
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCapturing and labeling CO2 in a jar: mechanochemical 17O-enrichment and ssNMR study of sodium and potassium (bi)carbonate salts †
Austin Peach *a,
Nicolas Fabreguea,
Célia Errea,
Thomas-Xavier Métroa,
David Gajanb,
Frédéric Mentink-Vigierc,
Faith Scottc,
Julien Tréboscd,
Florian Vorone,
Nicolas Patrisf,
Christel Gervaisg and
Danielle Laurencin*a
*a,
Nicolas Fabreguea,
Célia Errea,
Thomas-Xavier Métroa,
David Gajanb,
Frédéric Mentink-Vigierc,
Faith Scottc,
Julien Tréboscd,
Florian Vorone,
Nicolas Patrisf,
Christel Gervaisg and
Danielle Laurencin*a
aICGM, Univ. Montpellier, CNRS, ENSCM, Montpellier, France. E-mail: austin.peach@umontpellier.fr; danielle.laurencin@umontpellier.fr
bCRMN Lyon, UMR 5082 (CNRS, ENS Lyon, Université Lyon 1), Villeurbanne, France
cNational High Magnetic Field Laboratory (NHMFL), Florida State University, Tallahassee, Florida, USA
dUniversité de Lille, CNRS, INRAE, Centrale Lille, Université d'Artois FR2638–IMEC–Institut Michel Eugène Chevreul, Lille, France
eOSU OREME, UAR 3282, Université de Montpellier, CNRS, IRD, INRAE, Sète, Montpellier, France
fHydroSciences Montpellier, UMR 5151, CNRS, IRD, Université de Montpellier, Montpellier, France
gLCMCP, UMR 7574, Sorbonne Université, CNRS, Paris, France
First published on 21st April 2025
Abstract
With the rapid increase in temperatures around the planet, the need to develop efficient means to reduce CO2 emissions has become one of the greatest challenges of the scientific community. Many different strategies are being studied worldwide, one of which consists of trapping the gas in porous materials, either for its short- or long-term capture and storage, or its re-use for the production of value-added compounds. Yet, to further the development of such systems, there is a real need to fully understand their structure and properties, including at the molecular-level following the physisorption and/or chemisorption of CO2 (which can lead to various species, including carbonate and bicarbonate ions). In this context, 17O NMR naturally appears as the analytical tool of choice, because of its exquisite sensitivity to probe subtle differences in oxygen bonding environments. To date, it has scarcely been used, due to the very low natural abundance of 17O (0.04%), and the difficulty in purchasing or obtaining commercial 17O-labeled compounds adapted to such investigations (e.g., 17O–CO2(g), or 17O-enriched Na- and K-(bi)carbonate salts, which can be readily transformed into CO2). Herein, we demonstrate how, using mechanochemistry, it is possible to enrich with 17O a variety of Na- and K-(bi)carbonate salts in a fast, economical, scalable, and user-friendly way. The high enrichment levels enabled recording the first high-resolution 17O ssNMR spectra of these phases at different temperatures and magnetic fields. From these, the typical spectral signatures of (bi)carbonate ions could be obtained, showing their strong sensitivity to local environments and dynamics. Lastly, we show how thanks to the selective 17O-labeling, other aspects of the reactivity of carbonates in materials can be unveiled using in situ 17O ssNMR. In the long run, it is expected that this work will open the way to more profound investigations of the structure and properties of carbon capture and storage systems, and, more generally speaking, of functional materials containing carbonates.
1 Introduction
With temperatures constantly rising around the planet, and climate disasters increasing in frequency and intensity, it is urgent to rapidly develop ways of mitigating the causes of global warming, and notably anthropogenic carbon dioxide (CO2) emissions.1,2 In this context, much research has been dedicated to developing sustainable technologies for substantially reducing the net flow of CO2 in the atmosphere by capturing it directly at industrial sources, with the aim of storing it either permanently, or temporarily. Several different materials and processes have been investigated for this purpose.3,4 On one hand, solutions and sorbents involving amine functionalities have been studied for decades, with CO2 reacting to form ammonium carbamates or (bi)carbonates. However, their toxicity, corrosivity, and/or limited stability and recyclability were demonstrated to be problematic.5 On the other hand, the potential of mineral carbonation has also been widely studied, including at the industrial scale: it consists of injecting and sequestering CO2 into “reactive” natural rocks, so that it transforms into carbonate minerals (e.g., calcite and dolomite).3,6,7 Along the same line, it has been proposed to use simple metal oxides like CaO as CO2 sorbents by formation of CaCO3,8 with applications tested up to the pilot scale. Last but not least, several other classes of materials and sorbents have been investigated for CO2 capture,9 including metal organic frameworks (MOFs), which, depending on their structure, can incorporate CO2 in their pores by physisorption and/or chemisorption.10–12Although a large diversity of systems was studied and developed for CO2 capture, many aspects of the reactivity of this small molecule still deserve to be studied to ensure the optimal design and understanding of hitherto carbon capture systems. Indeed, beyond the identification of the CO2 speciation in the final material, which can involve either CO2 molecules upon physisorption, or (bi)carbonate/carbamate ionic entities upon chemisorption, numerous points are still obscure. Among these, the questions of the impact of temperature, pressure, and moisture on the binding modes of CO2, its speciation, and the local molecular motions it undergoes, still need to be investigated in detail, in view of helping conceive and improve, in the long run, the carbon-capture properties of these systems.
As a local analytical probe, NMR spectroscopy naturally appears as perfectly suited for studying CO2 environments within carbon-capture materials, and it has already been used in numerous investigations.13–15 To date, the vast majority of NMR studies have concerned carbon-13,15 due to the higher sensitivity of this spin-1/2 nucleus. Yet, oxygen-17, the only stable isotope of oxygen which can be analyzed by NMR, is also highly attractive:16 (i) its chemical shift range exceeds 1000 ppm (in comparison to only ca. 200 ppm for 13C); (ii) it is a spin-5/2 quadrupolar nucleus, meaning that complementary information on its local electronic environment can be derived from the quadrupolar parameters CQ and ηQ (which is not the case for 13C, as it is only spin-1/2); (iii) the 17O lineshape is exquisitely sensitive to molecular-level dynamics, making variable-temperature 17O NMR studies richly informative on local motions;17–21 (iv) while only one 13C resonance is expected per CO2 molecule or (bi)carbonate/carbamate species attached to a surface, 2 (or 3) distinct 17O resonances can potentially be observed, meaning that the latter can garner more detailed information on the bonding, reactivity, and adsorption modes of CO2.
To the best of our knowledge, 17O NMR has only been used a small number of times in the context of carbon capture.14,18,19,22–25 Indeed, the meagre natural abundance of 17O (0.04%, compared to 1.1% for 13C) is a real obstacle. In the previous works, only two were performed at natural abundance,19,23 by using (ultra)-high magnetic fields (i.e. 850 MHz to 1.0 GHz NMR instruments), relatively large amounts of sample, and long acquisition times (up to 42 h). However, in these conditions, only 1D NMR spectra could be recorded, which, in the case of ssNMR, hindered the deployment of high-resolution analyses (which are often required to produce robust results). In contrast, when the solid-state NMR investigations were performed on isotopically enriched species, with the initial 17O-labeling either on the CO2 gas,14,19,22 the material of interest,24 a labeled hydroxide salt,18 or the surrounding medium (H2O),25 more profound NMR studies were performed, leading to more extensive information on the structure and reactivity of the systems under investigation. Yet, with limited 17O-enriched precursors commercially available (the main two being 17O-enriched water and 17O–O2 gas), and limited availability of 17O-enriched CO2 gas, any prospect of a widespread application of 17O ssNMR for the engineering of materials and sorbents for CO2 capture appears restricted.
Spurred by this general context, we decided to focus on developing efficient and cost-effective routes for enriching in 17O carbonate and bicarbonate salts of sodium (i.e., Na2CO3·H2O, Na2CO3 and NaHCO3), and potassium (i.e., K2CO3·1.5H2O and KHCO3). Indeed, three key advantages can be seen in being able to enrich these compounds. First, they are known to decompose upon heat treatment or acidic exposure by releasing CO2,26–29 which makes them attractive sources for the production of 17O-enriched CO2. Second, they encompass a variety of local environments for (bi)carbonate ions, which can be representative of those expected within materials developed for carbon capture. Thus, acquiring high-resolution 17O ssNMR spectra of these model phases can be seen as a prerequisite for deriving sound conclusions on the structure and dynamics of CO2-related species within more complex materials. Last, beyond CO2-capture applications, Na- and K-(bi)carbonates are also key precursors for the synthesis of many different types of (bio)materials, meaning that their 17O enrichment could also be of value for the investigation of a gamut of compounds.
In this manuscript, we first demonstrate how, using mechanochemistry, it is possible to enrich the aforementioned Na- and K-(bi)carbonate salts in 17O, in a highly efficient way. Then, the high-resolution 17O ssNMR spectra of the enriched compounds are presented, revealing their strong dependency to the local structure around the (bi)carbonates, and to temperature. Finally, thanks to the high 17O-enrichment, we show how further aspects of the reactivity of carbonates can be brought to light by in situ 17O ssNMR, which should help propound investigations of the properties of carbon capture materials and/or functional materials containing carbonates.
2 Results and discussion
2.1 Mechanochemical isotopic enrichment
To the best of our knowledge, two main synthetic approaches have been used so far in the literature for the 17O-labeling of carbonate salts for ssNMR: (i) the equilibration of carbonate ions in the presence of labeled water (to eventually form enriched Ca- or mixed Ca, Mg-carbonates),30,31 and (ii) the quantitative reaction of CO2 gas with pre-labeled LiOH (to form enriched Li2CO3).18,32 In the former case, long reaction times (ca. 1 week at 90 °C)30 and/or an excess of expensive 17O-labeled water were used to ensure sufficient labeling. In the latter case, the synthesis was somewhat constraining, due to the manipulation of n-butyllithium in dry THF, and of CO2 gas at 100 °C.18,32 From what it appears, these procedures were not optimized in terms of synthetic yields and 17O-enrichment levels, and their scalability or transposability to other (bi)carbonate salts was not reported. Moreover, these protocols were not widely adopted by other synthetic chemists or spectroscopists, further demonstrating the need to develop more efficient enrichment procedures in terms of time, cost, and practicality.As an alternative, we looked into enriching (bi)carbonate salts by mechanochemistry, using microliter quantities of labeled water (H2*O), in a “liquid assisted grinding” type of approach.33–35 Initial attempts were performed by simply grinding Na2CO3 in presence of stoichiometric amounts of H2*O. Indeed, considering that mechanochemical reactions can help accelerate reactions, due to the highly concentrated conditions in the milling jar, we considered the possibility of having a direct isotopic enrichment, by an “equilibration” type of reaction. These first tests were carried out with 18O-enriched water (which is ca. 30-fold cheaper than 17O-enriched water, when considering their highest enrichments commercilaly available), and the ball-milling was performed for 30 to 90 minutes, with stainless steel jars and beads. The products recovered after milling were dried and analyzed by powder X-ray diffraction (pXRD) to confirm phase purity, followed by mass-spectrometry (MS) and/or IR spectroscopy to determine if 18O-labeling had occurred. In the case of IR, no clear isotope-shift was observed, suggesting highly inefficient labeling (if any). Quantitative Isotope-Ratio Mass Spectrometry (IRMS) analyses confirmed that the maximum 18O level was less than 0.6% (when starting from 99% 18O-enriched water), which corresponds to a mere 3-fold increase compared to the 18O natural abundance (0.2%), and remains well beneath the maximum value of ca. 40% (calculated for a full scrambling of the oxygen isotopes in the experimental conditions used – see ESI 1†). Although prolonged milling times or increased amounts of labeled water may help further enhance the enrichment level, such experiments were not attempted, as they would decrease the attractiveness of the procedure (i.e., due to the significantly-increased experimental times, cost-prohibitive 17O-labeling, and/or contaminations from the jar and beads upon longer milling).
A second synthetic approach was thus considered (Fig. 1), which consisted of performing a one-pot quantitative transformation by mechanochemistry. The general idea was to simultaneously introduce in the milling jar N,N′-carbonyl-diimidazole (CDI), 17O-labeled water, and an alkali metal base (e.g., NaOH or KOH), in order to hydrolyze the CDI precursor to form 17O-labeled CO2,36 and capture this gas with a base to form a (bi)carbonate salt, as CO2 is well known to react with bases (whether solvated in aqueous solutions, or present directly in their solid form).37,38 Initial tests were carried out with non-labeled water (to optimize the synthetic yield), before moving on to 18O-enriched water (to evaluate the extent of enrichment), and finally 17O-enriched water (in view of 17O NMR analyses). The amount of base was adapted to ensure the direct formation of either a bicarbonate or a carbonate salt in the jar. In all cases, full consumption of the NaOH (or KOH) and CDI precursors was observed after only 30 minutes of milling. This was attested by IR spectroscopy (see ESI, Fig. S1–S4†), through (i) the disappearance of the vibration bands characteristic of the reagents (especially the OH stretching band), and (ii) the appearance of the vibration bands of the products (especially the imidazole by-product). The latter was removed during a work-up step, by dissolution in ethanol. The final (bi)carbonate salts were dried, and subsequently characterized by pXRD, IR, and 13C ssNMR, confirming the formation of phase-pure forms of NaHCO3, KHCO3, Na2CO3·H2O, and K2CO3·1.5H2O (see ESI Fig. S5–S11†). A pure phase of anhydrous Na2CO3 was isolated by heat-treatment of Na2CO3·H2O for a few hours at 100 °C.
 | ||
| Fig. 1 17O-Labeling of Na and K (bi)carbonate salts by mechanochemistry, using a CDI-based procedure. | ||
Comparison of the IR spectra of samples prepared using non-labeled, 17O-labeled, and 18O-labeled water provided evidence of the successful labeling of the (bi)carbonate salts. This is illustrated in Fig. 2, which highlights some of the spectral regions with variations caused by 17O or 18O isotope shifts. Notably, regarding the carbonate salts, the splittings at ca. 1065 cm−1 for Na2CO3·H2O and K2CO3·1.5H2O (ν1 stretching mode), and at ca. 1775 cm−1 for Na2CO3 (tentatively assigned to the 2 × ν2 harmonic)42 show contributions from the different C16On*O3−n2− (n = 1, 2 and 3) isotopologues.43–46 In the case of 18O-labeled salts, the integration of the relative intensities of these IR bands enabled to estimate the 18O-enrichment level to be above 25% (when starting from 99% 18O-labeled water). Analyses of 13C isotope shifts in solution NMR confirmed this value: for an 18O-labeled Na2CO3 phase (prepared from 99% 18O-labeled water), the 18O-enrichment level was determined to be ca. 30% (see Fig. S12†). IRMS studies on 18O-labeled Na-carbonates were also performed, further attesting to a significant 18O-labeling when using the CDI-based protocol (see ESI S1†). Importantly, these measurements imply that an enrichment yield of ca. 75% can be achieved with this procedure (see Fig. S12†). Extrapolating to 17O, this means that when using 70%-17O-labeled water, an enrichment level of ca. 20% can be reached for the (bi)carbonates, which is 500 times more than natural abundance, and amply sufficient for ssNMR analyses, as will be shown below.
 | ||
| Fig. 2 Experimental IR spectra, zooming into regions of interest for unlabeled (blue), 17O-labeled (red), and 18O-labeled (green) Na- and K-(bi)carbonate salts, synthesized by mechanochemistry using the procedure described in Fig. 1. The IR spectra of Na2CO3, Na2CO3·H2O, and K2CO3·1.5H2O indicate the successful enrichment in 17O/18O by the splitting of the IR bands, due to the presence of different isotopologues in the sample (as illustrated in the bottom right corner, in which 17O/18O-enriched oxygens are shown in red). The dashed lines in black in the IR spectra of NaHCO3 and KHCO3 denote IR bands in the unlabeled purified product, which are shifted with respect to those labeled with either 17O/18O (assignments of these bands can be found in previous literature).39–41 The data shown here is for products which were mechanochemically enriched using H218O (99% 18O-labeled), or H217O (40% 17O-labeled for Na2CO3·H2O and NaHCO3, and 90% 17O-labeled for KHCO3 and K2CO3·1.5H2O). The Na2CO3 phase analyzed here was prepared by dehydration of a monohydrate phase, for which labeling had been done by the CDI-based ball-milling procedure, using 70% 17O-labeled water. | ||
Overall, the CDI-based synthesis proposed is particularly efficient for the production of 17O/18O-labeled Na- and K-(bi)carbonate salts, enabling to isolate phase-pure compounds in half a day (work-up included), with a high enrichment level and yield. The protocols are reproducible and user friendly (performed under ambient temperature and pressure), and do not require the use of highly toxic reagents or constraining procedures (vide supra). Herein, syntheses are described in quantities enabling the isolation of up to ca. 130 mg of product (Table S1†). Yet, experiments can be readily adapted to produce larger amounts of labeled products, by performing reactions simultaneously in two or more jars, increasing the amount of sample per jar, and/or by using larger volume reactors. In the course of our investigations, by simply tripling the initial amount of reagents, we were able to eventually obtain 400 mg of 17O-enriched Na2CO3 in just over half a day, thereby making the CDI-based 17O/18O-labeling far more attractive than the previously described carbonate-enrichment schemes.18,30,32 Although the full details of the enrichment mechanism would deserve further investigation (to rationalize the relative proportions between the isotopologues and the mode of formation of the triply labeled carbonates), at this stage our focus turned to pushing forward 17O ssNMR studies in view of the study of materials containing (bi)carbonate-related species.
2.2 High-resolution 17O ssNMR of 17O-labeled sodium and potassium (bi)carbonates
The 17O ssNMR spectra of the five Na- and K-(bi)carbonate salts were first acquired at 14.1 T (i.e., 600 MHz instrument) under magic angle spinning (MAS), using standard analytical conditions. For all phases, the successful enrichment enabled the observation of a 17O NMR signal in just one scan, with some of the spectra shown in Fig. 3A taking as little as 30 minutes (see Table S4† for experimental details). This is a major improvement in comparison to a recently reported work, where the natural abundance spectra of the two K salts (i.e., KHCO3 and K2CO3·1.5H2O) required more than one day to acquire at higher magnetic field.19 | ||
Fig. 3 Experimental 17O{1H} MAS NMR spectra acquired at (A) B0 = 14.1 T (600 MHz instrument) and (B) B0 = 28.2 T (1.2 GHz instrument) on enriched bicarbonates (NaHCO3 and KHCO3), carbonate hydrates (Na2CO3·H2O and K2CO3·1.5H2O), and anhydrous sodium carbonate (Na2CO3). Spinning rates and actual sample temperatures are indicated next to the spectra. In (A), the spectral regions expected for HCO3−/CO32− (200 to 40 ppm) and crystalline H2O (0 to −120 ppm) are shown in orange and light blue, respectively. In (B), only the HCO3−/CO32− spectral region is shown, in which the higher resolution achieved at 28.2 T enables the distinction between C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–OH signals. Spinning sidebands are denoted with an asterisk (*), 17O NMR signals arising from crystalline water with a dagger (†), and from satellite transitions with a cross (×). Further details on acquisition parameters are available in the ESI Tables S4 and S5.† O and C–OH signals. Spinning sidebands are denoted with an asterisk (*), 17O NMR signals arising from crystalline water with a dagger (†), and from satellite transitions with a cross (×). Further details on acquisition parameters are available in the ESI Tables S4 and S5.† | ||
17O NMR signals were observed in two key spectral regions: (i) between 200 and 40 ppm for all Na- and K-(bi)carbonate salts (Fig. 3A, orange zone), which is in the zone expected for carbonate-like environments;18,19,25,30 and (ii) between 0 and −120 ppm for the two hydrates (Fig. 3A, blue zone), which corresponds to the zone of crystallographic water.47–49 The latter signals suggest that some of the excess of enriched water used in the syntheses was incorporated into the hydrated crystal structures. When focussing on the carbonate zone (Fig. 3A, orange), the 17O NMR spectra appeared either as distorted gaussian lineshapes (Na2CO3·H2O and K2CO3·1.5H2O), or as broader asymmetric lineshapes featuring discontinuities typical of second-order quadrupolar central transition (CT) patterns (NaHCO3, KHCO3, and Na2CO3). Upon closer examination of the spectra of the two bicarbonates, “steps” could be observed on the left part of the signal, indicating an overlap of several 17O NMR resonances, as expected from the reported crystal structures (which both have 3 crystallographically distinct oxygen environments: 2 CO and 1 C–OH). Yet, no signature of the C–OH group of the HCO3− ions was resolved directly by 1D 17O ssNMR at this magnetic field.
To achieve better resolution in the bicarbonate/carbonate region, the 17O MAS ssNMR spectra of Na2CO3, K2CO3·1.5H2O, NaHCO3 and KHCO3 (Fig. 3B) were acquired at 28.2 T. Indeed, a significant gain in resolution can be achieved for half-integer quadrupolar nuclei when working at high magnetic fields (B0), since the broadening caused by the second-order quadrupolar interaction is inversely proportional to B0.50 For Na2CO3 and K2CO3·1.5H2O, the general appearance of the 17O NMR lineshapes at 28.2 T remained fairly similar to the one at 14.1 T, albeit much narrower. More importantly, for the bicarbonates, two spectral regions could be resolved (Fig. 3B), which can be assigned to the oxygens belonging to the carbonyl (CO) and hydroxyl (C–OH) oxygens of HCO3−. At this stage, we note that the C–OH resonance was not observed in the recently reported natural abundance 17O NMR spectrum of KHCO3, which may be due to the lack of sensitivity (non-labeled sample) and/or the measurement conditions used (i.e. difference in pulse sequence and/or temperature, see Fig. S13†).19
In order to extract typical 17O NMR parameters of bicarbonates (which are needed when interpreting the NMR data of more complex systems), the NMR spectra acquired at 28.2 T were fitted, considering three sites (2 CO and 1 C–OH, Fig. S14†). Regarding the CO region (Fig. S14†, green and blue-shaded resonances), the assignment of the resonances was made possible by using 1D and 2D 1H–17O HMQC/INEPT NMR experiments (Fig. S15 and S16†). The NMR parameters were then determined to be as follows, for KHCO3 and NaHCO3, respectively (reported in tabulated form in Table S10†):
*O1 (CO): δiso = 190.6 and 175.9 ppm, CQ = 7.30 and 7.21 MHz, and ηQ = 0.73 and 0.83;
*O2 (CO): δiso = 178.7 and 171.5 ppm, CQ = 6.75 and 6.58 MHz, and ηQ = 0.64 and 1.00;
*O3 (C–OH): δiso = 146.9 and 137.7 ppm, CQ = 7.30 and 7.65 MHz, and ηQ = 0.21 and 0.39.
Here, we note that an early 17O nuclear quadrupole resonance (NQR) study of NaHCO3 and KHCO3 at 291 K had reported similar CQ and ηQ values for O2 and O3, alongside the same assignment for these sites (see Table S10†).51 Yet, to the best of our knowledge, it is the first time that experimental values of δiso for C–OH groups in these bicarbonate salts are reported. Interestingly, the δiso values for C–OH and CO groups were all found to be higher for KHCO3 than NaHCO3. Such a trend is in line with an early computational study by Wong et al. on metal oxalates, where the 17O isotropic chemical shifts were found to globally increase along the alkali-metal series (i.e., Li+ < Na+ < K+ < Rb+ < Cs+).52
Taken together, these 17O NMR spectra of Na- and K-(bi)carbonatesalts demonstrate several significant points. On one hand, the efficient 17O-labeling using mechanochemistry enabled both improved sensitivity and spectral resolution, allowing the acquisition of high-quality 1D and 2D ssNMR spectra in minutes, and providing direct evidence of the spectral signature of the C–OH group in the bicarbonates. On the other hand, the full spectral assignments of KHCO3 and NaHCO3 enabled to demonstrate that the range of variation of 17O NMR parameters for (bi)carbonate anions extends beyond the recently reported values,19 especially in the case of C–OH groups (Fig. S17†), a point which was further supported by DFT calculations (Table S9†). Such features are highly promising for future studies on more complex (bi)carbonate containing materials by 17O ssNMR. Despite all the above advantages, only the spectra of the two bicarbonate salts were well resolved at 28.2 T, with the number of distinct 17O NMR resonances in agreement with the crystal structures. In stark contrast, for Na2CO3·H2O and K2CO3·1.5H2O, gaussian-like narrow spectra were obtained, suggesting the presence of molecular-level motions around the anions (Fig. 3). Moreover, for anhydrous Na2CO3, the two crystallographically inequivalent oxygen sites could not be resolved under the current measurement conditions, also suggesting the presence of molecular-level dynamics in this material.
To “freeze” these motions, 17O MAS NMR spectra of the Na- and K-carbonate salts were recorded at 14.1 T under ultra-low temperatures (i.e., sample temperatures ca. 105 K) as shown in Fig. S18† (blue spectra). In these conditions, all spectra now appear as well-defined 17O NMR second-order quadrupolar lineshapes, which is particularly noteworthy for Na2CO3·H2O and K2CO3·1.5H2O. This highlights that for materials developed for carbon-capture, ultra-low temperature 17O ssNMR analyses can provide additional means for identifying and resolving the distinct (bi)carbonate local environments. Moreover, it shows that a precise control of the actual sample temperature is needed, to enable robust comparisons of the (bi)carbonate local structure and motions between different samples. Based on these different observations, we then decided to study how 17O ssNMR may unveil other aspects of carbonate reactivity, in conditions closer to “real-life” application of the materials.
2.3 Reactivity of carbonates in the presence of water
In the development of novel systems for carbon capture, the study of the influence of water on the physi- and chemisorption of CO2 has been shown to be critical. This holds true not only for purely inorganic sorbents like zeolites and layered double hydroxides,53,54 but also for porous hybrid materials like MOFs.55,56 A wide variety of experimental and computational tools have thus been used to investigate the local structure and dynamics around H2O and CO2 (or (bi)carbonate ions), including using 17O NMR.19,25 Notably, some studies have shown 17O isotope exchanges taking place between CO2 (or carbonates) and the water present within the interlayer spacings, pores, or at the surface of the material.14,19,25 Yet, despite that such isotopic exchanges are widely studied in Earth sciences (essentially looking at the 18O isotopes),43–45 similar analyses are still underexplored in the investigations on carbon-capture materials. Given that the signatures of water and (bi)carbonates can be distinctly resolved by 17O NMR, we decided to perform in situ NMR analyses on the hydrated carbonates described above, to try to probe isotopic exchange processes between water and (bi)carbonates. More precisely, for the preliminary study described herein, we focused on the monohydrate salt Na2CO3·H2O. Selectively-labeled crystalline Na2CO3·H2O was synthesized by mechanochemistry, with 17O enrichment on the water only (see ESI S2† for details), to investigate the isotope transfer to the carbonates. The identity and purity of the starting sample were verified by pXRD and IR (Fig. S19†), and the lack of any substantial labeling of the initial carbonate was also confirmed by the latter technique.In situ variable temperature 17O NMR experiments were recorded on the freshly prepared Na2CO3·H217O phase (Fig. 4). A broad 17O NMR signal characteristic of crystalline H2O was observed between 0 and −120 ppm. No signals were present in the spectral region between 200 and 40 ppm, confirming the absence of any significant (bi)carbonate labeling (Fig. 4B, bottom spectrum). Following this, the sample temperature was increased in increments of 20 K, with spectra recorded at each step under identical conditions, up to a maximum sample temperature of 370 K (ca. 100 °C). A sub-set of these spectra is shown in Fig. 4, while the full range of 17O ssNMR experiments are in Fig. S20.† The 17O NMR spectrum recorded at 370 K was found to be nearly identical to the one initially recorded at ca. 310 K. However, when the sample was then “quenched” to ca. 290 K, we observed the appearance of a second signal centered around 119 ppm (i.e., in the region characteristic of (bi)carbonates). More specifically, this signal was found precisely at the resonance of the carbonate ions of Na2CO3·H2O at ambient temperature (Fig. S21†). When the sample was then reheated to 370 K, the disappearance of this carbonate peak was observed, and the only signal visible was that of crystalline H2O. Following these first NMR analyses (performed within a total timeframe of ca. 5 hours), the rotor was cooled back down to room temperature and weighed, showing that no significant weight loss had occurred. The sample was unpacked and re-analyzed by pXRD and IR, showing only the distinct fingerprints of the starting monohydrate salt (Fig. S19†).
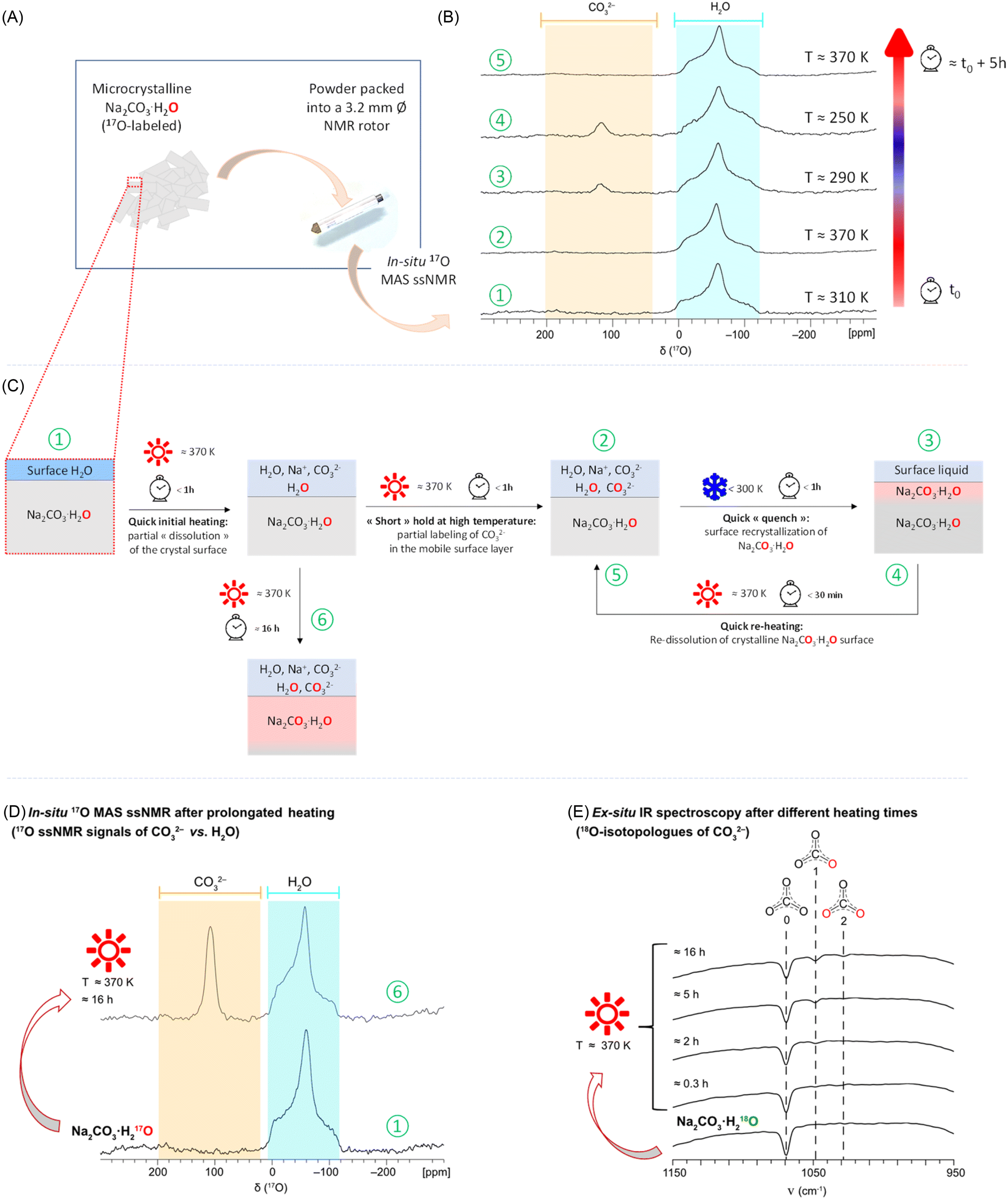 | ||
| Fig. 4 (A) Sample environment for the in situ 17O ssNMR study on Na2CO3·H217O (with initial selective labeling on the water). (B) Experimental VT 17O{1H} MAS NMR spectra acquired at B0 = 14.1 T (600 MHz instrument), using the DFS-echo sequence, with sample temperatures indicated on the right for each spectrum. The spectral regions for CO32− (200 to 40 ppm) and crystalline H2O (0 to −120 ppm) at this field are shown in orange and light blue, respectively. (C) Schematic representation of the phenomena occurring at the crystal surface accounting for the observations made, with the numbers circled in green referring to the spectra shown in (B) and (D). (D) 17O ssNMR spectra recorded in situ showing that when holding the sample at 370 K for ca. 16 hours, the carbonate signal is observed, indicating that a substantial amount of labeling has occurred by progressive dissolution/recrystallization of Na2CO3·H2O (see ESI Tables S11 and S12† for further details on acquisition parameters). (E) IR spectra acquired after different time-points on a sample of Na2CO3·H218O packed into a 3.2 mm rotor and heated in an oven at ca. 370 K, revealing the progressive increase in labeling of the carbonate overtime (see ESI† 3 for details). | ||
The 17O ssNMR spectra shown in Fig. 4B demonstrate that (i) an isotopic exchange between the enriched water and the initially non-labeled carbonates of Na2CO3·H2O has taken place under the experimental conditions used, and (ii) 17O-labeled carbonate ions may not always be observable in 17O MAS NMR (as shown here at 370 K), despite their presence in the sample. Complementary analyses were carried out to try to understand these observations, and see how they may be possibly due to the sample preparation and/or measurement conditions. First, when leaving the sample at ca. 370 K for a longer period of time (overnight, ≈16 hours), the 17O ssNMR spectrum revealed an increase in relative intensity of the signal centered at ca. 119 ppm (characteristic of enriched carbonates within the Na2CO3·H2O crystal structure), as shown in Fig. 4D. This demonstrates that the time spent at high temperature allows for a more significant isotope transfer from the labeled water towards the carbonates. This observation was further complemented by IR spectroscopy analyses, by heating up to ca. 370 K a freshly prepared Na2CO3·H218O sample (selectively labeled in 18O on the water, in order to be able to follow more distinctly the signals of the carbonate isotopologues), and then analyzing it by IR after different periods of time (see ESI S3† for the full experimental details). While no clear enrichment could be detected after only 20 minutes of heating, distinct carbonate vibration bands of the 18O-labeled isotopologues started to appear after 2 hours, which further increased over time (e.g., after 5 and 16 hours, Fig. 4E). In contrast, when the latter sample was left at lower temperatures overnight (e.g. 310 K), no such 18O-labeling was observed by IR spectroscopy (see Fig. S22†). Second, when analyzing by 17O NMR the phase which had been heated at ca. 370 K overnight, but using other NMR acquisition conditions (e.g. direct excitation analyses under MAS or static conditions, instead of an echo), resonances from more mobile 17O-enriched species became visible, namely H2O (at ca. 0 ppm), and solvated CO32− ions (at ca. 190 ppm)57 (Fig. S23†). Such resonances were not clearly visible in the initial in situ study (spectra shown in Fig. 4B).
From all the above observations, a possible explanation to the appearance/disappearance of the Na2CO3·H2O carbonate signals in Fig. 4B could be the following. Upon the first heating of the sample up to 370 K, a partial surface-dehydration of Na2CO3·H2O takes place, leading to the release of labeled water, which, in the confined environment of the NMR rotor, adds on to the thin water layer already present at the surface of the crystallites, in which the surface Na+ and CO32− ions dissolve. While holding at 370 K, an isotopic exchange process is able to take place in this “liquid-like” layer, leading to the formation of enriched carbonates within a few hours (as shown by IR spectroscopy, Fig. 4E). Because the concentration of these enriched carbonates is initially low (if the sample is only left for less than 30 minutes at 370 K), these are not directly visible at ca. 190 ppm in the 17O MAS NMR spectra obtained at high temperature, when the NMR analyses are performed using an echo type of sequence as in Fig. 4B. However, if the sample is then quenched back to low temperature, the dissolved sodium and carbonate ions recombine to form an enriched Na2CO3·H2O phase at the surface of the initial crystals, with not only H2O but also a carbonate enrichment now present (signal at 119 ppm). If rapidly heated back up to 370 K, this “enriched” surface layer redissolves, releasing the small amount of labeled CO32− ions in the liquid-like environment, making them become, again, difficult to detect under analysis conditions shown in Fig. 4B. Though, if the sample is maintained at 370 K overnight, a more significant labeling of the carbonates occurs, with resonances characteristic of enriched “core” carbonate ions (119 ppm) and solvated ones (190 ppm) now detectable by combining Hahn echo and direct-excitation 17O ssNMR experiments (see Fig. 4D and S23†).
Further investigations would be needed to pinpoint how the isotopic exchange occurs at the molecular scale, and to what extent it depends on the crystallinity and size-distribution of the particles composing the initial powder (as these parameters were found to have a significant effect on the dehydration of Na2CO3·H2O),58,59 as well as the sample packing and heating conditions. This would require more extensive analyses, including by variable-temperature 13C and 23Na NMR, but also variable-temperature pXRD and electron microscopy, which is beyond the scope of the present work.
Albeit preliminary, this in situ 17O NMR study has several implications. First, it shows that thanks to a selective labeling of the monohydrate, the existence of oxygen-isotopic exchange processes can be revealed, which could become a new handle to study CO2 and H2O interactions/reactivity and compare the properties of different materials developed for carbon capture. Importantly, this investigation clearly highlights the complementarity of in situ ssNMR and ex situ IR spectroscopy when studying the oxygen isotope-transfer processes, following the 17O and 18O isotopes, respectively (as shown in Fig. 4). Second, from a more practical perspective, our present 17O ssNMR study shows that precaution should be taken in the interpretation of the 17O MAS NMR spectra recorded for carbonate-containing phases in presence of water. Resonances relative to enriched (bi)carbonate ions may not be visible, depending on sample preparation and/or NMR acquisition conditions chosen (as shown in Fig. 4B for the 370 K data, and further illustrated in Fig. S13, S23 and S24†). Based on this observation, we would recommend performing analyses at various temperatures (bearing in mind that equilibria can be shifted upon changes in temperature), and with different 17O ssNMR sequences (e.g. Bloch decay and Hahn echo), in order to avoid missing out on chemical information. Lastly, beyond the study of materials for carbon capture applications, the observation of partial 17O-isotopic labeling of the carbonates of Na2CO3·H2*O at high temperature suggests that it may be possible to directly label hydrated carbonate salts using a “liquid-assisted grinding” approach, while including heating during the milling. With the increasing number of heating set-ups being developed for ball-milling equipment,60,61 the latter option appears as very valuable to help further expand the scope of carbonate-labeled precursors for other 17O NMR applications, which we are continuing to develop in our lab.
3 Conclusion
In this manuscript, we have described a new strategy for the 17O-enrichment of Na- and K-(bi)carbonate salts, using mechanochemistry. The synthetic approach is robust, user-friendly, and cost-effective, enabling the production of up to 400 mg of labeled Na2CO3 in just half a day of manipulation. The high enrichment levels achieved enabled the first high resolution 17O solid-state NMR analyses to be performed on these materials in short experimental times (as short as 8 minutes for some of the 2D measurements reported here). This enabled carrying out the studies not only at different magnetic fields, but also different temperatures, thereby shedding light on important features regarding the 17O NMR signatures of (bi)carbonates in solids, among which (i) broad ranges in the variation of the 17O NMR parameters of CO and C–OH groups of (bi)carbonates, with notably distinct signatures for the hydroxyl group in NaHCO3 and KHCO3; (ii) a strong sensitivity of their 17O NMR parameters to temperature (with direct impact on the quadrupolar lineshapes), especially for the hydrated phases, which appeared as “gaussian-like” resonances close to room temperature.
The high 17O-isotopic labeling achieved on the (bi)carbonate phases was shown to be critical not just to enable accurate spectral fitting, but also to avoid missing out on some resonances at natural abundance, and to help elucidate under-studied aspects of the reactivity of (bi)carbonate ions in solids, namely oxygen isotope-transfer processes. As such, the present work provides sound bases for future works on (bi)carbonate-based materials including those developed for CO2 capture, for which structure, reactivity, and speciation aspects are key for their rational design. Importantly, we have shown that the 17O NMR signatures, including during in situ investigations, are highly sensitive to temperature and NMR acquisition conditions, meaning that future studies of such materials will require not only an accurate control of the temperature and a careful choice of the pulse sequences, but also analyses at different temperatures, in order to avoid mis-interpretations (or over interpretations) of the data.
Beyond the aforementioned applications, the possibility of labeling with 17O Na- and K-(bi)carbonate salts opens new avenues to the study of many systems by 17O ssNMR, as these compounds are widely used as precursors in molecular and materials syntheses, for the preparation of functional (bio)materials, but also organic molecules.62,63 Moreover, the labeling CDI-based procedure is a priori applicable to the enrichment of other metal carbonates (including with transition-metal and lanthanide ions),64,65 which could then be engaged for the preparation of functional ceramics and glasses, for which 17O ssNMR is invaluable for establishing structure/property correlations. Last but not least, Na- and K-(bi)carbonates decompose thermally by release of CO2, with a temperature as low as ≈80 °C for NaHCO3. These reagents could therefore be used as simple, straightforward, and accessible sources for production of enriched CO2. The latter could then be used for pushing forward investigations on materials for carbon capture using high resolution 17O NMR. These are points we endeavour to look into, with on-going efforts in our research group.
Data availability
The data supporting this article have been included as part of the ESI:† (i) full description of syntheses, (ii) complementary IR, pXRD, IR-MS, and NMR analyses, including ssNMR acquisition parameters, (iii) computational details; and (iv) a video illustrating the release of CO2 during the reaction.Author contributions
AP, NF and DL conducted the majority of the research experiments (syntheses, general characterizations and ssNMR analyses). CE contributed to the early stages of the project, by performing the initial syntheses. TXM contributed to the discussion on the mechanochemical syntheses. AP performed the GIPAW-DFT calculations, in close collaboration with CG. CG participated in all discussions regarding computational results. FMV and FS carried out the low-temperature 17O ssNMR studies at 14.1 T, and DG participated to those at 18.8 T. JT assisted in the ultra-high field ssNMR analyses at 28.2 T. FV and NP performed the MS analyses on the 18O-labeled compounds. AP and DL wrote the first draft of the manuscript, and all authors contributed to the final preparation of the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This project is funded in part by the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (grant agreement no. 772204; 2017 ERC COG, MISOTOP project). Further financial support from the INFRANALYTICS FR2054 for conducting NMR experiments at the CRMN in Lyon and IMEC in Lille is acknowledged. NMR research was also conducted at the National High Magnetic Field Laboratory (NHMFL, Tallahassee) in Florida, which is supported by the National Science Foundation Cooperative Agreements (No. DMR-2128556), the State of Florida and a partial support by National Institutes of Health Grant (RM1-GM148766 and RM1-GM148556). This project has received support from the European Union's Horizon 2020 research and innovation programme under Grant Agreement 101008500 (PANACEA) and F. J. S. is supported by the postdoctoral scholar award from the Provost's Office at Florida State University. DFT computations were run using high performance computing resources from GENCI-IDRIS (grant no. 2024-AD010815148 and AD10-097535). Philippe Gaveau (ICGM, Montpellier) and Andrew Rankin (IMEC, Lille) are thanked for their experimental expertise and assistance on part of the ssNMR analyses. Dr Jessica Novák-Špačková is warmly acknowledged for early discussions related to this work, Dr Hugo Petitjean for exchanges about the IR data, and Dr Ieva Goldberga for assistance in some initial experiments.References
- K. Calvin, D. Dasgupta, G. Krinner, A. Mukherji, P. W. Thorne, C. Trisos, J. Romero, P. Aldunce, K. Barrett, G. Blanco, W. W. L. Cheung, S. Connors, F. Denton, A. Diongue-Niang, D. Dodman, M. Garschagen, O. Geden, B. Hayward, C. Jones, F. Jotzo, T. Krug, R. Lasco, Y.-Y. Lee, V. Masson-Delmotte, M. Meinshausen, K. Mintenbeck, A. Mokssit, F. E. L. Otto, M. Pathak, A. Pirani, E. Poloczanska, H.-O. Pörtner, A. Revi, D. C. Roberts, J. Roy, A. C. Ruane, J. Skea, P. R. Shukla, R. Slade, A. Slangen, Y. Sokona, A. A. Sörensson, M. Tignor, D. van Vuuren, Y.-M. Wei, H. Winkler, P. Zhai, Z. Zommers, J.-C. Hourcade, F. X. Johnson, S. Pachauri, N. P. Simpson, C. Singh, A. Thomas, E. Totin, A. Alegría, K. Armour, B. Bednar-Friedl, K. Blok, G. Cissé, F. Dentener, S. Eriksen, E. Fischer, G. Garner, C. Guivarch, M. Haasnoot, G. Hansen, M. Hauser, E. Hawkins, T. Hermans, R. Kopp, N. Leprince-Ringuet, J. Lewis, D. Ley, C. Ludden, L. Niamir, Z. Nicholls, S. Some, S. Szopa, B. Trewin, K.-I. van der Wijst, G. Winter, M. Witting, A. Birt and M. Ha, Climate Change 2023: Synthesis Report, Contribution of Working Groups I, II and III to the Sixth Assessment Report of the Intergovernmental Panel on Climate Change [Core Writing Team], ed. H. Lee and J. Romero, IPCC, Geneva, Switzerland, 2023 Search PubMed.
- H. D. Matthews and S. Wynes, Science, 2022, 376, 1404–1409 CrossRef CAS PubMed.
- S. Ó. Snæbjörnsdóttir, B. Sigfússon, C. Marieni, D. Goldberg, S. R. Gislason and E. H. Oelkers, Nat. Rev. Earth Environ., 2020, 1, 90–102 CrossRef.
- B. Dziejarski, J. Serafin, K. Andersson and R. Krzyżyńska, Mater. Today Sustain., 2023, 24, 100483 Search PubMed.
- L. B. Hamdy, C. Goel, J. A. Rudd, A. R. Barron and E. Andreoli, Mater. Adv., 2021, 2, 5843–5880 RSC.
- K. S. Lackner, C. H. Wendt, D. P. Butt, E. L. Joyce and D. H. Sharp, Energy, 1995, 20, 1153–1170 CrossRef CAS.
- H. S. Santos, H. Nguyen, F. Venâncio, D. Ramteke, R. Zevenhoven and P. Kinnunen, Inorg. Chem. Front., 2023, 10, 2507–2546 RSC.
- A. MacKenzie, D. L. Granatstein, E. J. Anthony and J. C. Abanades, Energy Fuels, 2007, 21, 920–926 CrossRef CAS.
- M. E. Zick, D. Cho, J. Ling and P. J. Milner, ChemNanoMat, 2023, 9, e202200436 CrossRef CAS.
- Z. Li, P. Liu, C. Ou and X. Dong, ACS Sustain. Chem. Eng., 2020, 8, 15378–15404 CrossRef CAS.
- M. Ding, R. W. Flaig, H.-L. Jiang and O. M. Yaghi, Chem. Soc. Rev., 2019, 48, 2783–2828 RSC.
- R. Vismara, S. Terruzzi, A. Maspero, T. Grell, F. Bossola, A. Sironi, S. Galli, J. A. R. Navarro and V. Colombo, Adv. Mater., 2024, 36, 2209907 CrossRef CAS PubMed.
- S. M. Pugh and A. C. Forse, J. Magn. Reson., 2023, 346, 107343 CrossRef CAS PubMed.
- A. H. Berge, S. M. Pugh, M. I. M. M. Short, C. Kaur, Z. Lu, J.-H. Lee, C. J. Pickard, A. Sayari and A. C. Forse, Nat. Commun., 2022, 13, 7763 CrossRef CAS PubMed.
- D. Pereira, R. Fonseca, I. Marin-Montesinos, M. Sardo and L. Mafra, Curr. Opin. Colloid Interface Sci., 2023, 64, 101690 CrossRef CAS.
- G. Wu, Prog. Nucl. Magn. Reson. Spectrosc., 2008, 52, 118–169 CrossRef CAS.
- X. Kong, L. A. O'Dell, V. Terskikh, E. Ye, R. Wang and G. Wu, J. Am. Chem. Soc., 2012, 134, 14609–14617 CrossRef CAS PubMed.
- M. T. Dunstan, J. M. Griffin, F. Blanc, M. Leskes and C. P. Grey, J. Phys. Chem. C, 2015, 119, 24255–24264 CrossRef CAS.
- B. J. Rhodes, L. L. Schaaf, M. E. Zick, S. M. Pugh, J. S. Hilliard, S. Sharma, C. R. Wade, P. J. Milner, G. Csányi and A. C. Forse, ChemPhysChem, 2025, 26, e202400941 CrossRef CAS PubMed.
- J. Beerwerth, R. Siegel, L. Hoffmann, L. S. Plaga, M. Storek, B. Bojer, J. Senker, W. Hiller and R. Böhmer, Appl. Magn. Reson., 2020, 51, 597–620 CrossRef CAS.
- M. Nava, N. Lopez, P. Müller, G. Wu, D. G. Nocera and C. C. Cummins, J. Am. Chem. Soc., 2015, 137, 14562–14565 CrossRef CAS PubMed.
- W. D. Wang, B. E. G. Lucier, V. V. Terskikh, W. Wang and Y. Huang, J. Phys. Chem. Lett., 2014, 5, 3360–3365 CrossRef CAS PubMed.
- M. Y. Hu, X. Deng, K. S. Thanthiriwatte, V. E. Jackson, C. Wan, O. Qafoku, D. A. Dixon, A. R. Felmy, K. M. Rosso and J. Z. Hu, Environ. Sci. Technol., 2016, 50, 12373–12384 CrossRef CAS PubMed.
- J.-H. Du, L. Chen, B. Zhang, K. Chen, M. Wang, Y. Wang, I. Hung, Z. Gan, X.-P. Wu, X.-Q. Gong and L. Peng, Nat. Commun., 2022, 13, 707 CrossRef CAS PubMed.
- P. Sahoo, S. Ishihara, K. Yamada, K. Deguchi, S. Ohki, M. Tansho, T. Shimizu, N. Eisaku, R. Sasai, J. Labuta, D. Ishikawa, J. P. Hill, K. Ariga, B. P. Bastakoti, Y. Yamauchi and N. Iyi, ACS Appl. Mater. Interfaces, 2014, 6, 18352–18359 CrossRef CAS PubMed.
- D. Paul and G. Skrzypek, Int. J. Mass Spectrom., 2007, 262, 180–186 CrossRef CAS.
- Y.-L. Wu and S.-M. Shih, Thermochim. Acta, 1993, 223, 177–186 CrossRef CAS.
- M. Hartman, K. Svoboda, B. Čech, M. Pohořelý and M. Šyc, Ind. Eng. Chem. Res., 2019, 58, 2868–2881 CrossRef CAS.
- J. M. McCrea, J. Chem. Phys., 1950, 18, 849–857 CrossRef CAS.
- M. E. Smith, S. Steuernagel and H. J. Whitfield, Solid State Nucl. Magn. Reson., 1995, 4, 313–316 CrossRef CAS PubMed.
- J. Ihli, J. N. Clark, N. Kanwal, Y.-Y. Kim, M. A. Holden, R. J. Harder, C. C. Tang, S. E. Ashbrook, I. K. Robinson and F. C. Meldrum, Chem. Sci., 2019, 10, 1176–1185 RSC.
- M. Leskes, A. J. Moore, G. R. Goward and C. P. Grey, J. Phys. Chem. C, 2013, 117, 26929–26939 CrossRef CAS PubMed.
- T.-X. Métro, C. Gervais, A. Martinez, C. Bonhomme and D. Laurencin, Angew. Chem., Int. Ed., 2017, 56, 6803–6807 CrossRef PubMed.
- C.-H. Chen, E. Gaillard, F. Mentink-Vigier, K. Chen, Z. Gan, P. Gaveau, B. Rebière, R. Berthelot, P. Florian, C. Bonhomme, M. E. Smith, T.-X. Métro, B. Alonso and D. Laurencin, Inorg. Chem., 2020, 59, 13050–13066 CrossRef CAS PubMed.
- J. Špačková, I. Goldberga, R. Yadav, G. Cazals, A. Lebrun, P. Verdié, T. Métro and D. Laurencin, Chem.–Eur. J., 2023, 29, e202203014 CrossRef PubMed.
- K. M. Engstrom, A. Sheikh, R. Ho and R. W. Miller, Org. Process Res. Dev., 2014, 18, 488–494 CrossRef CAS.
- L. Rincón, C. Ruiz, R. R. Contreras and J. Almarza, Environ. Sci.:Adv., 2023, 2, 957–966 Search PubMed.
- M. Yoo, S. J. Han and J. H. Wee, J. Environ. Manag., 2013, 114, 512–519 CrossRef CAS PubMed.
- A. Novak, P. Saumagne and L. Bok, J. Chim. Phys., 1963, 60, 1385–1395 CrossRef CAS.
- A. Bertoluzza, P. Monti, M. A. Morelli and M. A. Battaglia, J. Mol. Struct., 1981, 73, 19–29 CrossRef CAS.
- G. Lucazeau and A. Novak, J. Raman Spectrosc., 1973, 1, 573–586 CrossRef CAS.
- W. W. Rudolph, G. Irmer and E. Königsberger, Dalton Trans., 2008, 900–908 RSC.
- T. Geisler, C. Perdikouri, A. Kasioptas and M. Dietzel, Geochim. Cosmochim. Acta, 2012, 90, 1–11 CrossRef CAS.
- A. Landuyt, P. V. Kumar, J. A. Yuwono, A. H. Bork, F. Donat, P. M. Abdala and C. R. Müller, JACS Au, 2022, 2, 2731–2741 CrossRef CAS PubMed.
- S. Wang, K. Lu, T. Wang, J. Wu, H. Zheng and Y. Huang, Spectrochim. Acta, Part A, 2020, 241, 118648 CrossRef CAS PubMed.
- Q. Wu, N. Yang, M. Xiao, W. Wang and C. Cui, Nat. Commun., 2024, 15, 9145 CrossRef CAS PubMed.
- S. Nour, C. M. Widdifield, L. Kobera, K. M. N. Burgess, D. Errulat, V. V. Terskikh and D. L. Bryce, Can. J. Chem., 2016, 94, 189–197 CrossRef CAS.
- I. Goldberga, N. Patris, C.-H. Chen, E. Thomassot, J. Trébosc, I. Hung, Z. Gan, D. Berthomieu, T.-X. Métro, C. Bonhomme, C. Gervais and D. Laurencin, J. Phys. Chem. C, 2022, 126, 12044–12059 CrossRef CAS PubMed.
- E. G. Keeler, V. K. Michaelis, C. B. Wilson, I. Hung, X. Wang, Z. Gan and R. G. Griffin, J. Phys. Chem. B, 2019, 123, 3061–3067 CrossRef CAS PubMed.
- R. E. Wasylishen, S. E. Ashbrook and S. Wimperis, NMR of Quadrupolar Nuclei in Solid Materials, John Wiley & Sons Ltd, 2012 Search PubMed.
- I. J. F. Poplett and J. A. S. Smith, J. Chem. Soc., Faraday Trans. 2, 1981, 77, 761–796 RSC.
- A. Wong, G. Thurgood, R. Dupree and M. E. Smith, Chem. Phys., 2007, 337, 144–150 CrossRef CAS.
- D. G. Boer, J. Langerak and P. P. Pescarmona, ACS Appl. Energy Mater., 2023, 6, 2634–2656 CrossRef CAS.
- L. Santamaría, S. A. Korili and A. Gil, Chem. Eng. J., 2023, 455, 140551 CrossRef.
- I. Erucar and S. Keskin, Ind. Eng. Chem. Res., 2020, 59, 3141–3152 CrossRef CAS PubMed.
- A. Rajendran, G. K. H. Shimizu and T. K. Woo, Adv. Mater., 2024, 36, 2301730 CrossRef CAS PubMed.
- B. N. Figgis, R. G. Kidd and R. S. Nyholm, Proc. R. Soc. London, Ser. A, 1962, 269, 469–480 Search PubMed.
- S. Fukunaga, Y. Zushi, M. Hotta and N. Koga, Phys. Chem. Chem. Phys., 2025, 27, 3384–3400 RSC.
- Y. Zushi, S. Iwasaki and N. Koga, Phys. Chem. Chem. Phys., 2022, 24, 15736–15748 RSC.
- G. Félix, N. Fabregue, C. Leroy, T. X. Métro, C. H. Chen and D. Laurencin, Phys. Chem. Chem. Phys., 2023, 25, 23435–23447 RSC.
- N. Cindro, M. Tireli, B. Karadeniz, T. Mrla and K. Užarević, ACS Sustain. Chem. Eng., 2019, 7, 16301–16309 CrossRef CAS.
- F. Mele, A. Aquilini, A. M. Constantin, F. Pancrazzi, L. Righi, A. Porcheddu, R. Maggi, D. A. Cauzzi, G. Maestri, E. Motti, L. Capaldo and N. Della Ca', ChemRxiv, 2025, preprint, DOI:10.26434/chemrxiv-2024-zm9fw-v3.
- M. Sander, S. Fabig and L. Borchardt, Chem.–Eur. J., 2023, 29, e202202860 CrossRef CAS PubMed.
- P. Vishnu Vardhan, C. Jothilakshmi, U. Kamachi Mudali and S. Devaraj, Mater. Today: Proc., 2017, 4, 12407–12415 Search PubMed.
- P. Barai, X. Wang, M. Wolfman, J. Chen, A. Gutierrez, J. C. Garcia, J. Wen, T. Kinnibrugh, T. T. Fister, H. H. Iddir and V. Srinivasan, J. Mater. Chem. A, 2024, 12, 12835–12855 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc08491h |
| This journal is © The Royal Society of Chemistry 2025 |