
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Composite hydrogels of polyacrylamide and crosslinked pH-responsive micrometer-sized hollow particles†
Kyriaki
Pafiti
a,
Zhengxing
Cui
a,
Louise
Carney
a,
Anthony J.
Freemont
b and
Brian R.
Saunders
*a
aBiomaterials Research Group, School of Materials, The University of Manchester, MSS Tower, Manchester, M13 9PL, UK. E-mail: brian.saunders@manchester.ac.uk
bDivision of Regenerative Medicine, School of Medicine, Stopford Building, The University of Manchester, Oxford Road, Manchester, M13 9PT, UK
First published on 20th November 2015
Abstract
Whilst hydrogels and hollow particles both continue to attract much attention in the literature there are few examples of hydrogel composites containing hollow particles. Here, we study composite polyacrylamide (PAAm) hydrogels containing micrometer-sized pH-responsive shell-crosslinked hollow particles (abbreviated as HPXL) based on poly(methylmethacrylate-co-methacrylic acid) functionalised with glycidyl methacrylate (GMA). The HPXL particles were prepared using our scaleable emulsion template method and inclusion of GMA was found to promote spherical hollow particle formation. The pendant vinyl groups from GMA enabled shell-crosslinked hollow particles to be prepared prior to formation of the PAAm/HPXL composite gels. The morphologies of the particles and composite gels were studied by optical microscopy, confocal laser scanning microscopy and scanning electron microscopy. Dynamic rheology measurements for the composite gels showed that the modulus variation with HPXL concentration could be described by a percolation model with a HPXL percolation threshold concentration of 4.4 wt% and a scaling exponent of 2.6. The composite gels were pH-responsive and largely maintained their mechanical properties over the pH range 4.0 to 8.0. Because the composite gels had tuneable mechanical properties (with modulus values up to 530 kPa) and were pH-responsive they are potential candidates for future wound healing or membrane applications.
Introduction
Hydrogels have attracted considerable interest in the biomedical field,1 including drug1,2/gene delivery, stem cell and cancer research, cell therapy3 and regenerative medicine4 due to their tuneable structural, compositional and mechanical properties. Both naturally-derived hydrogels and synthetic hydrogels have been used widely in regenerative medicine due to their favourable biocompatibilities and mechanical property similarities with native tissues.4,5 Reinforcement strategies have been used to improve gel mechanical properties, which include the development of double networks.6–8 Workers have also investigated composite hydrogels containing inorganic nanoparticles. These nanocomposite gels combine high modulus, which is associated with the high surface area of the nanoparticles, with high extensibility.9–14 Nanocomposite gels have been prepared via incorporation of different species;15 these include mineral (organic or inorganic clays),16 metallic (gold and silver17 nanoparticles), magnetic18 (iron or gadolinium) and carbon-based (graphene oxide19,20 or carbon nanotubes21) species. Much less work has been reported for hydrogel composites containing micrometer-sized hollow particles, which is the focus of this study. We hypothesised that micrometer-sized particles would enable formation of composite gels containing high hollow particle concentrations without excessively large modulus values because of the low surface area-to-volume ratios of the particles. The pH-responsive micrometer-sized hollow particles used here offer potential to include larger sized species (e.g., proteins or cells) within composite gels and may provide new opportunities for triggered release of biological species.Hollow nano- and micrometer-sized spherical particles are of great technological importance because of their potential encapsulation utility in cosmetics, corrosion protection,22 paper coatings, delivery23 and as protective shells.24,25 For the preparation of hollow particles emulsion processing,26–28 multi-layer assembly29,30 and colloid templating methods (mainly based on silica particles)31,32 have been most commonly used. Here, we focus on the use of pH-responsive micrometer-sized hollow particles in order to investigate the effects they have on composite hydrogel morphological and mechanical properties. In this study the hollow particle structure and intra-shell crosslinking were controlled by including vinyl functionality within the hollow particle shells and crosslinking these groups (later).
Our hollow particles were prepared using a simple, scaleable, emulsion templating method that was facilitated by the amphiphilic nature of the pH-responsive vinyl-functionalised poly(methylmethacrylate-co-methacrylic acid) (PMMA-MAA) copolymer and its migration to the dichloromethane/water interface.33 The hollow particles studied here, which are pH responsive, differ considerably to the thermally responsive hollow poly(N-isopropyacrylamide)-based microgel particles reported elsewhere.34–36 In contrast to earlier work which focussed on dispersions and physical gels of hollow particles,37,38 here a study of poly(acrylamide) (PAAm) composite gels containing shell-crosslinked pH-responsive hollow particles is presented for the first time. After characterising the new hollow particles we investigate the morphology and mechanical properties of composite hydrogels. Inclusion of the hollow particles increased the composite gel modulus values and the data were successfully modelled using a percolation model. The mechanisms responsible for the mechanical properties of these new composites are discussed. We show that the composite gels are pH-responsive over the pH range of 4.0 to 8.0. The new composite hollow particle gels presented here may have potential applications as wound dressings or membranes.
Experimental section
Materials
Unless stated otherwise all materials were purchased from Sigma-Aldrich and used as received. The monomers used were methyl methacrylate (MMA, 99%), tert-butyl methacrylate (t-BuMA, 98%), glycidyl methacrylate (GMA, 97%), AAm (≥98%) and N,N′-methylene bisacrylamide (MBAAm, 99%). t-BMA and MMA contained 200 ppm and less than 30 ppm of inhibitor, respectively, based on supplier information. A preliminary study showed that the presence of the inhibitors did not have a discernible effect on copolymer and hollow particle properties and consequently the monomers were used without inhibitor removal. Azoisobutyronitrile (AIBN, 99%) was purchased from VWR. The other reagents and initiators were triphenylphosphine (99%), ammonium persulfate (APS, ≥98%) and N,N,N′,N′-tetramethylethylenediamine (TEMED, 99%), 2-hydroxy-1-(4-(2-hydroxyethoxy)phenyl)-2-methylpropan-1-one (98%, photoinitiator) and 2,2-diphenyl-1-picrylhydrazyl (DPPH). The chromophore used was Rhodamine B. The stabiliser employed was poly(vinyl pyrolidone) (PVP) with average molecular weight of 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1. The solvents used were tetrahydrofuran (THF, anhydrous, ≥99%), dimethylformamide (DMF, anhydrous, ≥99.8%), methanol (reagent grade), diethyl ether (reagent grade) and dichloromethane (DCM, reagent grade). All water used for this study was distilled and deionised. The copolymers employed to prepare the hollow particles are listed in Table 1 and the methods used for their synthesis are described in the ESI.† Note that in this work the MAA units within the copolymers were prepared by hydrolysis of copolymerised t-BMA units within PMMA-t-BuMA as described in Scheme S1 (ESI†).
000 g mol−1. The solvents used were tetrahydrofuran (THF, anhydrous, ≥99%), dimethylformamide (DMF, anhydrous, ≥99.8%), methanol (reagent grade), diethyl ether (reagent grade) and dichloromethane (DCM, reagent grade). All water used for this study was distilled and deionised. The copolymers employed to prepare the hollow particles are listed in Table 1 and the methods used for their synthesis are described in the ESI.† Note that in this work the MAA units within the copolymers were prepared by hydrolysis of copolymerised t-BMA units within PMMA-t-BuMA as described in Scheme S1 (ESI†).
| Hollow particle | Copolymer compositiona | mol% MAAa | Hollow particle details | Composite gelb |
|---|---|---|---|---|
| a Compositions determined by 1H NMR spectroscopy (see ESI). b The values of y is the concentration of hollow particles (wt%) used for composite gel preparation. All gels studied were prepared using 12 wt% AAm and 1 wt% of MBAAm. | ||||
| HPXL | PMMA-MAA0.20-GMA0.12 | 20 | Shell-crosslinked hollow particles vinyl functionalised with GMA. | PAAm/HPXL(y) |
| HPVF | PMMA-MAA0.20-GMA0.12 | 20 | Unreacted GMA groups present in hollow particles | PAAm/HPVF(y) |
| HP | PMMA-MAA0.35 | 35 | Hollow particles that were not functionalised with GMA | PAAm/HP(y) |
Hollow particle and composite gel identification
Three types of hollow particles and composite gels were studied and are identified in Table 1. The preparation methods for the hollow particles containing unreacted vinyl groups from GMA (abbreviated as HPVF) and shell-crosslinked hollow particles (abbreviated as HPXL) are depicted in Scheme 1. All hollow particles were prepared at 0 °C. Hollow particles that did not contain GMA groups (abbreviated as HP) were prepared using the same method as used to prepared HPVF. All PAAm/hollow particle gels were prepared using AAm (12 wt%) and MBAAm (1 wt%). The composite gels are identified as PAAm/HPXL(y), PAAm/HPVF(y) and PAAm/HP(y) where the values for y represent the hollow particle concentration used for gel preparation (in wt%). As an example, PAAm/HPXL(10) represents a composite gel prepared using AAm (12 wt%), MBAAm (1 wt%) and HPXL (10 wt%).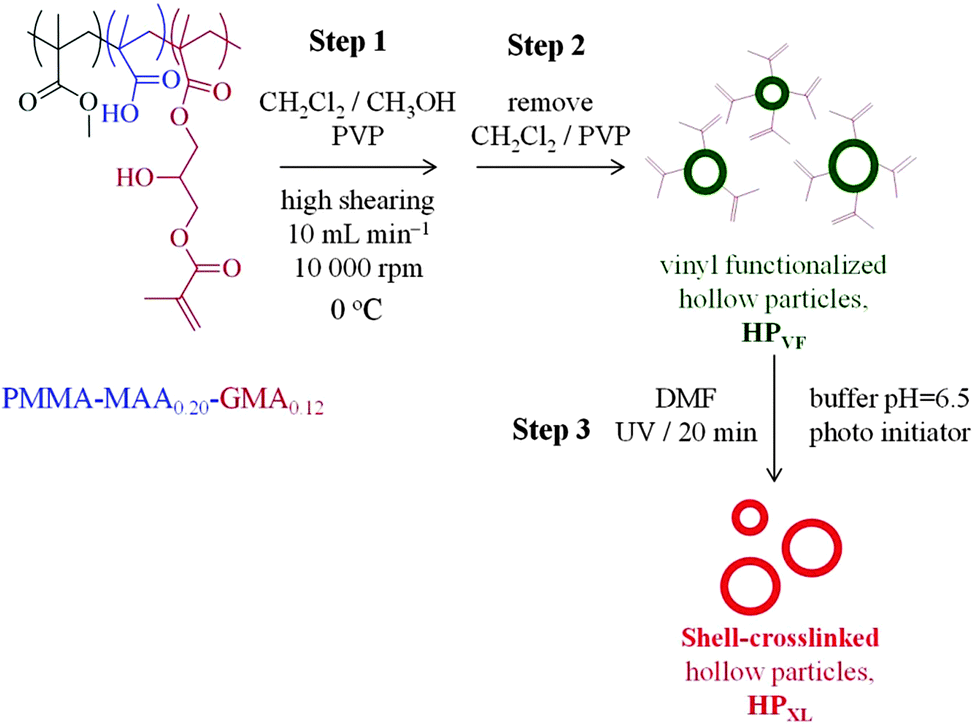 | ||
| Scheme 1 Hollow particle preparation. Depiction of preparation of hollow particles containing unreacted vinyl groups (HPVF) and shell-crosslinked hollow particles (HPXL). Hollow particles that did not contain pendant vinyl groups (HP) were also prepared. In that case PMMA-MAA0.35 was used as well as steps 1 and 2. | ||
Preparation of non-GMA functionalised hollow particles
HP dispersion preparation was performed using a similar emulsion templating method as reported previously.37 Briefly, PMMA-MAA (1.0 g) was dissolved in a cosolvent blend (22 mL) of DCM and methanol solvents (volume ratio, 8.4:1.6). The polymer solution was added into a PVP solution (60 mL, 4% w/w) over a constant rate of 10 mL min−1 which was sheared at 10000 rpm using a Silverson LR4 high speed mixer with cooling (0 °C). The emulsification continued for 30 s after polymer solution addition was complete. Afterwards, the emulsion was allowed to stir gently overnight to enable removal of DCM. Finally, the product was purified by repeated centrifugation and redispersion in water in order to remove excess PVP, followed by filtration using a 50 μm filter. The purified HP dispersion was dried in vacuum at room temperature.
Preparation of vinyl functionalised hollow particles
The preparation of HPVF was performed using PMMA-MAA0.20-GMA0.12 and followed the same procedure described above for the HP dispersion.Preparation of shell-crosslinked hollow particles
To prepare HPXL, the appropriate mass of HPVF particles was dispersed in buffer solution at pH 6.4 to give a HPVF concentration of 30 wt%, followed by the addition of the photoinitiator (3 wt%). To the polymer mixture a small amount of DMF was added to give a volume fraction of 10 vol%. Then, the mixture was sealed with a sheet of polypropylene to prevent evaporation and UV-irradiated for 30 min at 254 nm using a UV Crosslinker (Ultra-violet Products Ltd).Preparation of composite hydrogels
HP, HPVF and HPXL were used to prepare three series of PAAM composite hydrogels (see Table 1). The preparation of PAAm/HPXL(20) is described as an example. AAm (0.36 g, 5.06 mmol) and MBAAm (0.030 g, 0.19 mmol) were added to a dispersion (2.0 ml) containing HPXL (0.60 g) and stirred for 5.0 min. Water was added to adjust the final HPXL concentration to 20 wt%. APS (18.0 μL, 7.89 μmol) and TEMED (4.5 μL, 30 μmol) were added and the mixture purged with N2. The dispersion was heated at 60 °C until gelation occurred. The latter temperature was used in order to decrease the time for gelation and prevent significant hollow particle sedimentation. All the other composite gels were prepared using a similar method using the proper masses of HPXL, HPVF or HP. The pH of the composite gels studied in this work was ∼6.0 unless otherwise stated.Characterisation techniques
PMMA-t-BuMA copolymer was analysed using a GPC system equipped with a Shodex RI-101B refractive index detector at 35 °C. The mobile phase was THF, delivered at a flow rate of 1 mL min−1. The molecular weights were based on linear PMMA standards. The sample was dissolved in THF (0.2% w/v) and filtered prior to injection. The 1H NMR spectra of PMMA-t-BuMA (in CDCl3) as well as those of PMMA-MAA before and after vinyl functionalisation were obtained using a Bruker 400 MHz instrument (Fig. S1, ESI†). Potentiometric titration of the copolymers (1 wt% dispersion) was performed using a Mettler Toledo titration unit using NaOH (1.0 M).HPVF and HPVF-XL (0.1% w/w dispersion) optical images were obtained using an Olympus BX41 microscope and white transmitted light. Confocal laser scanning microscopy (CLSM) images were conducted using a Leica TCS SP5 broadband confocal instrument and Rhodamine B was used as the chromophore. A few drops of aqueous Rhodamine B solution (1 wt%) were mixed with the hollow particle dispersions and left for 24 h before obtaining CLSM images. The excitation wavelength used was 552 nm. SEM images of the freeze-dried gels (using liquid N2) were obtained using a Philips XL30 FEG SEM instrument. Dispersions of particles (10−5 wt%) were deposited on SEM stubs by evaporation at room temperature. The measurement of hollow particle size was performed using the Image J software. For rheology measurements a TA Instruments AR G2 temperature controlled rheometer which was equipped with an environmental chamber was used. For all gels a plate geometry (20 mm diameter) was employed. For measurements, the frequency sweep data were varied from 0.1 to 100 Hz, while the strain sweep data were run between 0.1 and 200%.
The composite gels were also characterised in terms of their degrees of swelling (DS) at pH values of 4.0 and 8.0. Two or three samples from each composite gel (approximate dimensions of 1.5 cm × 0.5 cm × 0.3 cm) were placed in small vials and were dried in a vacuum oven at room temperature for 72 h. Subsequently, aqueous buffer solutions at pH 4.0 and 8.0 (6.0 mL) were added to vials, and the hydrogels were left to swell. The buffers contained potassium dihydrogen phosphate and borax, respectively, and had nominal ionic strengths of 0.1 M. Periodically, the samples were reweighed (after carefully removing surface water) to determine their swollen mass. The DS values reported here were obtained after a swelling duration of 14 days. The DS values were calculated as the ratio of the average swollen network mass divided by the dry network mass determined gravimetrically.
Results and discussion
Synthesis and characterisation of hollow particles
The synthesis and characterisation of the copolymers used to prepare the hollow particles are described in the ESI.† Their compositions are shown in Table 1. The latter were obtained from 1H spectra (see Fig. S1, ESI†). The scaleable hollow particle preparation method used was straightforward and based on our earlier work.33,34 PMMA-MAA0.20-GMA0.12 was used for the preparation of hollow particles containing unreacted vinyl groups (HPVF) and also shell-crosslinked particles (HPXL). As a control system, PMMA-MAA0.35 was used to prepare non-GMA functionalised hollow particles (HP).The HPXL and HPVF hollow particles have not been previously reported. Potentiometric titration data for HP and HPVF dispersions were measured (Fig. S2, ESI†). The apparent pKa was determined from the pH corresponding to half the volume of NaOH added at the stoichiometric point. The latter and the mass of particles present were used to calculate the MAA content. The apparent pKa for the HP particles was 7.0 and the MAA content found by this method was 36.5 mol% The HPVF particle titration data gave an apparent pKa of 8.5 and the MAA and GMA contents determined from the data were 21.0 and 15.5 mol%, respectively. There was also a pH maximum when the neutralisation was ∼10 mol%. Related pH maxima were reported earlier for PMMA-MAA containing ∼20 mol% MAA and was attributed to a combination of pH-triggered shell expansion and subsequent pH buffering.39 The relatively high apparent pKa value for the HPVF particles is due to their lower MAA content. The MAA contents determined from the titration data for both HP and HPVF were within 1.5 mol% of the respective values determined using 1H NMR spectroscopy (Table 1), which shows that all of the MAA groups were accessible to OH−.
The morphologies of HPXL, HPVF and HP particles were probed using optical microscopy, CLSM and SEM. Optical microscopy (Fig. 1a, d and g) and CLSM images (Fig. 1b, e and h) of the HPXL and HPVF dispersions in water revealed spherical hollow particles. As noted earlier40 the HP dispersions contained hollow particles that were deformed (Fig. 1g and h). For all three systems the particles were polydisperse due to the turbulent flow present during the high shear emulsification method used to prepare the particles.
 | ||
| Fig. 1 Hollow particle morphologies. The identities of the hollow particles are shown in the left hand side of each row. Images from optical microscopy (left hand column) as well as CLSM (middle column) and SEM (right hand column) are shown. The pH of the dispersions was ∼5.6 for all systems. | ||
Analysis of the SEM images (Fig. 1c, f and i) gave number-average diameters for HPXL and HPVF of 4.2 ± 2.6 and 3.6 ± 1.8 μm, respectively, and they were not significantly different. In the case of HP the average equivalent spherical diameter was 4.4 ± 1.4 μm. CLSM images (Fig. 1b, e and h) indicate shell thicknesses in the range 1–2 μm. The SEM images of fragmented (Fig. 1c, inset) or folded (Fig. 1f, inset) hollow particles are consistent with this range. The average shell thickness can be estimated as ∼20% of the hollow particle diameter.
A new finding from this study is that the GMA functionalisation of the parent PMMA-MAA copolymer improved the spherical morphology of the hollow particles as can be seen by comparison of the SEM images for HPXL (Fig. 1c), HPVF (Fig. 1f) and the GMA-free HP system (Fig. 1i). We propose that GMA increased the hydrophobicity and decreased the total extent of hydrogen bonding between RCOOH groups during solvent evaporation and shell formation. Both factors should have favoured conformational rearrangement during phase separation and promoted the development of a smoother shell.
Composite gel morphology
HPXL, HPVF and HP were used for the preparation of the composite gels, PAAm/HPXL(y), PAAm/HPVF(y) and PAAm/HP(y), respectively (See Table 1 and Scheme 2). The latter two systems were used as controls to help elucidate the effects of HPXL shell crosslinking for the PAAm/HPXL(y) gels. Whilst HPVF contained unreacted GMA groups, the conditions used for preparation of the control PAAm/HPVF(y) gel (i.e., hydrophilic APS and pH much less than the pKa) deliberately favoured reaction of surface GMA groups for HPVF with the growing PAAm network and not the buried GMA groups. | ||
| Scheme 2 Composite gel preparation. Depiction of the procedure used for the preparation of PAAm/HPXL(y) composite gels. The same method was also used for the preparation of PAAm/HPVF(y) and PAAm/HP(y) gels. | ||
The composite gels formed free-standing gels (inset of Fig. 2h) and the morphologies of the composite gels were investigated using CLSM and SEM. Fig. 2a shows an CLSM image for PAAm/HPXl(10) and hollow particles are clearly evident and confirm that the hollow particles remained intact during PAAm network growth. SEM images of freeze-dried gel samples (Fig. 2b and c) show hollow particle cross sections. The hollow particles (arrows) had thick shells and may have fractured due to thermal stresses present during freeze-drying. CLSM images for PAAm/HPVF(20) gels (Fig. 2d) also showed spherical hollow particles which were also evident from SEM images (Fig. 2e and f). Whilst the PAAm/HP(20) gel also contained hollow particles (Fig. 2g), the particles were deformed, which was also apparent from the SEM images for PAAm/HP(10) (Fig. 2h and i).
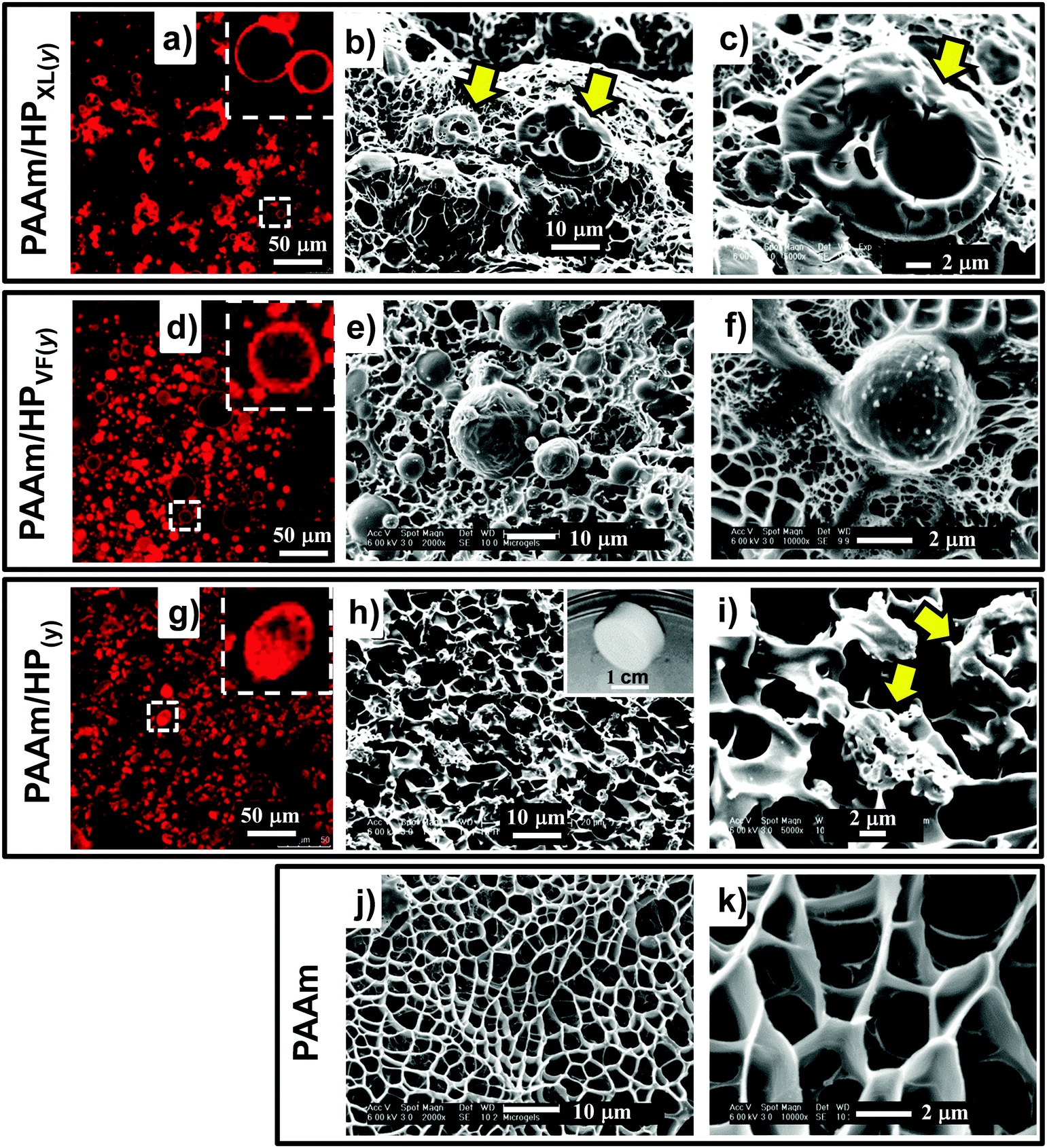 | ||
| Fig. 2 Composite gel morphologies. The identities of the gels are shown on the left hand side of each row. CLSM images are shown in (a), (d) and (g). The remaining images are from SEM of freeze-dried samples. The inset of (h) shows a free-standing gel. The values of y for all of the composite gels were 10 with the exception of (d) and (g), where the y values were 20. The arrows in (b), (c) and (i) identify hollow particles. | ||
The SEM images for PAAm are also shown for comparison (Fig. 2j and k). The morphology is similar to that commonly reported for AAm hydrogels and consisted of micrometer-sized pores that formed as a consequence of freeze-drying.41 In the hydrated state the pores would have contained expanded PAAm chains. This porous morphology can be seen from the SEM images for each of the composite gels (Fig. 2). Consequently, we conclude that the composite PAAm/hollow particle composite gel morphologies were combinations of those for PAAm gel (Fig. 2j and k) and the respective hollow particles (Fig. 1).
Composite gel mechanical properties
The composite gels studied here are unusual compared to most reported gel composites42,43 because of the high (hollow) particle concentrations present and the relatively large size of the hollow particles. The mechanical properties of the PAAm/HPXL(y) gels were studied using dynamic rheology. For this study PAAm/HPVF(y), PAAm/HP(y) and PAAm gel were used as controls. The frequency dependence of the storage modulus (G′) (Fig. 3a, c and e) increased with increasing hollow particle concentration (y). By contrast the G′ data for the PAAm gel (y = 0) had negligible frequency dependence. Fig. 3 also shows tanδ(=G′′/G′, where G′′ is the loss modulus) versus frequency data (Fig. 3b, d and f). The PAAm gel (y = 0) had tanδ values that were independent of frequency (Fig. 3b) indicating an ideal elastic response for the network.44 The frequency dependences of tanδ for PAAm/HPXL(y) (y = 2–20%) were also low (Fig. 3b). However, a pronounced frequency dependence was evident at the highest hollow particle concentration (y ≥ 25%) for all composite gels (Fig. 3b, d and f). It follows that for the composite gels with y ≤ 20% the PAAm network was able to accommodate the hollow particles without significant disruption of the PAAm network. At higher y values the elastic response was no longer ideal which is likely due to the dominance of the particle–particle network for stress distribution.
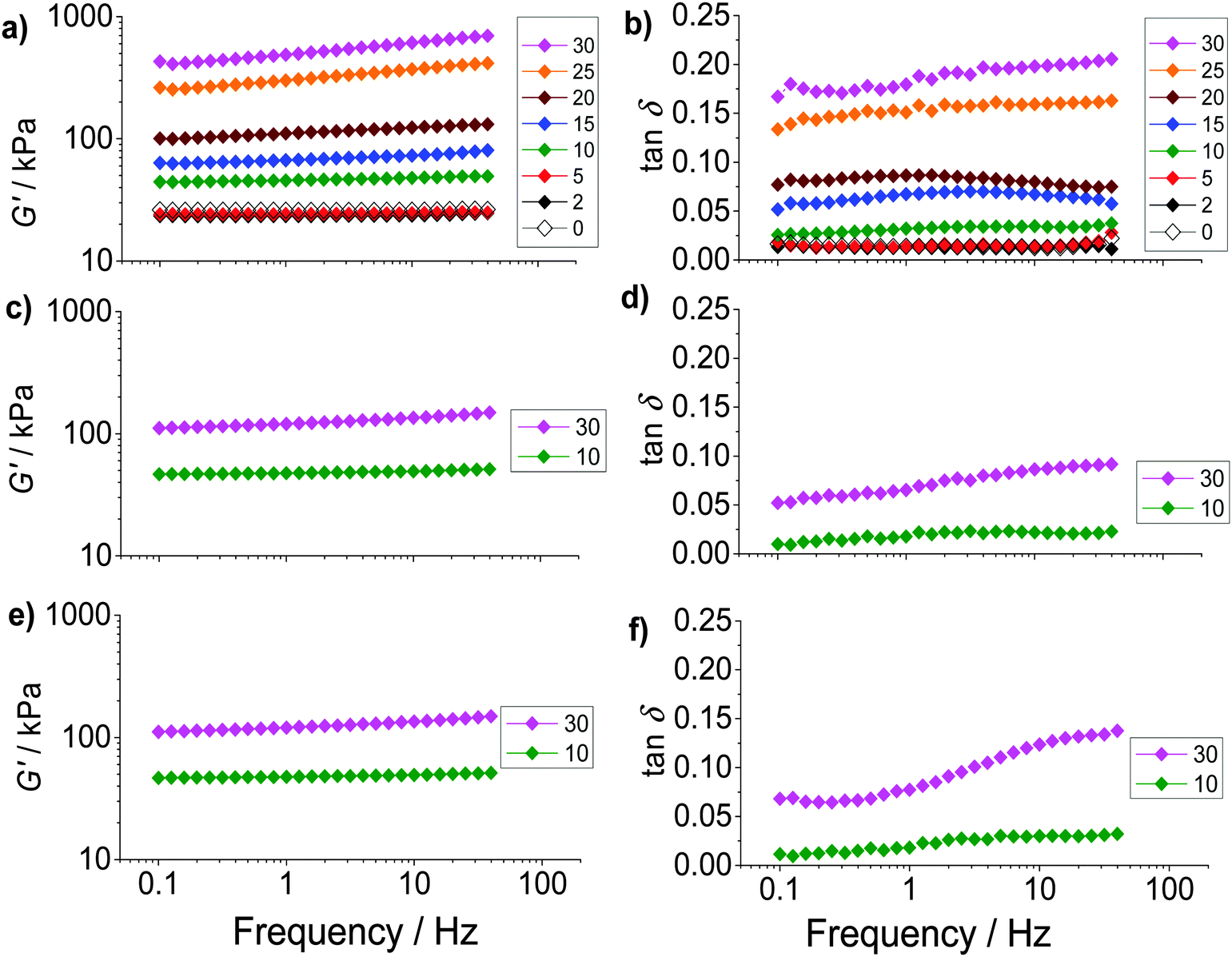 | ||
| Fig. 3 Frequency-sweep rheology study. The variations of G′ (storage modulus) and tanδ(=G′′/G′, where G′′ is the loss modulus) with frequency are shown in the left and right columns, respectively, for PAAm/HPXL(y) (a and b), PAAm/HPVF(y) (c and d) and PAAm/HP(y) (e and f). The legends show the values for y (%). | ||
The average G′ and tanδ values from the frequency-sweep data (corresponding to a frequency of ∼1.6 Hz) are shown as function of y in Fig. 4. The most striking feature of the data is the major increases of both G′ (Fig. 4a) and tanδ (Fig. 4b) that occur once y increases beyond 5%. The increase of tanδ with y (Fig. 4b) shows that the inclusion of hollow particles introduced additional inelastic deformation pathways within the composite gels and these were most pronounced for the PAAm/HPXL gels. A potential source of inelastic deformation is the relative movement of hollow particles under strain. Because the HPXL particles were exclusively shell-crosslinked one can imagine less efficient encapsulation of the HPXL particles near to the particle/PAAm interface and a greater chance for inelastic displacement.
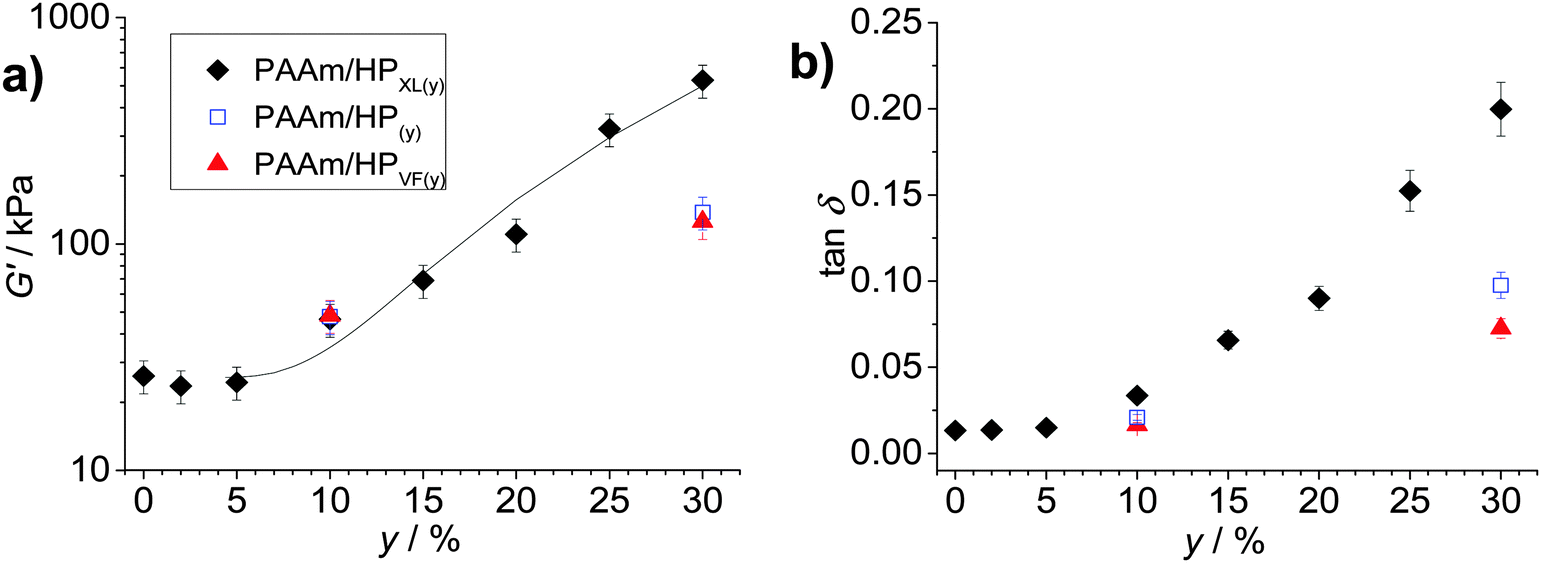 | ||
| Fig. 4 Effects of hollow particle concentration on gel mechanical properties. The variations of (a) G′ and (b) tanδ with particle concentration (y) are shown for the three composite gels. The data have been taken from Fig. 3. The legend applies to both graphs. The curve in (a) is the fit from eqn (1). The value for A was 0.10. | ||
What is the mechanism responsible for the variation of G′ with hollow particle concentration for the PAAm/HPXL gels shown in Fig. 4a? Supra-linear behaviour of the modulus with particle concentration is often observed for nanocomposite gels.45 The two primary mechanisms for modulus reinforcement for nanocomposite gels are interfacial interactions or percolation of the particles.46 Interfacial interactions are important for nanocomposite gels45 due to the high surface area-to-volume ratio of the nanoparticles. If we assume an average hollow particle diameter of ∼4 μm and that PAAm chains within 200 nm of the HP interface were constrained by contacts with the interface, which restricted their conformations,47 it can be shown that for PAAm/HPXL(20) less than 1% of the PAAm chains could have been affected by the hollow particle interfaces. By contrast hydrogels containing 18 nm silica nanoparticles had a maximum of about 40% of their chains adsorbed to the silica interface.48 Hence, interfacial interactions are not considered to have been significant for our PAAm/hollow particle gels. It was the use of low surface area-to-volume ratio micrometer-sized hollow particles that was hypothesised to enable high y values to be achieved without producing highly stiff, brittle, gels. The range of G′ values achieved for the gels (40–530 kPa) is wide (but not excessively high) and covers modulus values corresponding to skin49 and other soft tissue, which includes heart tissue.50
In order to test for percolation we applied the following equation which is an extension of particulate gel percolation models from van der Linden and Sagis51 and Mitsumata et al.52
| G′ = Gm′ + A(y − yc)n | (1) |
We next briefly consider the control gels. Whilst there is no significant difference between the G′ values for the PAAm/HPXL(y) data compared to those for the control samples when y = 10% (Fig. 4a), major differences were apparent for y = 30%. For those systems the G′ values decreased in the order: PAAm/HPXL(30) ≫ PAAm/HP(30) > PAAm/HPVF(30). We attribute the higher modulus for the former to shell-crosslinking for PAAm/HPXL, which increased the hollow particle modulus and hence the contribution of the hollow particle network to the overall gel modulus. The relatively low modulus values for PAAm/HPVF(30) and PAAm/HP(30) are ascribed to a lack (or absence) of intra-shell crosslinking. Another control composite PAAM/HP20 gel was also prepared which did not contain added MBAAm. The composite had a very low modulus (2.3 kPa), high tanδ value (0.84) at a frequency of 1.6 Hz and inelastically deformed when strained. Consequently, the composite gel had poor mechanical stability. Therefore, the roles of MBAAm were to increase matrix crosslinking (and matrix modulus) and composite mechanical stability.
To probe ductility and dissipation strain-sweep data were also obtained (Fig. 5). All of the composite gels showed linear viscoelastic regions for strain values less than ∼10%. The G′ values (Fig. 5a) decreased at higher strain values due to the onset of network breakdown. Fig. 5b shows the variation of G′′ with strain for PAAm/HPXL(y). The data show clearly that inclusion of the hollow particles strongly increased the G′′ values (and energy dissipation) at low strain (indicated by the arrow). A maximum was present at high strain for PAAm (y = 0), as well as for the composite gels, which is attributed to PAAm network breakdown. We propose that at low strain (e.g., ∼0.2–1.0%) the hollow particles consumed strain energy by deformation, fracture and inelastic particle movement. The latter probably included hollow particle-to-hollow particle bond breaking. At high strain (e.g., ∼20–70%) the PAAm network dissipated energy through strand breakage and network breakdown. As y increased the dissipation changed from being dominated by the PAAm network (e.g., y < 5%) to having a large contribution from the percolated hollow particle network (e.g., y ≥ 10%).
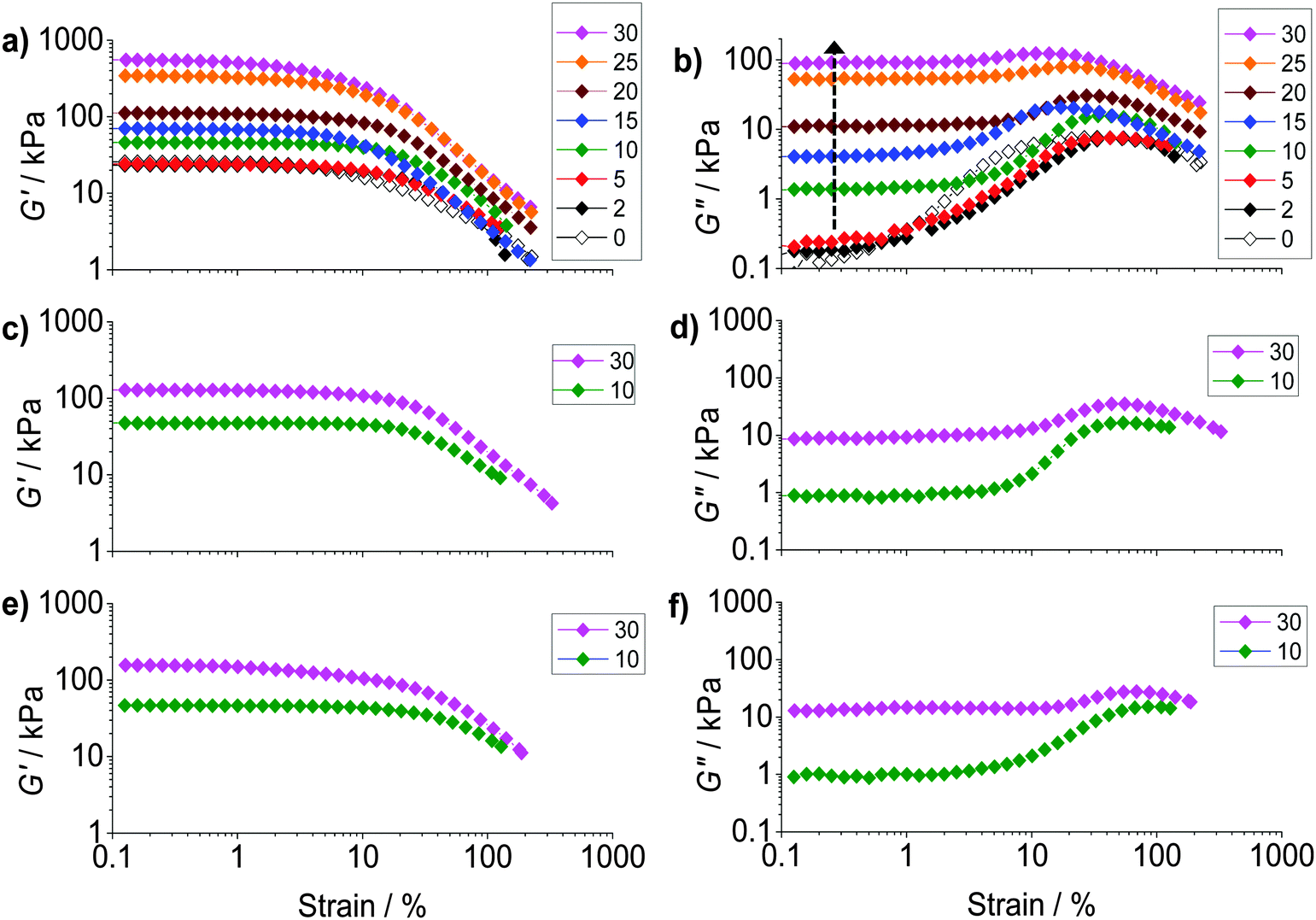 | ||
| Fig. 5 Strain-sweep rheology study. Data for G′ (left column) and G′′ (right column) are shown as a function of strain for PAAm/HPXL(y) (a and b), PAAm/HPVF(y) (c and d) and PAAm/HP(y) (e and f). The values for y are shown in the legends. The arrow in (b) highlights the increase for G′′ at low strain with increasing y. | ||
The strain dependence for G′′ is shown for PAAm/HPVF(y) and PAAm/HP(y) in Fig. 5d and f, respectively. Once again, the increase in the G′′ values at low strain can be seen as y increased. The extent of energy dissipation at low strain as evidenced by G′′ was about a factor of 10 lower when y = 30% for these two composite gels compared to that for PAAm/HPXL(30) (Fig. 5b). We speculate that for the former gels the hollow particles more easily fragmented (due to low or negligible shell crosslinking) and involved less large-scale particle movement (e.g., hollow particle-to-hollow particle bond breaking) under strain.
To further probe dissipation within the composite gels the normalised loss modulus, ΔGn′′(=(Gpeak′′ − Go′′)/Go′′) was plotted (Fig. 6a). The values for Gpeak′′ and Go′′ are the maximum G′′ value and the strain-independent value for G′′ measured at low strain, respectively, obtained from the strain-sweep data (Fig. 5). This representation for the G′′ data (which measures energy dissipated per unit strain) normalised the high strain energy loss (due to PAAm network breakdown) to that occurring at low strain (due to the hollow particles). It can be seen from Fig. 6a that the ΔGn′′ values for PAAm/HPXL(y) decreased exponentially with increasing y. Such a pronounced change in relative energy loss is attributed to dissipative relaxation processes associated with the percolated hollow particle network. For a percolated network it is reasonable to envisage that the proportion of inter-hollow particle contacts (and dissipative contacts) would increase very rapidly with increasing y.
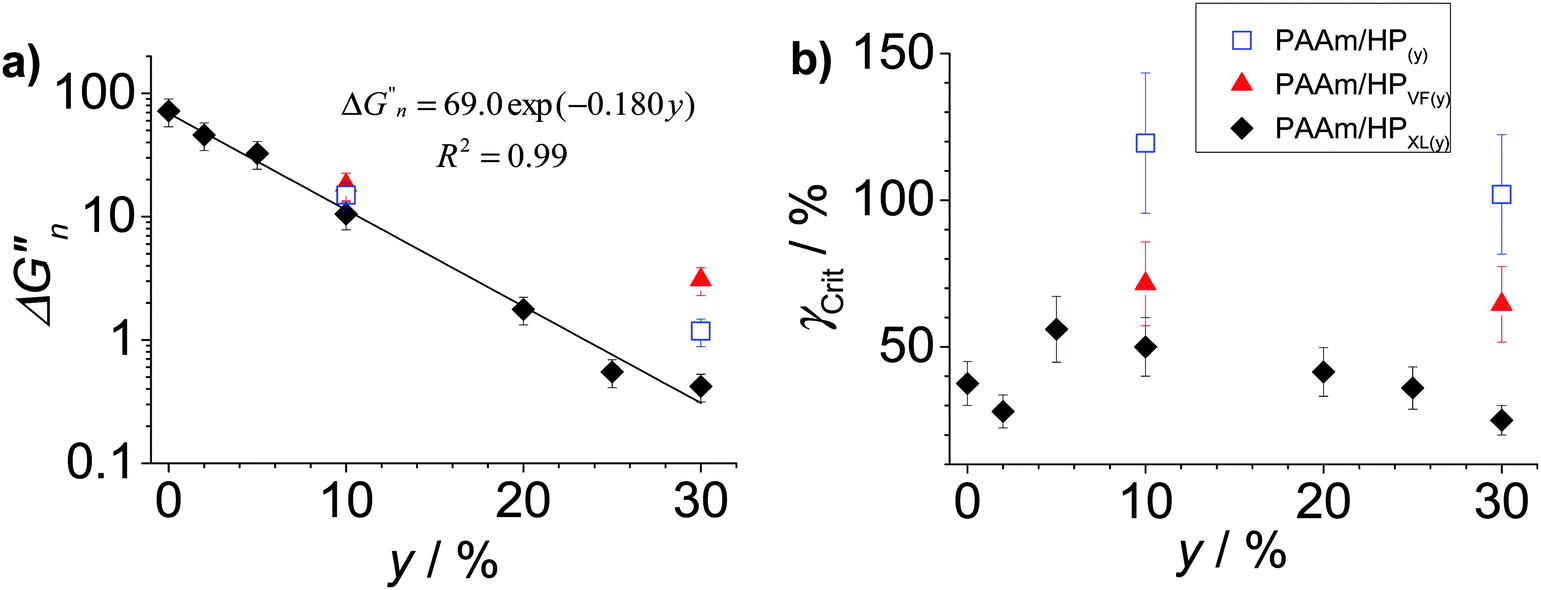 | ||
| Fig. 6 Effects of particle concentration on gel dissipation and ductility. (a) Variation of ΔGn′′((=(Gpeak′′ − Go′′)/Go′′) where Gpeak′′ and Go′′ are maximum G′′ and strain-independent G′′ values, respectively) and (b) Variation of γCrit with particle concentration (y) for the composite gels. The data have been taken from Fig. 5. | ||
The ductility of the composite gels was assessed from the critical strain (γCrit) which is the strain value at which G′ = G′′. The γCrit values were obtained from the data shown in Fig. 5 and are plotted as a function of y (Fig. 6b). Whilst there was scatter for the PAAm/HPXL(y) data it can be seen that the γCrit values increased for low values of y before decreasing at the highest y value (30%). Inclusion of HPXL was beneficial (or at the least was not detrimental) for composite gel ductility compared to PAAm as judged by the γCrit values. Interestingly, inclusion of HPVF and HP gave greater increases in composite gel ductility compared to HPXL. We propose that the HPVF and HP particles were less stiff (due to less or negligible intra-shell crosslinking) and were able to deform and/or fracture during strain. These inelastic processes dissipated fracture energy more uniformly, decreased crack propagation and increased overall composite gel ductility.
Effect of pH on the properties of PAAm/HPXL(y) gels
Hydrogels that contain pH-responsive hollow particles may have potential application for wound repair or membranes. In order to assess the effect of pH on the properties of PAAm/HPXL(y) composites we first investigated the response of the HPXL particles to pH variation. Optical micrographs were obtained for HPXL particles dispersed at pH values of 4.0 (Fig. 7a), 7.4 (Fig. 7b) and 12.3 (Fig. 7c). The hollow particles became noticeably more diffuse in appearance when the pH was greater than or equal to 7.4. The average size of the HPXL particles at pH 4.0, 7.4 and 12.3 were 7.4 ± 4.1, 8.7 ± 3.4 and 11.2 ± 3.2 μm, respectively. Those values are consistent with pH-triggered hollow particle swelling. Importantly, the images shown in Fig. 7b and c show that shell crosslinking was effective in preventing dissolution of the hollow particles. | ||
| Fig. 7 Shell-crosslinked hollow particle morphologies at different pH values. Optical micrographs for HPXL dispersions at pH values of (a) 4.0, (b) 7.4 and (c) 12.3. The scale bars in (b) also apply to (a) and (c). | ||
We next investigated the effects of pH on the morphology and mechanical properties of the PAAm/HPXL(y) gels. The pH values of 4.0 and 8.0 were selected in order to include the pH range for normal skin and a typical wound area.54 Optical micrographs for the as-made PAAm/HPXL(10) gel (Fig. 8a) and CLSM images for the gels after swelling at pH values of 4.0 (Fig. 8b) and 8.0 (Fig. 8c) show that the hollow particles were present within the composite gels at both pH values. The hollow particles can be seen from the insets and also the arrow in Fig. 8c. There is also evidence of aggregates of hollow particles within the composite gels (Fig. 8b and c) which is consistent with the percolated morphology proposed in Scheme 2.
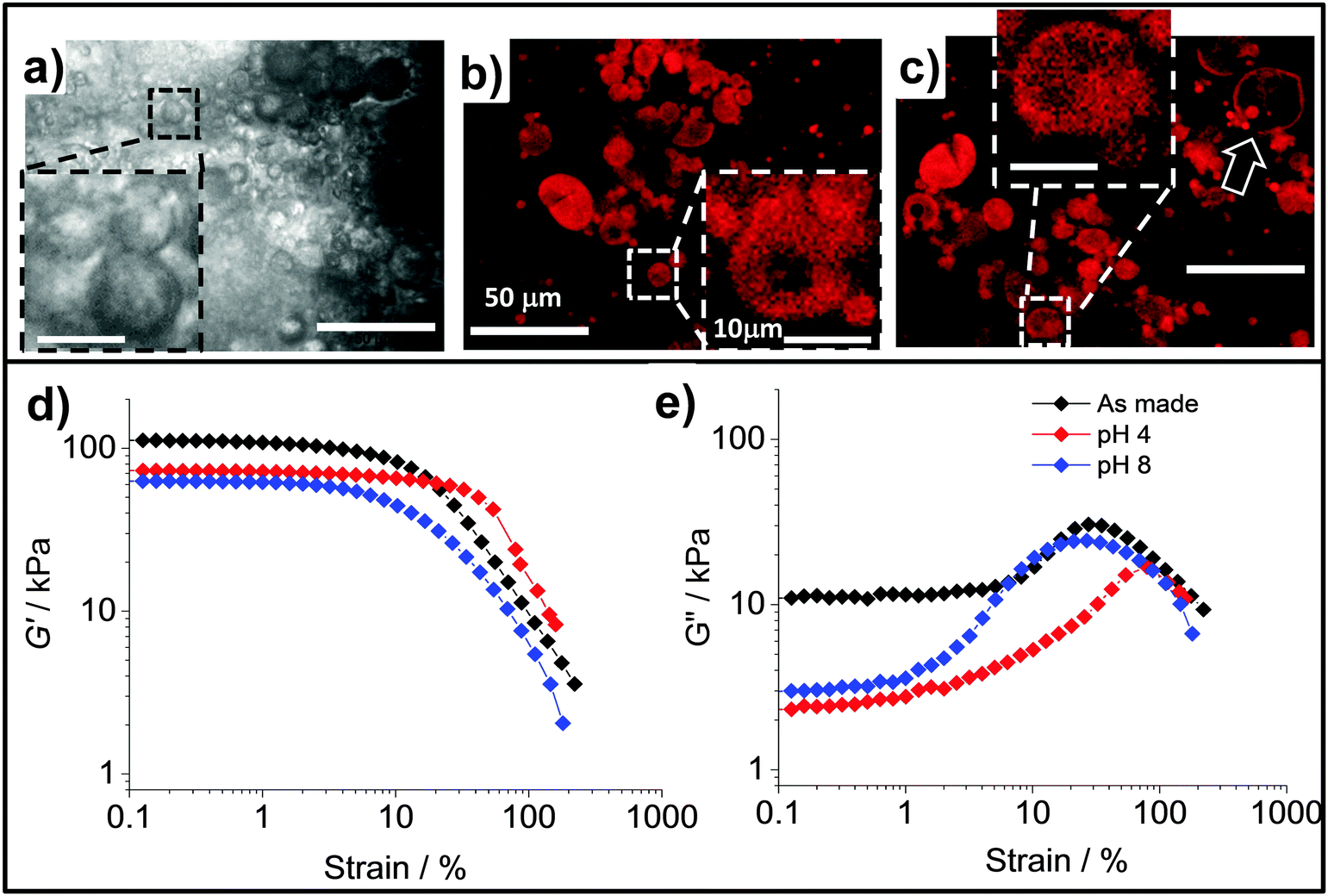 | ||
| Fig. 8 Morphology and mechanical properties at different pH values. Optical microscopy (a) and CLSM images (b and c) for as-made (a) and PAAm/HPXL(10) gels at pH 4.0 (b) and 8.0 (c), respectively. The scale bars in (b) also apply to (a) and (c). The arrow in (c) highlights a large hollow particle. (d and e) Show strain-sweep rheology data for PAAm/HPXL(20) gels before and after swelling equilibrium at pH 4.0 and 8.0. | ||
Strain-sweep rheology measurements were performed using PAAm/HPXL(20) gels which were swollen at pH 4.0 (degree of swelling, DS = 5.2 ± 0.2) and 8.0 (DS = 6.2 ± 0.1) and the data are shown in Fig. 8d and e. The initial DS for the as-made gel corresponds to 3.0. Consequently, swelling occurred at both pH values. A contribution to swelling originated from the osmotic pressure of the PAAm phase which became significant when the gels were no longer constrained by the limited amount of water used during their preparation. Compared to the modulus for the as-made gel, which had a G′ value of ∼110 kPa (Fig. 8d), the G′ values for the swollen gels decreased to 73 kPa (pH 4.0) and 63 kPa (pH 8.0), respectively. These decreases were due to increased swelling for the gels at both pH values. Taken together, both the DS and rheology data demonstrate stronger swelling for the gels at pH 8.0 and pH-responsiveness.
The values for γCrit determined from the strain-sweep data (Fig. 8d and e) obtained at pH 4.0 and 8.0 were 110 ± 22% and 30 ± 6.0%, respectively. The γCrit value at pH 8.0 (and hence ductility) had not significantly decreased compared to the as-made system (γCrit = 41.5 ± 8.3%) as a result of swelling. Whilst we currently do not have a simple explanation for the increased γCrit value at pH 4.0 the improved ductility compared to the as-made state is pleasing. These results show that the composite gels had good mechanical property robustness to swelling in the pH range of 4.0 to 8.0, which is potentially useful for membranes and wound healing applications.55
Conclusions
PAAm composite hydrogels containing micrometer-sized pH-responsive shell cross-linked hollow particles were prepared and studied for the first time. The hollow particles introduced here were vinyl functionalised and prepared by a scaleable method. A new finding for these particles was that the shell morphology became more uniform and spherical when the pH-responsive copolymer contained GMA. This result extends the range of functionalisation possibilities for this class of hollow particles33 and should increase their versatility for future use. PAAm/HPXL composite gels were found to have tuneable mechanical properties that were governed by percolation. The percolated hollow particle network increased the modulus in a predictable manner and the ductility for all of the dispersions was at least as high (or better) than that for the parent PAAm gel. Control hollow particles showed that shell-crosslinking was important for obtaining the highest modulus improvement through hollow particle inclusion. The PAAm/HPXL composite gels were pH-responsive and showed increased swelling at pH 8.0 compared to pH 4.0. Because the composites have tuneable modulus values, good mechanical properties in the swollen state and are pH-responsive they may enable future applications as pH-responsive wound dressings or membranes.Acknowledgements
The authors would like to thank the EPSRC for funding this work.References
- N. A. Peppas, J. Z. Hilt, A. Khademhosseini and R. Langer, Adv. Mater., 2006, 18, 1345–1360 CrossRef CAS.
- N. A. Peppas, P. Bures, W. Leobandung and H. Ichikawa, Eur. J. Pharm. Biopharm., 2000, 50, 27–46 CrossRef CAS PubMed.
- D. Seliktar, Science, 2012, 336, 1124–1128 CrossRef CAS PubMed.
- B. V Slaughter, S. S. Khurshid, O. Z. Fisher, A. Khademhosseini and N. A. Peppas, Adv. Mater., 2009, 21, 3307–3329 CrossRef PubMed.
- A. A. Amini and L. S. Nair, Biomed. Mater., 2012, 7, 1–3 Search PubMed.
- J. P. Gong, Y. Katsuyama, T. Kurokawa and Y. Osada, Adv. Mater., 2003, 15, 1155–1158 CrossRef CAS.
- Y. Tanaka, R. Kuwabara, Y. H. Na, T. Kurokawa, J. P. Gong and Y. Osada, J. Phys. Chem. B, 2005, 109, 11559–11562 CrossRef CAS PubMed.
- R. E. Webber, C. Creton, H. R. Brown and J. P. Gong, Macromolecules, 2007, 40, 2919–2927 CrossRef CAS.
- K. Haraguchi, Curr. Opin. Solid State Mater. Sci., 2007, 11, 47–54 CrossRef CAS.
- K. Haraguchi, R. Farnworth, A. Ohbayashi and T. Takehisa, Macromolecules, 2003, 36, 5732–5741 CrossRef CAS.
- K. Haraguchi and T. Takehisa, Adv. Mater., 2002, 14, 1120–1124 CrossRef CAS.
- K. Haraguchi, T. Takehisa and S. Fan, Macromolecules, 2002, 35, 10162–10171 CrossRef CAS.
- S. Miyazaki, T. Karino, H. Endo, K. Haraguchi and M. Shibayama, Macromolecules, 2006, 39, 8112–8120 CrossRef CAS.
- M. Shibayama, T. Karino, S. Miyazaki, S. Okabe, T. Takehisa and K. Haraguchi, Macromolecules, 2005, 38, 10772–10781 CrossRef CAS.
- A. K. Gaharwar, N. A. Peppas and A. Khademhosseini, Biotechnol. Bioeng., 2014, 111, 441–453 CrossRef CAS PubMed.
- O. Okay and W. Oppermann, Macromolecules, 2007, 40, 3378–3387 CrossRef CAS.
- Y. Xiang and D. Chen, Eur. Polym. J., 2007, 43, 4178–4187 CrossRef CAS.
- N. S. Satarkar and J. Z. Hilt, J. Controlled Release, 2008, 130, 246–251 CrossRef CAS PubMed.
- H. Bai, C. Li, X. Wang and S. Gaoquan, Chem. Commun., 2010, 46, 2376–2378 RSC.
- Z. Cui, A. H. Milani, P. J. Greensmith, J. Yan, D. J. Adlam, J. A. Hoyland, I. A. Kinloch, A. J. Freemont and B. R. Saunders, Langmuir, 2014, 30, 13384–13393 CrossRef CAS PubMed.
- S. R. Shin, S. M. Jung, M. Zalabany, K. Kim, P. Zorlutuna, S. B. Kim, M. Nikkhah, M. Khabiry, M. Azize, J. Kong, K.-T. Wan, T. Palacios, M. R. Dokmeci, H. Bae, X. S. Tang and A. Khademhosseini, ACS Nano, 2013, 7, 2369–2380 CrossRef CAS PubMed.
- T. H. Tran, A. Vimalanandan, G. Genchev, J. Fickert, K. Landfester, D. Crespy and M. Rohwerder, Adv. Mater., 2015, 27, 3825–3830 CrossRef CAS PubMed.
- L. H. Lim, A. Tan, S. Simovic and C. A. Prestidge, Int. J. Pharm., 2011, 409, 297–306 CrossRef CAS PubMed.
- S. H. Im, T. Herricks, Y. T. Lee and Y. Xia, Nat. Mater., 2005, 4, 671–675 CrossRef PubMed.
- G. Sukhorukov, A. Fery and H. Mohwald, Prog. Polym. Sci., 2005, 30, 885–897 CrossRef CAS.
- O. J. Cayre and S. Biggs, J. Mater. Chem., 2009, 19, 2724–2728 RSC.
- M. Sauer and W. Meier, Chem. Commun., 2001, 55–56 RSC.
- H. N. Yow and A. F. Routh, Soft Matter, 2006, 2, 940–949 RSC.
- I. Choi, S. T. Malak, W. Xu, W. T. Heller, C. Tsitsilianis and V. V. Tsukruk, Macromolecules, 2013, 46, 1425–1436 CrossRef CAS.
- W. Xu, P. A. Ledin, F. A. Plamper, C. V. Synatschke, A. H. E. Müller and V. V. Tsukruk, Macromolecules, 2014, 47, 7858–7868 CrossRef CAS.
- A. Zelikin, Q. Li and F. Caruso, Chem. Mater., 2008, 20, 2655–2661 CrossRef CAS.
- F. Caruso, R. A. Caruso and H. Mohwald, Science, 1998, 282, 1111–1114 CrossRef CAS PubMed.
- R. Bird, T. J. Freemont and B. R. Saunders, Chem. Commun., 2011, 47, 1443–1445 RSC.
- S. Nayak, D. Gan, M. J. Serpe and L. A. Lyon, Small, 2005, 4, 416–421 CrossRef PubMed.
- R. Contreras-Cáceres, L. Schellkopf, C. Fernández-López, I. Pastoriza-Santos, J. Pérez-Juste and M. Stamm, Langmuir, 2015, 31, 1142–1149 CrossRef PubMed.
- F. Zhang and C.-C. Wang, Colloid Polym. Sci., 2008, 286, 889–895 CAS.
- S. S. Halacheva, D. J. Adlam, T. J. Freemont, J. Hoyland and B. R. Saunders, Biomacromolecules, 2014, 15, 1814–1827 CrossRef CAS PubMed.
- S. S. Halacheva, T. J. Freemont and B. S. Saunders, J. Mater. Chem. B, 2013, 1, 4035–4036 RSC.
- R. Bird, T. Freemont and B. R. Saunders, Soft Matter, 2012, 8, 1047–1057 RSC.
- R. Bird, S. Tungchaiwattana, T. Freemont and B. R. Saunders, Soft Matter, 2012, 8, 3062 RSC.
- G. B. Trappmann, J. E. Connelly, D. G. T. Strange, Y. Li, M. L. Oyen, M. A. Cohen Stuart, H. Boehm, B. Li, V. Vogel, J. P. Spatz, F. M. Watt and W. T. S. Huck, Nat. Mater., 2012, 11, 642–649 CrossRef PubMed.
- J. R. Capadona, K. Shanmuganathan, S. Trittschuh, S. Seidel, S. J. Rowan and C. Weder, Biomacromolecules, 2009, 10, 712–716 CrossRef CAS PubMed.
- L. Carlsson, S. Rose, D. Hourdet and A. Marcellan, Soft Matter, 2010, 6, 3619–3631 RSC.
- H. H. Winter and F. Chambon, J. Rheol., 1986, 30, 367–382 CrossRef CAS.
- S. Rose, A. Dizeux, T. Narita, D. Hourdet and A. Marcellan, Macromolecules, 2013, 46, 4095–4104 CrossRef CAS.
- R. Perez-Aparicio, A. Vieyres, P.-A. Albouy, O. Sanseau, L. Vanel, D. R. Long and P. Sotta, Macromolecules, 2013, 46, 8694–8972 Search PubMed.
- A. D. Drozdov, Compos. Interfaces, 2013, 20, 673–692 CrossRef CAS.
- W.-C. Lin, W. Fan, A. Marcellan, D. Hourdet and C. Creton, Macromolecules, 2010, 43, 2554–2563 CrossRef CAS.
- A. Delalleau, G. Josse, J. M. Lagarde, H. Zahouani and J. M. Bergheau, Wear, 2008, 264, 405–410 CrossRef CAS.
- G. C. Engelmayr, M. Cheng, C. J. Bettinger, J. T. Borenstein, R. Langer and L. E. Freed, Nat. Mater., 2008, 7, 1003–1010 CrossRef CAS PubMed.
- E. van der Linden and L. M. C. Sagis, Langmuir, 2001, 17, 5821–5824 CrossRef CAS.
- T. Mitsumata, Y. Kosugi and S. Ouchi, Prog. Colloid Polym. Sci., 2009, 136, 163–170 CAS.
- M. C. Grant and W. B. Russel, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1993, 47, 2606 CrossRef CAS.
- S.-T. Oh, W.-R. Kim, S.-H. Kim, Y.-C. Chung and J.-S. Park, Fibers Polym., 2011, 12, 159–165 CrossRef CAS.
- A. M. Diez-Pascual and A. L. Diez-Vicente, Biomacromolecules, 2015, 16, 2631–2644 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5sm02521d |
| This journal is © The Royal Society of Chemistry 2016 |