
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Detection, quantification and derivation of number size distribution of silver nanoparticles in antimicrobial consumer products†
Claudia
Cascio
*a,
Otmar
Geiss
b,
Fabio
Franchini
a,
Isaac
Ojea-Jimenez
a,
François
Rossi
a,
Douglas
Gilliland
a and
Luigi
Calzolai
*a
aNanoBioSciences Unit, Institute for Health and Consumer Protection, European Commission-DG Joint Research Centre, Via Enrico Fermi 2749, Ispra, Italy. E-mail: claudia.cascio@jrc.ec.europa.au; luigi.calzolai@gmail.com; Tel: +39 0332 78 6561
bChemical Assessment and Testing Unit, Institute for Health and Consumer Protection, European Commission-DG Joint Research Centre Commission, Via Enrico Fermi 2749, Ispra, Italy
First published on 6th January 2015
Abstract
In 2011 the European Commission published its recommendation for a definition for the term nanomaterial which requires the materials to be characterized in terms of the number size distribution of their constituent particles. More recently, the definition has begun to be applied to the labelling of food and cosmetic products where any components present in the form of engineered nanomaterials must now be clearly indicated in the list of ingredients. The implementation of this definition requires that methods be developed and validated to accurately size particles with at least one external dimension in the range of 1–100 nm, and to quantify them on a ‘number-based’ particle size distribution. An in-house developed method based on Asymmetric Flow Field Flow Fractionation-Inductively Coupled Plasma Mass Spectrometry (AF4-ICP-MS) for the simultaneous detection and quantification of citrate-stabilised silver nanoparticles (AgNPs) in water, has been applied to real-world liquid antimicrobial consumer products based on colloidal silver. This transfer of the method from ideal model systems to real products was assessed in light of other techniques including Centrifugal Liquid Sedimentation (CLS), Dynamic Light Scattering (DLS) and Transmission Electron Microscopy (TEM). Five out of six analysed products were found to contain AgNPs in the nano-range by means of a number of techniques including AF4-ICP-MS. Comparative analysis shows that CLS has sufficient size resolution to size AgNPs in the consumer products while DLS was unsuccessful probably due to sample polydispersivity. Despite the silver nanoparticles having unknown surface properties and stabilisation agents which could have influenced the sizing with AF4, a relatively good agreement between TEM and AF4-ICP-MS was observed. The AF4-ICP-MS data could be converted from mass-based to number-based distributions; this transformation, despite the possibility of experimental artefacts being mathematically amplified, has shown promising results.
1. Introduction
The industry of nanotechnology-based ingredients, additives and food contact materials is expected to grow in the near future1 with alimentary supplements and food packaging nanomaterials already being used in several countries.2 These fast developments and their potential benefits have spurred not only scientific and commercial activity but also highlighted a need to undertake some regulatory harmonization of the nano-science field. In this context, the European Commission (EC) published in October 2011 its recommendation on the definition of nanomaterials. This recommendation defines a “Nanomaterial” as ‘a natural, incidental or manufactured material containing particles, in an unbound state or as an aggregate or as an agglomerate and where, for 50% or more of the particles in the number size distribution, one or more external dimensions is in the size range 1 nm–100 nm’.3 One important consequence of introducing this definition has been the need to create, develop and validate methods for detection and quantification measurement of nano-materials in food and consumer products. Furthermore, it has highlighted the limitations of current particle sizing methods when applied to measuring the number size distribution of particles both in and above the 1–100 nm size range.4Currently there is no single analytical method or instrument able to address the requirements of the definition but a variety of methods exist which, in combination, offer the possibility of tackle this problem.5,6
It is well recognised that among nanomaterials, silver nanoparticles (AgNPs) are the largest and fastest growing category in use in consumer products. In particular, their powerful antimicrobial properties7 and relatively low cost have led to silver nanoparticles being widely used in a number of applications including food contact materials,8 cosmetics, wall paints, textiles,9 laundry detergents, biocide sprays and medical devices.10 Over 430 consumer products are currently reported to be on the market11 with this number being expected to increase significantly in the coming years. Recently an Inventory of Nanotechnology applications in the agricultural, feed and food sector12 was published showing that silver (along with nano-encapsulates and titanium dioxide) is among the most exploited nano-technological materials and that food additives and food contact materials are the most common of the current applications.
From a toxicological point of view it is believed that the antibacterial properties13 of silver nano-particles are mainly due to the release of silver ions from AgNPs, with evidence of a size dependent in vitro AgNP toxicity associated with generation of reactive oxygen species, loss of mitochondrial activity and DNA damage.14–16 However there are still a number of critical points to be clarified, including possible in vivo transformation, accumulation and interaction with enzymes and biomolecules, before a definite conclusion about whether or not AgNPs are hazardous to humans can be established.17 In this scenario the development of methods for the simultaneous detection/quantification of AgNP from ionic and soluble species is crucial.
Among the techniques applicable to the detection and characterization of AgNPs in complex matrices, the use of Asymmetric Flow Field Flow Fractionation18 (AF4) coupled to Inductively Coupled Plasma-Mass Spectrometry (ICP-MS) appears extremely promising and applicable to a number of sample types including cell culture media19 and chicken meat.20 This approach combines the ability of AF4 to size-fractionate particles in the nano-range with the high sensitivity and element specificity of the ICP-MS, thus providing an ideal tool for the simultaneous detection and quantification of nanoparticles. Furthermore, being a technique for the analysis of liquid samples, AF4-ICP-MS allows the study of NP size distribution directly in their native dispersions, avoiding possible artefacts deriving from sample preparation and drying as it might happen in electron-microscopy analysis.4 Finally, AF4 is able to isolate AgNP from the ionic/soluble species, providing an appealing tool for ionic/particulate physico-chemical speciation. Single-particle ICP-MS and particle tracking analysis (PTA) are two techniques that can potentially provide the particle size distribution of silver nanoparticles. Both techniques have the advantage to count directly the number of particles, but have a lower limit of size detection of around 20 nm for AgNPs.21 In addition, at the moment, they are somewhat less robust than the AF4-ICP-MS combination and for PTA the measurements are highly operator-dependent.22
One of the disadvantages of AF4-ICP-MS is that method development can be time consuming leading to undesirable additional costs for testing laboratories.23 In order to reduce FFF method development time, in this work we applied a single AF4-ICP-MS method that has been previously optimised for citrate-stabilised AgNPs in the 10–110 nm range24 to a collection of commercial antimicrobial AgNPs samples. Thus, in contrast to other similar works published on the topic,25 our objective was not to adapt the AF4-ICP-MS method to individual products but rather to evaluate the results obtained from the straightforward application of a single standardized method to a range of real life samples. In doing this the performance of the AF4-ICP-MS method was compared to other sizing techniques such as Centrifugal Liquid Sedimentation (CLS), Dynamic Light Scattering (DLS) and Transmission Electron Microscopy (TEM). The data obtained was then used to verify whether or not the tested commercial samples contained nanomaterials according to the EC definition.
2. Materials and methods
2.1 Materials
Stock solutions of monomodal sodium citrate-stabilised silver nanoparticles with nominal sizes in the range between 20 nm and 110 nm (Ted Pella Inc., Redding, USA; Pelco Citrate NanoXact™, in 10 nm steps) were purchased as dispersions at a nominal concentration of 0.02 mg mL−1 (±5%); 10 nm citrate-stabilised AgNPs were purchased from Sigma Aldrich (Sigma-Aldrich Corp., St. Louis, USA). Bottles were opened in a nitrogen glove-box and divided into ready-to use sub aliquots in amber glass vials. These were then stored at 4 °C away from light to prevent oxidation and release of ionic silver from AgNPs.13 As a quality check immediately after delivery from the suppliers, the sizes of the AgNPs were verified by mean of CLS.Nitric acid used was 67–69% ultrapure for trace analysis (CARLO ERBA Reagents S.r.l., Italy); silver and rhodium ICP-MS standards, at a concentration of 1000 mg L−1 in 2% nitric acid were purchased from Absolute Standards (Absolute Standards INC, Hamden, USA); the ultrapure water used throughout the experiments was supplied from a Millipore Advantage System (Merck Millipore, © Merck KGaA, Darmstadt, Germany). Sucrose (puriss.) used to create the liquid density gradient in the CLS was purchased from Sigma Aldrich (Sigma-Aldrich Corp., St. Louis, USA); the AF4 eluent was ultrapure water adjusted to pH 9.2 with 0.1 mM NaOH-solution and was freshly prepared every day.
2.2 Consumer products
Six different colloidal silver based consumer products, sold as antimicrobial agent for external use were purchased via internet (Table 1). The products were delivered without any information leaflet but with the indication “for external use” on the label. All of them were in liquid form and declared to contain nano or colloidal silver and were delivered in amber or dark bottles (plastic or glass) with or without a nebulizer on top.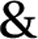 ) average of 3 replicates ± standard error
) average of 3 replicates ± standard error
| Product | Colour | Declared silver form | Declared concentration [mg L−1] | Measured total silver& [mg L−1] |
|---|---|---|---|---|
| A | Pale yellow | Colloidal/ionic | 15 | 11.63 ± 0.09 |
| B | Yellow | — | 10 | 9.3 ± 0.3 |
| C | Yellow | — | 20 | 29.3 ± 0.1 |
| D | No colour | Colloidal/ionic | 20 | 2.19 ± 0.07 |
| E | Yellow | Colloidal | 20 | 25.6 ± 0.3 |
| F | Yellow | Colloidal | 10 | 9.50 ± 0.03 |
2.3 Bench UV-VIS spectrophotometry
UV-VIS spectra were recorded at room temperature with a Thermo Nicolet Evolution 300 instrument (Thermo Fisher Scientific, Inc.) in the range 200–600 nm using 1 cm path length quartz cuvettes and 1.5 nm bandwidth. Recorded spectra were not smoothed. Raw data were exported and plotted in OriginPro 7.5 (OriginLab Corporation). Aliquots of consumer products were analysed both when opened and after 1 week to check their stability.2.4 DLS and Z-potential
Particle size distribution (PSD) was determined by Dynamic Light Scattering (DLS) using a Zetasizer Nano-ZS instrument (Malvern Instruments Ltd, UK) with temperature control (24.9 °C). Set parameters were the following: material refractive index 0.56, dispersant 1.330. Material absorption was set to 4.27, and viscosity to 0.8872 cP. Each sample was recorded in duplicate with an equilibration step of 120 s. Acquisition time was 80 s. Software was set to automatic acquisition mode. Hydrodynamic diameters were calculated using the internal software analysis. Z-potential was measured using the same instrument and recorded in a DTS1060C disposable cell with an equilibration time of 120 s. Measurements were done just after pH measurement. A Smulochowski model with a F(Ka) of 1.5 was used. All consumer product samples were analysed without any dilution.2.5 Particle size distribution by CLS
Particle size distribution (PSD) of undiluted consumer products were measured by Disc Centrifuge Photosedimentometer model DC24000UHR (CPS Instruments, Europe). The instrument was operated in line-start mode at a disc rotation speed of 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm using an aqueous sucrose gradient (8–24% w/w) capped with dodecane to prevent solvent evaporation. Each measurement was preceded by a calibration step done using an aqueous reference standard of 377 nm diameter PVC spheres. Consumer product samples were analysed fresh and without dilution.
000 rpm using an aqueous sucrose gradient (8–24% w/w) capped with dodecane to prevent solvent evaporation. Each measurement was preceded by a calibration step done using an aqueous reference standard of 377 nm diameter PVC spheres. Consumer product samples were analysed fresh and without dilution.
2.6 Total silver determination by ICP-MS
An Agilent ICP-MS 7700x (Agilent Technologies, Santa Clara, USA) equipped with platinum sampling and skimmer cones, MicroMist quartz nebuliser and a quartz Scott spray chamber was used for the determination of total silver in consumer products. Argon was used as nebulizer gas. The ICP-MS was operated in full quantification mode. Rhodium at a concentration of 50 μg L−1 in 1% nitric acid was added on-line as internal standard (ISTD), via a T-tube mounted before the nebuliser pump. Seven replicate readings per sample were performed, monitored signals included masses 107 and 109 for Ag and 103 for Rh, and integration time was 0.09 for Rh and 0.6 s for Ag isotopes. A total of 6 silver concentration standards (plus blank) were prepared in 2% nitric acid in the range 0.5–100 μg L−1. Calibration curves were read three times during the run. A total of 12 procedural blanks were analysed during the run. A number of silver spiked solutions were analysed along with the samples as well as Standard Reference Material 1643e trace elements in water (NIST) for quality control.Total silver in the consumer products was determined by ICP-MS following dilution in 2% nitric acid. A two-step dilution strategy (average total dilution factor 500×) was used in which pure nitric acid was firstly allowed to react with the consumer products for 3 hours at room temperature before dilution. All samples, quality controls and standards were prepared gravimetrically on a 4 digit scale. All samples and ionic silver spikes were prepared and analysed in triplicate. After preparation samples were promptly analysed for total silver concentration by ICP-MS.
2.7 On line AF4-UV-VIS-ICPMS
An Asymmetric Flow Field Flow Fractionation AF2000 MT Multiflow FFF, (Postnova Analytics, Germany) coupled to a UV-VIS detector (Postnova SPD-20AV) and ICP-MS were used for size fractionation and quantification of AgNPs and consumer products. AF4 eluent and elution conditions were carefully optimised for the separation mix of AgNPs citrate stabilised as reported elsewhere.24 Key details are provided in Table 2. Since the spectral response of silver nanoparticles varies as a function of their diameter it was not possible to set the UV-VIS detector at a detection wavelength which was optimum for all particle sizes. Instead, the UV-VIS detector was set to a wavelength of 420 nm which was judged to be a suitable compromise value able to detect silver nanoparticles across the size range of 10–110 nm. The AF4 channel was fitted with a regenerated cellulose membrane with a cut-off of 10 kDa (Postnova Analytics, Germany, part no. Z-MEM-AQU-527) which was substituted approximately every 50 injections. A 350 μm channel-spacer was used (Postnova Analytics, Germany, part no. Z-AF4-SPA-V-355). For the coupling to the ICP-MS, the outlet tube from the on-line UV-VIS spectrometer was connected to the sample feed tube of the ICP-MS. The outflow of the AF4 was set at 0.5 mL min−1 and aspirated by the peristaltic pump of the ICP-MS which was set at a flow of 0.6 mL min−1. The higher suction flow of the ICP-MS pump served to aspirate, via a T-inlet, from 1% nitric acid solution. Finally, Rh as internal standard was added in line with a T-inlet before reaching the ICP-MS detector. ICP-MS was run in chromatographic mode and m/z signal acquisition in Time Resolved Analysis (TRA). Platinum cones were used. Between each analysis run, the connection between AF4 and ICP-MS was manually removed, in order to allow an efficient purge of the AF4 and a flushing with 5% pure nitric acid of the ICP-MS, till a low, stable background for silver was reached. The consumer products were diluted in ultrapure water before injection to avoid overloading the ICP-MS detector or exceeding the highest calibration point. Raw data from the ICP-MS detector data was exported as counts per second versus time and the count ratio (107/103) versus time calculated and used for the quantification of silver in both calibrants and products; UV-VIS data at 420 nm were also exported and plotted versus time. OriginPro 7.5 (OriginLab Corporation) was used for further data treatment.| Parameter | Details |
|---|---|
| AF4 settings | |
| Permeation membrane | 10 kDa regenerated cellulose |
| Injection-loop volume (μL) | 50 |
| Wavelength (UV-VIS detector) | 420 nm |
| Eluent | basified ultrapure water |
| pH | 9.2 |
| Focusing time | 5 minutes |
| Applied cross flow (mL min−1) | From 1 to 0.1 |
| Gradient type | Linear decay in 35 minutes |
|
|
| ICP-MS settings | |
| RF-power (W) | 1550 |
| Reflected-power (V) | 12.2 |
| Plasma temperature (K) | 6500–8000 |
| Nebulizer | MicroMist (quartz) |
| Spray chamber | Scott (quartz) |
| Scan mode and resolution | Time resolved analysis |
| Integration time (s) | 0.3 (Ag), 0.120 (Rh) |
| Monitored masses | 107, 109, 103 |
2.8 TEM
TEM analysis of the commercial products was done following liquid spotting on copper support grids. A drop of undiluted product (4 μL) was placed onto ultrathin Formvar-coated 200-mesh copper grids (Tedpella Inc.) and left to dry in air at 4 °C. For each sample, at least 100 particles were measured to obtain the average and the size distribution. NPs were visualized using a transmission electron microscope (TEM) (JEOL 2100, Japan) at an accelerating voltage of 200 kV. Digital images were analysed with the ImageJ software and a custom macro performing smoothing (3 × 3 or 5 × 5 median filter), manual global threshold and automatic particle analysis provided by the ImageJ. The macro used is available at http://www.code.google.com/p/psa-macro. The circularity filter of 0.5 was used to exclude agglomerates that occurred during drying.3. Results and discussion
3.1 Total silver determination by ICP-MS
The measured values of total silver in the consumer products are shown in the last column of Table 1. The batch ICP-MS analysis of all six consumer products confirmed that they all contain some degree of silver, but with some discrepancies in the total silver content compared to the amount of nano or colloidal silver content declared by producers. For Product A, B, F the content of total silver is relatively close to the declared values. For Product C and E the content of total silver exceeds the declared value while for Product D the content of total silver was found to be far below the declared value. A comparative graph of declared versus measured values can be found in Fig. 1, while details on quality checks, Limit of Quantification (LoQ) and recovery are contained in ESI.†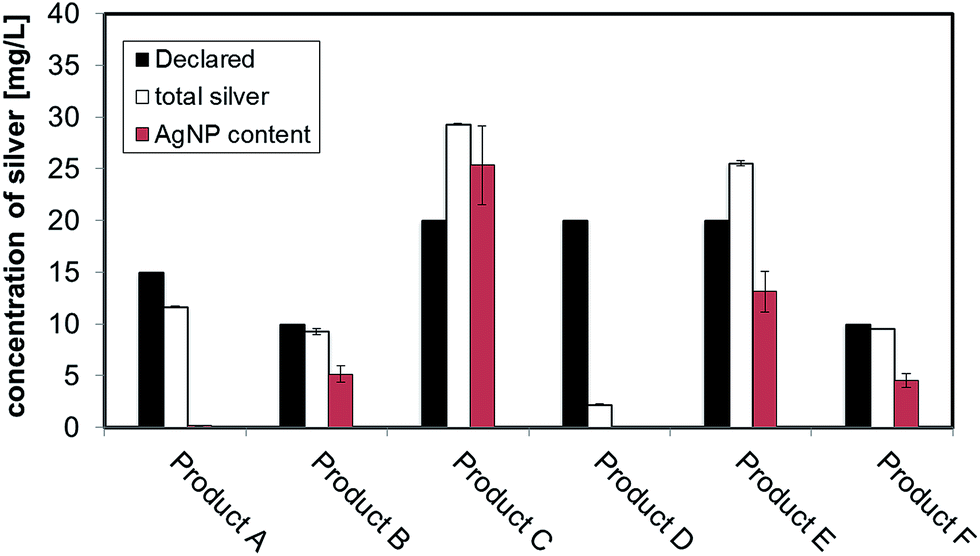 | ||
| Fig. 1 Declared content of nano/colloidal silver in the consumer products (black bars), total silver determined by batch ICP-MS in consumer products. Particulate silver in the nano-range detected in consumer products determined by AF4-ICP-MS are represented by red bars. | ||
3.2 Sizing strategies results: CLS, DLS, TEM
Preliminary UV-VIS analysis, allowed a quick and easy screening of consumer products, by identification of the localised Surface Plasmon Resonance (LSPR) peak typical of nano-silver.26 The analysis of freshly opened undiluted consumer products is shown in Fig. 2A and B: Products E, C, B and F showed a strong absorbance in the region of 400 nm that is characteristic of monomodal stocks of sodium citrate stabilised AgNPs in the size-range 10 to 50 nm (data not shown). Product A (Fig. 2B) showed a very weak peak suggesting the presence of a relatively low concentration of silver in the nano-range. From the analysis of Product D, no significant signal of the LSPR band was detected. | ||
| Fig. 2 (A) UV-VIS spectra of consumer products analysed on the day of opening without any further dilution; (B) magnification of UV-VIS spectra of Products A and D showing a small localised surface plasmon resonance band for Product A; (C) CLS analysis of consumer products analysed undiluted, dotted line shows the 100 nm size threshold; (D) Z-potential versus native pH of consumer products containing AgNPs. | ||
Centrifugal Liquid Sedimentation (CLS) revealed the presence of nanoparticles in 5 out of 6 products analysed (Fig. 2C and Table 3). CLS results confirmed the presence and relative quantities of the particle indicted by the UV-VIS analysis, while sample D did not show any detectable particles. Products B, C, F and E and A were all found to contain nanoparticles with 100% of the particle size distribution (in weight) below 100 nm. In particular, Products B, C, F and E showed very broad peaks centred below 15 nm, while Product A showed a small peak centred at 17.4 nm.
| Method | Product A | Product B | Product C | Product D | Product E | Product F |
|---|---|---|---|---|---|---|
| a Peak maximum provided by CLS software from particle size distribution in weight; (σ calculated from the peak half height width provided by CLS software dividing by 2.354). b Z average calculated from DLS software ±Z average × √PDI. c Distribution peak centre. d Average ± standard deviation. | ||||||
| CLSa | 17.4 (1.5) | 13.3 (7.3) | 12.1 (7.2) | — | 12.6 (6.8) | 13.6 (7.1) |
| TEMd | P1 15.1 ± 4.5 | 12.4 ± 5.7 | 11.9 ± 5.1 | — | 11.6 ± 4.4 | 13.6 ± 5.6 |
| P2 42.2 ± 11.6 | ||||||
| AF4-ICP-MSc (mass) | P1 33.8 | 16 | 16 | — | 18.5 | 17.4 |
| P2 47.3 | ||||||
| AF4-ICP-MSc (number) | P1 31.4 | 10.9 | 9 | — | 14.5 | 11.5 |
| P2 48.9 | ||||||
| DLS (Z-average)b | 224.7 ± 134.8 | 66.9 ± 25 | 53 ± 40.7 | 155.3 ± 80.7 | 36.8 ± 29.2 | 66.6 ± 43.2 |
| DLS (PdI) | 0.36 | 0.14 | 0.59 | 0.27 | 0.63 | 0.42 |
| Z-potential | −41.4 | −36.8 | −54 | — | −49.8 | −44.3 |
Batch DLS analysis was performed on undiluted consumer products (Table 3 and Fig. S.1 in ESI†). In the majority of cases, the polydispersivity index (PdI) of the products was quite large indicating low monodispersivity and/or possible matrix effects. For Product A, duplicate analysis on batch DLS produced poorly reproducible results and in one of the two measurements a bimodal distribution was detected (Fig. S.1.A of ESI†). All the detected Z-averages are greater than diameters generated by CLS probably due to a small number of larger particles or aggregates which, due to the highly non-linear variation of scattering intensity with particle size which, can introduce errors in DLS biasing the calculated mean size towards larger values.27
In contrast with DLS, CLS shows a greater ability to deal with heterogeneous dispersions, due to size fractionation before particle detection. CLS has proven to be considerably more suitable than DLS for the characterisation of multi-modal AgNP suspensions, and in particular to resolve bimodal mixtures of AgNPs.28 Nonetheless CLS, being a centrifugation based technique, depends on knowing a value of particle density which is normally assumed to correspond to the bulk metal one (10.49 g cm−3). While such an approximation may be valid for uncoated particles, it may create a systematic error with polymer or protein coated particles, and this effect might be especially significant on very small particles.29 Since it was not possible to assess the nature of any capping agent, coating or stabiliser in this study, some error related to the assumed particle density might ultimately affect the sizing by CLS.
TEM observations of the six products confirmed the presence of quasi-spherical nanoparticles in all samples with the exception of Product D (Fig. 3), which did not show any sign of nano- or macro-particles (Product D image not reported). Image analysis of Products B, C, E and F produced narrow size distributions with mean size values between 11–14 nm (Fig. 3, Table 3). However, Product A showed a more polydisperse distribution of NPs, which might be approximated as two populations: 1st with a mean size of 15 nm and a 2nd of 42.6 nm. However, this bimodal trend was not detected by CLS. For sample C, TEM observations showed a tail up to 30 nm, but the mean size is 11.9 nm. Indeed Product C showed a tendency to change colour from yellow to blue/grey when small aliquots were stored (1 week) in the fridge without rigorous exclusion of oxygen. This was confirmed as a shift of the LSPR peak in the UV-VIS absorbance spectrum (ESI†) which is assumed to be an indication of possible aggregation and/or surface oxidation.26,30
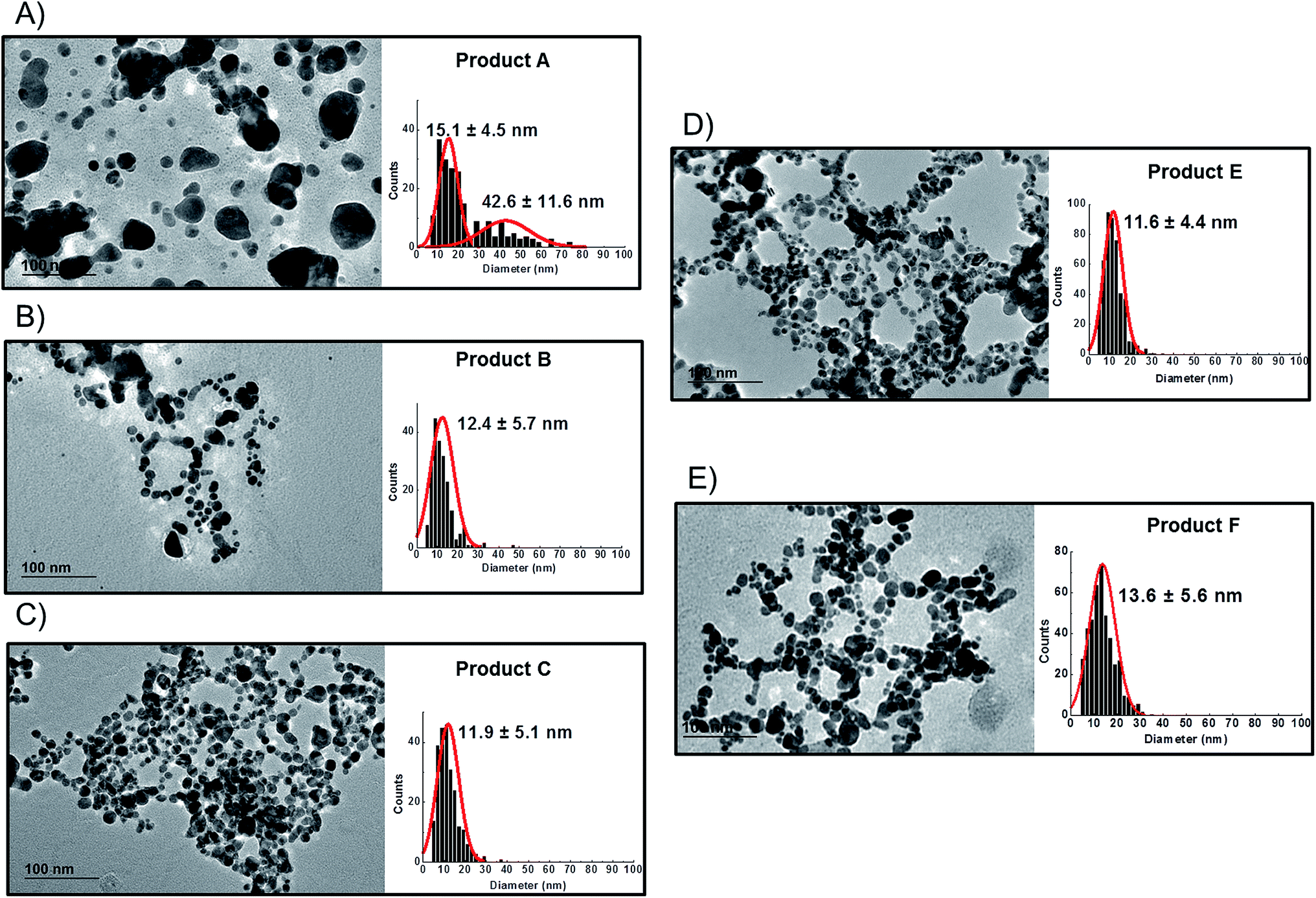 | ||
| Fig. 3 TEM images and particle size distributions of consumer products; (A): Product A; (B): Product B; (C): Product C; (D): Product E; (E): Product F. Product D did not show any presence of detectable silver nanoparticles on TEM analysis. | ||
3.3 Z-potential results
All the consumer products found to contain silver nanoparticles were further characterised to determine the Z-potential of particles (Fig. 2D). Initial pH was measured and analysis performed immediately. All the consumer products analysed showed a negative value (from −38.6 mV to −54 mV) at their original pH (ranging from 6.6 to 6.9). Z-potential values of this magnitude would normally be indicative of particles which carry sufficient charge as to be electrostatically stabilised and thus resistant to spontaneous aggregation. Average data are reported in Table 3 while further information is tabulated in Table S.2 of ESI.†3.4 Quantification of particulate silver with AF4-UV-VIS-ICP-MS
The previously developed AF4-UV-VIS-ICP-MS method optimised for citrate-stabilised AgNPs in the range between 10 nm and 110 nm,24 was applied to the consumer products.For mass calibration a method based on pre-channel injection was adopted22 in which accurately known quantities of three size AgNPs (nominal diameters 20 nm, 60 nm, 100 nm) on three concentration levels (0, 25 and 60 ng of total silver) were injected using a 50 μL loop before applying the standard elution profile. Three different calibration curves were built for each of three particle sizes respectively. This particular approach has both advantages and disadvantages – on one hand it has been previously demonstrated to be effective in compensating for incomplete particle channel recovery compared to ionic silver post-channel calibration24 but is hindered by the lack of AgNPs certified standards for size and concentration. To overcome this problem it was necessary to in-house determine the concentration of silver particles in the stock solutions.24
The ICP-MS on-line detector showed a Limit of Detection (LoD) (average blank + 3SD blank/slope) between 0.170–0.340 μg L−1 and a Limit of Quantification (LoQ) (average blank + 10SD blank/slope) in the range 0.566–1.133 μg L−1 for different particle sizes in line with previous results reported in literature.25
Fractograms obtained with both the UV-VIS detector and ICP-MS are shown in Fig. 4 which shows an example of the AgNPs mass calibration mixture together with data relative to the five consumer products. The five consumer products which showed a detectable amount of silver gave clear signals detectable both on UV-VIS and ICP-MS detectors with main peaks being well separated from the void peak. For the purpose of clarity, the UV-VIS detector signal collected at 420 nm (black-left axes) and ICP-MS (coloured lines, right axes) have been aligned by the void peak. After a preliminary run for each sample, dilution factors were chosen to avoid to overload the detector and to ensure quantification within the calibration curve. Exact dilution factors in ultrapure water before injection were the following: Product A: none; Product B: 10.97; Product C: 20.1; Product E: 16.7, Product F: 19.7.
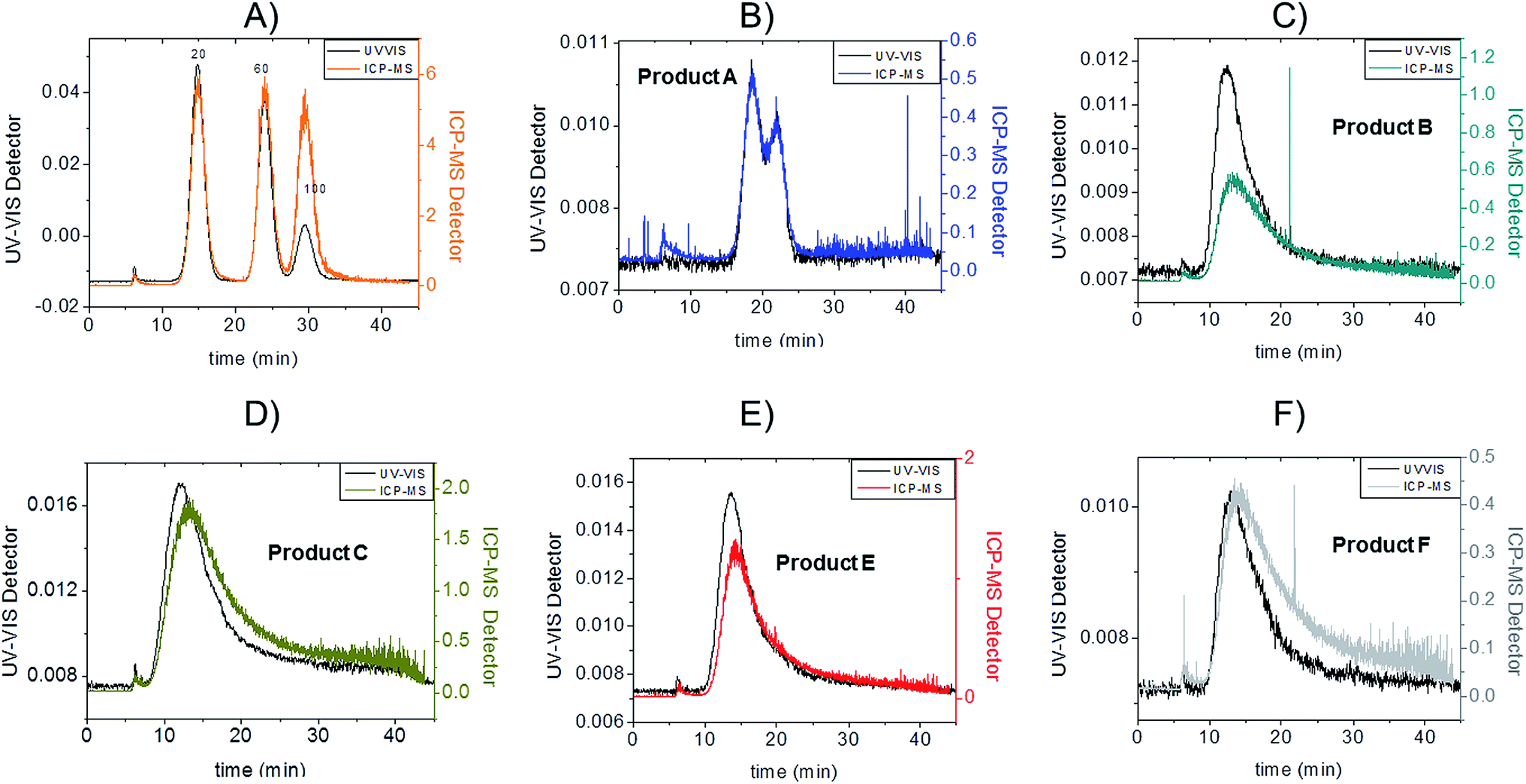 | ||
| Fig. 4 AF4-UV-VIS-ICP-MS fractograms. On the black left axis UV-VIS detector signal collected at fixed wavelength 420 nm (arbitrary unit); on the coloured right axes ICP-MS detector signal (count ratio between mass 107 and 103); (A): a mix of 20 nm 60 nm and 100 nm citrate coated AgNPs used for mass calibration; (B): Product A, injected without dilution; (C): Product B (dilution factor 10.97); (D): Product C (dilution factor 20.1); (E): Product E (dilution factor 16.7); (F): Product F (dilution factor:19.7). | ||
Channel recovery for real consumer products was evaluated by injecting samples with and without cross-flow and comparing the different peak areas obtained on the UV-VIS. Recovery was calculated as:
| %Recovery = 100 × peak area EL/peak area NC |
Channel recoveries were the following: 70% for Product A, 88% for Product B, 94% for Product C, 80% for Product F, 96% for E, and 85% for 20 nm AgNPs here used as standard. In our previous work the estimation of the size-related recovery was performed systematically on five replicates demonstrating the absence of significant difference for citrate-coated particles of 10, 20, 40 and 60 nm.24 In this context, considering the insignificant changes in detected particle size in the consumer products such a variation in recovery might be due more to surface chemistry and consequentially to membrane interaction.
To convert count ratio into concentration, the slope of the 20 nm AgNP calibration curve (count ratio versus area) was used. A curve fitting was done using OriginPro.7.5 and the ‘Gauss mod’ function (an exponentially modified Gaussian peak function for use in Chromatography) (details in Table S.3 of ESI†). Peak areas were used for quantification of the particulate silver (details in paragraph S.2 of ESI†) taking into account channel recovery rate and compared to total silver content determined by batch ICP-MS (Fig. 1, Table S.4 of ESI†). From these estimates, Product A was found to contain less than 2% of total silver as particulate silver; on the contrary Product C was found to contain over 85% of particulate silver. Fig. 1 (red bars) shows the AgNP content in the consumer products obtained by this approach; a similar trend was observed from CLS (peak height) and UV-VIS analysis with Product A being the one with the lowest particle concentrations and Product C being the highest, and with Product B and F having an intermediate content of particulate silver. For Product E, a qualitative analysis of UV-VIS and CLS data suggests a content of silver nanoparticles comparable to Product C (Fig. 2A and C), while the data from AF4-ICP-MS shows that it contains around 50% less silver NP than Product C (Table S.4†). The reason for this underestimation is unclear.
3.5 Sizing with AF4-UV-VIS-ICPMS and conversion from mass to number
For both the model mixture (composed of 20 nm, 60 nm and 100 nm citrate-stabilised AgNP) and the consumer products, the following strategy was used to convert the ICP-MS detector information (count ratio) into weight based and number based PSD (Fig. 5 and 6):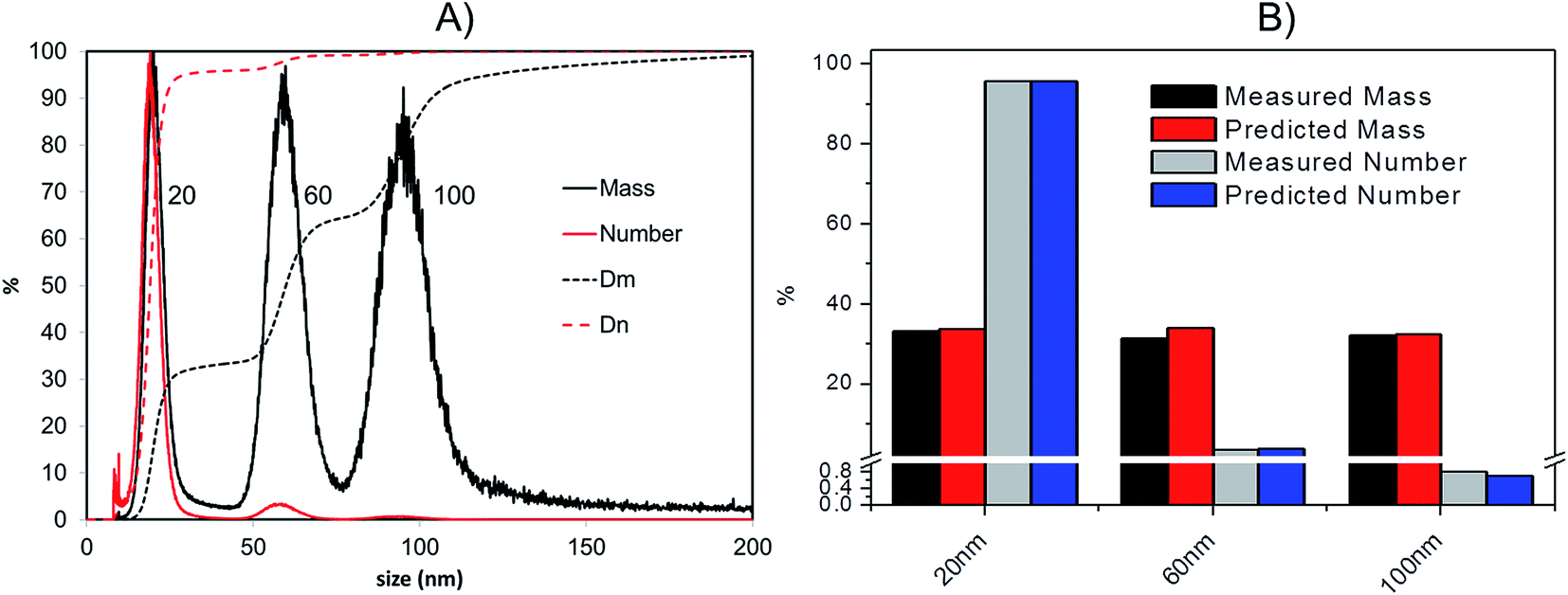 | ||
| Fig. 5 (A): Transformation of AF4-ICP-MS data into relative (continuous lines) and cumulant (dotted lines) distributions based on mass (black) and number (red) for a citrate-stabilised AgNPs mixture of 19.6 nm, 57.9 nm and 99.4 nm respectively (TEM diameter); Dm = cumulant distribution based on mass; Dn = cumulant distribution based on number; (B): predicted and measured (by AF4-ICP-MS) relative mass and number particle size distributions. | ||
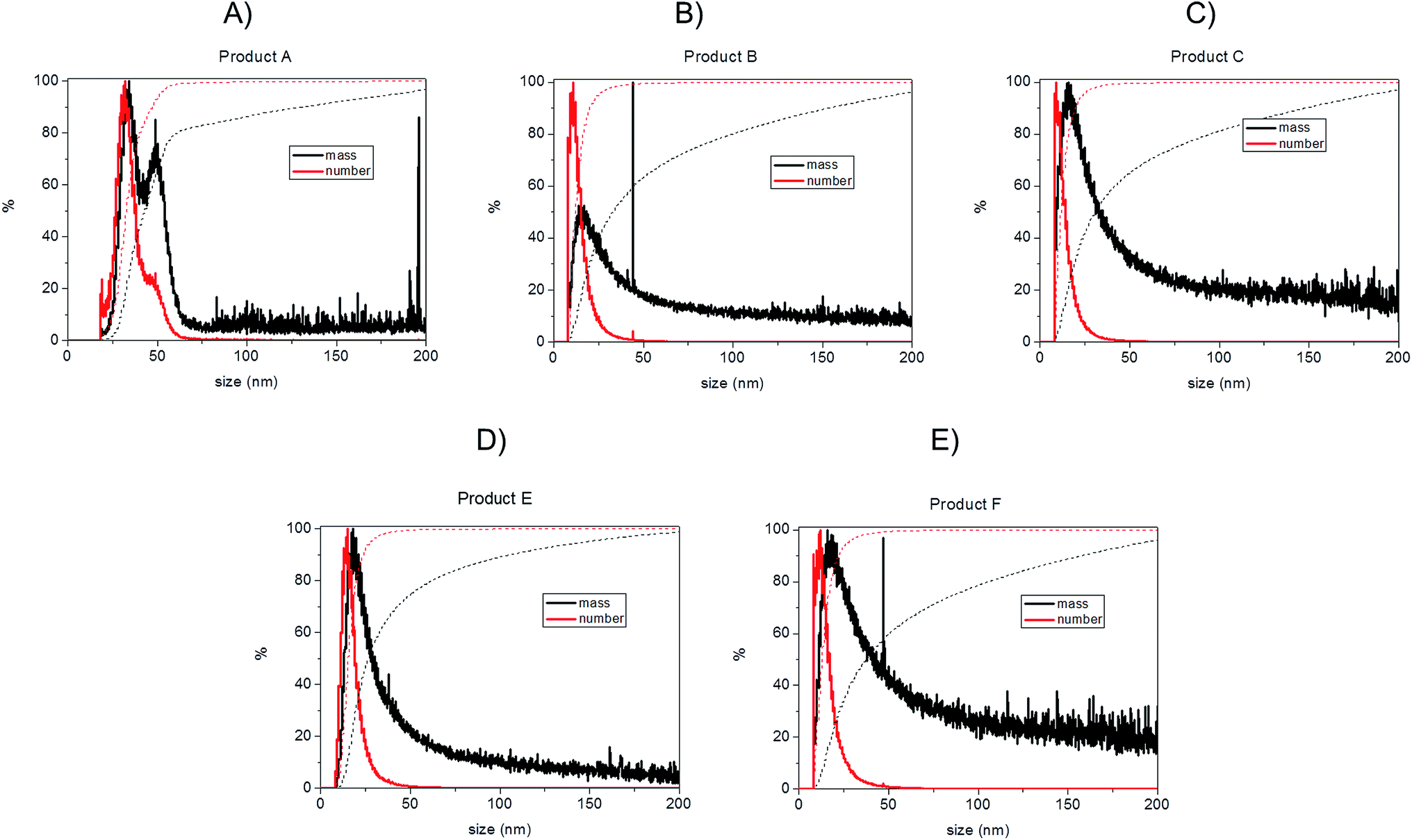 | ||
| Fig. 6 Transformation of AF4-ICP-MS data into relative (continuous lines) and cumulant (dotted lines) distributions based on mass (black) and number (red) for consumer products; (A): Product A; (B): Product B; (C): Product C; (D): Product E; (E): Product F. | ||
(i) The relationship between particle size and elution time when using the ICP-MS detector was determined as follows: a size vs. elution time calibration was performed daily with a mixture of citrate-stabilised AgNPs of known size; previous findings during method development showed elution times to be reproducible with a relative standard deviation generally below 1.5% on 14 replicate measurements performed on both intra-day and inter-day basis for different particle sizes.24 To do this a suspension containing 10 nm, 20 nm, 40 nm, 60 nm and 100 nm citrate stabilised AgNPs was run daily as a channel calibration mixture as in Fig. S.2.A of ESI.† The good reproducibility of the elution time–particle size relationship measured using the ICP-MS detector permitted it to be used for sizing silver nanoparticles in consumer products. An example of applied equation is
| Size = 0.027(elution time)2.38 |
(ii) The correspondence between counts and mass of silver in each ICP-MS sampling time was calculated from the integrated elution peak areas obtained from injections of known masses of 20 nm AgNPs.
(iii) By applying the correspondence determined in (ii) to the ICP-MS detector signal for unknown samples the number of counts accumulated in each sampling period was converted in a mass of silver in that time interval.
(iv) Since the mean particle size at each data point in the eluting profile is known from step (i), the volume and theoretical weight of the individual particles at each time point can be calculated by assuming the density of bulk silver of 10.49 g cm−331 and spherical geometry.
(v) From the total mass of silver in each given time interval and the corresponding mass expected for each particle the total number of particles in a certain time sampling period can be calculated.
As a confirmation of the quality of this sizing approach a mixture of 20 nm, 60 nm and 100 nm citrate-stabilised AgNPs was sized, providing satisfactory results of 20.3 (5.4) nm, 59.4 (10.4) nm and 95.1 (15.4) nm for peak centre (FWHM) when a gauss function was fit on data in OriginPro 7.5. In terms of mass, the 20 nm 60 nm and 100 nm mixture was analysed in a 1:1:1 mass ratio (Fig. 5A); the experimental values were compared to the expected ones, based on the known injected mass (459.35, 463.01 and 444.9 μg L−1 for 20, 60 and 100 nm respectively) and the measured TEM diameters (19.6, 57.4 and 99.4 nm respectively). The measured relative mass-PSD of 33.1%, 31.2%, 32.3% compares well with the expected values of 33.6%, 33.9% and 32.5% for the 20, 60, and 100 nm AgNP (Fig. 5B).
The above described strategy allowed converting the mass-based PSD into the number-based PSD. Such analysis gave an experimental relative number PSD of 95.8%, 3.4% and 0.8% for the small, medium and large AgNP, respectively. Such distribution compares well with the predicted number-PSD of 95.6%, 3.7% and 0.7%.
The effect of conversion from mass to number of particles clearly produces a great enhancement of the relative contribution from 20 nm compared to the 100 nm that very well reflects the predicted mass and number ratio calculated (respectively grey and blue bars in Fig. 5B). In performing this operation it is important that the lower size cut-off point be correctly selected to avoid errors from inclusion of materials in the void-peak. Indeed if the cut-off is fixed too early, the void peak will be included in the calculation and erroneously considered as small particles. This effect has been studied in detail for the model sample AgNPs 20, 60, 100 nm (Fig. S.3 in ESI†): the smaller the cut-off the higher the risk that a part of the void peak be incorrectly considered to be small sized particles with D50 for number PSD ranging from 3 nm up to 19 nm when going from a cut off of 2 up to 10 nm. For most of the samples analysed in this study the cut-off is set at 8 nm.
It should be noted that although the mass to number transformation is relatively simple to achieve, it suffers from a number of limitations, either intrinsically related to the analytical method or related to data conversion. For the latter case, the fact that the generically named as ‘silver nanoparticles’ present in consumer products could include a number of different surface chemistries; for example, silver–chitosan–poly(ethylene glycol) nanoparticles,32 and silver/poly(lactic acid)33 have been reported for antibacterial applications. Such ‘silver nanoparticles’ might show an unpredictable behaviour during channel elution, such as repulsion or attraction to the membrane potentially leading to variations in retention time and/or recovery. To some extent, the suitability of an unknown particle type could be verified by measuring its Z-potential under standard elution conditions and comparing it with that found for the citrate-stabilize particles. If the measured Z-potential values are substantially higher of lower than for the size-calibrants, extreme care should be taken in using elution time to derive the particle size since it might produce an erroneous size determination based on retention time only. In the extreme case of positively charged particles our method would likely be ineffective due to the strong interactions with the negatively charged membrane at the working pH of 9.2.
In addition, any inaccuracies in the experimentally determined weight-size distribution are disproportionately magnified by the mathematical conversion to the desired number-size distribution. In particular, the smaller the particles the greater is the potential error in number. Finally, the conversion from mass to number assumes that the particles have spherical geometry and that their density corresponds to that of bulk silver.
Bearing in mind the possible limitations discussed above the raw data for the five consumer products was elaborated to obtain mass and number size distributions together with their associated cumulant curves (Fig. 6). For the consumer samples analysed here a size cut-off value of 8 nm was chosen. This size corresponds to an elution time of 10.5 min with the ICP-MS detector and thus well away from the void peak occurring at around 7 min. For one sample only (Product A), the cut-off had to be set at 18 nm due to a broad void peak. The 8 nm cut-off chosen for the AF4-ICP-MS analysis could skew the particle size distribution, especially with respect to that one measured by TEM that is able to measure particles smaller than 8 nm. In any case the centres of the number distributions obtained by AF4-ICP-MS and TEM (Table 3) are remarkably similar.
For Products B, C, E and F the number based particle size distribution gives sizes (as measured by the centre of the PSD distribution) that are smaller than the mass-based PSD (as expected). In general, the number-based PSD obtained from the AF4-ICP-MS analysis is closer to the values measured by electron microscopy (Table 3) then the mass-based ones. For example, Product B had a mass-based PSD peak maximum at 16 nm and a number-based PSD peak maximum at 10.9 nm that compares quite well with the electron microscopy based PSD average of 12.4 nm. The cumulant analysis is also a very useful way to report the PSD and it is probably more relevant for the EC definition of nanomaterials. The calculated values of D50m (50% of the particles in the mass-based PSD) and D50n (50% of particles in the number-based PSD) were (32.8 nm, 12.1 nm), (30.6 nm, 11.6 nm), (26.2 nm, 15.4 nm) and (37.5 nm, 12.9 nm) for Products B, C, E and F respectively. Product A represented a slightly more complex case due the presence of a bimodal distribution both in TEM (Fig. 3A) and in AF4-ICP-MS analysis (Fig. 6A). Although a discrepancy is present between sizes of the two populations obtained with the two techniques (33.8, 48.9 nm by AF4-ICP-MS and 15.1, 42.2 nm by TEM) it is remarkable that AF4-ICP-MS is able to detected two populations and their relative number-based PSD (Fig. 6A, red line) is coherent with that obtained by electron microscopy (Fig. 3A).
The mass and number-based PSD obtained by AF4-ICP-MS analysis indicates that all the five consumer products by AF4-ICP-MS analysis contain more than 50% of particles in the size range below 100 nm (Table 3 and Fig. 6). On this basis the AF4-ICP-MS analysis would have classified the five products analysed as being ‘nanomaterials’ under the terms of the EU definition.
4. Conclusions
This work has contributed to address the urgent need to have testing methods for the simultaneous detection, sizing and characterisation of nanomaterials in consumer products by focusing on silver nanoparticles in antimicrobial liquid products intended for humans. The AF4-ICP-MS method previously developed for citrate-coated silver nanoparticles was successfully applied to the analysis of various commercial products based on colloidal silver and results were compared to other sizing strategies. The AF4-ICP-MS method tested here confirmed that five out of six consumer products contained detectable levels of silver nanoparticles and that this result was in general agreement with complementary analysis conducted by CLS, UV-VIS analysis and TEM observation. The developed methodology allowed the determination of both mass-based and number-based PSD and was able to correctly estimate the relative PSD in a mixture of silver nanoparticles of different sizes. The results obtained show the AF4-ICP-MS approach to be promising for the type of sample examined although a number of important technical issues remain to be resolved. The particle sizing strategy adopted for the AF4-ICP-MS produced results which were in good general agreement with other techniques for four of the five particulate containing materials. The methodology used here is based on a number of assumptions including the fact that the treated sample will behave similarly to citrate-stabilised NP during AF4 separation step. Since this usually cannot be guaranteed with unspecified commercial products it is recommended that a preliminary evaluation of unknown samples to be done by measuring Z-potential to verify charge compatibility with the method. Alternatively, the problem might also be overcome by including a size-measurement step before the ICP-MS detector such as on-line DLS detector.25,34 It should be noted that applicability of this solution, although attractive, may be limited by the fact that DLS in flow mode is less sensitive than ICP-MS, especially in the case of smaller nanoparticles. In the absence of on-line sizing, the greatest drawback of the method remains the lack of stable and certified standards of exact size and concentration to calibrate the AF4-ICP-MS both for size and for mass. Even considering all the possible limitations, the methodology described here shows encouraging results towards developing methods able to provide the number-based size distribution of silver nanoparticles. In the future, a more widespread use of such methods for regulatory application will require that validation procedures be undertaken to ensure that the methodologies can be transferred with acceptable precision, accuracy and reproducibility among control laboratories.Notes and references
- K. Tiede, A. B. Boxall, S. P. Tear, J. Lewis, H. David and M. Hassellov, Food Addit. Contam., 2008, 25, 795–821 CrossRef CAS PubMed.
- Q. Chaudhry, M. Scotter, J. Blackburn, B. Ross, A. Boxall, L. Castle, R. Aitken and R. Watkins, Food Addit. Contam., 2008, 25, 241–258 CrossRef CAS PubMed.
- EUROPEAN PARLIAMENT, in REGULATION (EU) no. 1169/2011, ed. E. PARLIAMENT, Official Journal of the European Commission, 2011, vol. L 304/18, p. 0040 Search PubMed.
- L. Calzolai, D. Gilliland and F. Rossi, Food Addit. Contam., Part A, 2012, 29, 1183–1193 CrossRef CAS PubMed.
- F. von Der Kammer, S. Legros, H. E. Larsen, K. Loeschner and T. Hofmann, Trends Anal. Chem., 2011, 30, 425–436 CrossRef CAS PubMed.
- T. Linsinger, G. Roebben, D. Gilliland, L. Calzolai, F. Rossi, P. Gibson and C. Klein, JRC Reference Report Requirements on measurements for the implementation of the European Commission definition of the term ‘nanomaterial’, JRC, 2012 Search PubMed.
- M. Rai, A. Yadav and A. Gade, Biotechnol. Adv., 2009, 27, 76–83 CrossRef CAS PubMed.
- G. Artiaga, K. Ramos, L. Ramos, C. Cámara and M. Gómez-Gómez, Food Chem., 2015, 166, 76–85 CrossRef CAS PubMed.
- E. Lombi, E. Donner, K. G. Scheckel, R. Sekine, C. Lorenz, N. Von Goetz and B. Nowack, Chemosphere, 2014, 111, 352–358 CrossRef CAS PubMed.
- X. Chen and H. J. Schluesener, Toxicol. Lett., 2008, 176, 1–12 CrossRef CAS PubMed.
- Woodrow Wilson International Center for Scholars and the Pew Charitable Trusts, The Project on Emerging Nanotechnologies, http://www.nanotechproject.org/cpi/browse/nanomaterials/silver-nanoparticle/, 2014.
- RIKILT and JRC, Inventory of Nanotechnology applications in the agricultural, feed and food sector, EFSA, 2014.
- Z.-m. Xiu, Q.-b. Zhang, H. L. Puppala, V. L. Colvin and P. J. J. Alvarez, Nano Lett., 2012, 12, 4271–4275 CrossRef CAS PubMed.
- S. Arora, J. Jain, J. M. Rajwade and K. M. Paknikar, Toxicol. Appl. Pharmacol., 2009, 236, 310–318 CrossRef CAS PubMed.
- P. V. AshaRani, G. L. K. Mun, M. P. Hande and S. Valiyaveettil, ACS Nano, 2009, 3, 279–290 CrossRef CAS PubMed.
- R. Foldbjerg, P. Olesen, M. Hougaard, D. A. Dang, H. J. Hoffmann and H. Autrup, Toxicol. Lett., 2009, 190, 156–162 CrossRef CAS PubMed.
- D. McShan, P. C. Ray and H. Yu, J. Food Drug Anal., 2014, 22, 116–127 CrossRef CAS PubMed.
- C. Giddings, Sep. Sci., 1966, 1, 123–125 CAS.
- E. Bolea, J. Jimenez-Lamana, F. Laborda, I. Abad-Alvaro, C. Blade, L. Arola and J. R. Castillo, Analyst, 2014, 139, 914–922 RSC.
- K. Loeschner, J. Navratilova, C. Købler, K. Mølhave, S. Wagner, F. Von Der Kammer and E. H. Larsen, Anal. Bioanal. Chem., 2013, 405, 8185–8195 CrossRef CAS PubMed.
- F. Laborda, E. Bolea and J. Jiménez-Lamana, Anal. Chem., 2013, 86, 2270–2278 CrossRef PubMed.
- J. A. Gallego-Urrea, J. Tuoriniemi and M. Hassellöv, TrAC, Trends Anal. Chem., 2011, 30, 473–483 CrossRef CAS PubMed.
- D. M. Mitrano, A. Barber, A. Bednar, P. Westerhoff, C. P. Higgins and J. F. Ranville, J. Anal. At. Spectrom., 2012, 27, 1131–1142 RSC.
- O. Geiss, C. Cascio, D. Gilliland, F. Franchini and J. Barrero-Moreno, J. Chromatogr. A, 2013, 1321, 100–108 CrossRef CAS PubMed.
- H. Hagendorfer, R. Kaegi, M. Parlinska, B. Sinnet, C. Ludwig and A. Ulrich, Anal. Chem., 2012, 84, 2678–2685 CrossRef CAS PubMed.
- V. Amendola, O. Bakr and F. Stellacci, Plasmonics, 2010, 5, 85–97 CrossRef CAS.
- L. Calzolai, D. Gilliland, C. Pascual Garcìa and F. Rossi, J. Chromatogr. A, 2011, 1218, 4234–4239 CrossRef CAS PubMed.
- C. Cascio, D. Gilliland, F. Rossi, L. Calzolai and C. Contado, Anal. Chem., 2014, 86, 12143–12151 CrossRef CAS PubMed.
- R. Capomaccio, I. Ojea-Jimenez, P. Colpo, D. Gilliland, G. Ceccone, F. Rossi and L. Calzolai, in preparation.
- C.-N. Lok, C.-M. Ho, R. Chen, Q.-Y. He, W.-Y. Yu, H. Sun, P.-H. Tam, J.-F. Chiu and C.-M. Che, JBIC, J. Biol. Inorg. Chem., 2007, 12, 527–534 CrossRef CAS PubMed.
- J. Ristimäki, A. Virtanen, M. Marjamäki, A. Rostedt and J. Keskinen, J. Aerosol Sci., 2002, 33, 1541–1557 CrossRef.
- M. Ahmad, K. Shameli, M. Tay, M. Hussein and J. Lim, Res. Chem. Intermed., 2014, 40, 817–832 CrossRef.
- K. Shameli, M. B. Ahmad, W. Md Zin Wan Yunus, N. A. Ibrahim, R. A. Rahman, M. Jokar and M. Darroudi, Int. J. Nanomed., 2010, 5, 573–579 CrossRef CAS.
- D. Conduto-Antonio, C. Cascio, D. Gilliland and L. Calzolai, in preparation.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ja00410h |
| This journal is © The Royal Society of Chemistry 2015 |