
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMachine learning-assisted benign transformation of three zinc states in zinc ion batteries†
Jianbo
Dong‡
a,
Guolang
Zhou‡
 ab,
Wenhao
Ding
b,
Jiayi
Ji
a,
Qing
Wang
a,
Tianshi
Wang
a,
Lili
Zhang
ab,
Xiuyang
Zou
*a,
Jingzhou
Yin
*a and
Edison Huixiang
Ang
*c
ab,
Wenhao
Ding
b,
Jiayi
Ji
a,
Qing
Wang
a,
Tianshi
Wang
a,
Lili
Zhang
ab,
Xiuyang
Zou
*a,
Jingzhou
Yin
*a and
Edison Huixiang
Ang
*c
aJiangsu Key Laboratory for the Chemistry of Low-Dimensional Materials, Jiangsu Engineering Laboratory for Environmental Functional Materials, School of Chemistry and Chemical Engineering, Huaiyin Normal University, Huai’an 223300, P. R. China. E-mail: xyzou@hytc.edu.cn; jingzhouyin@hytc.edu.cn
bKey Laboratory for Soft Chemistry and Functional Materials of Ministry of Education, Nanjing University of Science and Technology, Nanjing 210094, P. R. China
cNatural Sciences and Science Education, National Institute of Education, Nanyang Technological University, Singapore 637616, Singapore. E-mail: edison.ang@nie.edu.sg
First published on 22nd April 2025
Abstract
The applications of aqueous zinc-ion batteries (ZIBs) are limited by challenges such as zinc dendrite formation and hydrogen evolution. However, the traditional trial-and-error approach to designing interface layer structures has proven to be both inefficient and costly. To address this issue, a fully connected neural network model has been developed to analyze the relationship between the structure and stability across 168![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 interface layer candidates. Leveraging the power of machine learning, a cerium-iron bimetallic metal–organic framework interface layer has been successfully designed. This layer contains ion-sieving channels, polar –CN organic ligands, and a zincophilic Ce cation, which play crucial roles in reducing the desolvation energy barrier for the conversion of Zn(H2O)n2+ to Zn2+. Coordination channels ensure selective ion transport and facilitate uniform Zn2+–Zn transformation, which prevents zinc dendrite formation. As a result, this continuous transformation among Zn(H2O)n2+, Zn2+, and Zn states leads to long-term stability. The MOF@Zn‖MOF@Zn configuration attains an impressive cycle life of over 4300 hours at 1 mA cm−2 (1 mA h cm−2). Furthermore, the MOF@Zn‖Cu configuration exhibits an average coulombic efficiency of 99.8% over 1400 cycles at 2 mA cm−2 (1 mA h cm−2). This study proposes a straightforward and effective protective layer strategy for significantly improving the stability of the zinc anode.
000 interface layer candidates. Leveraging the power of machine learning, a cerium-iron bimetallic metal–organic framework interface layer has been successfully designed. This layer contains ion-sieving channels, polar –CN organic ligands, and a zincophilic Ce cation, which play crucial roles in reducing the desolvation energy barrier for the conversion of Zn(H2O)n2+ to Zn2+. Coordination channels ensure selective ion transport and facilitate uniform Zn2+–Zn transformation, which prevents zinc dendrite formation. As a result, this continuous transformation among Zn(H2O)n2+, Zn2+, and Zn states leads to long-term stability. The MOF@Zn‖MOF@Zn configuration attains an impressive cycle life of over 4300 hours at 1 mA cm−2 (1 mA h cm−2). Furthermore, the MOF@Zn‖Cu configuration exhibits an average coulombic efficiency of 99.8% over 1400 cycles at 2 mA cm−2 (1 mA h cm−2). This study proposes a straightforward and effective protective layer strategy for significantly improving the stability of the zinc anode.
Broader contextAs the global demand for efficient energy storage systems increases, aqueous zinc-ion batteries (ZIBs) have emerged as a viable alternative due to their abundant resources, low cost, and safety. However, challenges such as zinc dendrite formation and hydrogen evolution during charging hinder their practical application. A fully connected neural network model comprising 168000 interface layer configurations was developed to systematically investigate the structure–stability relationship. Through advanced machine learning algorithms, a cerium-iron bimetallic metal–organic framework (MOF) interface layer that enhances ZIB performance and stability was synthesized. The MOF architecture features ion-sieving channels and zincophilic Ce cations, significantly reducing the desolvation energy barrier, which minimizes reactive water and suppresses hydrogen evolution reactions. This mechanism facilitates selective ion transport and promotes uniform zinc deposition, preventing dendrite formation. Achieving a cycle life of over 4300 hours and a coulombic efficiency of 99.8%, this research provides a straightforward and effective protective layer strategy, contributing to the advancement of sustainable and reliable energy storage solutions and paving the way for practical ZIB implementation in various applications.
|
Introduction
Aqueous zinc-ion batteries (ZIBs) represent a cutting-edge electrochemical energy storage technology that utilizes zinc ions as charge carriers. They have garnered significant attention due to their exceptional safety, low cost, and remarkable environmental friendliness.1–3 However, during the dissolution of Zn to form Zn2+, hydration leads to the generation of Zn(H2O)n2+. Upon the deposition of Zn(H2O)n2+, desolvation occurs, converting Zn(H2O)n2+ to Zn2+ while releasing a substantial amount of reactive H2O molecules.4 This process can trigger severe hydrogen evolution reactions (HER). Subsequently, as Zn2+ is transformed into Zn, zinc ions preferentially deposit at surface micro-defects, gradually forming zinc dendrites that can puncture the separator and cause battery short circuits.5,6 Notably, the hydrogen evolution reaction and the growth of zinc dendrites mutually exacerbate each other, leading to a significant reduction in the lifespan of the zinc anode.To enhance the stability of aqueous zinc-ion batteries, various strategies have been proposed, including the introduction of electrolyte additives,7–10 the design of separators11–14 and the modification of the anode interface.15–17 Among these strategies, electrolyte additives function by forming strong coordination with zinc ions, thereby reducing the amount of active water in the solvation structure and inhibiting hydrogen evolution reactions. Furthermore, the additive molecules adsorbed onto the zinc surface can guide the uniform deposition of zinc ions, which helps prevent dendrite growth. Separator modifications aim to suppress the growth of zinc dendrites and hydrogen evolution by adjusting the interfacial electric field. However, since the hydration of zinc ions preferentially occurs at the interface, the irregular growth of zinc dendrites typically initiates there. In contrast, anode interface modification directly enhances the interface structure, providing a more efficient means of inhibiting various side reactions.
Organic materials (e.g., ZnTAPP-NTCDA-POP,18 MNC,19 PVA,20 PANZ,21etc.) and inorganic materials (e.g., ZnO,22 Nb2O5,23 CaF2,24 SiO2,25etc.) have proven to be simple and effective choices for anode interface modification. Organic coatings can significantly enhance zinc ion transport through strategic design configurations and the incorporation of various functional groups, thereby optimizing performance. Among these, metal–organic frameworks (MOFs) are particularly popular for organic material coatings on anodes. Their abundant pore structures not only facilitate the uniform deposition of zinc ions, but also enable orderly ion screening, which reduces the disruption of water molecules and subsequently lowers the desolvation energy barrier. The structure of Ti-MOF nanosheets is rich in desolvation sites, effectively shortening the ion transport pathway. This characteristic plays a crucial role in regulating the solvation structure of Zn2+, enabling rapid diffusion and uniform deposition of zinc ions while reducing the hydrogen evolution reaction (HER) response.26 For instance, MOF-801 nanoparticles, with a pore diameter of 6.0 Å, are uniformly coated onto zinc foil to form a selective ion transport layer, facilitating the uniform flux of zinc ions.27 Furthermore, modulating the organic ligands and metal centers within the MOF structure can enhance the ability of the MOF to inhibit hydrogen evolution reactions and irregular dendrite growth.28
The discovery of materials with optimal chemical structures is often hindered by the complexity of spatial arrangements.29 High-throughput screening provides a method to identify a limited number of MOFs with desired properties. However, this approach faces challenges when attempting to screen MOF materials based on the specific performance requirements of zinc-ion batteries. Traditional theoretical calculations, such as density functional theory (DFT) and molecular dynamics (MD) simulations, can help elucidate the structure–activity relationship between MOF structures and zinc-ion battery performance.30 Despite their utility, these methods are constrained by high computational costs and time demands, which limit the rapid exploration of unexplored chemical spaces. In contrast, machine learning offers a promising alternative, enabling efficient correlation between the chemical structures of materials and their properties. This capability accelerates the identification of high-performance material candidates, facilitating the design of novel material structures. For example, Yan et al. employed unsupervised learning to screen 420000 candidate materials for highly chemically stable anion exchange membranes for fuel cell testing, achieving membrane chemical stability with a degradation rate of less than 5% within 1000 h.31
In this study, fully connected neural network (FCNN) models were trained to explore a chemical space containing over 168000 candidates, identifying promising interface layers with high stability, as shown in Fig. 1. Guided by the FCNN, cerium-iron Prussian blue analog (Ce–Fe MOF, CeK[Fe(CN)6]·4H2O) is proposed as a modification layer for the zinc anode in aqueous zinc-ion batteries (ZIBs) to address challenges like dendrite formation and side reactions. The Ce–Fe MOF features unique ion-sieving pore channels (∼7 Å) that block sulfate ions and highly reactive water molecules from directly accessing the zinc surface. These channels, combined with strongly polar cyano (–CN) ligands and a zincophilic cerium (Ce) metal center, create an effective coordination environment for zinc ions, reducing the desolvation energy of Zn(H2O)n2+ and enabling a smooth, continuous conversion to Zn2+ under dynamic equilibrium conditions. The open three-dimensional framework of the Ce–Fe MOF further regulates zinc ion flux, ensuring uniform zinc deposition and consistent transformation from Zn2+ to metallic Zn. This framework leverages its long-range ordered structure, distinctive pore channels, organic ligands, and the cerium cation to promote gradient transformations among Zn(H2O)n2+, Zn2+, and Zn states. Experimental results and molecular simulations confirm that the Ce–Fe MOF layer significantly inhibits hydrogen evolution reactions (HER), suppresses irregular dendrite growth, and minimizes side reactions during extended cycling. This study introduces an innovative and efficient protective layer strategy, greatly enhancing the stability and performance of zinc anodes in ZIBs.
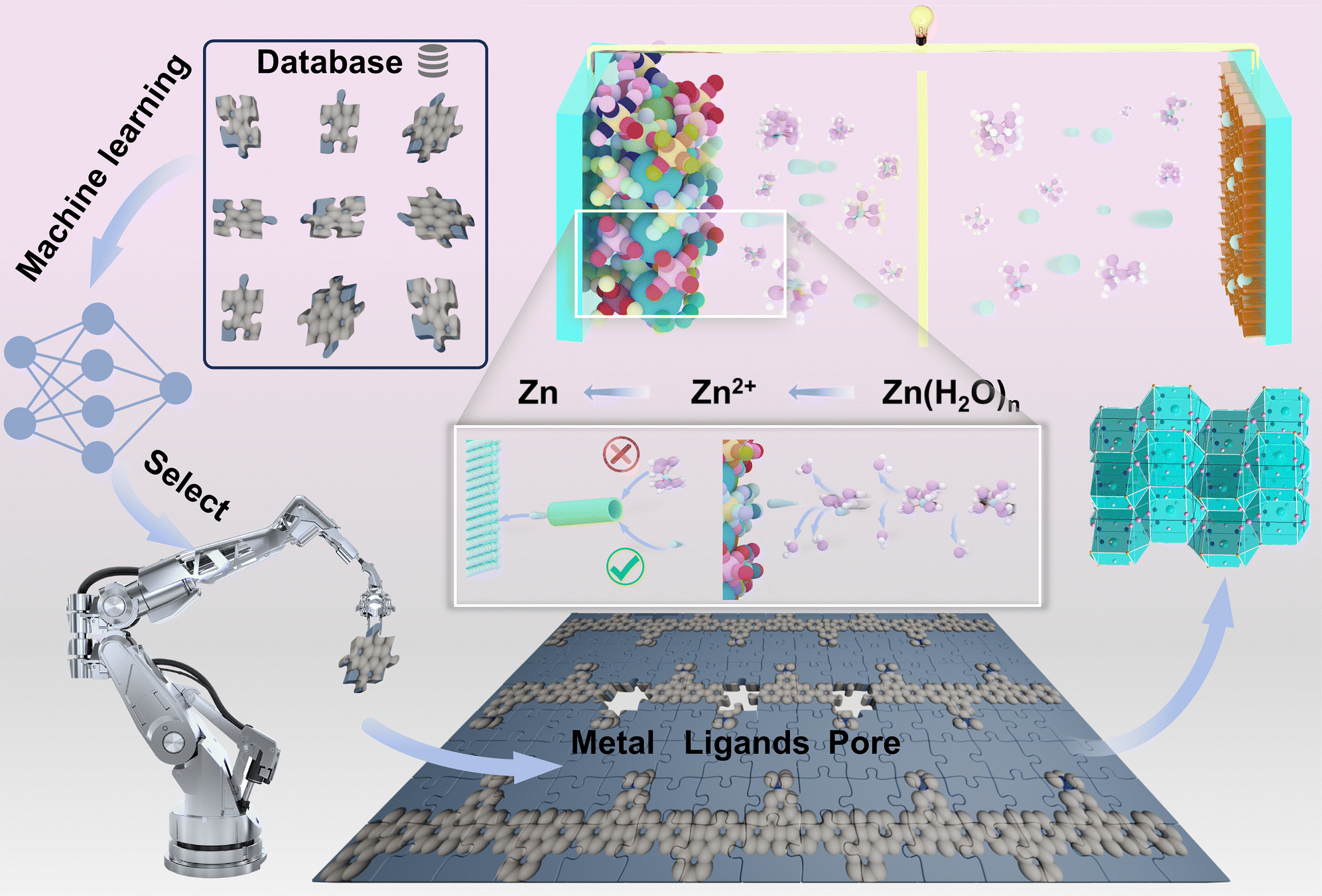 | ||
| Fig. 1 Machine learning-enhanced design was employed to create a highly stable interface layer. A trained FCNN model analyzed approximately 168000 interface layer candidates, enabling the identification of MOF structures with high stability. Additionally, MD simulation analysis was conducted on the transmission channels of the MOF. | ||
Results and discussion
Machine learning-assisted structural design
Regardless of the machine learning algorithm used to construct the MOF-based zinc electrode stability model, a comprehensive database of input and output data is essential. Published literature studies serve as a valuable source for building ML models. In this study, the database was compiled from published sources. To enhance the accuracy of the machine learning algorithm, we implemented several optimization strategies. One key approach involved analyzing the importance of input features by focusing on cases with high stability (exceeding 3000 hours). Specifically, we conducted feature selection at the input layer, identifying 24 molecular descriptors with the greatest impact on stability from a total of 155 descriptors for each metal type and organic linker. Among these, the 10 features with the highest intermediate influence rates are highlighted and visualized in Fig. 2a. The features are ranked in order of importance, from high to low: NumValenceElectrons of metal (NVE of metal), SlogP_VSA3 of metal (SVSA of metal), largest cavity diameter (LCD of Sam), qed of metal, void fraction of Sam, NumValenceElectrons of organic linker (NVE of organic linker), volumetric surface area (VSA), pore limiting diameter (PLD), NumHBA of the organic linker (NHBA of OL), and MaxAbsPartialCharge (MAPC of OL). Battery stability is significantly influenced by the electrostatic interactions between the metal type and organic linker, as well as the steric hindrance effect of metal–organic frameworks (MOFs), as demonstrated in numerous experiments. Key features that describe the charge, electrostatic potential, molecular accessible surface area, and polarity of the metal type and organic linker are critical in this context. The feature selection process reveals that the machine learning algorithm effectively identifies and emphasizes these critical features, aligning well with experimental findings. This suggests that the algorithm accurately recognizes how the structure of the metal type and organic linker in the interface layer affects stability. | ||
| Fig. 2 (a) Top ten molecular descriptors affecting stability. (b) Impact of the number of hidden layers and the number of neurons in the hidden layers on the root mean square error (RMSE) in the FCNN model. (c) Loss curves on the training set and the split training set used for validation in the training process of the FCNN model. (d) Influence of the number of hidden layers and the number of neurons in the hidden layers on the computation time in the FCNN model. (e) Influence of the number of hidden layers and the number of neurons in the hidden layers on the computation time in the FCNN model. (f) Organic linker structure and stability of the unlabeled dataset labeled by the trained FCNN. (g) Schematic illustration of pore size. (h) Cross-sectional view of MOF@Zn along with its primary element distribution map. (i) XRD diffraction patterns of the bare Zn, Ce–Fe MOF and MOF@Zn. | ||
Using the previously established labeled dataset, we generated training and test sets to train and evaluate four machine learning algorithms: Gaussian process regression (GPR), support vector regression (SVR), random forest regression (RFR), and XGboost. The results of these algorithms are shown in Fig. S1 and S2 (ESI†). The coefficient of determination (R2) and root mean square error (RMSE) values for each algorithm are as follows: GPR (R2 = 0.1939, RMSE = 2247.59), SVR (R2 = 0.6908, RMSE = 899.07), RFR (R2 = 0.4939, RMSE = 968.72), and XGboost (R2 = 0.7317, RMSE = 874.11). The performance of a machine learning model is typically assessed using two key metrics: the coefficient of determination (R2) and the root mean square error (RMSE). A model exhibits a better fit when its R2 value approaches 1 and its RMSE is minimized. Among the models tested, the fully connected neural network (FCNN) algorithm demonstrated the best fit, making it the most effective for this task. The FCNN algorithm, designed to simulate the transmission of neural signals in the human brain, comprises an input layer, multiple hidden layers, and an output layer. These layers are all connected to each other by many neurons, enabling algorithms to model nonlinear and complex relationships. This is especially important as many real-world relationships exhibit intricate, non-linear patterns. Moreover, the FCNN has the ability to generalize and infer relationships within unknown datasets, enhancing its predictive accuracy. As shown in Fig. 2b and Fig. S3 (ESI†), the FCNN achieved optimal performance when configured with three hidden layers and 240 neurons. However, increasing the number of hidden layers to 8 resulted in overfitting. To improve computational efficiency while maintaining accuracy, we reduced the number of hidden layers and neurons in our models. A grid search method was used to optimize the hyperparameters of each algorithm, resulting in satisfactory models. Despite these adjustments, the FCNN still faced overfitting, which affected its accuracy. Notably, at 1810 epochs, the validation set‘s loss began to rise, indicating overfitting and diminishing the model's generalization ability. Therefore, the optimal validation performance for the FCNN was achieved at 1810 epochs (Fig. 2c). To further enhance FCNN performance, we optimized its architecture, balancing the number of hidden layers and neurons. Excessive layers or neurons can lead to generalization errors and increased computational cost (Fig. 2d). Following optimization, the R2 value of the FCNN improved to 0.8705, and the RMSE decreased to 586.60, as illustrated in Fig. 2e. In this figure, the horizontal axis represents the actual stability values from the literature, while the vertical axis shows the predicted stability values generated by the optimized FCNN model. A virtual dataset comprising 168000 metal–organic framework (MOF) structures was created by randomly combining two key components: metal sources and ligands. This dataset was screened using commercially available or previously reported ligands, leading to the identification of 220 MOF structures, as shown in Fig. 2f. Notably, Prussian blue was chosen as the ligand for the MOF.
Preparation of the Ce–Fe MOF and MOF@Zn
The Ce–Fe MOF was synthesized using a chemical coprecipitation method and subsequently collected after drying, as depicted in Fig. S4 (ESI†). The addition of trace amounts of ammonium hydroxide created an alkaline environment that facilitated the coordination of acetylacetone (AcAc) with cerium ions. Concurrently, competitive coordination between the precipitant potassium ferrocyanide and acetylacetone directed the formation of a Ce–Fe MOF, which exhibited a distinctive dodecahedral morphology (Fig. S5, ESI†) and a pore size of approximately 7 Å (Fig. 2g). Following the synthesis, the Ce–Fe MOF was combined with polyvinylidene fluoride (PVDF) in a water–ethanol solvent system to create a homogeneous suspension. This suspension was then uniformly applied to zinc foil through a blade coating technique, as illustrated in Fig. 2h and Fig. S6 (ESI†), resulting in the successful preparation of a zinc anode featuring a Ce–Fe MOF modification layer with an approximate thickness of 6 μm.To confirm the successful synthesis of the Ce–Fe MOF and its integration with MOF@Zn, a series of detailed characterization studies were performed. As illustrated in Fig. 2i, XRD analyses were conducted on bare Zn, Ce–Fe MOF powder samples, and MOF@Zn. The newly observed diffraction peaks for MOF@Zn and the powder samples, which appeared between 15° and 35°, exhibited a strong correlation with the simulated pattern of CeK[Fe(CN)6]·4H2O. Additionally, the diffraction peaks for MOF@Zn and bare Zn between 40° and 75° also displayed corresponding relationships. Furthermore, as shown in Fig. S7 (ESI†), the Fourier transform infrared (FT-IR) spectrum revealed a distinct peak at 2050 cm−1, indicative of the stretching vibration associated with the –CN functional group.32 As shown in Fig. S8 (ESI†), Raman spectroscopy reveals peaks at 2074 cm−1 and 2115 cm−1, further confirming the presence of the –CN group.33 X-Ray photoelectron spectroscopy (XPS) was employed to analyze the chemical environments and bonding states of the elements within the Ce–Fe MOF and MOF@Zn. The XPS spectra presented in Fig. S9 (ESI†) show distinct photoelectron signals corresponding to Ce 3d, Fe 2p, O 1s, N 1s, C 1s, and K 2p, thereby confirming the presence of each element. The Ce 3d spectrum shown in Fig. S10 (ESI†) reveals peaks at 916.4 eV, 904 eV, 900.8 eV, 898.1 eV, 885.5 eV, and 882.3 eV, which correspond to the characteristic peaks of Ce4+ in the 3d5/2 and 3d3/2 states. The peaks at 903.1 eV and 883.4 eV are associated with the Ce3+ 3d5/2 and 3d3/2 states.34 Additionally, the prominent peak at 397.5 eV in the N 1s spectrum, as shown in Fig. S11 (ESI†), provides direct evidence of the –CN group.35 This finding supports the earlier conclusions, offering robust evidence for the successful synthesis of the Ce–Fe MOF and its integration into MOF@Zn. Moreover, the nearly overlapping elemental maps of Ce, Fe, K, C, N, O, and Zn obtained from energy-dispersive X-ray spectroscopy (EDS) in Fig. S12 (ESI†) not only confirm the successful integration of MOF@Zn but also demonstrate the uniform distribution of its chemical components across the surface of the zinc foil.
Dynamic regulation role of the MOF@Zn interface structure
To assess the ion sieving capability at the MOF@Zn interface, molecular dynamics (MD) simulations were performed, incorporating a specific ratio of SO42−, H2O, and Zn2+ to replicate their movement toward and through the MOF interface. As shown in Fig. 3a and Fig. S13 (ESI†), the nanometer-scale pore structure of MOF@Zn facilitates ion sieving, where the selective interaction of the highly polar organic ligand cyanide with Zn2+, coupled with the influence of the zincophilic metal center Ce, leads to significantly lower diffusion coefficients for SO42− and H2O compared to Zn2+. This finding indicates that SO42− and H2O experience limited diffusion ranges and higher resistance, enhancing the transport efficiency of Zn2+ while minimizing interference from impurity ions during transport and storage. Fig. S14 (ESI†) depicts the initial state at 0 ps, where SO42− ions are randomly distributed above the MOF@Zn interface. By 5 ps and 100 ps, it becomes evident that the SO42− gradually approach the MOF layer under the influence of an electric field. At 500 ps, the size sieving effect prevents SO42− from penetrating the MOF@Zn surface, causing them to accumulate. Similarly, as shown in Fig. S15 (ESI†), the system is initially filled with numerous water molecules at 0 ps. By 5 ps and 100 ps, H2O molecules begin to converge toward the MOF layer. At 500 ps, while a small fraction of H2O molecules manage to pass through the MOF layer to reach the Zn surface, the majority are impeded at the MOF layer and travel at an exceedingly slow rate. This restriction significantly reduces the direct contact between H2O and the Zn surface, thereby mitigating the competitive electronic interactions between H+ and Zn2+. This facilitates the efficient conversion of Zn(H2O)n2+ to Zn2+ and diminishes the likelihood of hydrogen evolution reactions. As shown in Fig. 3b, the MOF layer does not obstruct Zn2+ ; instead, the ions can freely intercalate and deintercalate, enabling normal charge–discharge processes. The pore channels of the MOF interfacial layer create a relatively stable environment, optimizing the diffusion path for Zn2+. This arrangement allows for more ordered and efficient migration of Zn2+ between electrodes, inhibiting dendrite formation and reducing unnecessary energy loss and polarization phenomena. Ultimately, this leads to the ideal conversion of Zn2+ to Zn. The MOF@Zn system demonstrates critical capabilities in sieving various transport ions and optimizing ion transport pathways, effectively suppressing side reactions such as hydrogen evolution and dendrite formation, thereby enhancing the cycle stability of the battery.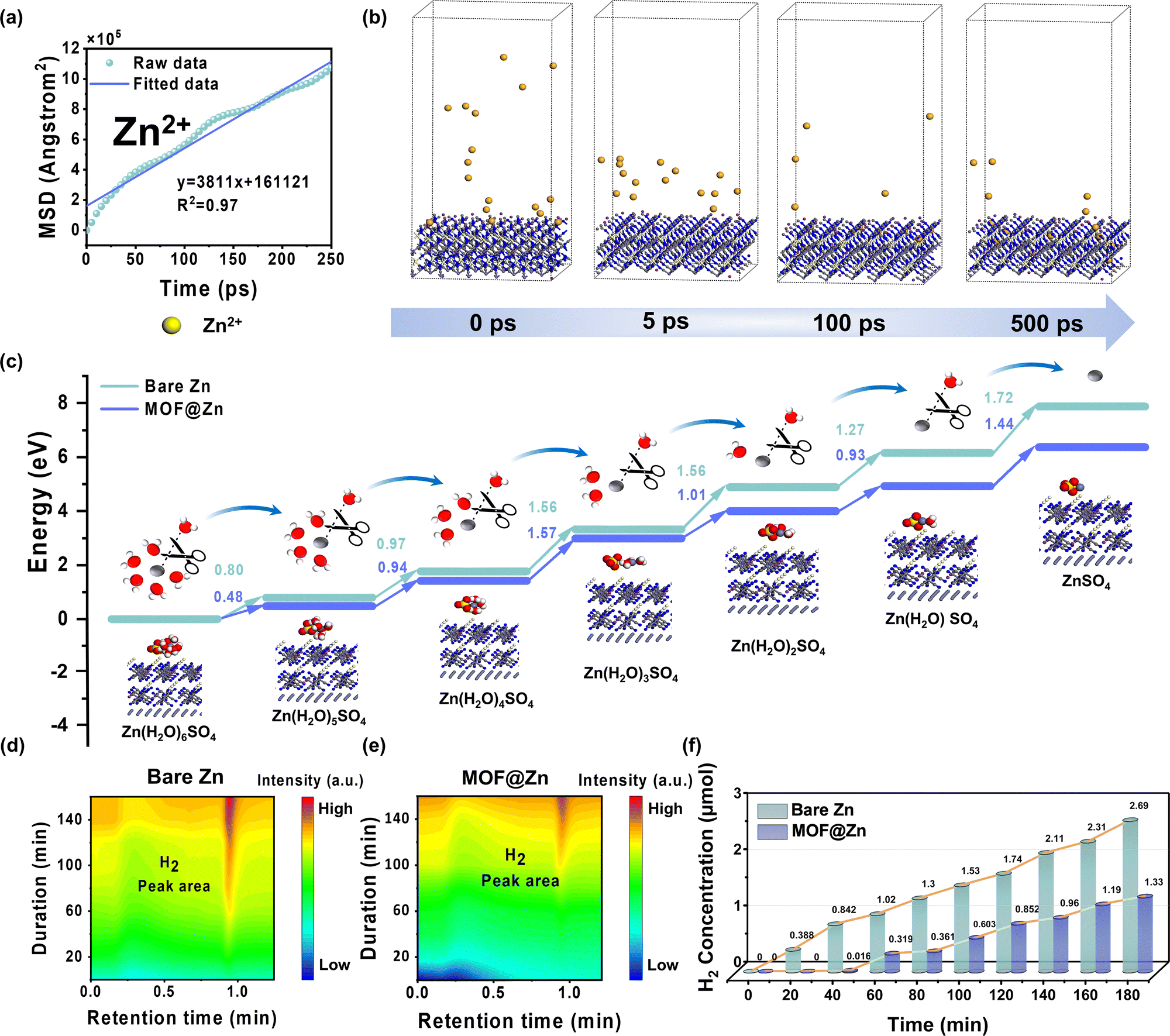 | ||
| Fig. 3 (a) Diffusion coefficients for Zn2+ within the MOF@Zn framework. (b) Side views of molecular dynamics simulations depicting in MOF@Zn. (c) DFT calculations of the energy changes of Zn(H2O)n2+ under various desolvation states at the MOF@Zn interface. (d) Gas chromatograms for bare Zn (e) and MOF@Zn. (f) In situ electrochemical gas chromatography (EC-GC) measurement of H2 production during the electrodeposition process. | ||
Ion–dipole interactions between zinc ions and water molecules in the solvent, combined with ongoing thermal motion, lead to the formation of the common solvation structure Zn(H2O)n2+. During the desolvation process at the electrode surface, free water molecules are released. These highly reactive and thermodynamically unstable water molecules can undergo hydrolysis or ionization, producing H+ and OH−. Consequently, Zn2+ can react with OH− to form the undesirable corrosion byproduct Zn4SO4(OH)6·3H2O. To evaluate the corrosion resistance of MOF@Zn, both bare Zn and MOF@Zn samples were sealed and immersed in a zinc sulfate electrolyte for a duration of 7 days. As shown in Fig. S16 (ESI†), bare Zn underwent significant degradation, losing its original metallic luster and becoming covered with numerous metallic particles post-immersion. In contrast, MOF@Zn showed no visible metallic particle formation on its surface even after prolonged immersion in the zinc sulfate electrolyte. Scanning electron microscopy (SEM) observations revealed an abundance of hexagonal platelets randomly distributed across the surface of bare Zn (Fig. S17, ESI†), corresponding to the undesired Zn4SO4(OH)6·3H2O byproduct. No such hexagonal platelets were detected on MOF@Zn before or after immersion (Fig. S18, ESI†). X-Ray diffraction (XRD) analyses, presented in Fig. S19 (ESI†), demonstrated distinct peaks associated with the Zn4SO4(OH)6·3H2O byproduct in the diffraction pattern of immersed bare Zn within the 5° to 20° range, while no corresponding peaks were observed for MOF@Zn. This further highlights the superior corrosion resistance of MOF@Zn. Additionally, roughness comparisons made using a three-dimensional confocal microscope corroborate this conclusion (Fig. S20, ESI†). The lower corrosion current and the positive shift in corrosion potential observed for the MOF@Zn anode quantitatively demonstrate a reduced corrosion rate (Fig. S21, ESI†). The remaining H+ ions readily undergo preferential reduction to produce H2, leading to the undesired hydrogen evolution reaction (HER). As shown in Fig. S22 (ESI†) and Fig. 3c, density functional theory (DFT) simulations were conducted to assess the energy required for the detachment of each water molecule during the desolvation process of Zn(H2O)n2+ on the surfaces of bare Zn and MOF@Zn. Notably, MOF@Zn demonstrates a lower desolvation energy barrier, facilitating a more rapid desolvation process. As a result, during the charging and discharging cycles of the battery, more energy is dedicated to zinc storage and release, thereby reducing energy consumption associated with overcoming the desolvation energy barrier. Moreover, an efficient desolvation process contributes to the formation of a stable solid electrolyte interphase (SEI) film, which protects the electrode from direct corrosion by the electrolyte and minimizes the production of excessively reactive water molecules, effectively inhibiting the HER. Fig. S23 (ESI†) shows that the electrochemical system with the MOF interfacial layer exhibits a higher hydrogen evolution overpotential compared to bare Zn for achieving the same current density. This increase in polarization during the HER demonstrates a significantly enhanced inhibitory effect of MOF@Zn against the HER. In electrochemical systems, the desolvation capability of electrodes is evaluated by comparing the activation energy (Ea) using the Arrhenius equation,36 which is consistent with the earlier findings (Fig. S24 and S25, ESI†). Fig. 3d and e provide direct evidence of HER inhibition by MOF@Zn through in situ EC-GC measurements of H2 production during electrodeposition. Over a 3-hour plating period with sampling intervals of 20 minutes, the H2 peak area for MOF@Zn (with a retention time of approximately 0.936 minutes) shows a gradual increase, contrasting sharply with the rapid increase in the H2 peak area observed for bare Zn. As depicted in Fig. 3f, the total amount of H2 released by bare Zn consistently exceeds that of MOF@Zn by more than twofold throughout the 3-hour electrolysis process. Additionally, Fig. S26 (ESI†) illustrates that MOF@Zn exhibits certain hydrophobic properties in a 2 M ZnSO4 electrolyte, which are beneficial for modulating interfacial water molecule behavior and hydrogen bonding interactions, thus inhibiting water molecule dissociation.37 Collectively, these simulations and experimental results robustly demonstrate the superior ability of MOF@Zn to lower the desolvation energy barrier and inhibit the HER.
As shown in Fig. 4a, the unique three-dimensional pore structure of MOF@Zn facilitates spatial confinement and molecular sieving during zinc deposition, providing uniform nucleation sites for zinc growth. This structural advantage leads to a reduced nucleation barrier for the formation of new zinc phase nuclei, resulting in a lower nucleation overpotential of 22.6 mV compared to 41.1 mV for bare Zn. Consequently, this promotes more uniform deposition of zinc ions while effectively inhibiting the formation of zinc dendrites. Furthermore, Fig. S27 (ESI†) and Fig. 4b present the results of COMSOL theoretical simulations, which analyze the electric field distribution and changes in zinc ion concentration over time for bare Zn and MOF@Zn. The physical defects on the surface of bare Zn create an uneven electric field distribution due to the tip effect, causing zinc ions to preferentially deposit at the tips. In contrast, MOF@Zn significantly mitigates this tip effect, resulting in a more uniform electric field. The simulated deposition results reflect these differences, revealing distinct concentration patterns after 30 minutes; zinc ions accumulate at the protrusions on the bare Zn surface, whereas MOF@Zn exhibits uniform deposition across its surface without localized enrichment. This disparity becomes even more pronounced at the 60-minute mark. To visualize the zinc deposition process more clearly, in situ optical microscopy was employed to observe the real-time changes in deposition behavior for both bare Zn and MOF@Zn over time (Fig. 4c). These observations align closely with the simulation results, confirming the expected deposition characteristics.
 | ||
| Fig. 4 (a) Nucleation overpotential of bare Zn and MOF@Zn. (b) Illustration of the COMSOL simulation depicting the concentration field for deposition with varying time gradients. (c) In situ optical microscopy observations of the Zn anode surface (top) and MOF@Zn anode surface (bottom) during deposition in a ZnSO4 electrolyte. (d) Ion transference numbers for bare Zn and MOF@Zn (e) chronoamperograms (CAs) for bare Zn and MOF@Zn. | ||
Two-dimensional diffusion is widely recognized as a critical factor influencing zinc deposition and the subsequent formation of zinc dendrites. Consequently, achieving a rapid transition to and stabilization of three-dimensional diffusion is a vital strategy for maintaining charge balance, mitigating the HER caused by charge accumulation, and enhancing the coulombic efficiency and long-term stability of batteries.38 A higher ion transference number is instrumental in reducing concentration polarization, allowing zinc ions to distribute uniformly throughout the electrode and minimizing the risk of dendriteformation associated with excessive local ion concentration. As shown in Fig. 4d, MOF@Zn exhibits a significantly higher ion transference number compared to bare Zn. This indicates that the three-dimensional structure and specialized ion channels within the MOF layer not only provide uniform nucleation sites but also act as an ion sieve, limiting the entry of sulfate ions and most water molecules. This design reduces the diffusion barrier and markedly accelerates ion transport.39Fig. 4e demonstrates the use of constant potential chronoamperometry (CA) at an applied voltage of −250 mV to monitor current changes over time, revealing the diffusion modes of zinc ions in both bare Zn and MOF@Zn. Unlike the prolonged two-dimensional diffusion observed in bare Zn, MOF@Zn facilitates a more rapid transition from two-dimensional to three-dimensional diffusion. Further insights are provided by scanning electron microscopy (SEM) observations depicted in Fig. S28 (ESI†), which show the surfaces of bare Zn and MOF@Zn after 0, 10, 20, and 30 cycles. In stark contrast to the significantly roughened surface of bare Zn, MOF@Zn maintains a dense zinc layer, indicative of uniform deposition. This evidence provides a clear visual demonstration of efficacy of MOF@Zn in inhibiting dendrite growth while promoting even deposition.
Battery performance of the MOF@Zn anode
To assess the commercial viability of MOF@Zn, we first performed electrochemical impedance spectroscopy (EIS, Fig. S29, ESI†) and cyclic voltammetry (CV, Fig. S30, ESI†) on assembled Zn‖Zn and MOF@Zn‖MOF@Zn symmetric batteries, as well as Zn‖Cu and MOF@Zn‖Cu asymmetric batteries. The results demonstrated that MOF@Zn exhibited a significantly lower charge transfer resistance (Rct)40 and a higher response current, along with a reduced surface nucleation overpotential. These findings suggest that MOF@Zn possesses enhanced kinetic properties compared to bare Zn, indicating improved stability and reversibility of the MOF@Zn anode as demonstrated by the assembly of Zn‖Zn of the MOF@Zn anode. Further validation of the exceptional stability of MOF@Zn‖MOF@Zn symmetric batteries was carried out by performing cyclic galvanostatic tests. As shown in Fig. 5a, MOF@Zn exhibited an exceptionally stable cycling life of approximately 4300 hours at a current density of 1 mA cm−2 with a capacity of 1 mA h cm−2. In contrast, the lifespan of bare Zn was less than one-fiftieth that of MOF@Zn, with MOF@Zn maintaining consistent polarization voltages over extended periods. Fig. S31–S33 (ESI†) further demonstrate that MOF@Zn exhibits remarkable stability even at elevated current densities and areal capacities of 2 mA cm−2 at 2 mA h cm−2, 5 mA cm−2 at 5 mA h cm−2, and 10 mA cm−2 at 5.5 mA h cm−2, respectively. In Fig. S34 (ESI†) and Fig. 5b, rate capability tests were conducted at a capacity of 1 mA h cm−2, covering low current densities ranging from 0.1 to 1.5 mA cm−2 and high current densities from 1 to 15 mA cm−2. The bare Zn electrode exhibited significant voltage instability during both low and high current density tests. In contrast, the MOF@Zn electrode successfully completed cycling tests across all current densities, demonstrating its superior electrochemical stability. | ||
| Fig. 5 (a) Voltage distribution of Zn‖Zn and MOF@Zn‖MOF@Zn symmetric batteries operating at a current density of 1 mA cm−2 and a capacity of 1 mA h cm−2. (b) Rate capability at 1 mA h cm−2. (c) Coulombic efficiency of Zn‖Cu and MOF@Zn‖Cu asymmetric batteries at a current density of 1 mA cm−2 and a capacity of 1 mA h cm−2. (d) Comparison of charge capacity (CPC), coulombic efficiency (CE), depth of discharge (DOD), and current density between MOF@Zn‖Cu and MOF@Zn‖MOF@Zn batteries, alongside relevant literature reports. | ||
To assess the superior reversibility of MOF@Zn, asymmetric batteries comprising Zn‖Cu and MOF@Zn‖Cu were constructed, and their coulombic efficiencies (CE) were evaluated at current densities of 1 mA cm−2 with a capacity of 1 mA h cm−2 (Fig. S35, ESI†) and 2 mA cm−2 with a capacity of 1 mA h cm−2. As illustrated in Fig. 5c, the bare Zn anode exhibited significant fluctuations in CE after 60 cycles at 2 mA cm−2 and 1 mA h cm−2, primarily due to detrimental processes such as dendrite formation, which ultimately led to the deactivation of the electrochemical system. In contrast, MOF@Zn demonstrated remarkable durability, sustaining over 1400 cycles with an average CE of up to 99.8% and maintaining a stable polarization voltage platform. These findings indicate that MOF@Zn not only exhibits exceptional stability but also outstanding reversibility. Furthermore, as shown in Fig. 5d, the stability and reversibility of MOF@Zn are notably superior compared to other reported materials.20,27,41–46
To further validate the significant commercial potential of MOF@Zn, electrochemical systems were constructed by integrating manganese dioxide (MnO2) as the cathode with both bare Zn and MOF@Zn as the anodes, as depicted in Fig. S36 and S37 (ESI†).
A comprehensive evaluation of their practical application capabilities was conducted by assembling MOF@Zn‖MnO2 and Zn‖MnO2 full batteries, illustrated in Fig. S38 (ESI†). As shown in Fig. 6a, the full battery utilizing the MOF@Zn anode exhibited a lower charge transfer resistance (Rct) and a higher Zn2+ diffusion coefficient (Fig. S39, ESI†),18,47 suggesting its exceptional rate capability. Fig. S40 (ESI†) and Fig. 6b present the rate capabilities of the full batteries tested across various current densities ranging from 0.1 A g−1 to 0.25 A g−1 and 1.5 A g−1 to 3 A g−1. As shown in Fig. S40 (ESI†), at low current densities, MOF@Zn not only delivers a higher capacity than bare Zn but also achieves an impressive capacity retention of 89.67%, significantly surpassing the 79.44% retention of bare Zn. Furthermore, at high current densities, as depicted in Fig. 6b, MOF@Zn maintains a capacity retention of 93.34%, markedly exceeding the 74.05% retention of bare Zn. These results from both low and high current density rate capability tests provide strong evidence supporting the conclusions mentioned above. CV tests were performed at a scan rate of 0.1 mV s−1, as illustrated in Fig. 6c, revealing that the battery incorporating MOF@Zn exhibited a significantly larger response area. This finding reflects a considerable enhancement in zinc storage capacity and electrochemical reactivity. Additionally, as depicted in Fig. 6d, the cycling stability of the full batteries with MOF@Zn and bare Zn was assessed at a high current density of 3 A g−1. After 1000 cycles, the MOF@Zn retained a specific capacity of 75.9 mA h g−1, in contrast to 47.25 mA h g−1 for bare Zn, demonstrating the robust stability of the MOF@Zn anode. Furthermore, as illustrated in Fig. 6e, multiple pouch cells were connected in series to illuminate LED lights of various colors, underscoring the vast potential of MOF@Zn for practical applications.
 | ||
| Fig. 6 (a) Nyquist plot. (b) Rate capability. (c) CV curve at a scan rate of 0.1 mV s−1. (d) Cycling performance of Zn‖MnO2 and MOF@Zn‖MnO2 full batteries. (e) Application diagram of LED color lights utilizing MOF@Zn. | ||
Conclusions
Guided by machine learning, we developed an FCNN-based algorithm to efficiently analyze the 168000 interface layer candidates and correlate them with their stability. Using this approach, this study introduces a cerium-iron bimetallic Prussian blue-based MOF interfacial layer on zinc anodes to address key issues such as hydrogen evolution and dendrite formation. The layer incorporates distinctive ion-sieving channels (∼7 Å) that effectively separate transport ions and enhance ion mobility, creating optimized ion transport pathways. Strongly polar cyano (–CN) organic ligands, along with the zincophilic cerium (Ce) cation, play crucial roles in facilitating the continuous and dynamic transformation between Zn(H2O)n2+, Zn2+, and metallic zinc states. Experimental findings reveal that the zinc anode modified with the cerium-iron bimetallic MOF interfacial layer demonstrates an impressive cycling life of nearly 4300 hours at a current density of 1 mA cm−2 with a capacity of 1 mA h cm−2. Furthermore, it achieves a high average coulombic efficiency of 99.8% after 1400 cycles at 2 mA cm−2 and 1 mA h cm−2. Notably, at a high current density of 3 A g−1, the MOF@Zn retains a specific capacity of 75.9 mA h g−1 after 1000 cycles. This research highlights an innovative and streamlined approach to protective-layer design, aimed at significantly enhancing the stability of zinc anodes.
Author contributions
Jianbo Dong: writing – review & editing, writing – original draft, data curation. Guolang Zhou: writing – review & editing, data curation. Wenhao Ding: writing – review & editing. Jiayi Ji: writing – original draft. Qing Wang: data curation. Tianshi Wang: investigation. Lili Zhang: data curation. Xiuyang Zou: writing – review & editing. Jingzhou Yin: funding acquisition, formal analysis, writing – review & editing. Edison Huixiang Ang: funding acquisition, writing – review & editing, supervision, formal analysis. Jianbo Dong and Guolang Zhou contributed equally to this work.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 52102250 and 22405093), the Natural Science Foundation of Jiangsu Province (No. BK20210951), the Natural Science Foundation of the Jiangsu Higher Education Institutions of China (No. 21KJB480008 and 24KJB150006), the Science and Technology Plan Basic Research Project of Huaian (HAB2024057), the National Institute of Education, Singapore, under its Academic Research Fund (RI 1/21 EAH and RI 3/23 EAH) and Nanyang Technological University Research Scholarship.References
- Y. Shang and D. Kundu, Joule, 2023, 7, 244–250 CrossRef
.
- Y. Zhu, G. Liang, X. Cui, X. Liu, H. Zhong, C. Zhi and Y. Yang, Energy Environ. Sci., 2024, 17, 369–385 RSC
- J. Wei, P. Zhang, J. Sun, Y. Liu, F. Li, H. Xu, R. Ye, Z. Tie, L. Sun and Z. Jin, Chem. Soc. Rev., 2024, 53, 10335–10369 RSC
- W. Ma, S. Wang, X. Wu, W. Liu, F. Yang, S. Liu, S. C. Jun, L. Dai, Z. He and Q. Zhang, Energy Environ. Sci., 2024, 17, 4819–4846 RSC
- M. Zhang, W. Xu, X. Han, H. Fan, T. Chen, Y. Yang, Y. Gao, C. Zheng, Y. Yang, T. Xiong, Y. W. Zhang, W. S. V. Lee, W. Wang, H. Pan, Z. G. Yu and J. Xue, Adv. Energy Mater., 2023, 14, 2303737 CrossRef
- Q. Zou, Z. Liang, W. Wang, D. Dong and Y.-C. Lu, Energy Environ. Mater., 2023, 16, 6026–6034 RSC
- S. Guo, L. Qin, T. Zhang, M. Zhou, J. Zhou, G. Fang and S. Liang, Energy Storage Mater., 2021, 34, 545–562 CrossRef
- H. Ren, S. Li, B. Wang, Y. Gong, H. Zhang, J. Wang, Q. Lv, D. Wang, H. Liu and S. Dou, Energy Storage Mater., 2024, 68, 103364 CrossRef
- Y. Xia, R. Tong, J. Zhang, M. Xu, G. Shao, H. Wang, Y. Dong and C. A. Wang, Nano-Micro Lett., 2024, 16, 82 CrossRef CAS PubMed
- Y. Zhou, X. Ni, B. Hao, X. Zhou, C. Yan, J. Zhou and T. Qian, Energy Storage Mater., 2024, 66, 103227 CrossRef
- Y. Zong, H. He, Y. Wang, M. Wu, X. Ren, Z. Bai, N. Wang, X. Ning and S. X. Dou, Adv. Energy Mater., 2023, 13, 2300403 CrossRef CAS
- L. Yang, M. Zhou, Y. Xie, X. Shen, S. Liang and G. Fang, Energy Storage Mater., 2024, 67, 103271 CrossRef
- Z. Hao, Y. Dai, X. Xu, X. Zhao, Y. Cong, X. Wu and W. Zhou, J. Mater. Chem. A, 2023, 11, 11031–11047 RSC
- Y. Song, P. Ruan, C. Mao, Y. Chang, L. Wang, L. Dai, P. Zhou, B. Lu, J. Zhou and Z. He, Nano-Micro Lett., 2022, 14, 218 CrossRef CAS PubMed
- J. Yang, B. Yin, Y. Sun, H. Pan, W. Sun, B. Jia, S. Zhang and T. Ma, Nano-Micro Lett., 2022, 14, 42 CrossRef CAS PubMed
- G. Qian, G. Zan, J. Li, S. J. Lee, Y. Wang, Y. Zhu, S. Gul, D. J. Vine, S. Lewis, W. Yun, Z. F. Ma, P. Pianetta, J. S. Lee, L. Li and Y. Liu, Adv. Energy Mater., 2022, 12, 2200255 CrossRef CAS
- Y. Pan, Z. Zuo, Y. Jiao and P. Wu, Adv. Mater., 2024, 36, e2314144 CrossRef PubMed
- X. Zhang, Y. Liu, P. Shen, L. Ren, D. Han, M. Feng and H. G. Wang, Adv. Funct. Mater., 2024, 34, 2400032 CrossRef CAS
- C. Guo, J. Zhou, Y. Chen, H. Zhuang, J. Li, J. Huang, Y. Zhang, Y. Chen, S. L. Li and Y. Q. Lan, Angew. Chem., Int. Ed., 2023, 62, e202300125 CrossRef CAS PubMed
- X. Chen, W. Li, S. Hu, N. G. Akhmedov, D. Reed, X. Li and X. Liu, Nano Energy, 2022, 98, 107269 CrossRef CAS
- P. Chen, X. Yuan, Y. Xia, Y. Zhang, L. Fu, L. Liu, N. Yu, Q. Huang, B. Wang, X. Hu, Y. Wu and T. van Ree, Adv. Sci., 2021, 8, e2100309 CrossRef PubMed
- W. Zhao, I. P. Perera, H. S. Khanna, Y. Dang, M. Li, L. F. Posada, H. Tan and S. L. Suib, ACS Appl. Energy Mater., 2024, 7, 1172–1181 CrossRef CAS
- S. So, Y. N. Ahn, J. Ko, I. T. Kim and J. Hur, Energy Storage Mater., 2022, 52, 40–51 CrossRef
- Y. Feng, Y. Wang, L. Sun, K. Zhang, J. Liang, M. Zhu, Z. Tie and Z. Jin, Small, 2023, 19, e2302650 CrossRef PubMed
- X. Han, H. Leng, Y. Qi, P. Yang, J. Qiu, B. Zheng, J. Wu, S. Li and F. Huo, Chem. Eng. J., 2022, 431, 133931 CrossRef CAS
- C. Wang, Q. Xie, T. Guo, M. Fang, W. Mao, Y. Zhang, H. Wang, X. Ma, Y. Wu, S. Li and J. Han, Nano Lett., 2023, 23, 10930–10938 CrossRef CAS PubMed
- Y. H. Lee, Y. Jeoun, J. H. Kim, J. Shim, K. S. Ahn, S. H. Yu and Y. E. Sung, Adv. Funct. Mater., 2023, 34, 2310884 CrossRef
- T. Zhao, H. Wu, X. Wen, J. Zhang, H. Tang, Y. Deng, S. Liao and X. Tian, Coord. Chem. Rev., 2022, 468, 214642 CrossRef CAS
- Y. Xiao, L. J. Miara, Y. Wang and G. Ceder, Joule, 2019, 3, 1252 CrossRef CAS
- W. B. Tu, S. Liang, L. N. Song, X. X. Wang, G. J. Ji and J. J. Xu, Adv. Funct. Mater., 2024, 34, 2316137 CrossRef CAS
- X. Zou, G. Xu, P. Fang, W. Li, Z. Jin, S. Guo, Y. Hu, M. Li, J. Pan, Z. Sun and F. Yan, Angew. Chem., Int. Ed., 2023, 62, e202300388 CrossRef CAS PubMed
- J.-G. Wang, Z. Zhang, X. Zhang, X. Yin, X. Li, X. Liu, F. Kang and B. Wei, Nano Energy, 2017, 39, 647–653 CrossRef CAS
- C. Shi, S. Ye, X. Wang, F. Meng, J. Liu, T. Yang, W. Zhang, J. Wei, N. Ta, G. Q. M. Lu, M. Hu and J. Liu, Adv. Sci., 2021, 8, 2001987 CrossRef CAS PubMed
- H. Xie, L. Mao and J. Mao, Chem. Eng. J., 2021, 421, 127826 CrossRef CAS
- H. Ma, M. Jiang, Z. Hou, T. Li, X. Zhang, Y. Gao, J. Peng, Y. Li and J.-G. Wang, Energy Storage Mater., 2024, 70, 103411 CrossRef
- Y. Chu, S. Zhang, S. Wu, Z. Hu, G. Cui and J. Luo, Energy Environ. Sci., 2021, 14, 3609–3620 RSC
- G. Zhou, W. Ding, Y. Guan, J. Dong, T. Wang, C. Liu, C. Zhao, L. Zhang and J. Zhu, Energy Storage Mater., 2024, 72, 103756 CrossRef
- Z. Yang, C. Lv, W. Li, T. Wu, Q. Zhang, Y. Tang, M. Shao and H. Wang, Small, 2022, 18, e2104148 CrossRef PubMed
- W. Ding, G. Zhou, X. Guo, C. Liu, T. Wang, Y. Fu, J. Yin, L. Zhang and E. H. Ang, Chem. Eng. J., 2024, 481, 148544 CrossRef CAS
- D. Sha, C. Lu, R. Hu, Z. Bao, L. Pan and Z. Sun, Energy Storage Mater., 2024, 66, 103228 CrossRef
- L. Hong, X. Wu, L. Y. Wang, M. Zhong, P. Zhang, L. Jiang, W. Huang, Y. Wang, K. X. Wang and J. S. Chen, ACS Nano, 2022, 16, 6906–6915 CrossRef CAS PubMed
- R. Wang, S. Xin, D. Chao, Z. Liu, J. Wan, P. Xiong, Q. Luo, K. Hua, J. Hao and C. Zhang, Adv. Funct. Mater., 2022, 32, 2207751 CrossRef CAS
- J. Zhou, M. Xie, F. Wu, Y. Mei, Y. Hao, R. Huang, G. Wei, A. Liu, L. Li and R. Chen, Adv. Mater., 2021, 33, e2101649 CrossRef PubMed
- J. Hao, X. Li, S. Zhang, F. Yang, X. Zeng, S. Zhang, G. Bo, C. Wang and Z. Guo, Adv. Funct. Mater., 2020, 30, 2001263 CrossRef CAS
- Z. Wang, P. Zhang, J. Zhang, K. Tang, J. Cao, Z. Yang, S. Qin, J. M. Razal, W. Lei and D. Liu, Energy Storage Mater., 2024, 67, 103298 CrossRef
- Y. Pu, Y. Zhang, K. Zhan, X. Zeng, W. Yang, Y. Zhang and X. Li, Chem. Eng. J., 2024, 494, 153002 CrossRef CAS
- H. Sun, Y. Huyan, N. Li, D. Lei, H. Liu, W. Hua, C. Wei, F. Kang and J. G. Wang, Nano Lett., 2023, 23, 1726–1734 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: Additional analysis details, Fig. S1–S40 as described in the main text. See DOI: https://doi.org/10.1039/d5ee00650c |
| ‡ Jianbo Dong and Guolang Zhou contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |