
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent advances in light-driven synthesis of complex trifluoromethylated aliphatic amines and derivatives
Chi Wai Cheung
 *,
Xiao-Tong Wang
and
Fuk Yee Kwong
*
*,
Xiao-Tong Wang
and
Fuk Yee Kwong
*
State Key Laboratory of Synthetic Chemistry and Department of Chemistry, The Chinese University of Hong Kong, Shatin, New Territories, Hong Kong 999077, P. R. China. E-mail: cw.cheung@cuhk.edu.hk; fykwong@cuhk.edu.hk
First published on 12th August 2025
Abstract
Trifluoromethylated aliphatic amines, particularly those with structurally complex architectures, have emerged as valuable motifs in drug discovery due to their unique capacity to enhance metabolic stability, membrane permeability, and affinity for biological targets. However, the selective and efficient synthesis of these compounds remains a significant challenge, limiting their broader incorporation into pharmaceutical agents. Recent advances in light-driven organic synthesis, employing mild and sustainable visible-light-mediated conditions, have enabled modular and practical strategies for constructing these privileged structures from simple, readily available starting materials. A variety of photoinduced reactions, photocatalytic methods, and metallaphotoredox-catalyzed transformations have been developed, facilitating the direct installation of trifluoromethyl (CF3) groups and CF3-containing synthons onto diverse aliphatic frameworks and amine precursors. This review presents a comprehensive overview of recent progress in the photoinduced synthesis of trifluoromethylated aliphatic amines and their derivatives, emphasizing key methodological innovations, representative pharmaceutical targets, and mechanistic insights, with the goal of guiding future efforts in the design and synthesis of fluorine-rich bioactive molecules.
 Chi Wai Cheung | Chi Wai Cheung received his PhD from CUHK under Prof. Kin Shing Chan, followed by postdoctoral training with Prof. Stephen L. Buchwald at MIT and Prof. Xile Hu at EPFL. He subsequently joined Tianjin University as an associate professor in collaboration with Prof. Jun-An Ma. In 2024, he relocated to Hong Kong as a research assistant professor in Prof. Fuk Yee Kwong's group. His research focuses on photocatalysis, base-metal catalysis, inert-bond activation, and cross-coupling reactions for organic synthesis. |
 Xiao-Tong Wang | Xiao-Tong Wang received her MSc in Medical Sciences from The University of Hong Kong in 2023. She is currently pursuing her PhD at The Chinese University of Hong Kong under the joint supervision of Prof. Fuk Yee Kwong and Prof. Chi Wai Cheung. Her research focuses on photocatalysis and base-metal catalysis. |
 Fuk Yee Kwong | Fuk Yee (Michael) Kwong is Head and Professor of Chemistry at CUHK and Director of the Shenzhen Center of Novel Functional Molecules and the Shenzhen Municipal Key Laboratory of Chemical Synthesis of Medicinal Organic Molecules. He earned his PhD at CUHK in 2000 and was a Croucher postdoctoral fellow with Stephen L. Buchwald at MIT (2001–2003), later group manager for Prof. Albert S. C. Chan (2003–2004). Elected to the Hong Kong Young Academy of Science in 2020, his research focuses on phosphine ligand design for cross-coupling, C–H functionalization, and enantioselective transformations. |
1. Introduction
In drug discovery, the strategic incorporation of heteroatoms profoundly alters the electronic, steric, and structural properties of molecules, enhancing their biological efficacy and potential for therapeutic application.1 Among various heteroatoms, fluorine and fluoroalkyl groups have gained prominence due to their capacity to improve pharmacokinetic properties such as metabolic stability, membrane permeability, and target-binding affinity, significantly influencing ADMET (absorption, distribution, metabolism, excretion, and toxicity) profiles.2 Particularly, the introduction of trifluoromethyl (CF3) groups into bioactive small molecules has proven to be a robust strategy for constructing hit compounds, clinical candidates, and marketed pharmaceuticals.2,3 Indeed, CF3-substituted arenes and heterocycles are common structural motifs in many commercially important drugs (Scheme 1A(i)), as exemplified by sitagliptin (antidiabetic), apalutamide (anticancer), and celecoxib (neurology). Remarkably, within the top 200 best-selling pharmaceuticals in 2024, fourteen drugs contain C(sp2)–CF3 bonds.4 | ||
| Scheme 1 The significance, synthetic challenges, and recent developments in trifluoroalkyl amines and their derivatives. (A) Distribution of C(sp2)–CF3 and C(sp3)–CF3 scaffolds in pharmaceuticals. (B) Representative amine-based drugs bearing complex CF3-substituted aliphatic skeletons. (C) Conventional synthetic approaches to CF3-substituted aliphatic amines and their derivatives. (D) Modern synthetic methodologies for complex CF3-substituted aliphatic amines and derivatives (this review). | ||
In contrast, CF3 groups directly attached to aliphatic carbons (C(sp3)–CF3) are far less common in pharmaceutical compounds, with only a few examples such as the 2,2,2-trifluoroethyl fragment (three instances, Scheme 1A(ii)) and α-CF3-substituted quaternary centers (three instances, Scheme 1A(iii)), despite their steadily increasing prevalence in recent years.4–6 More structurally intricate trifluoromethylated aliphatic (trifluoroalkyl) motifs—particularly those featuring remote CF3 groups embedded within saturated, three-dimensional aliphatic frameworks—remain significantly underdeveloped (Scheme 1A(iv)). This scarcity arises predominantly from synthetic challenges associated with selectively constructing these complex trifluoroalkyl frameworks, which typically require multiple synthetic steps, sensitive reagents, and harsh reaction conditions.7 However, molecules bearing such sophisticated trifluoroalkyl scaffolds present substantial opportunities for modulating physicochemical properties through multiple mechanisms, including the distinct contributions of the CF3 group,2,3 and those arising from enhanced fraction of sp3-hybridized carbons,8 methyl effects,9 and quaternary centers10 introduced by complex aliphatic architectures. Collectively, these features may significantly accelerate the discovery and optimization of bioactive compounds.
On the other hand, amines and their derivatives constitute indispensable structural components in drug design, as nearly all best-selling pharmaceuticals contain at least one amine group (C(sp2)–N and C(sp3)–N).4–6 Structurally complex trifluoroalkyl amines, specifically, appear in several notable drugs such as efavirenz, alpelisib, mapracorat, masilukast, and enpatoran, underscoring their potential value in medicinal chemistry11 (Scheme 1B). The electron-withdrawing nature of fluoroalkyl groups lowers both the basicity and reducing power of amines, thereby enhancing the site selectivity and oxidative stability of the resulting drug molecules.7,12 Nevertheless, the medicinal application of trifluoroalkyl-substituted amines significantly lags behind that of their CF3-substituted aromatic or heteroaromatic counterparts. The primary factor hindering their broader adoption is the difficulty associated with synthesizing complex trifluoroalkyl precursors, which often entails lengthy, inefficient synthetic routes with limited structural diversity, thereby impeding the streamlined construction of structurally intricate trifluoroalkyl amines and derivatives 1A7 (Scheme 1C).
Addressing these synthetic bottlenecks, light-driven synthesis has emerged in recent years as a versatile and powerful approach for assembling complex molecules from simple substrates under mild, environmentally benign conditions.13,14 Photocatalytic methods harness visible-light energy to enable single-electron transfer (SET)13 and energy-transfer (EnT)14 processes, generating reactive radical intermediates capable of forging otherwise challenging chemical bonds. Photoinduced strategies (operating without photocatalysts or metal catalysts), photocatalysis (employing a single photoexcited metal complex or photocatalyst), and metallaphotoredox catalysis (combining transition metal catalysis with photocatalysis) have opened new avenues for the direct installation of CF3 and other trifluoroalkyl groups onto aliphatic frameworks (Scheme 1D). These methods typically employ readily accessible starting materials, including trifluoromethylating reagents, simple amines, alkyl precursors, nitroarenes, and unsaturated substrates, enabling direct construction of C(sp3)–CF3, C(sp3)–N, and C(sp3)–C(sp3) bonds. Consequently, these modular photocatalytic strategies15 have greatly expanded synthetic accessibility to structurally complex trifluoromethylated amines and their derivatives, including cyclic amines, CF3-substituted imines, amino acids, and medicinally relevant heterocycles.
In this review, we highlight recent advances in light-driven strategies for the synthesis of trifluoromethylated aliphatic amines and their derivatives, organized according to key mechanistic pathways, including alkene difunctionalization, metallaphotoredox radical cascades, inert bond functionalization, as well as photocatalyst-free and stereoselective approaches. We highlight key methodological innovations, structural motifs of pharmaceutical relevance, and mechanistic insights. Our goal is to provide a clear and concise foundation for understanding current advances and to inspire future directions in the design of trifluoroalkyl amines and the development of fluorine-rich amine-containing pharmaceuticals.
2. Photocatalytic alkene difunctionalization
The simultaneous incorporation of trifluoromethyl and nitrogen-based functionalities across alkenes represents a powerful strategy for constructing complex trifluoromethylated amines from simple olefins. Photoinduced difunctionalization offers modular and regioselective access to these valuable motifs under mild conditions, typically through radical–polar crossover or radical–radical coupling pathways. This section highlights recent advances in photocatalytic simultaneous construction of C–CF3, C–N, C–C, and C–heteroatom bonds within the unsaturated compounds to offer complex trifluoroalkyl amines and derivatives.2.1 Simultaneous C–CF3 and C–N bond formation
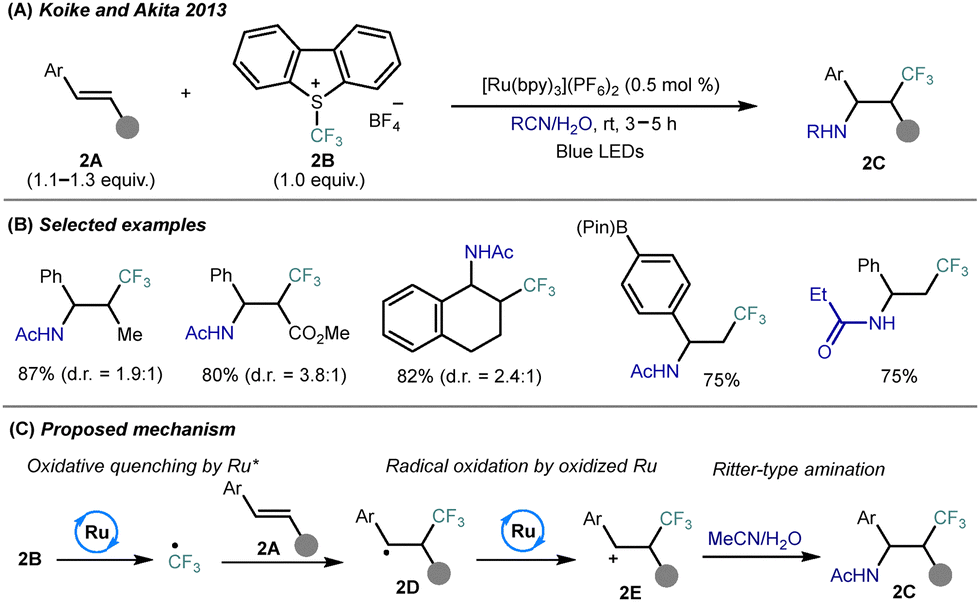 | ||
| Scheme 2 Photocatalytic amino-trifluoromethylation of alkenes using nitriles as nitrogen sources. (A) General reaction conditions. (B) Representative substrate scope. (C) Proposed mechanism. | ||
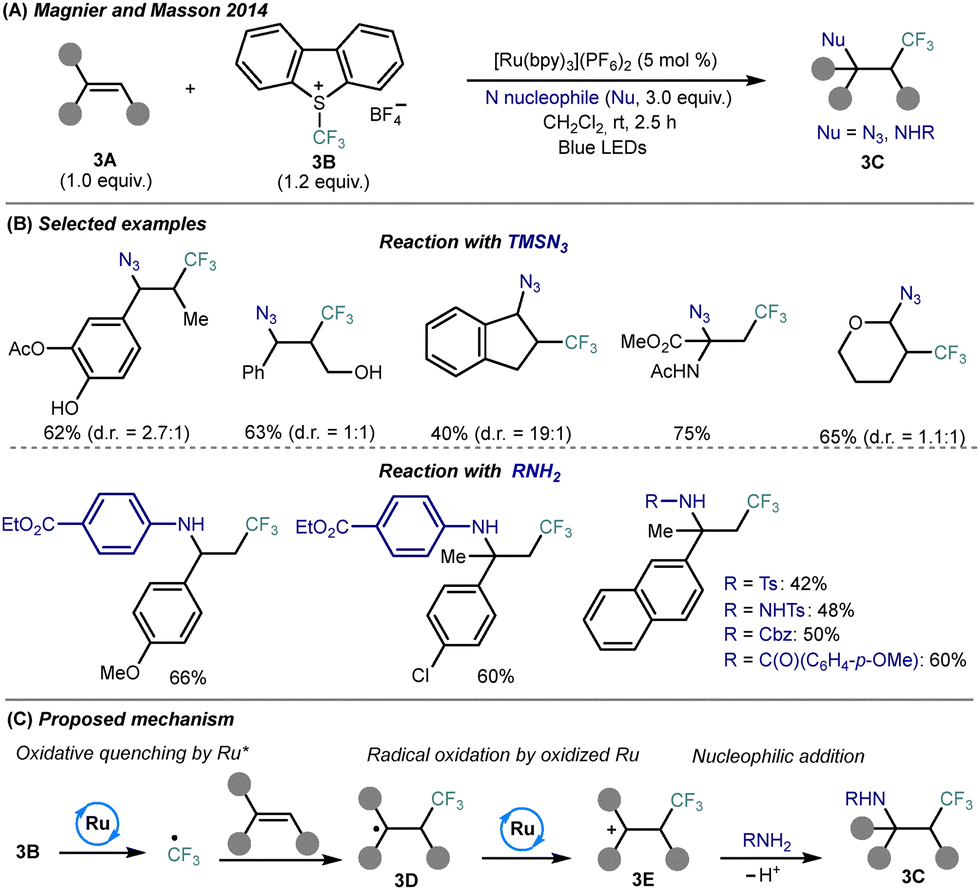 | ||
| Scheme 3 Photocatalytic azido- and amino-trifluoromethylation of alkenes using nitrogen nucleophiles. (A) General reaction conditions. (B) Representative substrate scope. (C) Proposed mechanism. | ||
Mechanistic studies support a radical/cationic sequence: photoexcited Ru(bpy)32+* initiates SET reduction of Umemoto reagent to generate a CF3˙ radical, which adds regioselectively to the alkene to form a trifluoroalkyl radical 3D (Scheme 3C). Subsequent oxidation by Ru(bpy)33+ affords a β-CF3 carbocation 3E, which undergoes nucleophilic trapping by TMSN3 or RNH2, completing the difunctionalization and regenerating the Ru(II) photocatalyst. This protocol represents a rare example of successful amino-trifluoromethylation involving direct use of diverse amine classes, thus providing access to structurally complex CF3-substituted building blocks under operationally simple and scalable conditions.
 | ||
| Scheme 4 Photocatalytic azo-trifluoromethylation of alkenes with aryldiazonium salts. (A) General reaction conditions. (B) Representative substrate scope. (C) Proposed mechanism. (D) Synthetic utility and applications. | ||
 | ||
| Scheme 5 Photocatalytic amino-trifluoromethylation of styrenes with phenylhydrazines. (A) General reaction conditions. (B) Representative substrate scope. (C) Proposed mechanism. | ||
Mechanistic studies support a radical pathway involving the photocatalytic single-electron oxidation of phenylhydrazines 5B to aryl hydrazinyl radical 5E, followed by aerobic oxygenation, rearrangement, and N–N bond cleavage to generate nitrogen-centered N-aryl aminyl radical 5F (Scheme 5C). Concurrently, the Togni reagent 5C undergoes photoredox activation to form a CF3 radical, which adds to the styrene to yield a benzylic radical intermediate 5G. The resulting radical–radical coupling between the aminyl 5F and benzylic radical 5G furnishes the trifluoroalkylamine product 5D. Alternatively, a stepwise pathway involving oxidation of the benzylic radical to a carbocation 5H, followed by nucleophilic attack by the hydrazine and subsequent N–N bond cleavage, may also operate to afford 5D. This transformation represents a rare example of intermolecular amino-trifluoromethylation utilizing aryl hydrazines as nitrogen sources.
 | ||
| Scheme 6 Photocatalytic trifluoromethyl-imination of alkenes with bifunctional sulfonamide reagent. (A) General reaction conditions. (B) Representative substrate scope. (C) Proposed mechanism. | ||
The transformation proceeds via EnT from the excited-state photosensitizer to the sulfonamide 6D to form the photoexcited species 6D*, inducing homolytic N–S bond cleavage and subsequent SO2 extrusion to generate a CF3 radical and a persistent diphenyliminyl radical (Scheme 6C). The CF3 radical adds to the terminal carbon of the alkene 6A, forming a trifluoroalkyl radical intermediate 6G. This intermediate is intercepted by the diphenyliminyl radical to yield the β-CF3 imine product 6E. The addition of KOH facilitates the conversion of SO2 into sulfite ions (SO32−), thereby suppressing its coupling with alkyl radicals and minimizing the formation of undesired side products.
Notably, the methodology also accommodates 1,1-trifluoromethyl-imination of alkyne 6F, offering access to CF3-decorated cyclopentyl imines (Scheme 6B, bottom right). Furthermore, the hydrolysis of trifluoroalkyl imine product 6H offers trifluoroalkyl amine 6I (Scheme 6D). This strategy provides a modular approach for vicinal and remote C–C and C–N bond construction using a single bifunctional reagent, expanding the toolkit for photochemical olefin difunctionalization and enabling access to trifluorinated imine architectures under mild, sustainable conditions.
2.2 Simultaneous C–CF3 and C–C bond formation
 | ||
| Scheme 7 Photocatalytic carbo-trifluoromethylation of alkenes with glycine derivatives. (A) General reaction conditions. (B) Representative substrate scope. (C) Synthetic utility and applications. (D) Proposed mechanism. | ||
Mechanistically, Eosin Y is photoexcited under visible-light irradiation to its singlet excited state EY*, which promotes single-electron reduction of the trifluoromethyl thianthrenium salt 7C to generate the electrophilic CF3 radical (Scheme 7D). Concurrently, the glycine derivative 7B undergoes single-electron oxidation by the resulting EY+˙ species to afford an α-amino radical cation 7H, which upon deprotonation forms the corresponding glycine-derived carbon-centered radical 7I. The CF3 radical adds to the diaryl alkene 7A, yielding a stabilized tertiary trifluoroalkyl radical intermediate 7J, which subsequently undergoes cross-coupling with the glycine-derived radical to furnish the trifluoromethylated glycine product 7D. Notably, control experiments reveal that moderate product formation can still occur in the absence of photocatalyst, likely proceeding through a photoactive electron donor–acceptor (EDA) complex 7K formed between the glycine derivative and the CF3-thianthrenium salt. Upon direct photoexcitation, this EDA complex facilitates simultaneous generation of both CF3 and glycine-based radicals, which then engage the alkene substrate to deliver the desired product 7D. This transformation represents a rare example of a photoredox-neutral alkyl–trifluoromethylation strategy, broadening the scope of late-stage peptide diversification and offering a streamlined approach to fluorinated α-amino acid derivatives.
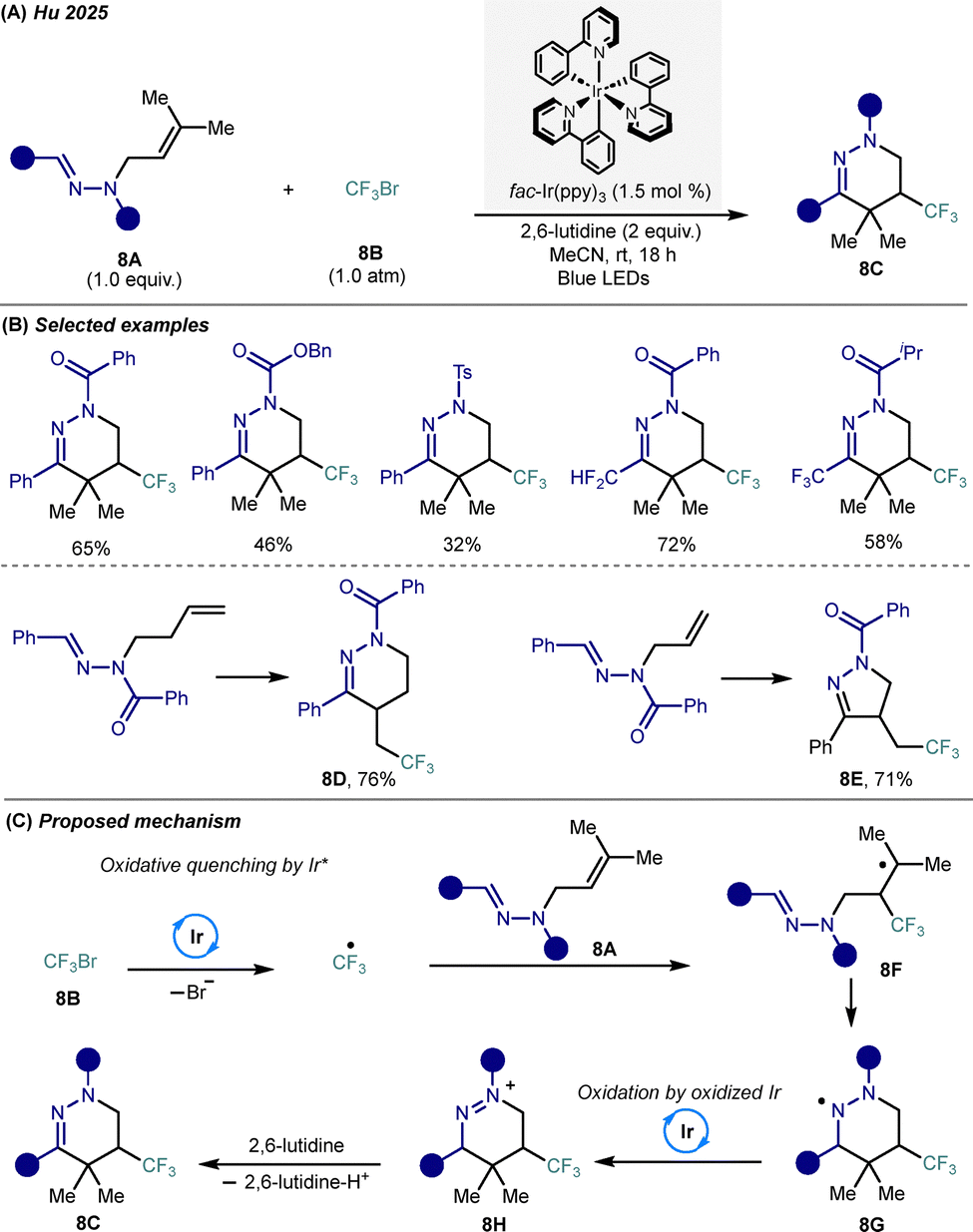 | ||
| Scheme 8 Photocatalytic carbo-trifluoromethylation for the construction of trifluoromethylated amino-based azaheterocycles. (A) General reaction conditions. (B) Representative substrate scope. (C) Proposed mechanism. | ||
Mechanistically, photoexcitation of Ir(ppy)3 under blue light generates the excited-state Ir(ppy)3*, which engages in SET to reduce CF3Br, generating a CF3 radical. This electrophilic species adds to the terminal alkene of the N-isopentenyl aldehyde hydrazone 8A, forming a trifluoroalkylated, stabilized tertiary carbon-centered radical 8F intermediate, which subsequently undergoes an intramolecular 6-endo-trig cyclization to afford a nitrogen-centered radical azaheterocycle 8G. Subsequent oxidation by the resulting Ir(IV) species generates the cationic intermediate 8H, which undergoes deprotonation by 2,6-lutidine to yield the trifluoromethylated tetrahydropyridazine product 8C. DFT calculations support the kinetic preference for this 6-endo-trig pathway over competing 5-endo-trig or C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N-targeted cyclizations. This transformation offers a streamlined strategy for the direct incorporation of trifluoromethyl groups into N-heterocyclic frameworks via visible-light photocatalysis, expanding synthetic access to medicinally relevant tetrahydropyridazine architectures under operationally simple conditions.
N-targeted cyclizations. This transformation offers a streamlined strategy for the direct incorporation of trifluoromethyl groups into N-heterocyclic frameworks via visible-light photocatalysis, expanding synthetic access to medicinally relevant tetrahydropyridazine architectures under operationally simple conditions.
2.3 Simultaneous C–CF3 and C-heteroatom bond formation
The groups of Magnier and Masson reported an alternative three-component strategy for the synthesis of complex and functionalized β-trifluoromethylated amine derivatives 9C, employing N-alkenyl carbamates 9A, Togni reagent 9B, and a variety of nucleophiles25 (Scheme 9A). In the presence of methanol, α-oxy-β-trifluoromethylated aminals are formed, while the use of cyanide or azide delivers α-cyano-β-CF3 and α-azido-β-CF3 carbamates, respectively (Scheme 9B). In the oxytrifluoromethylation reaction, a side reaction leading to an iodo-trifluoromethylated byproduct (<10%) is observed, likely due to in situ iodine generation from Togni reagent. | ||
| Scheme 9 Photocatalytic oxy-, cyano-, and azido-trifluoromethylation of N-alkenyl carbamates. (A) General reaction conditions. (B) Representative substrate scope. (C) Proposed mechanism. | ||
The reaction proceeds via a likely radical/cationic pathway (Scheme 9C). Photoexcitation of the Ru photocatalyst facilitates single-electron reduction of the Togni reagent to generate a CF3 radical, which adds regioselectively to the N-alkenyl carbamate to form an α-amidoalkyl radical 9D. Subsequent oxidation by Ru(III) affords an N-acyliminium ion 9E, which is then intercepted by O-, C-, or N-based nucleophiles to furnish the corresponding difunctionalized products 9C. This protocol thus offers a versatile and operationally simple platform for the synthesis of β-trifluoromethylated amine derivatives with broad functional diversity. This protocol represents one of the earliest examples of intermolecular carbotrifluoromethylation of N-alkenyl carbamates and offers modular access to structurally diverse CF3-functionalized amines under mild conditions.
2.4 Simultaneous C–N and C–C bond formation
Aiming to develop an operationally simple and economical route to trifluorinated amines, Zhang and co-workers reported a visible-light-induced, copper-catalyzed three-component carboamination of alkenes 10A with amines 10B and 1,1,1-trifluoro-2-iodoethane (10C)26 (Scheme 10A). This method utilizes inexpensive CuCl as both photocatalyst and cross-coupling catalyst, as well as racemic BINOL as ligand, enabling the regioselective introduction of trifluoroethyl and amino groups across styrenes under mild, base-promoted conditions to create trifluoroalkyl amines 10D. Broad amine compatibility was demonstrated, including primary and secondary anilines, cyclic amines, and indolines, with high functional group tolerance (Scheme 10B). While the amino-trifluoroethylation reaction proceeds efficiently with styrenes, the use of unactivated aliphatic alkenes results in significantly reduced reactivity and low product yields.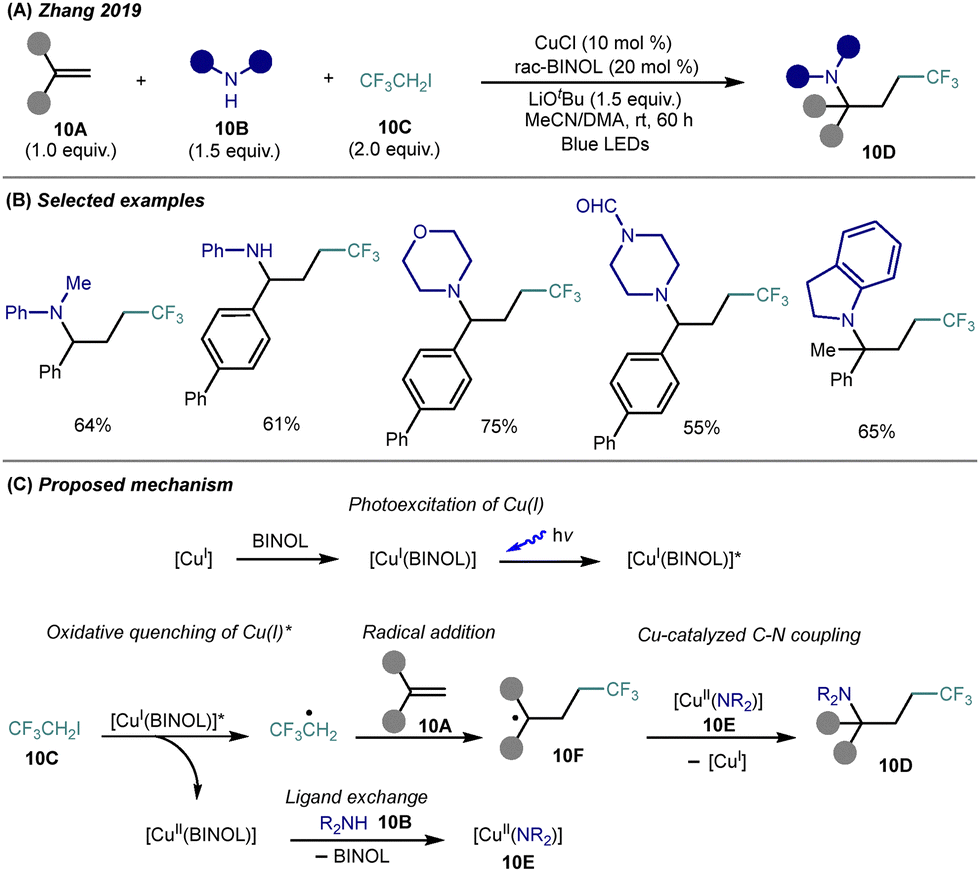 | ||
| Scheme 10 Photoinduced Cu-catalyzed amino-trifluoroethylation of alkenes. (A) General reaction conditions. (B) Representative substrate scope. (C) Proposed mechanism. | ||
Mechanistic investigations suggest that in situ formed CuI(BINOL) serve as the photoactive species, which upon visible light excitation reduce 1,1,1-trifluoro-2-iodoethane (10C) to generate trifluoroethyl radical (Scheme 10C). This radical adds to the alkene, and the resulting carbon-centered radical 10F is intercepted by a Cu(II)-bound amine species 10E—formed via ligand exchange between the resulting CuII(BINOL) complex and the amine—to forge the C–N bond through either reductive elimination or radical recombination, affording the trifluoroalkyl amine 10D. This transformation represents a rare example of copper-catalyzed, photoinduced intermolecular amino-fluoroalkylation, enabling the efficient construction of structurally complex γ-trifluoromethylated alkylamines.
3. Radical cross-couplings via metallaphotoredox catalysis
Metallaphotoredox catalysis has enabled a new class of convergent multicomponent reactions that merge electrophilic and nucleophilic radical species to construct C(sp3)–CF3 and C(sp3)–N bonds in a programmable fashion. These transformations combine the unique reactivity of photocatalytically generated radicals with the selectivity of transition metal catalysis, allowing for precise construction of trifluoroalkyl amines from simple, feedstock-compatible precursors. This section highlights recent advances in multicomponent synthetic methodologies that leverage dual photoredox and transition metal catalysis to access structurally complex fluoroalkyl amine derivatives.3.1 Dual copper/photoredox-catalyzed deoxytrifluoromethylation of amino-alcohols
Alcohols are abundant and structurally diverse feedstocks that serve as versatile building blocks in complex organic synthesis, particularly in radical C–C coupling reactions.27 MacMillan and co-workers developed a copper/photoredox-catalyzed deoxytrifluoromethylation protocol that converts amino alcohols into trifluoromethylated alkylamines28 (Scheme 11A). In this metallaphotocatalytic strategy in the presence of an iridium photocatalyst and a copper(II) terpyridine complex [Cu(terpy)Cl2], amino alcohols 11A function as nucleophilic alkyl radical precursors, while dimesityl(trifluoromethyl)sulfonium trifluoromethanesulfonate (dMesSCF3(OTf), 11B) serves as the electrophilic CF3 radical source. This platform accommodates a wide variety of acyclic, cyclic, spirocyclic, and heterocyclic amino alcohols, enabling the streamlined construction of three-dimensional, densely functionalized, and heterocycle-decorated trifluoromethylated alkylamine scaffolds 11C (Scheme 11B). Electron-rich heterocycles, such as furan and N-alkyl indole, when present in primary alcohols, are prone to oxidative degradation, leading to low to moderate yields under standard conditions. Sterically hindered tertiary alcohols also perform poorly, likely due to the high kinetic barriers associated with radical capture and the subsequent C–CF3 reductive elimination. These limitations pose significant challenges to the efficient diversification of fluoroalkyl amines. | ||
| Scheme 11 Dual copper/photoredox-catalyzed deoxytrifluoromethylation of amino-alcohols. (A) General reaction conditions. (B) Representative substrate scope. (C) Proposed mechanism. | ||
Mechanistically, the transformation proceeds via in situ activation of amino alcohols with benzoxazolium salts (NHC), forming NHC–alcohol adducts 11D (Scheme 11C). Photoexcitation of the iridium catalyst initiates single-electron oxidation of 11D, followed by deprotonation to 11E and exothermic β-scission to generate an aminoalkyl radical 11F. Concurrently, single-electron reduction of 11B by the photocatalyst generates an electrophilic CF3 radical. Reduction of Cu(terpy)Cl2 by the photocatalyst furnishes a Cu(I) species, which reacts with the CF3 radical to form a Cu(II)–CF3 intermediate 11G. Subsequent interception of 11F yields a high-valent Cu(III) species 11H that undergoes reductive elimination to forge the C(sp3)–CF3 bond, affording the trifluoroalkyl amine 11C. Importantly, the addition of tetrabutylammonium chloride (TBACl) was found to modulate the coordination sphere of the Cu(I) complex and suppress off-cycle reduction of CF3 radical to fluoroform (CHF3), thereby improving reaction efficiency. This protocol offers a general, modular, and operationally simple approach to access structurally complex trifluoromethylated alkylamines from native alcohols, providing versatile building blocks for applications in organic synthesis and drug discovery.
3.2 Dual nickel/photoredox-catalyzed methyl–trifluoromethylation of amino-substituted alkenes
To address the persistent challenge of constructing C(sp3)–C(sp3) bonds via intermolecular alkene difunctionalization,29 MacMillan and co-workers introduced a triple-radical sorting strategy that enables trimolecular 1,2-dialkylation of unactivated alkenes through bimolecular homolytic substitution (SH2) catalysis30 (Scheme 12A). This methodology achieves the regioselective union of electrophilic and nucleophilic alkyl radicals across unactivated olefins by leveraging steric and electronic differentiation to facilitate selective radical coupling. The reaction operates under dual catalysis, employing an iridium photocatalyst and an in situ-formed nickel catalyst based on Ni(acac)2 in combination with sterically hindered potassium tris(pyrazolyl)borohydride (KTp*). Under visible-light metallaphotoredox conditions, methanol 12B serves as a nucleophilic methyl radical precursor, while dimesityl(trifluoromethyl)sulfonium trifluoromethanesulfonate [dMesSCF3(OTf)] (12C) acts as the electrophilic CF3 radical source. This platform tolerates a wide range of structurally complex amino-substituted alkenes 12A, enabling access to three-dimensional and densely functionalized trifluoromethyl-substituted alkylamine scaffolds 12D (Scheme 12B), including drug derivatives (Scheme 12C).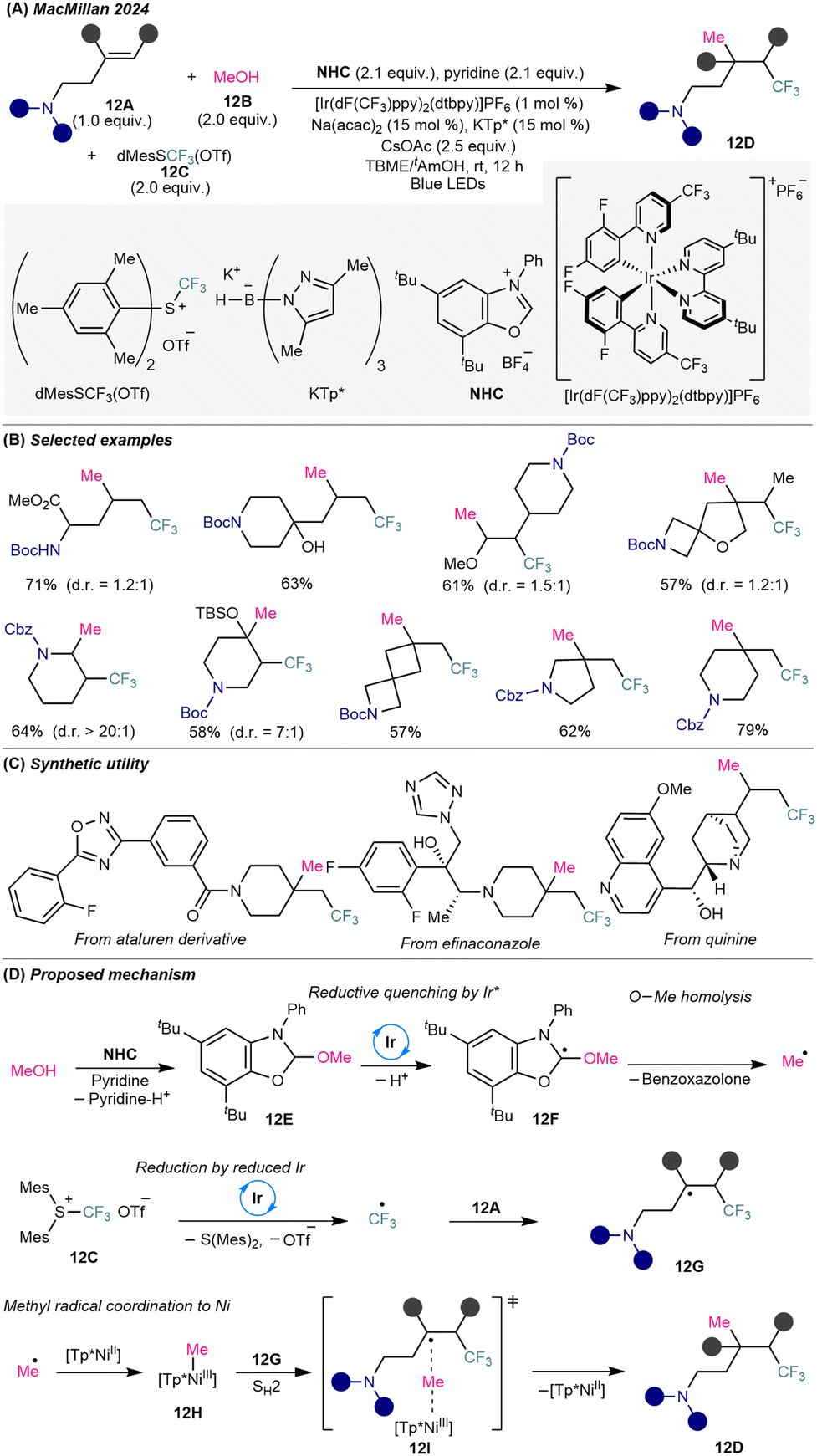 | ||
| Scheme 12 Dual nickel/photoredox-catalyzed deoxygenative methyl–trifluoromethylation of amino-substituted alkenes. (A) General reaction conditions. (B) Representative substrate scope. (C) Proposed mechanism. | ||
Mechanistically, methanol condenses with benzoxazolium salt (NHC) to form NHC-alcohol adduct 12E, triggering SET transfer via reductive quenching by Ir(III) photocatalyst, deprotonation to species 12F, and β-scission to release a methyl radical (Scheme 12D). In parallel, SET reduction of MesSCF3(OTf) 12C by the resulting Ir(II) photocatalyst forms an electrophilic CF3 radical that adds to the electron-rich amino-alkene 12A, generating a sterically and electronically distinct secondary or tertiary trifluoroalkyl radical 12G. The small-sized methyl radical is intercepted by a NiII(KTP*) to form a Ni(III)–methyl intermediate 12H, which undergoes outer-sphere SH2 coupling with bulky radical 12G via the transition state 12I, forging the methyl–alkyl bond and offer the complex trifluoroalkyl amine 12D. Only unactivated alkenes are employed in the methyl–trifluoromethylation reaction, presumably due to the enhanced reactivity of the resulting secondary or tertiary trifluoromethylated alkyl radicals, which facilitates their coupling with the Ni-bound methyl radical via SH2 substitution. This deoxygenative strategy showcases the potential of radical sorting to overcome the selectivity and reactivity challenges in three-component alkene difunctionalization and opens a general path toward C(sp3)-rich trifluoroalkylamine architectures.
3.3 Photocatalytic generation of trifluoromethylated iron carbenes for synthesis of trifluoroalkyl amines
Trifluoromethylated carbenes are highly reactive intermediates that offer significant potential for constructing compact, fluorine-rich scaffolds with enhanced physicochemical and pharmacokinetic properties.31 However, traditional diazo-based methods for accessing these species often suffer from limited substrate scope and safety issues. To overcome these limitations, MacMillan and co-workers developed a general metallaphotoredox platform that generates trifluoromethylated iron carbenes from bench-stable redox-active esters (RAE) 13B derived from 2,2,2-trifluoroethanol, enabling the [2+1] cycloaddition of amino-substituted alkenes 13A to access CF3-cyclopropyl-embedded alkylamines 13C32 (Scheme 13A). Using [Ir(dF(CF3)ppy)2(dtbpy)]PF6 as an iridium photocatalyst, iron(II) porphyrin Fe(TMPP)Cl as a catalyst, and Hantzsch ester (HE) as a reductant, a wide array of amino-substituted styrenes and alkenes, acrylic esters, and drug-like alkenes were efficiently converted into trifluoromethylated cyclopropylamines in good to excellent yields (Scheme 13B, top). Diastereoselectivity varies significantly across reaction substrates, with particularly low stereocontrol observed in reactions involving certain amino-substituted alkenes, potentially limiting the utility of this method in stereoselective synthesis. In addition, the same iron–carbene species enabled σ-bond insertion into N–H bonds, providing access to diverse N-2,2,2-trifluoroethylated amines (Scheme 13B, bottom).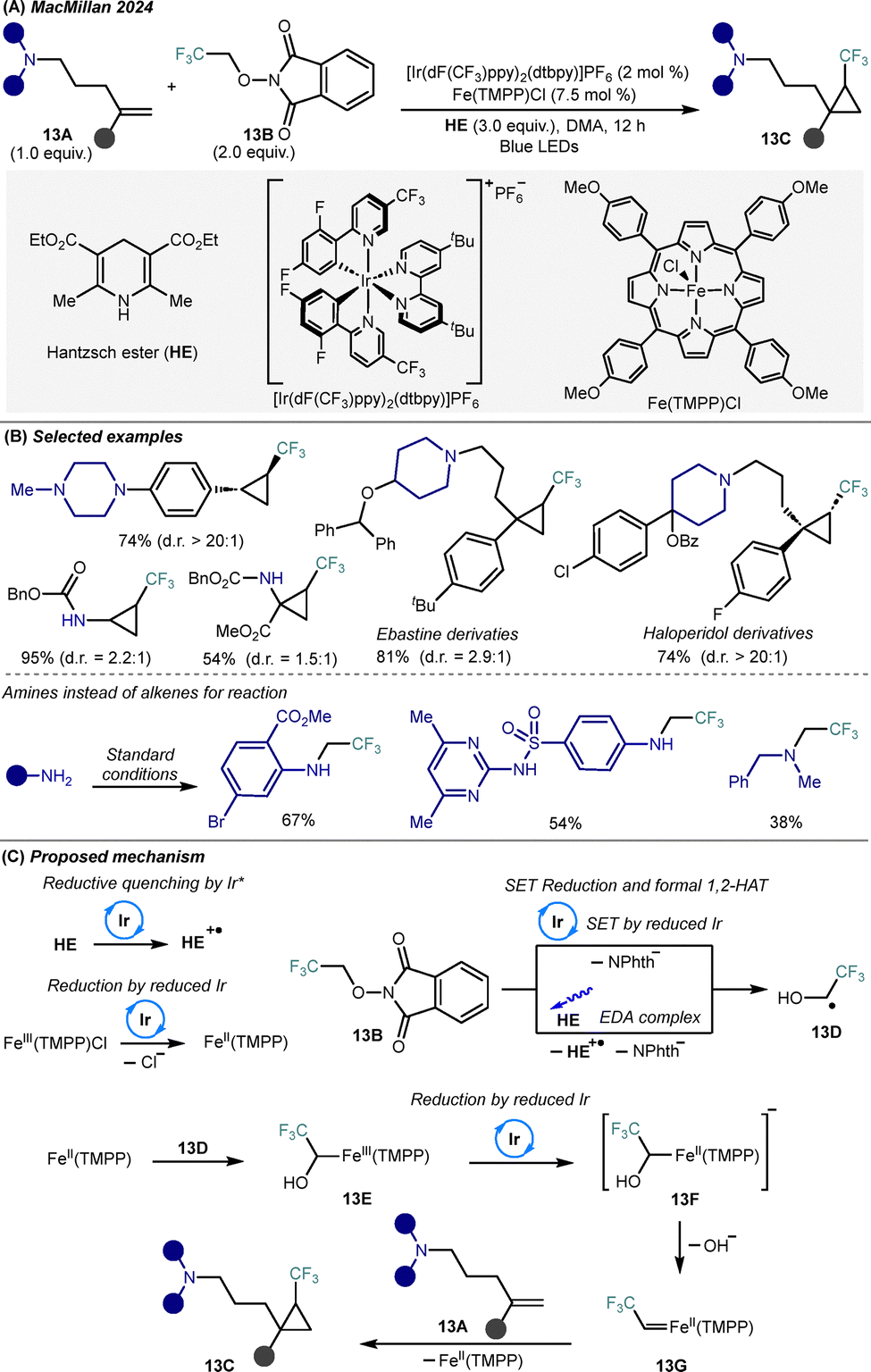 | ||
| Scheme 13 Photocatalytic generation of trifluoromethylated iron carbenes for cyclopropanation and N–H insertion reactions. (A) General reaction conditions. (B) Representative substrate scope. (C) Proposed mechanism. | ||
Mechanistically, photoexcitation of the Ir(III) catalyst followed by reductive quenching by Hantzsch ester generates a strongly reducing Ir(II) species, which reduces Fe(III) porphyrin to Fe(II) species (IrII(TMPP), Scheme 13C). Simultaneously, the RAE 13B is reduced either by Ir(II) or through the formation of a photoactive EDA complex with Hantzsch ester, leading to decarboxylation and generation of a 2-hydroxytrifluoroethyl radical 13D. This radical is intercepted by Fe(II) species to form an Fe(II)–(1-hydroxy-2,2,2-trifluoroethy) intermediate 13E, which undergoes a second photocatalytic single-electron reduction to give a nucleophilic alkyl-Fe(II) porphyrin species 13F. α-Elimination from this species ejects a hydroxide ion and furnishes the reactive α-CF3-carbene-substituted Fe(II) porphyrin 13G. This carbene intermediate engages in cyclopropanation of alkene 13A to deliver structurally complex CF3-substituted alkylamines 13C. Overall, this platform provides a mild, modular, and diazo-free approach for generating fluorinated carbene species, expanding the synthetic toolbox for late-stage functionalization of complex molecules under visible light.
3.4 Dual nickel/photoredox-catalyzed methyl-trifluoroethylation of amino-substituted alkyl alkenes
To expand the scope of C(sp3)–C(sp3) bond formation via alkene difunctionalization, Koh and co-workers developed a photoredox/nickel dual-catalyzed platform that enables trimolecular 1,2-dialkylation of unactivated alkenes through a SH2 mechanism33 (Scheme 14A). This strategy allows for the simultaneous installation of electrophilic and nucleophilic alkyl radicals across unactivated amino-substituted alkenes without the need for directing auxiliaries. Under optimized conditions, the reaction combines 1,1,1-trifluoro-2-iodoethane (14B), a hypervalent iodine(III) reagent (PhI(OAc)2, 14C), and a variety of mono- and 1,1-disubstituted amino-substituted alkenes 14A in the presence of Ni(acac)2 catalyst, KTp* ligand, and the organic photocatalyst 4CzIPN, affording complex trifluoroalkyl amine derivatives 14D bearing tertiary or quaternary carbon centers in good to excellent yields with high chemo- and regioselectivity. Similar to the MacMillan metallophotocatalytic system (Scheme 12), alkyl-substituted alkenes are required to enhance reactivity and productivity for accessing trifluoroalkylamine products.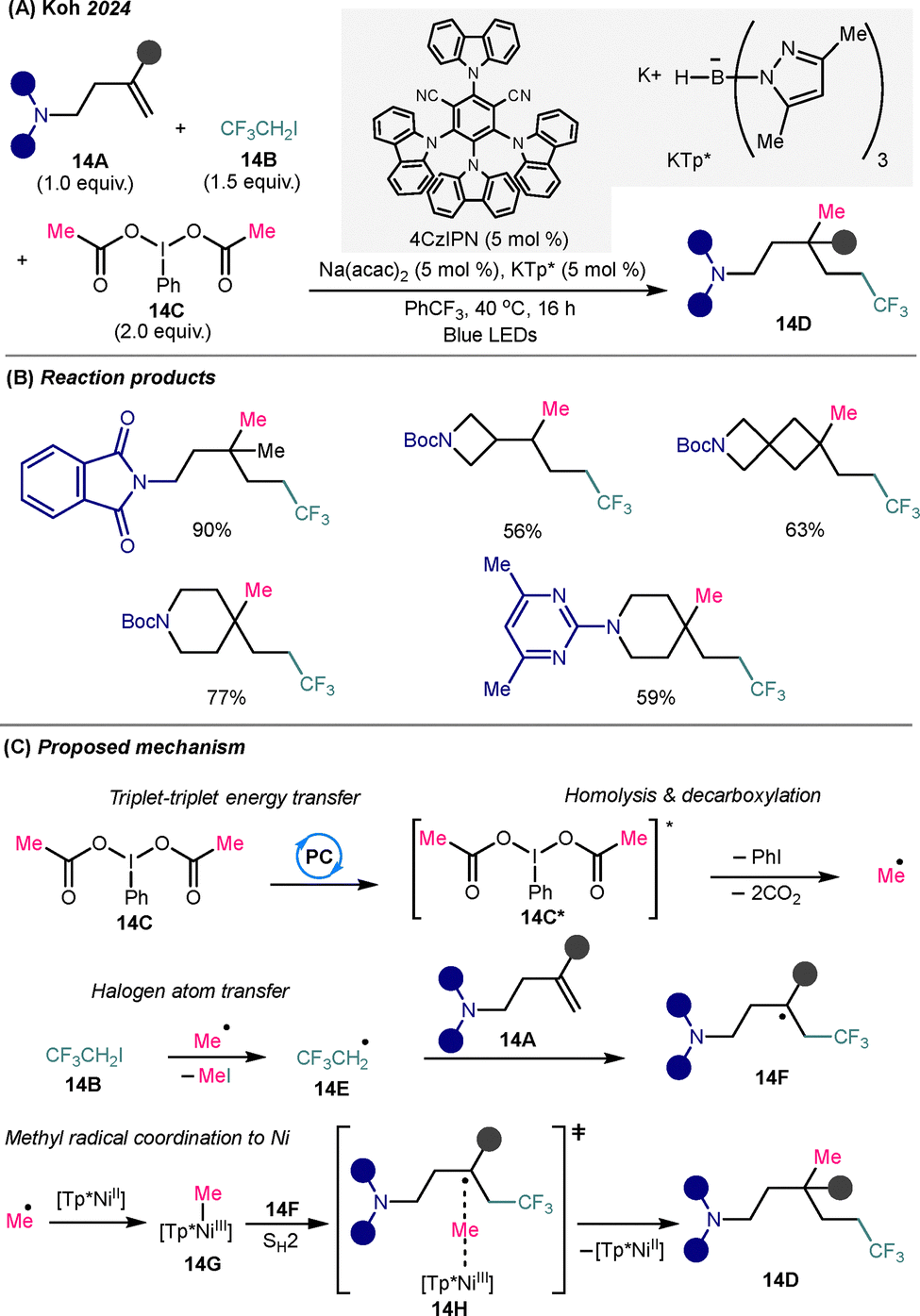 | ||
| Scheme 14 Dual nickel/photocatalytic methyl-trifluoroethylation of amino-substituted alkyl alkenes. (A) General reaction conditions. (B) Representative substrate scope. (C) Proposed mechanism. | ||
Mechanistic studies suggest that a nucleophilic methyl radical is generated via O–I bond homolysis of photoexcited PhI(OAc)2* (14C*), triggered by triplet–triplet energy transfer from the photoexcited 4CzIPN. The nucleophilic methyl radical selectively initiates halogen atom transfer (XAT) with electron-deficient 1,1,1-trifluoro-2-iodoethane to produce an electrophilic trifluoroethyl radical 14E, which subsequently undergoes a polarity-matched addition to the electron-rich amino-alkene substrate (14A), forming the amino-trifluoroalkyl radical 14F. The methyl group also binds with NiII(KTp*), forming the Me-NiIII(KTp*) species 14G. The resulting bulky secondary trifluoroalkyl radical 14F is subsequently intercepted through an outer-sphere SH2 coupling event with the Ni(KTp)-bound methyl radical via the transition state 14H, forging the second C(sp3)–C(sp3) bond to deliver the trifluoroalkyl amine 14D. This transformation represents a significant advance in site-selective dialkylation of unactivated alkenes, offering a modular strategy for constructing structurally complex, three-dimensional, carbon-rich trifluoroalkylamine scaffolds.
3.5 Dual nickel/photoredox-catalyzed synthesis of masked arylaminomethyl trifluoromethyl ketones
Structurally elaborate trifluoromethyl ketones (TFMKs) bearing aminomethyl groups are attractive intermediates in pharmaceutical and agrochemical synthesis, yet their direct construction remains challenging due to the inherent reactivity mismatch between nucleophilic amines and electrophilic trifluoroacetyl moieties.34 To address this, Cheung, Ma, and co-workers reported a modular metallaphotoredox-catalyzed three-component coupling of nitroarenes 15A, tertiary alkylamines 15B, and 2-(trifluoromethyl)-1,3-dioxolane-2-carboxylic acid (15C)—serving as a masked trifluoroacetyl radical precursor34 (Scheme 15A). Employing an iridium photocatalyst in combination with a phenanthroline–nickel complex as the catalytic system, this strategy enables direct access to structurally diverse N-(CF3-dioxolanyl)methyl anilines 15D as protected surrogates of aminomethyl TFMKs, offering enhanced chemoselectivity and functional group tolerance (Scheme 15B). The resulting acetal-masked products can be unmasked to access otherwise unstable aminomethyl TFMKs or derivatized into valuable fluorinated azaheterocycles. | ||
| Scheme 15 Dual nickel/photoredox-catalyzed synthesis of masked arylaminomethyl trifluoromethyl ketones from nitroarenes, tertiary alkylamines, and CF3-dioxolanyl precursors. (A) General reaction conditions. (B) Representative substrate scope. (C) Proposed mechanism. (D) Synthetic utility and applications. | ||
Mechanistically, photoexcitation of [Ir(dF(CF3)ppy)2(dtbpy)]PF6 initiates an oxidative quenching process with the tertiary alkylamine 15B, generating an α-aminoalkyl radical 15E (Scheme 15C). Concurrently, CF3-dioxolanyl-carboxylic acid 15C is deprotonated by excess tertiary alkylamine to its anionic form, which undergoes photocatalytic SET oxidation to afford radical species 15F, followed by facile decarboxylation to yield the CF3-dioxolanyl radical 15G. The resulting Ir(II) species reduces a nitroarene, furnishing nitrogen-centered intermediates (e.g., nitrosoarenes or diarylhydrazines), which ultimately form the N-aryl aminyl radical 15H through sequential reduction pathways. In parallel, the Ni(II) catalyst is photochemically reduced to low-valent Ni(I) and Ni(0) species. The nucleophilic α-aminoalkyl radical 15E couples with the electrophilic N-aryl aminyl radical 15H to form aminal intermediate 15I, which undergoes Ni-mediated dealkylation to generate the N-arylaminomethyl–Ni(II) species 15J. Crucially, the final C(sp3)–C(sp3) bond formation proceeds either via an inner-sphere Ni-mediated reductive elimination through a Ni(III) complex 15K, or via an outer-sphere SH2 mechanism through transition state 15L between the Ni(II)–alkyl intermediate 15J and the sterically bulky CF3-dioxolanyl radical 15E, ultimately affording the masked TFMK product 15D with high selectivity.
The synthetic utility of this method is underscored by its late-stage diversification potential and compatibility with downstream transformations (Scheme 15D). For instance, product 15M undergoes BBr3-promoted deprotection to yield the annulated product, CF3-substituted dihydrooxazine 15P. The corresponding trifluoromethyl ketone (TFMK) 15N and its hydrated form 15O—detectable only by high-resolution mass spectrometry—were found to be unstable, thereby significantly limiting the general applicability of this method for the synthesis and further study of arylamino TFMK compounds. Nevertheless, 15P serves as a valuable synthetic intermediate, undergoing condensation with hydroxylamine to furnish CF3-substituted piperazine 15Q and reduction to generate N-(2-hydroxy-3,3,3-trifluoropropyl)anilines 15R. Collectively, this work expands the synthetic toolbox for the construction of complex, functionalized trifluorinated amines and their derivatives, offering novel compounds with potential applications in organic synthesis, drug discovery, and agrochemical development.
3.6 Dual Ni/photoredox-catalyzed synthesis of complex N-trifluoroalkyl anilines
Structurally complex CF3-incorporated N-alkyl anilines represent a valuable class of fluorinated scaffolds with promising applications in drug discovery. However, conventional approaches typically require multistep prefunctionalization to prepare reactive trifluoroalkyl electrophiles for coupling with anilines, thus limiting molecular diversity and synthetic efficiency. To overcome these challenges, Ma, Cheung, and co-workers developed a metallaphotoredox-catalyzed three-component amination strategy that achieves formal C–N transposition and C(sp3)–H alkylation of tertiary alkylamines 16B with nitroarenes 16A and RAEs (16C) derived from CF3-containing carboxylic acids35 (Scheme 16A). The reaction employs [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 as the photocatalyst, a bathophenanthroline-ligated Ni(II) complex as the transition metal co-catalyst, and Hantzsch ester (HE) as the reductant, operating under mild, redox-neutral conditions. Two examples of trifluoroalkyl aniline products 16D are demonstrated (Scheme 16B). In contrast, the use of electron-deficient nitroarenes and branched tertiary alkylamines typically resulted in diminished yields, likely due to reduced reactivity and increased steric hindrance. | ||
| Scheme 16 Photocatalytic synthesis of N-trifluoroalkyl anilines from nitroarenes, alkylamines, and CF3-substituted redox-active esters. (A) General reaction conditions. (B) Representative substrate scope. (C) Proposed mechanism. | ||
Mechanistically, visible-light excitation of the Ir(III) photocatalyst triggers SET with triethylamine 16B′ to form an α-aminoalkyl radical 16E via reductive quenching and deprotonation (Scheme 16C). Simultaneously, Ir(III) undergoes SET with Hantzsch ester to generate a highly reducing Ir(II) species, which sequentially reduces both the nitroarene 16A (in the presence of triflic acid (HOTf)) to form an N-aryl aminyl radical 16F and the CF3–alkyl RAE 16C to produce a trifluoroalkyl radical 16G. Ni(II) salt is also photocatalytically reduced to low-valent Ni(I) or Ni(0) species. The nucleophilic α-aminoalkyl radical 16E couples with the electrophilic N-aryl aminyl radical 16F—either directly or via Ni-mediated C–N bond formation—to furnish an aminal intermediate 16H. This intermediate undergoes Lewis acidic Ni(II)-promoted deamination to generate an N-aryl imine 16I, which is then trapped by the trifluoroalkyl radical 16G (potentially stabilized by Ni(I) or Ni(0)) to form a trifluoroalkylated aminyl radical 16J. Finally, this radical undergoes photoreduction or HAT by the Hantzsch ester radical cation, furnishing the N-trifluoroalkyl aniline product 16D. Although demonstrated in a limited number of examples, this strategy provides a modular and efficient route to access structurally intricate trifluoroalkylated N-alkyl anilines that are otherwise difficult to synthesize using classical two-component methods.
3.7 Dual nickel/photoredox-catalyzed four-component synthesis of complex trifluoroalkyl anilines
Structurally elaborate N-trifluoroalkyl anilines are valuable motifs in pharmaceutical research due to their improved metabolic stability, lipophilicity, and biological activity. However, conventional strategies for accessing such scaffolds often suffer from limited efficiency and structural diversity. Four-component reactions offer a powerful synthetic platform for the rapid construction of complex trifluoroalkylated amines from simple precursors. By integrating multiple radical coupling events and bond-forming processes in a single operation, these approaches enable direct access to densely substituted, functionally rich trifluoroalkyl amines, highlighting the potential of photocatalytic radical chemistry in medicinal compound development.In this context, Ma, Cheung, and co-workers developed a modular metallaphotoredox-catalyzed four-component amination strategy that merges nitroarenes 17A, tertiary alkylamines 17B, 3,3,3-trifluoropropene 17C, and RAEs 17D derived from aliphatic carboxylic acids7 (Scheme 17A). This platform enables streamlined access to N-trifluoroalkyl anilines 17E, bearing both CF3 units and distal tertiary or quaternary carbon centers along the alkyl chain. The reaction employs [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 as the photocatalyst, a bathophenanthroline-ligated Ni(II) complex as the transition metal co-catalyst, Hantzsch ester (HE) as a sacrificial reductant, and HOTf as an additive, operating under blue light irradiation at approximately 80 °C. A broad range of functionalized nitroarenes, tertiary alkylamines, and structurally diverse RAEs—including primary, secondary, and tertiary alkyl derivatives—are well tolerated, enabling the synthesis of a diverse array of N-trifluoroalkyl aniline products (Scheme 17B). In contrast, the use of electron-deficient nitroarenes and RAEs derived from primary and secondary carboxylic acids resulted in lower product yields, likely due to the instability of the corresponding nitrogen- and carbon-centered radicals, which tend to undergo competing two- or three-component side reactions, thereby impeding efficient four-component bond formation.
 | ||
| Scheme 17 Nickel/photoredox-catalyzed synthesis of N-trifluoroalkyl anilines from nitroarenes, tertiary amines, redox-active esters, and 3,3,3-trifluoropropene. (A) General reaction conditions. (B) Representative substrate scope. (C) Proposed mechanism. | ||
Mechanistically, photoexcitation of the Ir(III) photocatalyst initiates single-electron reduction of the nitroarene 17A to generate an N-aryl aminyl radical 17F, while concurrently reducing the RAE 17D to furnish an alkyl radical 17G and the ligated Ni(II) salt to low-valent Ni(I) and Ni(0) species (Scheme 17C). Concurrently, both Hantzsch ester and tertiary alkylamine 17B undergo oxidative quenching with the resulting Ir(IV) species to generate Hantzsch ester radical cation and α-aminoalkyl radical 17H, respectively. The nucleophilic alkyl radical 17G adds to the electron-deficient 3,3,3-trifluoropropene 17C to generate a trifluoroalkyl radical 17I. Meanwhile, the highly nucleophilic α-aminoalkyl radical 17H couples with the highly electrophilic N-aryl aminyl radical 17F to form an aminal intermediate 17J, which likely undergoes Ni(0)-mediated dealkylation to furnish an N-arylaminomethyl–Ni(II) complex 17K. This complex subsequently releases an N-arylaminomethyl radical 17L, which is intercepted by the trifluoroalkyl radical 17I to deliver the final N-trifluoroalkyl aniline product 17E. Low-valent nickel species (Ni0/NiI) are proposed to play key roles in stabilizing the trifluoroalkyl radical 17I and facilitating both C–C and C–N bond-forming events, thereby enhancing overall reaction efficiency. This work represents a significant advance in multicomponent trifluoromethylation chemistry, offering an efficient and programmable route to highly architecturally complex N-trifluoroalkyl aniline scaffolds that are otherwise difficult to access using conventional approaches.
3.8 Scandium/photoredox-catalyzed amido-, amino-, and azido-trifluoromethylation of alkenes
In an effort to develop highly efficient trifluoromethylative difunctionalization of alkenes, Shen and co-workers reported a synergistic Lewis acid and photoredox-catalyzed method employing a selenium ylide-based trifluoromethylating reagent 18C36 (Scheme 18A). Using fac-Ir(ppy)3 as photocatalyst and Sc(OTf)3 as a Lewis acid co-catalyst, a broad range of aromatic and heterocyclic alkenes 18A underwent smooth amino-trifluoromethylation reaction with various nitrogen-based nucleophiles 18B, such as sulfonamides, anilines and azide, under blue light irradiation (Scheme 18B). A variety of β-trifluoromethylated alkylamines and azo compounds 18D decorated with diverse functional groups are formed. In contrast, electron-deficient styrenes, alkyl-substituted alkenes, and Cbz-protected amines failed to afford the desired difunctionalized products under the optimized conditions. | ||
| Scheme 18 Synergistic scandium/photoredox-catalyzed amido-, amino-, and azido-trifluoromethylation of alkenes. (A) General reaction conditions. (B) Representative substrate scope. (C) Proposed mechanism. | ||
Mechanistic investigations reveal that Sc(OTf)3 forms a Lewis acid–base complex 18E with the electrophilic ylide reagent 18C (Scheme 18C). 18E significantly enhances the SET reactivity with the photoexcited Ir(ppy)3 by lowering the LUMO energy and raising the reduction potential. Cyclic voltammetry and DFT studies suggest that 18E is more readily reduced than the free ylide 18C, facilitating SET event and CF3 radical generation. The CF3 radical adds to the alkene to generate trifluoroalkyl radical 18F, which undergoes photocatalytic oxidation to form cationic intermediate 18G. Subsequent nucleophilic trapping by a nitrogen nucleophile completes the 1,2-difunctionalization, affording the trifluoroalkylamine product 18D. This study highlights the power of Lewis acid modulation in tuning radical generation from electrophilic reagents and establishes a general platform for constructing CF3-bearing molecules with high efficiency and functional diversity.
4. Inert bond functionalization for direct C(sp3)–CF3 bond formation
Direct C(sp3)–H and C(sp3)–N functionalization strategies obviate the need for prefunctionalized substrates, providing streamlined access to trifluoromethylated aliphatic amines from native feedstocks. Visible-light-mediated approaches, leveraging either HAT or deaminative radical generation, enable regioselective and late-stage incorporation of CF3 groups. This section highlights representative photoinduced methodologies that achieve site-selective trifluoromethylation of unactivated aliphatic amines via C–H or C–N activation.4.1 Copper/decatungstate-catalyzed C–H trifluoromethylation
C(sp3)–H trifluoromethylation provides a direct strategy for the synthesis of fluorinated molecules; however, achieving regioselective C–H activation in the absence of directing groups remains a longstanding challenge.37 To address this challenge, MacMillan and co-workers developed a dual catalytic platform combining decatungstate photocatalysis with copper catalysis, enabling the direct trifluoromethylation of both strong, unactivated aliphatic C–H bonds and weaker benzylic C–H bonds in simple alkylamine feedstocks 19A38 (Scheme 19A). This strategy employs bench-stable, commercially available Togni reagent (19B) as the CF3 source, sodium decatungstate (NaDT) as a potent HAT photocatalyst, Cu(II) salt as co-catalyst, and sulfuric acid (H2SO4) along with sodium chloride (NaCl) as key additives. The method exhibits broad substrate scope, accommodating linear, cyclic, and aromatic alkylamines (Scheme 19B), as well as amine-containing bioactive compounds, natural products, and pharmaceuticals (Scheme 19C), without the need for prefunctionalization or directing groups, delivering trifluoromethylated alkylamines 19C in modest to good yields. However, for aliphatic amines bearing multiple types of C(sp3)–H bonds with similar bond strengths, the regioselective C–H trifluoromethylation is typically modest to moderate, underscoring the challenge of achieving high site selectivity in such systems. | ||
| Scheme 19 Copper/decatungstate photocatalytic trifluoromethylation of aliphatic C(sp3)–H bonds in alkylamines. (A) General reaction conditions. (B) Representative substrate scope. (C) Synthetic utility and applications. (D) Proposed mechanism. | ||
Notably, the regioselectivity of C–H trifluoromethylation is governed by both steric and electronic factors (Scheme 19D). Upon protonation of the amine nitrogen—such as in pyrrolidine (19D)—by H2SO4, the corresponding ammonium salt (19E) is formed, which converts the typically hydridic α-C–H bond into an acidic one. Under near-UV irradiation, the photoexcited decatungstate ion ([W10O32]4−*), bearing an electrophilic W–O˙ radical, selectively abstracts a hydrogen atom from the electron-neutral and sterically accessible C(sp3)–H bond to generate an alkyl radical (e.g., β-C–H abstraction of protonated pyrrolidine to form a pyrrolidinyl radical 19F). Deprotonation of the resulting H[W10O32]5− species to form [W10O32]5− significantly enhances its reducing power, enabling SET to the Togni reagent 19B and subsequent generation of a CF3 radical. Simultaneously, CuCl2 is reduced by [W10O32]5− to Cu(I), which interacts with the CF3 radical to form a Cu(II)–CF3 species 19G. Capture of the alkyl radical 19F and subsequent reductive elimination from (Alkyl)(CF3)Cu(III) species 19H furnishes the trifluoromethylated alkylamine product 19I. The NaCl additive likely suppresses oxidative degradation pathways in sensitive alkylamines and may serve as an additional source of chloride radicals under photooxidative conditions, further facilitating selective HAT from protonated amines. This work represents a significant advance in non-directed C(sp3)–H trifluoromethylation and underscores the potential of metallaphotoredox catalysis for site-selective, late-stage molecular editing to access trifluoroalkylamine architectures.
4.2 Deaminative trifluoromethylation of primary alkylamines
Alkylamines are not only versatile feedstocks in synthesis and industrial processing, but also transformable groups to allow structural modifications.39 To address the underdeveloped yet challenging area of deaminative functionalizations, Li, Wang, and co-workers developed a site-specific, radical-mediated trifluoromethylation of aliphatic primary amines under visible light using N-anomeric amide (Levin reagent, 20C) and copper(III) tris(trifluoromethyl) complex (Grushin reagent, CuIII(bipy)(CF3)3, 20B)40 (Scheme 20A). This transformation enables direct conversion of diamine scaffolds 20A (e.g., amino-substituted carbamates, amides, and cyclic amines) into trifluoroalkyl amine derivatives 20D (Scheme 20B), including amino acids, dipeptides, and drug derivatives (Scheme 20C), without requiring preactivation of native amino groups or harsh conditions. More sterically bulky α-branched substrates, such as α-secondary carbon-based primary amines, also underwent the reaction; however, only low yields of the trifluoromethylated products were obtained, likely due to increased steric hindrance that hampers the formation of key reactive intermediates. | ||
| Scheme 20 Photoinduced deaminative trifluoromethylation of alkylamines.(A) General reaction conditions. (B) Representative substrate scope. (C) Synthetic utility and applications. (D) Proposed mechanism. | ||
The protocol proceeds via in situ generation of an intermediate 20E from the diamine 20A and Levin reagent 20C, followed by facile decomposition to isodiazene 20F (Scheme 20D). Upon blue-light irradiation, the photolysis of the Grushin reagent 20B takes place to form both CF3 radical and CuII(bipy)(CF3)2 species 20B'. The CF3 radical engages in HAT event from 20F to generate the diazenyl radical 20G. Alternatively, 20F reacts directly with the CF3 groups in the Grushin reagent to form 20G. 20G then undergoes N2 elimination to yield an aminoalkyl radical 20H, which interacts with CuII(bipy)(CF3)2 to furnish a high-valent Cu(III) species that undergoes reductive elimination to construct the C(sp3)–CF3 bond, creating the complex trifluoroalkyl amine product 20D. This strategy significantly expands the toolbox for direct amine-to-CF3 conversion and offers a general approach for site-specific alkyl trifluoromethylation in complex molecular settings.
5. Photoinduced catalyst-free synthesis of trifluoroalkyl amines
The elimination of catalysts in trifluoromethylation chemistry aligns with the growing demand for sustainable and pharmaceutically compatible synthetic methodologies. Catalyst-free, light-driven transformations offer notable advantages, including operational simplicity, lower cost, and the avoidance of residual metal contaminants. This section highlights a recent catalyst-free protocol for the construction of trifluoroalkyl amines via a radical pathway, employing readily available substrates under ambient conditions.Gaunt and co-workers developed a visible-light-mediated, multicomponent carbonyl trifluoromethylative amination protocol that directly couples aldehydes 21A, secondary amines 21B, and the Ritter trifluoroiodomethane–DMSO reagent (CF3I·DMSO, 21C) to afford β-trifluoromethylated tertiary alkylamines 21D41 (Scheme 21A). Notably, this transformation proceeds under mild conditions without the need for a photocatalyst or transition metal, relying solely on visible light. The method exhibits broad substrate scope, accommodating a wide range of functionalized aliphatic aldehydes and secondary alkylamines, including medicinally relevant scaffolds, thus highlighting its potential for late-stage modification and lead optimization in drug discovery (Scheme 21B). In contrast, α-substituted aldehydes fail to deliver the corresponding trifluoromethylated amines, likely due to steric hindrance that impedes the radical addition step.
 | ||
| Scheme 21 Photoinduced carbonyl trifluoromethylative amination of aldehydes. (A) General reaction conditions. (B) Representative substrate scope. (C) Proposed mechanism. | ||
Mechanistically, the reaction initiates via condensation of the aldehyde and amine to generate an iminium ion 21E, which undergoes tautomerization to the corresponding enamine 21F. Upon visible-light promoted conditions, CF3I is converted to CF3 radical, which undergoes regioselective addition to the electron-rich enamine to form a transient α-amino radical 21G. This intermediate is proposed to undergo either single-electron oxidation by CF3I, or XAT with CF3I to iodo-trifluoroalkyl amine intermediate 21H followed by iodide elimination, to generate a β-trifluoromethylated iminium species 21I. Subsequent reductive workup with sodium triacetoxyborohydride furnishes the desired β-trifluoromethylated tertiary alkylamine 21D. This methodology provides a simple and efficient strategy for constructing structurally intricate trifluoroalkylamines that are otherwise challenging to access using conventional synthetic approaches.
6. Stereoselective and asymmetric approaches
Achieving stereocontrol in trifluoromethylative difunctionalization reactions remains a formidable challenge due to the high reactivity of CF3 radicals and aminyl intermediates. Recent progress in asymmetric photocatalysis has demonstrated that regio- and enantioselective amino-trifluoromethylation of dienes is feasible through judicious design of chiral catalysts and radical coupling pathways. This section highlights an emerging strategy for the enantioselective synthesis of chiral trifluoroalkyl amines via radical-based difunctionalization, illustrating the potential of stereochemically defined fluorinated scaffolds in medicinal chemistry.Despite the synthetic appeal of enantioselective aminative difunctionalization of 1,3-dienes, previous efforts have been largely limited to two-component systems or 1,2-selective processes, with limited success in achieving 1,4-regioselectivity—particularly when employing anilines, which are weak nucleophiles and prone to catalyst deactivation via coordination. The development of a regio- and enantioselective three-component protocol that directly couples simple anilines, 1,3-dienes, and CF3 radicals thus poses both a significant synthetic challenge and a conceptual advance in radical C–N bond-forming methodologies. A recent study by Li, Zhang, and co-workers has addressed this challenge by developing a copper-catalyzed, enantioselective three-component 1,4-trifluoroalkylamination of 1,3-dienes 22A, employing anilines 22B as the nitrogen source and a Togni reagent 22C as the CF3 source under mild conditions42 (Scheme 22A). This transformation enables highly regio- and stereoselective access to a structurally diverse library of chiral trifluoromethylated allylic anilines 22D. A wide range of aryl-substituted 1,3-dienes and drug-like frameworks were converted to the desired products in high yields and good enantioselectivities (Scheme 22B). Both electron-rich and electron-deficient primary and secondary anilines prove compatible under the optimized conditions. Conversely, trifluoromethylamination with aliphatic 1,3-dienes (e.g., 1-benzyl-1,3-diene) and aliphatic amines (e.g., dibenzylamine, benzylamine, morpholine, and N-methyl benzylamine) proved unproductive under the standard conditions. Selective oxidation of product 22E afforded the highly stereoselective dihydroxylated trifluoroalkyl aniline 22F, thereby increasing the molecular complexity (Scheme 22C).
 | ||
| Scheme 22 Cu-catalyzed enantioselective radical 1,4-trifluoroalkylamination of 1,3-dienes. (A) General reaction conditions. (B) Representative substrate scope. (C) Synthetic utility and applications. (D) Proposed mechanism. | ||
Mechanistically, the reaction proceeds via copper-mediated SET from the chiral ligand-coordinated Cu(I) complex 22G to the Togni reagent, generating a CF3 radical and a Cu(II) benzoate species (Scheme 22D). The CF3 radical undergoes regioselective addition to the terminal position of the 1,3-diene, forming a trifluoromethylated allylic radical 22H. Simultaneously, ligand exchange between the Cu(II) benzoate and aniline yields a Cu(II)–aminyl species, existing as an aminyl radical-associated Cu(I) complex 22I. The enantioselective C–N bond formation arises from radical–radical cross-coupling between the allylic radical 22H and the chiral Cu(I)-bound aminyl radical, affording thec trifluoromethylated allyl aniline 22D in an enantiomerically enriched form. The high enantioselectivity arises from noncovalent interactions between the chiral bisoxazoline ligand's aromatic rings, the C–F bond of the CF3 radical 22H, and the methyl C–H bond of the aminyl radical derived from secondary anilines in species 22I. This work showcases a rare yet powerful platform for stereocontrolled radical difunctionalization of dienes, opening new avenues for the synthesis of fluorinated allylic amines with high regio- and stereoselectivity. It represents a significant advancement in asymmetric radical chemistry and enriches the toolkit for constructing fluorine-containing molecules of pharmaceutical relevance.
7. Conclusion
In this review, we summarize recent advances in the light-driven synthesis of trifluoromethylated aliphatic amines, organized by reaction strategy and disconnection logic. Key developments include direct formation of C(sp3)–CF3 bonds from inert precursors, alkene difunctionalization via radical–polar crossover or radical trapping, and metallaphotoredox-catalyzed cascades that forge C–CF3, C–N, and C–C bonds in a convergent fashion. Additionally, emerging examples of photocatalyst-free and asymmetric strategies have begun to expand the synthetic toolbox. Collectively, these approaches underscore the power of photocatalysis to construct structurally complex, fluorine-rich amines from simple building blocks under mild and modular conditions.Despite these advances, several challenges and opportunities remain. Most reported transformations proceed in a racemic manner; therefore, the development of enantioselective variants represents a critical yet underexplored frontier. The design of highly efficient chiral Brønsted acids, Lewis acids, and transition-metal catalysts, along with the strategic use of chiral auxiliaries on substrates, will be pivotal for enabling asymmetric bond formation and accessing chiral analogues. Emerging photoenzymatic radical-based coupling reactions may also pave the way toward highly stereoselective synthesis. In addition, many current protocols rely on photocatalysts or transition-metal complexes, which can limit their pharmaceutical applicability due to concerns over cost, regulatory hurdles, and residual metal contamination. In this context, the growing use of electron donor–acceptor (EDA) complexes provides a promising metal-free alternative for initiating radical processes, particularly when targeting drug-like or sensitive substrates. Furthermore, while photoredox catalysis has demonstrated impressive reactivity on the laboratory scale, scalability remains a major bottleneck for industrial translation. The development of more efficient, scalable, and continuous-flow-compatible photochemical systems will be essential to unlock the full potential of these methodologies for large-scale synthesis. Looking ahead, future innovations that integrate asymmetric catalysis, metal-free or photocatalyst-free radical generation, and process intensification are expected to significantly expand the practical utility of light-driven synthesis across both academic and industrial settings.
In conclusion, the light-driven synthesis of trifluoromethylated aliphatic amines constitutes a rapidly evolving and highly promising field, offering powerful strategies to access molecular architectures of significant pharmaceutical relevance. Continued innovation in photocatalysis is poised to enable increasingly sophisticated synthetic methodologies, thereby expanding chemical space for drug discovery and facilitating the streamlined synthesis of novel, fluorine-rich bioactive compounds.
Conflicts of interest
There are no conflicts to declare.Data availability
No primary research results, software or code have been included and no new data were generated or analyzed as part of this review.Acknowledgements
F. Y. K. thank the Research Grants Council of Hong Kong, General Research Fund (GRF 14309123). C. W. C. thanks the Research Assistant Professorship Scheme (CUHK, 3130197) and the National Natural Science Foundation of China (no. 21971186 and 22271216) for financial support. We thank the financial support from Innovation and Technology Commission (HKSAR, China) to the State Key Laboratory of Synthetic Chemistry.Notes and references
- (a) R. D. Taylor, M. MacCoss and A. D. G. Lawson, Rings in drugs, J. Med. Chem., 2014, 57, 5845–5859 CrossRef CAS PubMed; (b) M. A. M. Subbaiah and N. A. Meanwell, Bioisosteres of the phenyl ring: recent strategic applications in lead optimization and drug design, J. Med. Chem., 2021, 64, 14046–14128 CrossRef CAS PubMed; (c) J. Shearer, J. L. Castro, A. D. G. Lawson, M. MacCoss and R. D. Taylor, Rings in clinical trials and drugs: present and future, J. Med. Chem., 2022, 65, 8699–8712 CrossRef CAS PubMed; (d) N. A. Meanwell, Applications of bioisosteres in the design of biologically active compounds, J. Agric. Food Chem., 2023, 71, 18087–18122 CrossRef CAS PubMed.
- (a) N. A. Meanwell, Fluorine and fluorinated motifs in the design and application of bioisosteres for drug design, J. Med. Chem., 2018, 61, 5822–5880 CrossRef CAS PubMed; (b) M. Inoue, Y. Sumii and N. Shibata, Contribution of organofluorine compounds to pharmaceuticals, ACS Omega, 2020, 5, 10633–10640 CrossRef CAS PubMed; (c) C. Zhang, Fluorine in medicinal chemistry: in perspective to COVID-19, ACS Omega, 2022, 7, 18206–18212 CrossRef CAS PubMed.
- (a) W. Zhu, J. Wang, S. Wang, Z. Gu, J. L. Acena, K. Izawa, H. Liu and V. A. Soloshonok, Recent advances in the trifluoromethylation methodology and new CF3-containing drugs, J. Fluorine Chem., 2014, 167, 37–54 CrossRef CAS; (b) A. S. Nair, A. K. Singh, A. Kumar, S. Kumar, S. Sukumaran, V. P. Koyiparambath, L. K. Pappachen, T. M. Rangarajan, H. Kim and B. Mathew, FDA-approved trifluoromethyl group-containing drugs: a review of 20 years, Processes, 2022, 10, 2054 CrossRef CAS.
- H. M. Leatherwood and J. T. Njardarson, Top 200 Small Molecule Drugs by Retail Sales in 2024, The Njardarson Group, University of Arizona, 2024, https://sites.arizona.edu/njardarson-lab/top200-posters/(accessed June 1, 2025) Search PubMed.
- R. E. Williams, H. M. Leatherwood and J. T. Njardarson, Top 200 Small Molecule Drugs by Retail Sales in 2023, The Njardarson Group, University of Arizona, 2023, https://sites.arizona.edu/njardarson-lab/top200-posters/(accessed June 1, 2025) Search PubMed.
- R. E. Williams and J. T. Njardarson, Top 200 Small Molecule Drugs by Retail Sales in 2022, The Njardarson Group, University of Arizona, 2022, https://sites.arizona.edu/njardarson-lab/top200-posters/(accessed June 1, 2025) Search PubMed.
- T. Zhou, Z.-W. Zhang, J. Nie, F. Y. Kwong, J.-A. Ma and C. W. Cheung, Metallaphotocatalytic triple couplings for modular synthesis of elaborate N-trifluoroalkyl anilines, Nat. Commun., 2024, 15, 9926 CrossRef CAS PubMed.
- F. Lovering, J. Bikker and C. Humblet, Escape from flatland: increasing saturation as an approach to improving clinical success, J. Med. Chem., 2009, 52, 6752–6756 CrossRef CAS.
- (a) H. Scoherr and T. Cernak, Profound methyl effects in drug discovery and a call for new C–H methylation reactions, Angew. Chem., Int. Ed., 2013, 52, 12256–12267 CrossRef PubMed; (b) E. J. Barreiro, A. E. Kümmerle and C. A. M. Fraga, The methylation effect in medicinal chemistry, Chem. Rev., 2011, 111, 5215–5246 CrossRef CAS PubMed; (c) C. S. Leung, S. S. F. Leung, J. Tirado-Rives and W. L. Jorgensen, Methyl effects on protein–ligand binding, J. Med. Chem., 2012, 55, 4489–4500 CrossRef CAS PubMed; (d) S. Sun and J. Fu, Methyl-containing pharmaceuticals: methylation in drug design, Bioorg. Med. Chem. Lett., 2018, 28, 3283–3289 CrossRef CAS PubMed.
- (a) T. T. Talele, Opportunities for tapping into three-dimensional chemical space through a quaternary carbon, J. Med. Chem., 2020, 63, 13291–13315 CrossRef CAS PubMed; (b) I. P. Silvestri and P. J. J. Colbon, The growing importance of chirality in 3D chemical space exploration and modern drug discovery approaches for hit-ID, ACS Med. Chem. Lett., 2021, 12, 1220–1229 CrossRef CAS PubMed; (c) J. Tsien, C. Hu, R. R. Merchant and T. Qin, Three-dimensional saturated C(sp3)-rich bioisosteres for benzene, Nat. Rev. Chem., 2024, 8, 605–627 CrossRef CAS PubMed.
- DrugBank, https://go.drugbank.com (accessed June 1, 2025).
- (a) A. T. Brusoe and J. F. Hartwig, Palladium-catalyzed arylation of fluoroalkylamines, J. Am. Chem. Soc., 2015, 137, 8460–8468 Search PubMed; (b) R. T. McGuire, A. A. Yadav and M. Stradiotto, Nickel-catalyzed N-arylation of fluoroalkylamines, Angew. Chem., Int. Ed., 2021, 60, 4080–4084 Search PubMed; (c) S. Liu, J. Zhou, L. Yu, Y. Liu, Y. Huang, Y. Ouyang, G.-K. Liu, X.-H. Xu and N. Shibata, Nitrogen-based organofluorine functional molecules: synthesis and applications, Chem. Rev., 2025, 125, 4603–4764 CrossRef CAS PubMed.
- (a) J. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, The merger of transition metal and photocatalysis, Nat. Rev. Chem., 2017, 1, 0052 CrossRef CAS; (b) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Visible light photoredox catalysis with transition metal complexes: applications in organic synthesis, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed; (c) A. Y. Chan, I. B. Perry, N. B. Bissonnette, B. F. Buksh, G. A. Edwards, L. I. Frye, O. L. Garry, M. N. Lavagnino, B. X. Li, Y. Liang, E. Mao, A. Millet, J. V. Oakley, N. L. Reed, H. A. Sakai, C. P. Seath and D. W. C. MacMillan, Metallaphotoredox: the merger of photoredox and transition metal catalysis, Chem. Rev., 2022, 122, 1485–1542 CrossRef CAS PubMed; (d) N. Holmberg-Douglas and D. A. Nicewicz, Photoredox-catalyzed C–H functionalization reactions, Chem. Rev., 2022, 122, 1925–2016 CrossRef CAS PubMed.
- (a) F. Strieth-Kalthoff, M. J. James, M. Teders, L. Pitzer and F. Glorius, Energy transfer catalysis mediated by visible light: principles, applications, directions, Chem. Soc. Rev., 2018, 47, 7190–7202 RSC; (b) F. Strieth-Kalthoff and F. Glorius, Triplet energy transfer photocatalysis: unlocking the next level, Chem, 2020, 6, 1888–1903 CrossRef CAS; (c) S. Dutta, J. E. Erchinger, F. Strieth-Kalthoff, R. Kleinmans and F. Glorius, Energy transfer photocatalysis: exciting modes of reactivity, Chem. Soc. Rev., 2024, 53, 1068–1089 RSC.
- (a) E. Ruijter, R. Scheffelaar and R. V. A. Orru, Multicomponent reaction design in the quest for molecular complexity and diversity, Angew. Chem., Int. Ed., 2011, 50, 6234–6246 CrossRef CAS; (b) M. J. Buskes, A. Coffin, D. M. Troast, R. Stein and M.-J. Blanco, Accelerating drug discovery: synthesis of complex chemotypes via multicomponent reactions, ACS Med. Chem. Lett., 2023, 14, 376–385 CrossRef CAS PubMed.
- (a) S. Barata-Vallejo, B. Lantano and A. Postigo, Recent advances in trifluoromethylation reactions with electrophilic trifluoromethylating reagents, Chem. – Eur. J., 2014, 20, 16806–16829 CrossRef CAS PubMed; (b) R. T. Kareem, B. Azizi, M. Asnaashariisfahani, A. Ebadid and E. Vessally, Vicinal halo-trifluoromethylation of alkenes, RSC Adv., 2021, 11, 14941–14955 RSC.
- (a) T. E. Muller, K. C. Hultzsch, M. Yus, F. Foubelo and M. Tada, Hydroamination: direct addition of amines to alkenes and alkynes, Chem. Rev., 2008, 108, 3795–3892 CrossRef PubMed; (b) P. Kalck and M. Urrutigoity, Tandem hydroaminomethylation reaction to synthesize amines from alkenes, Chem. Rev., 2018, 118, 3833–3861 CrossRef CAS PubMed; (c) R. Y. Liu and S. L. Buchwald, CuH-catalyzed olefin functionalization: from hydroamination to carbonyl addition, Acc. Chem. Res., 2020, 53, 1229–1243 CrossRef CAS PubMed; (d) H. Jiang and A. Studer, Intermolecular radical carboamination of alkenes, Chem. Soc. Rev., 2020, 49, 1790–1811 RSC; (e) K. Kwon, R. T. Simons, M. Nandakumar and J. L. Roizen, Strategies to generate nitrogen-centered radicals that may rely on photoredox catalysis: development in reaction methodology and applications in organic synthesis, Chem. Rev., 2022, 122, 2353–2428 Search PubMed; (f) C. Pratley, S. Fenner and J. A. Murphy, Nitrogen-centered radicals in functionalization of sp2 systems: generation, reactivity, and applications in synthesis, Chem. Rev., 2022, 122, 8181–8260 CrossRef CAS PubMed.
- Y. Yasu, T. Koike and M. Akita, Intermolecular aminotrifluoromethylation of alkenes by visible-light-driven photoredox catalysis, Org. Lett., 2013, 15, 2136–2139 CrossRef CAS PubMed.
- G. Dagousset, A. Carboni, E. Magnier and G. Masson, Photoredox-induced three-component azido- and aminotrifluoromethylation of alkenes, Org. Lett., 2014, 16, 4340–4343 CrossRef CAS PubMed.
- X.-L. Yu, J.-R. Chen, D.-Z. Chen and W.-J. Xiao, Visible-light-induced photocatalytic azotrifluoromethylation of alkenes with aryldiazonium salts and sodium triflinate, Chem. Commun., 2016, 52, 8275–8278 RSC.
- L. Zheng, L. Cai, X. Zhuo, L. Deng, Y. Wu, B. Yang, Y. Wang and W. Guo, Direct intermolecular three-component aminotrifluoromethylation of styrenes by visible-light-photoredox catalysis, Org. Chem. Front., 2022, 9, 6498–6504 RSC.
- Y. Zheng, Z. Liao, Z. Xie, H. Chen, K. Chen, H. Xiang and H. Yang, Photochemical alkene trifluoromethylimination enabled by trifluoromethylsulfonylamide as a bifunctional reagent, Org. Lett., 2023, 25, 2129–2133 CrossRef CAS PubMed.
- C. Zhou, X. Wang, P. Kong, D. Ji, P. Zhou and C. Huo, Alkyl-Trifluoromethylation of Glycine Derivatives, Org. Lett., 2025, 27, 7126–7131 CrossRef CAS.
- Y. Ren, Y. Zhou, K.-H. Wang, J. Wang, D. Huang and Y. Hu, A visible light promoted regioselective cascade radical trifluoromethylation/cyclization of N-isopentenyl aldehyde hydrazones: access to trifluoromethylated tetrahydropyridazine derivatives, Org. Lett., 2025, 27, 7033–7038 CrossRef CAS PubMed.
- A. Carboni, G. Dagousset, E. Magnier and G. Masson, Photoredox-induced three-component oxy-, amino- and carbotrifluoromethylation of enecarbamates, Org. Lett., 2014, 16, 1240–1243 CrossRef CAS PubMed.
- Y. Xiong, X. Ma and G. Zhang, Copper-catalyzed intermolecular carboamination of alkenes induced by visible light, Org. Lett., 2019, 21, 1699–1703 CrossRef CAS PubMed.
- (a) J. A. Rossi-Ashton, A. K. Clarke, W. P. Unsworth and R. J. K. Taylor, Phosphoranyl radical fragmentation reactions driven by photoredox catalysis, ACS Catal., 2020, 10, 7250–7261 CrossRef CAS PubMed; (b) T. Mandal, S. Mallick, M. Islam and S. De Sarkar, Alcohols as alkyl synthons enabled by photoredox-catalyzed deoxygenative activation, ACS Catal., 2024, 14, 13451–13496 CrossRef CAS; (c) W. Luo, Z. Chen and B. Yu, Photocatalytic deoxygenative alkylation with alcohols via xanthates or thionocarbonates, Asian J. Org. Chem., 2025, 14, e202400627 CrossRef CAS.
- N. E. Intermaggio, A. Millet, D. L. Davis and D. W. C. MacMillan, Deoxytrifluoromethylation of alcohols, J. Am. Chem. Soc., 2022, 144, 11961–11968 CrossRef CAS PubMed.
- (a) C. Lei and Q. Zhou, Advances on nickel-catalyzed C(sp3)–C(sp3) bond formation, Acta Chim. Sin., 2020, 78, 1017–1029 CrossRef; (b) Q.-Y. Huang and M. Shi, Recent advancements in Ni/photoredox dual catalysis for C(sp3)–C(sp3) cross-coupling reactions, Org. Chem. Front., 2024, 11, 4913–4925 RSC.
- J. Z. Wang, W. L. Lyon and D. W. C. MacMillan, Alkene dialkylation by triple radical sorting, Nature, 2024, 628, 104–109 CrossRef CAS PubMed.
- (a) P. K. Mykhailiuk, 2,2,2-Trifluorodiazoethane (CF3CHN2): a long journey since 1943, Chem. Rev., 2020, 120, 12718–12755 CrossRef CAS PubMed; (b) P. K. Mykhailiuk, Fluorinated pyrazoles: from synthesis to applications, Chem. Rev., 2021, 121, 1670–1715 CrossRef CAS PubMed.
- B. T. Boyle, N. W. Dow, C. B. Kelly, M. C. Bryan and D. W. C. MacMillan, Unlocking carbene reactivity by metallaphotoredox α-elimination, Nature, 2024, 631, 789–795 CrossRef CAS PubMed.
- F. Cong, G.-Q. Sun, S.-H. Ye, R. Hu, W. Rao and M. J. Koh, A bimolecular homolytic substitution-enabled platform for multicomponent cross-coupling of unactivated alkenes, J. Am. Chem. Soc., 2024, 146, 10274–10280 CrossRef CAS PubMed.
- Y.-W. Jiang, Z.-W. Zhang, C. W. Cheung and J.-A. Ma, Metallaphotocatalytic modular synthesis of masked arylaminomethyl trifluoromethyl ketones, Cell Rep. Phys. Sci., 2025, 6, 102564 CrossRef CAS.
- Z.-W. Zhang, Z. Feng, J.-A. Ma and C. W. Cheung, Metallaphotocatalytic synthesis of anilines through tandem C–N transposition and C–H alkylation of alkylamines, Nat. Synth, 2023, 2, 1171–1183 CrossRef CAS.
- H. Ge, B. Wu, Y. Liu, H. Wang and Q. Shen, Synergistic Lewis acid and photoredox-catalyzed trifluoromethylative difunctionalization of alkenes with selenium ylide-based trifluoromethylating reagent, ACS Catal., 2020, 10, 12414–12424 CrossRef CAS.
- (a) G. Choi, G. S. Lee, B. Park, D. Kim and S. H. Hong, Direct C(sp3)–H trifluoromethylation of unactivated alkanes enabled by multifunctional trifluoromethyl copper complexes, Angew. Chem., Int. Ed., 2021, 60, 5467–5474 CrossRef CAS PubMed; (b) J. He, T. N. Nguyen, S. Guo and S. P. Cook, Csp3–H trifluoromethylation of unactivated aliphatic systems, Org. Lett., 2021, 23, 702–705 CrossRef CAS PubMed.
- P. J. Sarver, V. Bacauanu, D. M. Schultz, D. A. DiRocco, Y. Lam, E. C. Sherer and D. W. C. MacMillan, The merger of decatungstate and copper catalysis to enable aliphatic C(sp3)–H trifluoromethylation, Nat. Chem., 2020, 12, 459–467 CrossRef CAS PubMed.
- (a) Y. Pang, D. Moser and J. Cornell, Pyrylium salts: selective reagents for the activation of primary amino groups in organic synthesis, Synthesis, 2020, 489–503 CAS; (b) K. J. Berger and M. D. Levin, Reframing primary alkyl amines as aliphatic building blocks, Org. Biomol. Chem., 2021, 19, 11–36 RSC; (c) B. Saxena, R. I. Patel and A. Sharma, Recent advances in electron donor–acceptor (EDA) complex reactions involving quaternary pyridinium derivatives, Adv. Synth. Catal., 2023, 365, 1538–1564 CrossRef CAS; (d) J. U. Kim, E. S. Lim, J. Y. Park, D. Jung and S. Lee, Radical approaches for C(sp3)–N bond cleavage in deaminative transformations, Chem. Commun., 2025, 61, 6997–7008 RSC; (e) S. Singh, D. Gambhir and R. P. Singh, Photoinduced stereoselective reactions using pyridinium salts as radical precursors, Chem. Commun., 2025, 61, 3436–3446 RSC.
- J.-H. Xue, Y. Li, Y. Liu, Q. Li and H. Wang, Site-specific deaminative trifluoromethylation of aliphatic primary amines, Angew. Chem., Int. Ed., 2024, 63, e202319030 CrossRef CAS PubMed.
- K. Kolahdouzan, R. Kumar and M. J. Gaunt, Visible-light mediated carbonyl trifluoromethylative amination as a practical method for the synthesis of β-trifluoromethyl tertiary alkylamines, Chem. Sci., 2020, 11, 12089–12094 RSC.
- X. Shangguan, L. Zhu, Y. Zhang, Y. Li and Q. Zhang, Copper-catalyzed enantioselective three-component radical 1,4-perfluoroalkylamination of 1,3-dienes, Nat. Commun., 2025, 16, 4939 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |