
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Computational design of polaronic conductive Li-NASICON mixed ionic–electronic conductors†
Jiawei
Lin
 ab,
KyuJung
Jun
*ab and
Gerbrand
Ceder
*ab
ab,
KyuJung
Jun
*ab and
Gerbrand
Ceder
*ab
aDepartment of Materials Science and Engineering, University of California, Berkeley, USA. E-mail: gceder@berkeley.edu; kjun@berkeley.edu
bMaterials Science Division, Lawrence Berkeley National Laboratory, USA
First published on 25th April 2025
Abstract
Li-NASICONs featuring three-dimensional corner-sharing frameworks have been experimentally and theoretically demonstrated to be fast Li-ion conductors, with high room-temperature ionic conductivities. In this study, we conduct first-principles calculations to investigate the polaronic conduction in Li-NASICONs comprising different transition metal (TM) elements (Ti, V, Cr, Mn, Fe, Mo) and evaluate their potential as mixed electron-ion conductors. We find that the electron polaron conductivity of octahedral–octahedral (oct–oct) hopping in Li-NASICONs is strongly correlated with the Jahn–Teller activity on the TM site. While LiMn2(PO4)3 and Li3Cr2(PO4)3 have a low predicted electron polaron conductivity owing to the J–T distortion resulting from the polaron formation, LiFe2(PO4)3 is predicted to have the most facile polaron transport, which stems from the elimination of its intrinsic J–T distortion upon polaron formation. We also find that polaron hopping between octahedral and tetrahedral sites contributes limited electronic conductivity because of the large difference in the polaron formation energies on these sites. For polaron transport along purely tetrahedral sites, we find limited transport through the V5+/V4+ pair in LiGe2(VO4)3 and LiTi2(VO4)3 but better conductivity through the Mo6+/Mo5+ pair in LiTi2(MoO4)3. Our research suggests that Li-NASICONs with TM elements involving no J–T distortion upon polaron formation on octahedral sites and Mo on tetrahedral sites are promising candidates for mixed ionic electronic conductors (MIECs).
1. Introduction
Many electrochemical energy storage devices require the addition of carbon as a conductive additive to the cathode composite. While carbonaceous materials are inexpensive, they can be the source of degradation phenomena in several new technologies. For example, in solid-state batteries, carbon has been identified as the source of electrochemical breakdown of sulfide solid-state conductors.1,2 In Li–O2 batteries, porous carbon cathodes and organic carbonate electrolytes in non-aqueous Li–O2 batteries are prone to degradation during cycling.3 The carbonaceous components react with the Li2O2 discharge product and form insulating Li2CO3 (ref. 4) and reduce the capacity of the cell.5 In addition, the incorporation of lithium as lithium carbonate at the Li2O2–electrolyte interface leads to an increased overpotential during cycling.6To address these issues, attempts have been made to replace carbon in cathodes with other electron-conducting materials,7 such as noble metals and alloys,8–11 titanium compounds (oxides,12,13 carbides,14 and nitrides15), and perovskites in nonaqueous Li–O2 batteries.16,17 In addition, solid-state Li–O2 batteries with Li-NASICON,18,19 perovskite,20 and zeolite21 inorganic electrolytes have also been proposed to avoid Li2CO3 formation.
One feasible approach to develop MIECs is to introduce electronic conductivity in crystals that already exhibit high Li-ion conductivity, such as perovskite, garnet, and Li-NASICON. For instance, Ma et al. investigated the perovskites LixLayMO3−z with various TM elements (M = Ti, Cr, Mn, Fe, and Co) and demonstrated that Li0.34La0.55MnO3−z exhibits high electronic conductivity (2.04 × 10−3 S cm−1) and Li-ion conductivity (8.53 × 10−5 S cm−1).22 This remarkable electronic conductivity was attributed to the favorable formation of oxygen vacancies, which result in a higher mobile electron concentration in the compound. Kim et al.23 developed a MIEC cathode consisting of electron-conducting RuO2 and Li-ion conducting La2LiRuO6−x to enhance the cyclability of solid-state Li–O2 batteries. Jin et al.24 introduced diverse TM elements (Mn, Fe, Co, Ni) into the Li and Zr sites of garnets and discovered that garnet with Fe in Li sites exhibits high mixed ionic–electronic conductivity ranging from 10−5 to 10−4 S cm−1. The electronic conduction was attributed to polaronic conduction through the Fe2+–O–Fe3+ pathway. Recently, Li-NASICONs with compositions of LiTi2(PO4)3−x(VO4)x and Li1.3Al0.3Ti1.7(PO4)3−x(VO4)x, where x = 0.1, 0.2, 0.3, and 0.4, were shown to display mixed ionic–electronic conductivity ranging between 10−5 and 10−4 S cm−1, with the enhanced electronic conductivity being linked to polaronic conduction through the V5+/V4+ pair.25 Previous theoretical work has also reported low polaron hopping barriers in doped SrCeO3 perovskite26 and Li-Garnet compounds,27 highlighting the possibility for electronic conduction in wide-band-gap transition metal oxides. Since Li-NASICONs can be synthesized with a diversity of TM on the octahedral and tetrahedral sites,28–30 we evaluate in this paper the effect of each chemistry on the electronic conductivity.
Polarons are quasiparticles that involve the coupling of excess carriers with lattice vibrations in polarizable materials.31 Within polarons, holes or electrons are trapped in a local structural distortion. Numerous materials, including inorganic crystals (commonly oxides) and polymers, have been reported to contain polarons induced from a variety of sources, such as oxygen vacancies, photoexcitation, and extrinsic doping.31 Polarons contribute to electronic conductivity via a hopping mechanism, where a localized charge transfers from one atom to another assisted by the transport of the lattice distortion and the tunneling of the charge between two atomic sites.
In this study, we investigate polaronic conduction in Li-NASICONs with different transition metal (TM) elements (Ti, V, Cr, Mn, Fe, Mo) using first-principles calculation. We first compute the polaron formation energies in Li-NASICONs and find that all Li-NASICONs except Li3Fe2(PO4)3 have negative electron polaron-formation energies, indicating the favorable existence of electron polarons in the compounds. Next, we identify percolating conduction paths in Li-NASICONs and calculate their rate-determining hopping barriers for oct–oct, oct–tet, and tet–tet polaronic conduction. We use the computed hopping barriers and the electronic coupling between the initial and final polaronic state to estimate the room-temperature polaronic conductivity of Li-NASICONs. Our study suggests that Li-NASICONs with TM elements involving no J–T distortion upon polaron formation on octahedral sites or with Mo on tetrahedral sites to be promising candidates for MIECs.
2. Computational methodology
To localize an electron polaron in electronic structure methods, self-interaction needs to be fully or partially removed. Density functional theory (DFT) with a Hubbard-U correction or hybrid functionals are commonly adopted to reduce the charge self-interaction32–37 for systems with d- or f-band electrons. The DFT+U scheme requires an appropriate selection of the Hubbard-U value for a reliable description of the d- or f-band electronic structure, which typically involves fitting theoretically predicted physical properties against their experimental counterparts. This process renders a different set of +U potentials for different physical properties and compounds, and the calculated polaron formation energy or hopping barrier is susceptible to change with +U potentials.38,39 In contrast, hybrid functionals involve fewer empirical parameters to obtain localized polaronic solutions40 and have been reported to be more successful in obtaining localized solutions than the DFT+U scheme.41 Hence, in this study, we adopt the HSE06 hybrid functional42 for all calculations, including the optimization of the polaron structure, its hopping barrier, and the projected crystal orbital Hamilton population (p-COHP) of the polaronic orbitals on the neighboring TM atoms. All the calculations are performed using the Vienna ab initio simulation package (VASP),43–45 and the input generation is performed using the Pymatgen library.46To model the formation of a polaron, lattice-parameter optimizations for all neutral Li-NASICONs are first conducted. Subsequently, we add an extra electron to a Li-NASICON supercell containing 72 oxygens and perform geometrical optimization of the supercell with an excess electron with the lattice parameters of the cell fixed to those of the neutral cell. To more easily converge to a polaronic state, we introduce a structural perturbation to a TM–O6 octahedron and a TM–O4 tetrahedron by elongating each of the six and four TM–O bonds by 10%. This bond-distortion approach has been extensively used to break the symmetry of a crystal and facilitates electronic density localization on a polaronic site.47–54 We note that initial bond distortions are only applied on the TM-site with an electron polaron. In addition, to promote the localization of an electron at the specified TM-site, we initialize the magnetic moment of the TM atom containing the polaron to be 1μB higher than those of the other TM atoms. To obtain a delocalized electronic structure, we add one excess electron to each Li-NASICON compound without bond distortion and initialization of the magnetic moments for TM atoms, which facilitates the optimization of the non-polaronic structure. For all DFT calculations, an energy cutoff of 520 eV is used for the plane-wave basis set, and a gamma-point-only K-point grid55 is adopted. The convergence criteria for electronic and ionic steps are set to 10−5 eV for energy and 0.01 eV Å−1 for force, respectively.
Polaron transport is studied using a linear-interpolation method to describe the transfer of lattice distortion56–62 between two sites. In this approach, the intermediate images of the hopping trajectory {qx} are obtained by linearly interpolating the initial {qi} and final {qf} optimized polaronic structures, i.e., {qx} = (1 − x){qi} + x{qf}, where 0 < x < 1. In this study, seven intermediate images are used to locate the transition state and determine the kinetic barrier of each hop. We also initialize the magnetic moments of the two TM atoms that participate in a hop in the seven intermediate images as a linear interpolation between the magnetic moments of the two TM atoms in the initial and final polaronic structures for Fe- and Mn-containing Li-NASICONs. This magnetic moment initialization serves only to guide the convergence of the electronic structure of the intermediate images along the minimum energy pathways and does not constrain the magnetic moments during the optimization of the reaction pathway.
Although the linear interpolation approach may not always be valid for long-range polaron hops, we expect that it remains a reasonable approximation for simulating such hops in Li-NASICONs. To validate this, we perform a CI-NEB calculation for the longest hop (Hop IV) in LiTi2(PO4)3, which has a hopping distance of 6.02 Å, using seven images and a force convergence criterion of 0.02 eV Å−1. Notably, the hopping barrier obtained from the CI-NEB method is 0.27 eV, closely matching the 0.28 eV barrier obtained using the linear interpolation approach for the same hop. This suggests that the linearly interpolated images are already close to the minimum energy path for the hop. Therefore, we believe this approach reasonably captures the lattice distortion geometry during long-range hopping in Li-NASICON, although a more accurate hopping barrier can be obtained using the CI-NEB method at the expense of significantly higher computational cost.
We investigate the electron polaron transport for Li-NASICONs in the well-studied structure of LiTi2(PO4)3, which has a trigonal lattice with space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) C. The chemical space is explored by replacing different elements in octahedral or tetrahedral sites. Although Li-NASICONs can form in other structures (monoclinic,63 triclinic,64 and orthorhombic65), the topologies of the corner-sharing framework consisting of MO6 octahedra and PO4 tetrahedra are similar to those of trigonal Li-NASICONs.66
C. The chemical space is explored by replacing different elements in octahedral or tetrahedral sites. Although Li-NASICONs can form in other structures (monoclinic,63 triclinic,64 and orthorhombic65), the topologies of the corner-sharing framework consisting of MO6 octahedra and PO4 tetrahedra are similar to those of trigonal Li-NASICONs.66
In the three-dimensional corner-sharing framework of Li-NASICONs, the octahedral sites can accommodate various TMs, including Ti, V, Cr, Mn, Fe, and Co. In contrast, the tetrahedral sites are usually occupied by main group cations (e.g. P, Si), though some fully oxidized TM such as V and Mo can occasionally also be accommodated on these sites. We study eleven different Li-NASICONs, eight of which contain one Li atom with the other three containing three Li atoms per formula unit in the RC and R space group, respectively. We exclude Co-containing Li-NASICONs in this work as we are not able to localize an electron polaron to form a Co3+ in Co4+-containing Li-NASICON, which may be related to the ability of Co4+ to become metallic in oxides.67,68 In this study, we only consider electron polarons as a previous work by Kaur et al.69 found in V-oxides that electron and hole polarons behave similarly.
3. Results
3.1. Polaron formation in Li-NASICONs
The localization of a polaron causes the TM–O bonds of the TM in the polaronic site to elongate and the magnetic moment on the TM to increase by approximately 1μB. The TM–O bond lengths and the magnetic moments of the TM atoms before and after polaron formation in Li-NASICONs are shown in Fig. S1.† The computed polaron formation energies (EP) of each Li-NASICON, as well as the space group and the source of each crystal model are listed in Table 1. EP is calculated as the energy difference between the polaronic and delocalized solution, i.e., EP = Elocalized − Edelocalized. This value is negative for all the Li-NASICONs considered (Table 1), indicating favorable polaron formations in all compounds, consistent with our expectation. For Li3Fe2(PO4)3, we successfully find a localized solution, indicating that the formation of a polaron in this structure is feasible, although the formation energy could not be determined as we are unable to converge a delocalized solution for Li3Fe2(PO4)3. The formation energy of an electron polaron on Ti4+ is among the least negative ones, consistent with it being an early transition metal with more extended d-orbitals and its tendency in oxides to often become metallic when partially reduced. The electrochemical stability windows of Li-NASICONs, computed with the previous defined formalism,71–73 are listed in the Table S3.† These values are calculated without considering the possibility of Li (de)intercalation, which may occur due to the mixed ionic–electronic conductivity. Details of the methodology are provided in the ESI.†| Polaronic site | Valence state | E P (eV) | S. G. | Crystal model source | Energy above the hull (eV per atom) | |
|---|---|---|---|---|---|---|
| LiTi2(PO4)3 | Octahedral | Ti4+/Ti3+ | −0.42 |
RC |
mp – 18640 | 0 |
| LiV2(PO4)3 | Octahedral | V4+/V3+ | −0.52 |
RC |
mp – 26712 | 0.002 |
| LiCr2(PO4)3 | Octahedral | Cr4+/Cr3+ | −0.54 |
RC |
mp – 25838 | 0.044 |
| LiMn2(PO4)3 | Octahedral | Mn4+/Mn3+ | −0.60 |
RC |
Replace Ti with Mn in LiTi2(PO4)3 | 0.012* |
| LiFe2(PO4)3 | Octahedral | Fe4+/Fe3+ | −1.71 |
RC |
mp – 26581 | 0.079 |
| Li3Ti2(PO4)3 | Octahedral | Ti3+/Ti2+ | −0.16 |
R |
mp – 26883 | 0.064 |
| Li3Cr2(PO4)3 | Octahedral | Cr3+/Cr2+ | −0.33 |
R |
mp – 26971 | 0 |
| Li3Fe2(PO4)3 | Octahedral | Fe3+/Fe2+ | — |
R |
mp – 19430 | 0.010 |
| LiTi2(VO4)3 | Octahedral | V5+/V4+ | −0.21 |
RC |
Replace P with V in LiTi2(PO4)3 | 0.005* |
| LiTi2(VO4)3 | Tetrahedral | V5+/V4+ | −0.61 |
RC |
Replace P with V in LiTi2(PO4)3 | |
| LiGe2(VO4)3 | Tetrahedral | V5+/V4+ | −0.71 |
RC |
Replace Ti with Ge in LiTi2(VO4)3 | 0.008* |
| LiTi2(MoO4)3 | Tetrahedral | Mo6+/Mo5+ | −0.39 |
RC |
Replace P with Mo in LiTi2(PO4)3 | 0.132* |
3.2. Percolating paths for polaronic conduction in Li-NASICONs
Polarons must have percolating paths through the entire structure to enable overall conduction. The Li-NASICONs which contain one Li per formula unit, namely Li(TM/Ge)2((P/V/Mo)O4)3, contain only one symmetrically distinct octahedral (12c) and tetrahedral site (18e) to accommodate TM atoms. In these cases, we find that an electron polaron can be localized on each TM atom. However, for Li-NASICONs with compositions Li3TM2(PO4)3, there are two symmetrically distinct octahedral TM sites in the R space group. We find that the less distorted octahedral TM site (6c1) (edge-sharing with Li–O4 tetrahedra) is able to accommodate localized charge, whereas the more distorted octahedral TM site (6c2) (corner-sharing with Li–O4 tetrahedra) cannot accommodate a polaron. Consequently, fewer polaron hops are available in Li3TM2(PO4)3 than in LiTM2(PO4)3. The octahedral TM sites that can accommodate polaron are visualized in Fig. 1b.
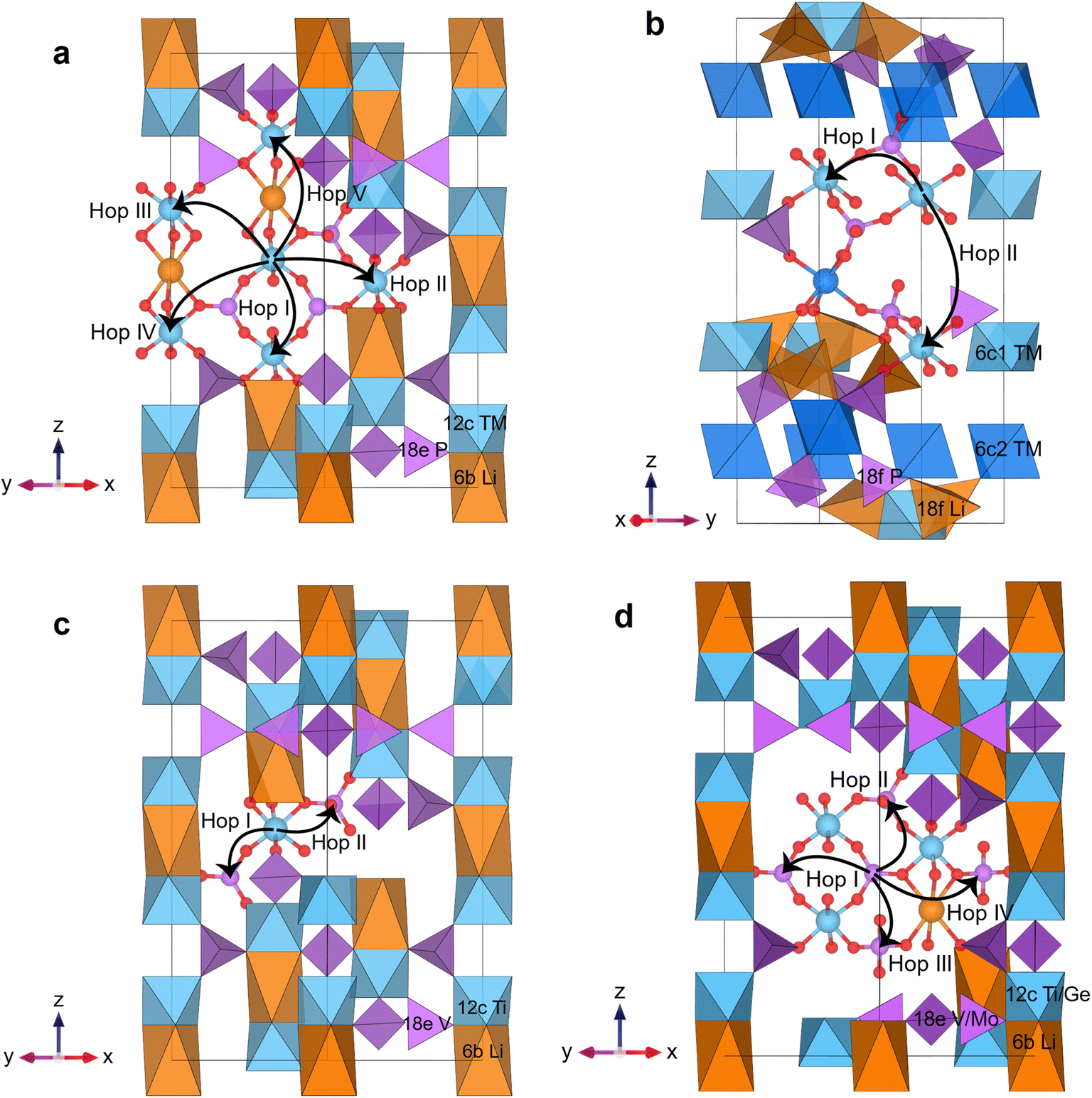 | ||
| Fig. 1 Electron polaron hops of (a) oct–oct hopping in LiTM2(PO4)3, TM = Ti/V/Cr/Mn/Fe; (b) oct–oct hopping in Li3TM2(PO4)3, TM = Ti/Cr/Fe; (c) oct–tet hopping in LiTi2(VO4)3; and (d) tet–tet hopping in LiTi2(TMO4)3, TM = V/Mo and LiGe2(VO4)3. Only atoms near each constitutive hop are shown. The orange spheres denote Li atoms, the blue spheres denote atoms (TMs or Ge) in octahedral sites, the purple spheres denote atoms (P, V, or Mo) in tetrahedral sites, and the red spheres denote O atoms. All constitutive electron polaron hops are represented by the arrows from the center TM atom. For Li3TM2(PO4)3 in (b), the dark blue octahedra denote 6c2 sites which cannot accommodate polarons. The hops we consider for LiTi2(MoO4)3 are the hop I and hop IV in (d). | ||
In this work, we first determine the polaron hops between two neighboring TMs with hopping distances of less than 7 Å. This includes hops between octahedral sites, octahedral-to-tetrahedral, as well as between tetrahedral sites. We exclude hops with hopping distances of more than 7 Å, as these hops are combinations of shorter hops. In addition, the tunneling probability of a hop between two TM atoms exponentially decays with respect to the hopping distance. All the hops considered are either perpendicular or parallel to [001] direction, which aligns with the z-axis of the Li-NASICON crystal structure, as shown in Fig. 1. While most ions in our compounds have reasonable valence states for the coordination they occupy, LiTi2(MoO4)3 with both Mo6+ and Mo5+ on the tetrahedral site is introduced as a more hypothetical compound, as Mo5+ is unlikely to be found on a tetrahedral site.74,75 The ground-state electronic structure of LiTi2(MoO4)3 exhibits magnetic moments of 0.90μB on each Ti ion, suggesting a valence +3. To maintain charge neutrality, the average valence of Mo atoms is then +17/3, which contain one-third Mo5+ and two-thirds Mo6+, consistent with the magnetic moments of 0.78μB and 0.09μB found in the calculations. We find that an electron polaron in this compound preferentially localizes on Mo6+, as evidenced by the electron polaron formation energy of −0.39 eV on Mo6+ and −0.34 eV on Mo5+. Therefore, we only consider hops between Mo6+ ions.
As shown in Fig. 1, the possible polaron sites lead to five distinct oct–oct hops in LiTM2(PO4)3, two distinct oct–oct hops in Li3TM2(PO4)3, two distinct oct–tet and four distinct tet–tet hops in LiTi2(VO4)3 and in LiGe2(VO4)3. For LiTi2(MoO4)3 we only considered two tet–tet hops through the Mo6+/Mo5+ pair perpendicular to [001], as the other two hops parallel to [001] create Mo4+ ions. For Li3TM2(PO4)3, we only investigate the oct–oct polaronic transport as P5+ is unlikely to accommodate a polaron.
The possible hops join to form percolating conduction paths. In this study, we classify the percolating paths into two types: perpendicular to [001] or parallel to [001]. For LiTi2(MoO4)3, we only consider percolating paths perpendicular to [001]. The oct–tet hopping in LiTi2(VO4)3 only leads to a percolating path perpendicular to [001]. The directions of all percolating paths, possible hops, and the polaron Wyckoff site are listed in Table 2.
| LiTM2(PO4)3, TM = Ti,V,Cr,Mn,Fe Oct–oct hopping | Li3TM2(PO4)3, TM = Ti,Cr,Fe Oct–oct hopping | ||||
|---|---|---|---|---|---|
| Direction | Constitutive hop | Wyckoff site | Direction | Constitutive hop | Wyckoff site |
| //[001] | Hop I–Hop V | 12c | //[001] | Hop I–Hop II | 6c1 |
| ⊥[001] | Hop II | 12c | ⊥[001] | Hop I | 6c1 |
| ⊥[001] | Hop III | 12c | |||
| ⊥[001] | Hop IV | 12c | |||
| LiTi2(VO4)3 Oct–tet hopping | LiTi2(VO4)3 and LiGe2(VO4)3 Tet–tet hopping | ||||
|---|---|---|---|---|---|
| Direction | Constitutive hop | Wyckoff site | Direction | Constitutive hop | Wyckoff site |
| //[001] | Hop II–Hop III | 18e | ⊥[001] | Hop I–Hop II | 12c (Oct) and 18e (Tet) |
| ⊥[001] | Hop I–Hop IV | 18e | |||
| ⊥[001] | Hop IV | 18e | |||
| LiTi2(MoO4)3 Tet–tet hopping | ||
|---|---|---|
| Direction | Constitutive hop | Wyckoff site |
| ⊥[001] | Hop I–Hop IV | 18e |
3.3. Rate-determining polaron hopping barriers of Li-NASICONs
To determine the rate-determining polaron hopping barriers in each compound, we first identify the hop with the highest barrier for each percolating conduction path. The path with the lowest maximum barrier is the preferential conduction path to realize percolating conduction, and the lowest maximum barrier is identified as the rate-determining hopping barrier for the polaronic conduction in a Li-NASICON. The barriers for all hops in each compound are shown in Table 3. For LiTi2(MoO4)3, only the barriers of hop I and IV perpendicular to [001] with the Mo6+/Mo5+ pair are calculated. The rate-determining hopping barriers are used to estimate the polaronic conductivity of Li-NASICONs.| Oct–oct hopping | Tet–tet hopping | |||||||
|---|---|---|---|---|---|---|---|---|
| LiTi2(PO4)3 | LiV2(PO4)3 | LiCr2(PO4)3 | LiMn2(PO4)3 | LiFe2(PO4)3 | LiTi2(VO4)3 | LiGe2(VO4)3 | LiTi2(MoO4)3 | |
| Hop I | 0.16 | 0.28 | 0.26 | 0.43 | 0.14 | 0.40 | 0.51 | 0.26 |
| Hop II | 0.22 | 0.26 | 0.23 | 0.36 | 0.12 | 0.39 | 0.54 | — |
| Hop III | 0.27 | 0.27 | 0.19 | 0.43 | 0.13 | 0.49 | 0.58 | — |
| Hop IV | 0.28 | 0.33 | 0.26 | 0.35 | 0.09 | 0.40 | 0.52 | 0.29 |
| Hop V | 0.30 | 0.38 | 0.33 | 0.47 | 0.25 | — | — | — |
| Oct–oct hopping | Oct–tet hopping | |||
|---|---|---|---|---|
| Li3Ti2(PO4)3 | Li3Cr2(PO4)3 | Li3Fe2(PO4)3 | LiTi2(VO4)3 | |
| Hop I | 0.17 | 0.43 | 0.20 | 0.41 |
| Hop II | 0.13 | 0.39 | 0.17 | 0.45 |
The rate-determining hopping barriers of oct–oct hopping in different Li-NASICONs range from less than 0.1 eV to more than 0.4 eV, with the lowest barrier of 0.09 eV through the Fe4+/Fe3+ pair and much higher hopping barriers of 0.35 and 0.43 eV through the Mn4+/Mn3+ and Cr3+/Cr2+ pairs, respectively. The oct–oct hopping through the Ti4+/Ti3+, V4+/V3+, Cr4+/Cr3+, Ti3+/Ti2+, and Fe3+/Fe3+ have barriers between 0.15 and 0.3 eV. These results indicate element-dependent oct–oct polaron transport in Li-NASICONs. The tet–tet hopping also depends on the nature of the TM present on that site. Although hopping through the V5+/V4+ pair has an energy barrier of more than 0.4 eV in LiTi2(VO4)3 and LiGe2(VO4)3, through the Mo6+/Mo5+ pair in LiTi2(MoO4)3 a lower barrier of 0.29 eV has to be crossed. In LiTi2(VO4)3, the oct–tet hopping has a high barrier of 0.45 eV through the Ti4+/V4+ pair. The rate-determining polaron hopping barriers for all Li-NASICONs are shown in Fig. 2a. All rate-determining hops occur along the percolating paths perpendicular to [001].
 | ||
| Fig. 2 (a) Rate-determining polaron hopping barriers of Li-NASICONs and (b) polaron formation energy in Li-NASICONs. | ||
3.4. Polaronic conductivity in Li-NASICONs
We estimate the room-temperature polaronic conductivity of Li-NASICONs from the calculated rate-determining hopping barriers using the Marcus theory framework, which formulates the rate of charge-transfer across molecules or atoms in compounds.76–78 In Marcus theory, a small polaron hopping is described by a parabolic free-energy curve for the polaron on each site, and their crossing point gives the hopping activation energy. When a polaron transfers from one site to another, its energy rises and reaches a maximum when the polaronic distortion is distributed between the two sites. At this crossing point, the polaron hopping rate depends on the tunneling probability. A tunneling probability close to identity indicates an adiabatic charge transfer, whereby a polaron hops to the final state once the lattice distortion reaches the transition-state configuration. In contrast, a limited tunneling probability results in a diabatic charge transfer, and the polaron may go back to the initial state even when the transition state has been reached. The tunneling probability is given by78| Ptunnel = 1 − exp[−(Ve2/hυn)(π3/λkBT)1/2], | (1) |
We estimate polaron hopping frequencies to obtain the diffusivities in Li-NASICONs. The adiabatic and diabatic polaron hopping frequencies are given by78
fadiabatic = υn![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp[−Ea/kBT] exp[−Ea/kBT] | (2) |
 | (3) |
| D = fa2g (diffusivity) | (4) |
| u = eD/kBT (charge mobility). | (5) |
| σ = neu. | (6) |
| V e (eV) | n (#/cm3) | a (Å) | E a (eV) | P tunnel | Transfer type | Conductivity at 300 K (S cm−1) | |
|---|---|---|---|---|---|---|---|
| LiTi2(PO4)3 | 0.23 | 7.626 × 1020 | 5.03 | 0.22 | 1 | Adia; oct–oct | 2.42 × 10−2 |
| LiV2(PO4)3 | 0.38 | 7.863 × 1020 | 4.99 | 0.26 | 1 | Adia; oct–oct | 5.24 × 10−3 |
| LiCr2(PO4)3 | 0.30 | 8.067 × 1020 | 5.37 | 0.19 | 1 | Adia; oct–oct | 9.31 × 10−2 |
| LiMn2(PO4)3 | 0.33 | 8.229 × 1020 | 5.85 | 0.35 | 1 | Adia; oct–oct | 2.32 × 10−4 |
| LiFe2(PO4)3 | 0.13 | 8.430 × 1020 | 5.85 | 0.09 | 1 | Adia; oct–oct | 5.51 |
| Li3Ti2(PO4)3 | 0.20 | 7.171 × 1020 | 4.89 | 0.17 | 1 | Adia; oct–oct | 1.49 × 10−1 |
| Li3Cr2(PO4)3 | 0.23 | 7.599 × 1020 | 4.83 | 0.43 | 1 | Adia; oct–oct | 6.65 × 10−6 |
| Li3Fe2(PO4)3 | 0.18 | 7.405 × 1020 | 4.86 | 0.20 | 1 | Adia; oct–oct | 4.75 × 10−2 |
| LiTi2(VO4)3 | 0.41 | 6.650 × 1020 | 3.43 | 0.45 | 1 | Adia; oct–tet | 1.35 × 10−6 |
| LiTi2(VO4)3 | 0.50 | 6.650 × 1020 | 4.51 | 0.40 | 1 | Adia; tet–tet | 1.62 × 10−5 |
| LiGe2(VO4)3 | 0.30 | 6.994 × 1020 | 5.54 | 0.52 | 1 | Adia; tet–tet | 2.48 × 10−7 |
| LiTi2(MoO4)3 | 0.40 | 5.999 × 1020 | 5.82 | 0.29 | 1 | Adia; tet–tet | 1.70 × 10−3 |
Ultimately, for adiabatic and diabatic polaron hopping, the conductivity can be formulated as
| σadiabatic = (ne2vna2g/kBT) × exp[−Ea/kBT] | (7) |
 | (8) |
All predicted characteristic properties of the rate-determining hops in Li-NASICONs are listed in Table 4.
Although the polaronic conductivity is determined by the hopping barrier and tunneling probability, for Li-NASICONs, most of the hops are adiabatic with tunneling probabilities of identity. Even the most diabatic hop, which is found in LiMn2(PO4)3, has a tunneling probability of 0.41, suggesting a feasible polaron transfer when the transition state is reached, as shown in Table 5. Therefore, for Li-NASICONs the polaronic conductivity is mainly dominated by the hopping barrier and a percolating path with the lowest maximum barrier is the path with the highest polaronic conductivity. Amongst all Li-NASICONs in the current study, polaron transport via the Fe4+/Fe3+ pair on octahedral sites in LiFe2(PO4)3 has the highest calculated room-temperature conductivity of 5.51 S cm−1, whereas the transport through the Cr3+/Cr2+ pair on octahedral sites in Li3Cr2(PO4)3 and through the V5+/V4+ pair on tetrahedral sites in LiTi2(VO4)3 and LiGe2(VO4)3 has much lower predicted room-temperature conductivity of 10−7 to 10−5 S cm−1. The oct–tet polaron transport in LiTi2(VO4)3 is also predicted to be poor (near 10−6 S cm−1). All rate-determining hops of Li-NASICONs are adiabatic.
| E a (eV) | V e (eV) | P tunneling | Hopping type | ||
|---|---|---|---|---|---|
| LiTi2(PO4)3 | Hop I | 0.16 | 0.28 | 1 | Adiabatic |
| Hop II | 0.22 | 0.23 | 1 | Adiabatic | |
| Hop III | 0.27 | 0.13 | 1 | Adiabatic | |
| Hop IV | 0.28 | 0.13 | 1 | Adiabatic | |
| Hop V | 0.30 | 0.03 | 0.480 | Diabatic | |
| LiV2(PO4)3 | Hop I | 0.28 | 0.25 | 1 | Adiabatic |
| Hop II | 0.26 | 0.38 | 1 | Adiabatic | |
| Hop III | 0.27 | 0.23 | 1 | Adiabatic | |
| Hop IV | 0.33 | 0.18 | 1 | Adiabatic | |
| Hop V | 0.38 | 0.03 | 0.444 | Diabatic | |
| LiCr2(PO4)3 | Hop I | 0.26 | 0.18 | 1 | Adiabatic |
| Hop II | 0.23 | 0.40 | 1 | Adiabatic | |
| Hop III | 0.19 | 0.30 | 1 | Adiabatic | |
| Hop IV | 0.26 | 0.23 | 1 | Adiabatic | |
| Hop V | 0.33 | 0.03 | 0.466 | Diabatic | |
| LiMn2(PO4)3 | Hop I | 0.43 | 0.13 | 1 | Adiabatic |
| Hop II | 0.36 | 0.28 | 1 | Adiabatic | |
| Hop III | 0.43 | 0.18 | 1 | Adiabatic | |
| Hop IV | 0.35 | 0.33 | 1 | Adiabatic | |
| Hop V | 0.47 | 0.03 | 0.413 | Diabatic | |
| LiFe2(PO4)3 | Hop I | 0.14 | 0.05 | 0.909 | Diabatic |
| Hop II | 0.12 | 0.08 | 0.997 | Adiabatic | |
| Hop III | 0.13 | 0.05 | 0.915 | Diabatic | |
| Hop IV | 0.09 | 0.13 | 1 | Adiabatic | |
| Hop V | 0.25 | 0.05 | 0.852 | Diabatic | |
| Li3Ti2(PO4)3 | Hop I | 0.17 | 0.20 | 1 | Adiabatic |
| Hop II | 0.13 | 0.03 | 0.610 | Diabatic | |
| Li3Cr2(PO4)3 | Hop I | 0.43 | 0.23 | 1 | Adiabatic |
| Hop II | 0.39 | 0.03 | 0.440 | Diabatic | |
| Li3Fe2(PO4)3 | Hop I | 0.20 | 0.18 | 1 | Adiabatic |
| Hop II | 0.17 | 0.03 | 0.569 | Diabatic | |
| LiTi2(VO4)3 (oct–tet) | Hop I | 0.41 | 0.51 | 1 | Adiabatic |
| Hop II | 0.45 | 0.41 | 1 | Adiabatic | |
| LiTi2(VO4)3 (tet–tet) | Hop I | 0.40 | 0.50 | 1 | Adiabatic |
| Hop II | 0.39 | 0.23 | 1 | Adiabatic | |
| Hop III | 0.49 | 0.18 | 1 | Adiabatic | |
| Hop IV | 0.40 | 0.48 | 1 | Adiabatic | |
| LiGe2(VO4)3 | Hop I | 0.51 | 0.45 | 1 | Adiabatic |
| Hop II | 0.54 | 0.23 | 1 | Adiabatic | |
| Hop III | 0.58 | 0.18 | 1 | Adiabatic | |
| Hop IV | 0.52 | 0.30 | 1 | Adiabatic | |
| LiTi2(MoO4)3 | Hop I | 0.26 | 0.40 | 1 | Adiabatic |
| Hop IV | 0.29 | 0.40 | 1 | Adiabatic | |
| LiTi2(VO4)1/9(PO4)8/9 | Hop I | 0.38 | 0.53 | 1 | Adiabatic |
| Hop II | 0.38 | 0.58 | 1 | Adiabatic |
4. Discussion
Owing to the corner-sharing framework that alleviates the coulombic repulsion between Li ions and positively charged TM ions,83 Li-NASICONs have low ionic migration barriers and high room-temperature ionic conductivities of 10−4 to 10−3 S cm−1.84,85 However, intrinsic wide band gaps (several eVs) and narrow bands prohibit band conduction in the structures.85 For instance, LiTi2(PO4)3 has a band gap of 2.50 eV (ref. 86) and a low room-temperature electronic conductivity of 10−8 S cm−1.87 To improve their electronic transport, attempts have been made to exploit mixed-valency in Li-NASICONs to generate localized charge carriers such that electronic conduction is activated via a polaron hopping mechanism. Specifically, Sharma and Dalvi25 doped vanadium into LiTi2(PO4)3 and Li1.3Al0.3Ti1.7(PO4)3 to generate LiTi2(VO4)x(PO4)3−x and Li1.3Al0.3Ti1.7(VO4)x(PO4)3−x and improved its room-temperature electronic conductivity to 10−5 S cm−1.4.1. Polaronic conduction in LiTi2(VO4)x(PO4)3−x
To validate our computational results, we compare the predicted hopping barrier and the room-temperature polaronic conductivity of LiTi2(VO4)x(PO4)3−x with published experimental results.25 We replace two P atoms with V atoms in LiTi2(PO4)3 to approximate the experimental composition of LiTi2(VO4)0.3(PO4)2.7 (Fig. S5†). In this V-doped Li-NASICON, the polaron formation is more favorable on V atoms than on Ti atoms, as the charge only localizes on V atoms even when a structural distortion and an initialization of the Ti magnetic moment are imposed on a Ti–O6 octahedron. The preferential localization of an electron polaron on a V atom in a tetrahedral site is consistent with the experimental observation of V5+/V4+ pairs in the V-doped Li-NASICON. The computed tet–tet hopping barrier via the V5+/V4+ pair is 0.38 eV, comparable but somewhat higher than the experimental hopping barrier of 0.20 eV. The computed room-temperature polaronic conductivity is 3.62 × 10−5 S cm−1, in reasonable agreement with the experimental conductivity of 4.46 × 10−6 S cm−1. In our calculations, the polaron concentration is fixed at one polaron per supercell containing 72 oxygen atoms.4.2. Correlation between J–T distortion and rate-determining hopping barrier of oct–oct polaron transport in Li-NASICONs
We classify the oct–oct polaron transport into three classes depending on the presence of any Jahn–Teller (J–T) activity with or without the electron polaron present: only the state with polaron is J–T active (Class 1: Mn4+/Mn3+ and Cr3+/Cr2+); both the polaronic and pristine state of the TMs are J–T inactive (Class 2: Ti4+/Ti3+, V4+/V3+, Cr4+/Cr3+, Ti3+/Ti2+ and Fe3+/Fe2+); and only the state without the polaron is J–T active (Class 3: Fe4+/Fe3+). We note that only cations with a doublet degeneracy in the eg band (Exe J–T coupling) involving a static J–T distortion are classified as J–T active, whereas cations with either a triplet degeneracy in the t2g band (Tx(t + e) coupling) involving a dynamic J–T effect, or cations with no degeneracy, are classified as J–T inactive. For Class 1, the J–T distortion is stabilized by the electron polaron formation and results in a relatively high hopping barrier as the lattice distortion that must be migrated is largely caused by the J–T coupling with the polaron. This leads to low polaronic conduction in LiMn2(PO4)3 and Li3Cr2(PO4)3. For Class 2, no J–T distortion is involved during polaron hopping, resulting in lower hopping barriers than those of class one. In Class 3, the J–T distortion vanishes upon the formation of an electron polaron leading to a lower lattice distortion, while the TM sites without the extra electron are “pre-distorted” and possess multiple degrees of freedom by which they can distort at low cost in energy (i.e., the J–T modes).88 Hence, the polaronic conductivity in LiFe2(PO4)3 is the highest with a very low barrier of 0.09 eV. This obvious correlation between hopping barriers and J–T distortion in Li-NASICONs reflects the adiabatic nature of polaron transport, which is phonon-assisted and dominated by the transport of the lattice distortion. The TM–O bond lengths of the J–T active species are shown in Table S1.†With the assumption that polaron transport can be approximated as adiabatic hopping at room temperature in most oxides,69,89,90 other materials involving TM–O6 octahedra might also exhibit a correlation between high conductivity and J–T distortion in the non-polaronic TM ion. For instance, in olivine LiMPO4 structured phosphates, the hole polaron hopping barrier via the Mn2+/Mn3+ pair is more than 0.3 eV, higher than those through the Fe2+/Fe3+ and Co2+/Co3+ pairs with no J–T distortion during hopping,59 consistent with the fact that Mn3+ is J–T active. In addition, both the hole polaron hopping through the Co3+/Co4+ pair and the electron polaron hopping through the Co3+/Co2+ pair involve no J–T distortion in LiCoO2 (ref. 60) and have energy barriers roughly between 0.1 and 0.3 eV, which are similar to the hopping barriers in class two. Furthermore, the electron polaron transport through the Mo6+/Mo5+ J–T inactive pair in MoO3 ranges from 0.11 to 0.35 eV,61 similar to the hopping barriers in class two. Notably, the electron polaron hopping barrier through the Mn3+/Mn2+ pair in MnPO4 is only two-thirds of the hole polaron hopping barrier through the Mn2+/Mn3+ pair in LiMnPO4.62 The applicability of J–T distortion-correlated polaron hopping barriers to other oxides of TMs underpins the 3d electronic configuration as an indicator for polaronic conduction. Despite the hopping barriers of the oct–oct polaronic transport being quite different among various mixed-valency pairs, a majority of Li-NASICONs have predicted room-temperature polaronic conductivity above 10−4 S cm−1, including those with the Ti4+/Ti3+, Ti3+/Ti2+, V4+/V3+, Cr4+/Cr3+, Fe4+/Fe3+, and Fe3+/Fe2+ mixed-valency pairs. Polaronic conduction through the oct–oct pathway in Li-NASICONs may open up the possibility of Li-NASICON mixed ionic electronic conductors.
Previous study on polaron hopping in AgCl compound found that the hopping barrier depends on the orientation of the J–T axis in the octahedra.91 To investigate whether the orientation of the J–T axis influences the polaron hopping barrier in Li-NASICONs, we examine whether polaronic states can be stabilized along all three principal axes. We reorient the elongated axis of the original extrinsic J–T distorted octahedron of Mn3+ and Cr2+ along the other two principal axes and then perform geometry optimizations. Our results show that, in both Mn3+ and Cr2+ octahedra, the elongated axis reverts back to the original principal axis, indicating that J–T distortion is only stable along a single principal axis in Li-NASICONs. This finding rules out the possibility that the polaron hops by first changing direction of the JT axis and then hopping.
4.3. The effect of the Li charge state on polaron hopping in Li-NASICONs
Another factor that may contribute to the oct–oct polaron transport in the Ti-based Li-NASICONs is the amount of Li present in the NASICON. The additional Li ions in Li3Ti2(PO4)3 introduce attractive coulombic interactions with the polaron during a hop, potentially leading to a smoother hopping energy landscape and a lower hopping barrier. This effect is reflected in the two hopping barriers of 0.13 eV and 0.17 eV in Li3Ti2(PO4)3 compared to the hopping barriers in LiTi2(PO4)3 which are mostly above 0.2 eV as shown in the Table 3. In contrast, for Cr-based NASICONs, although the additional Li+ ions in Li3Cr2(PO4)3 could facilitate polaron hopping, their effect may be overpowered by the J–T active Cr2+ upon polaron formation resulting in a higher barrier. Hence, unlike in Ti-based NASICONs where increasing the Li content lowers the hopping barriers, Li3Cr2(PO4)3 has higher hopping barriers than in LiCr2(PO4)3, as shown in the Table 3.4.4. Oct–tet and tet–tet polaron transport in Li-NASICONs
Oct–tet polaron transport is kinetically less favorable than oct–oct polaron transport in the only compound for which we evaluate this because of the difference in the polaron formation energy on a Ti and V atom in LiTi2(VO4)3. Although a polaron on Ti has a formation energy of −0.21 eV, it is more stable on V with a formation energy of −0.61 eV, which leads to an asymmetric hopping energy landscape that creates a high barrier of 0.45 eV, as shown in Fig. 3. Oct–tet polaron hopping may also have high barriers in other systems due to the difference in the polaron formation energy in the different coordinated environments. Similar results were reported in a previous study on oct–tet electron polaron hopping in Co3O4.92 | ||
| Fig. 3 Asymmetric polaron hopping energy curve of oct–tet polaron transport in LiTi2(VO4)3. | ||
As fewer TM elements (V and Mo) have been reported to occupy tetrahedral sites in Li-NASICONs, tet–tet polaron hopping may be of less practical importance for application in MIECs. Nonetheless, the room-temperature polaronic conductivity via the Mo6+/Mo5+ pair on tetrahedral sites in LiTi2(MoO4)3 is predicted to be on the order of 10−3 S cm−1, comparable to the room-temperature ionic conductivities in Li-NASICONs reported in previous studies.93–95 This result indicates that molybdenum-based Li-NASICONs may also exhibit efficient electronic conductivity via polaronic conduction.
4.5. The effect of lattice volume and polaronic orbital overlap on polaronic conduction in Li-NASICONs
Aside from the J–T distortion, other factors have also been proposed to account for the different hopping barriers in a compound. For instance, Kweon et al.52 and Moradabadi et al.90 independently pointed at an increase in the polaron hopping barrier as a result of lattice expansion for BiVO4 and LiCoO2, respectively. In our study, we also find a similar trend in the hopping barriers of LiTi2(PO4)3 and LiGe2(VO4)3, as shown in Fig. 4. Lattice expansions elongate the hopping distance between two TM atoms, which is accompanied by the increase in the hopping barrier, consistent with the previous studies. However, the volume changes arising from the chemistry differences in Li-NASICONs cannot explain the barrier variation seen with chemistry. Fig. 5 shows the oct–oct hopping barrier as a function of the volume of each LiTM2(PO4)3 unit cell. Although the lattice volume of Li-NASICONs monotonically increases from Ti to Fe, the hopping barriers of oct–oct polaronic transport via their +4/+3 mixed-valency TMs show no such monotonic change. | ||
| Fig. 4 Polaron hopping barrier versus lattice parameter variation of (a) LiGe2(VO4)3 and (b) LiTi2(PO4)3. The lattice parameter variation is the percentage of the homogeneous compression or elongation of the lattice parameters in all three dimensions, i.e., −1% denotes compressing all three lattice parameters by 1%; 1% denotes elongating all three lattice parameters by 1%. Numbers near squares denote the polaron hopping distance. | ||
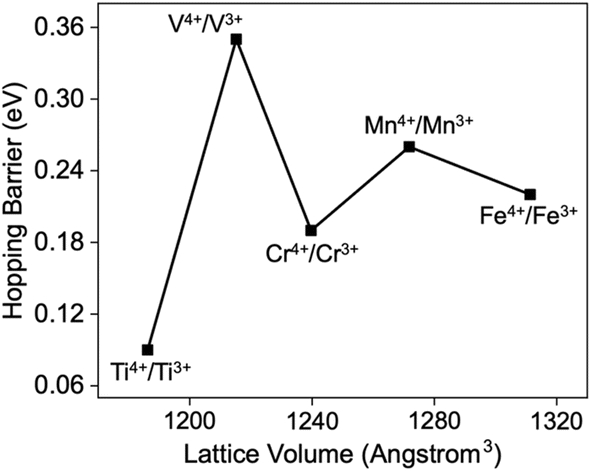 | ||
| Fig. 5 Oct–oct polaron hopping barrier as a function of the lattice volume of LiTM2(PO4)3, TM = Ti, V, Cr, Mn, and Fe. | ||
Another possible factor reported by Garcia-Lastra et al.96 to affect polaron hopping barrier is the extent of polaronic orbital overlap. These authors found that both hole polaron and electron polaron hopping barriers exhibit differences along different hopping directions in Li2O2 stemming from the different extent of polaronic orbital overlap in the transition states. Nevertheless, these differences in hopping barriers were found to be less than 0.1 eV in Li2O2 and may have limited effect on the polaronic transport. The polaronic orbital overlap is quantified by the integration of the projected crystal orbital Hamilton population (pCOHP) of the polaronic state with respect to energy up to the highest occupied state (the Fermi level);97 namely, the integrated projected crystal orbital Hamilton population (IpCOHP). We compute the IpCOHP of the polaronic 3d orbitals of two TM atoms participating in the rate-determining hop in the transition states of various Li-NASICONs, and plot them in Fig. 6. There appears to be no correlation between the overlaps and the hopping barriers.
 | ||
| Fig. 6 Polaron hopping barrier as a function of the polaronic orbital overlap between the 3d orbitals of two TMs participating in the rate-determining hop in the transition state of Li-NASICONs. The blue data points denote oct–oct hopping, the pink data point denotes oct–tet hopping, and the purple data points denote tet–tet hopping. | ||
5. Conclusion
We examine polaronic conduction in Li-NASICONs using first-principles calculations and demonstrate that significantly enhanced electronic conductivity may be achieved from polaron hopping if a high enough carrier concentration can be generated. Polaronic conductivities through the Ti4+/Ti3+, Ti3+/Ti2+, V4+/V3+, Cr4+/Cr3+, Fe4+/Fe3+, Fe3+/Fe2+, and Mo6+/Mo5+ pairs are predicted to be over 10−4 S cm−1 at room temperature, with the highest conductivity of 5.51 S cm−1 achieved through the Fe4+/Fe3+ pair in LiFe2(PO4)3. Our calculations indicate that in Li-NASICONs, the hopping barrier of oct–oct polaron transport is correlated with the J–T distortion associated with polaron formation, whereas the lattice volume and the polaronic orbital overlap have minor effects on polaronic conduction. Our understanding of polaronic conduction in Li-NASICONs may assist in identifying effective strategies to develop Li-NASICON MIECs for electrochemical energy storage devices.Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work is supported by the Samsung Advanced Institute of Technology (SAIT). The computational analysis is performed using computational resources sponsored by the Department of Energy's Office of Energy Efficiency and Renewable Energy located at the National Renewable Energy Laboratory (NREL) under the saepssic and ahlssic allocations. Computational resources are also provided by the Advanced Cyberinfrastructure Coordination Ecosystem: Services & Support (ACCESS) program, which is supported by National Science Foundation grants #2138259, #2138286, #2138307, #2137603, and #2138296. K. Jun gratefully acknowledges support from the Kwanjeong Educational Foundation scholarship.References
- Y. Kyungho, K. JungJoon, S. WonMo, L. MyeongHwan and K. Kisuk, Investigation on the interface between Li10GeP2S12 electrolyte and carbon conductive agents in all solid-state lithium battery, Sci. Rep., 2018, 8, 10366 CrossRef PubMed.
- X. Yihan, W. Yan, B. ShouHang, K. JaeChul, J. M. Lincoln and G. Ceder, Understanding interface stability in solid-state batteries, Nat. Rev. Mater., 2020, 5, 105–126 Search PubMed.
- F. Li, T. Zhang and H. Zhou, Challenges of non-aqueous Li-O2 batteries: electrolytes, catalysts, and anodes, Energy Environ. Sci., 2013, 6, 1125–1141 RSC.
- B. M. Gallant, et al., Chemical and Morphological Changes of Li-O2 Battery Electrodes upon Cycling, J. Phys. Chem. C, 2012, 116, 20800–20805 CrossRef CAS.
- M. M. O. Thotiyl, S. A. Freunberger, Z. Peng and P. G. Bruce, The Carbon Electrode in Nonaqueous Li-O2 Cells, J. Am. Chem. Soc., 2013, 135, 494–500 CrossRef PubMed.
- B. D. McCloskey, et al., Twin Problems of Interfacial Carbonate Formation in Nonaqueous Li-O2 Batteries, J. Phys. Chem. Lett., 2012, 3, 997–1001 CrossRef CAS PubMed.
- D. Cao, S. Zhang, F. Yu, Y. Wu and Y. Chen, Carbon-Free Cathode Materials for Li-O2 Batteries, Batteries Supercaps, 2019, 2, 428–439 CrossRef CAS.
- Z. Peng, S. A. Freunberger, Y. Chen and P. G. Bruce, A Reversible and Higher-Rate Li-O2 Battery, Science, 2012, 337, 563–566 CrossRef CAS PubMed.
- W. Luo, et al., Binder-Free and Carbon-Free 3D Porous Air Electrode for Li-O2 Batteries with High Efficiency, High Capacity, and Long Life, Small, 2016, 12, 3031–3038 CrossRef CAS PubMed.
- F. Li, et al., Li-O2 Battery Based on Highly Efficient Sb-Doped Tin Oxide Supported Ru Nanoparticles, Adv. Mater., 2014, 26, 4659–4664 CrossRef CAS PubMed.
- F. Li, et al., Ru/ITO: A Carbon-Free Cathode for Nonaqueous Li-O2 Battery, Nano Lett., 2013, 13, 4702–4707 CrossRef CAS PubMed.
- J. Kang, et al., Breathable Carbon-Free Electrode: Black TiO2 with Hierarchically Ordered Porous Structure for Stable Li-O2 Battery, Adv. Energy Mater., 2017, 7, 1700814 CrossRef.
- G. Zhao, R. Mo, B. Wang, L. Zhang and K. Sun, Enhanced Cyclability of Li-O2 Batteries Based on TiO2 Supported Cathodes with No Carbon or Binder, Chem. Mater., 2014, 26, 2551–2556 CrossRef CAS.
- M. M. O. Thotiyl, et al., A stable cathode for the aprotic Li-O2 battery, Nat. Mater., 2013, 12, 1050–1056 CrossRef PubMed.
- Y. Chang, et al., A Carbon- and Binder-Free Nanostructured Cathode for High-Performance Nonaqueous Li-O2 Battery, Adv. Sci., 2015, 2, 1500092 CrossRef PubMed.
- J. Cheng, et al., Perovskite La0.6Sr0.4Co0.2Fe0.8O3 as an effective electrocatalyst for non-aqueous lithium air batteries, Electrochim. Acta, 2016, 191, 106–115 CrossRef CAS.
- Q. Xu, et al., Ba0.9Co0.7Fe0.2Nb0.1O3-δ Perovskite as Oxygen Electrode Catalyst for Rechargeable Li-Oxygen Batteries, Electrochim. Acta, 2016, 191, 577–585 CrossRef CAS.
- Y. Liu, et al., Fabrication and Performance of All-Solid-State Li-Air Battery with SWCNTs/LAGP Cathode, ACS Appl. Mater. Interfaces, 2015, 7, 17307–17310 CrossRef CAS PubMed.
- X. B. Zhu, T. S. Zhao, Z. H. Wei, P. Tan and G. Zhao, A novel solid-state Li-O2 battery with an integrated electrolyte and cathode structure, Energy Environ. Sci., 2015, 8, 2782–2790 RSC.
- H. T. T. Le, et al., Highly efficient and stable solid-state Li-O2 batteries using a perovskite solid electrolyte, J. Mater. Chem. A, 2019, 7, 3150–3160 RSC.
- X. Chi, et al., A highly stable and flexible zeolite electrolyte solid-state Li-air battery, Nature, 2021, 592, 551–557 CrossRef CAS PubMed.
- S. B. Ma, et al., Mixed Ionic–Electronic Conductor of Perovskite LixLayMO3-δ toward Carbon-Free Cathode for Reversible Lithium-Air Batteries, Adv. Energy Mater., 2020, 10, 2001767 CrossRef CAS.
- M. Kim, et al., Carbon-free high-performance cathode for solid-state Li-O2 battery, Sci. Adv., 2022, 8, eabm8584 CrossRef CAS PubMed.
- Z. Jin, et al., Garnet-type solid-state mixed ionic and electronic conductor, Energy Storage Mater., 2023, 59, 102788 CrossRef.
- N. Sharma and A. Dalvi, Vanadium substituted Li+-NASICON systems: Tailoring electronic conductivity for electrode applications, J. Alloys Compd., 2021, 861, 157954 CrossRef CAS.
- Q. Bai, Y. Zhu, X. He, E. Wachsman and Y. Mo, First principles hybrid functional study of small polarons in doped SrCeO3 perovskite: towards computation design of materials with tailored polaron, Ionics, 2018, 24, 1139–1151 CrossRef CAS.
- C. E. Schwarz, R. S. Saravanan, N. M. Borodin, Y. Liu, E. D. Wachsman and Y. Mo, Polaron-Based Electronic Conduction in Mixed Ionic-Electronic Conducting Lithium Garnets, ACS Energy Lett., 2024, 9(11), 5334–5340 CrossRef CAS.
- K. Zhang, et al., Molybdenum-based NASICON Li2M2(MoO4)3 (M = Zn, Cu): Understanding structural evolution and lithium storage mechanism, J. Alloys Compd., 2022, 890, 161884 CrossRef CAS.
- K. Feng, F. Wang, H. Zhang, X. Li and H. Zhang, Li3Cr(MoO4)3 : a NASICON-type high specific capacity cathode material for lithium ion batteries, J. Mater. Chem. A, 2018, 6, 19107–19112 RSC.
- S. R. S. Prabaharan, A. Fauzi, M. S. Michael and K. M. Begam, New NASICON-type Li2Ni2(MoO4)3 as a positive electrode material for rechargeable lithium batteries, Solid State Ionics, 2004, 171, 157–165 CrossRef CAS.
- C. Franchini, M. Reticcioli, M. Setvin and U. Diebold, Polarons in materials, Nat. Rev. Mater., 2021, 6, 560–586 CrossRef CAS.
- P. Huang and E. A. Carter, Advances in Correlated Electronic Structure Methods for Solids, Surfaces, and Nanostructures, Annu. Rev. Phys. Chem., 2008, 59, 261–290 CrossRef CAS PubMed.
- G. Pacchioni, Modeling doped and defective oxides in catalysis with density functional theory methods: Room for improvements, J. Chem. Phys., 2008, 128, 182505 CrossRef PubMed.
- M. V. Ganduglia-Pirovano, A. Hofmann and J. Sauer, Oxygen vacancies in transition metal and rare earth oxides: Current state of understanding and remaining challenges, Surf. Sci. Rep., 2007, 62, 219–270 CrossRef CAS.
- A. I. Liechtenstein, V. I. Anisimov and J. Zaanen, Density-functional theory and strong interactions: Orbital ordering in Mott-Hubbard insulators, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 52, R5467–R5470 CrossRef CAS PubMed.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys and A. P. Sutton, Electron-energy-loss spectra and the structural stability of nickel oxide: An LSDA+U study, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505–1509 CrossRef CAS.
- V. I. Anisimov, F. Aryasetiawan and A. I. Lichtenstein, First-principles calculations of the electronic structure and spectra of strongly correlated systems: the LDA+ U method, J. Phys.: Condens. Matter, 1997, 9, 767 CrossRef CAS.
- M. Setvin, et al., Direct View at Excess Electrons in TiO2 Rutile and Anatase, Phys. Rev. Lett., 2014, 113, 086402 CrossRef CAS PubMed.
- T. D. Pham and N. A. Deskins, Efficient Method for Modeling Polarons Using Electronic Structure Methods, J. Chem. Theory Comput., 2020, 16, 5264–5278 CrossRef CAS PubMed.
- P. Deák, B. Aradi and T. Frauenheim, Polaronic effects in TiO2 calculated by the HSE06 hybrid functional: Dopant passivation by carrier self-trapping, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 155207 CrossRef.
- E. Finazzi, C. D. Valentin, G. Pacchioni and A. Selloni, Excess electron states in reduced bulk anatase TiO2: Comparison of standard GGA, GGA+U, and hybrid DFT calculations, J. Chem. Phys., 2008, 129, 154113 CrossRef PubMed.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, Hybrid functionals based on a screened Coulomb potential, J. Chem. Phys., 2003, 118, 8207–8215 CrossRef CAS.
- G. Kresse and J. Furthmüller, Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Ab initio molecular dynamics for liquid metals, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- S. P. Ong, et al., Python Materials Genomics (pymatgen): A robust, open-source python library for materials analysis, Comput. Mater. Sci., 2013, 68, 314–319 CrossRef CAS.
- D. M. Ramo, A. L. Shluger, J. L. Gavartin and G. Bersuker, Theoretical Prediction of Intrinsic Self-Trapping of Electrons and Holes in Monoclinic HfO2, Phys. Rev. Lett., 2007, 99, 155504 CrossRef PubMed.
- Z. Wang, C. Brock, A. Matt and K. H. Bevan, Implications of the DFT+U method on polaron properties in energy materials, Phys. Rev. B, 2017, 96, 125150 CrossRef.
- T. Shibuya, K. Yasuoka, S. Mirbt and B. Sanyal, A systematic study of polarons due to oxygen vacancy formation at the rutile TiO2(110) surface by GGA + U and HSE06 methods, J. Phys.: Condens. Matter, 2012, 24, 435504 CrossRef PubMed.
- S. Chrétien and H. Metiu, Electronic Structure of Partially Reduced Rutile TiO2(110) Surface: Where Are the Unpaired Electrons Located?, J. Phys. Chem. C, 2011, 115, 4696–4705 CrossRef.
- T. Liu, Q. Zhao, C. Li, Y. Lyu and M. Dupuis, Photocatalytic Facet Selectivity in BiVO4 Nanoparticles: Polaron Electronic Structure and Thermodynamic Stability Considerations for Photocatalysis, J. Phys. Chem. C, 2019, 123, 20142–20151 CrossRef CAS.
- K. E. Kweon, G. S. Hwang, J. Kim, S. Kim and S. Kim, Electron small polarons and their transport in bismuth vanadate: a first principles study, Phys. Chem. Chem. Phys., 2014, 17, 256–260 RSC.
- R. Defrance, B. Sklénard, M. Guillaumont, J. Li and M. Freyss, Ab initio study of electron mobility in V2O5 via polaron hopping, Solid-State Electron., 2022, 198, 108455 CrossRef CAS.
- A. R. Elmaslmane, M. B. Watkins and K. P. McKenna, First-Principles Modeling of Polaron Formation in TiO2 Polymorphs, J. Chem. Theory Comput., 2018, 14, 3740–3751 CrossRef CAS PubMed.
- H. J. Monkhorst and J. D. Pack, Special points for Brillouin-zone integrations, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13, 5188–5192 CrossRef.
- J. J. Plata, A. M. Márquez and J. F. Sanz, Electron Mobility via Polaron Hopping in Bulk Ceria: A First-Principles Study, J. Phys. Chem. C, 2013, 117, 14502–14509 CrossRef CAS.
- C. W. M. Castleton, A. Lee and J. Kullgren, Benchmarking Density Functional Theory Functionals for Polarons in Oxides: Properties of CeO2, J. Phys. Chem. C, 2019, 123, 5164–5175 CrossRef CAS.
- S. P. Ong, Y. Mo and G. Ceder, Low hole polaron migration barrier in lithium peroxide, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 081105 CrossRef.
- M. D. Johannes, K. Hoang, J. L. Allen and K. Gaskell, Hole polaron formation and migration in olivine phosphate materials, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 115106 CrossRef.
- K. Hoang and M. D. Johannes, Defect chemistry in layered transition-metal oxides from screened hybrid density functional calculations, J. Mater. Chem. A, 2014, 2, 5224–5235 RSC.
- H. Ding, et al., Computational Investigation of Electron Small Polarons in α-MoO3, J. Phys. Chem. C, 2014, 118, 15565–15572 CrossRef CAS.
- S. P. Ong, V. L. Chevrier and G. Ceder, Comparison of small polaron migration and phase separation in olivine LiMnPO4 and LiFePO4 using hybrid density functional theory, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 075112 CrossRef.
- A. B. Bykov, et al., Superionic conductors Li3M2(PO4)3 (M = Fe, Sc, Cr): Synthesis, structure and electrophysical properties, Solid State Ionics, 1990, 38, 31–52 CrossRef CAS.
- M. Catti, S. Stramare and R. Ibberson, Lithium location in NASICON-type Li+ conductors by neutron diffraction. I. Triclinic α′-LiZr2(PO4)3, Solid State Ionics, 1999, 123, 173–180 CrossRef CAS.
- F. Sudreau, D. Petit and J. P. Boilot, Dimorphism, phase transitions, and transport properties in LiZr2(PO4)3, J. Solid State Chem., 1989, 83, 78–90 CrossRef CAS.
- N. Anantharamulu, et al., A wide-ranging review on Nasicon type materials, J. Mater. Sci., 2011, 46, 2821–2837 CrossRef CAS.
- M. Ménétrier, I. Saadoune, S. Levasseur and C. Delmas, The insulator-metal transition upon lithium deintercalation from LiCoO2: electronic properties and 7Li NMR study, J. Mater. Chem., 1999, 9, 1135–1140 RSC.
- C. A. Marianetti, G. Kotliar and G. Ceder, A first-order Mott transition in LiXCoO2, Nat. Mater., 2004, 3, 627–631 CrossRef CAS PubMed.
- J. Kaur, M. L. H. Chandrappa, C. Chen and S. P. Ong, Polaron-induced metal-to-insulator transition in vanadium oxides from density functional theory calculations, Phys. Rev. B, 2023, 107, 125162 CrossRef CAS.
- A. Jain, et al., Commentary: The Materials Project: A materials genome approach to accelerating materials innovation, APL Mater., 2013, 1, 011002 CrossRef.
- Y. Xiao, Y. Wang, S. H. Bo, J. C. Kim, L. J. Miara and G. Ceder, Understanding interface stability in solid-state batteries, Nat. Rev. Mater., 2020, 5, 105–126 CrossRef CAS.
- Y. Zhu, X. He and Y. Mo, First principles study on electrochemical and chemical stability of solid electrolyte–electrode interfaces in all-solid-state Li-ion batteries, J. Mater. Chem. A, 2016, 4, 3253–3266 RSC.
- W. D. Richards, L. J. Miara, Y. Wang, J. C. Kim and G. Ceder, Interface Stability in Solid-State Batteries, Chem. Mater., 2015, 28, 266–273 CrossRef.
- A. Manthiram and J. B. Goodenough, Lithium Insertion into Fe2(MO4)3 Frameworks: Comparison of M = W with M = Mo, J. Solid State Chem., 1987, 71, 349–360 CrossRef CAS.
- D. MiKhailova, et al., Li3V(MoO4)3: A New Material for Both Li Extraction and Insertion, Chem. Mater., 2010, 22, 3165–3173 CrossRef CAS.
- R. A. Marcus and N. Sutin, Electron transfers in chemistry and biology, Biochim. Biophys. Acta, Rev. Bioenerg., 1985, 811, 265–322 CrossRef CAS.
- R. A. Marcus, Electron transfer reactions in chemistry. Theory and experiment, Rev. Mod. Phys., 1993, 65, 599–610 CrossRef CAS.
- N. A. Deskins and M. Dupuis, Electron transport via polaron hopping in bulk TiO2: A density functional theory characterization, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 195212 CrossRef.
- J. R. Reimers and N. S. Hush, Electronic Properties of Transition-Metal Complexes Determined from Electroabsorption Mononuclear Complexes of Ruthenlum(II), J. Phys. Chem., 1991, 95, 9773–9781 CrossRef CAS.
- N. Adelstein, J. B. Neaton, M. Asta and L. C. D. Jonghe, Density functional theory based calculation of small-polaron mobility in hematite, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 245115 CrossRef.
- R. Nelson, et al., LOBSTER: Local orbital projections, atomic charges, and chemical-bonding analysis from projector-augmented-wave-based density-functional theory, J. Comput. Chem., 2020, 41, 1931–1940 CrossRef CAS PubMed.
- S. Maintz, V. L. Deringer, A. L. Tchougréeff and R. Dronskowski, LOBSTER: A tool to extract chemical bonding from plane-wave based DFT, J. Comput. Chem., 2016, 37, 1030–1035 CrossRef CAS PubMed.
- K. Jun, et al., Lithium superionic conductors with corner-sharing frameworks, Nat. Mater., 2022, 21, 924–931 CrossRef CAS PubMed.
- B. Lang, B. Ziebarth and C. Elsässer, Lithium Ion Conduction in LiTi2(PO4)3 and Related Compounds Based on the NASICON Structure: A First-Principles Study, Chem. Mater., 2015, 27, 5040–5048 CrossRef CAS.
- X. Zhao, et al., Computational screening and first-principles investigations of NASICON-type LixM2(PO4)3 as solid electrolytes for Li batteries, J. Mater. Chem. A, 2018, 6, 2625–2631 RSC.
- H. Wang, et al., Rational design and synthesis of LiTi2(PO4)3-xFx anode materials for high-performance aqueous lithium ion batteries, J. Mater. Chem. A, 2016, 5, 593–599 RSC.
- J.-Y. Luo, L.-J. Chen, Y.-J. Zhao, P. He and Y.-Y. Xia, The effect of oxygen vacancies on the structure and electrochemistry of LiTi2(PO4)3 for lithium-ion batteries: A combined experimental and theoretical study, J. Power Sources, 2009, 194, 1075–1080 CrossRef CAS.
- C. A. Marianetti, D. Morgan and G. Ceder, First-principles investigation of the cooperative Jahn-Teller effect for octahedrally coordinated transition-metal ions, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 224304 CrossRef.
- A. J. E. Rettie, W. D. Chemelewski, D. Emin and C. B. Mullins, Unravelling Small-Polaron Transport in Metal Oxide Photoelectrodes, J. Phys. Chem. Lett., 2016, 7, 471–479 CrossRef CAS PubMed.
- A. Moradabadi and P. Kaghazchi, Effect of Strain on Polaron Hopping and Electronic Conductivity in Bulk LiCoO2, Phys. Rev. Appl., 2017, 7, 064008 CrossRef.
- S. Loftager, P. Garcia Fernandez, J. A. Aramburu, M. Moreno and J. M. Garcia-Lastra, Stability and Polaronic Motion of Self-Trapped Holes in Silver Halides: Insight through DFT+U Calculations, J. Phys. Chem. C, 2016, 120(16), 8509–8524 CrossRef CAS.
- C. Ku and P. H.-L. Sit, Ab Initio Study of Electron and Hole Polaron Transport in Cobalt(II,III) Oxide Using Oxidation-State Constrained Density Functional Theory, J. Phys. Chem. C, 2023, 127, 13266–13275 CrossRef CAS.
- A. Rossbach, F. Tietz and S. Grieshammer, Structural and transport properties of lithium-conducting NASICON materials, J. Power Sources, 2018, 391, 1–9 CrossRef CAS.
- P. Zhang, et al., Water-stable lithium ion conducting solid electrolyte of the Li1.4Al0.4Ti1.6-x Gex(PO4)3 system (x=0∼1.0) with NASICON-type structure, Solid State Ionics, 2013, 253, 175–180 CrossRef CAS.
- S. Xuefu, et al., High Lithium-Ion-Conducting NASICON-Type Li1+xAlxGeyTi2-x-y(PO4)3 Solid Electrolyte, Front. Energy Res., 2016, 4, 12 Search PubMed.
- J. M. Garcia-Lastra, J. S. G. Myrdal, R. Christensen, K. S. Thygesen and T. Vegge, DFT+U Study of Polaronic Conduction in Li2O2 and Li2CO3: Implications for Li–Air Batteries, J. Phys. Chem. C, 2013, 117, 5568–5577 CrossRef CAS.
- V. L. Deringer, A. L. Tchougréeff and R. Dronskowski, Crystal Orbital Hamilton Population (COHP) Analysis As Projected from Plane-Wave Basis Sets, J. Phys. Chem. A, 2011, 115, 5461–5466 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ta00367a |
| This journal is © The Royal Society of Chemistry 2025 |