
Sr3Zr2Cu4Q9 (Q = S and Se): two novel layered quaternary mixed transition metal chalcogenides†
Sayani
Barman
 a,
Sweta
Yadav
a,
Akshay K.
Ray
a,
Swati
a,
M.
Deepa
a,
Manish K.
Niranjan
b and
Jai
Prakash
*a
a,
Sweta
Yadav
a,
Akshay K.
Ray
a,
Swati
a,
M.
Deepa
a,
Manish K.
Niranjan
b and
Jai
Prakash
*a
aDepartment of Chemistry, Indian Institute of Technology Hyderabad, Kandi, Sangareddy, Telangana 502284, India. E-mail: jaiprakash@chy.iith.ac.in
bDepartment of Physics, Indian Institute of Technology Hyderabad, Kandi, Sangareddy, Telangana 502284, India
First published on 17th December 2024
Abstract
Depending on their bandgaps, mixed metal layered chalcogenides are potential candidates for thermoelectric and photovoltaic applications. Herein, we reported the exploratory synthesis of Sr–Zr–Cu–Q (Q = S/Se) systems, resulting in the identification of two novel quaternary chalcogenides: Sr3Zr2Cu4S9 and Sr3Zr2Cu4Se9. These isoelectronic compounds (Sr3Zr2Cu4Q9) crystallized in two different structural types. The Sr3Zr2Cu4S9 structure (space group: P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) adopted the Ba3Zr2Cu4S9 structure type with eighteen unique atomic sites: 3 × Sr, 2 × Zr, 4 × Cu, and 9 × S. In contrast, the Sr3Zr2Cu4Se9 structure (P) represented a unique structure type with nineteen unique atomic positions including one additional Cu site compared to the Sr3Zr2Cu4S9 structure. The sulfide structure was stoichiometric, whereas the selenide structure was found to be non-stoichiometric with three partially occupied Cu positions. The Sr3Zr2Cu4Q9 structures consisted of
) adopted the Ba3Zr2Cu4S9 structure type with eighteen unique atomic sites: 3 × Sr, 2 × Zr, 4 × Cu, and 9 × S. In contrast, the Sr3Zr2Cu4Se9 structure (P) represented a unique structure type with nineteen unique atomic positions including one additional Cu site compared to the Sr3Zr2Cu4S9 structure. The sulfide structure was stoichiometric, whereas the selenide structure was found to be non-stoichiometric with three partially occupied Cu positions. The Sr3Zr2Cu4Q9 structures consisted of  layers with the Sr2+ cations occupying the interstitial spaces. In both structures, the Zr atoms occupied distorted octahedral positions. A striking difference between the two structures resulted from the distinct bonding interactions between the Cu and Q atoms. The optical bandgap of polycrystalline Sr3Zr2Cu4S9 was 1.7(1) eV. Interestingly, resistivity measurements of polycrystalline Sr3Zr2Cu4Se9 revealed metallic/degenerate semiconducting behavior at low temperatures. The photovoltaic performance of semiconducting Sr3Zr2Cu4S9 demonstrated ∼24% increment in power conversion efficiency when incorporated into a TiO2/CdS photoanode due to its narrower bandgap, which increased the light-harvesting ability of the cell. We also explored the theoretical electronic structures, COHP, and Bader charges of the Sr3Zr2Cu4Q9 structures using DFT calculations.
layers with the Sr2+ cations occupying the interstitial spaces. In both structures, the Zr atoms occupied distorted octahedral positions. A striking difference between the two structures resulted from the distinct bonding interactions between the Cu and Q atoms. The optical bandgap of polycrystalline Sr3Zr2Cu4S9 was 1.7(1) eV. Interestingly, resistivity measurements of polycrystalline Sr3Zr2Cu4Se9 revealed metallic/degenerate semiconducting behavior at low temperatures. The photovoltaic performance of semiconducting Sr3Zr2Cu4S9 demonstrated ∼24% increment in power conversion efficiency when incorporated into a TiO2/CdS photoanode due to its narrower bandgap, which increased the light-harvesting ability of the cell. We also explored the theoretical electronic structures, COHP, and Bader charges of the Sr3Zr2Cu4Q9 structures using DFT calculations.
1. Introduction
Inorganic multinary chalcogenide compounds, featuring two or more types of metals/metalloids and chalcogen elements (Q), such as sulfur, selenium, and tellurium, represent a fascinating class of materials. Their tunable compositions and structural flexibility enable the tailoring of physical properties for a wide range of technological applications. Therefore, chalcogenides have emerged as potential materials for a variety of applications, such as high-temperature superconductivity,1,2 photovoltaics,3,4 catalysis,5,6 thermoelectric properties,7,8 and non-linear optics.9–12 Unlike binary counterparts, multinary chalcogenides allow the incorporation of multiple metals and chalcogen atoms, opening a vast design space to tailor their properties. This flexibility allows for fine-tuning of the energy difference between the electron-filled and empty states, thereby influencing their electrical and thermal conductivity, light absorption, and emission. Many chalcogenides are also well known for their sensitivity to light and other stimuli, making them ideal candidates for use in advanced sensors and imaging technologies.13–16Copper-containing chalcogenides, especially sulfides and selenides, are emerging materials for photovoltaic and thermoelectric (TE) applications due to their non-toxicity and earth abundance.4,17–20 The high mobility of Cu1+ ions in these multinary chalcogenide structures is well known in compounds such as Cu2−xSe,21 Cu7PSe6,22 CuCrSe2,23 and AgCuSe.24 However, some of these structures are unstable at higher temperatures due to the migration of Cu ions within their crystal lattices (e.g., Cu2−xSe).25,26 Unlike copper oxides, the Cu atoms in chalcogenides exist in the monovalent state and are mostly four-coordinated with a tetrahedral coordination geometry. Notable examples of such compounds include BaCu6−xSeTe6,27 BaLnCuS3 (Ln = Pr, Sm, Dy, Ho, Yb),28,29 and Cu4SnS4.30 In these structures, alkali (A) and alkaline-earth (Ak) metals function as electron donors and exhibit higher coordination numbers than the transition metals. The A1+ or Ak2+ ions in these compounds can be replaced with lower or higher valent chemical dopants to control the number of charge carriers. Also, many of these multinary chalcogenides offer low intrinsic thermal conductivity values due to their complex structures and chemical bonding. Therefore, the Cu-containing multinary chalcogenides with narrow bandgaps are actively pursued for their potential thermoelectric applications. Examples of such compounds are ACuZrQ3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 31 and BaSc1−xGdxCuTe3.32
31 and BaSc1−xGdxCuTe3.32
Moreover, the tunable bandgaps of the Cu/Ag-containing chalcogenides also make them potential candidates for solar cells, light-emitting diodes (LEDs), and photocatalysis applications. As discussed earlier, thanks to the high mobility of copper or silver atoms in these semiconducting chalcogenides, additional electronic levels close to the valence band are present in these compounds, making them efficient in electron transfer rather than hole recombination on sunlight absorption.33,34 BaCu2SnS4−xSex,35 CuGaSe2,36 CuSbS2,37 BaCeCuS3,38 Cu2ZnSnQ4 (Q = S, Se),39 and CuInxGa1−x(SxSe1−x)240 are a few examples of such chalcogenide materials with excellent photovoltaic power conversion efficiencies. CuSbS237 is one of the chalcogenides actively explored for photovoltaics and thermoelectric applications.
Therefore, many research groups worldwide have been actively synthesizing new Cu/Ag-containing multinary chalcogenides with semiconducting properties to identify new potential materials for solar cells and TE applications. We have produced several novel complex metal chalcogenides in our quest to develop new semiconducting compounds for TE and solar cell applications; these include 12-BaBi2S4,41 Ba8Zr2Se11(Se2),42 and BaCeCuS3.38 In this work, we report the synthesis of two novel compounds, namely, Sr3Zr2Cu4S9 and Sr3Zr2Cu4Se9, with layered structures. Sr3Zr2Cu4S9 is the first quaternary phase in the Sr–Zr–Cu–S system, while Sr3Zr2Cu4Se9 is the second member of the Sr–Zr–Cu–Se system for which only the quaternary phase, Sr0.5CuZrSe3,43 is reported. Sr3Zr2Cu4Se9 represents a new structure type. We have studied the structural aspects and physical properties of Sr3Zr2Cu4Q9 compounds. Thermoelectric properties of Sr3Zr2Cu4Se9, a semimetal/degenerate semiconductor, are explored in detail. The Sr3Zr2Cu4S9 sulfide, a semiconductor with an optical bandgap of 1.7(1) eV, is explored for photovoltaic applications. Incorporating the quaternary title sulfide phase in a TiO2/CdS photoanode shows a 24% increment in the power conversion efficiency of the fabricated solar cell. The electronic band structures of the Sr3Zr2Cu4Q9 phases are studied using the DFT method.
2. Experimental section
2.1 Chemicals used and synthetic methodologies
Sr3Zr2Cu4S9 and Sr3Zr2Cu4Se9 phases were synthesized using S/Se powder, Cu powder, Zr rod, and Sr granules. All these elements were procured from Sigma Aldrich with purities ≥99.5%. The Sr granules were cut into smaller pieces, and the zirconium rod was filed into powder for the reactions. Unless stated otherwise, all the chemicals are handled inside a glove box filled with argon gas.We used the SnO2:F (FTO) coated glass substrates (Pilkington) with a sheet resistance of ∼25 Ω cm−2 to fabricate solar cells for photovoltaic studies. Other chemicals (reagent grade) used for preparing the solar cells and photovoltaic measurements are the following: TiO2 P25 powder, fumed silica and Ni-foam were procured from Evonik, Cabosil, and Gelon, respectively, and cadmium acetate, sodium sulfide, titanium tetrachloride, methanol, toluene, Triton X-100, acetylacetone, multi-walled carbon nanotubes (MWCNTs), polyvinylidene fluoride (PVdF), carbon black (CB), and N-methyl-2-pyrrolidone (NMP) were purchased from Merck and used directly.
Similarly, a vacuum-sealed tube containing the elements strontium (37.28 mg, 0.43 mmol), zirconium (25.88 mg, 0.28 mmol), copper (36.05 mg, 0.57 mmol), and selenium (100.79 mg, 1.28 mmol) was also heated in the furnace to prepare single crystals of Sr3Zr2Cu4Se9.
The tubes were heated in the furnace using the temperature profile described in section S1 of the ESI.† After completion of the reactions, dark reddish and black-colored melted lumps were found for the reactions targeting the sulfide and selenide, respectively. These lumps were further fractured, and a few crystals were chosen for their semiquantitative elemental energy-dispersive X-ray spectroscopic (EDX) analyses using a field-emission scanning electron microscope (FE-SEM) (Model: JSM 7610F) and an X-ray spectrometer (octane elite, EDAX Inc., USA).
Most of the red-colored crystals selected from the sulfide reaction product showed Sr:Zr:Cu:S ≈ 3:2:4:9, consistent with the loaded Sr3Zr2Cu4S9 composition (Fig. 1a). A few crystals turned out to be binary SrS (1:1). The crystals of Sr3Zr2Cu4Se9 were black in color, and their average composition was also consistent with the loaded ratio of Sr:Zr:Cu:Se ≈ 3:2:4:9 (Fig. 1a inset).
 | ||
| Fig. 1 (a) The EDX spectrum of a typical Sr3Zr2Cu4S9 crystal. The inset shows the EDX spectrum of a Sr3Zr2Cu4Se9 crystal. (b) The Le Bail refinement plots of the powder X-ray diffraction (PXRD) patterns of Sr3Zr2Cu4S9 and Sr3Zr2Cu4Se9. | ||
A few of these EDX-analyzed red and black-colored crystals were further used for structural characterization at room temperature (RT) using the single-crystal X-ray diffraction (SCXRD) method.
Solid state method. Phase pure bulk polycrystalline Sr3Zr2Cu4Q9 (Q = S, Se) phases were prepared by first heating the respective elements using the temperature profile discussed in section S1 of the ESI† and then reheating the homogenized products in the form of compressed pellets inside the vacuum-sealed fused silica tubes in two steps.
The elements, strontium (266.03 mg, 3.04 mmol), zirconium (184.65 mg, 2.02 mmol), copper (257.25 mg, 4.05 mmol), and sulfur (292.07 mg, 9.11 mmol), were sealed in a fused silica tube under vacuum and heated inside the furnace to synthesize polycrystalline Sr3Zr2Cu4S9.
Similarly, a vacuum-sealed fused silica tube containing strontium (186.41 mg, 2.13 mmol), zirconium (129.38 mg, 1.41 mmol), copper (180.26 mg, 2.84 mmol), and selenium (503.95 mg, 6.38 mmol) was heated in the furnace to prepare the polycrystalline Sr3Zr2Cu4Se9 using the heating profile discussed in section S1 of the ESI.†
The products of the reactions were homogenized into fine powders and were compressed individually into cylindrical-shaped thin pellets. These pellets were again encapsulated inside evacuated fused silica tubes and annealed at 1023 K for two days. This step is required to make high-quality polycrystalline Sr3Zr2Cu4Q9 (Q = S, Se) samples. The heat-treated pellets were ground again, and the products were characterized using the PXRD studies.
We also attempted to synthesize polycrystalline Sr3Zr2Cu4S9 by heating stoichiometric amounts of metal oxides/carbonates under N2 and CS2 atmosphere at 1073 K for 9 h. The details of this CS2 method are given in section S2 of the ESI.† The ease of making bulk samples and the scalability of the method without the need for expensive fused silica tubes were the motivations behind attempting the CS2 method. However, we failed to synthesize the phase pure material via the CS2 method. Further optimization of the reaction temperature and time is required to find suitable reaction conditions to synthesize the Sr3Zr2Cu4S9 phase in the future.
The PXRD data sets of the powdered samples mentioned above were collected with the help of a PANalytical X'pert pro-X-ray diffractometer (Cu-Kα, λ = 1.5406 Å) at RT. The TOPAS V6 software program was used to refine the unit cell parameters of the polycrystalline samples, Sr3Zr2Cu4Q9, using the Le Bail refinement method (Fig. 1b).44
2.2 Single-crystal X-ray diffraction studies
Two SCXRD datasets for the Sr3Zr2Cu4S9 and Sr3Zr2Cu4Se9 crystals at RT under a normal atmosphere were recorded using a Bruker D8 Venture diffractometer. We used EDX-characterized crystals for the SCXRD studies. The diffractometer was fitted with a Photon III mixed-mode X-ray detector. Graphite was utilized to monochromatize the X-ray radiations from a Mo-Kα radiation source (λ = 0.71073 Å). The intensity data for the Sr3Zr2Cu4Q9 (Q = S, Se) crystals were collected using a series of ω and φ scans with 0.5° frame width and 2 s per frame of the exposure time with the help of the APEX3 software.45The crystal structures for the Sr3Zr2Cu4Q9 compounds were determined and refined in the centrosymmetric triclinic space group P with the help of the SHELXTL46 program suite. For the sulfide structure, all the atomic positions were easily assigned to respective elements, which resulted in the formula Sr3Zr2Cu4S9. However, the reliability factor values were poor: R(F) = 8.87%, Rw(Fo2) = 26.72%, and the maximum positive residual electron density was 13.3 e− Å−3. Later, the CELLNOW45 program was used to check the presence of any twinning in the dataset, which identified two twin domains for the crystal. Also, absorption correction was done afterward using the TWINABS.47 The second twin domain was found to be rotated from the first domain with an angle of about 180° along the [1 0 1] direction. After these steps, the refinement parameters were significantly improved with final R(F) = 4.74% and Rw(Fo2) = 12.38%. The Sr3Zr2Cu4S9 structure is stoichiometric and isostructural to the Ba3Zr2Cu4S948 (space group: P) structure. The final structure solution identified a total of 18 independent sites, out of which three sites were identified as Sr, two as Zr, four sites as Cu, and the remaining nine as S sites. The STRUCTURE TIDY49 program was used to standardize the atomic positions of the Sr3Zr2Cu4Q9 structure.
We also found multiple twin domains for the Sr3Zr2Cu4Se9 dataset using the CELLNOW45 program. Similar to the sulfide structure, for this structure as well, the two domains are related with a rotation angle of about 180° with the [1 0 1] direction as the rotation axis. The APEX345 was used to integrate the SCRXD dataset using two twin domains in the triclinic setting. The TWINABS47 was used for the X-ray absorption corrections of the observed reflections. The final structure solution of the Sr3Zr2Cu4Se9 with the P space group was comprised of nineteen crystallographically independent sites. The structure was refined considering 100% occupancy for all the atoms. The reliability parameters R(F) and Rw(Fo2) were 6.68% and 17.56%, respectively. The anisotropic displacement parameters (ADPs) of the Cu atoms were abnormally large, indicating some vacancies at these sites. Subsequently, the fractional occupancy factor (SOF) values of these sites were allowed to be refined freely, resulting in an improvement of the reliability parameters: R(F) = 5.34% and Rw(Fo2) = 11.82%. The freely refined SOF values of the Cu1, Cu3, and Cu4 atomic sites are 0.598, 0.856, and 0.533. The final refined formula, Sr3Zr2Cu3.99(1)Se9, is close to the electrically neutral composition, Sr3Zr2Cu4Se9. Thus, we refer to the chemical formula of the selenide as Sr3Zr2Cu4Se9 in this study.
We finally validated the symmetry of the Sr3Zr2Cu4S9 and Sr3Zr2Cu4Se9 structures using the ADDSYM program of the PLATON50 software. Further crystallographic details are given in Tables 1, 2 and Tables S1, S2, S3, and S4 in the ESI.†
| a λ = 0.71073 Å, T = 298(2) K. b R(F) = ∑||Fo| − |Fc||/∑|Fo| for Fo2 > 2σ(Fo2). c R w(Fo2) = {∑[w(Fo2 − Fc2)2]/∑wFo4}1/2. For Fo2 < 0, w = 1/[σ2(Fo2) + (mP)2 + nP]; where P = (Fo2 + 2Fc2)/3. For the sulfide structure, m = 0.0574 and n = 6.3137, and for the selenide structure, m = 0 and n = 27.2292. | ||
|---|---|---|
| Sr3Zr2Cu4S9 | Sr3Zr2Cu4Se9 | |
| Space group |
P |
|
| a (Å) | 6.6907(13) | 6.9779(14) |
| b (Å) | 10.312(2) | 10.685(2) |
| c (Å) | 11.929(2) | 12.302(3) |
| α (°) | 109.49(3) | 109.18(3) |
| β (°) | 104.54(3) | 105.38(3) |
| γ (°) | 96.73(3) | 96.56(3) |
| V (Å3) | 732.6(3) | 814.6(3) |
| Z | 2 | |
| ρ (g cm−3) | 4.479 | 5.745 |
| μ (mm−1) | 19.12 | 36.13 |
| R(F)b | 0.047 | 0.053 |
| R w(Fo2)c | 0.124 | 0.118 |
| Sr3Zr2Cu4S9 | Sr3Zr2Cu4Se9 | |
|---|---|---|
| Zr–Q (Å) | 2.490(2)–2.694(2) | 2.6342(19)–2.8083(18) |
| Tetrahedral Cu–Q (Å) | 2.309(2)–2.372(2) | 2.386(4)–2.682(5) |
| Trigonal Cu–Q (Å) | 2.234(2)–2.312(2) | — |
| Sr–Q (Å) | 2.924(3)–3.628(2) | 3.0655(19)–3.4277(19) |
2.3 Solid-state ultraviolet-visible (UV-vis)-near-infrared (NIR) spectroscopy
Diffuse reflectance datasets of the polycrystalline Sr3Zr2Cu4S9 and Sr3Zr2Cu4Se9 samples were recorded at RT using a spectrophotometer JASCO V-770. A dry sample of BaSO4 was used for calibration. The Kubelka–Munk equation51 was utilized to obtain absorption data from the reflectance data.2.4 Thermal conductivity (κtot) measurements
Sr3Zr2Cu4S9 and Sr3Zr2Cu4Se9 powders were first pressed into thin cylindrical pellets (dia. 8 mm) and then sintered at 973 K (for 24 h) using the sealed tube method. A hydraulic press was used to compact the powders into hard pellets under ca. 8 tons of pressure. The density (d) values of the resulting compact pellets of Sr3Zr2Cu4S9 and Sr3Zr2Cu4Se9 were determined using the Archimedes method.52 The experimental density values were about 92% of the theoretical density.The thermal diffusivity (D) values of Sr3Zr2Cu4S9 and Sr3Zr2Cu4Se9 were measured using a LINSEIS XFA 500 instrument. The details of the measurements are given elsewhere.41 The Dulong–Petit law53 was employed to estimate the heat capacity (Cp) values of the Sr3Zr2Cu4S9 and Sr3Zr2Cu4Se9 samples. Finally, the total thermal conductivity (κtot = D × d × Cp) values were calculated. The final κtot value has an uncertainty of around ±10%.
2.5 Transport property measurements for Sr3Zr2Cu4S9 and Sr3Zr2Cu4Se9
The sintered rectangular parallelepiped-shaped Sr3Zr2Cu4S9 and Sr3Zr2Cu4Se9 pellets were obtained using the method described in section 2.4. The electrical resistivity (ρ) values of the Sr3Zr2Cu4Q9 (Q = S, Se) pellets were measured at low temperatures using the standard four-contact method. The pellets were cooled using a Janis SHI-950 closed-cycle cryostat, and the four contacts on each of the pellets were made using the Cu-probes (0.05 mm diameter) and Ag-paint. A nanovoltmeter (Keithley, Model: 2182A) and a current source (Keithley, Model: 6221) were used for the study.The Sr3Zr2Cu4Se9 pellet was further used for the simultaneous determination of electrical resistivity and Seebeck coefficient (S) values at higher temperatures (323 K–573 K) with the help of a ULVAC-RIKO ZEM-3 equipment under a low-pressure He-atmosphere. No hysteresis between the heating and cooling cycles of the measured data was observed.
2.6. Photovoltaic study of Sr3Zr2Cu4S9
A photoanode, FTO/TiO2/CdS, was prepared using the method described elsewhere.38 The powdered Sr3Zr2Cu4S9 was dispersed in toluene at 1 mg mL−1 concentration by ultrasonication for 2 h and then drop-casted over the FTO/TiO2/CdS electrode and dried under vacuum for two days to obtain a FTO/TiO2/CdS/Sr3Zr2Cu4S9 photoanode.A counter electrode (CE) was fabricated by coating MWCNTs over a Ni foam substrate. CB, PVdF, and MWCNTs were mixed in a 1:1:8 weight ratio, ground well, and converted into a slurry using NMP. The slurry was coated on the Ni-foam and dried. A polysulfide gel was prepared by adding 5 wt% of fumed silica in an aqueous suspension of (1 M Na2S + 1 M S) under constant stirring until it became a uniform gel.
Finally, we made the solar cells, FTO/TiO2/CdS/S2−, S/MWCNTs/Ni-foam and FTO/TiO2/CdS/Sr3Zr2Cu4S9/S2−, and S/MWCNTs/Ni-foam, by sandwiching the polysulfide gel in between the photoanodes and the CE using binder clips. These solar cells were subjected to I–V measurements with the help of a Newport Oriel 3A solar simulator coupled with a Keithley 2420 source meter. A 450 W Xenon arc lamp was used as the light source with a light intensity of 100 mW cm−2 and Air Mass (AM) 1.5G illumination; the spatial uniformity of irradiance was determined by calibrating with a 2 cm × 2 cm Si reference cell and confirmed using a Newport power meter.
2.7 Theoretical and computational methodologies
We employed density functional (DFT) calculations54 to investigate the electronic structures and estimate the structural parameters of relaxed Sr3Zr2Cu4S9 and Sr3Zr2Cu4Se9 unit cells. The Kohn–Sham equations within the DFT framework are solved using the plane wave basis expansion with the help of the VASP package.55 Further details of the theoretical calculations are given in the ESI (see Section S3).† We used the Sr3Zr2Cu4Se9 unit cell with half-occupied (50%) Cu1 and Cu4 atomic sites for the calculations.3. Results and discussion
3.1 Syntheses
The Sr3Zr2Cu4S9 and Sr3Zr2Cu4Se9 crystals were prepared by the reaction of elements at high temperatures (Tmax = 1223 K) and slow cooling of the reaction mixtures to RT. The Sr3Zr2Cu4S9 crystals were red in contrast to the black-colored crystals of the Sr3Zr2Cu4Se9 phase.Furthermore, we used two methods to prepare bulk polycrystalline samples of Sr3Zr2Cu4S9: (a) the direct reaction of elements at high temperatures and (b) the CS2 method. The CS2 method failed to produce the targeted Sr3Zr2Cu4S9 phase. Instead, the cubic SrS phase (space group: Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m)56 was formed as the major phase according to the PXRD analysis (see Fig. S1 of the ESI†). Presumably, the phases containing Zr and Cu are amorphous in the powdered sample obtained by the CS2 reaction. On the other hand, the high-temperature reaction of elements with one intermittent grinding and reheating produced a monophasic Sr3Zr2Cu4S9 sample. The polycrystalline Sr3Zr2Cu4Se9 sample was prepared using heating conditions similar to those employed for the sulfide sample.
m)56 was formed as the major phase according to the PXRD analysis (see Fig. S1 of the ESI†). Presumably, the phases containing Zr and Cu are amorphous in the powdered sample obtained by the CS2 reaction. On the other hand, the high-temperature reaction of elements with one intermittent grinding and reheating produced a monophasic Sr3Zr2Cu4S9 sample. The polycrystalline Sr3Zr2Cu4Se9 sample was prepared using heating conditions similar to those employed for the sulfide sample.
The PXRD patterns of the Sr3Zr2Cu4Q9 (Q = S, Se) phases are shown in Fig. 1b. The refined unit cell parameters of Sr3Zr2Cu4S9 and Sr3Zr2Cu4Se9, calculated from the PXRD data using the Le Bail refinement method, are a = 6.695(2) Å, b = 10.322(1) Å, c = 11.941(2) Å, α = 109.60(1)°, β = 104.54(1)°, γ = 96.67(1)°, and a = 6.986(2) Å, b = 10.695(2) Å, c = 12.272(1) Å, α = 109.33(1)°, β = 105.35(1)°, γ = 96.57(1)°, respectively. The cell parameters of Sr3Zr2Cu4Q9 (Q = S, Se) obtained from the powder and single crystal XRD studies are in good agreement.
3.2 Crystal structure description
The quaternary sulfide Sr3Zr2Cu4S9 is the first quaternary compound found in the Sr–Zr–Cu–S system, whereas Sr3Zr2Cu4Se9 is the second member of the Sr–Zr–Cu–Se system for which a Sr-deficient phase Sr0.5ZrCuSe343 is known in the literature.
The single-crystal XRD study showed that, at RT, Sr3Zr2Cu4S9 crystallizes in the triclinic Ba3Zr2Cu4S948 structure. As expected, the unit cell volume of the centrosymmetric triclinic Sr3Zr2Cu4S9 structure (space group: P) with the refined cell parameters a = 6.6907(13) Å, b = 10.312(2) Å, c = 11.929(2) Å, α = 109.49(3)°, β = 104.54(3)°, and γ = 96.73(3)°, is smaller than the Ba3Zr2Cu4S9 cell. These experimental lattice parameters augur well with those obtained from theoretical calculations (see section 3.7). The asymmetric unit of the Sr3Zr2Cu4S9 structure contains nine sulfur and nine metal sites (3 × Sr sites, 2 × Zr sites, and 4 × Cu sites). As seen in Fig. 2a, the  layers of the Sr3Zr2Cu4S9 structure are separated by the Sr2+ cations. The anionic
layers of the Sr3Zr2Cu4S9 structure are separated by the Sr2+ cations. The anionic  layer features distorted ZrS6 octahedra, CuS4 tetrahedra (Cu2 and Cu4), and trigonal planar CuS3 units (Cu1 and Cu3), which serve as the main motifs of the structure.
layer features distorted ZrS6 octahedra, CuS4 tetrahedra (Cu2 and Cu4), and trigonal planar CuS3 units (Cu1 and Cu3), which serve as the main motifs of the structure.
 | ||
| Fig. 2 The unit cells of (a) Sr3Zr2Cu4S9 and (b) Sr3Zr2Cu4Se9 crystal structures along the b-axis. | ||
The Sr3Zr2Cu4Se9 structure is isoelectronic to the related sulfide. Interestingly, its structure is different from the Sr3Zr2Cu4S9 structure. The selenide adopts the original structure type with triclinic symmetry (space group: P). The refined cell constants of the Sr3Zr2Cu4Se9 structure are a = 6.9779(14) Å, b = 10.685(2) Å, c = 12.302(3) Å, α = 109.18(3)°, β = 105.38(3)°, and γ = 96.56(3)°. In contrast to the sulfide structure, the Sr3Zr2Cu4Se9 phase is non-stoichiometric with partially vacant Cu sites. Sr3Zr2Cu4Se9 is best described as the pseudo-2D structure, like the sulfide structure, as shown in Fig. 2b.
The selenide structure comprises nineteen independent atoms (3 × Sr, 2 × Zr sites, 5 × Cu sites, and 9 × Se sites), i.e., one additional Cu site in reference to the sulfide structure. The three Cu sites, Cu1, Cu3, and Cu4, have free SOF values of 0.598(8), 0.856(7), and 0.532(8), respectively, in the Sr3Zr2Cu4Se9 structure. The composition of the infinite layers present in the selenide structure is identical to the sulfide structure, i.e.,  and their negative charges are counterbalanced by the Sr2+ ions. The Zr and Cu atoms in the selenide structure are octahedrally and tetrahedrally coordinated with the Se atoms, respectively. The short Cu1⋯Cu1 and Cu4⋯Cu4 distances (<2.2 Å) in the Sr3Zr2Cu4Se9 structure are the result of their occupancy disorder and imply that these Cu atoms do not coexist simultaneously close to each other in the structure.
and their negative charges are counterbalanced by the Sr2+ ions. The Zr and Cu atoms in the selenide structure are octahedrally and tetrahedrally coordinated with the Se atoms, respectively. The short Cu1⋯Cu1 and Cu4⋯Cu4 distances (<2.2 Å) in the Sr3Zr2Cu4Se9 structure are the result of their occupancy disorder and imply that these Cu atoms do not coexist simultaneously close to each other in the structure.
Next, we compare the layers of the Sr3Zr2Cu4S9 and Sr3Zr2Cu4Se9 structures in Fig. 3.
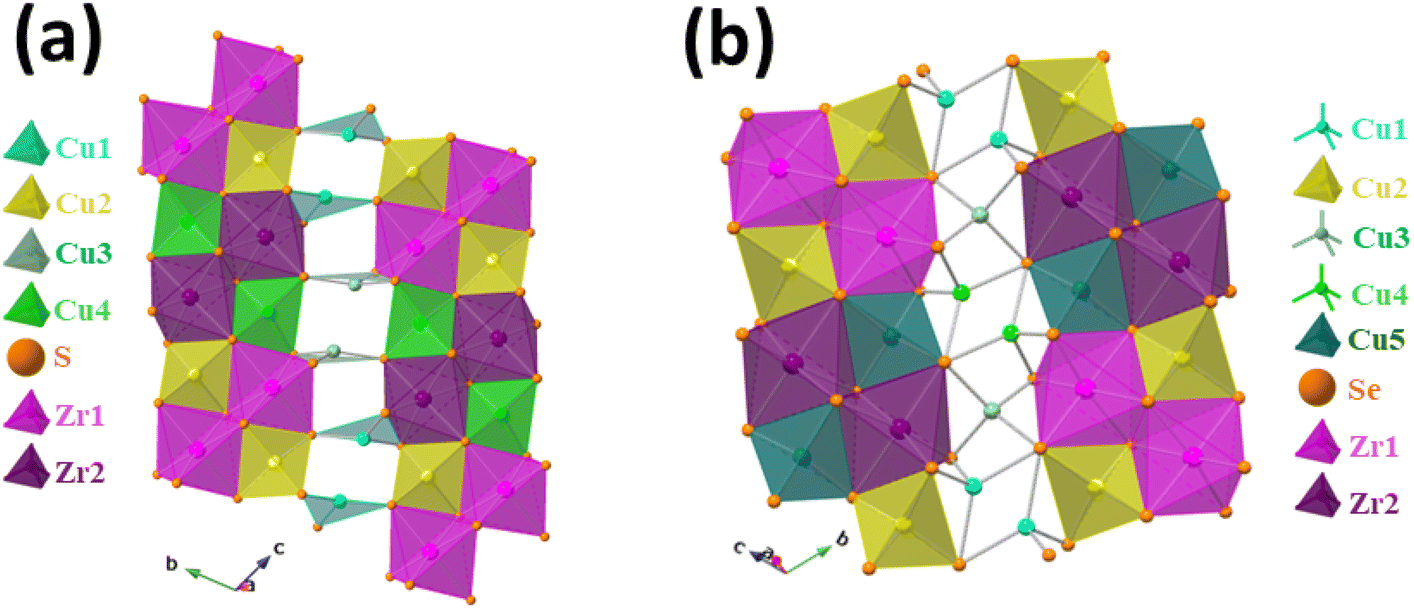 | ||
Fig. 3 (a) The  chains that are bridged by the Cu1 and Cu3 atoms in the Sr3Zr2Cu4S9 and (b) chains that are bridged by the Cu1 and Cu3 atoms in the Sr3Zr2Cu4S9 and (b)  chains of the Sr3Zr2Cu4Se9 that are bridged by the Cu1, Cu3, and Cu4 atoms to form infinite layers of chains of the Sr3Zr2Cu4Se9 that are bridged by the Cu1, Cu3, and Cu4 atoms to form infinite layers of  . . | ||
The layers of both the structures have edge and vertex-sharing ZrQ6 octahedra (see Fig. 3). The oppositely oriented Cu2S4 and Cu4S4 tetrahedra share edges and vertices with the ZrS6 octahedra to form chains of  (see Fig. 3a). Similarly, the connectivity of ZrSe6 octahedra, Cu2Se4, and Cu5Se4 tetrahedra in the Sr3Zr2Cu4Se9 structure creates
(see Fig. 3a). Similarly, the connectivity of ZrSe6 octahedra, Cu2Se4, and Cu5Se4 tetrahedra in the Sr3Zr2Cu4Se9 structure creates  chains (see Fig. 3b). Finally, the
chains (see Fig. 3b). Finally, the  layers in both structures are formed by filling of Cu atoms in the structures, which hold together the
layers in both structures are formed by filling of Cu atoms in the structures, which hold together the  chains, as shown in Fig. 3. The main difference between the layers of the title structures is the bonding environments of the additional Cu atoms, Cu1 and Cu3, of the sulfide structure and Cu1, Cu3, and Cu4 atoms in the selenide structure. The Cu atoms (Cu1 and Cu3) in the sulfide structure are only three-coordinated in contrast to the four-coordinated (tetrahedral) Cu atoms of the selenide structure.
chains, as shown in Fig. 3. The main difference between the layers of the title structures is the bonding environments of the additional Cu atoms, Cu1 and Cu3, of the sulfide structure and Cu1, Cu3, and Cu4 atoms in the selenide structure. The Cu atoms (Cu1 and Cu3) in the sulfide structure are only three-coordinated in contrast to the four-coordinated (tetrahedral) Cu atoms of the selenide structure.
The important interatomic distances of the Sr3Zr2Cu4Q9 structures are provided in Table 2. The Zr–S distances of Sr3Zr2Cu4S9 (see Table 2) match well with the corresponding distance of the Ba3Zr2Cu4S9 (2.525(1) Å–2.704(1) Å)48 structure. The Cu–S distances of the CuS3 unit (2.234(2) Å–2.312(2) Å) are shorter than the tetrahedral CuS4 units (2.309(2) Å–2.372(2) Å) in the title sulfide structure. These distances compare well with those found in Ba3Zr2Cu4S9 (2.241(2) Å–2.364(2) Å)48 and the BaCeCuS3 (2.351(1) Å–2.461(2) Å)38 structures.
The Cu–Se (2.386(4) Å–2.682(5) Å) distances of Sr3Zr2Cu4Se9 are consistent with those found in the BaCu2Se2 (2.456(1) Å to 2.618(3) Å)57 and BaNdCuSe3 (2.460(4) Å to 2.567(3) Å)58 structures. The Zr–Se bond distances of ZrSe6 octahedra vary from 2.6342(19) Å to 2.8083(18) Å in the Sr3Zr2Cu4Se9 structure. These distances are in good agreement with those reported for the Ba0.5ZrCuSe3 (2.661(7) Å to 2.707(5) Å)43 and Ba8Zr2Se11(Se2) (2.6337(7) Å to 2.7940(7) Å)42 structures.
Another interesting feature of Sr3Zr2Cu4Q9 (Q = S, Se) is the Cu⋯Cu interactions, which are intermediate to the single Cu–Cu bond (∼2.556 Å)59 and the van der Waals distance of 2.80 Å (ref. 60) between two Cu atoms. These interactions are shown in Fig. 4 for the Sr3Zr2Cu4Q9 (Q = S, Se) structures. The Cu⋯Cu interactions are shorter in the Sr3Zr2Cu4Se9 structure (2.621(5) Å to 2.711(4) Å) than in the Sr3Zr2Cu4S9 structure (2.753(3) Å to 2.845(3) Å). Similar Cu⋯Cu interactions are also known for the Cu5FeS4 (2.587 Å–2.989 Å),61 K2Cu3AlS4 (2.734(3) Å–2.736(3) Å),62 and Cu3SbSe3 (2.665(2) Å)63 structures. The chalcogen atoms of Sr3Zr2Cu4Q9 structures make distorted bicapped trigonal prism-like geometries around the Sr atoms, as shown in Fig. S2 in the ESI.†
 | ||
Fig. 4 A fragment of the  layers showing the short Cu⋯Cu interactions in (a) Sr3Zr2Cu4S9 and (b) Sr3Zr2Cu4Se9 structures. layers showing the short Cu⋯Cu interactions in (a) Sr3Zr2Cu4S9 and (b) Sr3Zr2Cu4Se9 structures. | ||
To the best of our knowledge, only monovalent copper species are known for the chalcogenide structures. Also, the Sr3Zr2Cu4Q9 structures are traditional chalcogenides (not polychalcogenides) with more stable Q2− species. Therefore, these two structures can be charge balanced with the Sr2+, Zr4+, Cu1+, and Q2− species, i.e., the charge partitioned formula of the title phases is (Sr2+)3(Zr4+)2(Cu1+)4(Q2−)9.
3.3 Structural relationship between Ak3Zr2Cu4S9 (Ak = Ba and Sr), Sr3Zr2Cu4Se9, and Sr0.5ZrCuSe3 structures
We next correlate the structural aspects of the three structure types: Ak3Zr2Cu4S9 (Ak = Ba and Sr),48 Sr3Zr2Cu4Se9, and Sr0.5ZrCuSe343 structures. A common feature of these structure types is the presence of anionic layers built up of the transition metals and Q (S/Se) atoms. These anionic frameworks are counterbalanced and separated by the Ak2+ cations (Ak = Ba and Sr) (see Fig. 5). Among these structures, Sr0.5ZrCuSe343 is stabilized with the highest symmetry (space group: Cmcm) and is isotypic with the KCuZrS3 (Cmcm)64 structure type. In the literature, a large number of quaternary chalcogenides are known to adopt the KCuZrS364 type structure.
 | ||
| Fig. 5 Unit cells of the (a) Ak3Zr2Cu4S9 (Ak = Ba, Sr) (P) and (b) Sr0.5ZrCuSe3 (Cmcm) structures. | ||
The Sr3Zr2Cu4Se9 (P) type is closely related to Sr0.5ZrCuSe343 in terms of the coordination environments of the transition metals, i.e., Zr and Cu atoms, which occupy distorted octahedral and tetrahedral sites, respectively. The CuS3 units with a triangular planar geometry make the triclinic Ak3Zr2Cu4S9 (Ak = Ba and Sr) structure type (P)48 unique to the other two structure types. No short Cu⋯Cu interactions exist in the Sr0.5ZrCuSe343 structure. In contrast, the Ak3Zr2Cu4S9 (Ak = Ba and Sr) and Sr3Zr2Cu4Se9 structure types feature a variety of short Cu⋯Cu interactions.
Although the Ak3Zr2Cu4S9 (Ak = Ba, Sr) and Sr3Zr2Cu4Se9 structures are isoelectronic and layered, the intralayer connections are entirely different in these two structure types, as described above (see Fig. 3). While the stoichiometric sulfur compound has two Cu sites as the bridging elements, the non-stoichiometric selenide compound has three Cu atoms to link the  chains, as shown in Fig. 3 and 4. Thus, the flexibility of copper atoms to stabilize different polyhedra in the title sulfide structure can be considered as the main contrasting feature between the Ak3Zr2Cu4S9 and Sr3Zr2Cu4Se9 types.
chains, as shown in Fig. 3 and 4. Thus, the flexibility of copper atoms to stabilize different polyhedra in the title sulfide structure can be considered as the main contrasting feature between the Ak3Zr2Cu4S9 and Sr3Zr2Cu4Se9 types.
3.4 Optical bandgap and electrical resistivity
We used the polycrystalline samples of Sr3Zr2Cu4Q9 (Q = S, Se) for physical property measurements. The Tauc plot of the Sr3Zr2Cu4S9 powder shows a sharp drop in absorption coefficient (α) value below 2 eV, as shown in Fig. 6a. | ||
| Fig. 6 (a) Tauc plots of the finely ground polycrystalline Sr3Zr2Cu4S9 and Sr3Zr2Cu4Se9 samples. (b) The electrical resistivity plot of the Sr3Zr2Cu4Se9 compound below RT. | ||
A direct bandgap energy value of 1.7(1) eV is calculated for the Sr3Zr2Cu4S9 phase from the Tauc plot, which agrees with the reddish-brown color of the powder. The theoretical bandgap of 1.78 eV for the Sr3Zr2Cu4S9 structure is in good agreement with the experimental value (see section 3.7 for details). On the other hand, Sr3Zr2Cu4Se9, which is black in color, does not show any sharp transition in the Tauc plot, indicating that this selenide has a narrow bandgap energy (Eg < 0.6 eV), or it is a metal/semimetal.
The sulfide sample, Sr3Zr2Cu4S9, is electrically insulating, having resistance values of the order of MΩ at RT in contrast to the Sr3Zr2Cu4Se9, which has electrical resistivity (ρ) values of about 80 mΩ cm (at RT). The ρ-value further drops to ∼20 mΩ cm on cooling the Sr3Zr2Cu4Se9 sample to 3 K, as shown in Fig. 6b. A kink in the resistivity plot was observed at ∼225 K, and this effect was reproducible. The origin of the kink observed in the resistivity plot is not clear. The resistivity values of Sr3Zr2Cu4Se9 are several orders higher than coinage metals like Cu (1.7 × 10−3 mΩ cm at RT) and Ag (1.6 × 10−3 mΩ cm at RT). Thus, Sr3Zr2Cu4Se9 can be described as a poor metal or a degenerate semiconductor. The detailed theoretical electronic structure of Sr3Zr2Cu4Se9 is discussed in section 3.7.
3.5 High-temperature transport studies: thermal conductivity (κtot), Seebeck coefficient (S), and electrical resistivity (ρ)
Both Sr3Zr2Cu4S9 and Sr3Zr2Cu4Se9 are relatively good heat conductors in comparison to related layered quaternary structures such as the RbZrCuTe3 (0.49 W mK−1 at 323 K to 0.35 W mK−1 at 575 K)31 and BaCeCuS3 (0.62 W mK−1 at 323 K to 0.32 W mK−1 at 773 K).38 The Sr3Zr2Cu4Se9 compound shows a κtot of 3.5 W mK−1 at 323 K, which is almost twice the value observed for Sr3Zr2Cu4S9 (1.82 W mK−1 at 323 K). As expected, the κtot values gradually decreased as the samples were heated from 323 K to higher temperatures (573 K). The κtot value drops to 2.5 W mK−1 and 1.4 W mK−1 for Sr3Zr2Cu4Se9 and Sr3Zr2Cu4S9, respectively, at 573 K (Fig. 7a).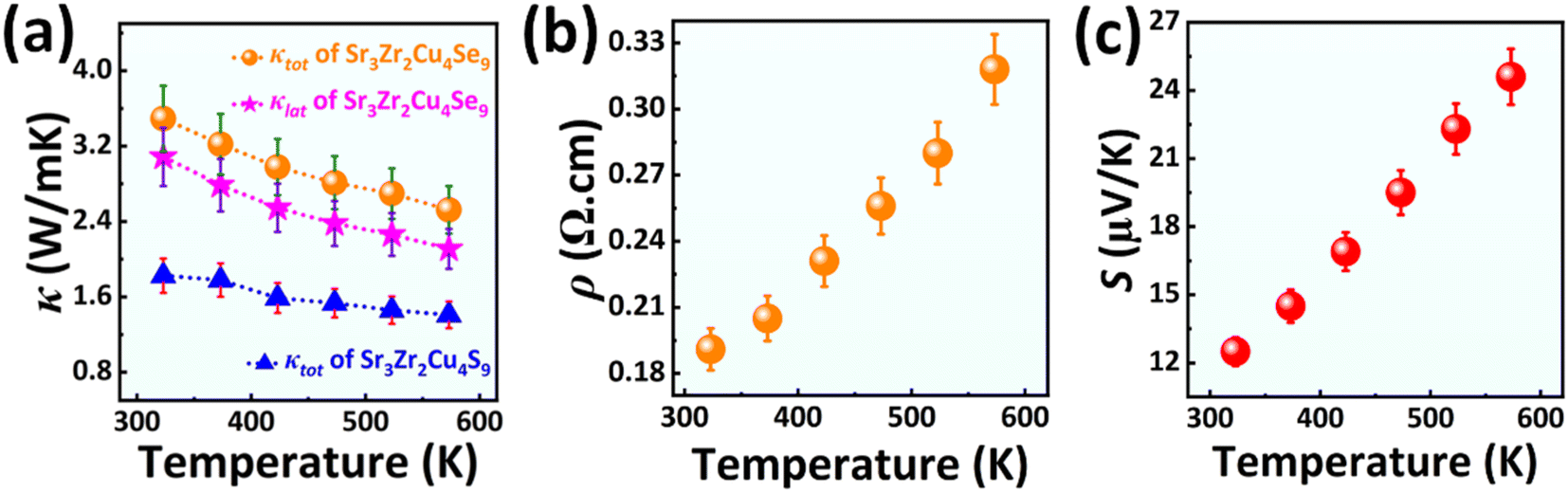 | ||
| Fig. 7 (a) Temperature-dependent thermal conductivity plots of Sr3Zr2Cu4Q9 (Q = S, Se) compounds. (b) Electrical resistivity and (c) Seebeck coefficient as a function of temperature for the Sr3Zr2Cu4Se9 compound. | ||
The κtot value for each sample is the sum of the electronic (κele) and lattice thermal conductivities (κlat) of the crystals. Since Sr3Zr2Cu4Se9 is metallic, in contrast to the insulating Sr3Zr2Cu4S9, the free charge carriers in the former compound contribute to the total thermal conductivity. On the contrary, the κele value of the insulating Sr3Zr2Cu4S9 is negligible in comparison to the κlat values. We calculated the κele value of Sr3Zr2Cu4Se9 using the Wiedemann–Franz model65 (refer to the caption of Fig. S3 in the ESI†) to compare the κlat values of the two compounds. The κlat value is also higher for Sr3Zr2Cu4Se9 (3.08 W mK−1 to 2.1 W mK−1 in the range of 323 K to 573 K) than for the isoelectronic sulfide (see Fig. 7a). The higher thermal conductivity values for Sr3Zr2Cu4Q9 (Q = S, Se) make them unsuitable for thermoelectric applications.
Next, we discuss the trends of the Seebeck coefficient and resistivity data obtained for Sr3Zr2Cu4Se9 as a function of temperature. The electrical resistivity plot shown in Fig. 7b is consistent with metal-like behavior with a gradual increase in ρ-values on increasing the sample's temperature. The ρ-value of Sr3Zr2Cu4Se9 elevated from 190 mΩ cm at 323 K to 320 mΩ cm at 573 K. The p-type nature of Sr3Zr2Cu4Se9 is confirmed by the positive sign of the S-values, as shown in Fig. 7c. As expected, the S-value increases at higher temperatures as the sample becomes more electrically resistive. Unfortunately, the low S values (12.51 μV K−1 at 323 K to 24.60 μV K−1 at 573 K) combined with the relatively higher thermal conductivity values of the Sr3Zr2Cu4Se9 sample make it unsuitable for thermoelectric applications.
The maximum thermoelectric figure of merit, zT, is almost zero (16.5 × 10−7), as can be seen from the zT plot in Fig. S3 in the ESI.† Further improvement of the zT value by optimization of the charge carrier concentration seems unlikely due to the poor S-value of Sr3Zr2Cu4Se9.
3.6 Photovoltaic studies of the Sr3Zr2Cu4S9 polycrystalline compound
The current density (J)–voltage (V) measurements of the solar cells were performed under one sun illumination. The J–V plot of the solar cell, FTO/TiO2/CdS/S2−, S/MWCNTs/Ni-foam, presented in Fig. 8, shows the open circuit voltage (VOC) = 723 mV, short circuit current density (JSC) = 13.00 mA cm−2, fill factor (FF) = 58.83%, and the power conversion efficiency (PCE or η) = 5.59%. | ||
| Fig. 8 The J–V characteristics of the solar cells based on different photoanodes, S2−, S gel electrolyte, and MWCNTs/Ni-foam as CE under 1 sun (AM 1.5G, 100 m cm−2). | ||
The J–V plot of the FTO/TiO2/CdS/Sr3Zr2Cu4S9/S2−, S/MWCNTs/Ni-foam shows: VOC = 868 mV, JSC = 13.28 mA cm−2, FF = 60.11%, and the efficiency = 6.93%, as seen in Table 3. From the Tauc plot, shown in Fig. 6a, the bandgap of Sr3Zr2Cu4S9 was estimated to be 1.7 eV, which is much lower than the bandgaps of TiO2 (3.2 eV) and CdS (2.3 eV) used in our solar cell. By the inclusion of Sr3Zr2Cu4S9 in the photoanode, the overall efficiency was increased by ∼24% because Sr3Zr2Cu4S9, a narrow band gap semiconductor, serves as a co-sensitizer. This implies that in the co-sensitized cell, upon illumination, in addition to the CdS, the Sr3Zr2Cu4S9 phase also undergoes an electron–hole separation. Hence, in the co-sensitized cell, additional charge carriers are available for circulation, improving the VOC as well as the JSC, thus increasing the efficiency of the cell. Furthermore, co-sensitization also reduces back electron transfer to the electrolyte during cell operation, which maximizes charge separation and enhances efficiency.
| Photoanode | VOC (mV) | JSC (mA cm−2) | FF (%) | PCE (%) |
|---|---|---|---|---|
| FTO/TiO2/CdS | 723 | 13.00 | 58.83 | 5.59 |
| FTO/TiO2/CdS/Sr3Zr2Cu4S9 | 868 | 13.28 | 60.11 | 6.93 |
3.7 Band structures of Sr3Zr2Cu4S9 and Sr3Zr2Cu4Se9
We estimated the structural parameters of the optimized unit cells and explored the electronic structures of Sr3Zr2Cu4S9 and Sr3Zr2Cu4Se9. The results are tabulated in Table 4 and are found to be aligned with the experimental values. As can be seen, the GGA (LDA) schemes slightly overestimate (underestimate) the cell parameters as expected. In general, the GGA (LDA) XC schemes slightly overestimate (underestimate) the cell parameters, respectively, for most of the material systems.66 The GGA bandgap energy for Sr3Zr2Cu4S9 is computed to be 1.37 eV. As expected, the computed GGA bandgap value for the sulfide is lower as compared to the experimental value of ∼1.7 eV since the GGA (LDA) schemes are known to underestimate the bandgaps as much as by ∼50%. The bandgap of 1.78 eV for Sr3Zr2Cu4S9 computed using the GGA-1/2 scheme is in better agreement with the experimental value. The GGA-1/2 scheme is expected to provide better bandgap estimates since it partially corrects the self-interaction error in local and semi-local XC functionals for extended systems.| a (Å) | b (Å) | c (Å) | α | β | γ | E g (eV) | |||
|---|---|---|---|---|---|---|---|---|---|
| Cal. | GGA | 6.719 | 10.321 | 11.937 | 109.41 | 104.79 | 96.80 | 1.37 | |
| Sr3Zr2Cu4S9 | LDA | 6.539 | 9.953 | 11.713 | 109.97 | 104.21 | 96.99 | 1.29 | |
| GGA-1/2 | — | — | — | — | — | — | 1.78 | ||
| Exp. | 6.662 | 10.232 | 11.939 | 109.69 | 104.84 | 96.39 | 1.7(1) | ||
| Cal. | GGA | 7.044 | 10.697 | 12.333 | 109.31 | 105.68 | 96.50 | 1.17 | |
| Sr3Zr2Cu4Se9 | LDA | 6.830 | 10.402 | 12.006 | 109.33 | 105.60 | 96.56 | 1.09 | |
| GGA-1/2 | — | — | — | — | — | — | 1.56 | ||
| Exp | 6.9779 | 10.685 | 12.302 | 109.18 | 105.38 | 96.56 | 0 | ||
The GGA and GGA-1/2 computed bandgap values of Sr3Zr2Cu4Se9 are 1.17 eV and 1.56 eV, respectively. These computed bandgap energies disagree with the experimental result that suggests Sr3Zr2Cu4Se9 is metallic/degenerate semiconducting in nature. This disagreement may be partially attributed to the slightly different occupancy values of Cu1, Cu3, and Cu4 sites in the unit cell used to model the Sr3Zr2Cu4Se9 structure as compared to the experimental unit cell content obtained from the SCXRD study. In our theoretical model, the Cu1 and Cu4 sites are half occupied (50%), whereas the Cu3 site is fully occupied (100%). However, in the experimental unit cell, the Cu1, Cu3, and Cu4 sites have occupancies of 59.8%, 85.8%, and 53.3%, respectively.
Fig. 9 shows the density of states (DOS) and the band structure plots for Sr3Zr2Cu4S9. The energies in the DOS plot are scaled in such a way that the valence band maximum (VBM) lies at 0 (see Fig. 9a). As can be seen, the top of the valence band (VB) (∼−5.5 eV < E < 0) is primarily comprised of Cu-3d, Zr-4d, and S-3p states. The VB at ∼−12 eV is mainly contributed by the S-3s localized states, with some minor contributions from the Sr-4s states. The conduction band (CB) close to the Fermi energy (∼1.1 eV < E < 2.2 eV) is primarily made up of the antibonding Zr-4d states. The contributions to the CB in the higher energy range (2.9 eV < E < 4.5 eV) are from the Zr-4d and Sr-4d states. The band structure of Sr3Zr2Cu4S9, as shown in Fig. 9b, indicates that the bandgap is indirect. However, the difference between the indirect and direct bandgap is minute (∼0.07 eV). The DOS and band structure plots for Sr3Zr2Cu4Se9 are found to be qualitatively similar to that for Sr3Zr2Cu4S9, as shown in Fig. S4 of the ESI.†
 | ||
| Fig. 9 (a) The total and projected density of states (DOS) and (b) the band structure plot along high symmetry directions in the Brillouin zone for Sr3Zr2Cu4S9. The high symmetry k-points are Y ≡ (0, ½, 0), Γ ≡ (0, 0, 0), X ≡ (½, 0, 0), C ≡ (½, ½, 0), and Z ≡ (0, 0, ½). | ||
The projected crystal orbital Hamilton populations (COHP)67 are computed for the title structures to estimate the bonding type between the constituent atoms and their strength. Fig. 10 shows the −COHP (E) and integrated-COHP plots for Sr3Zr2Cu4S9. The −ICOHP values suggest that the bond strength between atom pairs varies in the order Zr–S > Cu–S > Cu–Zr > Sr–S. The COHP and ICOHP plots for Sr3Zr2Cu4Se9 are presented in Fig. S5 in ESI† and indicate a similar order for bonding strengths.
 | ||
| Fig. 10 (a) The crystal orbital Hamilton populations (COHP) and (b) the integrated COHP (ICOHP) values for the bonding atoms in Sr3Zr2Cu4S9. | ||
We next discuss the computed Bader charges (QB) of the atoms in the Sr3Zr2Cu4Q9 structures. In general, the magnitudes of the Bader charges are smaller than the formal oxidation states of atoms since the latter values are calculated by considering the full charge transfer from metals (cations) to non-metals (anions), i.e., considering the crystalline inorganic solids as perfect ionic compounds. The QB values provide estimates of the relative charge transfer between atom pairs and the degree of ionic/covalent character of the bonds.68 For Sr3Zr2Cu4S9, these charges are +1.54, +2.02, +0.39, and −1.16 for Sr, Zr, Cu, and S ions, respectively. Likewise, in Sr3Zr2Cu4Se9, QB values of +1.49, +1.87, +0.35, and −1.10 were found for the Sr, Zr, Cu, and Se ions, respectively. Indeed, the magnitudes of the computed QB values are smaller than the formal oxidation states +II, +IV, +I, and −II for the Sr, Zr, Cu, and S (Se) ions. The deviation of QB values of the atoms, especially Zr, Cu, and S, in the Sr3Zr2Cu4Q9 structures from their formal oxidation numbers indicate the partial covalent character of the metal–chalcogen bonds due to the overlapping of orbitals, as discussed earlier.
4. Conclusions
The single crystals and polycrystals of two new quaternary mixed metal chalcogenides, Sr3Zr2Cu4S9 and Sr3Zr2Cu4Se9, were successfully prepared by reacting elemental reactants at high temperatures under vacuum. The SCXRD studies of the prepared quaternary crystals are carried out at RT, revealing that Sr3Zr2Cu4Se9 adopts a new structure type. The selenide phase is non-stoichiometric due to Cu-deficiency in the structure. On the contrary, the isoelectronic sulfide, Sr3Zr2Cu4S9, is stoichiometric and crystallizes in the Ba3Zr2Cu4S9 structure type. The triclinic structures (space group: P) of both Sr3Zr2Cu4Q9 phases are pseudo-two-dimensional, containing layers of  . The two structures differ in the coordination geometries of their Cu atoms. For the sulfur analog, there are tetrahedral CuS4 units and trigonal CuS3 units, and for the selenide structure, only tetrahedrally coordinated Cu atoms are present in the structure. The optical absorption study of the red-colored Sr3Zr2Cu4S9 sample yields a direct bandgap of 1.7(1) eV. The resistivity study of the selenide, Sr3Zr2Cu4Se9, shows a temperature dependence typical for a semimetal/degenerate semiconductor. Also, the p-type nature of the Sr3Zr2Cu4Se9 sample is established from the Seebeck study. The photovoltaic behavior of the Sr3Zr2Cu4S9 powder was studied by using it as a co-sensitizer in a CdS-based solar cell, which increased the overall efficiency by ∼24%. The DFT studies were used to calculate the electronic structures of Sr3Zr2Cu4Q9. The theoretical analysis of the band structure of the Sr3Zr2Cu4S9 phase shows the direct bandgap nature of the sulfide and it is consistent with the experimental studies. The COHP, ICOHP, and Bader charge calculation studies suggest the bonding interactions between Zr/Cu and S atoms are stronger than the Sr–S interactions.
. The two structures differ in the coordination geometries of their Cu atoms. For the sulfur analog, there are tetrahedral CuS4 units and trigonal CuS3 units, and for the selenide structure, only tetrahedrally coordinated Cu atoms are present in the structure. The optical absorption study of the red-colored Sr3Zr2Cu4S9 sample yields a direct bandgap of 1.7(1) eV. The resistivity study of the selenide, Sr3Zr2Cu4Se9, shows a temperature dependence typical for a semimetal/degenerate semiconductor. Also, the p-type nature of the Sr3Zr2Cu4Se9 sample is established from the Seebeck study. The photovoltaic behavior of the Sr3Zr2Cu4S9 powder was studied by using it as a co-sensitizer in a CdS-based solar cell, which increased the overall efficiency by ∼24%. The DFT studies were used to calculate the electronic structures of Sr3Zr2Cu4Q9. The theoretical analysis of the band structure of the Sr3Zr2Cu4S9 phase shows the direct bandgap nature of the sulfide and it is consistent with the experimental studies. The COHP, ICOHP, and Bader charge calculation studies suggest the bonding interactions between Zr/Cu and S atoms are stronger than the Sr–S interactions.
Data availability
Electronic supplementary information (ESI) is available, providing additional crystallographic details of the Sr3Zr2Cu4S9 and Sr3Zr2Cu4Se9 structures. The CIF files for these structures have been deposited with the joint CCDC/FIZ Karlsruhe deposition service under CSD numbers 2390675 and 2390676, respectively.†Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this article.Acknowledgements
JP thanks DST-SERB, the Government of India (GOI) for the financial support under the core research grant (Grant Number: CRG/2021/0003641), and IIT Hyderabad for research facilities. SB and SY thank MOE and GOI for the Prime Minister Research Fellowship (PMRF). AKR thanks DST, India, for the INSPIRE fellowship, and Swati thanks CSIR for the Senior Research Fellowship. The authors are thankful to DST-FIST(SR/FST/PSI-215/2016) for computational resources. The authors also acknowledge Dr Surya Jammalamadaka from the Department of Physics at IIT Hyderabad for his help with electrical resistivity data collection.References
- C. D. Malliakas, D. Y. Chung, H. Claus and M. G. Kanatzidis, J. Am. Chem. Soc., 2016, 138, 14694–14698 CrossRef PubMed.
- Y. Kamihara, T. Watanabe, M. Hirano and H. Hosono, J. Am. Chem. Soc., 2008, 130, 3296–3297 CrossRef PubMed.
- A. Le Donne, V. Trifiletti and S. Binetti, Front. Chem., 2019, 7, 297 CrossRef PubMed.
- D. Shin, T. Zhu, X. Huang, O. Gunawan, V. Blum and D. B. Mitzi, Adv. Mater., 2017, 29, 1606945 CrossRef.
- M. Chauhan, K. Soni, P. E. Karthik, K. P. Reddy, C. S. Gopinath and S. Deka, J. Mater. Chem. A, 2019, 7, 6985–6994 RSC.
- J. Deng, W. Yuan, P. Ren, Y. Wang, D. Deng, Z. Zhang and X. Bao, RSC Adv., 2014, 4, 34733–34738 RSC.
- Y. Shi, A. Assoud, S. Ponou, S. Lidin and H. Kleinke, J. Am. Chem. Soc., 2018, 140, 8578–8585 CrossRef PubMed.
- A. Assoud, N. Soheilnia and H. Kleinke, Intermetallics, 2007, 15, 371–376 CrossRef.
- V. V. Atuchin, F. Liang, S. Grazhdannikov, L. I. Isaenko, P. G. Krinitsin, M. S. Molokeev, I. P. Prosvirin, X. Jiang and Z. Lin, RSC Adv., 2018, 8, 9946–9955 RSC.
- Z.-H. Kang, J. Guo, Z.-S. Feng, J.-Y. Gao, J.-J. Xie, L.-M. Zhang, V. Atuchin, Y. Andreev, G. Lanskii and A. Shaiduko, Appl. Phys. B, 2012, 108, 545–552 CrossRef.
- A. H. Reshak, V. V. Atuchin, S. Auluck and I. V. Kityk, J. Phys.: Condens. Matter, 2008, 20, 325234 CrossRef.
- V. P. Sachanyuk, G. P. Gorgut, V. V. Atuchin, I. D. Olekseyuk and O. V. Parasyuk, J. Alloys Compd., 2008, 452, 348–358 CrossRef.
- Y. Han, H. Yi, Y. Wang, Z. Li, X. Chu and J.-H. Jiang, ACS Nano, 2022, 16, 19053–19066 CrossRef PubMed.
- V. Singh, K. S. Rawat, S. Mishra, T. Baghel, S. Fatima, A. A. John, N. Kalleti, D. Singh, A. Nazir, S. K. Rath and A. Goel, J. Mater. Chem. B, 2018, 6, 3366–3371 RSC.
- A. Rydosz, Coatings, 2018, 8, 425 CrossRef.
- Z. Ibupoto, K. Khun, V. Beni, X. Liu and M. Willander, Sensors, 2013, 13, 7926–7938 CrossRef PubMed.
- B. S. Sengar, V. Garg, A. Kumar, V. Awasthi, S. Kumar, V. V. Atuchin and S. Mukherjee, Opt. Mater., 2018, 84, 748–756 CrossRef.
- V. Garg, B. S. Sengar, G. Siddharth, S. Kumar, V. V. Atuchin and S. Mukherjee, Surf. Interfaces, 2021, 25, 101146 CrossRef.
- R. Mulla and M. H. K. Rabinal, Energy Technol., 2019, 7, 1800850 CrossRef.
- O. V. Parasyuk, V. V. Atuchin, Y. E. Romanyuk, L. P. Marushko, L. V. Piskach, I. D. Olekseyuk, S. V. Volkov and V. I. Pekhnyo, J. Cryst. Growth, 2011, 318, 332–336 CrossRef.
- H. Liu, X. Shi, F. Xu, L. Zhang, W. Zhang, L. Chen, Q. Li, C. Uher, T. Day and G. J. Snyder, Nat. Mater., 2012, 11, 422–425 CrossRef PubMed.
- K. S. Weldert, W. G. Zeier, T. W. Day, M. Panthöfer, G. J. Snyder and W. Tremel, J. Am. Chem. Soc., 2014, 136, 12035–12040 CrossRef PubMed.
- J. L. Niedziela, D. Bansal, A. F. May, J. Ding, T. Lanigan-Atkins, G. Ehlers, D. L. Abernathy, A. Said and O. Delaire, Nat. Phys., 2019, 15, 73–78 Search PubMed.
- X. Wang, P. Qiu, T. Zhang, D. Ren, L. Wu, X. Shi, J. Yang and L. Chen, J. Mater. Chem. A, 2015, 3, 13662–13670 RSC.
- C. Sturm, N. Boccalon, A. Assoud, T. Zou, J. Kycia and H. Kleinke, Inorg. Chem., 2021, 60, 12781–12789 CrossRef PubMed.
- T. P. Bailey, S. Hui, H. Xie, A. Olvera, P. F. P. Poudeu, X. Tang and C. Uher, J. Mater. Chem. A, 2016, 4, 17225–17235 RSC.
- O. Mayasree, C. R. Sankar, Y. Cui, A. Assoud and H. Kleinke, Eur. J. Inorg. Chem., 2011, 2011, 4037–4042 CrossRef.
- N. O. Azarapin, A. S. Aleksandrovsky, V. V. Atuchin, T. A. Gavrilova, A. S. Krylov, M. S. Molokeev, S. Mukherjee, A. S. Oreshonkov and O. V. Andreev, J. Alloys Compd., 2020, 832, 153134 CrossRef.
- N. O. Azarapin, V. V. Atuchin, N. G. Maximov, A. S. Aleksandrovsky, M. S. Molokeev, A. S. Oreshonkov, N. P. Shestakov, A. S. Krylov, T. M. Burkhanova, S. Mukherjee and O. V. Andreev, Mater. Res. Bull., 2021, 140, 111314 CrossRef.
- A. Choudhury, S. Mohapatra, H. Yaghoobnejad Asl, S. H. Lee, Y. S. Hor, J. E. Medvedeva, D. L. McClane, G. E. Hilmas, M. A. McGuire, A. F. May, H. Wang, S. Dash, A. Welton, P. Boolchand, K. P. Devlin, J. Aitken, R. Herbst-Irmer and V. Petříček, J. Solid State Chem., 2017, 253, 192–201 CrossRef.
- C. C. Laing, B. E. Weiss, K. Pal, M. A. Quintero, H. Xie, X. Zhou, J. Shen, D. Y. Chung, C. Wolverton and M. G. Kanatzidis, Chem. Mater., 2022, 34, 8389–8402 CrossRef.
- X. Yu, Z. Wang, P. Cai, K. Guo, J. Lin, S. Li, J. Xing, J. Zhang, X. Yang and J.-T. Zhao, Inorg. Chem., 2024, 63, 20093–20101 CrossRef PubMed.
- C. Coughlan, M. Ibáñez, O. Dobrozhan, A. Singh, A. Cabot and K. M. Ryan, Chem. Rev., 2017, 117, 5865–6109 CrossRef PubMed.
- R. Xu, S. Lai, Y. Zhang and X. Zhang, Nanomaterials, 2024, 14, 832 CrossRef PubMed.
- D. Shin, B. Saparov, T. Zhu, W. P. Huhn, V. Blum and D. B. Mitzi, Chem. Mater., 2016, 28, 4771–4780 CrossRef.
- W. Arndt, H. Dittrich and H. W. Schock, Thin Solid Films, 1985, 130, 209–216 CrossRef CAS.
- B. Yang, L. Wang, J. Han, Y. Zhou, H. Song, S. Chen, J. Zhong, L. Lv, D. Niu and J. Tang, Chem. Mater., 2014, 26, 3135–3143 CrossRef.
- O. Shahid, S. Yadav, D. Maity, M. Deepa, M. K. Niranjan and J. Prakash, New J. Chem., 2023, 47, 5378–5389 RSC.
- S. Chander, S. K. Tripathi, I. Kaur and A. K. De, Mater. Today Sustain., 2024, 25, 100662 Search PubMed.
- P. Jackson, D. Hariskos, R. Wuerz, O. Kiowski, A. Bauer, T. M. Friedlmeier and M. Powalla, Phys. Status Solidi RRL, 2015, 9, 28–31 CrossRef.
- K. Srivastava, O. Shahid, A. K. Ray, M. Deepa, M. K. Niranjan and J. Prakash, J. Phys. Chem. Solids, 2024, 192, 112085 CrossRef.
- K. Srivastava, O. Shahid, A. K. Ray, M. Deepa, M. K. Niranjan and J. Prakash, J. Solid State Chem., 2023, 328, 124344 CrossRef.
- S. Maier, J. Prakash, D. Berthebaud, O. Perez, S. Bobev and F. Gascoin, J. Solid State Chem., 2016, 242, 14–20 CrossRef.
- A. A. Coelho, J. Appl. Crystallogr., 2018, 51, 210–218 CrossRef.
- APEX3: Program for Data Collection on Area Detectors, Bruker AXS Inc., Madison, WI, USA, 2016 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., 2008, 64, 112–122 Search PubMed.
- G. M. Sheldrick, SADABS, Department of Structural Chemistry, University of Göttingen, Göttingen, Germany, 2008 Search PubMed.
- S. Barman, S. Jana, G. Panigrahi, S. Yadav, M. K. Niranjan and J. Prakash, New J. Chem., 2022, 46, 15976–15986 RSC.
- L. M. Gelato and E. Parthé, J. Appl. Crystallogr., 1987, 20, 139–143 CrossRef.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef.
- G. Kortüm, in Reflectance Spectroscopy, Springer Berlin Heidelberg, Berlin, Heidelberg, 1969, pp. 170–216 Search PubMed.
- R. Piazza, S. Buzzaccaro, E. Secchi and A. Parola, Soft Matter, 2012, 8, 7112 Search PubMed.
- E. I. Andritsos, E. Zarkadoula, A. E. Phillips, M. T. Dove, C. J. Walker, V. V. Brazhkin and K. Trachenko, J. Phys.: Condens. Matter, 2013, 25, 235401 Search PubMed.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133–A1138 Search PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B:Condens. Matter Mater. Phys., 1996, 54, 11169–11186 Search PubMed.
- W. Primak, H. Kaufman and R. Ward, J. Am. Chem. Soc., 1948, 70, 2043–2046 Search PubMed.
- J. Huster and W. Bronger, Z. Anorg. Allg. Chem., 1999, 625, 2033–2040 CrossRef.
- A. V. Ruseikina, A. N. Pinigina, M. V. Grigoriev and D. A. Safin, Cryst. Growth Des., 2024, 24, 2485–2492 CrossRef.
- H. M. Otte, J. Appl. Phys., 1961, 32, 1536–1546 CrossRef.
- S. O. Long, A. V. Powell, S. Hull, F. Orlandi, C. C. Tang, A. R. Supka, M. Fornari and P. Vaqueiro, Adv. Funct. Mater., 2020, 30, 1909409 CrossRef.
- Z. Shu, C. Shen, A. Lu, X. Gu and Z. Liu, Crystals, 2021, 11, 1495 CrossRef.
- F. Alahmari, S. Dey, A.-H. Emwas, B. Davaasuren and A. Rothenberger, J. Alloys Compd., 2019, 776, 1041–1047 CrossRef.
- A. Pfitzner, Z. Anorg. Allg. Chem., 1995, 621, 685–688 CrossRef.
- M. F. Mansuetto, P. M. Keane and J. A. Ibers, J. Solid State Chem., 1992, 101, 257–264 CrossRef.
- A. Yadav, P. Deshmukh, K. Roberts, N. Jisrawi and S. Valluri, J. Phys. Commun., 2019, 3, 105001 CrossRef.
- R. M. Martin, Electronic Structure: Basic Theory and Practical Methods, Cambridge University Press, 2020 Search PubMed.
- V. L. Deringer, A. L. Tchougréeff and R. Dronskowski, J. Phys. Chem. A, 2011, 115, 5461–5466 CrossRef PubMed.
- W. Tang, E. Sanville and G. Henkelman, J. Phys.: Condens. Matter, 2009, 21, 084204 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional crystallographic details of the Sr3Zr2Cu4S9 and Sr3Zr2Cu4Se9 structures. CCDC 2390675 and 2390676. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt02928c |
| This journal is © The Royal Society of Chemistry 2025 |