifestyle and dietary data was collected from the volunteers who were then tested for total As and AsIII, AsV, MMA, DMA and AB using HPLC-ICP-MS. AB and DMA were detected in 96.8% of the study participants (LOD 1.0 μg L−1 for AB and 1.9 μg L−1 for DMA). MMA and AsIII were detected in 5.6% (LOD 1.8 μg L−1) and 1.6% (LOD 1.4 μg L−1) of the participants, respectively; AsV was not detected (LOD 1.4 μg L−1). AB and DMA were the main contributors to total urinary As levels and urinary DMA was positively associated with AB. The total As levels observed in the Spanish population sample were higher than those reported in other European studies. The most recurrent urinary As species was AB, followed by DMA, probably attributable to the high Spanish consumption of seafood. The sum of urinary iAs, MMA, and DMA concentrations are often used for the biological monitoring of occupational iAs exposure. Although DMA is a major metabolite of iAs, it is an inadequate index because high DMA levels are present in urine after seafood consumption. To study this, Hata et al.79 have estimated the urinary iAs + MMA concentration corresponding to iAs exposure. Data was collected from speciation analyses (HPLC-ICP-MS) of urine samples from 330 Bangladeshi with oral iAs exposure and 172 Japanese workers without occupational iAs exposure using. The results indicated that iAs, MMA, and DMA, but not AB could be detected in the urine of the Bangladeshi subjects. The regression relationship between iAs + MMA + DMA and iAs + MMA was obtained as log(iAs + MMA) = 1.038log(iAs + MMA + DMA) + 0.658. Using the regression formula, the iAs + MMA value was calculated as 2.15 and 7.5 μg As L−1, corresponding to 3 and 10 μg As m3 of exposures, respectively. In the urine of the Japanese workers, As was mostly excreted as AB. The authors used the 95th percentile of iAs + MMA (12.6 μg As L−1) as the background value. The sum of the calculated and background values was proposed as a biological indicator of iAs exposure. However, other studies have questioned the use of As metabolites patterns for prediction. AsIII in solution has been shown to be the most active single agent in combating acute promyelocytic leukemia (APL). It is metabolized and excreted via urine as MMA, DMA and AsV, along with excess AsIII. In a study by Slejkovec et al.80 eight APL patients were treated (intravenously) with 0.15 mg As2O3 per kg per day. During the therapy, AsIII and its metabolites were followed in pre- and post-infusion urine using HPLC-HG-AFS. Five patients had a normal excretion pattern of residual As compounds in morning pre-infusion urine, with 15–25% of AsIII, 35–55% of DMA, 25–30% of MMA and 1–5% of AsV, while three patients showed unexpected exceptions from typical excretion patterns of As compounds (i) a high DMA/MMA ratio (factor 5.3), (ii) severe AsIII oxidation (10.2% AsIII converted to AsV) or (iii) the presence of an excessive amount of AsIII (average 30.4% of total As). An intriguing finding was the occurrence of post-infusion oxidation of AsIII to AsV observed in almost all patients and being especially high (>40%) in patient with increased residual AsV. The results indicated that As metabolites patterns can be unpredictable. Observed high levels of un-metabolised AsIII are a warning signal for side effects and for routine determination of As metabolites during first days of treatment. High or low percentages of MMA or DMA did not show any observable effect on treatment results, while clear presence of post-infusion AsV supports theoretical claims of in vivo oxidation (detoxification) of AsIII to AsV associated with various metabolic processes. Chen et al.81 have also studied the use of AsIII in treating acute promyelocytic leukemia, in this case constructing a metabolomics profile of the effect of AsIII on the gastric carcinoma cell line SGC7901. The global metabolite profiles were monitored by metabolomics analysis using GC-MS and LC-MS-MS. A total of 281 certified metabolites were detected.
Arsenic toxicokinetics are important for disease risks in exposed populations, but genetic determinants are not fully understood. The Strong Heart Study82 has examined urine As species patterns (% iAs, % MMA, and % DMA) measured by HPLC-ICP-MS among 2189 participants 18 years of age and older. The genome-wide linkage study also suggested genetic determinants of As toxicokinetics to be identified by future fine-mapping, and illustrated the utility of PCA as a novel approach that considers % As species. To improve our understanding of the metabolism of iAs in animals, Chen et al. conducted an animal feeding study with an emphasis on identifying new As metabolites.83 Female F344 rats were given 0, 1, 10, 25, 50, and 100 μg g−1 of AsIII in the diet. Arsenic species in rat urine were then determined using HPLC-ICP-MS and ES-MS-MS detection. Nine As species were detected in the urine of the rats. Seven of these As species were consistent with previous reports, AsIII, MMA, DMA, TMAO, monomethylmonothioarsonic acid, and dimethylmonothioarsinic acid. Two new methyldithioarsencals, monomethyldithioarsonic acid (MMDTAV) and dimethyldithioarsinic acid (DMDTAV), were reported. MMDTAV has not been identified previously in biological samples of animals, and DMDTAV has not been reported as a metabolite of iAs in rats. The concentrations of both MMDTAV and DMDTAV in the rat urine were dependent on the dosage of AsIII in diet. The concentration of DMDTAV was approximately 5 times higher than that of MMDTAV. A study to evaluate the influencing factors of As metabolism in children has been reported.84 Urinary concentrations of iAs, MMA, and DMA measured using HPLC-HG-ICP-MS were found for 488 children (9 years old), whose mothers had previously participated in a study on As exposure during pregnancy in rural Bangladesh. The methylation efficiency was assessed by the relative amounts (%) of the metabolites and the impact of factors such as maternal urinary metabolite pattern, As exposure, gender, socioeconomic status, season of sampling, and nutritional factors, including erythrocyte selenium (Ery-Se), and plasma folate and vitamin B12 were evaluated. Children had higher % DMA and lower % iAs in urine compared to their mothers, unrelated to their lower exposure (median urinary As 53 vs. 78 μg L−1). The plasma folate was inversely associated with % iAs and positively associated with % DMA. The children methylated As more efficiently than their mothers, although the subject warrants further research. Dietary predictors of low-level iAs exposure in 357 children (5–8 year old) from Montevideo, Uruguay have also been reported.85 328 children collected morning urine samples, and 317 had two 24 h dietary recalls. Urinary As metabolites (iAs, MMA and DMA) were measured using HPLC-HG-ICP-MS as a function of food groups, dietary patterns, nutrient intake, and nutritional status. Exposure to As was low (median 9.9 μg L−1) and household water (water As: median 0.45 μg L−1) was not a major contributor to exposure. Children with higher consumption of rice had higher urinary As but lower % iAs, % MMA, and higher % DMA. Children with higher meat consumption had lower % iAs and higher % DMA. Higher scores on “nutrient dense” dietary pattern were related to lower % iAs and % MMA, and higher % DMA. Higher intake of dietary folate was associated with lower % MMA and higher % DMA. Overweight children had lower % MMA and higher % DMA than normal-weight children. In summary, rice was an important predictor of exposure to iAs and DMA. Higher meat and folate consumption, diet rich in green leafy and red-orange vegetables and eggs, and higher BMI contributed to higher As methylation capacity. The possible association between iAs exposure and semen quality (semen volume, sperm concentration, and sperm motility) has been explored in male partners of couples (n = 42) who visited a gynecology clinic in Tokyo for infertility consultation.86 Semen parameters were measured according to WHO guideline at the clinic, and urinary iAs, MMA, and DMA concentrations were determined by LC-ICP-MS. Biological attributes, dietary habits, and exposure levels to other chemicals with known effects on semen parameters were also taken into consideration as covariates. Multiple regression analyses and logistic regression analyses did not find the iAs exposure as significant contributor to semen parameters.
Studies on the mobility of As species through the terrestrial landscape have been fewer in number this year. The study of alluvial aquifers contaminated by geogenic As often lack direct solid-phase speciation data, hindering efforts to understand landscape-scale spatial heterogeneity and controls on As mobility. Johnston et al.87 have examined solid-phase As and Fe speciation of alluvial aquifer, river and hyporheic-seep sediments along a topographic gradient at the Himalayan foothills/upper-Ganges floodplain margin. They employed a combination of XAS, selective extracts, electronmicroscopy and XRF. Source-river sediments were found to contain a mixture of solid-phase AsV and AsIII species, while vertically heterogeneous bore-hole sediments also contained a solid-phase As-sulfide species. In general, the abundance of reduced As species [AsIII + As-sulfide] increased with depth below ground surface, as well as down the topographic gradient from the foothills to the floodplain. Although FeIII oxides diminished with depth, goethite-rich nodules were present above the seasonal water table minima and contained 10–100 fold more As than the corresponding bulk sediments (50–800 mg kg−1 mainly as AsV). In contrast, organic-rich clay layers below the seasonal water table contained abundant As-sulfide species (30–80% of solid-phase As), as well as authigenic pyrite enriched in As (up to 8500 mg kg−1). The authors report that the aquifer sediments contained discrete facies that are strongly enriched in As due to post-depositional diagenetic coupling between As retention and redox-dependent Fe mineralization. In contrast with the lower Gangetic plain, where large-scale floodplain sedimentary features are a key control on solid-phase As and Fe speciation, in these alluvial sediments solid-phase As and Fe speciation appeared to be controlled mainly by physiographic properties (elevation; depth; seasonal water table) that influence local redox conditions. A study to elucidate the role of cyanobacteria in the biotransformation of As oxyanions in mining areas has been reported.88 The EC50 for AsIII and AsV were calculated and the biotransformation of As by Synechococcus sp. evaluated using HPLC-ICP-MS and XAFS. Synechococcus sp. exhibited a higher sensitivity to AsIII with an EC50, 96 h of 6.64 mg L−1 that was approximately 400-fold lower than that for AsV. Even though the cells were exposed to concentrations of AsIII (6 mg L−1) approximately 67-fold lower than those of AsV (400 mg L−1), similar intracellular concentrations of As (60.0 μg g−1) were observed after 30 days. AsV was the predominant intracellular As species followed by AsIII, although MMA and DMA were observed in higher proportions after exposure to AsIII. The differential toxicity among As oxyanions indicated that determining the redox state of As in the environment is fundamental to estimate toxicity risks to aquatic organisms.
Among the more unusual applications reported this year for As speciation was the visualization of AsIII and AsV distributions in degraded paint micro-samples from Baroque- and Rococo-era paintings.89 The As sulfide pigments orpiment and realgar are used for their vivid yellow and red-orange hues, but both potential degrade with exposure to light. This often results in whitening or transparency in the painted surfaces. While conventional techniques such as μRaman and μFTIR spectroscopies can allow a quick and relatively easy identification of the pigments, the identification and distributions of the degradation products is more challenging due to an unfavourable LOD and low spectral resolution. This restricts the conclusions that can be drawn regarding the conservation state of valuable works of art. In this study the authors used SR-μXRF mapping on a visually degraded orpiment-containing paint stratigraphy revealing that the As is distributed throughout the entire cross-section, while XANES demonstrated that the As is present in both AsIII and AsV forms. The latter compounds, were not only located at the surface of large and partially altered grains of As sulfide but also spread throughout the entire paint stratigraphy. Their presence and distribution are attributed either to the complete degradation of smaller As sulfide grains or to migration of the AsV within the paint layer away from their original location of formation.
Table 2 shows applications of As speciation presented in the literature during the time period covered by this ASU.
3.3 Chromium
The numbers of papers describing Cr speciation analysis published in the current review period has almost doubled (to 34) from the numbers published in the period covered by last year’s update. Only one of the current papers is a review article. Trzonkowska et al.106 have reviewed recent advances in on-line extraction methods for speciation analysis of environmental matrices. The bulk of the 65 articles (with titles) cited describe SPE methods, which the authors have categorized into eight different schemes depending on how any columns (one or two) and which specie(s) is/are retained. The nature of the extractant materials is also described in some detail, as are developments in miniaturization and automation. There is a shorter section devoted to LLE, a technique that is harder to adapt to the on-line format. The reviewers conclude by pointing out that microfluidic systems, which promote green chemistry principles, are likely to be further developed for measurements in the field. They highlight some recent developments of new materials with greater improved selectivities and capacities, which they consider will help with achieving the LODs needed to determine CrVI at concentrations that are set by some regulatory agencies (e.g. 20 ng L−1), which many published methods did not achieve. They also point out that although the availability of CRMs is limited, their use should be an integral part of the evaluation of accuracy and laboratory performance, concluding that, unfortunately, in many reports of method development this aspect of the validation is neglected.The number of reports of methods based on HPLC separation has increased over that of the previous review period. It is possible that Cr will join the group of elements, exemplified by As, for which there would appear to be an ever-increasing number of chromatographic separations of species of interest with no sign of convergence on one particular procedure. Many of these procedures have been applied to samples other than waters, and so the methods contain an extraction step, which although increasing the complexity may also be an opportunity for additional selectivity. However, there are at least three reports of Cr speciation in waters by HPLC with ICP-MS detection. Markiewicz et al.107 measured total Cr in two CRM waters, but were unable to detect CrVI in either of these; nor could they detect either species in five drinking water samples. Results are presented for the development of three methods, two of which involve ID with 53CrVI. Much of the article is taken up with a discussion of the preparation and use of the isotopically enriched standard material, whereas there is minimal discussion of the chromatography, which was carried out isocratically on a C-8 Brownlee cartridge column (PerkinElmer, Pecosphere, 3 μm diameter particles, 4.6 mm i.d. × 33 mm length) with a mobile phase of 1.2 mmol L−1 EDTA + 0.8 mmol L−1 tetrabutyl ammonium hydroxide. The LOD values for CrVI of the two HPLC procedures were 20 and 100 ng L−1 for the ID procedure and the non-ID procedure respectively. For total Cr, the LOD was 9 ng L−1. For all the methods, the instrument was operated in DRC mode with ammonia as the reaction gas. Recoveries of spikes of CrVI at concentrations of 2 and 12 μg L−1 are reported, but percentages are not calculated. There is considerable discussion of the calculation of the overall uncertainty in the results. Jin et al.108 devised a method for the analysis of waste waters in which the Cr species were separated by ‘low-pressure IC’ and detected by ICP-MS. The procedure is really selective SPE of CrVI on a styrene divinylbenzene material modified with quaternary ammonium synthesized in-house. No details of this procedure are given, nor is there a reference to previous work. A 50 μL sample was injected and the CrIII detected directly by the spectrometer; the retained CrVI was washed off with dilute sodium nitrate solution at pH 7–8 flowing at 2 mL min−1. The LOD values ranged from 3 to 10 ng L−1. Validation was by (a) spike recovery from three tannery effluent samples and (b) by comparing the results obtained for the same samples with those obtained by a procedure in which total Cr was determined by ICP-MS and CrVI by spectrophotometry with diphenylcarbazide (and hence CrIII by difference). Jia et al.109 improved the LOD of an HPLC-ICP-MS method with pre-concentration by SPE on poly(chloromethyl styrene-co-styrene) functionalized with N,N-bis(2-aminoethyl)ethane-1,2-diamine, 180 mg of which was contained in a small column mounted in the loop of the injection valve. Samples were prepared by filtering (0.22 μm) and adding EDTA to convert the cationic CrIII to the anionic complex so that both species were retained. The species were washed off the pre-concentration column with mobile phase (10 mM TBAH, pH 3.5) and separated by AEC (Aichrom C-18 reversed phase column 50 mm × 4.6 mm id, 5 μm packing material) in 100 s. Interferences were decreased by the use of a reaction cell with He as the gas and by not using methanol in the mobile phase. Enrichment factors for a 30 mL sample volume (loaded at 8 mL min−1) of 105 and 128 for CrVI and CrIII, respectively were obtained leading to LODs of 0.007 and 0.004 ng mL−1. The developed method was validated by analyzing a seawater CRM GSBZ50027-94, which contains CrVI at a concentration of 120 ng mL−1 and by spike recoveries of between 0.5 and 5.0 ng mL−1 from three wastewater samples. All of which contained measureable concentrations of both species.
A “water-like” matrix of spiked exhaled breath condensate has also been analysed110 by an HPLC-ICP-MS method in which EDTA was added to the samples to convert CrIII to an anion, which was then separated by LC along with CrVI, on a short column of Dionex IONPAC AG7 (4 mm × 50 mm i.d. 10 μm particles), which has both cation and anion exchange capabilities, due to the presence of sulfonate and alkyl quaternary ammonium functional groups, respectively. Recoveries of 5 μg L−1 spikes, measured at four intervals over a six-week storage period, were 91% and 95% for CrIII and CrVI, respectively. For a 500 μL sample volume, the LODs were 6 and 2 ng L−1. Stability studies showed that CrVI was not stable, even when frozen, in aqueous standards, but was stable in the presence of EDTA as long as the pH did not drop below 8. It is not clear whether the researchers investigated the effect of freezing on the stability of CrVI spiked into the six exhaled breath condensate samples collected. No results were presented for the Cr content of the samples, and so it is concluded that it was not possible to detect Cr in these samples. The instrument was operated with a collision cell into which a mix of helium (7%) and hydrogen was introduced, and the isocratic mobile phase was described as ‘4% v/v ammonia solution + 3.2% v/v nitric acid, pH 1.8–2,’ which is presumably equivalent to a mixture of nitric acid and ammonium nitrate. The instrumentation was also described as a ‘micro-LC system,’ but the mobile phase flow rate was 1 mL min−1 and the separation was completed in 5 min. The method was also validated by the satisfactory analysis of a diluted (1000-times) proficiency test material from Sigma Aldrich (UK) for CrVI in drinking water (WS13-4 lot LRAA1427), which had a target value of 2.81 μg L−1 and an acceptable range of 2.28–3.28 μg L−1. Calibration standards, samples and quality control material, which were diluted 10-fold with 0.5 mmol L−1 EDTA solution adjusted to pH 8 with 10% v/v ammonia solution, were stable for 6 weeks when stored refrigerated. In a study of the Cr speciation in tobacco, including smoke condensate, Cuello et al.111 separated the Cr species by ion-pair reversed-phase chromatography on an Agilent PLRP-S polymeric reversed phase column made of PEEK (150 mm × 4.6 mm, 3 μm) with a mobile phase containing 0.18 mM tetrapropylammonium bromide (ion-pair agent) and 1 mmol L−1 EDTA at pH 8. The injection volume was 50 μL at 40 °C and a flow rate was 0.8 mL min−1 with a run time of 20 min. Prior to injection, EDTA was added and the sample heated to 70 °C for 30 min to ensure the formation of CrIII–EDTA. The spectrometer was operated in collision cell mode with He as the collision gas with a microflow quartz concentric nebulizer and Scott-type double-pass spray chamber cooled to 2 °C. The LOD values for CrIII and CrVI in the solution injected were 0.1 and 0.08 ng g−1, respectively. Chloride based interferences, such as that due to 35Cl16O1H+, were not a problem as chloride eluted in the void volume. Cut tobacco was also analyzed and the method was validated by spike recoveries. For the determination of total Cr, the procedure was validated by the analysis of NIST SRM1575 (pine needles), which contains 2.6 μg g−1. Additional characterization of the Cr species in cigarette tobacco was made by fractionation based on a variety of extractants and it was concluded that 12% of the total Cr was associated with silicates. In addition, in situ XANES analysis showed that the tobacco and the smoke condensates contained only CrIII, though spiking studies showed that CrVI was not stable in the latter sample.
A number of studies of Cr speciation in foodstuffs have been undertaken. For the analysis of edible animal oils, Lin et al.112 separated the Cr species by reversed-phase ion-pair HPLC on a Perkin-Elmer C8 silica based column (3.0 mm × 33 mm, 3 μm) with isocratic elution by 0.5 mmol L−1 tetrabutylammonium phosphate (TRAP) and 0.3 mmol L−1 EDTA in 1% methanol at pH 6.9. The injection volume was 100 μL with an elution flow rate was 1 mL min−1 and a separation time of 110 s. The instrument was run in DRC mode with ammonia as the reaction gas, and the LODs were 0.04 and 0.05 ng mL−1 for CrIII and CrVI, respectively. The method, which was validated by the analysis of a CRM (NRCC SLRS-3 Riverine Water) which contains 0.33 ng mL−1 total Cr, was applied to three edible animal oils, from which the Cr species were extracted by microwave heating with 0.4% v/v HF + 2% Triton X-100 + mobile phase. No CrVI was detected, but a large peak close to the void volume was interpreted as a possible Cr fatty acid complex. Spike studies showed that CrVI was not stable in the oils. Unceta et al.113 extracted the Cr species from dietary supplements by hot 50 mmol L−1 EDTA and, following centrifuging and filtering, separated them by AEC with ICP-MS detection. The column was a Metrosep A Supp 5 250 (250 mm × 4.0 mm, 5 μm), the injection volume was 100 μL and the mobile phase was 2 mmol L−1 EDTA at pH11 at a flow rate of 0.7 mL min−1. The separation time was 10 min. The LOQ in solution was 0.4 ng g−1, corresponding to 80 ng g−1 in the solid samples. A study of calibration procedures showed that external calibration by monitoring 52Cr gave results not significantly different from those obtained by ssIDMS, in which 50CrIII and 53CrVI spikes were added. Total Cr was determined following microwave-assisted digestion with concentrated nitric acid. The procedure was applied to 11 dietary supplement samples, in five of which CrVI was detected. The spiking studies showed that there was no interspecies conversion, apart from the reduction of CrVI at very high concentrations of CrVI, which are not expected in dietary supplements. To determine Cr in traditional Chinese medicines, Li et al.114 separated the extracted species by AEC with ICP-MS detection. The column was an Agilent Bio WAX, (250 mm × 4.6 mm, 5 μm), the injection volume was 50 μL (or maybe 100 μL, both values are given), and the mobile phase was 75 mmol L−1 ammonium nitrate at pH 7 flowing at 1 mL min−1. Preliminary experiments with a reversed-phase ion-pair system did not give satisfactory results, mainly because of the high background signal from 40Ar12C+, which could not be alleviated by the use of the octopole reaction system (with He). Despite the fact that most Chinese traditional medicines are “taken in the form of decoction in water,” (meaning that the active ingredients are extracted by boiling water), samples were extracted at room temperature with a solution containing magnesium chloride, phosphate buffer, sodium hydroxide and sodium carbonate (pH not given), but assumed to be alkaline. The researchers investigated both the possible reduction of CrVI and oxidation of CrIII and chose conditions to minimize both. The LOD was 0.1 μg L−1, which would correspond to 5 μg kg−1 in the solid, assuming 100% extraction. The chromatographic fate of CrIII is not shown; the only chromatograms presented contain a peak at around 6 min, attributed to CrVI, and a peak at about 2 min, attributed to “interferences”. Neither of the two samples tested contained measurable concentrations of CrVI. Spiking of the extracts showed that CrVI was stable for up to 60 h. The authors conclude that the alkaline extraction procedure used yielded quantitative recovery of CrVI in the present of a large amount (sic) of CrIII, but no results are presented to support this. Also of possible interest, is the development of a method for the determination of Cr species in mineral mixtures and animal feeds by HPLC.115 The procedure involved derivatization of both CrIII and CrVI with APDC, and as the detection was by UV spectrophotometry (injection volume of 20 μL), the LOD values were 200 and 70 μg L−1. Separation was achieved on a Phenomenex polymer-based reversed-phase column (Kinetex, 250 mm × 4.5 mm, 5 μm) at 50 °C with a mobile phase of acetonitrile and water (7 + 3), which is incompatible with ICP-MS detection. A similar comment might be made about 5% methanol in 25 mmol L−1 phosphate buffer at pH 6.5, which was the mobile phase in an AEC method with UV detection.116 The stationary phase was silica functionalized with N-methylimidazolium-chloride ionic liquid on which CrIII was separated by chromatography as the anionic chelate with potassium hydrogen phthalate (CrVI was assumed to be present as an anion). The LODs were 10 and 200 μg L−1 for CrIII and CrVI, respectively. The method was applied to one tap water and one groundwater sample. Cr was not detected in either sample which could, possibly be as a consequence of the dilution involved in sample preparation and the 20 μL sample volume. Results for spike recoveries of up to 500 mg L−1 are included. Readers of Chinese may be interested in an account of an HPLC-ICP-MS method for the determination of CrIII and CrVI in “duplicate diet,” from which the species were extracted by hot 5% tetrabutyl ammonium hydroxide solution.117 No details of the chromatography are given in the abstract, but the LOD in the solid samples were 0.3 and 0.4 μg kg−1 for CrIII and CrVI, respectively. Both species were detected in all 60 samples analyzed.
Saxena and co-workers have continued their development of procedures based on the on-line SPE of CrIII.118,119 Their method is based on the modification of Amberlite XAD-16 resin with a chelating agent capable of retaining CrIII, which is then desorbed by dilute acid and quantified by FAAS. For 1,10-phenanthroline and 120 s accumulation, the LOD was 0.09 μg L−1,118 and for α-benzoin oxime and 300 s pre-concentration, the LOD was 0.1 μg L−1. The total Cr was determined after reduction of CrVI with hydroxylamine hydrochloride, and hence CrVI was determined by difference. The methods were validated by the analysis of NIST SRM 1643e (trace elements in water) and applied to (the same) three industrial water samples, all of which contained measurable concentrations of both analytes. The samples were acidified on collection, filtered prior to analysis, and the pH adjusted to 5.0. No details of the pH adjustment steps are given in either paper. A similar procedure has been devised by Lesniewska et al.120 in which CrIII was retained at pH 9.0 on a material containing 1,5-diphenylcarbazone embedded in a polymeric matrix. Following elution with EDTA solution the Cr was determined by FAAS. The LOD was 30 μg L−1 and the procedure was applied to the analysis of a CRM wastewater (RES 10.2) and to two sewage samples, neither of which contained CrIII at measurable concentrations. No details were given of how CrVI was reduced to CrIII, or of the effect of sample volume.
Two methods based on dispersive SPE have been described for the analysis of waters. In the first121 CrIII was selectively retained on a single-use chitosan or poly(vinyl alcohol) film loaded with silver nanoparticles (synthesized in-house) in a batch extraction from 20 mL of sample at pH 8.5. Measurement of the Cr concentration before (total) and after (CrVI) by ICP-MS allowed the CrIII to be calculated by difference. The LOD was 0.02 μg L−1 for both species. The method was validated by the accurate determination of CrVI in two CRMs [CrVI WS from Sigma-Aldrich and CrVI in sea water (Fluka® Analytical)] and by spike recoveries from six real samples, five of which contained measureable concentrations. In the second method,122 Pytlakowska determined Cr by energy dispersive XRF spectrometry following extraction of CrIII on graphene oxide NPs, purchased from an external supplier. To a 50 mL sample solution, 1 mL of a 500 μg mL−1 suspension of graphene oxide was added. The pH was adjusted to 6, the mixture stirred at 700 rpm for 5 min and filtered (no details given, other than the diameter was 5 mm). The filter was then dried under an IR lamp. Calibration over the range 1–150 ng mL−1 was done by taking standards through the same procedure. The LOD was 0.06 ng mL−1, and CrVI was determined after reduction to CrIII with acidified ethanol. The procedure was validated by the analysis of NIST SRM 1640a (trace elements in natural water) and by spike recoveries from five real samples. Of the 10 possible analyses, 7 were below the LOD of the method.
Several methods based on DLLME have been developed, one of which features quantification by TXRF123 following separation by selective extraction, from a 7.5 mL sample volume, of the CrVI complex with APDC into 30 μL of carbon tetrachloride (in methanol as the disperser solvent) at pH 2; whereas at pH 6, both the CrVI and CrIII complexes were extracted. Complete formation of the CrIII complex was achieved by doubling the APDC concentration and heating to 80 °C for 30 min. No details of how the pH was adjusted to 6 are provided. The organic phase was separated by centrifuging at 3500 rpm, and 10 μL was mixed with 10 μL of a 10 mg kg−1 Mo internal standard solution in carbon tetrachloride and 10 μL was dried on a pre-heated quartz glass sample carrier with an IR lamp. The procedure was calibrated by taking standards through the entire procedure. The LOD was 0.8 μg L−1 and the working range was from 5 to 4000 μg L−1. The method was applied to five water samples, but CrIII and CrVI were only detected in the wastewater; the concentrations in the mineral and river waters were below the LOD. The method was validated by spike recoveries from these samples. Gaubeur et al. detected124 the Cr by surface-enhanced LIBS (described in earlier publications from this research group) from an Al substrate to which 10 μL of the organic solvent had been applied and then dried. There are no details of this stage of the procedure, though the rest of the LIBS system is described in considerable detail. The CrVI complex with DDTC was extracted into 50 μL of undecanol (with ethanol as the disperser solvent). A mass of 1000 mg of sample was taken to which was added 522 mg of a 1% DDTC solution and 900 mg of 0.1 M sulfuric acid followed by dilution to 9000 mg. In a separate sample, CrIII was oxidized by acidified permanganate, and total Cr was determined. Calibration was performed by taking standards through the procedure. The only validation was the somewhat inaccurate analysis of a hard water CRM (ERM CA011a from LGC Standards), which contains 49.3 μg L−1 CrIII (and has density of 1 g cm−3). The LOD was 3 μg kg−1, which, while better than that of LIBS analysis of the dried water samples, is considerably worse than those of many previously published procedures. The researchers concede this is most likely due to, the poor precision of the LIBS technique. It is difficult to see that this approach has much merit. Meeravali et al. determined125 Cr in the trichloroethylene extracts by ETAAS after mixing with methanol containing 2% (v/v) nitric acid. For CrVI, the pH was adjusted to 2 (at this pH Cr is present as the anionic HCrO4), and the ion-pair with cationic quaternary ammonium Aliquat-336 was extracted; whereas CrIII (present as cation) was extracted as the ion-pair with anionic dioctylsulfosuccinate. For total Cr determination, both ion-pair agents were added. The LODs for CrVI, CrIII and total Cr were 0.5, 0.6 and 0.7 pg mL−1, respectively. The method was validated by the accurate analysis of CRMs (BCR 714 industrial influent, BCR 715 industrial effluent and NIST 1640 natural water), and was applied to the analysis of two river waters, two lake waters, and three tannery effluents. In all samples, both Cr species were detected at concentrations above the LOD of the method. Sadhegi and Moghaddam126 also determined Cr by ETAAS, following extraction of the APDC complexes in the ionic liquid 1-hexyl-3-methylimidazolium hexafluorophosphate, which was formed in the presence of the sample by the sequential addition to the buffered sample solution of 1-hexyl-3-methylimidazolium tetrafluoroborate followed by potassium hexafluorophosphate. The quantities were such that only about 20 μL of the extractant phase was collected at the bottom of the vessel. After removal of the aqueous phase, the extractant was diluted to 100 μL with ethanol and 20 μL transferred to the electrothermal atomizer. The mechanism of discrimination is not entirely clear: CrVI was extracted first under a given set of conditions of reagent concentration (0.2% m/v) and heating time (5 min). The CrIII was extracted by increasing the reagent concentration to 0.5% and the heating time to 25 min. For both species the pH was 5 (phosphate buffer) and the temperature 45 °C. The LODs were 4 ng L−1 for CrIII and 2 ng L−1 for CrVI. The method was applied to the determination of CrIII and CrVI in one tap water and one groundwater, in which both species were detected. Results are also presented for spike recoveries. Much of the report is taken up with a description of the optimization strategy, which consisted of several steps: (1) screening the procedure variables and their experimental domains using a two-level fractional factorial design, (2) a central composite design with response surface methodology to develop a model to estimate the relationships between the responses and the significant variables, and (3) use of the desirability function to select the experimental variables to maximize the extraction of one Cr species, while minimizing that of the other species. Shirkhanloo and coworkers have devised two methods for the determination of Cr species in blood.127,128 In the first procedure, CrVI was selectively extracted at pH 7.5–8.0 as an ion-pair into what the researchers call “task specific ionic liquids,” two of which (1-(3-aminopropyl)-3-methylimidazolium hexafluorophosphate and 1-butyl-3-methylimidazolium tetrafluoroborate) were needed. The disperser solvent was acetone and Triton X-100 was added as an “anti-sticking agent”. The dispersion/extraction processes were aided by ultrasound (no details given), the droplets were aggregated by centrifugation, and following removal of the aqueous phase, the Cr was back extracted into 0.2 mL of 2 M nitric acid and determined without matrix modification. Total Cr was determined following oxidation of Cr with hydrogen peroxide. The LOD was 5 ng L−1 and the method was validated by the accurate analysis of NIST SRM (bovine serum), which contains 0.14 μg L−1 of Cr and by spike recoveries of each species from four whole blood and one serum sample. In all samples, both species were detected. In the second method,128 CrIII was selectively extracted, at pH 5–8, as the complex with (2R)-2-acetamido-3-sulfanylpropanoic acid (also known as acetyl cysteine) into 1-hexyl-3-methylimidazolium hexafluorophosphate. Acetone was again the disperser solvent and Triton X-100 was also added. Finally the Cr was extracted into a small volume of dilute nitric acid. The LOD was again 5 ng L−1, and the method was validated by the accurate analysis, for total Cr, of three CRMs, GBW (E) 90006 bovine serum; NIST CRM 2668 frozen human urine; LGC-CRM sero-210105 human whole blood, and by the spike recoveries from two blood, two urine and two serum samples. For the CRMs validation was by the sum of the species detected. The researchers were also able to conclude that cysteine, produced in the intestine when acetyl cysteine is administered orally as a pharmaceutical compound, can decrease the concentration of CrVI in blood.
Several methods based on selective LLE have been developed. Fakhriyan et al.129 selectively extracted CrVI as the complex with diethyl dithiocarbamate, from a 25 mL sample, into a drop of octanol placed into the vortex of the stirred solution. Of the original 173 μL, about 70 μL was collected to which 30 μL of nitric acid + ethanol was added. The drop was subsequently analyzed by FAAS with introduction by a home-made micro-sampling device, for which some details are provided. Total Cr was determined after oxidation of CrIII with acidic permangate solution and CrIII determined by difference. The LOD values were 1 and 2 ng mL−1 for CrIII and CrVI, respectively. The method was applied to the analysis of one tap and one river water sample. CrIII was not detected in the river water sample. The method was validated by spike recoveries from these two samples and by the analysis of a mineral CRM (JR-1 from The National Institute of Advanced Industrial Science and Technology in Japan), which contains, according to the supplier’s website, 2.83 μg g−1 of Cr. The sample preparation involves dissolving 1 g in a mixture of nitric and hydrochloric acids with dilution to 100 mL, so it is hard to follow the authors’ claim that this CRM contains 5.48 μg L−1. Sadeghi and Moghaddam130 selectively extracted CrIII with the ionic liquid 1-octyl-3-methylimidazolium salicylate, only 220 μL of which was added to 10 mL of sample. After phase separation, the extractant was diluted to 500 μL with a (1 + 1) mixture of ethanol and water, and the Cr content determined by ETAAS. Following reduction with ascorbic acid, total Cr was determined and hence the CrVI by difference. The LOD for CrIII was 8 μg L−1, and the method was applied to the analysis of a tap water, a groundwater, and two urine samples. Neither species was detected in any of the samples. Spike recoveries appeared (from visual examination of the data presented in the relevant table) to be not significantly different from 100%. A procedure in which the Cr species were extracted as different complexes but into water-soluble solvents has been developed.131 Two-phases were created by the addition of ammonium sulfate. The CrIII was extracted as the complex with 4-(2-pyridylazo)resorcinol and the CrVI was extracted as the complex with 1,5-diphenylcarbazide. The details of each extraction procedure indicate that the acidities for each are different: pH 8.3 for CrIII and pH 1 for CrVI. The solvent was polyethylene glycol, ethanol, or isopropanol. The Cr in the organic phase was quantified by ETAAS (and spectrophotometry). The researchers found that formation of the CrIII complex was catalysed by bicarbonate. The LOD was 0.2 μg L−1 for both species. The procedure was applied to the analysis of two waste waters and two mine waters in which both species were detected, though one sample was first pre-concentrated 10-times by evaporation. Finally, readers of Japanese might be interested in a (probably malodorous) “micro-extraction” procedure, in which the species were extracted with the same reagent and solvent system, but at different pH values.132 The extractant, dioctyldithiocarbamic acid, was first synthesized by mixing carbon disulfide and dioctylamine in ethanol. The complex with CrVI was selectively extracted at pH 3; whereas both species were extracted at pH 6.5. After the phase separation, the organic phase was dissolved in a small volume of ethanol and the Cr content determined by ETAAS. The LOD for CrVI was 5 ng L−1 and for total Cr was 10 ng L−1. The method was applied to the analysis of river water samples. There is no mention of validation in the abstract of the article.
Table 3 shows other applications of Cr speciation presented in the literature during the time period covered by this ASU.
| Analyte species | Technique | Matrix | Sample treatment | Separation | LOD | Validation | Reference |
|---|---|---|---|---|---|---|---|
| CrIII, CrVI | FAAS, XRF, spectrophotometry | Leather | Soaked in pH 8 phosphate buffer | Total by AAS. CrVI by spectrophotometry | FAAS 0.07 mg L−1 | Pseudo-RM | 133 |
| CrVI, total Cr | Continuum source-ET-AAS | Bread and breakfast cereals | Leached 0.25 g with 25 mL 0.1 mol L−1 Na2CO3, boiled 20 min diluted filtered | Selective leaching | 0.045 μg g−1 | NCS DC 73349 (bush branches and leaves), SQC012 (CrVI in soil) | 134 |
| CrIII, CrVI | ICP-OES, spectrophotometry | Leather | Extracted, various solvents and pH | Total by OES. CrVI by spectrophotometry | Not given | None | 135 |
| CrIII, polymeric CrIII, CrVI | HPLC-ICP-MS O2 reaction gas | Wood leachate, waters | Wood: field leaching station, rainfall. Waters: waste, natural filtered | RP ion-pair, PerkinElmer Spheri-5, 5% (v/v) methanol + 0.005 mol L−1 EDTA + 0.001 mol L−1 TBAH, pH 7.2 | 0.9 μg L−1 | None | 136 |
| CrIII, CrVI | HPLC-ICP-MS kinetic energy discrimination He | Cereals, dairy products | UAE NH4OH. Centrifuged, filtered | AEC. IonPac CS5A, gradient (program given) 2.0 M HNO3 water | CrIII, 0.07 μg L−1 CrVI 0.01 μg L−1 | Spike recovery ERM-CE278k (mussel tissue) | 137 |
| CrIII, CrVI | HPLC-ICP-MS SSID 53CrVI | Soils | MAE 0.15 g + 5 mL of 0.05 mol L−1 EDTA (pH 10) + 0.15 g of 5 μg g−1 53CrVI enriched solution + 0.05 mol L−1 Na2EDTA 80 °C for 5 min centrifuged diluted | Previously published method (citation provided) | Not given | NIST 2701 (hexavalent chromium in contaminated soil) | 138 |
| CrIII, CrVI | HPLC tandem MS, SSID 50CrIII 53CrVI | Environmental waters | Preserved (details given) and filtered on site | IC Dionex, AS20 gradient KOH 0.03 mol L−1 (0–6 min), 0.06 mol L−1 (8–13 min), 0.03 mol L−1 (16–20 min) at 0.3 mL min−1 combined at T-junction with methanol at 0.2 mL min−1 | CrVI, 0.007 μg L−1 | Spiked proficiency test material | 139 |
| CrIII, CrVI | HPLC-ICP-MS | River water and sediment | Waters, preserved with Na2EDTA or HNO3 sediments frozen, thawed, air-dried, sieved, UAE various extractants | AEC, Dionex Ion Pack AG7 gradient (a) 0.1 mol L−1 NH4NO3 pH 4 (b) 0.8 mol L−1 HNO3 | 0.2–0.4 μg L−1 | China National Analysis Center for Iron and Steel DC 73309 (stream sediment) | 36 |
| Various CrIII species | Preparative LC ICP-MS, multi-collector ICP-MS | Various acid solutions | None | IEC | Not relevant | None | 140 |
3.4 Gadolinium
The content of different Gd based MRI contrast agents in water samples by HILIC-ICP-MS has been recently reported.141 An YMC-Triart diol HILIC column, operated at 30 °C, with a mobile phase of 50 mmol L−1 ammonium formate adjusted to pH 3.7 as eluent A and acetonitrile as eluent B, was used to separate Gd-BOPTA, Gd-BT-DO3A, Gd-DOTA and GD-DTPA within 15 minutes. The mobile phase flow rate was 0.8 mL min−1 in isocratic mode with 25% A and 75% B. An ultrasonic nebuliser was interfaced between the column outlet and the ICP-MS to improve sensitivity and O2 was added to the plasma to oxidise the high organic content mobile phase. Water samples from six waterworks in Germany were analysed, covering different methods of drinking water purification from surface water, were analysed using the developed HILIC-ICP-MS method. The respective LOQ values (signal to noise = 10) were 26 pmol L−1 for Gd-BT-DO3A, 37 pmol L−1 for Gd-DOTA and 46 pmol L−1 for Gd-DTPA. Of the four Gd compounds studied Gd-DTPA was detected in all water samples, at concentrations ranging from 82–167 pmol L−1, whilst Gd-BT-DO3A and Gd-DOTA were only detected in some samples and Gd-BOPTA was not detected in any sample demonstrating the direct anthropogenic impact of the use of Gd based MRI contrast agents on surface and drinking waters in the areas under study.3.5 Halogens
New developments in methods for the separation of different halogen species based on IC-ICP-MS have included improvements to species separation and also the removal of interferences prior to detection. In two related papers142,143 a group from the China University of Geosciences, Wuhan, used a short anion-exchange column (Dionex AG-19) coupled to ICP-MS in combination with an isocratic eluent containing KOH to separate inorganic I and Br species. Different gases were used in the reaction cell to remove the difficult interferences both elements suffer. In the first paper a fast separation of less than one minute was combined with a sensitive detection method achieving LOD values of 0.005 and 0.006 μg L−1 for IO3− and I− respectively. This was applied to their measurement in groundwater. A five-fold improvement in analytical sensitivity for I, which is a difficult element by ICP-MS due to the high first ionization potential (10.08 eV), was facilitated by the addition of CH4 to the Ar plasma. The analytical results for three SRMs were in agreement with the certified values and the method was applied to the determination of I− and IO3− in seven shallow groundwater samples collected from Northern China. The second paper used a similar approach but for the measurement of a different halogen, in this case the carcinogen BrO3−, formed during the sterilization of drinking water using ozone. The potentially interfering polyatomic ions 38Ar40Ar1H+ and 39K40Ar+ at the main Br ion at m/z 79 were significantly reduced by oxidization of 79Br+ to the interference free mass of m/z 95 (79Br16O+) by reaction with an unusual gas (N2O) in the CRC. The issue of reduced signal intensity due to the high-salt containing eluent, was alleviated by use of an online argon aerosol dilution technique, which improved stability for long runs (>27 h). The LOD was 0.013 μg L−1 for BrO3− and the short term precision (RSD) for five injections of 0.5 μg L−1 BrO3− was in the range of 2.1–4.5%. The analytical results for three water SRMs were in agreement with the certified values. The technique was applied to the determination of BrO3− in fifteen bottled water samples and two tap water samples.Complex halogenated organic compounds have continued to be the focus for the development of new analytical approaches, most recently utilising GC coupled to either MS or ICP-MS detectors. These methods have now started to be applied to persistent organic environmental contaminants such as brominated organic compounds (BOCs).144 Compound-specific stable Br isotope ratio analysis was used for the investigation of the environmental transformations of BOCs and demonstrated that precise Br isotope analysis can be successfully performed by GC-MS systems that are widely available. Comparison of the results obtained by GC-MS and GC-ICP-MS with a magnetic sector mass analyser for 1,2-dibromoethane and 3-bromophenol with different Br isotope composition was carried out to evaluate the analytical performance of the method. The precision (RSD) for sample amounts in the range of 10–1000 pmol was between 0.2–0.3%. Good correlation between the results obtained by GC-MS and GC-ICP-MS for 1,2-dibromoethane and 3-bromophenol (R2 > 0.98) was achieved. Due to the observed dependence of the measured isotope ratios on the amount of the analyte and the calculation scheme applied, normalization of the results versus appropriate standards was required for source attribution applications. Following on from some work reviewed last year in this ASU, a collaboration between different European centres has developed a reference method for the measurement of PBDEs (fire retardants) in surface waters145 at concentrations appropriate for the EU Water Frame Directive (WFD). An annual average environmental quality standard for inland surface waters of 0.5 ng L−1 for PBDE congeners 28, 47, 99, 100, 153 and 154 has been established. Within a recent European metrology project, different approaches for the traceable quantification of PBDE, based on LLE or SPE and analysis using single or triple quadrupole GC-ICP-MS were investigated and the related LOQ values obtained and expanded uncertainties compared. All the developed analytical procedures could serve as reference measurement procedures for monitoring the six priority PBDEs and an LOQ of 0.026 ng kg−1 with an associated expanded uncertainty of 0.002 ng kg−1 were achieved. Another method146 for the measurement of the same 6 priority PBDE congeners in environmental samples, this time sewage sludge, where up to 90% of PBDEs are accumulated because of their low aqueous solubility, also utilised GC-ICP-MS. Different extraction agents including: methanol (MeOH); acetic acid:MeOH (3:1 v/v); and 0.1 mol L−1 HCl in MeOH, followed by the addition of a Tris–citrate buffer and iso-octane, were assessed using mechanical shaking and microwave- and ultrasound-assisted extraction. Mechanical shaking and microwave-assisted extraction were the most effective at removing the PBDEs from the complex sludge matrix. The PBDEs in the organic phase were quantified using a standard addition calibration approach. Spike recovery using ssIDMS GC-ICP-MS confirmed the accuracy of the developed analytical procedure achieving recoveries between 95–104%. The procedural LOD values for the 6 PBDEs congeners ranged between 0.2 and 0.3 ng g−1, repeatability and reproducibility RSDs were 2.2 and 5.7%. The last publication147 in this area compared a range of different MS techniques this time for the determination of the polychlorinated biphenyls (PCBs). Four different MS techniques: ICP-QMS; ICP-QQQ-MS; NCI-Q-MS and EI-MS/MS, for the detection of PCBs in environmental samples after GC separation were compared using a new IDMS approach. A mixture of twelve priority PCBs labelled with 37Cl were employed as the ssIDMS internal standard. The 37Cl-labelled PCBs enable the use of both molecular and elemental ionization sources, because the heteroatom is detectable by both techniques. The comparison was carried out by assessing the capabilities of all the instruments to measure Cl isotope ratios and determine the isotopic enrichment of the labelled analogues. The analysis of the marine sediment NIST SRM 1941b, which contains PCBs in the low ng g−1, range was carried out. Elemental ionization sources such as ICP and NCI combined with QMS provided Cl specific detection and high sensitivity for higher chlorinated compounds but suffered from high background signals from other Cl-containing, co-eluting compounds in the sample which prevented the accurate measurement of PCB-specific Cl isotope ratios. However, the use of GC-MS/MS in the SRM mode provided selective and accurate measurements, but suffered from lower sensitivity for higher chlorinated compounds.
A range of different halogens, halogenated biomolecules and pharmaceuticals in clinical samples such as blood and blood plasma have been measured using LC coupled to various detectors including ICP-QMS, ICP-QQQ-MS and ES-MS. A method148 for the determination of the Cl species chlorate and chlorite in animal blood was developed using IC-ICP-MS. The suitability of the method was only evaluated using spiking experiments giving LOD values of approximately 0.5 mg L−1 for chlorite and 1.0 mg L−1 for chlorate in blood. In order to overcome the interferences from 17O17OH+ and 36ArH+ ions, a ICP-QQQ-MS instrument operated in MS/MS mode with hydrogen as reaction gas was used. However, the signal-to-background ratios and hence the LOD could not be improved due to a constant Cl background. However, with appropriate sample preparation, the method was shown to be suitable for the determination of anionic Cl compounds in blood. A fast, accurate and precise method149 for the measurement of the total content of drug-related Cl and Br in human blood plasma, based on HPLC-ICP-QQQ-MS has been developed to overcome the drawbacks of the current approaches involving the use of radio-labelled drug molecules. Separation by HPLC was used to determine the drug-related by-products from the substantial amount of iCl present in the samples and allow for the interference-free determination of 35Cl using ICP-QQQ-MS with H2 as the reaction gas and monitoring 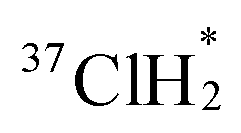 at m/z 37 and Br “on mass” at m/z 79. The method was found to be sufficiently precise (repeatability < 10% RSD) and accurate (recovery between 95 and 105%), with a good linear dynamic range (R2 better than 0.990) and an LOD for Cl and Br of 0.05 and 0.01 mg L−1 in blood plasma, respectively. Quantification using either external or internal standard calibration procedures produced reliable results for both elements. As a proof-of-concept, human blood plasma samples from a clinical study involving a newly developed pharmaceutical ingredient containing Cl- and Br were analysed and the total drug exposure was successfully measured. In another clinical study150 the identification and quantification of electrochemically generated metabolites of thyroxine (T-4) by LC-ES-MS and counter-gradient HPLC-ICP-MS were measured. The electrogenerated metabolites were separated by LC, subsequently mixed with a compensating counter-gradient, and then introduced into an ICP-MS instrument. The gradient compensation was achieved using an additional pump module which generated a reversed gradient to the analytical gradient used for the separation. This setup enabled a constant composition of LC eluent to flow into the plasma so that stable plasma conditions and a uniform response over the complete elution time could be achieved. Combined with identification information from online-coupled EC to LC-ES-MS, robust and reliable quantification of T-4 and its oxidation products was accomplished with an LOD of 33 nM of I. Another paper151 featuring the speciation of I containing species 3-monoiodotyrosine (MIT), iodide, 3,5-diiodotyrosine (DIT), 3,5-diiodothyronine (3,5-T2),3,5,3′-triiodothyronine (T3), and thyroxine (T4) also used HPLC-ICP-MS to measure these species in dog foods and treats. The method utilised both AE and RP and was evaluated by the analysis of the I species concentrations in twelve dog foods and treats following enzymatic digestion. The concentrations of MIT, iodide, DIT, T3, and T4 in the samples ranged from <MDL – 59.5 μg g−1, depending on the species. An additional analysis for DIT, T3, and T4 in the enzyme digest by LC-MS/MS was used to cross validate the results obtained by LC-ICP-MS and both methods were in good agreement.
at m/z 37 and Br “on mass” at m/z 79. The method was found to be sufficiently precise (repeatability < 10% RSD) and accurate (recovery between 95 and 105%), with a good linear dynamic range (R2 better than 0.990) and an LOD for Cl and Br of 0.05 and 0.01 mg L−1 in blood plasma, respectively. Quantification using either external or internal standard calibration procedures produced reliable results for both elements. As a proof-of-concept, human blood plasma samples from a clinical study involving a newly developed pharmaceutical ingredient containing Cl- and Br were analysed and the total drug exposure was successfully measured. In another clinical study150 the identification and quantification of electrochemically generated metabolites of thyroxine (T-4) by LC-ES-MS and counter-gradient HPLC-ICP-MS were measured. The electrogenerated metabolites were separated by LC, subsequently mixed with a compensating counter-gradient, and then introduced into an ICP-MS instrument. The gradient compensation was achieved using an additional pump module which generated a reversed gradient to the analytical gradient used for the separation. This setup enabled a constant composition of LC eluent to flow into the plasma so that stable plasma conditions and a uniform response over the complete elution time could be achieved. Combined with identification information from online-coupled EC to LC-ES-MS, robust and reliable quantification of T-4 and its oxidation products was accomplished with an LOD of 33 nM of I. Another paper151 featuring the speciation of I containing species 3-monoiodotyrosine (MIT), iodide, 3,5-diiodotyrosine (DIT), 3,5-diiodothyronine (3,5-T2),3,5,3′-triiodothyronine (T3), and thyroxine (T4) also used HPLC-ICP-MS to measure these species in dog foods and treats. The method utilised both AE and RP and was evaluated by the analysis of the I species concentrations in twelve dog foods and treats following enzymatic digestion. The concentrations of MIT, iodide, DIT, T3, and T4 in the samples ranged from <MDL – 59.5 μg g−1, depending on the species. An additional analysis for DIT, T3, and T4 in the enzyme digest by LC-MS/MS was used to cross validate the results obtained by LC-ICP-MS and both methods were in good agreement.
3.6 Manganese
The speciation analysis and fractionation of manganese in a wide variety of sample types, including soils, sediments, waters aerosols, plants and clinical samples, has been reviewed this year covering 113 references.152 The techniques covered include full speciation methods, such as HPLC-ICP-MS, CE-ICP-MS and AE-ICP-MS, and those whereby speciation is inferred by difference e.g. SPE-FAAS from MnII and MnVII in waters. A discussion, and often interpretation of the results obtained, is included for each sample type reviewed. The authors conclude that most Mn speciation studies are undertaken for liquid samples, that problems with species integrity can occur during the extraction of Mn species from solid samples and that the sensitivity of most of the direct methods for solid samples, such as X-ray based analysis, is not yet sufficient for most sample types.3.7 Mercury
Two review papers covering Hg speciation have been published during the period this ASU covers.The first of these, with 90 references, focuses on the various stages involved in elucidating Hg speciation in environmental samples.153 Sample preservation and storage are discussed as are the numerous methods available for pre-concentrating Hg species from water samples, both off and on-line. In this respect the title is a bit misleading as only water samples are covered in the review. A detailed discussion of the various separation techniques commonly in use, HPLC, GC and CE, and a table of the various non-chromatographic techniques, are also included. The section on detectors currently in use mainly deals with AFS and AAS with a small section on IDMS. Newer areas, such as the use of mobile phones as detectors for Hg species and other sensor techniques are also briefly discussed. The second review paper, 43 references, summarises the current status of the application of Hg speciation and the isotopic signature of Hg at the biomolecular level in living organisms, and discusses potential future uses of this combination of techniques.154
Mercury speciation methods and applications continue to develop in a wide variety of matrices. One research group has studied the interaction of Hg species with human erythrocytes to investigate possible high molecular mass binding partners for Hg species.155 Diluted human blood haemolysate was spiked with MeHg to a final concentration of 50 μmol L−1, the pH adjusted to 7.4 and incubated overnight at 37 °C. In addition, a 5 μmol L−1 solution of carbonic anhydrase (CA), which has been shown to be inhibited in the presence of Hg, was spiked with MeHg and thiomersal to give final concentrations of 2.5 μmol L−1 of each species, and, after pH adjustment to 7.4, also incubated overnight at 37 °C. This mixture was then subjected to an enzymatic digestion with either trypsin or chytotrypsin for 1 h at 37 °C. Each of these sample types was subsequently analysed by HPLC-ES-TOS-MS and HPLC-ICP-MS. A C18 column was used to separate the components of the haemolysate samples with a binary gradient of 0.1% formic acid and acetonitrile flowing at 300 μL min−1 with O2 added to the plasma for the HPLC-ICP-MS analyses. A C5 column was used to separate components of the enzymatically digested CA samples. For the spiked haemolysate samples mono MeHg adducts were detected for the haemoglobin α-chain and mono and di MeHg adducts for the haemoglobin β-chain. For the CA samples, MeHg and EtHg, from the added thiomersal, were found to be bound to cysteine groups of the enzyme. In order to study Hg speciation in HepG2 cells (incubated with HgCl2 or MeHgCl2) an online microfluidic chip (MFC) based magnetic SPME-μHPLC-ICP-MS system has been developed.156 The MFC contained two cell lysis units and two SPME columns packed with γ-mercaptopropyltrimethoxysilane (γ-MPTS). Mercury species were extracted from the HepG2 cells in the cell lysing channels, with a solution of 1% m/v sodium dodecyl sulfate and 1% m/v Tris–HCl at pH 8.0 flowing at 10 μL min−1, retained on the SPME columns followed by elution, with 10 μL of 0.01 mol L−1 HNO3 containing 4% (m/v) thiourea flowing at 1 μL min−1, into a sample loop followed by injection onto the μHPLC C18 column. The HPLC mobile phase consisted of 0.1% mercaptoethanol in 30 mmol L−1 NH4 acetate at pH 7.0 and increasing amounts of MeOH in a gradient programme flowing at 40 μL min−1. The LOD values obtained ranged between 13 and 42 ng L−1 for four Hg species, Hg2+, MeHg+, EtHg+ and PhHg+ which were separated in 24 minutes. For the incubated HepG2 cells only Hg2+ was detected in those spiked with Hg2+ whilst both Hg2+ and MeHg+ was detected in those spiked with MeHg+. Using test solutions and the MFC-SPME-μHPLC-ICP-MS system only no conversion of MeHg+ to Hg+ was observed and it was concluded that the observed demethylation of Hg occurred in the HepG2 cells. It was also found that the HepG2 cells accumulated five to eight times more MeHg+ than Hg+ depending on the length of the incubation period. A labels and separation free bioassay for single-strand DNA, thrombin and double-strand DNA, using Hg as a beacon with detection by CV-AFS, has been developed.157 The bioassay utilises a molecular beacon possessing several thymine bases at each end, which, in the presence of Hg2+, forms into a ‘hairpin’ shape. When the target DNA interacts with the molecular probe Hg2+ ions are released and, after reduction with SnCl2, detected by the AFS system. The probe DNA was incubated for 6 hours at 37 °C in the presence of Hg2+ ions, in the presence of phosphate buffer saline (PBS) at pH 7.4 followed by the addition of the target analyte and hybridisation for 1 further hour releasing Hg2+ ions back into solution. After a 10 fold dilution ‘free’ Hg2+ in the sample was reduced to Hg0 with 0.1% SnCl2 in an “intermittent flow manner” and swept into the AFS instrument. The stoichiometric binding ratio of the probe DNA:Hg was 6:1 so to ensure all of the added Hg was bound to the probe the DNA:Hg ratio in the incubation solution was 1:5. The LOD value obtained was 0.2 nmol L−1 with an RSD of 3.7% for single stranded DNA. For thrombin the LOD value ranged between 0.08 and 0.03 nmol L−1 depending on the aptamer used as the probe. Selectivity for thrombin was demonstrated in the presence of several other proteins, including bovine and human serum albumins and immunoglobulin G. Subsequently, thrombin was measured in spiked human serum samples with recoveries ranging from 87–104% at a spiking level of up to 100 nmol L−1 of thrombin. The authors suggest that the “proposed AFS-based protocol has a promising future in the analysis of complex biological samples”.
Two papers report new pre-concentration/extraction methods for Hg species in waters and biota this year. The first paper describes the use of a nanosorbent based on mesoporous silica functionalized with 1,5-bis(2-pyridyl) methylene thiocarbohydrazide.158 The system was based on chelating retention of Hg2+ and MeHg+ ions on a mini-column filled with the nanosorbent and their sequential elution with 0.2% HCl for MeHg+ and 0.1% thiourea in 0.5% HCl for Hg2+. Followed by CV generation with NaBH4 and ICP-MS detection. Under the optimum conditions and a 120 s pre-concentration time, the enrichment factors were 4.7 and 11.0, the detection limits (3 sigma) were 0.002 and 0.004 μg L−1, and the RSD values (n = 10 at 2 μg L−1 standard of both species) were 2.8 and 2.6% for MeHg+ and Hg2+ respectively. The method was validated with two CRMs, LGC 6016 estuarine water (adjusted to pH 5) and SRM 2976 mussel tissue (after species extraction with 4 mol L−1 HNO3 and MAE) and recoveries of 95% or greater were obtained for both species in each CRM. The method was also applied to the speciation of mercury in sea-water samples collected in the Malaga Bay. Only Hg2+ was detected in these samples, ranging from 25–28 ng L−1. Spike recoveries for both Hg species in the seawater samples were 95% or better. The second paper covered here describes the synthesis of a molecularly imprinted polymer (MIP) for the extraction of MeHg.159 Methylmercury chloride was used as the template, phenobarbital as the ligand, methacrylic acid (MMA) as a monomer, and ethylene glycoldimethacrylate (EDMA) as a cross-linking agent. The method was validated using two CRMs, BCR 464 (Tuna) and TORT-2 (lobster hepatopancreas). Mercury species were extracted from the CRMs with 6 mol L−1 HCl and then transferred to toluene with UAE for 30 minutes at 60 °C. The MIP was loaded into SPE cartridges, conditioned with NH3/NH4+ buffer at pH 8, the toluene extracts loaded at a flow rate of 0.5 mL min−1 and the retained MeHg eluted with 1 mol L−1 thiourea in 1 mol L−1 HCl. The Hg in the collected eluate was then quantified using CV HR continuum source AAS. The LOD of the method was 6.6 μg kg−1 and MeHg recoveries from the CRMS were 103% for BCR 464 and 102% for TORT-2.
Two papers that report the determination of Hg species in seafoods are described here. An interlaboratory study for the determination of MeHg in marine origin food stuffs has been carried out. The laboratories represented commercial laboratories, academic institutions, and government agencies, as well as research and development laboratories.160 Species specific ID-GC-ICP-MS was the analytical method chosen for the study. To extract the Hg species an Me201Hg enriched spike was added to 0.2 g of sample followed by complete sample dissolution with 3 mL of 25% TMAH, pH adjustment with 1 mL of a sodium acetate/acetic acid buffer at pH 5 and 600 μL concentrated HNO3. After the addition of 1 mL of hexane and 500 μL of 2% NaBEt4 the mixture was mixed for 10 minutes before centrifugation to separate the phases. Method validation was by the analysis of three CRMs, SRM 1566b (Oyster), ERM-CE464 (Tuna) and DOLT-4 (Dogfish liver) and a further 6 seafood samples, Cod muscle, Halibut muscle, Squid muscle, Whale meat and Crab claw meat were also analysed for MeHg content. The MeHg content of the samples under test ranged from 0.035–3.6 mg kg−1 and the author concludes that the methodology was fit for purpose and produced robust and reliable results, with HorRat scores of <1 for all samples except the mussel tissue, which had a score of 1.6 due to the low MeHg mass fraction of 0.035 mg kg−1. The paper gives a very detailed description of the method in a simple to follow format and is recommended reading for workers in the field. The second paper reviewed here covers the use of an in-house fabricated electrochemical (EC) flow cell coupled with CV-AFS for the determination of MeHg in seafood extracts. An acidic leaching procedure, 2.5 g of sample and 10 mL of HCl followed by transfer of extracted Hg species into DCM and back extraction into water. The extracted MeHg was reduced to Hg0 on the surface of a glassy carbon cathode in the EC flow cell detected by AFS with the analysis taking one minute per sample. Under optimal conditions linearity was in the range of 0.2–5 ng mL−1 as Hg and the LOD for MeHg was 1.9 × 10−3 ng mL−1 with an RSD of 2.0% (n = 6) at 2 ng mL−1. The MeHg content in locally bought seafoods, ranged between 3.7–45.8 ng g−1. Recoveries for spiked samples were in the range of 87–103%.
An unusual coupled technique, GC-dielectric barrier discharge (DBD)-OES, has been reported for the determination of Hg species in rice extracts.161 A methanolic KOH based method was used to extract the Hg species from rice samples and three CRMs, SRM 1568b (rice flour), TORT-3 (lobster hepatopancreas) and DORM-3 (Dogfish muscle) followed by pH adjustment to 5 with a sodium acetate/acetic acid buffer, phenylation with NaBPh4 and headspace extraction onto a carboxen/PDMS or PDMS/DVB fibre. Argon was used both as the GC carrier gas and the discharge gas in the DBD device. Under optimal conditions the LOD values for iHg, MeHg and EtHg were 0.16, 0.24 and 0.34 ng respectively. The RSD, for a 15 pg mL−1 solution of each species ranged between 3.7 and 5.2%. The found values for iHg and MeHg and total Hg sum of the species were all in statistical agreement (t test) with the certified values for the three CRMs used. The concentrations of iHg and MeHg ranged from <LOD to 73 μg kg−1 and <LOD to 15 μg kg−1 respectively. These values were in agreement with those found by HPLC-ICP-MS and the sum of the species agreed with the values obtained by ICP-MS after a MAE acid digestion procedure.
Three papers reviewed here have used Hg isotope ratio measurements to investigate Hg methylation and demethylation in different environmental compartments. The first of these determined the isotopic composition of iHg and MeHg in Beluga whales and seals.162 The compounds of interest were extracted from the tissue samples suing 25% TMAH and MAE. The isotope ratios of the extracted iHg and MeHg were measured using GC-ICP-MS, after compound derivatisation, and these Hg species were quantified by ssID-GC-ICP-MS, again after derivatisation with method validation by the analysis of BCR CRM 464 (Tuna fish muscle) and NRCC DOLT-4 (Dogfish liver). The full methodology is given in a cited reference. A mass balance approach was also used for method validation and was stated to be ‘conservative’ within uncertainty limits. The authors suggest that in vivo demethylation of MeHg is the main process in mammals producing mass dependent fraction of Hg isotopes during trophic transfer and that, by analogy, similar studies of Hg in human tissues could help identify pathways of iHg and MeHg exposure in humans. The second study covered here used the same methodology as the previous reference to determine iHg and MeHg dietary pathways in Zebra fish exposed to differing amounts of iHg and MeHg via the diet.162 The authors conclude that “noticeable differences on the isotopic and speciation patterns between fish exposed to iHg and MeHg enriched food are observed as a reflex of the dissimilar metabolization of both Hg species” and that the results were in excellent agreement with a companion study (reference cited in the paper). The final paper on this theme investigated the effect of nutrient loading on Hg speciation in the pelagic zone using sediment/water mesocosms to which nutrients (NO3 and PO42−) and isotopically enriched iHg or MeHg tracers.163 The concentrations of iHg and MeHg in sediments and waters were determined by ssID-GC-ICP-MS with the full details given in a cited reference. The authors conclude that the study ‘demonstrated a causal positive relationship between pelagic biomass production and the short-term HgII methylation rate in sediments’. The study showed that the rate of MeHg formation, which was induced by the nutrient loading, increased when iHg was not complexed with natural organic matter e.g. via thiol groups and MeHG formation was controlled by a combination of microbial activity and the speciation of iHg.
Table 4 shows other applications of Hg speciation presented in the literature during the time period covered by this ASU.
| Analyte species | Technique | Matrix | Sample treatment | Separation | LOD | Validation | Reference |
|---|---|---|---|---|---|---|---|
| CH3Hg+, Hg+ | Purge and trap ID-GC-ICP-MS | Faecal matter | Homogenisation in 1 mL of 1% cysteine, 2 mL 0.9% saline. Homogenate added to 2 mL 45% NaOH heated @ 90 °C for 10 minutes, cooled, 5 mL 0.9% saline added. ID after addition of NaBEt4 | Details given in a separate referenced article | Not given | None stated | 164 |
| CH3Hg+, Hg+ | Purge and trap GC-CV-AFS, GC-AFS | Waters, grey mullet eyes, sediments | Waters: US EPA method 1631. Complexation with C5H9NS2·NH3, distillation, ethylation with NaBEt4 | Details given in a separate referenced article | Not given | MESS-2, IAEA-405 and BCR-580, DORM-4. Spiking experiments to evaluate species transformations | 165 |
| Fish: lyophilisation and homogenisation, KOH/KBr/CuSO4/DCM extraction. Back extraction into sulfide solution then into DCM | |||||||
| Sediments: KOH/MeOH extraction, transfer to DCM, transfer to aqueous sulfide solution, further extraction into DCM | |||||||
| CH3Hg+, Hg+, Hg0 | GC-ICP-MS | Liquid hydrocarbons | Helium purge to remove Hg0. OrganoHg measured using GC-ICP-MS after purging. Inorganic Hg extracted into 100 mg L−1 NaCl solution. Organo Hg and Hg derivatised with either BuMgCl or NaBPh4 | HP-5 (30 m × 0.32 mm, 0.25. μm) column. 60 to 280 °C@25 °C per minute. Carrier gas 100 mg L−1 Xe in He, 2 mL min−1 | 0.2–0.5 μg L−1 | Japanese language paper so not known | 166 |
| CH3Hg+, Hg+ | HPLC-ICP-MS | Waters | Extraction with SiO2-SH coated Fe3O4 NPs. pH range of 3–9. Magnetic separation. Desorption with 2.5 mL of 1% thiourea in 0.5 mol L−1 HCl and sonication for 20 s | Shim-pack CLC-ODS column (15 cm × 6 mm × 5 μm). MP: 1 g L−1L-cysteine, 0.06 mol L−1 ammonium acetate, 1.0 mL min−1 | CH3Hg+, 0.3 ng L−1 and Hg+ 1.0 ng L−1 | Spike recoveries only, range 96–104% | 167 |
| CH3Hg+, EtHg, Hg+ | ID-GC-ICP-MS | Spiked bovine and human blood stability tests | Isotopically enriched Hg species added to spiked blood followed by 500 μL TMAH in 25% v/v MeOH. Heated for 24 h 80 °C. SPME in presence of 7.7 mL of 0.1 mol L−1 CH3CH2Na. Derivatisation with NaBPr4 | Elite-5 (30 m × 0.25 mm × 0.25 μm) | Not given | None stated | 168 |
| CH3Hg+, Hg+ | ICP-MS | Hair, water | Hair: 4 mL of 5 mol L−1, 0.1 g sample, sonication | CH3Hg+, 1.6, Hg+ 1.9 ng L−1 | DORM-2 Dogfish muscle CRM | 169 | |
| Water: filtered, 0.45 μm. Hg species extracted with γ-MPTS modified Fe3O4: SiO2 NPs. MeHg and total Hg extracted with 1.5 mol L−1 HCl containing 0.01% and 3% thiourea (m/v), respectively. iHg estimated by difference | |||||||
| Hg, organoHg | CV-AAS | Fish, squid, sea urchins | Total Hg: 0.1% cysteine, 5 mol L−1 NaOH, 80 °C, 1–2 hours. Fat removal with MIBK and hexane | None. Selective extraction procedure | Not given | NMIJ CRMs 7402-a (Cod) and 7403-a (Swordfish). NIES CRM No 13 (hair) | 170 |
| OrganoHg: As for total Hg then HBr/CuCl2/toluene added to extract organoHg. Toluene layer removed and organoHg back extracted into 0.2% cysteine, 2% sodium acetate | |||||||
| CH3Hg+, Hg+, alkyltins, alkyllead | ssID-GC-ICP-MS | Sediments, waters | Sediments: acetic acid/MeOH MAE, spiking addition of IDMS spike, in situ propylation. Full method in cited reference | MXT I column (30 m × 0.28 mm i.d., 1 μm) | Waters: LOQ between 0.01 and 1.0 ng L−1. Sediments: LOQ between 0.08 and 1.4 ng g−1 | Sediments: BCR 405, 462 and 646 | 171 |
| Waters: addition of IDMS spike, in situ propylation. Full method in cited reference | Waters: spike recoveries | ||||||
| CH3Hg+, CH3CH2Hg+, Hg+ | HPLC-ICP-MS | Sediments | Extraction into CuSO4 (1 mol L−1)/HNO3 (3 mol L−1), transfer into CH2Cl2 then back extraction into water | RP C18 column (15 cm × 4.6 mm id), 0.1% L-cysteine, 0.1% HCl + 0.1%, L-cysteine at 0.8 mL min−1 | ≤0.02 ng L−1 for all three species | ERMCC580 and NRCC Tort-2. Spike recoveries | 172 |
| CH3Hg+, Hg+ | HPLC-ICP-MS | Rice plants | Extraction with 6 mol L−1 HCL, 45 minutes UAE then overnight shaking | RP C18 column (15 cm × 3.9 mm id), 60 mmol L−1 ammonium acetate/5% MeOH/3% 2-mercaptoethanol at pH 6.7, 1.0 mL min−1 | 173 | ||
| CH3Hg+, CH3CH2Hg+, Hg+ | CE-ICP-MS | Waters | Solid phase extraction onto thiol cotton particles (pre-treated with mercaptoacetic acid (MAA) and HCl). Elution with 0.5 mL 3% MAA in 0.001 mol L−1 HCl | 90 cm length × 75 mm id × 375 μm od fused silica capillary. Running buffer 50 mmol L−1 H3BO3/12.5 mmol L−1 Na2B4O7 (pH 9.20) | ≤0.11 ng mL−1 for all species | Not given | 174 |
| OrganoHg | AAS | Sushi | Selective extraction of organoHg: toluene/HCl (30%) MAE. Transfer from toluene to 2.5% L-cysteine | None | 3.8 μg kg−1 | NRCC TORT-2 (lobster hepatopancreas) and DORM-4 (dogfish muscle) | 175 |
| CH3Hg+, Hg+ | ssID-GC-ICP-MS | Zebra fish organs | Extraction with TMAH followed by derivatization. Details given in cited reference | Details given in cited reference | Not given | Not given | 176 |
| OrganoHg | AAS | Raw and cooked fish | Extraction with 18% KBr/5% H2SO4, transfer into toluene, back-extracted into aqueous sodium thiosulfate solution | None | Not given | IAEA-407 (fish tissue) | 177 |
3.8 Selenium
During the past year, two reviews have appeared. Se speciation in biota by HPLC-ICP-MS has been presented.178 A range of topics such as extraction protocols (aqueous, acid, basic and enzymatic hydrolysis) and separation mechanisms (ionic-exchange, reversed-phase and size exclusion) are included. There are also extensive tables of special interest for the readers summarizing details and characteristics of analytical methodologies for determining Se species in different biological samples. Special effort has been made in compiling recovery values when analyzing SeMet and total Se in certified reference materials by applying different extraction procedures. The review includes 134 references. The second review focus on different methods for Se supplementation.179 The reviewers include an overview on supplementation methods and new approaches for improving Se status. Additional information on analytical methodologies for total Se, Se-species and Se bioaccessibility determination is also presented.Several nanomaterials have been used for Se speciation in samples of different nature. Silica-coated magnetic nanoparticles (SMNPs) modified by 5-sulfosalicylic acid (SSA) were used to pre-concentrate Se species enzymatically extracted from Radix puerariae (a root frequently used in traditional Chinese medicine) prior HPLC-ICP-MS.180 The adsorption was based on the interaction between –SO3H on the surface of SSA-SMNPs and Se species. Best extraction conditions were obtained when adjusting solution pH at a value of 4.0 with glacial HAc. The resulting Se-loaded nanoparticles were separated from the solution with an external magnetic field followed by a treatment with 0.5 mL of 0.5 mol L−1 of Na2CO3 subsequently, Se species were separated and detected by HPLC Hamilton PRP-X100, (20 mm length × 2.1 mm i.d., 10 μm particle size) on line coupled to ICP-MS. The use of the nanomaterial improved the detection limits of the method by 10 times with values of 0.0015, 0.0016, 0.0043 and 0.0023 mg L−1 for SeVI, SeIV, SeMet and SeCys2, respectively. SeVI was the Se dominating species when applying the methodology to Radix puerariae extracts. A mass balance approach, which compared the sum of the Se species detected with the total Se found in root (0.2500 mg kg−1) by a MAE digestion procedure, gave quantitative recoveries about of 95%. A dispersive solid phase extraction method (SDSPME) for inorganic Se speciation based on the use of alumina nanoparticles functionalized with Aliquat 336 and ICP-OES has been presented.181 Most of the paper is devoted to the optimization of the extraction process by using a multivariate approach. SeIV was selectively and quantitatively retained (>90%) in the Al2O3NPs in the pH range of 5.0–7.0. After extraction, samples were submitted to centrifugation to separate the adsorbent from sample solution. Subsequently, the adsorbed SeIV was eluted with 3 mL of 0.1 M HNO3 and measured by ICP-OES. The LOD value was determined to be 1.4 ng L−1 of Se for SeIV. The enrichment factors and RSDs were 850 and lower than 5%, respectively. The proposed method was successfully applied for the determination of Se in a CRM (SRM, 1643e from NIST, trace elements in water). The method was further employed to the speciation analysis of Se in three different river water samples where SeIV was the main Se specie found. In this line, a novel method based on the combined used of magnetic solid phase microextraction (MSPE) and hollow fiber liquid microextraction (HFLLME) was used for the extraction and pre-concentration of Se-aminoacids from rice samples before HPLC-ICP-MS measurements.182 Se compounds extraction by MSPE was achieved by employing Cu-loaded Fe3O4@SiO2–polyaniline–graphene oxide NPs as adsorbent. Se compounds from aqueous extracts of rice were first isolated by MSPE at pH 7 and then eluted by applying 0.1 mol L−1 ethylenediamine as eluent. Subsequently, the isolated Se compounds were preconcentrated by HF-LLME using 1-octanol as extraction solvent, ionic liquid trioctylmethylammonium chloride ([MTOA]+[Cl]) as carrier for the extraction and aqueous NaNO3 as the acceptor solution. Selenium species were further separated on a PAK C18 (5 μm, 4.6 × 250 mm) column operating at 55 °C and using 25 mmol L−1 ammonium formate–formic acid, at pH 3.0 and methanol/water (3/97, v/v) as mobile phase. The enrichment factor was reported to be 150–280. The LOD values for the selenoamino acids tested were as low as 0.0075–0.013 μg L−1. The method was validated using SELM-1 (selenized yeast, certified in SeMet). The determined value (3388 ± 72 μg per SeMet g) was in a good agreement with the certified value (3389 ± 173 μg per SeMet g). No selenoamino acids were detected when applying the method to rice aqueous extracts probably due to the low efficiency of hot water for extraction for this matrix. An interesting approach based on the use of a magnetic effervescent tablet was employed for the speciation of inorganic selenium based on ionic liquid dispersive liquid–liquid microextraction (ILLLME) and GFAAS analysis.183 The magnetic effervescent table is composed of Na2CO3 as source of CO2, ionic liquids and Fe3O4 magnetic nanoparticles (MNPs). The proposed method seems to have many practical advantages such as the combined use of extractant dispersion and magnetic recovery in a single step. Most of the paper is devoted to the optimization of the parameters influencing the microextraction step, such as pH of the sample solution, volume of ionic liquid, amount of MNPs, concentration of the chelating agent, salt effect and matrix effect. The LOD values were determined to be for SeIV 0.021 ng L−1. The method was validated by spike recoveries and accurate analysis of the standard reference materials (GBW10016 tea, GBW10017 milk powder, GBW10043 Liaoning rice, GBW10046 Henan wheat, GBW10048 celery). The developed methodology was applied to the speciation analysis of SeIV and SeVI in samples of different nature such black tea, milk powder, mushroom, soybean, bamboo shoots, energy drink, bottled water, carbonated drink and mineral water.
Selenium speciation has also been focused on Se nanoparticles as a new chemical form of Se. Palomo-Siguero et al.184 combined different independent bio(analytical) approaches to carry out a comparison between the effects of chitosan-modified SeNPs (CS-SeNPs) and selenite on lactic bacteria (Lactobacillus bulgaricus). Flow cytometry measurements using dead/live tests evidenced a decreases in bacteria cell viability when adding 10 μg Se mL−1. This effect is increased when Se was supplemented as SeVI. Under these conditions, SeNPs killed approximately 20% of Lactobacillus after 24 h while the percentage increased up to 60% of bacteria when SeIV was applied. Additionally, analysis of the bacteria pellet extracts by Hamilton PRP X-100 (150 mm × 4.6 mm, 10 μm) and Zorbax C8 (250 × 4.60 mm, 5 μm) columns coupled to the ICP-MS revealed that most of the CS-Se NPs in the culture media were transformed by the bacteria to organoselenocompounds while SeIV remains almost unaltered. The authors attributed the differences observed in bacteria viability to the different efficiency of Lactobacillus in transforming SeIV and CS-SeNPS into Se organic compounds. The results were in agreement with TEM images where CS-Se NPs were located inside the bacteria cells without compromising the integrity of the bacteria. The results evidenced the importance of biological system in transforming NPs as a key factor to be taken into account when assessing NPs toxicity.
Several papers have appeared this year on Se determination in animal tissues. Se speciation in CRMs and fish tissues has been reported by utilizing HPLC-ICP-MS.185 Total Se concentrations in the fish samples measured by ICP-MS prior acid digestion were found between 2.6 to 7.1 mg kg−1. Isolation of Se species was performed by using several sample treatment methodologies such as: aqueous leaching, enzymatic hydrolysis and acid hydrolysis, and by employing ultrasound-, microwave-assisted extraction and conventional heating. Enzymatic hydrolysis with protease by means of conventional heating at 37 °C for 20 h and microwave assisted heating at 45 °C for 45 min gave the best results with recovery values ranged from 72 to 90% of the total Se. Se species were further separated and detected by using two methodologies depending on the Se species involved. Enzymatic hydrolysis with protease XIV followed by separation using anion exchange (PRP-X100) or cation exchange (Supercosil LC) was used for assessing SeMet concentration. In contrast, SeCys measurements involved a carboxymethylation derivatisation step prior to the enzymatic hydrolysis to protect SeCys and subsequent separation using size exclusion (for urea removing) followed of C8-RP chromatography. The mean concentration of SeCys in CRMs and fish tissues was 0.50 ± 0.40 mg kg−1 indicating that a low amount of SeCys is present in CRMs and fish tissues. In all fish samples, the main Se species found was SeMet with values ranging from 0.7 to 5.4 mg kg−1. Based on these results, a mass balance approach, which compared the sum of the Se species detected with the total Se found gave recoveries of 33 ± 5% of Se in CRMs and 76 ± 18% of Se in fish. The authors suggest resolubilisation of the freeze dried extracts and/or filtration as the main causes of Se losses. In other paper186 the effect of microwave acid digestion on the integrity of 6 Se species usually found in animal tissues and urine (SeCys, SeMet, TMSe+ and three selenosugars, SeSug1, SeSug2 and SeGalNAc) was evaluated by HPLC-ICP-MS. Selenium standards (10 μg as Se) were digested in 3 mL of HNO3 at 160 °C for 30 min. The pH of extracts was subsequent adjusted with KOH to 6 and diluted before analysis. Se species in the extracts were separated and detected by HPLC-ICP-MS using a Hamilton PRP-X100 anion-exchange column (250 mm × 4.1 mm) and an aqueous 20 mM NH4H2PO4 at pH 6, flowing at 1.5 mL min−1 as the mobile phase. Data showed that microwave assisted digestion at 160 °C using closed vessels does not quantitatively convert all Se species to inorganic Se. SeCys was completely degraded to selenious acid while SeMet and the three selenosugars were transformed to different Se species such as selenious acid and methylseleninic acid. In contrast, TMSe+ remained stable after submitting to microwave treatment. These results are of interest when using Se species-dependent techniques such as hydride generation.
The metabolisms of Se in cattle beef when supplementing with several feeds containing Se (as selenized yeast) alone or in combination with canola oil and/or vitamin E was evaluated by Oliveira et al.187 Selenium speciation in the resulting products (feed and beef muscle) was achieved by using a Zorbax SB-C18 (4.6 × 250 mm), column on line coupled to ICP-MS. A combination of enzymes (protease type XIV, proteinase K and pancreatin) in a buffer (pH 7.5) containing NaCl, Na3PO4 and ammonium citrate and subsequent incubation at 37 °C for 24 h was tested for releasing Se from samples. Besides the laborious and time consuming sample treatment applied, an extraction yield of only 60% was attained. RP-ICP-MS chromatograms from beef samples without Se supplementation and with and without canola oil to their diet show SeCys2 and SeMet as the only Se species, however the identity of the former was not fully confirmed. Canola oil addition did not significantly alter the Se content in beef. It is worth noting that although both non-enriched and enriched feed types contained SeMet as the sole primary Se species (the former at lower Se levels), only the non-enriched diet produced a Se-containing peak in beef extracts with a retention time that matches the retention time of SeCys2 standard. The effect of Se deficiency on metal containing proteins in mouse brain was evaluated by Abiko et al.188 For this purpose, 82SeIV and 82SeMet were intravenously injected in mice fed with either a Se deficient diet or a diet with adequate levels of Se. Analysis of brain by HPLC-ICP-MS evidenced that supplemented Se did not alter the SelP levels whereas the levels of antioxidant proteins such as cellular glutation peroxidase (cGPx), superoxide dismutase (SOD) and metallothionein (MT) were increased. The highest enhancement of antioxidant proteins was detected when supplementing SeIV in Se deficient rats. Finally, it is worth mentioning the detection of volatile Se compounds in urine of male golden hamsters.189 For this purpose, head space Se volatile compounds from intact and 48 hour aged urine were extracted with SPME (30 μm carboxen, 50 μm divinylbenzene, polydimethylsiloxane) during 30 min at 37 °C followed by GC-MS analysis using a Supelcowax® 10 column (30 m × 0.25 mm with 0.50 μm film thickness). Four volatile organic Se compounds were detected and identified in the samples (dimethyl selenenylsulfide, dimethyl diselenide, dimethyl bis(thio)selenide, and dimethyl selenodisulfide). Increasing concentrations of dimethyl selenodisulfide were detected when analyzing 48 h urine. The presence of dimethyl bis(thio)selenide and dimethyl selenodisulfide as urine metabolites was reported for the first time and the authors speculate whether these metabolites are formed by hamster and/or by the hamster’s gut flora.
Two papers describe the investigations of Se speciation in vegetable oils An interesting paper on the speciation of Se in a mixture of selenite triglycerides produced by the reaction of SeIV and sunflower oil (SeOl) was reported by Bierla et al.190 Selenium species from samples of either SeOl dissolved in isopropanol or methanolic extracts of SeOl were separated by using a Zorbax Eclipse XDB-C18 (4.6 mm × 150 mm × 5 μm) column at 50 °C by using a non-aqueous mobile phase consisted of isopropanol/acetonitrile. Variations of the plasma conditions during elution due to the use of a non-aqueous mobile phase were compensated by a post-run mathematical correction of the chromatograms. Chromatography revealed up to 14 Se containing species in sunflower oil dissolved in isopropanol of which 11 were also detected in methanol extracts. These correspond to more 60% of the total Se in the oil. Subsequently, HPLC-ES-MS/MS was applied to identify the Se-compounds detected by HPLC-ICP-MS. A total of 11 Se-containing triglycerol derivatives resulting from the oxidation of one or two double bonds of linoleic acid and analogous derivatives of glycerol-mixed linoleate(s)/oleinate(s) were identified for first time. Undoubtedly, the detection of selenite triglycerides opens a new perspective on the role of these compounds in Se metabolisms. In a separate study191 a 2D chromatography approach (2-D SEC-RP-ICP-MS) was used to determine selenoamino acids in protein fraction of olive oil. The optimized conditions for isolating proteins in 5 g of olive oil was achieved by using a two-step approach consisting of a protein precipitation step with 10 mL of n-hexane/acetone followed by a redissolution of the protein pellet with a mixture of water:methanol (80:20). Proteins were screened on a SEC column (TSK gel G3000SW 7.5 × 300 mm 10 μm) and a RP column Zorbax SB C18 (150 mm 4.6 mm, 5 μm). SEC measurements revealed that Se was bound to two molecular mass fractions of 443 kDa and 66 kDa. Se containing fractions were further collected and submitted to microwave-assisted acid hydrolysis with 15% (HCl) and analysed by RP-ICP-MS. MeSeCys and SeCys were found in the olive oil samples with values ranging from 1.03–2.03 ± 0.2 μg kg−1 and 1.47 ± 0.1 μg kg−1, respectively. ANOVA showed significant variation of Se and S concentration in the protein fractions from different olive tree varieties which could be of interest as a tool for cultivar origin differentiation.
Methods for the determination of Se in various body fluids have been also developed during the period covered by this update. Two papers from Jager et al.192,193 cover the influence of TMSe levels on the Se metabolic pathway in humans orally supplemented with SeIV, SeVI and selenized yeast. The study was conducted with a group of healthy volunteers classified as TMSe eliminators and non-TMSe eliminators. Data from ICP-MS and HPLC-ICP-MS measurements revealed important differences of Se metabolism between TMSe eliminators and non-TMSe eliminators. TMSe was not detected in non-TMSe eliminator individuals supplemented with selenite and selenate remaining both inorganic Se species unaltered during the metabolic pathway. In contrast, in TMSe eliminator individuals, TMSe was the main Se metabolite found in urine along with high levels of Se-containing carbohydrates. Selenized yeast exposure produces SeSug1 as the main metabolite in all urine samples, whereas TMSe was another main metabolite in TMSe eliminators’ urine. Flouda et al.194 developed a method for the determination of low molecular weights Se metabolites in human plasma. The optimized methodology includes separation and detection of Se-species by RP-HPLC-ICP-MS and quantification by species-unspecific isotope dilution. Plasma samples were first filtered using a 3000 Da molecular weight cut-off membrane, and further centrifuged at 14000 × g during 30 min. Se-species in the resulting filtrates were determined using reversed phase Gemini 5 μm C18 (250 × 2 mm) and ICP-MS detection. Additionally, LC-ES/MS/MS analysis was employed to confirm the identity of each Se compound by the characteristic fragmentation profiles. The method allowed the separation and identification of Se-compounds in a set of plasma samples from cancer patients receiving selenite treatment in a clinical trial. Three Se-compounds were detected in the samples, two of these were identified as SeGal and SeIV while the third minor metabolite was not identified. Finally, the levels of glutathione peroxidase (GPx) were determined in human plasma samples of both healthy and diabetic mellitus patients by using affinity chromatography-post column IDA-ICP-MS along with the enzymatic activity of GPx.195 No significant differences in GPx levels in plasma from healthy and diabetic patients were found while GPx activity was significantly lower in diabetic patients compared with controls, suggesting GPx activity as a better biomarker for diabetics.
Two papers dealing with the quantification of selenoprotein P (SEPP1) have appeared this year. In an outstanding study by Deitrich et al.196 have reported the quantification of SEPP1 based on the use of double SSIDA in combination with HPLC-ICP-MS/MS via Se-specific detection at the peptide level. Isotopically enriched Se-containing peptides specific to SEPP1, including ENLPSLCSUQGLR (ENL) and AEENITESCQUR (AEE), were synthesized and characterized for their Se content, Se distribution, and isotopic composition. Confirmation of the structural identity of the alkylated Se peptides was performed by HPLC hyphenated in parallel to ES-MS and ICP-MS. The addition of peptide spikes to the plasma sample was followed by tryptic digestion, alkylation, and isotope ratio determination using HPLC-ICP-MS. The method allowed SePP1 quantification in human plasma samples without the need of extensive cleanup or pre-concentration steps. Based on their own experimental data, the authors underline the applicability of the method as a valuable alternative to classical proteomics or antibody-based assays in routine clinical medical research. In a second paper, [Sec-to-Cys]SEPP1 and [Sec-to-Cys]GPx3 recombinant proteins, both in non-labelled and stable isotope labelled forms, for quantitative selenoproteomics were synthesized by Konopka et al.197 by using a using a cell-free E. coli protein expression system. For this purpose, an amino acid mix containing all proteinogenic amino acids (except Met) was prepared and added to the cell-free E. coli synthesis medium. SeMet and 76SeMet were used for the preparation of 197SEPP1 and [Sec-to-Cys]GPx3 standards and [Sec-to-Cys]SEPP1 and [Sec-to-Cys]GPx3 spikes, respectively. The cell-free protein synthesis was carried out for 24 h at 30 °C with shaking at 1000 rpm. Subsequently, the synthesized [Sec-to-Cys]selenoproteins were purified by SDS-PAGE and triptically digested. The resulting peptides were characterized by UPLC-ES-MS/MS and submitted for database searching by Mascot. Matched peptides confirmed the presence of [Sec-to-Cys]GPx3 and [Sec-to-Cys] SEPP1. The [Sec-to-Cys]SEPP1 standard containing 78SeMet was successfully applied for the quantification of SEPP1 in human serum/plasma reference materials (BCR-637 and SRM 1950) by SSID-ICP-MS using double affinity high performance liquid chromatography separation.
Two papers report on the Hg–Se interaction by SEC-ICP-MS. Hg and Se binding biomolecules were detected in liver and kidney of reed deer and wild board from a Hg-rich mining area (Almadén, Spain) using two columns; a BioSep-SEC-S2000 GL 300–1 kDa; and a Superdex 75 10/300 GL 70–3 kDa.198 By means of this methodology, Se and Hg containing biomolecules were isolated from the samples using Tris–HCl (100 mM at pH 7.4) as extractant. Unfortunately, the extraction efficiencies reported by the authors are far from being satisfactory with values ranged from 19% to 5% for Se and Hg, respectively. Selenium and Hg fractionation by separation with SEC showed that a Se-containing peak and a Hg-containing peak coelute at high-medium molecular weight area (60–80 kDa). Based on data reported in the literature, the authors suggest SelP as the compound responsible of the Se-containing peak and subsequently speculate on the role of SelP in Hg–Se interaction. However, as the authors also mentioned, the use of complementary techniques to establish the structure of Se-containing peak is required to correctly address the role of Se compounds on Hg toxicity. In the second paper199 the protective effect of Se in MeHg+ poisoned rats after SeIV treatment was evaluated. SEC-ICP-MS data revealed that Hg was mainly distributed in serum samples in molecular mass fractions of 21, 40 and 75 kDa whereas Se was mostly associated to 40 and 75 molecular fraction. Authors suggested the 75 kDa – Hg and Se binding protein as the responsible of the decreased urinary Hg excretion in MeHg+ poisoned rats after Se treatment. A 75 kDa protein was further identified as albumin by structural characterization with MALDI-TOF-MS.
Studies on the conversions and transformations of Se in plants by HPLC-ICP-MS and HPLC-ES-MS/MS have been a topic of much research in the year under the review. Concentration and speciation of Se in mug bean sprouts exposed at different SeIV concentration levels (from 1 to 12 mg L−1) were evaluated by GFAAS and HPLC-ICP-MS.200 The maximum Se concentration in mug bean sprouts was of 7275.0 mg kg−1 as measured by GFAAS. Selenium compounds were isolated from the samples by using a multi-step approach. Proteins were first precipitated from the samples with HCl at pH 4.5 and then redissolved by applying Tris–HCl at pH 7.5. The resulting solution was treated with trypsin during 4 h at 50 °C followed by protease and further incubation at 50 °C for another 4 hours. For the separation of the species, a Zorbax 300A SB-C18 (100 mm × 2.1 mm i.d., 3.5 mm) column was employed and formic acid 0.1% m/v:acetonitrile (70:30) was used as mobile phase. The eluate was analysed by ES-MS/MS. The HPLC/MS parameters such as capillary fragmentation voltage, collision energy and mobile phase composition were optimized in detail and Se species were monitored by using multi reaction monitoring (MRM) mode. The identification of selenoamino acids was performed by comparing the MS spectra of the sample with those of standards at the corresponding retention times. Unfortunately, MeSeCys was the only Se-species identified which accounted for only 3.9% of the total Se. A similar approach was used for Se species determination in Se-enriched Heriacum erinaceus (lion’s mane mushroom) using RP-HPLC-ICP-MS and HPLC-ES-QTOF-MS.201 Se enriched mushroom were growth in presence of 300 mg of SeIV. Determination of Se species was achieved by reversed-phase HPLC-ICP-MS before enzymatic hydrolysis with protease. The main Se-species found were SeMet (20.7 mg g−1 d.w.) and MeSeCys (0.3 mg g−1 d.w.), representing 50% of total Se. Compared to other mushrooms, the amount of organoseleno compounds accumulated by Heriacum erinaceus was very high and similar to that of selenized-yeast. With the aim of detecting similarities in Se metabolisms between both fungi, a methodology already employed for Se-yeast was applied for detecting Se-adenosyl compounds (a selenometabolite usually found in Se-yeast) in Se-enriched Heriacum erinaceus. For this purpose, samples were submitted to water extraction and the extracts were further purified by SEC-RP-ICP-MS. Finally, low molecular weight fractions were collected and analysed by HPLC-ESI-QTOF-MS. Five Se species were identified in the selenized mushroom H. erinaceus. Four of them (SeMet, MeSeCys, Se-methyl-5-selenoadenosine and Se-methyl-5-selenoadenosine Se-oxide) are also found in selenized S. cerevisiae. In another study, Se-species in Se-enriched strawberries were determined by HPLC-thermoreduction (TR)-HG-AFS.202 Se-enriched strawberries were hydroponically growth and exposed to SeIV, SeVI and SeMet by either irrigation or foliar application at concentrations between 0.5–500 mg L−1. Once collected, the samples were submitted to enzymatic treatment by using a mixture of protease and lipase followed by 2 minutes of tip-sonication. Separation and detection of Se species was performed by anion exchange (Hamilton PRP-X100, 4.1 × 250 mm) chromatography, followed by on-line Se species conversion into volatile hydrides by means of a thermoreduction step by adding a KBr solution prepared in HCl at 150 °C. Subsequently, hydride generation and detection by AFS was performed. LODs of 0.4 μg L−1, 2.6 μg L−1, 0.2 μg L−1, 0.5 μg L−1 and 2.6 μg L−1 were obtained for SeIV, SeVI, SeCys, MetSeCys and SeMet, respectively which were low enough to detect either species in samples. A Certified Reference Material (SELM-1 Se-enriched yeast) was employed for analytical method validation. Among methods tested, foliar spray of SeIV was found to be the best procedure for performing biosynthesis of selenoamino acids. Under these conditions, SeCys, MetSeCys, SeIV, SeMet, and SeVI, were detected and their concentration accounted an 84% of the total Se content. Unfortunately, details about SeCys integrity preservation during sample treatment are not given which make it difficult to properly evaluate the validity of the reported data. Additionally, identification of SeCys by means of using a PRPX-100 column should be properly checked by other separation techniques or by employing structural assignment techniques such as ESI/MS/MS. The production of Se species203 by chickpea plants cultivated in different amounts, (10, 25, 50, and 100 μmol L−1) of SeIV and at different times of exposure was explored by HPLC-ICP-MS. SeMet, SeCys, SeIV, SeVI and GPx were detected in varying concentrations in shoots and roots at concentration levels directly correlated with the amount of Se supplemented and the time of exposure. Finally, the concentration and bioaccessible fraction of Cu, Co and Se in Cape gooseberry was assessed by combining an in vitro gastro intestinal digestion method with SEC-ICP-MS.202 With the aim of evaluating the content of metal in the soluble fraction, Cape gooseberry samples were treated with different extracting solutions: 10 mM ammonium acetate (pH 5.5); 30 mM Tris–HCl (pH 7.4) and 2% sodium dodecyl sulfate (SDS) in water (pH 7.4). The resulting filtrates were analysed by SEC-ICP-MS. The extraction efficiency was highly dependant on the type of metal/metalloid and the extracting solution employed. The best efficiency of extraction of Co was obtained for ammonium acetate (56%) and Tris–HCl (60%); for Cu was obtained for SDS (66%) and for Se the best extraction efficiency was achieved after applying SDS (48%). The SEC-ICP-MS analysis of the extracts revealed that metals are distributed mostly in medium and low molecular mass range between 44000 and 1000 Da. The berries were subsequently subjected to in vitro gastrointestinal digestion assay using pepsine and pancreatine. The resulting fractions were analyzed to identify the metal species that become bioavailable for human body functions. The metal bioaccessible fraction accounted for 90% of the total metal content. The SEC-ICP-MS analysis of the gastrointestinal extracts and subsequent confirmation with reference standards confirmed the presence of vitamin B12 and SeMet as the forms of elements available for human organisms. However, structural assignment using ES-MS/MS should be performed to obtain a better characterization of the compounds detected in the bioaccesible fraction.
Once again a report has focused on Se speciation in Se-enriched food and supplements. The Se species in Se-yeast were determined using a non-conventional approach consisting of HPLC-HG coupled to microwave induced plasma (MIP) sustained by nitrogen.204 Selenium species were quantitatively released from Se-yeast by adding to 100 mg of sample 2 mL of 4 mol L−1 methanesulfonic acid and then heating at 120 °C during 16 h. Se species were further separated on a reversed phase column Gemini C18 (50 × 3 mm, 5 mm) by using HFBA 0.08% m/v: methanol (92:8) as mobile phase. Subsequently, the effluent from the column was passed through a 100 cm reaction coil submerged in a water bath at 60 °C which was treated with an alkaline solution (6% m/v K2S2O8 and 3% m/v NaOH) to transform the Se species into SeIV prior to hydride generation (NaBH4 2% m/v, NaOH 0.3%) and detection by MIP. The results showed that the method offered good recovery values (100%) for Se-enriched yeast, with limit of quantification (LOQ) values for SeIV and SeMet of 11.9 μg Se g−1 and 104 μg Se g−1, respectively. The results were consistent with those provided when using HPLC-ICP-MS.
The number of papers focused on Se speciation in environmental samples has been scarce this year. The use of ultrasonic assisted cloud extraction to isolate Se and Sb prior to their determination by HGAAS has been reported.33 The SeIV and SbIII were chelated with 3-amino-7-dimethylamino-2-methylphenazine hydrochloride (Neutral red, NRH) in presence of pyrogallol and cetyltrimethylammonium bromide (CTAB) at pH 6.0. The mixture was sonicated (300 W, 40 kHz) at 35 °C for 10 minutes and then centrifuged for 5 min at 4000 rpm. The SeIV and SbIII content in the surfactant-enriched phase was measured by HGAAS. The LOD values obtained were of 2.45 and 3.60 ng L−1 with enrichment factors of 155 and 120 SeIV and SbIII, respectively. SeVI remained in the aqueous phase. Total Se was determined after reduction with L-cysteine and tartaric acid. The concentration of SeVI was calculated by difference between total iSe and SeIV. Validation using CRMs (SRM 1573a tomato leaves and SRM 1643e simulated fresh water-trace elements) was performed and good agreement was found. The method was successfully applied to water, food and fruit juices of a different nature with good recovery values (93–100%). Chen et al.205 described a novel method for the speciation of inorganic Se based on solidified floating organic drop microextraction (SFODME) followed by electrothermal vaporization inductively coupled plasma mass spectrometry (ETV-ICP-MS). APDC was used as both, the chelating reagent in SFODME, and the chemical modifier in ETV. SeIV and SeVI were separated at the pH range of 2.0–5.0. Total Se was determined after the reduction of SeVI to SeIV by using 5 mol L−1 HCl. SeVI concentration was obtained by subtracting SeIV from the total Se concentration. This approach offers a LOD for SeIV and SeVI of 0.26 and 0.34 pg mL−1. The developed method was also applied to Se speciation in spiked natural water samples with recovery values in the range of 93.2–106%. Finally, SeMet SeOMet, SeIV and SeVI were simultaneously determined in high-ionic-strength waters using anion exchange HPLC-on-line coupled to ICP-MS and ES-MS/MS.206 All Se species were separated in a single AEC run on a Dionex AS16 (analytical 25 cm 10/32 with guard 4 cm 10/32). The paper contains detailed discussions of different experimental parameters affecting AEC when coupling with molecular mass spectrometry for structural elucidation and confirmation. A LOD value as low as 5 ng L−1 was obtained for all Se species either using ICP-MS or ES-MS/MS as detectors. The proposed method was successfully applied to Se-species determination in industrial water effluents. Water samples were first submitted to an extensive sample treatment consisting of a cation-exchange solid-phase extraction with a DOWEX 50WX8 50–100 mesh cation-exchange resin to remove the high concentrations of sulfate and chloride from industrial process waters. The developed method allowed authors to determine SeMet and SeOMet at nanogram-per-litre concentrations in effluent collected at a coal-fired power plant’s biological remediation site. It is the first time that these species are found in water samples, outside of a biological identity.
3.9 Sulfur
Two papers have reported ssIDMS methods for the determination of biomolecules on the basis of their sulfur content. In the first of these 34S-labelled methionine was obtained from yeast, grown in a medium enriched with 34S (>91%) in the form of sulfate as species-specific spike.207 The methionine was extracted from yeast using enzymatic digestion with protease XIV and extracted for use as a species specific spike via preparative reverse phase HPLC. The 34S-labelled methionine standard solutions obtained were characterised, both in terms of 34S isotope enrichment (82.7 ± 0.6%) and total sulfur concentration (396 ± 6 μg g−1) by reverse ID-HPLC-MC-ICP-MS. This was by the use of a natural abundance methionine standard solution and the MC-ICP-MS instrument was operated in the pseudo-high resolution mode to avoid spectral interference from e.g.16O2. A RP wide pore C18 column was used for these separations with a mobile phase of 75 mmol L−1, at a pH of 7.4, and 2% MeOH. The identity of the 34S-labelled methionine isolated by preparative HPLC and the 34S isotopic enrichment was confirmed by GC-MS after derivatisation, with N-tert-butyldimethylsilyl-N-methyltrifluoroacetamide and 1% tert-butyldimethylchlorosilane as a catalyst for this procedure. Human blood plasma samples were spiked with the 34S-labelled methionine spike, the plasma proteins precipitated with trifluoroacetic acid, centrifuged and the supernatant collected. Following ssID-HPLC-ICP-MS the recoveries, for samples spiked at different concentration levels, ranged between 98.4 and 100.5%. The LOD and LOQ values were evaluated by repeat analysis of the blank solution and were calculated to be 0.07 and 0.2 μg g−1 respectively. Finally, 13 unfortified human plasma samples were analysed by both ssID-HPLC-ICP-MS and a routine amino acid analyser based method. For all 13 samples the results obtained by each method were in very good agreement for methionine concentrations ranging between 14 and 35 μmol L−1. The second paper covered here describes a ssID-AE-HPLC-ICP-MS method utilising 34S enriched methionine and cysteic acid, which were also extracted from a yeast culture (Pichia pastoris).208 Two different instruments were initially used as a detector for the HPLC separations, a single quadrupole ICP-MS and a triple quadrupole ICP-MS. In each case O2 was used as a reaction gas so that the S content in the samples was measured as SO. The LOD, assessed using a 60 μmol L−1 standard, for 32S16O was similar for both instruments (0.45 μmol L−1 single Q and 0.14 μmol L−1 triple Q) but much improved for 34S16O by the triple Q instrument (0.56 μmol L−1) compared with the single Q instrument (16 μmol L−1). The standard uncertainty of the measured isotope amount ratio was also significantly lower for the triple Q instrument compared with the single Q instrument, 0.6 and 2.8% respectively. In the light of this only the triple Q instrument was used for the remainder of the study. This allowed the 34S enriched methionine sulfate and cysteic acid to be characterised by reverse ssIDMS with expanded relative uncertainties (k = 2) of 12 and 8% respectively. Finally, ceruloplasmin was quantified using each spike material with relative standard uncertainty of 5% in each case. The authors conclude that in principle any protein containing either methionine and/or cysteine can be quantified using the developed methodology and that this could overcome reported poor comparability of results from the different measurement platforms currently in use. Both papers are recommended reading for those workers wishing to work in this field.3.10 Thallium
Two papers report on Tl speciation methods this year. In the first of these TlIII, as a complex with diethylenetriaminepentaacetate (DTPA), was selectively retained on an alumina column modified with sodium dodecyl sulfate (SDS) and eluted with 40% HNO3.209 The unretained Tl, assumed to be TlI, was measured in the samples after passage through the alumina-SDS column. All Tl species were then quantified using ICP-MS. The method LOD values were 25 pg of TlI and 160 pg of TlIII in 10 mL samples. The method proved suitable for measuring the TlIII content in the presence of an excess of TlI of up to 4000 ng. No interference from other metal ions, such as a 500-fold excess of Pb(II) and Cd(II), and a 2000-fold excess of Zn(II), respectively, was observed. The second paper covered here, by workers of the same research group, reports an HPLC-ICP-MS method for Tl speciation using an AE column.209 The mobile phase was 100 mMol L−1 ammonium acetate, 5 mMol L−1 DTPA at pH 6.2 flowing at 1.5 mL min−1 and both Tl species were baseline separated in 500 s. The study focused on the extraction and determination of Tl species in white mustard plants, and includes a detailed discussion of the processes involved, finding that species stability was only possible when Tl was extracted from fresh plant tissues and only short-term storage of an extract from fresh plant tissues was possible.3.11 Tin
Two papers report methods aimed at achieving the Environmental Quality Standard (EQS) for TBT in whole water samples, 0.2 ng L−1 as Sn, of the EU Water Framework Directive. The first of these methods reports the quantification of TBT in whole water matrices containing SPM and humic acid fractions using ssID-GC-ICP-MS.210 To develop the method, water samples containing BCR 646, added to give TBT concentrations ranging between 0.8 and 1.25 ng L−1, and humic acids (5 mg L−1). A 119Sn enriched TBT spike was added to these solutions to allow ssIDMS to be performed. Three different strategies, 25% KOH in MeOH, 0.5 mol L−1 tartaric acid in 20% v/v MeOH, and acetic acid/MeOH, all followed by pH adjustment to 5, derivatisation and extraction into n-hexane, to extract the TBT from the SPM/humic acid/water mixture were evaluated. Recoveries of the TBT, added via the BCR 646, ranged between 75 and 85% with RSDs of up to 5% with the highest recovery obtained for the acetic acid/MeOH procedure. This procedure was investigated further and the addition of 20 mL of 1:1 acetic acid/MeOH to one L of water sample and shaking for 30 minutes proved to be the optimal conditions for low SPM (equivalent to 0.237 ng L−1 TBT) and humic acid concentration (5 mg L−1) samples, giving recoveries of the added TBT of between 95 to 105%. The expanded uncertainty of the method (k = 2) was 15% which is considerably lower than the WFD limit of <50%. The method was then applied to two water samples, containing SPM and humic acids, from an interlaboratory comparison. The results obtained were 108 and 111% recoveries compared to the robust mean of the interlaboratory comparison. However, the method was not suitable for samples with a high SPM and humic acid content. For this type of sample the method was heavily modified by adjusting the sample pH to 2 with HCL, shaking for 30 min, filtration and separate analysis of the filtrate and separated particulates. Incomplete sample/spike equilibration was observed if the IDMS spike was added before filtration and each fraction was therefore spiked after filtration. After this modification recoveries of TBT from model high SPM/humic acid samples ranged between 87 and 100%. The second paper on this topic evaluated two approaches, ssIDMS and either head space (HS) SPE or LLE, followed by derivatisation, extraction into hexane and analysis by GC-ICP-MS211 The authors report that HS-SPE method gave poor repeatability, with an RSD > 20%, and that the multistep analytical process led to an increased risk of contamination and the extraction time of 40 minutes was too time consuming. For the LLE method 1 L water samples were spiked with 119Sn enriched TBT followed by pH adjustment to 5 with 10 mL of an acetate buffer, derivatisation with 1% NaBEt,4 extraction into 1 mL of hexane and analysis of the extract. The LOQ of the optimized method, 0.06 ng L−1, met that required by the WFD and thus is suitable for use to ascertain if water samples meet the EQS. A full uncertainty budget was estimated for the LLE procedure, and is discussed in detail, and ranged between 12 and 27% at the EQS TBT concentration and between 28 and 57% at the LOQ concentration. Both of these papers are recommended reading for workers in this field.
Finally in this section an HPLC-ICP-MS method for the determination of DPhT, DBT, TPhT and TBT has been reported.212 The OTCs were separated in 30 minutes on a RP column using a gradient elution programme with the mobile phase consisting of MeOh and 10% acetic acid in water and adjusted to pH 3 with trimethylamine. The LOD values ranged between 0.28 to 0.59 μg L−1 for the four OTCs under study. The method was applied to determine the OTC content in vegetable oils, after a low temperature precipitation process in which 1 g of oil was added to 10 mL of MeOH, vortex mixed for 10 minutes and then refrigerated for 2 hours at −20 °C. The MeOH layer was then removed, the extraction repeated and the MeOH layers combined followed by solvent evaporation, dissolution of the residue in 1 mL of MeOH, clean-up with carbon black and filtration before injection onto the HPLC column. Spike recoveries of the OTCs from various vegetable oil types ranged from 73 to 99%, with RSDs of between 4.3 and 9.5%. Finally, the method was applied to locally purchased vegetable oil samples of which some contained DBT, 3.1 to 12.1 μg kg−1, and TBT, 13 to 29 μg kg−1.
3.12 Vanadium
A method for the determining the content of V-porphyrins in crude oil by HPLC-FI-ICP-MS has been reported this year.213 The crude oil sample was first fractionated by preparative liquid chromatography and the porphyrin containing fractions, eluted in either MeOH or 50:50 v/v MeOH/toluene, collected. Subsequently, the porphyrin fractions were then separated on a 250 mm RP column and eluted with two different mobile phases, either MeOH or MeOH:toluene (80:20; v/v) depending on the sample solvent, flowing at 0.8 mL min−1. To allow direct coupling of the HPLC eluent to the ICP-MS the spray chamber was cooled (temperature not given) to reduce solvent loading on the plasma, the torch injector diameter was 0.8 mm and O2 was added at 100 mL min−1 to prevent C build up on the sample and skimmer cones. A vanadyl octaethylporphine standard, prepared in the same solvent as the mobile phase, and injected post-column directly into the plasma was used to quantify the V-porphyrins. An internal standard, 20 μg L−1 Ge in methanol, was teed into the HPLC eluent. The minimal detectable concentration, 3 standard deviations of the baseline V signal, was 0.4 μg L−1 as V. The concentrations of V as V-porphyrins were between 2.7 and 11 mg kg−1 in the fractions, which were close to the total concentration of V in the porphyrin fractions of the studied crude oil.
3.13 Zinc
A study into the combined effects of N, S and Zn fertilization on the molecular speciation and tissue compartmentation of Zn in Durum wheat grains has been published this year.214 Durum wheat plants were grown in soil fertilised with N at two concentrations (50 and 400 mg N kg−1 soil) and Zn at three concentrations (0.5, 5.0 and 50 mg Zn kg−1 soil plus foliar feeding at the highest concentration). The whole wheat grains where then harvested and the outer grain, embryo and endosperm separated. The determination of total Fe, P, S and Zn content was by ICP-MS, after an HNO3/H2O2 digestion process, whilst the N content was measured using a combustion IR method. The endosperm samples were subjected to an ammonium acetate extraction protocol (100 mmol L−1, pH 7.5 at 0 °C) followed by centrifugation and filtration through a 0.45 μm filter and a 100 kDa ultrafilter. Subsequently, the extracts were analysed by SEC-ICP-MS (no mobile phase details are given) and, after further processing including freeze drying, re-dissolution and digestion with trypsin, by HPLC-ES-MS. Two size fractions of Zn containing proteins, of 10–30 and 50–100 kDa, were identified by SEC-ICP-MS and the HPLC-ES—MS results showed that these fractions consisted of 57 and 161 Zn-proteins respectively. The type of Zn-proteins detected, Zn uptake to the grain, internal transfer mechanisms and Zn storage and homeostasis in then discussed in detail. The authors conclude that the results of the study will prove useful in breeding programmes aimed at producing cereals with a high endosperm Zn content.The speciation of Zn in human breast milk has been investigated using SEC-ICP-MS.215 The mobile phase was a Tris–HCl buffer at a pH of 7 flowing at 0.5 mL min−1 with Ga used as an internal standard. Ultrafiltration was used to separate the whey fraction from other milk components, such as fat and casein, prior to analysis. Zinc was found to be mainly associated with immunoglobulins and citrate in human milk whey whilst in formula milk Zn was mainly in the citrate form with only very low amounts of Zn associated with proteins. No significant differences in Zn speciation were found between pre-term and full-term milk samples.
4 Biomolecular speciation analysis
An established authority in the use of inorganic mass spectrometry for the measurement of macromolecular biomolecules has produced a useful summary216 covering the different ways that ICP-MS is now being used in this field. Although the title ““heteroatom-tagged” quantitation of proteins”, is slightly misleading, as it implies addition of an atom detectable by ICP-MS when in fact the heteroatom is part of the molecular structure. It is probably better to classify the application areas covered as being “direct” i.e. measuring the molecules via the hetero-atoms present and “indirect” i.e. tagging the molecule with a moiety detectable by ICP-MS. The paper describes the measurement of biomolecules via their naturally occurring hetero-atoms, focusing mainly on S and P but also identifying less frequently targeted atoms such as I and Se that could also be used and the use of methods to label or tag molecules that would otherwise be unresponsive by ICP-MS with metals that make them responsive. It makes the point that the use of ICP-MS, whilst complementary to organic MS, has a number of advantages such as: less sensitivity to matrix effects; ability to perform absolute quantitation without an authentic molecular standard; and high detectability due to the inherent sensitivity of the instrumentation. Finally, the article notes the importance of triple quadrupole instruments, which have improved the measurement of S and P to such an extent compared to conventional quadrupole ICP-MS, due to the LOD values that they deliver, that it is now feasible to measure macromolecules containing these elements in real world applications and not just model or spiked systems. All in all, a useful update on the state-of the art in this area. A review summarising the analysis of metallothioneins (MTs) has also been published this year.217 The use of ICP-MS and ES-MS as well as matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF)-MS as detectors are covered. The combination of CE or HPLC with ICP-MS and ESI-MS for the analysis of MTs isoforms is also discussed as is the use of four different HPLC separation modes, SEC, AEC, RPLC and HILIC. The coupling of RPLC with ICP-MS and ESI-MS in parallel for the analysis of MT sub-isoforms is also highlighted.4.1 Direct macromolecular analysis
The ability to measure S to accurately quantify proteins and some peptides without having to use an authentic molecular standard is a clear advantage of inorganic MS compared to molecular MS. Whilst it is necessary to identify the molecule to translate the mass of quantified S for each chromatographic peak into the individual molecular mass, this is much easier than potentially having to obtain a pure standard for every compound being studied. This is similarly true for the analysis of DNA fragments and nucleotides by the measurement of the P heteroatom present in the phosphate group. In an unusual application area, the measurement of proteins in snake venom,218 was used as a model system to show the capabilities of using ICP-QQQ-MS to measure S and provide absolute quantitation via IDMS using enriched 34S, as well as compound identification using ES-MS detection. The method used a reversed phase narrow bore (150 × 0.3 mm, 3.2 μm particle size) BIOshell A400 C4 column operated at 80 °C separate proteins in the mass range 7 to 47 kDa. The column was coupled via a capillary LC total consumption nebuliser interface to ICP-QQQ-MS and the following gradient elution conditions used under different flow programmes to separate the proteins: formic acid 0.2% (v/v): water to formic acid 0.2% (v/v): acetonitrile. The methodology was tested using a mixture of 4 standard proteins, N-(tert-butoxycarbonyl)-L-methionine (BOC-met), cytochrome-C, transferrin and BSA, achieving excellent baseline resolution and quantitative column recoveries in the range 96–99%. The combination of post-column addition of isotopically enriched 34S and a generic S-containing internal standard (BOC-met) spiked into the sample, provided compound independent detector response and thus protein quantification without the need for authentic standards. The accuracy of the method was established by comparison with commercially available standard proteins and the results were statistically indistinguishable, with LOD values ranging from 7 to 15 fmol depending on the protein. The suitability of the strategy for real sample analysis was illustrated for the quantification of the individual proteins in a snake venom sample. Parallel LC-ES-QTOF analysis was required for identification and to allow for full quantitation of the proteins. Unfortunately, due to the complexity of the sample the resolution achieved using the developed HPLC separation was not optimal and certainly did not meet the specification set by the authors that “so, as long as species are separated and peaks well resolved, absolute quantification can be accomplished in one chromatographic run, just requiring one generic sulfur-containing standard”. Additionally, although the ICP-MS peaks appeared to correspond well with those detected by ES-MS, due to the differences in response for the two detectors the profiles appear markedly different. The data presented showed that of the 27 peaks in the venom sample, 12 were identified, with 7 there was some equivocation of the identity and the rest remained unidentified. Clearly, with further development and verification work the analytical platform will become a powerful tool in protein studies. The quantification of a single pharmaceutical peptide using HPLC-ICP-MS and detection via the heteroatom S as 48SO+ has been reported.219 A RP separation on a C18 narrow bore 2.1 mm i.d. column required the use of a multi-step gradient elution with acetonitrile. As pointed out by the authors the change in plasma conditions as the composition of the eluent changes can lead to different sensitivities for the separated species that elute at different times. To overcome these effects two approaches were used: a membrane desolvation system, which reduced the organic load on the plasma; and calibration by suIDMS using post-column addition of isotopically enriched 34S as sulfate. Two different ICP-MS systems were used to overcome the many interferences on the S isotopes monitored. The first used a reactive gas in the cell to convert S to SO whilst the second was a triple quadrupole instrument, also using oxygen as the reaction gas. Interestingly the LODs for both instruments were almost identical at approximately 0.5–0.6 pmol, which was attributed to the use of the membrane desolvation system on the single quadrupole instrument, which allowed for more of the analyte to enter the plasma. The system was ultimately applied to the measurement of a single peptide, penetratin, one of the new group of cell-penetrating peptides that facilitate uptake of different entities into cells, in the presence of other peptides, including: desmopressin, substance P, parathyroid hormone and insulin. The main issue with the work was the low recovery of the analyte with each of the methods used to isolate the peptide from plasma proteins. In the case of the precipitation method that was finally chosen it was 70%, compared to 30% using a molecular weight cut-off filter. This drawback could presumably be overcome with further optimization work, or perhaps the use of species-specific IDMS.The measurement of P by ICP-MS as a direct method to quantify different macromolecules, including post-translationally modified proteins and DNA, has been reported. In a wide ranging review220 of the use of elemental mass spectrometry in phosphoproteomic applications (covering 79 papers) Maes et al. highlight the advantages of elemental MS compared to molecular MS techniques such as MALDI-MS and ES-MS/MS. Although identification and relative quantification of phosphopeptides is straightforward with these techniques, absolute quantification is more complex and usually requires specific isotopically enriched standards. However, the use of elemental mass spectrometry, and in particular ICP-MS, in phosphoproteomics-based experiments, makes absolute quantification possible. The paper covers the very basics of ICP-MS and then expands into the coupling of chromatography, including CE, with ICP-MS as well as the use of LA-ICP-MS and how they have been applied specifically to the challenging area of phosphorylated proteins. It shows that one of the most beneficial developments by far in this area is the variety of phosphopeptide enrichment strategies, which include immunoaffinity assays, where antibodies raised against a protein or against a specific phosphosite, are used to isolate phosphopeptides or a certain subset of them. Enrichment methods based on metal-assisted affinity chromatography, such as immobilized metal affinity chromatography or metal-oxide materials (TiO2) are applied even more often. In another application, ssIDMS was used by the National Metrology Institute of Japan221 for the quantification of DNA but this time using LC-MS/MS rather than HPLC-ICP-MS for the measurement. Digestion of the DNA by formic acid was used to form the nucleobases which were then quantified against the 13C and 15N labelled analogues. The digestion conditions (time and temperature) were optimized so that the reaction products were not further decomposed. The developed method could quantify lambda DNA with an expanded uncertainty of 4.6% using 10 fmol of sample. The results were validated using SF-ICP-MS in high resolution mode to measure P in the digested samples and the results showed good agreement with each other. Another method for the quantitation of DNA, this time synthetic structurally-modified oligonucleotides in drug discovery, used a high-throughput FI-ICP-MS approach to provide rapid and accurate quantification of P content in synthetic small interfering RNAs.222 Conventional quadrupole ICP-MS was used and the isobaric interferences on 31P+ were avoided by monitoring the 31P16O+ ion, which was formed in the plasma, not by addition of O2 but by “optimization” of the plasma conditions (carrier gas flow rate, sample depth, nebuliser flow rate, extraction lens voltage and plasma power). By using a well-plate autosampler the sample injection cycle time was reduced and a throughput of 39 s per sample was possible. The method required minimal sample consumption of 1 to 100 μL making oligonucleotide screening a practical possibility. Other advantages included excellent sensitivity (an LOD of 136 ng L−1 as P), wide linearity range and reduced solvent consumption. The FI-ICP-MS method was capable of accurate quantitation of a large number of oligonucleotide samples with significantly improved sample analysis throughput compared to conventional methods, such as UV spectroscopy.
4.2 Tagging and labelling for macromolecular analysis
In the area of indirect analysis of biomolecules by inorganic MS, work on tagging and labelling methods divides into two main areas of focus: the development of new tagging reagents with some enhanced utility, either analytically or mechanistically; and the application of tagging to specific measurement problems. This area of research is now sufficiently developed that review papers detailing the advantages of the approach and potential future areas of application are being published.The use of lanthanides as part of the tagging reagent detectable by ICP-MS is a well established approach which has recently been the focus of two short review papers.223,224 One,223 from the authors of the first work in the area, summaries the progress from using radioimmunoassay for the measurement of biomolecules via a radioactive tag, through the development of ELISA, fluorescent immunoassay, chemiluminescent immunoassay and electrochemiluminescent immunoassay, to the current developments using atomic spectroscopy as the detector (covering 58 papers). They make the point that the initial methods, despite great success, suffered from drawbacks such as: the requirement for radioactive tags; limited spectral window capacity for multiplex detection; and inability to provide absolute quantification of biomolecules. The use of ICP-MS to detect the tagged biomolecules has been shown to have a number of advantages for various applications, including the measurement of peptides, proteins, nucleobases, DNA, RNA as well as more disease specific biomarkers e.g. prostate specific antigen (PSA). The most important advantage over the earlier methods is the possibility for a greater level of multiplexing, due to the higher resolution between the signal for each tag, compared to the spectroscopic methods which suffer from spectral overlap limiting the number of elements that can be measured in a single sample. This was illustrated by reviewing the development and application of the mass cytometer, which fully exploits the multiplexing potential of metal stable isotope tagging. It allows for the simultaneous detection of many parameters in single cells, accurate immunophenotyping in cell populations and the modelling of intracellular signalling networks. Other advantages of using atomic spectroscopy include high sensitivity, wide dynamic linear range, and possibility for absolute quantification. The second review224 also covers the use of lanthanide tags for the analysis of proteins, DNA and RNA (57 papers were included), but focuses less on the development from the older methods and more on the common conjugation approaches and lanthanide-chelating tags used in the current applications. Along with their high sensitivity in ICP-MS the authors make the point that: lanthanides are low-abundance elements in nature and consequently the background levels in biological sample matrices are low, resulting in favourable LOD values; polyatomic interferences are rarely significant; the ionization efficiency of lanthanides is high due to low first ionization potentials. However this is not completely accurate as lanthanide elements do have significant interference due to the formation of oxides, which both reduce the sensitivity to some extent, but more importantly cause interference with other lanthanides in the series at m/z +16, which means some thought needs to go into the experimental design if multiplexing is required.
Some interesting developments in the area of new tagging reagents with enhanced functionality have been reported. All the work has potential applications in the clinical area, with two papers relating to analytical methods and the last interesting from a conceptual perspective, as it identifies how tagging reagents are developing, but rather than a specific analytical method it relates to monitoring uptake, tumour imaging and therapy. In an exciting new development Tang and co-workers225 have developed a sophisticated multifunctional tagging reagent based on Ga that has been designed to aid the generic pre-concentration of a class of post-translationally modified proteins, but also enables the measurement of a specific phosphorylated protein, in the presence of other such altered proteins. The inclusion of a magnetic nanoparticle in the tag allowed pre-concentration of the phosphorylated proteins and the presence of a linkage cleavable using a specific enzyme, tyrosine phosphatase, made it so specific that it was possible to identify and measure phosphotyrosine, without interference from the other Ga-tagged phosphorylated proteins. The clever rationale behind the developed tag stems from the stability of the peptide phosphate ester bond during collision-activated dissociation ES-MS/MS, which is improved by the use of a dinuclear Ga-complex for protection, suggesting a highly specific interaction between Ga and the phosphate ester group. Furthermore, Ga has a low first ionization potential (5.99 eV) and a low biological background, which makes for excellent analytical properties in the ICP-MS plasma. Using suID-HPLC-ICP-MS with isotopically enriched 71Ga, a method detection limit for phosphotyrosine of 30 amol was achieved with a short term precision of 5.7% (n = 5 at pmol level). The specificity of the method was demonstrated using a range of synthetic phosphorylated peptide sequences including: VIVQIGTLSEpYIK; VNQIGTLpSEpYIK; and extracellular regulated protein kinase 1 peptide (–pTEpY–) achieving a recovery >96% (n = 5). To demonstrate the application of the method it was used for the absolute quantification of phosphotyrosine in human breast cancer MCF-7 cells. The second clinical application paper226 was based on a sandwich type ICP-MS immunoassay. In this approach a magnetic nanoparticle with a Cs-doped core was coated with antibodies to “capture” the antigens under investigation, any remaining active sites were pacified with BSA and the functionalised particle was reacted with the serum sample. A second silica based “reporter” nanoparticle containing a metal and dye molecule were coated with antibodies, such that there was a different metal/dye composition for each antigen. The composition of the three different nanoparticles were: Y/RhBITC (rhodamine B isothiocyanate) plus antibody for C-reactive protein a biomarker for inflammation; Cd/RhBITC plus antibody for alpha-fetoprotein a cancer biomarker; and Au/X-rhodamine-5-(and-6)-isothiocyanate plus antibody for neuron-specific enolase a biomarker of neuroendocrine tumours. The ICP-MS response for each of the metals was used to determine the concentration of the biomarker. The multifunctional magnetic nanoparticle was used not only for magnetic extraction of the target analytes but the Cs signal was also used to correct the response for any loss of the particles during the process. However, it wasn’t clear in the paper how excess Cs doped particles, that did not react with the second nanoparticle or which were not fully reacted with the biomarkers, were removed prior to analysis. Although not evident in the results, without this step the correction would lead to a low result. The LOD for the three biomarkers was between 0.35 and 0.77 μg L−1 and recoveries of 83–125% were obtained for spiked serum samples. Although the authors claim that the method is rapid with a short analysis time, the fact that the samples were reacted with the first magnetic nanoparticle for 2 hours, followed by another 2 hours reaction with the reporter nanoparticle, means that the method would be of little real clinical utility, considering that the current immunochemical methods for these analytes take a matter of minutes. The final novel tagging reagent227 with multi-functional properties is based on labelling a surface modified magnetoferritin protein “cage” with radioactive Rh. In this way it is possible to transport a radionuclide for cancer treatment, which also has MRI contrast agent properties for imaging the tumour. This allows for the progress of the treatment to be monitored non-invasively using MRI.
4.3 Elemental and molecular imaging
Although not focusing specifically on elemental speciation a review focusing on recent new developments in the design of rapid response ablation cells for LA-ICP-MS (covering 165 papers) will help to improve understanding of the options available for such speciation studies and high-resolution bioimaging in general.228 Covering cell designs, transport connections and the characteristics of different ICP-MS mass analysers required for high-resolution bioimaging, the review focused on papers written in English over the last 5 years, which report cell designs supported by data that demonstrate a characteristic pulse response width of 100 ms or less. From a LA perspective the optimal figures of merit for the technique including sample-throughput, sensitivity and spatial resolution, depend to a large degree on the ability of the ablation set-up to rapidly remove the aerosol from the ablation site to the plasma, in a manner that minimizes the mass loss and the dispersion induced in the aerosol cloud. The review describes the most recent ablation cell designs which have reduced the pulse response duration for a single laser shot to <10 ms, using the criterion of the full peak width at 1% of the height of the maximum signal intensity. The impact of the technical developments in LA-ICP-MS systems on emerging application areas, including multiplexed metal-tagged antibody detection (for immunohistochemistry), nanoparticle and compound hypo- and hyper-accumulation and (intra-) cellular/histological studies, are also discussed in some detail. However it is clear that for LA systems to be useful to the clinical community for diagnostic purposes there are still serious hurdles to be surmounted, in particular whether the applications can be sufficiently clinically validated for use, whether these validated methods can be carried out in a suitable time frame and most importantly what additional clinical information do they provide. The example in the review of a paper from 2012 (ref. 229) that constructed a 46-plate reference atlas of the quantified 3D distribution of Fe, Cu and Zn in a mouse brain, shows a high degree of ambition to develop a highly difficult and sophisticated workflow, encompassing high quality analytical measurements and data analysis. Using a 80 μm spot size and 160 μm s−1 scan speed each of the 46 slices with dimensions of 30 μm thick × 10 mm2 area were ablated and the trace elements measured. The analytical run took 158 h (22 working days) and must have amassed a large data file of points for the three analytes. However, this also goes to demonstrate the short comings of the current state of the art from a diagnostic perspective: the time taken only making it feasible for research groups to use; the difficulty in using experimental designs with replication, due to the time to measure each individual sample; the lack of molecular information to put the trace elements in context (although the review does discuss methods where this is possible); the issues around analytical validation when so few standards and quality control materials are available, particularly with such long run times; and the not insignificant clinical validation requirements. Clearly, there is huge potential for imaging using the analytical hardware as described in the review, but are the application areas currently being discussed really feasible as far as clinical and medical diagnostics are described? Or are they only useful to demonstrate what, in its most useful form of LA-ICP-TOF-MS, this highly expensive instrumentation can achieve for “model” applications?The potential for mapping the elemental and speciation distributions in single cells rather than a whole colony or community of cells is now being realised in some work and two recent papers show the range of instrumental approaches possible. In the first Malucelli et al.230 review (covering 103 papers) the indirect detection techniques based on labelling with fluorescent dyes, and label free direct methods, such as PIXE, proton backscattering spectrometry, scanning transmission ion spectrometry, nano-SIMS, XRF microscopy, complemented by X-ray imaging. It is clear from the review that the development of new fluorescent probes for the chemical sensing of metal ions has only partially been successful. A number of highly-selective chemosensors have been synthesized, showing the capability to detect single elements specifically visualizing their content within cells and in a few cases, accurately quantifying the amount. However, none of these probes allow the assessment of the intracellular spatial distribution of the elemental concentration in single cells, in terms of concentration and location. Direct or label-free imaging methods, which make use of mechanisms exploiting interactions with charged particles, photons, and atoms have been successful. In this regard, X-ray microscopy based on a synchrotron light source has gained increasing attention in the last few years due to its high sensitivity and the improvements that have been achieved in X-ray sources and focusing optics. In particular, XRF microscopy is a highly sensitive method for mapping trace element (Cd, Co, Cr, Cu, Fe, Mn, Ni, and Zn) distribution in single cells at nanoscale resolution. However, the low reproducibility and repeatability of these measurements is one of the major difficulties in achieving statistical significance in single cell analysis. The use of ICP-MS for single cell analysis231 based on labelling techniques for biomolecules has also been reviewed. The paper covers both the ICP-MS-based methodology and selected applications in immunoassay, disease detection, drug screening and nanoanalysis at a single cell level. The authors compare the MS based labelling methods to flow cytometry, fluorescence microscopy, CE and microfluidic chips. In these methods, single cells are usually stained by a fluorescent label (e.g. fluorescein, quantum dots, green fluorescent protein, etc.) and then detected via the fluorescent signal. However, simultaneous analysis of multiple parameters in a single cell is challenging because of the overlap in fluorescent spectrum. In addition, the linear range is relatively narrow, making it difficult for accurate quantification, especially when comparing signals which differ considerably.
Abbreviations
| AB | Arsenobetaine |
| AC | Arsenocholine |
| AE | Anion exchange |
| AEC | Anion exchange chromatography |
| AF | Atomic fluorescence |
| AFS | Atomic fluorescence spectrometry |
| APDC | Ammonium pyrrolidine dithiocarbamate |
| ASU | Atomic spectrometry update |
| CE | Capillary electrophoresis |
| cGPx | Cellular glutation peroxidase |
| CPE | Cloud point extraction |
| CRM | Certified reference material |
| CV | Cold vapour |
| CZE | Capillary zone electrophoresis |
| DBD | Dielectric barrier discharge |
| DBT | Dibutyltin |
| DCM | Dichloromethane |
| DDTC: | Diethyldithiocarbamate |
| DLLME | Dispersive liquid–liquid microextraction |
| DMA | Dimethylarsinic (include oxidation state if known) |
| DMDTAV | Dimethyldithioarsinic acid |
| DNA | Deoxyribonucleic acid |
| DPhT | Diphenyltin |
| DTPA | Diethylenetriaminepentaacetate |
| DTPA | Diethylenetriaminepentaacetic acid |
| EDTA | Ethylenediaminetetraacetic acid |
| EQS | Environmental quality standard |
| ERM | European reference material |
| ES-MS | Electrospray mass spectrometry |
| ET-AAS | Electrothermal atomic absorption spectrometry |
| EU | European Union |
| FAAS | Flame atomic absorption spectrometry |
| FI | Flow injection |
| GC | Gas chromatography |
| Gd-BOPTA | Gadobenate |
| Gd-BT-DO3A | Gadobutrol |
| Gd-DOTA | Gadoterate |
| GD-DTPA | Gadopentetate |
| GFAAS | Graphite furnace atomic absorption spectrometry |
| HAS | Human albumin serum |
| HFLLME | Hollow fiber liquid microextraction |
| HG: | Hydride generation |
| HGB | Haemoglobin |
| HILIC | Hydride generation |
| HILIC | Hydrophilic interaction liquid |
| HPLC | High performance liquid chromatography |
| IAEA | International atomic energy agency |
| iAs | Inorganic arsenic |
| ICP-MS | Inductively coupled plasma mass spectrometry |
| ICP-OES | Inductively coupled plasma optical emission spectrometry |
| Id | Internal diameter |
| ID | Isotope dilution |
| iHg | Inorganic mercury |
| ILLLME: | Ionic liquid liquid liquid microextraction |
| LC | Liquid chromatography |
| LLE | Liquid–liquid extraction |
| LOD | Limit of quantification |
| LOQ | Limit of detection |
| MAE | Microwave assisted extraction |
| MALDI | Matrix-assisted laser desorption ionization |
| MeHg | Methylmercury |
| MeOH | Methanol |
| MIP | Microwave induced plasma |
| MMA | Monomethylarsenic |
| MMDTAV | Monomethyldithioarsonic acid |
| MNP | Magnetic nanoparticle |
| MSPE | Magnetic solid phase microextraction |
| MT | Metallothionein |
| NP | Nanoparticle |
| OTC | Organotin compounds |
| PBDE | Polybrominated diethyl ether |
| PCB | Polychlorinated biphenyl |
| PIXE | Particle-induced X-ray emission |
| RNA | Ribonucleic acid |
| RP | Reverse phase |
| RSD | Relative standard deviation |
| SDS | Sodium dodecyl sulfate |
| SEC | Selenosugar |
| SeCys | Selenocysteine |
| SeCys2 | Selenocysteine |
| SeGal | Silica-coated magnetic nanoparticle |
| SelP | Solidified floating organic drop microextraction |
| SelP | Selenoprotein |
| SeMet | Selenomethionine |
| SeSug | Sodium dodecyl sulfate |
| SFODME | Solidified floating organic drop microextraction |
| SMNP | Silica-coated magnetic nanoparticles |
| SOD | Superoxide dismutase |
| SPE | Solid phase extraction |
| SPM | Suspended particulate material |
| SR-μXRF | Synchrotron radiation |
| ssIDMS | Species specific isotope dilution mass spectrometry |
| suIDMS | Species unspecific isotope dilution mass spectrometry |
| TBAH | Tetrabutyl ammonium hydroxide |
| TBT | Tributyltin |
| TEM | Transmission electron microscopy |
| TMAH | Tetramethylammonium hydroxide |
| TMAO | Trimethylarsine oxide |
| TMSe | Trimethylselenium |
| TPhT | Triphenyltin |
| TRF | Transferrin |
| USE | Ultrasound-assisted extraction |
| UV | Ultraviolet |
| WFD | Water framework directive |
| WHO | World health organisation |
| XAFS | X-ray absorption fine structure spectrometry |
| XANES | X-ray absorption near-edge structure |
| XRF | X-ray fluorescence |
References
- R. Clough, C. F. Harrington, S. J. Hill, Y. Madrid and J. F. Tyson, J. Anal. At. Spectrom., 2016, 31(7), 1330–1373 RSC.
- O. T. Butler, W. R. L. Cairns, J. M. Cookc and C. M. Davidsond, J. Anal. At. Spectrom., 2017, 32(1), 11–57 RSC.
- A. Taylor, N. Barlow, M. P. Day, S. Hill, M. Patriarca and M. White, J. Anal. At. Spectrom., 2017, 32(3), 432–476 RSC.
- E. H. Evans, J. Pisonero, C. M. M. Smith and R. N. Taylor, J. Anal. At. Spectrom., 2016, 31(5), 1057–1077 RSC.
- M. West, A. T. Ellis, P. J. Potts, C. Streli, C. Vanhoof and P. Wobrauschek, J. Anal. At. Spectrom., 2016, 31(9), 1706–1755 RSC.
- S. Carter, A. Fisher, R. Garcia, B. Gibson, S. Lancaster, J. Marshallf and I. Whiteside, J. Anal. At. Spectrom., 2015, 30(11), 2249–2294 RSC.
- B. Michalke, in Metallomics: Analytical Techniques and Speciation Methods, ed. B. Michalke, John Wiley-VCH, Schwaben, 2016 Search PubMed.
- Analytical and Practical Aspects of Drug Testing in Hair, ed. P. Kintz, CRC Press, Boca Raton, 2016 Search PubMed.
- J. Delafiori, G. Ring and A. Furey, Talanta, 2016, 153, 306–331 CrossRef CAS PubMed.
- R. Milacic, T. Zuliani, J. Vidmar and J. Scancar, J. Anal. At. Spectrom., 2016, 31(9), 1766–1779 RSC.
- J. S. Barin, P. A. Mello, M. F. Mesko, F. A. Duarte and E. M. M. Flores, Anal. Bioanal. Chem., 2016, 408(17), 4547–4566 CrossRef CAS PubMed.
- H. Holtkamp, G. Grabmann and C. G. Hartinger, Electrophoresis, 2016, 37(7–8), 959–972 CrossRef CAS PubMed.
- H. Y. Cheng, P. Li, J. H. Liu and M. Y. Ye, J. Anal. At. Spectrom., 2016, 31(9), 1780–1810 RSC.
- C. H. Li, Z. Long, X. M. Jiang, P. Wu and X. D. Hou, TrAC, Trends Anal. Chem., 2016, 77, 139–155 CrossRef CAS.
- D. J. Butcher, Appl. Spectrosc. Rev., 2016, 51(5), 397–416 CrossRef CAS.
- A. D’Ulivo, Spectrochim. Acta, Part B, 2016, 119, 91–107 CrossRef.
- V. N. Postnov, V. Rodinkov, L. N. Moskvin, A. G. Novikov, A. S. Bugaichenko and A. Krokhina, Russ. Chem. Rev., 2016, 85(2), 115–138 CrossRef CAS.
- I. Karadjova, I. Dakova, T. Yordanova and P. Vasileva, J. Anal. At. Spectrom., 2016, 31(10), 1949–1973 RSC.
- M. Trojanowicz and K. Kolacinska, Analyst, 2016, 141(7), 2085–2139 RSC.
- J. R. Lawrence, G. D. W. Swerhone, J. J. Dynes, D. R. Korber and A. P. Hitchcock, J. Microsc., 2016, 261(2), 130–147 CrossRef CAS PubMed.
- Y. F. Li, J. T. Zhao, Y. Qu, Y. X. Gao, Z. H. Guo, Z. L. Liu, Y. L. Zhao and C. Y. Chen, Nanomedicine, 2015, 11(6), 1531–1549 CrossRef CAS PubMed.
- E. M. Kroukamp, T. Wondimu and P. B. C. Forbes, TrAC, Trends Anal. Chem., 2016, 77, 87–99 CrossRef CAS.
- A. K. Das, J. Indian Chem. Soc., 2016, 93(7), 809–814 CAS.
- P. Kumkrong, B. Thiensong, P. M. Le, G. McRae, A. Windust, S. Deawtong, J. Meija, P. Maxwell, L. Yang and Z. Mester, Anal. Chim. Acta, 2016, 943, 41–49 CrossRef CAS PubMed.
- C. Swart and N. Jakubowski, J. Anal. At. Spectrom., 2016, 31(9), 1756–1765 RSC.
- W. Quiroz, F. Astudillo, M. Bravo, F. Cereceda-Balic, V. Vidal, M. R. Palomo-Marin, F. Rueda-Holgado and E. Pinilla-Gil, Microchem. J., 2016, 129, 111–116 CrossRef CAS.
- R. Tisarum, J. H. Ren, X. L. Dong, H. Chen, J. T. Lessl and L. N. Q. Ma, Talanta, 2015, 144, 1171–1175 CrossRef CAS PubMed.
- C. Barrera, S. Lopez, L. Aguilar, L. Mercado, M. Bravo and W. Quiroz, Anal. Bioanal. Chem., 2016, 408(11), 2937–2944 CrossRef CAS PubMed.
- J. L. Ren, X. Z. Zhang, Y. X. Sun, S. M. Liu, D. Huang and J. Zhang, Deep Sea Res., Part II, 2016, 124, 29–42 CrossRef CAS.
- J. O. Vinhal, A. D. Goncalves, G. F. B. Cruz and R. J. Cassella, Talanta, 2016, 150, 539–545 CrossRef CAS PubMed.
- N. Altunay and R. Gurkan, Anal. Methods, 2015, 7(23), 9850–9860 RSC.
- R. Gurkan and M. Eser, J. Iran. Chem. Soc., 2016, 13(9), 1579–1591 CrossRef CAS.
- N. Altunay and R. Gurkan, Talanta, 2016, 159, 344–355 CrossRef CAS PubMed.
- M. Marcinkowska, I. Komorowicz and D. Baralkiewicz, Talanta, 2015, 144, 233–240 CrossRef CAS PubMed.
- M. Marcinkowska, I. Komorowicz and D. Baralkiewicz, Anal. Chim. Acta, 2016, 920, 102–111 CrossRef CAS PubMed.
- M. Jablonska-Czapla and S. Szopa, Water Sci. Technol.: Water Supply, 2015, 16(2), 354–361 CrossRef.
- H. Y. Cheng, P. Li, J. H. Liu and Z. G. Xu, J. Anal. At. Spectrom., 2016, 31(9), 1869–1876 RSC.
- X. Y. Jia, D. R. Gong, J. N. Wang, F. Y. Huang, T. C. Duan and X. Zhang, Talanta, 2016, 160, 437–443 CrossRef CAS PubMed.
- D. Sanchez-Rodas, A. M. S. de la Campa and L. Alsioufi, Anal. Chim. Acta, 2015, 898, 1–18 CrossRef CAS PubMed.
- F. B. Ono, R. Tappero, D. Sparks and L. R. G. Guilherme, Environ. Sci. Pollut. Res., 2016, 23(1), 638–647 CrossRef CAS PubMed.
- D. R. S. Middleton, M. J. Watts, E. M. Hamilton, E. L. Ander, R. M. Close, K. S. Exley, H. Crabbe, G. S. Leonardi, T. Fletcher and D. A. Polya, Sci. Rep., 2016, 6, 11 CrossRef PubMed.
- I. Komorowicz and D. Baralkiewicz, Environ. Monit. Assess., 2016, 188(9), 22 CrossRef PubMed.
- M. Jablonska-Czapla, S. Szopa, P. Zerzucha, A. Lyko and R. Michalski, Environ. Sci. Pollut. Res., 2015, 22(20), 15731–15744 CrossRef CAS PubMed.
- R. Michalski, M. Jablonska-Czapla, S. Szopa, A. Lyko and K. Grygoyc, Int. J. Environ. Anal. Chem., 2016, 96(7), 682–693 CrossRef CAS.
- B. G. Wei, J. P. Yu, H. R. Li, L. S. Yang, Y. J. Xia, K. G. Wu, J. W. Gao, Z. W. Guo and N. Cui, Biol. Trace Elem. Res., 2016, 170(2), 300–308 CrossRef CAS PubMed.
- S. Hong, H. O. Kwon, S. D. Choi, J. S. Lee and J. S. Khim, Mar. Pollut. Bull., 2016, 108(1–2), 155–162 CrossRef CAS PubMed.
- R. D. Foust, A. M. Bauer, M. Costanza-Robinson, D. W. Blinn, R. C. Prince, I. J. Pickering and G. N. George, Coord. Chem. Rev., 2016, 306, 558–565 CrossRef CAS.
- M. S. Podder and C. B. Majumder, Biochem. Eng. J., 2016, 105, 114–135 CrossRef CAS.
- L. Pillay and A. Kindness, S. Afr. Med. J., 2016, 69, 9–14 CAS.
- W. A. Maher, E. Duncan, G. Dilly, S. Foster, F. Krikowa, E. Lombi, K. Scheckel and P. Girguis, Deep Sea Res., Part I, 2016, 116, 41–48 CrossRef CAS.
- Y. Gao, R. E. Sturgeon, Z. Mester, X. D. Hou and L. Yang, Anal. Chim. Acta, 2015, 901, 34–40 CrossRef CAS PubMed.
- B. E. S. Costa, L. M. Coelho, C. S. T. Araujo, H. C. Rezende and N. M. M. Coelho, J. Chem., 2016, 11 Search PubMed.
- E. Bralatei, S. Lacan, E. M. Krupp and J. Feldmann, Anal. Chem., 2015, 87(22), 11271–11276 CrossRef CAS PubMed.
- Y. Fang, Y. S. Pan, P. Li, M. Xue, F. Pei, W. J. Yang, N. Ma and Q. H. Hu, Food Chem., 2016, 213, 609–615 CrossRef CAS PubMed.
- L. Ma, Z. G. Yang, J. Tang and L. Wang, J. Sep. Sci., 2016, 39(11), 2105–2113 CrossRef CAS PubMed.
- L. Ma, L. Wang, J. Tang and Z. G. Yang, Food Chem., 2016, 204, 283–288 CrossRef CAS PubMed.
- L. Ma, L. Wang, Y. Y. Jia and Z. G. Yang, Sci. Total Environ., 2016, 557, 438–444 CrossRef PubMed.
- Z. L. Yang, H. Y. Peng, X. F. Lu, Q. Q. Liu, R. F. Huang, B. Hu, G. Kachanoski, M. J. Zuidhof and X. C. Le, Environ. Sci. Technol., 2016, 50(13), 6737–6743 CrossRef CAS PubMed.
- U. Arroyo-Abad, M. Pfeifer, S. Mothes, H. J. Staerk, C. Piechotta, J. Mattusch and T. Reemtsma, Environ. Pollut., 2016, 208, 458–466 CrossRef CAS PubMed.
- U. Arroyo-Abad, Z. H. Hu, M. Findeisen, D. Pfeifer, J. Mattusch, T. Reemtsma and C. Piechotta, Eur. J. Lipid Sci. Technol., 2016, 118(3), 445–452 CrossRef CAS.
- E. R. Pereira, J. F. Kopp, A. Raab, E. M. Krupp, J. D. Menoyo, E. Carasek, B. Welz and J. Feldmann, J. Anal. At. Spectrom., 2016, 31(9), 1836–1845 RSC.
- W. Zhang, W. X. Wang and L. Zhang, Environ. Sci. Technol., 2016, 50(5), 2413–2423 CrossRef CAS PubMed.
- S. D. Choi, H. S. Son, M. Choi and M. K. Park, Ocean Sci. J., 2015, 50(4), 741–750 CrossRef CAS.
- P. K. Krishnakumar, M. A. Qurban, M. Stiboller, K. E. Nachman, T. V. Joydas, K. P. Manikandan, S. A. Mushir and K. A. Francesconi, Sci. Total Environ., 2016, 566, 1235–1244 CrossRef PubMed.
- B. A. Sadee, M. E. Foulkes and S. J. Hill, Food Addit. Contam., Part A, 2016, 33(3), 433–441 CrossRef CAS PubMed.
- L. Ma, Z. G. Yang, Q. Kong and L. Wang, Food Chem., 2017, 217, 524–530 CrossRef CAS PubMed.
- I. Pizarro, M. Gomez-Gomez, J. Leon, D. Roman and M. A. Palacios, Sci. Total Environ., 2016, 565, 557–563 CrossRef CAS PubMed.
- M. Mleczek, P. Niedzielski, P. Rzymski, M. Siwulski, M. Gasecka and L. Kozak, J. Environ. Sci. Health, Part B, 2016, 51(7), 469–476 CrossRef CAS PubMed.
- V. F. Taylor and B. P. Jackson, Chemosphere, 2016, 163, 6–13 CrossRef CAS PubMed.
- B. A. Sadee, M. E. Foulkes and S. J. Hill, Food Chem., 2016, 210, 362–370 CrossRef CAS PubMed.
- P. Y. Shuai, X. J. Yang, Z. Q. Qiu, X. H. Wu, X. Zhu, G. R. Pokhrel, Y. Y. Fu, H. M. Ye, W. X. Lin and G. D. Yang, J. Sep. Sci., 2016, 39(16), 3239–3245 CrossRef CAS PubMed.
- N. V. Campos, T. O. Araujo, S. Arcanjo-Silva, L. Freitas-Silva, A. A. Azevedo and A. Nunes-Nesi, Physiol. Plant., 2016, 157(2), 135–146 CrossRef CAS PubMed.
- J. Tremlova, I. Vasickova, J. Szakova, W. Goessler, O. Steiner, J. Najmanova, T. Horakova and P. Tlustos, Environ. Exp. Bot., 2016, 123, 108–115 CrossRef CAS.
- T. Iwai, K. Chiba and T. Narukawa, Anal. Sci., 2016, 32(9), 957–962 CrossRef CAS PubMed.
- A. C. Grijalba, E. F. Fiorentini, L. D. Martinez and R. G. Wuilloud, J. Chromatogr., 2016, 1462, 44–54 CrossRef PubMed.
- S. Stice, G. L. Liu, S. Matulis, L. H. Boise and Y. Cai, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2016, 1009, 55–65 CrossRef PubMed.
- R. Garla, R. Ganger, B. P. Mohanty, S. Verma, M. P. Bansal and M. L. Garg, Toxicology, 2016, 366, 68–73 CrossRef PubMed.
- I. N. Serrano, M. T. L. Ballesteros, S. S. F. Pacheco, S. I. Alvarez and J. L. L. Colon, Sci. Total Environ., 2016, 571, 164–171 CrossRef PubMed.
- A. Hata, H. Kurosawa, Y. Endo, K. Yamanaka, N. Fujitani and G. Endo, J. Occup. Health, 2016, 58(2), 196–200 CrossRef CAS PubMed.
- Z. Slejkovec, H. Podgornik, P. Cernelc and I. Falnoga, BioMetals, 2016, 29(1), 107–118 CrossRef CAS PubMed.
- Z. Q. Chen, H. N. Zhang, L. N. Yang, H. W. Jiang, S. J. Guo, Y. Li and S. C. Tao, ABBS, 2016, 48(5), 474–481 Search PubMed.
- M. O. Gribble, V. S. Voruganti, S. A. Cole, K. Haack, P. Balakrishnan, S. L. Laston, M. Tellez-Plaza, K. A. Francesconi, W. Goessler, J. G. Umans, D. C. Thomas, F. Gilliland, K. E. North, N. Franceschini and A. Navas-Acien, Toxicol. Sci., 2015, 148(1), 89–100 CrossRef CAS PubMed.
- B. W. Chen, X. F. Lu, L. L. Arnold, S. M. Cohen and X. C. Le, Chem. Res. Toxicol., 2016, 29(9), 1480–1487 CrossRef CAS PubMed.
- H. S. Loveborn, M. Kippler, Y. Lu, S. Ahmed, D. Kuehnelt, R. Raqib and M. Vahter, Toxicol. Sci., 2016, 152(1), 29–39 CrossRef PubMed.
- K. Kordas, E. I. Queirolo, N. Manay, F. Peregalli, P. Y. Hsiao, Y. Lu and M. Vahter, Environ. Res., 2016, 147, 16–23 CrossRef CAS PubMed.
- T. Oguri, J. Yoshinaga, H. Toshima, Y. Mizumoto, S. Hatakeyama and S. Tokuoka, J. Environ. Sci. Health, Part A: Toxic/Hazard. Subst. Environ. Eng., 2016, 51(6), 463–466 CrossRef CAS PubMed.
- S. G. Johnston, J. Diwakar and E. D. Burton, Chem. Geol., 2015, 419, 55–66 CrossRef CAS.
- M. W. Franco, F. A. G. Ferreira, I. F. Vasconcelos, B. L. Batista, D. G. F. Pujoni, S. M. S. Magalhaes, F. Barbosa and F. A. R. Barbosa, Environ. Sci. Pollut. Res., 2015, 22(23), 18607–18615 CrossRef CAS PubMed.
- M. Vermeulen, G. Nuyts, J. Sanyova, A. Vila, D. Buti, J. P. Suuronen and K. Janssens, J. Anal. At. Spectrom., 2016, 31(9), 1913–1921 RSC.
- T. D. Ciftci and E. Henden, Bull. Environ. Contam. Toxicol., 2016, 97(2), 272–278 CrossRef CAS PubMed.
- B. S. Pinheiro, L. L. S. Gimenes, A. J. Moreira, C. D. Freschi and G. P. G. Freschi, At. Spectrosc., 2016, 37(2), 83–89 CAS.
- Y. I. Coskun, T. D. Ciftci and E. Henden, Desalin. Water Treat., 2016, 57(43), 20411–20421 CrossRef.
- A. Oliveira, M. H. Gonzalez, H. M. Queiroz and S. Cadore, Food Chem., 2016, 213, 76–82 CrossRef CAS PubMed.
- A. Khaligh, H. Z. Mousavi, H. Shirkhanloo and A. Rashidi, RSC Adv., 2015, 5(113), 93347–93359 RSC.
- A. Maratta, L. D. Martinez and P. Pacheco, Microchem. J., 2016, 127, 199–205 CrossRef CAS.
- X. A. Yang, X. P. Lu, L. Liu, M. B. Chi, H. H. Hu and W. B. Zhang, Talanta, 2016, 159, 127–136 CrossRef CAS PubMed.
- S. S. Arain, T. G. Kazi, H. I. Afridi, Naemullah, K. D. Brahman, F. N. Talpur, A. H. Panhwar, J. Ali and F. Shah, Microchem. J., 2016, 124, 290–295 CrossRef CAS.
- A. Tunceli, G. Ocak, O. Acar and A. R. Turker, Int. J. Environ. Anal. Chem., 2015, 95(14), 1395–1411 CrossRef CAS.
- M. Murakami, M. Hirano, T. Shibahara and T. Kubota, Anal. Lett., 2016, 49(13), 2119–2131 CrossRef CAS.
- K. Marschner, S. Musil and J. Dedina, Anal. Chem., 2016, 88(7), 4041–4047 CrossRef CAS PubMed.
- S. Z. Chen, Y. Y. He, S. P. Zhu and D. B. Lu, At. Spectrosc., 2015, 36(6), 247–253 CAS.
- J. Y. Choi, N. Khan, E. Y. Nho, H. Choi, K. S. Park, M. J. Cho, H. J. Youn and K. S. Kim, Anal. Lett., 2016, 49(12), 1926–1937 CrossRef CAS.
- J. Moreda-Pineiro, A. Cantarero-Roldan, P. Hermelo-Herbello, A. Moreda-Pineiro and P. Bermejo-Barrera, At. Spectrosc., 2016, 36(6), 237–246 Search PubMed.
- A. Terol, M. Marcinkowska, F. Ardini and M. Grotti, Anal. Sci., 2016, 32(8), 911–915 CrossRef CAS PubMed.
- M. Santoso, D. D. Lestiani, E. Damastuti, S. Kurniawati, J. W. Bennett, J. J. Leani, M. Czyzycki, A. Migliori, J. Osan and A. G. Karydas, J. Radioanal. Nucl. Chem., 2016, 309(1), 413–419 CrossRef CAS.
- L. Trzonkowska, B. Lesniewska and B. Godlewska-Zylkiewicz, Crit. Rev. Anal. Chem., 2016, 46(4), 305–322 CrossRef CAS PubMed.
- B. Markiewicz, I. Komorowicz and D. Baralkiewicz, Talanta, 2016, 152, 489–497 CrossRef CAS PubMed.
- H. C. Jin, X. S. Zhang, X. Wan and J. W. Wang, J. Soc. Leather Technol. Chem., 2016, 100(4), 182–185 Search PubMed.
- X. Y. Jia, D. R. Gong, B. Xu, Q. Q. Chi and X. Zhang, Talanta, 2016, 147, 155–161 CrossRef CAS PubMed.
- E. Leese, J. Morton, P. H. E. Gardiner and V. A. Carolan, J. Anal. At. Spectrom., 2016, 31(4), 924–933 RSC.
- S. Cuello, J. Entwisle, J. Benning, C. Liu, S. Coburn, K. G. McAdam, J. Braybrook and H. Goenaga-Infante, J. Anal. At. Spectrom., 2016, 31(9), 1818–1829 RSC.
- Y. A. Lin, S. J. Jiang, A. C. Sahayam and Y. L. Huang, Microchem. J., 2016, 128, 274–278 CrossRef CAS.
- N. Unceta, M. Astorkia, Z. Abrego, A. Gomez-Caballero, M. A. Goicolea and R. J. Barrio, Talanta, 2016, 154, 255–262 CrossRef CAS PubMed.
- P. Li, L. M. Li, J. Xia, S. Cao, X. Hu, H. Z. Lian and S. Ji, J. Sep. Sci., 2015, 38(23), 4043–4047 CrossRef CAS PubMed.
- B. Umesh, R. M. Rajendran and M. T. Manoharan, Poult. Sci., 2015, 94(11), 2805–2815 CrossRef CAS PubMed.
- S. Sadeghi and A. Z. Moghaddam, Environ. Monit. Assess., 2015, 187(12), 12 CrossRef PubMed.
- D. Chen, X. W. Wang, R. Zhao, B. Shao, Z. Wang, G. H. Wu and Y. Xue, Chin. J. Anal. Chem., 2015, 43(12), 1901–1905 CAS.
- N. Sharma, S. Tiwari and R. Saxena, RSC Adv., 2016, 6(13), 10775–10782 RSC.
- S. Tiwari, N. Sharma and R. Saxena, New J. Chem., 2016, 40(2), 1412–1419 RSC.
- B. Lesniewska, A. Jeglikowska and B. Godlewska-Zylkiewicz, Water, Air, Soil Pollut., 2016, 227(8), 10 CrossRef PubMed.
- L. Djerahov, P. Vasileva and I. Karadjova, Microchem. J., 2016, 129, 23–28 CrossRef CAS.
- K. Pytlakowska, J. Anal. At. Spectrom., 2016, 31(4), 968–974 RSC.
- Z. Bahadir, V. N. Bulut, M. Hidalgo, M. Soylak and E. Margui, Spectrochim. Acta, Part B, 2016, 115, 46–51 CrossRef CAS.
- I. Gaubeur, M. A. Aguirre, N. Kovachev, M. Hidalgo and A. Canals, J. Anal. At. Spectrom., 2015, 30(12), 2541–2547 RSC.
- N. N. Meeravali, K. Madhavi and S. J. Kumar, J. Anal. At. Spectrom., 2016, 31(8), 1582–1589 RSC.
- S. Sadeghi and A. Z. Moghaddam, J. Iran. Chem. Soc., 2015, 13(1), 117–130 CrossRef.
- H. Shirkhanloo, A. A. M. Beigi, M. M. Eskandari and B. Kalantari, J. Anal. Chem., 2015, 70(12), 1448–1455 CrossRef CAS.
- H. Shirkhanloo, M. Ghazaghi and H. Z. Mousavi, J. Pharm. Biomed. Anal., 2016, 118, 1–8 CrossRef CAS PubMed.
- G. Fakhriyan, H. Z. Mousavi and S. M. Sajjadi, Anal. Methods, 2016, 8(25), 5070–5078 RSC.
- S. Sadeghi and A. Z. Moghaddam, J. Mol. Liq., 2016, 221, 798–804 CrossRef CAS.
- T. N. Simonova, V. A. Dubrovina and A. B. Vishnikin, J. Serb. Chem. Soc., 2016, 81(6), 645–659 CrossRef.
- M. Sasaki, T. Shimizu and N. Uehara, Bunseki Kagaku, 2016, 65(8), 433–438 CrossRef CAS.
- Y. S. Hedberg and C. Liden, Contact Dermatitis, 2016, 75(2), 82–88 CrossRef CAS PubMed.
- M. W. Mathebula, K. Mandiwana and N. Panichev, Food Chem., 2017, 217, 655–659 CrossRef CAS PubMed.
- K. Ogata, Y. Kumazawa, Y. Koyama, K. Yoshimura and K. Takahashi, J. Soc. Leather Technol. Chem., 2015, 99(6), 293–296 CAS.
- L. G. Hu, Y. Cai and G. B. Jiang, Chemosphere, 2016, 156, 14–20 CrossRef CAS PubMed.
- F. Hernandez, F. Seby, S. Millour, L. Noel and T. Guerin, Food Chem., 2017, 214, 339–346 CrossRef CAS PubMed.
- L. Guidotti, S. Q. Abad, P. Rodriguez-Gonzalez, J. I. G. Alonso and G. M. Beone, Environ. Sci. Pollut. Res., 2015, 22(22), 17569–17576 CrossRef CAS PubMed.
- S. Madler, A. Todd, H. M. Kingston, M. Pamuku, F. R. Sun, C. Tat, R. J. Tooley, T. A. Switzer and V. I. Furdui, Talanta, 2016, 156, 104–111 CrossRef PubMed.
- K. K. Larsen, D. Wielandt, M. Schiller and M. Bizzarro, J. Chromatogr., 2016, 1443, 162–174 CrossRef CAS PubMed.
- M. Birka, C. A. Wehe, O. Hachmoller, M. Sperling and U. Karst, J. Chromatogr., 2016, 1440, 105–111 CrossRef CAS PubMed.
- W. Guo, Y. X. Wang, J. X. Li, Y. E. Peng, L. L. Jin, Q. H. Guo and S. H. Hu, At. Spectrosc., 2016, 37(1), 7–12 CAS.
- Y. E. Peng, W. Guo, J. Zhang, Q. H. Guo, L. L. Jin and S. H. Hu, Microchem. J., 2016, 124, 127–131 CrossRef CAS.
- Y. Zakon, L. Halicz, O. Lev and F. Gelman, Rapid Commun. Mass Spectrom., 2016, 30(17), 1951–1956 CrossRef CAS PubMed.
- C. Swart, F. Gantois, P. Petrov, J. Entwisle, H. Goenaga-Infante, M. Nousiainen, M. Bilsel, B. Binici, A. Gonzalez-Gago, D. Profrock and A. C. Goren, Talanta, 2016, 152, 251–258 CrossRef CAS PubMed.
- P. Novak, T. Zuliani, R. Milacic and J. Scancar, Anal. Chim. Acta, 2016, 915, 27–35 CrossRef CAS PubMed.
- L. Somoano-Blanco, P. Rodriguez-Gonzalez, D. Profrock, A. Prange and J. I. G. Alonso, Anal. Methods, 2015, 7(21), 9068–9075 RSC.
- A. M. Schwan, R. Martin and W. Goessler, Anal. Methods, 2015, 7(21), 9198–9205 RSC.
- B. Klencsar, E. Bolea-Fernandez, M. R. Florez, L. Balcaen, F. Cuyckens, F. Lynen and F. Vanhaecke, J. Pharm. Biomed. Anal., 2016, 124, 112–119 CrossRef CAS PubMed.
- C. K. Mak, C. A. Wehe, M. Sperling and U. Karst, J. Chromatogr., 2015, 1419, 81–88 CrossRef CAS PubMed.
- R. A. Wilson, E. G. Yanes and R. J. Kemppainen, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2016, 1022, 183–190 CrossRef CAS PubMed.
- E. Grygo-Szymanko, A. Tobiasz and S. Walas, TrAC, Trends Anal. Chem., 2016, 80, 112–124 CrossRef CAS.
- M. Amde, Y. G. Yin, D. Zhang and J. F. Liu, Chem. Speciation Bioavailability, 2016, 28(1–4), 51–65 CrossRef.
- Z. Pedrero, O. F. X. Donard and D. Amouroux, Anal. Bioanal. Chem., 2016, 408(11), 2641–2648 CrossRef CAS PubMed.
- J. Hogeback, M. Schwarzer, C. A. Wehe, M. Sperling and U. Karst, Metallomics, 2015, 8(1), 101–107 RSC.
- H. Wang, B. B. Chen, S. Q. Zhu, X. X. Yu, M. He and B. Hu, Anal. Chem., 2015, 88(1), 796–802 CrossRef PubMed.
- P. P. Chen, P. Wu, J. B. Chen, P. F. Yang, X. F. Zhang, C. B. Zheng and X. D. Hou, Anal. Chem., 2016, 88(4), 2065–2071 CrossRef CAS PubMed.
- I. S. Trujillo, E. V. Alonso, J. M. C. Pavon and A. G. de Torres, J. Anal. At. Spectrom., 2015, 30(12), 2429–2440 RSC.
- R. Rodriguez-Fernandez, E. Pena-Vazquez and P. Bermejo-Barrera, Talanta, 2015, 144, 636–641 CrossRef CAS PubMed.
- S. Valdersnes, P. Fecher, A. Maage and K. Julshamn, Food Chem., 2015, 194, 424–431 CrossRef PubMed.
- Y. Lin, Y. Yang, Y. X. Li, L. Yang, X. D. Hou, X. B. Feng and C. B. Zheng, Environ. Sci. Technol., 2016, 50(5), 2468–2476 CrossRef CAS PubMed.
- V. Perrot, J. Masbou, M. V. Pastukhov, V. N. Epov, D. Point, S. Berail, P. R. Becker, J. E. Sonke and D. Amouroux, Metallomics, 2016, 8(2), 170–178 RSC.
- L. N. Van, S. Jonsson, U. Skyllberg, M. B. Nilsson, A. Andersson, E. Lundberg and E. Bjorn, Environ. Sci. Technol., 2016, 50(13), 6983–6990 CrossRef PubMed.
- M. D. Rand, D. Vorojeikina, E. van Wijngaarden, B. P. Jackson, T. Scrimale, G. Zareba, T. M. Love, G. J. Myers and G. E. Watson, Toxicol. Sci., 2016, 149(2), 385–395 CrossRef CAS PubMed.
- R. Pereira, S. Guilherme, F. Brandao, J. Raimundo, M. A. Santos, M. Pacheco and P. Pereira, Mar. Environ. Res., 2016, 113, 70–79 CrossRef CAS PubMed.
- M. Kawasaki, M. Otsuka, J. Yamada and A. Kobayashi, J. Jpn. Pet. Inst., 2016, 59(5), 235–240 CrossRef CAS.
- S. X. Zhang, H. Luo, Y. Y. Zhang, X. Y. Li, J. S. Liu, Q. Xu and Z. H. Wang, Microchem. J., 2016, 126, 25–31 CrossRef CAS.
- Y. L. Sommer, C. D. Ward, Y. Pan, K. L. Caldwell and R. L. Jones, J. Anal. Toxicol., 2016, 40(3), 222–228 CrossRef CAS PubMed.
- S. S. Ma, M. He, B. B. Chen, W. C. Deng, Q. Zheng and B. Hu, Talanta, 2016, 146, 93–99 CrossRef CAS PubMed.
- K. Yoshimoto, H. T. V. Anh, A. Yamamoto, C. Koriyama, Y. Ishibashi, M. Tabata, A. Nakano and M. Yamamoto, J. Toxicol. Sci., 2016, 41(4), 489–500 CrossRef CAS PubMed.
- J. Cavalheiro, C. Sola, J. Baldanza, E. Tessier, F. Lestremau, F. Botta, H. Preud’homme, M. Monperrus and D. Amouroux, Water Res., 2016, 94, 32–41 CrossRef CAS PubMed.
- S. Le Roux, P. Baker and A. Crouch, S. Afr. J. Chem., 2016, 69, 124–131 CrossRef CAS.
- X. Xu, J. Zhao, Y. Li, Y. Fan, N. Zhu, Y. Gao, B. Li, H. Liu and Y. F. Li, Environ. Pollut., 2016, 210, 113–120 CrossRef CAS PubMed.
- Y. Q. Chen, X. Cheng, F. Mo, L. M. Huang, Z. J. Wu, Y. N. Wu, L. J. Xu and F. F. Fu, Electrophoresis, 2016, 37(7–8), 1055–1062 CrossRef CAS PubMed.
- E. L. de Paiva, J. C. Alves, R. F. Milani, B. S. Boer, K. D. Quintaes and M. A. Morgano, Food Control, 2016, 69, 115–123 CrossRef.
- S. Gentes, R. Maury-Brachet, C. Y. Feng, Z. Pedrero, E. Tessier, A. Legeay, N. Mesmer-Dudons, M. Baudrimont, L. Maurice, D. Amouroux and P. Gonzalez, Environ. Sci. Technol., 2015, 49(24), 14560–14569 CrossRef CAS PubMed.
- C. L. Mieiro, J. P. Coelho, M. Dolbeth, M. Pacheco, A. C. Duarte, M. A. Pardal and M. E. Pereira, Food Control, 2016, 60, 575–581 CrossRef CAS.
- R. Jagtap and W. Maher, Microchem. J., 2016, 124, 422–529 CrossRef CAS.
- M. Bodnar, M. Szczyglowska, P. Konieczka and J. Namiesnik, Crit. Rev. Food Sci. Nutr., 2016, 56(1), 36–55 CrossRef CAS PubMed.
- Y. P. Cao, L. Z. Yan, H. L. Huang and B. Y. Deng, Spectrochim. Acta, Part B, 2016, 122, 172–177 CrossRef CAS.
- L. Nyaba, J. M. Matong, K. M. Dimpe and P. N. Nomngongo, Talanta, 2016, 159, 174–180 CrossRef CAS PubMed.
- X. Q. Guo, M. He, K. Nan, H. Yan, B. B. Chen and B. Hu, J. Anal. At. Spectrom., 2016, 31(2), 406–414 RSC.
- X. J. Wang, L. Wu, J. Q. Cao, X. C. Hong, R. Ye, W. J. Chen and T. Yuan, Food Addit. Contam., Part A, 2016, 33(7), 1190–1199 CrossRef CAS PubMed.
- M. Palomo-Siguero, A. M. Gutierrez, C. Perez-Conde and Y. Madrid, Microchem. J., 2016, 126, 488–495 CrossRef CAS.
- R. Jagtap, W. Maher, F. Krikowa, M. J. Ellwood and S. Foster, Microchem. J., 2016, 128, 248–257 CrossRef CAS.
- W. Maher, F. Krikowa and S. Foster, Microchem. J., 2016, 126, 92–95 CrossRef CAS.
- A. F. Oliveira, J. Landero, K. Kubachka, A. R. A. Nogueira, M. A. Zanetti and J. Caruso, J. Anal. At. Spectrom., 2016, 31(4), 1034–1040 RSC.
- T. Abiko, K. Kobayashi, T. Matsukawa, A. Shinohara and N. Furuta, Bunseki Kagaku, 2016, 65(7), 371–378 CrossRef CAS.
- J. Kwak, S. A. Ohrnberger and T. G. Valencak, Anal. Bioanal. Chem., 2016, 408(18), 4927–4934 CrossRef CAS PubMed.
- K. Bierla, A. Flis-Borsuk, P. Suchocki, J. Szpunar and R. Lobinski, J. Agric. Food Chem., 2016, 64(24), 4975–4981 CrossRef CAS PubMed.
- S. Torres, R. Gil, M. F. Silva and P. Pacheco, Food Chem., 2016, 197, 400–405 CrossRef CAS PubMed.
- T. Jager, H. Drexler and T. Goen, Arch. Toxicol., 2016, 90(5), 1069–1080 CrossRef PubMed.
- T. Jager, H. Drexler and T. Goen, Arch. Toxicol., 2016, 90(1), 149–158 CrossRef PubMed.
- K. Flouda, J. M. Dersch, C. Gabel-Jensen, S. Sturup, S. Misra, M. Bjornstedt and B. Gammelgaard, Anal. Bioanal. Chem., 2016, 408(9), 2293–2301 CrossRef CAS PubMed.
- R. G. de Vega, M. L. Fernandez-Sanchez, J. C. Fernandez, F. V. A. Menendez and A. Sanz-Medel, J. Trace Elem. Med. Biol., 2016, 37, 44–49 Search PubMed.
- C. L. Deitrich, S. Cuello-Nunez, D. Kmiotek, F. A. Torma, M. E. D. Busto, P. Fisicaro and H. Goenaga-Infante, Anal. Chem., 2016, 88(12), 6357–6365 CrossRef CAS PubMed.
- A. Konopka, D. Winter, W. Konopka, M. E. D. Busto, S. Nunez, H. Goenaga-Infante, P. Fisicaro and W. D. Lehmann, J. Anal. At. Spectrom., 2016, 31(9), 1929–1938 RSC.
- M. J. P. Ropero, N. R. Farinas, E. Krupp, R. Mateo, J. J. B. Nevado and R. C. R. Martin-Doimeadios, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2016, 1022, 159–166 CrossRef CAS PubMed.
- Y. Y. Li, Y. Q. Fan, J. T. Zhao, X. H. Xu, H. Jing, L. H. Shang, Y. X. Gao, B. Li and Y. F. Li, BioMetals, 2016, 29(5), 893–903 CrossRef CAS PubMed.
- M. Tie, Y. Gao, Y. L. Xue, A. N. Zhang, Y. Yao, J. F. Sun and S. Xue, Anal. Methods, 2016, 8(15), 3102–3108 RSC.
- O. Egressy-Molnar, L. Ouerdane, J. Gyorfi and M. Dernovics, LWT--Food Sci. Technol., 2016, 68, 306–312 CrossRef CAS.
- J. Wojcieszek and L. Ruzik, J. Trace Elem. Med. Biol., 2016, 34, 15–21 CAS.
- L. Lyubenova, X. Sabodash, P. Schroder and B. Michalke, Environ. Sci. Pollut. Res., 2015, 22(21), 16978–16986 CrossRef CAS PubMed.
- E. Y. Barrientos, K. Wrobel, J. C. T. Guzman and A. R. C. Escobosa, J. Anal. At. Spectrom., 2016, 31(1), 203–211 RSC.
- S. Z. Chen, S. P. Zhu and D. B. Lu, At. Spectrosc., 2015, 36(3), 128–133 CAS.
- K. L. LeBlanc, J. Ruzicka and D. Wallschlager, Anal. Bioanal. Chem., 2016, 408(4), 1033–1042 CrossRef CAS PubMed.
- O. G. San Blas, F. M. Sanz, P. H. Espilez, B. P. Garcia, F. V. A. Menendez, J. M. Marchante-Gayon and J. I. G. Alonso, J. Anal. At. Spectrom., 2016, 31(9), 1885–1894 RSC.
- G. Hermann, L. H. Moller, B. Gammelgaard, J. Hohlweg, D. Mattanovich, S. Hann and G. Koellensperger, J. Anal. At. Spectrom., 2016, 31(9), 1830–1835 RSC.
- E. Biadun, M. Sadowska, N. Ospina-Alvarez and B. Krasnodebska-Ostrega, Microchim. Acta, 2016, 183(1), 177–183 CrossRef CAS PubMed.
- J. Richter, I. Fettig, R. Philipp, N. Jakubowski, U. Panne, P. Fisicaro and E. Alasonati, J. Chromatogr., 2016, 1459, 112–119 CrossRef CAS PubMed.
- E. Alasonati, I. Fettig, J. Richter, R. Philipp, R. Milacic, J. Scancar, T. Zuliani, M. Tunc, M. Bilsel, A. C. Goren and P. Fisicaro, Talanta, 2016, 160, 499–511 CrossRef CAS PubMed.
- Y. X. Liu, Y. Q. Ma, Y. Q. Wan, L. Guo and X. F. Wan, J. Sep. Sci., 2016, 39(12), 2380–2387 CrossRef CAS PubMed.
- F. G. Wandekoken, C. B. Duyck, T. C. O. Fonseca and T. D. Saint’Pierre, Spectrochim. Acta, Part B, 2016, 119, 1–9 CrossRef CAS.
- D. P. Persson, T. C. de Bang, P. R. Pedas, U. B. Kutman, I. Cakmak, B. Andersen, C. Finnie, J. K. Schjoerring and S. Husted, New Phytol., 2016, 211(4), 1255–1265 CrossRef CAS PubMed.
- S. Fernandez-Menendez, M. L. Fernandez-Sanchez, B. Fernandez-Colomer, R. R. D. St Remy, G. D. C. Cotallo, A. S. Freire, B. F. Braz, R. E. Santelli and A. Sanz-Medel, J. Chromatogr., 2016, 1428, 246–254 CrossRef CAS PubMed.
- A. Sanz-Medel, Anal. Bioanal. Chem., 2016, 408(20), 5393–5395 CrossRef CAS PubMed.
- B. Hu, L. Peng, M. He and B. B. Chen, Appl. Spectrosc. Rev., 2015, 51(2), 94–116 CrossRef.
- F. Calderon-Celis, S. Diez-Fernandez, J. M. Costa-Fernandez, J. R. Encinar, J. J. Calvete and A. Sanz-Medel, Anal. Chem., 2016, 88(19), 9699–9706 CrossRef CAS PubMed.
- L. H. Moller, A. Macherius, T. H. Hansen, H. M. Nielsen, C. Cornett, J. Ostergaard, S. Sturup and B. Gammelgaard, J. Anal. At. Spectrom., 2016, 31(9), 1877–1884 RSC.
- E. Maes, K. Tirez, G. Baggerman, D. Valkenborg, L. Schoofs, J. R. Encinar and I. Mertens, Mass Spectrom. Rev., 2016, 35(3), 350–360 CrossRef CAS PubMed.
- S. Shibayama, S. Fujii, K. Inagaki, T. Yamazaki and A. Takatsu, J. Chromatogr., 2016, 1468, 109–115 CrossRef CAS PubMed.
- Q. Tu, E. N. Guidry, F. Y. Meng, T. B. Wang and X. Y. Gong, Microchem. J., 2016, 124, 668–674 CrossRef CAS.
- R. Liu, S. X. Zhang, C. Wei, Z. Xing, S. C. Zhang and X. R. Zhang, Acc. Chem. Res., 2016, 49(5), 775–783 CrossRef CAS PubMed.
- T. C. de Bang and S. Husted, TrAC, Trends Anal. Chem., 2015, 72, 45–52 CrossRef CAS.
- N. N. Tang, Z. X. Li, L. M. Yang and Q. Q. Wang, Anal. Chem., 2016, 88(20), 9890–9896 CrossRef CAS PubMed.
- J. A. Ko and H. B. Lim, Anal. Chim. Acta, 2016, 938, 1–6 CrossRef CAS PubMed.
- T. N. Aslan, E. Asik and M. Volkan, RSC Adv., 2016, 6(11), 8860–8869 RSC.
- S. J. M. Van Malderen, A. J. Managh, B. L. Sharp and F. Vanhaecke, J. Anal. At. Spectrom., 2016, 31(2), 423–439 RSC.
- D. J. Hare, J. K. Lee, A. D. Beavis, A. van Gramberg, J. George, P. A. Adlard, D. I. Finkelstein and P. A. Doble, Anal. Chem., 2012, 84(9), 3990–3997 CrossRef CAS PubMed.
- E. Malucelli, M. Fratini, A. Notargiacomo, A. Gianoncelli, L. Merolle, A. Sargenti, C. Cappadone, G. Farruggia, S. Lagomarsino and S. Iotti, Analyst, 2016, 141(18), 5221–5235 RSC.
- J. W. Shi, X. Y. Zhang, L. Li, W. Y. Feng, H. F. Wang and M. Wang, Pharmacol., Biochem. Behav., 2016, 43(8), 739–746 Search PubMed.
| This journal is © The Royal Society of Chemistry 2017 |