DOI:
10.1039/C6RA00524A
(Paper)
RSC Adv., 2016,
6, 45475-45481
A Zn-MOF constructed from electron-rich π-conjugated ligands with an interpenetrated graphene-like net as an efficient nitroaromatic sensor†
Received
12th January 2016
, Accepted 21st April 2016
First published on 25th April 2016
Abstract
A novel zinc-based luminescent metal–organic framework (Zn-MOF) has been successfully constructed based on a designed flexible and electron-rich N-involved linker (HL = 4-(bis(4-(pyridin-4-yl)phenyl)amino)benzoic acid). The framework of this Zn-MOF exhibits a 2-fold interpenetrated network which is composed of (3,3)-c sheets. The Zn-MOF has a strong solid state emission at 512 nm. The luminescence signal of the Zn-MOF can be quenched efficiently by trace amounts of electron-deficient nitroaromatics, especially 2,4,6-trinitrophenol (TNP). The quenching constant (Ksv) for TNP is 2.11 × 104 M−1, indicating that this framework can be employed as an excellent chemical sensor for identifying and quantifying TNP. This work highlights a strategy for designing a N-involved π-electron-rich enhanced ligand with nucleophilic properties for MOF-based materials as sensors. It also paves the way toward exploring other more efficient MOF materials as sensors for determining electron-deficient nitroaromatics.
Introduction
Metal–organic framework (MOF) materials have received much attention due to their tuneable open-framework structures and pore sizes. The physicochemical properties of MOF materials can be tuned easily through organic ligand design or modification. These materials have attracted broad interest due to their potential diverse applications, for instance, gas storage,1,2 separation,3–5 luminescence,6–10 and catalysis.11,12 Recently, MOF materials have been explored intensively as chemosensors or biosensors.13,14 When using different MOF materials within chemical sensors, the host–guest properties of the MOF material can be transferred to the device for achieving detectable changes in the light-emission colour or luminescence characteristics.13,15–17 Moreover, the different tuneable porosities, functional pore surfaces and bulk conjugated backbones of the MOF materials could also introduce very promising device performances with diverse sensitivity, selectivity and response time.
Explosive materials such as nitroaromatics, including different nitrophenols, nitrobenzenes and nitrotoluenes, are primary components of industrial explosives and environmentally poisonous substances. Due to their crucial effects on the safety of the environment, the effective detection of nitroaromatics is of paramount importance and has aroused burgeoning interest worldwide. Among all these nitroaromatics, 2,4,6-trinitrophenol (TNP) has the strongest explosive capacity and a low safety coefficient. However, it has been widely used in many fields, such as in the dye, explosives, matches, fireworks and pharmaceutical industries.18 Although there are many different types of materials which can be used to detect TNP and its derivatives, MOF based fluorescence detection shows great advantages, because of the electronic tunability, feasible portability and ease of operation. Moreover, the organic ligands involved in MOF materials can be designed for a specific use, based on the oxidative quenching mechanism. As reported in the literature,19–22 when electron-rich small molecules or MOFs interact with TNP and its derivatives, upon photo-excitation the electrons or energy will be transferred from electron donors such as small molecules or organic ligands to the electron deficient TNP derivatives, which could lead to oxidation of the excited state and luminescence quenching.
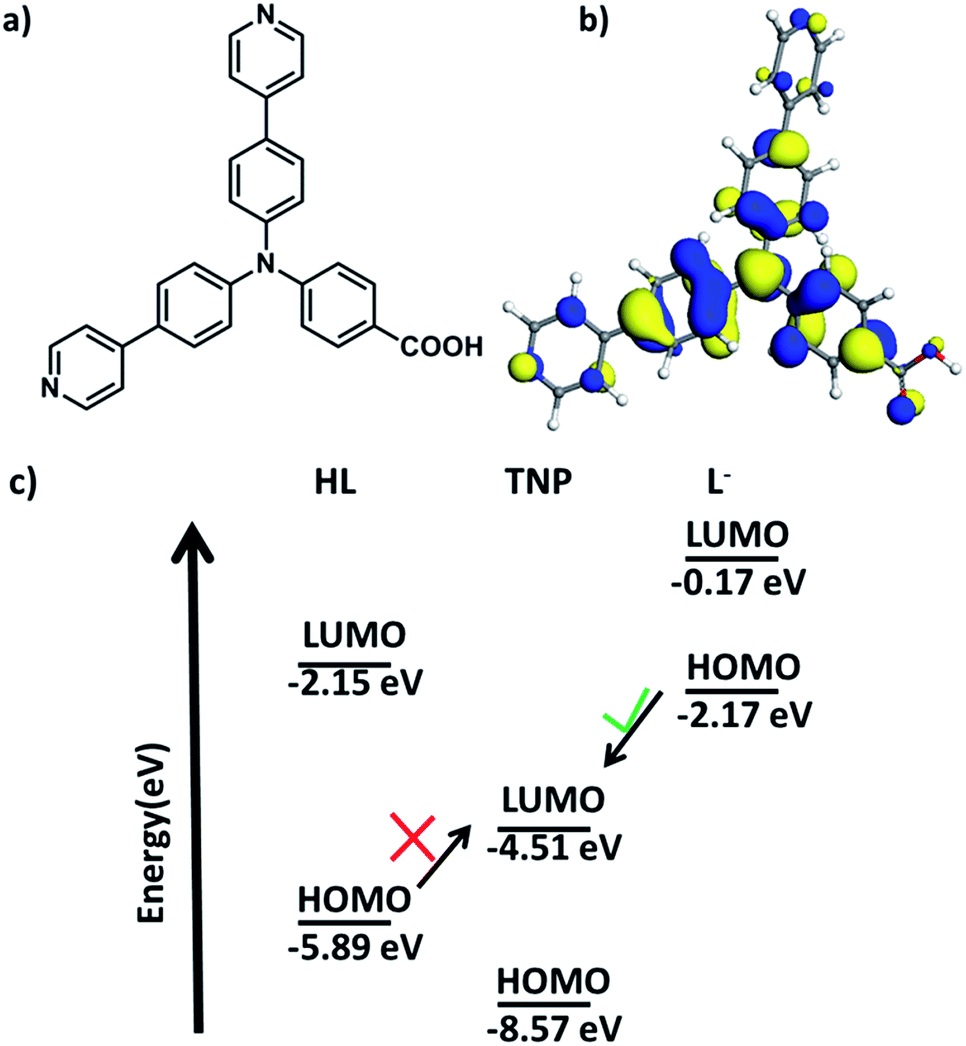
Inspired by this knowledge, the design of electron-rich π-conjugated fluorescent ligands is a promising way to fabricate effective MOF materials with nucleophilic features and appropriate pore sizes and shapes for application as efficient luminescent sensors. Herein, we design a novel non-rigid π-electron-rich organic ligand HL, 4-(bis(4-(pyridin-4-yl)phenyl)amino)benzoic acid (Fig. 1a), which is a triangular ligand with an electron-rich N atom located at the centre. The introduced nitrogen atom with lone pair electrons is expected to enrich the electron density of the conjugated π-electrons of the three benzene or pyridine rings. The optimized structure geometry and its highest occupied molecular orbital (HOMO) with an energy level of −5.89 eV are also shown in Fig. 1b and c, and based on quantum chemical calculations, a rich electron density population is observed around the N atom. Moreover, the HOMO and LUMO levels of the deprotonated 4-(bis(4-(pyridin-4-yl)phenyl)amino)benzoic acid were also calculated as shown in Fig. 1c. The energy levels of the deprotonated ligand are increased compared to HL, energy transfer is infeasible from HL to TNP but feasible from deprotonated HL to TNP. Inspired by these calculation results, herein, using this ligand, we successfully synthesize a novel zinc-based MOF material which exhibits excellent luminescence properties. This new MOF material can be used as a highly efficient sensor for the detection of nitroaromatics, especially TNP.
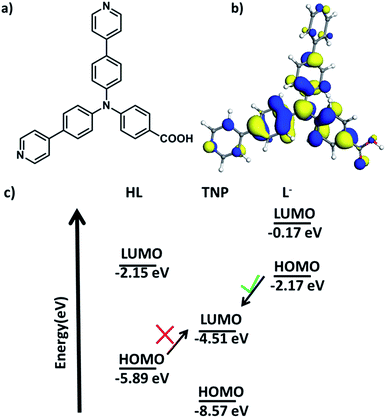 |
| | Fig. 1 (a) Structure of 4-(bis(4-(pyridin-4-yl)phenyl)amino)benzoic acid (HL); (b) its HOMO configuration; (c) energy level diagram of HL, TNP and deprotonated HL. | |
Experimental section
Materials
All the reagents and solvents were purchased from different companies, and directly used as received without further purification, except N,N-dimethylformamide (DMF). DMF was distilled over CaH2 under reduced pressure.
The details of the reagents used are as follows: zinc nitrate hexahydrate (AR, 99.0%, Xilong Chemical Co., Ltd.), dimethylformamide (DMF) (AR, 99.5%, Sinopharm Chemical Reagent Co., Ltd.), triphenylamine (AR, 99%, Energy Chemistry Co., Ltd.), phosphorus oxychloride (POCl3) (AR, 98%, Zhengzhou alpha Chemical Co., Ltd.), N-bromosuccinimide (NBS) (AR, 98%, Zhengzhou alpha Chemical Co., Ltd.), pyridin-4-yl-boronic acid (AR, 98%, Neiqiu county huaxing chemical Co., Ltd.), potassium permanganate (AR, 98%, Zhengzhou Huawen Chemical Co., Ltd.), ethyl acetate (EA) (99%, Aladdin Industrial Corporation), petroleum ether (PE) (99%, Wuxi zhongkun biochemical technology Co., Ltd.), magnesium sulfate (AR, 98%, Shanghai meryer chemical technology Co., Ltd.), aluminum oxide (Al2O3) (AR, 99%, Accelerating Scientific and Industrial Development Serving Humanity), tetrahydrofuran (THF) (AR, 99%, Aladdin Industrial Corporation), potassium carbonate (K2CO3) (AR, 99%, Shanghai Macklin Biochemical Co., Ltd), 1,4-dioxane (AR, 99%, Shanghai meryer chemical technology Co., Ltd.), hydrochloric acid (99%, Shenzhen Weichangshun Chemical Co., Ltd.), and acetonitrile (99%, Meryer (Shanghai) Chemical Technology Co., Ltd.).
Synthesis of 4-(bis(4-(pyridin-4-yl)phenyl)amino)benzoic acid (HL)
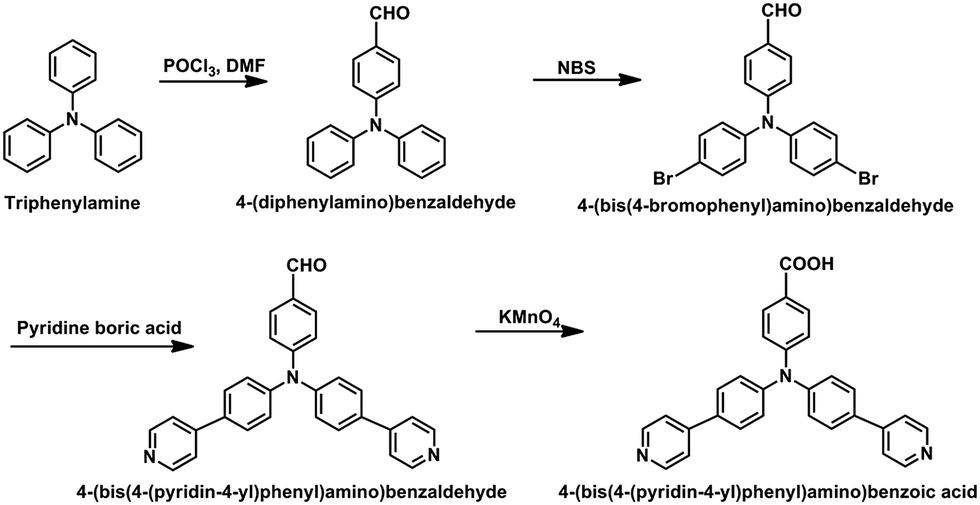
The HL ligand was synthesized according to previously reported literature by combing different reaction steps from different literature references.23–31 A full representation of the synthesis procedure is illustrated in Scheme 1.
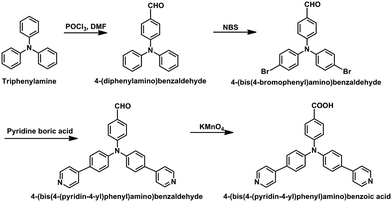 |
| | Scheme 1 Synthesis routine of 4-(bis(4-(pyridin-4-yl)phenyl)amino)benzoic acid (HL). | |
4-(Diphenylamino)benzaldehyde
Phosphorus oxychloride (POCl3) (31.5 mL, 0.34 mol) was added dropwise to DMF (70 mL 0.90 mol) at 0 °C under N2. The reaction mixture was stirred at 0 °C for 1 h. Afterwards, triphenylamine (10.0 g, 40.8 mol) was added and stirred for another 0.5 h at 0 °C. Then the whole mixture was stirred at 95 °C for 24 h. After cooling to room-temperature, the mixture was transferred into ice-water slowly with stirring, and 1 M NaOH was added until a brown solid precipitated completely. The crude product was purified using column chromatography (PE![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EA, 4:1, v/v) with Al2O3 gel to yield a uniform yellow solid (32.2%, 3.6 g).
:EA, 4:1, v/v) with Al2O3 gel to yield a uniform yellow solid (32.2%, 3.6 g).
4-(Bis(4-bromophenyl)amino)benzaldehyde
Into a double-neck round bottom flask and under N2, 4-(diphenylamino)benzaldehyde (2.2 g, 8.05 mmol) and N-bromosuccinimide (NBS) (3.58 g, 20.13 mmol) were charged, and 40 mL tetrahydrofuran (THF) was slowly added to the reaction flask. The mixture was stirred at room temperature for 2 h, then for another 4 h at 65 °C. After cooling to room temperature, the reaction was quenched using 20 mL water and extracted with 30 mL EA three times. The organic layer was dried with anhydrous MgSO4. The organic solution was filtered and the solvent was removed by vacuum evaporation. The crude solid product was purified using column chromatography (PE:EA, 5:1, v/v) with Al2O3 gel, and 2.9 g of a yellow solid was obtained with a yield of 82.6%.
4-(Bis(4-(pyridin-4-yl)phenyl)amino)benzaldehyde
A mixture of 4-(bis(4-bromophenyl)amino)benzaldehyde (2.3 g, 5.3 mmol), pyridin-4-yl-boronic acid (1.97 g, 15.9 mmol) and K2CO3 (3.6 g, 26.5 mmol) was added to a double-neck round bottom flask. Afterwards, Pd(PPh3)4 (317 mg, 0.276 mmol) was added quickly, and the mixture was stirred under N2. Water (20 mL) and 1,4-dioxane (30 mL) were injected into the flask and the reaction mixture was heated at 90 °C for 72 h. The 1,4-dioxane was removed by vacuum evaporation. The residuum was extracted using EA (3 × 30 mL). The organic layer was dried with anhydrous MgSO4. The solution was filtered and the solvent was removed by vacuum evaporation. The obtained solid product was further purified using column chromatography (EA) with Al2O3 gel, and 2.0 g of a yellow solid was harvested with a yield of 87.7%.
4-(Bis(4-(pyridin-4-yl)phenyl)amino)benzoic acid
4-(Bis(4-(pyridin-4-yl)phenyl)amino)benzaldehyde (1.3 g, 3.04 mmol), KMnO4 (1.44 g, 9.12 mmol) and the solvent (40 mL, acetone:H2O, 3:1, v/v) were added into a flask. The mixture was heated at 60 °C for 24 h. The reaction mixture was filtered to remove the generated manganese oxides. The water phase was obtained by evaporating the acetone and 3 M HCl was added into it until a yellow solid precipitated completely. A yellow product (1 g) was filtered off and washed with cold water, providing a yield of 76.9%. IR (Fig. S1†): 3032 (w), 1698 (w), 1583 (s), 1467 (s), 1409 (w), 1327 (m), 1270 (s), 1200 (m), 1165 (m), 1096 (w), 1050 (w), 1016 (w), 796 (s), 750 (m), 691 (w), 563 (w), 506 (s), 448 (w), 401 (w). 1H NMR (500 MHz, DMSO-d6, 25 °C, Fig. S2†): δ 8.68 (m, 4H), 7.91–7.83 (m, 10H), 7.28 (d, J = 8.3 Hz, 4H), 7.14 (d, J = 8.4 Hz, 2H). MS (Fig. S3†) m/z calcd for (C29H21N3O2): 443.16, found: 444.33 ([M + H]+).
Synthesis of [Zn(L)(HCOO)·H2O]n (Zn-MOF)
The Zn-MOF was prepared by mixing Zn(NO3)2·6H2O (29.7 mg, 0.1 mmol) and HL (44.6 mg, 0.1 mmol) in a 10 mL mixed solvent of acetonitrile, DMF and distilled H2O with a volume ratio of 2:2:1. The final mixture was completely dissolved and placed in a 15 mL Teflon-lined stainless steel vessel and heated at 95 °C for 3 days. Yellowish block crystals were obtained in 46% yield based on Zn. Anal. calcd for C30H23N3O5Zn: C, 63.06%; H, 4.03%; N, 7.36%. Observed: C, 61.05%; H, 4.23%; N, 7.18%. IR (Fig. S4†): 3424 (w), 3053 (w), 2845 (w), 1594 (s), 1479 (s), 1397 (m), 1328 (s), 1282 (m), 1224 (m), 1165 (m), 1060 (w), 865 (w), 829 (s), 730 (m), 703 (w), 679 (w), 575 (m), 506 (s).
Sensor experiments for nitroaromatic compounds
To prepare the Zn-MOF in H2O suspension, a procedure was adopted from the literature.32 1.0 mg of Zn-MOF powder was well dispersed in 3.0 mL H2O using ultrasonic agitation for 120 minutes. Different NAC solutions were prepared using DMF (5 mM) and added into a quartz cuvette containing 2 mL of the Zn-MOF in H2O suspension for luminescence detection. For quantitative analysis, a wide range of different concentrations of TNP (0–169 μM) were investigated using the same procedure.
Single crystal X-ray crystallography
A block-like single crystal of Zn-MOF was sealed in a capillary tube under an optical microscope with some mother liquor inside in order to prevent desolvation of the sample. Single crystal X-ray diffraction data was obtained using a Bruker smart Apex CCD diffractometer at 293 K using graphite monochromated Mo Kα radiation (λ = 0.71073 Å). Data reduction was performed with a Bruker Saint Program. Semi-empirical absorption correction was applied using SADABS.33 The crystal structure was solved using direct methods and refined with a full-matrix least square technique using a SHELXTL package.34 Non-hydrogen atoms were refined with anisotropic displacement parameters during the final cycles. Hydrogen atoms were placed in calculated positions. Crystal data and further information on the structure determination are summarized in Table S1.† Topological analysis was done using a TOPOS program.35
Other characterization
IR absorption spectra of the complex and HL ligand were recorded in the range of 400–4000 cm−1 on a Varian FT-IR spectrometer. 1H NMR spectra were obtained using a Bruker 500 MHz NMR spectrometer. Electrospray ionization mass spectrometry (ESI-MS) spectra were recorded using a Finnigan MAT SSQ 710 mass spectrometer in the mass range 120–1000 m/z. Powder X-ray diffraction (PXRD) patterns were collected using a Bruker D8 Advance X-ray diffractometer using Cu Kα radiation (λ = 1.5418 Å) at room temperature. Thermogravimetric analysis (TGA) was performed with a Perkin Elmer TGA7 at a heating rate of 5 °C min−1. Luminescence spectra were recorded using a Perkin Elmer LS55 fluorescence spectrophotometer at room temperature. The molecular structure optimization and molecular orbital calculations were conducted using density functional theory (DFT) calculations with a B3LYP functional and 6-31G* basis set.
Results and discussion
Description of crystal structure
The obtained Zn-MOF crystals had crystallized in space group P21/n, with lattice parameters of a = 9.861(2) Å, b = 28.306(6) Å, c = 10.024(2) Å, and β = 102.863(4)°. The asymmetric unit of the structure is composed of one Zn(II) ion, one deprotonated HL ligand and one deprotonated coordinated formic acid molecule as shown in Fig. S5.† The formic acid originates from partial decomposition of DMF during the hydro(solvo)thermal process. Similar phenomena have been reported in other MOF synthesis related literature.36–38 The zinc cation is located in a twisted triangular bipyramid configuration as shown in Fig. 2a, of which the triangular plane consists of two oxygen atoms O1 and O4, and one nitrogen atom N1 from the pyridine ring of another symmetrically related L ligand. The distances of Zn–O1, Zn–O4 and Zn–N1 are 2.048 Å, 1.977 Å, and 2.067 Å, respectively. The apical positions of the ZnO3N2 configuration are occupied by N2 from the third L ligand with a Zn–N2 distance of 2.067 Å, and O2, with a distance of 2.404 Å. It is worth noting that O3 is from the formic acid in the asymmetric unit, with a Zn–O3 distance of 2.693 Å, which is exceptionally large compared to most of the Zn–O coordination bond distances in small molecules;39 herein we don’t ascribe this distance to any Zn–O bonding.
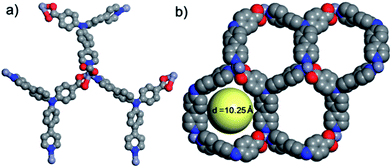 |
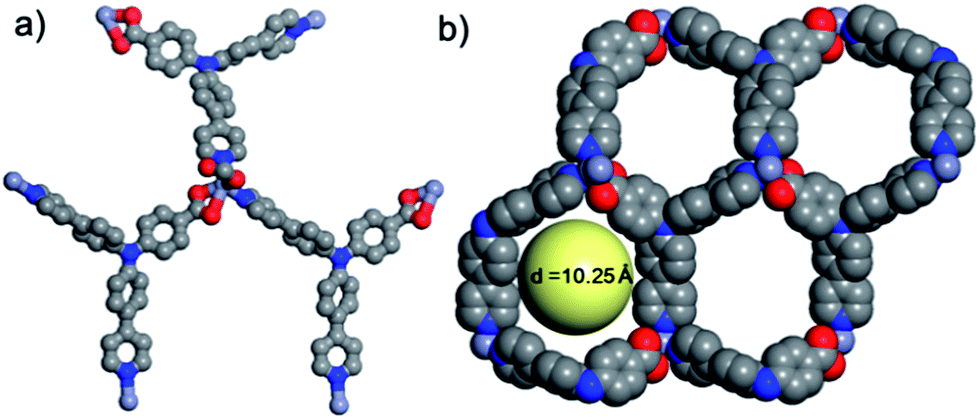
| | Fig. 2 (a) Coordination environment of the zinc atom, and (b) pore shape and size in a 2D layer. | |
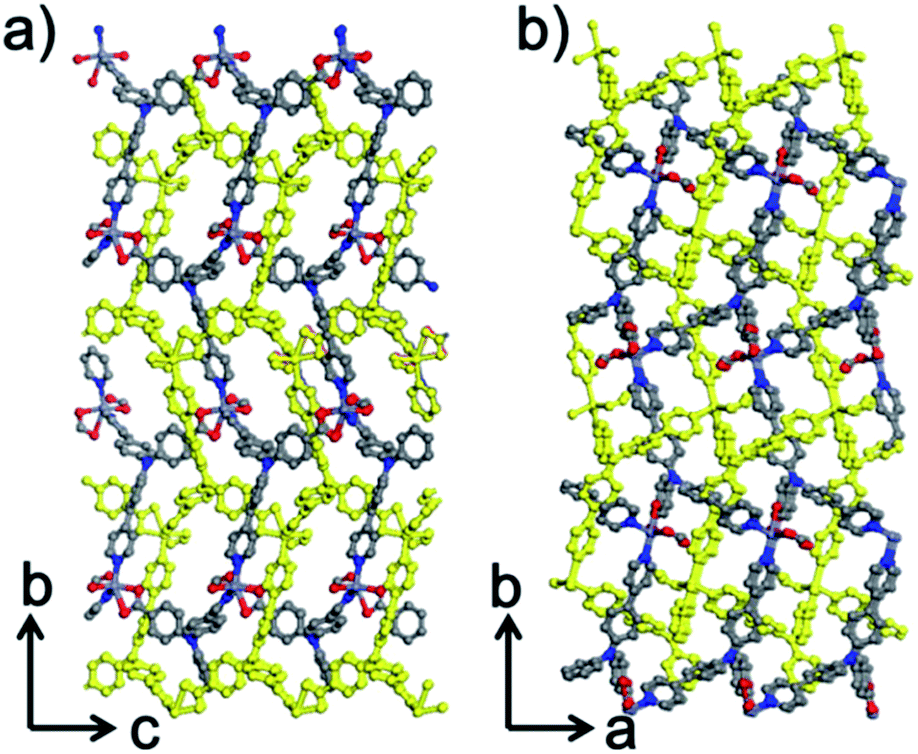
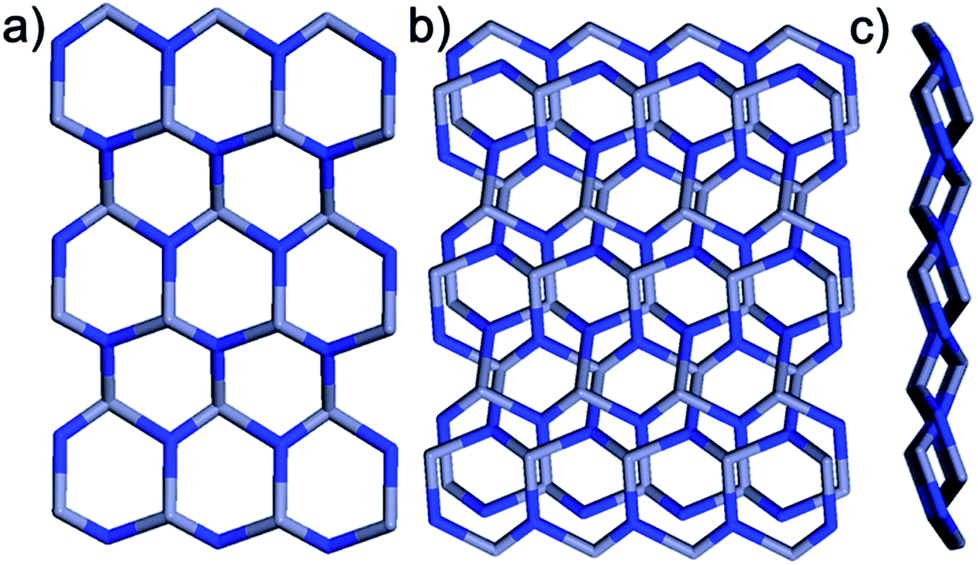
Within the framework, the Zn atom is connected to three HL ligands extending in a planar configuration, and each ligand is also connected to three zinc cations. Based on this recurring pattern, an infinite two-dimensional network is formed as shown in Fig. 2b. Moreover, if we consider the van der Waals radius of carbon to be 1.70 Å, the pore size within the 2D network is around 10.25 Å. Furthermore, with two identical networks following the 21 chiral symmetric axis interpenetrated together, an interpenetrated 2D network of Zn-MOF is achieved. Although the interpenetration causes partial blocking of the pore compared to the single 2D network, there is still some open pore space in the framework as shown in Fig. 3a and b, which is occupied by solvent water molecules in the crystal. Topologically, if we consider the N3 and zinc atom as nodes, and the benzoic/pyridine rings as edges, the single 2D network can be simplified as a zig-zag graphene-like net as shown in Fig. 4a. The front and side views of the interpenetrated 2D network are shown in Fig. 4b and c. Through intramolecular hydrogen bonds, the interpenetrated 2D nets are assembled into 3D crystals.
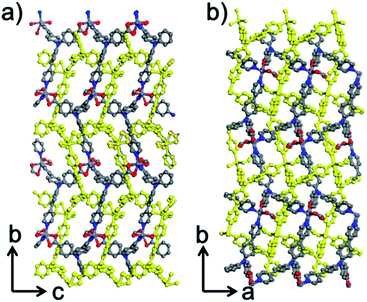 |
| | Fig. 3 Structural view of the framework along (a) the a-axis and (b) the c-axis. The yellow framework represents the second interpenetrated layer. | |
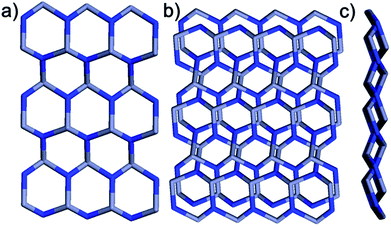 |
| | Fig. 4 Topological structure of the framework. (a) Zig-zag graphene-like layer. (b) Top view and (c) side view of two fold interpenetrated zig-zag graphene-like layers. | |
Purity and thermal stability
In order to explore application of the obtained Zn-MOF material, the purity and thermal stability of the obtained material were investigated. As shown in Fig. S6,† the obtained PXRD pattern of the prepared material is well matched with the simulated PXRD pattern based on single crystal X-ray diffraction data, which indicates a high purity of the obtained Zn-MOF. Moreover, the chemical stability of the material was also investigated. After immersing the Zn-MOF in H2O for 3 days, the PXRD pattern (Fig. S7†) is preserved and is the same as the as-synthesized one, which indicates that the Zn-MOF is stable under H2O. The thermal stability was investigated as shown in Fig. S8.† The first weight loss of 3.61% was observed from 25 °C to 93 °C, which corresponds to the removal of surface water molecules.40 The second weight loss of 2.52% between 93 °C and 254 °C is attributed to the loss of one lattice H2O molecule (calc. 3.15%). The third weight loss of 6.18% from 254 °C to 314 °C corresponds to the loss of coordinating formic acid (calc. 7.87%). Afterwards, the curve has a small plateau, up to 380 °C, where a sharp weight loss represents the structure collapsing. The weight percentage remains at 19.75% from 624 °C to 800 °C.
Luminescence properties
The ligand showed a strong photoluminescence (PL) emission maxima at 529 nm upon excitation at 395 nm in the solid state, which is attributed to n–π* or π–π* transitions of the conjugated ligand. When the ligand has been incorporated into the Zn-MOF material, the characteristic luminescence band is preserved with a peak maximum at 497 nm upon excitation at 387 nm (Fig. S9†). The slight blue-shift of the emission maxima of Zn-MOF compared to the bare ligand is most probably due to substantial electronic coupling between neighbouring ligand functional groups and Zn2+ ions.
Luminescent sensing of nitroaromatic compounds
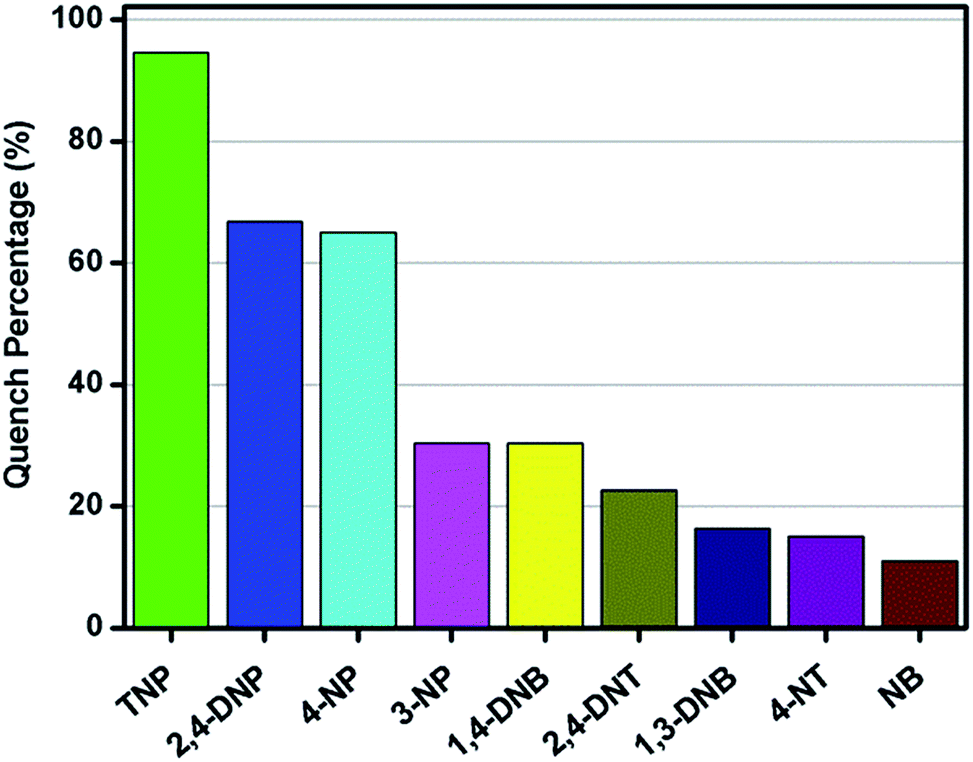
Motivated by the green-yellow luminescence of the obtained Zn-MOF material, together with the π-electron rich ligand and the nucleophilic features of the tertiary amine group involved in the ligand, we investigated its potential application in the sensing of various electron deficient nitroaromatics (NACs). The π-electron and nucleophilic features of the ligand are thought to enhance the electron density of the Zn-MOF material. The addition of a low concentration of different NACs into a dispersion of Zn-MOF quenches the photoluminescence intensities of Zn-MOF. In this study, PL spectra were used to investigate and monitor the sensitivity of the Zn-MOF in H2O solution to the corresponding NACs. Several different NACs with different electron-deficient properties and with a molecular volume to fit the void space of this Zn-MOF have been used in our experiments. The reason for careful selection of the molecular volume is that, big sized molecules will definitely not fit in the pore void of the Zn-MOF, which may only cause a crystal surface sensing effect which is outside of our main interest. PLATON analysis reveals that the close packed Zn-MOF contains a void of 284.5 Å3, which represents 10.4% of the volume of the unit cell when all the solvent in the channels is removed. Considering each non-hydrogen atom has a volume of around 17.5 Å3, only molecules for which the number of non-hydrogen atoms is less than 17 will possibly interact with the pore volume in the Zn-MOF. So, only those nitroaromatics with a number of non-hydrogen atoms that is less than 17 were selected for this study. The chemical structures are shown in Fig. S10† for 2,4,6-trinitrophenol (TNP), 2,4-dinitrophenol (2,4-DNP), 4-nitrophenol (4-NP), 3-nitrophenol (3-NP), 1,4-dinitrobenzene (1,4-DNB), 2,4-dinitrotoluene (2,4-DNT), 1,3-dinitrobenzene (1,3-DNB), 4-nitrotoluene (4-NT) and nitrobenzene (NB). In order to gain a deep understanding of the capability of the obtained Zn-MOF to probe NACs, PL quenching titrations were performed, with different volumes of a 5 mM NAC–DMF solution added into a Zn-MOF–H2O dispersion. The PL spectra were recorded and are shown in Fig. S11–18.† The adding of 50 μL of a 5 mM NAC solution leads to different degrees of fluorescence quenching (Fig. 5). Although the emission peak of Zn-MOF can be quenched by all of the nine NACs, the phenol derivative analytes, especially TNP, are the most effective quenchers and TNP can effectively quench the emission of Zn-MOF with a quench percentage (QP) of 94.57%, which is about 30% larger than that of the second best quencher (2,4-DNP with a QP of 66.84%). Other than for 4-NP, the QPs of the other non-phenol derivative quenchers, including nitrobenzene or nitrotoluene derivatives, are less than 30%. This observation is somewhat consistent with the electron deficiency order of the analytes. It is expected that with more strongly electrophilic substitution groups on the benzoic ring, the π-electrons of the ring will be delocalized to satisfy its own charge deficiency. This may have a strong effect on the ligand-to-metal charge transfer (MLCT) of the π-electron rich Zn-MOF. Thus, it causes a high quench percentage.
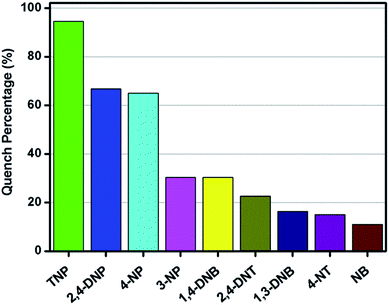 |
| | Fig. 5 Quench percentage for the luminescence emission of the Zn-MOF in H2O suspension in the presence of different NACs (λex = 387 nm). | |
Mechanism of the luminescence quenching
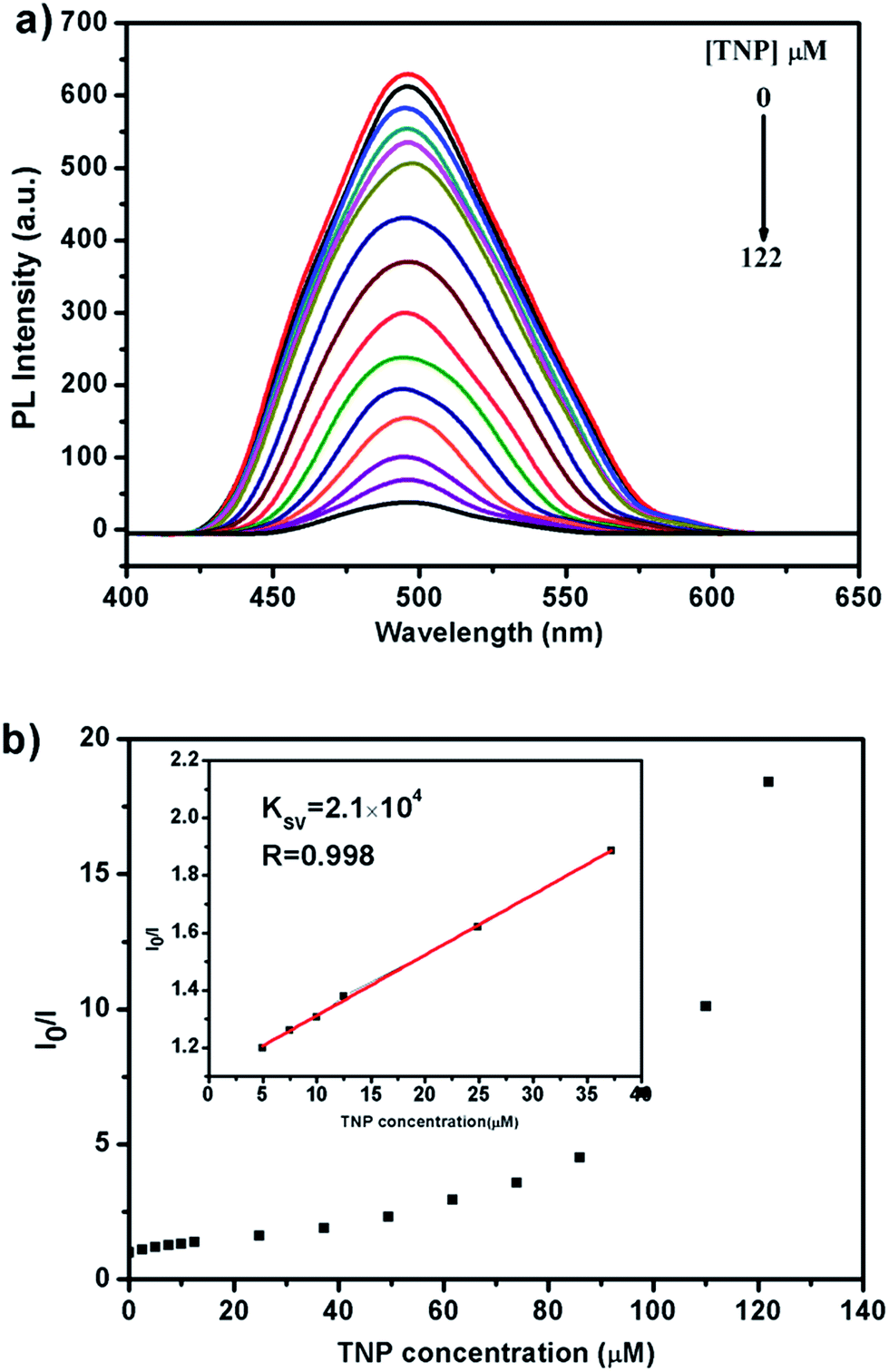
In order to understand the mechanism of the sensing process in detail, emission spectra of the Zn-MOF in H2O suspension with different concentrations of TNP were carefully recorded. The luminescence intensity decreases rapidly with the increased TNP concentration as shown in Fig. 6a. The fluorescence was quenched with a QP ca. 94.57% when the concentration of TNP in the Zn-MOF–H2O suspension reached 122 μM. The Stern–Volmer equation I0/I = 1 + Ksv × [M] was used to rationalize the luminescence titration results. As shown in Fig. 6b, a linear relationship is observed with a quenching constant (Ksv) of 2.11 × 104 M−1 when the TNP concentration is lower than 40 μM. This value is comparable with other reported MOFs or MOF-involved composite materials.41–44 With increasing of the TNP concentration up to 50 μM, a nonlinear curve feature develops, and this phenomenon indicates that the quenching mechanism is dominated by a combination of dynamic and static quenching mechanisms, which is also observed for other MOF materials.32,45,46 The static quenching mechanism dominates in the formation of a ground-state non-fluorescent complex between the electron-deficient TNP and the nucleophilic ligand 4-(bis(4-(pyridin-4-yl)phenyl)amino)benzoic acid. The dynamic quenching mechanism may be attributed to diffusive collisions of the TNP into the channel of Zn-MOF. Moreover, the Ksv of 2,4-NP and 4-NP is 3.0 × 104 M−1 and 2.4 × 104 M−1, respectively (Fig. S19 and S20†).
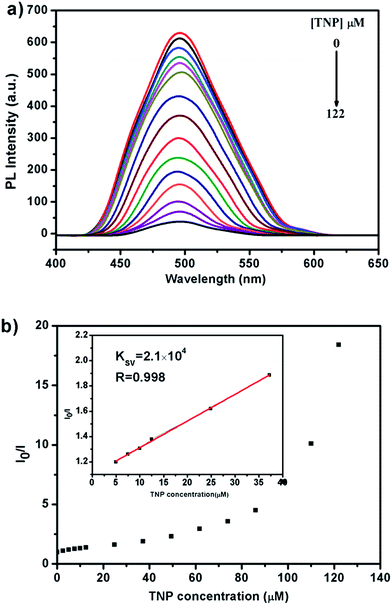 |
| | Fig. 6 (a) Luminescence quenching of the Zn-MOF dispersed in H2O with the gradual addition of a 5 mM TNP–DMF solution. (b) Stern–Volmer plot of I0/I versus TNP concentration for the Zn-MOF–H2O suspension (insert: enlarged view of a selected area). | |
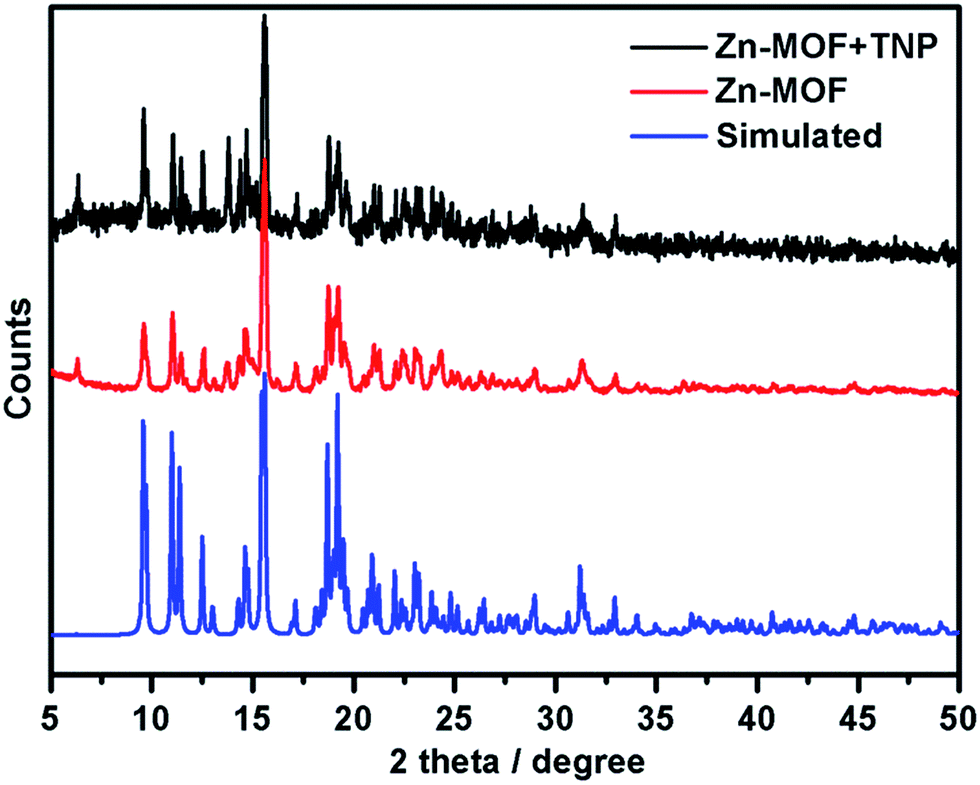
To further confirm the material stability upon interaction with the investigated analytes, PXRD was used to study the structural stability of the MOF material in each sensing step. As shown in Fig. 7, the PXRD pattern of the recycled sample after a 1 h sensing experiment is consistent with the as-synthesized one, which confirmed that the structure of the Zn-MOF is well preserved during the sensing process. The functional sensor is Zn-MOF, instead of possibly the decomposed ligand. However, similar to other MOF materials47 used as sensors, no obvious structure transformation was observed during the sensing process with our Zn-MOF as evidenced by PXRD. But the relative intensity change of the PXRD pattern may indicate that there are weak host–guest interactions. Moreover, only a slight change was observed in the FT-IR spectra before and after the TNP sensing experiment for 1 h as shown in Fig. S19,† which could also be ascribed to the trace amount of analyte adsorbed by the Zn-MOF material.
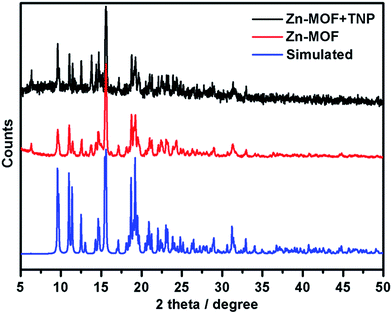 |
| | Fig. 7 PXRD patterns of the as-synthesized Zn-MOF before and after immersing in a TNP solution for 1 h, compared with the simulated PXRD pattern based on single crystal X-ray diffraction results. | |
Conclusions
In conclusion, a tridentate π-electron rich ligand has been successfully designed based on a ligand strategy and employed in the construction of a novel Zn-MOF [Zn(L)(HCOO)·H2O]n material. It possesses a 3,3-connected and 2-fold interpenetrated graphene-like topological structure, which is constructed by mononuclear zinc clusters and deprotonated 4-(bis(4-(pyridin-4-yl)phenyl)amino)benzoic acid linkers. Due to the appropriate void space and nucleophilic features of the designed ligand involved in the Zn-MOF, it can be used as an efficient sensor material for the detection of low concentrations of phenol, nitrobenzene and nitrotoluene derivatives. Among all the studied analytes, the Zn-MOF material shows high sensitivity to TNP with a quenching constant of 2.11 × 104 M−1. The Zn-MOF material is promising for further sensing and biosensing applications.
Acknowledgements
This project is supported by the Swedish Governmental Agency for Innovation Systems (VINNOVA) and the Swedish Research Council (VR), the Domestic Senior Visiting Scholar Funded Projects of Jiangsu province, China (2014FX007), the Industry-Academia-Research Prospective Joint Research Project of Jiangsu Province, China (BY2015032-01) and the Jiangsu Overseas Research & Training Program for university prominent young & middle-aged teachers and presidents. We are grateful to the Berzelii EXSELENT Centre on porous materials. We are thankful to Professor He-gen Zheng, Prof. Junliang Sun, Prof. Licheng Sun, Dr Shu-guang Chen and Dr Chuan-lei Zhang for their suggestions and help.
Notes and references
- J. Liu, P. K. Thallapally, B. P. McGrail, D. R. Brown and J. Liu, Chem. Soc. Rev., 2012, 41(6), 2308–2322 RSC.
- B. Chen, X. Zhao, A. Putkham, K. Hong, E. B. Lobkovsky, E. J. Hurtado, A. J. Fletcher and K. M. Thomas, J. Am. Chem. Soc., 2008, 130(20), 6411–6423 CrossRef CAS PubMed.
- J.-R. Li, R. J. Kuppler and H.-C. Zhou, Chem. Soc. Rev., 2009, 38(5), 1477–1504 RSC.
- J. Li, J. Sculley and H. Zhou, Chem. Rev., 2012, 112, 869–932 CrossRef CAS PubMed.
- B. Van de Voorde, B. Bueken, J. Denayer and D. De Vos, Chem. Soc. Rev., 2014, 43(16), 5766–5788 RSC.
- M. D. Allendorf, C. A. Bauer, R. K. Bhakta and R. J. T. Houk, Chem. Soc. Rev., 2009, 38(5), 1330–1352 RSC.
- Y. Cui, Y. Yue, G. Qian and B. Chen, Chem. Rev., 2012, 112(2), 1126–1162 CrossRef CAS PubMed.
- M. S. Wang, S. P. Guo, Y. Li, L. Z. Cai, J. P. Zou, G. Xu, W. W. Zhou, F. K. Zheng and G. C. Guo, J. Am. Chem. Soc., 2009, 131(38), 13572–13573 CrossRef CAS PubMed.
- F. Xu, H. Wang, S. J. Teat, W. Liu, Q. Xia, Z. Li and J. Li, Dalton Trans., 2015, 44(3), 20459–20463 RSC.
- P. Ghosh, K. Saha and A. Roychowdhury, Eur. J. Inorg. Chem., 2015, 2851–2857 CrossRef CAS.
- L. Ma, C. Abney and W. Lin, Chem. Soc. Rev., 2009, 38(5), 1248–1256 RSC.
- J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38(5), 1450–1459 RSC.
- Y. Li, S. Zhang and D. Song, Angew. Chem., Int. Ed., 2013, 52(2), 710–713 CrossRef CAS PubMed.
- Z. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43(16), 5815–5840 RSC.
- Y. Cui, H. Xu, Y. Yue, Z. Guo, J. Yu, Z. Chen, J. Gao, Y. Yang, G. Qian and B. Chen, J. Am. Chem. Soc., 2012, 134(9), 3979–3982 CrossRef CAS PubMed.
- B. Chen, L. Wang, F. Zapata, G. Qian and E. B. Lobkovsky, J. Am. Chem. Soc., 2008, 130(21), 6718–6719 CrossRef CAS PubMed.
- J.-S. Qin, S.-R. Zhang, D.-Y. Du, P. Shen, S.-J. Bao, Y.-Q. Lan and Z.-M. Su, Chem.–Eur. J., 2014, 20(19), 5625–5630 CrossRef CAS PubMed.
- G. He, H. Peng, T. Liu, M. Yang, Y. Zhang and Y. Fang, J. Mater. Chem., 2009, 19(39), 7347–7375 RSC.
- A. Chowdhury and P. S. Mukherjee, J. Org. Chem., 2015, 80(8), 4064–4075 CrossRef CAS PubMed.
- S. Shanmugaraju and P. S. Mukherjee, Chem. Commun., 2015, 51, 16014–16032 RSC.
- J. Ye, L. Zhao, R. F. Bogale, Y. Gao, X. Wang, X. Qian, S. Guo, J. Zhao and G. Ning, Chem.–Eur. J., 2015, 21(5), 2029–2037 CrossRef CAS PubMed.
- Z.-Q. Shi, Z.-J. Guo and H.-G. Zheng, Chem. Commun., 2015, 51(39), 8300–8303 RSC.
- Y. Fan, B. Lin, Y. Sun, X. Gong, H. Yang and X. Zhang, Polym. Chem., 2013, 4, 4245–4255 RSC.
- C. Hua, P. Turner and D. M. D’Alessandro, Dalton Trans., 2013, 42(18), 6310–6313 RSC.
- J. Huang, B. G. Sumpter, V. Meunier, Y. H. Tian and M. Kertesz, Chem. Mater., 2011, 23(3), 874–885 CrossRef CAS.
- H. Liu, R. Bo, H. Liu, N. Li, Q. Xu, H. Li, J. Lu and L. Wang, J. Mater. Chem. C, 2014, 2(28), 5709 RSC.
- J. Z. Patel, T. Parkkari, T. Laitinen, A. A. Kaczor, S. M. Saario, J. R. Savinainen, D. Navia-Paldanius, M. Cipriano, J. Leppänen, I. O. Koshevoy, A. Poso, C. J. Fowler, J. T. Laitinen and T. J. Nevalainen, Med. Chem., 2013, 56(21), 8484–8496 CrossRef CAS PubMed.
- B. Hu, X. Chen, Y. Wang, P. Lu and Y. Wang, Chem.–Asian J., 2013, 8(6), 1144–1151 CrossRef CAS PubMed.
- J.-F. Lefebvre, X.-Z. Sun, J. A. Calladine, M. W. George and E. A. Gibson, Chem. Commun., 2014, 50(40), 5258–5260 RSC.
- K. Neumann, C. Schwarz, A. Köhler and M. Thelakkat, J. Phys. Chem. C, 2014, 118(1), 27–36 CAS.
- C.-H. Yang, S.-H. Liao, Y.-K. Sun, Y.-Y. Chuang, T.-L. Wang, Y.-T. Shieh and W.-C. Lin, J. Phys. Chem. C, 2010, 114(49), 21786–21794 CAS.
- C. Zhang, Y. Yan, Q. Pan, L. Sun, H. He, Y. Liu, Z. Liang and J. Li, Dalton Trans., 2015, 44(29), 13340–13346 RSC.
- A. C. T. North, D. C. Phillips and F. S. Mathews, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1968, 24(2), 351–359 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64(1), 112–122 CrossRef CAS PubMed.
- V. A. Blatov, IUCr Computing Commission Newsletter, 2006, 7, 4–38 Search PubMed.
- F. Dai, P. Cui, F. Ye and D. Sun, Cryst. Growth Des., 2010, 10(4), 1474–1477 CAS.
- A. Aijaz, E. Barea and P. K. Bharadwaj, Cryst. Growth Des., 2009, 9(10), 4480–4486 CAS.
- B. Liu, W.-P. Wu, L. Hou and Y.-Y. Wang, Chem. Commun., 2014, 50(63), 8731–8734 RSC.
- M. M. Harding, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2001, 57, 401–411 CAS.
- C.-L. Zhang, L. Qin, Z.-Z. Shi and H.-G. Zheng, Dalton Trans., 2015, 44(9), 4238–4245 RSC.
- S. Khatua, S. Goswami, S. Biswas, K. Tomar, H. Sekhar Jena and S. Konar, Chem. Mater., 2015, 27, 5349–5360 CrossRef CAS.
- C. Zhang, L. Sun, Y. Yan, J. Li, X. Song, Y. Liu and Z. Liang, Dalton Trans., 2015, 44(1), 230–236 RSC.
- J. J. Qian, L. G. Qiu, Y. M. Wang, Y. P. Yuan, A. J. Xie and Y. H. Shen, Dalton Trans., 2014, 43(10), 3978–3983 RSC.
- X. Jiang, Y. Liu, P. Wu, L. Wang, Q. Wang, G. Zhu, X. Li and J. Wang, RSC Adv., 2014, 4(88), 47357–47360 RSC.
- X.-H. Zhou, L. Li, H.-H. Li, A. Li, T. Yang and W. Huang, Dalton Trans., 2013, 42(34), 12403–12409 RSC.
- X.-Z. Song, S.-Y. Song, S.-N. Zhao, Z.-M. Hao, M. Zhu, X. Meng, L.-L. Wu and H.-J. Zhang, Adv. Funct. Mater., 2014, 24(26), 4034–4041 CrossRef CAS.
- X.-Y. Dong, R. Wang, J.-Z. Wang, S.-Q. Zang and T. C. W. Mak, J. Mater. Chem. A, 2015, 3(2), 641–647 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: IR, PL, XRD, MS and NMR data. CCDC 1444088. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra00524a |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.