
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
In situ electrochemical regeneration of active 1,4-NADH for enzymatic lactic acid formation via concerted functions on Pt-modified TiO2/Ti†
Nada H. A.
Besisa
a,
Ki-Seok
Yoon
b and
M.
Yamauchi
 *abcde
*abcde
aDepartment of Chemistry, Graduate School of Science, Kyushu University, Fukuoka 819-0395, Japan. E-mail: yamauchi@ms.ifoc.kyushu-u.ac.jp
bInternational Institute for Carbon-Neutral Energy Research (WPI-I2CNER), Kyushu University, Fukuoka 819-0395, Japan
cInstitute for Materials Chemistry and Engineering (IMCE), Kyushu University, Fukuoka 819-0395, Japan
dAdvanced Institute for Materials Research (WPI-AIMR), Tohoku University, Sendai 980-8577, Miyagi, Japan
eResearch Center for Negative Emissions Technologies (K-Nets), Kyushu University, Fukuoka 819-0395, Japan
First published on 16th January 2024
Abstract
Nicotinamide adenine dinucleotide (NAD+) and its reduced form (NADH) are key cofactors serving as essential hydrogen acceptors and donors to facilitate energy and material conversions under mild conditions. We demonstrate direct electrochemical conversion to achieve highly efficient regeneration of enzymatically active 1,4-NADH using a Pt-modified TiO2 catalyst grown directly on a Ti mesh electrode (Pt-TOT). Spectral analyses revealed that defects formed by the inclusion of Pt species in the lattice of TiO2 play a critical role in the regeneration process. In particular, Pt-TOT containing approximately 3 atom% of Pt exhibited unprecedented efficiency in the electrochemical reduction of NAD+ at the lowest overpotential to date. This exceptional performance led to the production of active 1,4-NADH with a significantly high yield of 86 ± 3% at −0.6 V vs. Ag/AgCl (−0.06 V vs. RHE) and an even higher yield of 99.5 ± 0.4% at a slightly elevated negative potential of −0.8 V vs. Ag/AgCl (−0.2 V vs. RHE). Furthermore, the electrochemically generated NADH was directly applied in the enzymatic conversion of pyruvic acid to lactic acid using lactate dehydrogenase.
Introduction
Enzymatic technology provides a highly efficient and environmentally friendly approach to the synthesis of a wide range of value-added chemicals, including chiral compounds, pharmaceuticals, and food additives, among other applications.1–4 Active enzymes are often accompanied by cofactors such as nicotinamide adenine dinucleotide (NAD), particularly its reduced form (NADH), which serves as a critical electron or hydrogen donor in both nature and industry.3,5–7NADH is normally recycled through complex processes involving an electron transport chain such as glycolysis and the Krebs cycle, which has been a bottleneck preventing the widespread application of enzymatic systems. Therefore, more efficient cofactor recycling methods are highly desirable.4 Among various approaches, electrochemical hydrogenation of NAD+ to regenerate enzymatically active 1,4-NADH (NADH-reg, Scheme 1) has attracted considerable attention due to its simplicity and environmental friendliness.7–10 Indirect electrochemical systems often require mediators to facilitate electron transfer, resulting in lower overpotentials and avoidance of electrode fouling.3 However, these mediator systems suffer from the mediator instability and toxicity, affecting the final enzymatic product.3,4 In contrast, recent research on direct electrochemical hydrogenation systems for NADH-reg has shown an important advantage in eliminating the need for mediators.3,5,11–16
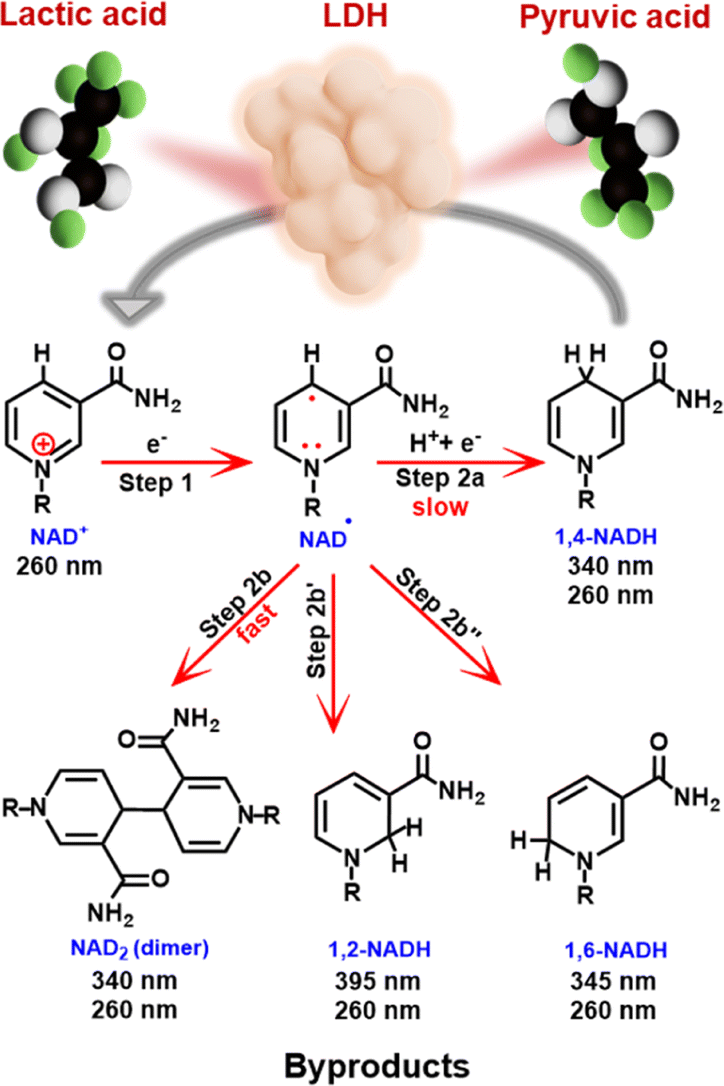 | ||
| Scheme 1 Mechanism of electrochemical reduction of NAD+, possible products and wavelengths of observable UV absorption peaks for each product. Application of the enzymatically active 1,4-NADH for lactic acid formation is also illustrated. | ||
Both bare and modified electrodes have been employed for NADH-reg, but the yields remain unsatisfactorily low due to the preferential production of the enzymatically inactive forms (Scheme 1).9 NADH-reg proceeds in two steps:8,17 first, NAD+ is reduced to an NAD-free radical, and second, the free radical can either undergo protonation and further reduction to yield enzymatically active 1,4-NADH (Step 2a), or it can rapidly dimerize to form inactive products such as NAD2 (Step 2b) and other side products such as 1,2-NADH (Step 2b′) and 1,6-NADH (Step 2b′′). The kinetics of Step 2a for the formation of 1,4-NADH is significantly slower than that of the dimerization process.8 Therefore, efforts have been made to accelerate the second pathway, emphasizing the need for a smooth electrode-driven hydrogenation of the NAD free radical to achieve selective production of 1,4-NADH (Scheme S1a†). In this regard, the use of catalysts with high hydrogen evolution reaction (HER) capability has been a strategy to find a good electrocatalyst for NADH-reg.8 In addition, previous studies have reported high overpotentials and usage of unaffordable and high-cost materials,8,10 highlighting the demand for more practical electrode materials that can efficiently produce active 1,4-NADH at lower overpotentials.
Titanium dioxide (TiO2) has received much attention due to its abundance, non-toxicity, good stability, affordable cost.18,19 In addition, it has high activity in various applications, including electrochemistry.20–24 Recently, TiO2 has been found to exhibit high selectivity for the production of valuable products such as alcohols and amino acids from organic acids.24–27 Furthermore, sufficient controllability of its electronic states by doping with other elements is another advantage as an electrode material.28–31 Previous studies, including ours, have suggested that the unique performance of TiO2-based electrodes results from their ability to interact with carboxy groups of the reactants.26,27,32,33 Considering the polar nature of the amide moiety in NADs, analogous to the carboxy group, the TiO2 surface is expected to show feasible interactions with NADs and contribute to enhancing the selectivity for the production of NADH (Scheme 2a). Therefore, in this study, we chose TiO2 as an affordable and promising electrocatalyst to efficiently trap NAD+ and incorporation of Pt species, which shows excellent HER capability, and further promotes NAD+ hydrogenation to achieve highly selective and efficient electrochemical NADH-reg (Scheme 2b) for the advancement of sustainable and cost-effective enzymatic processes.
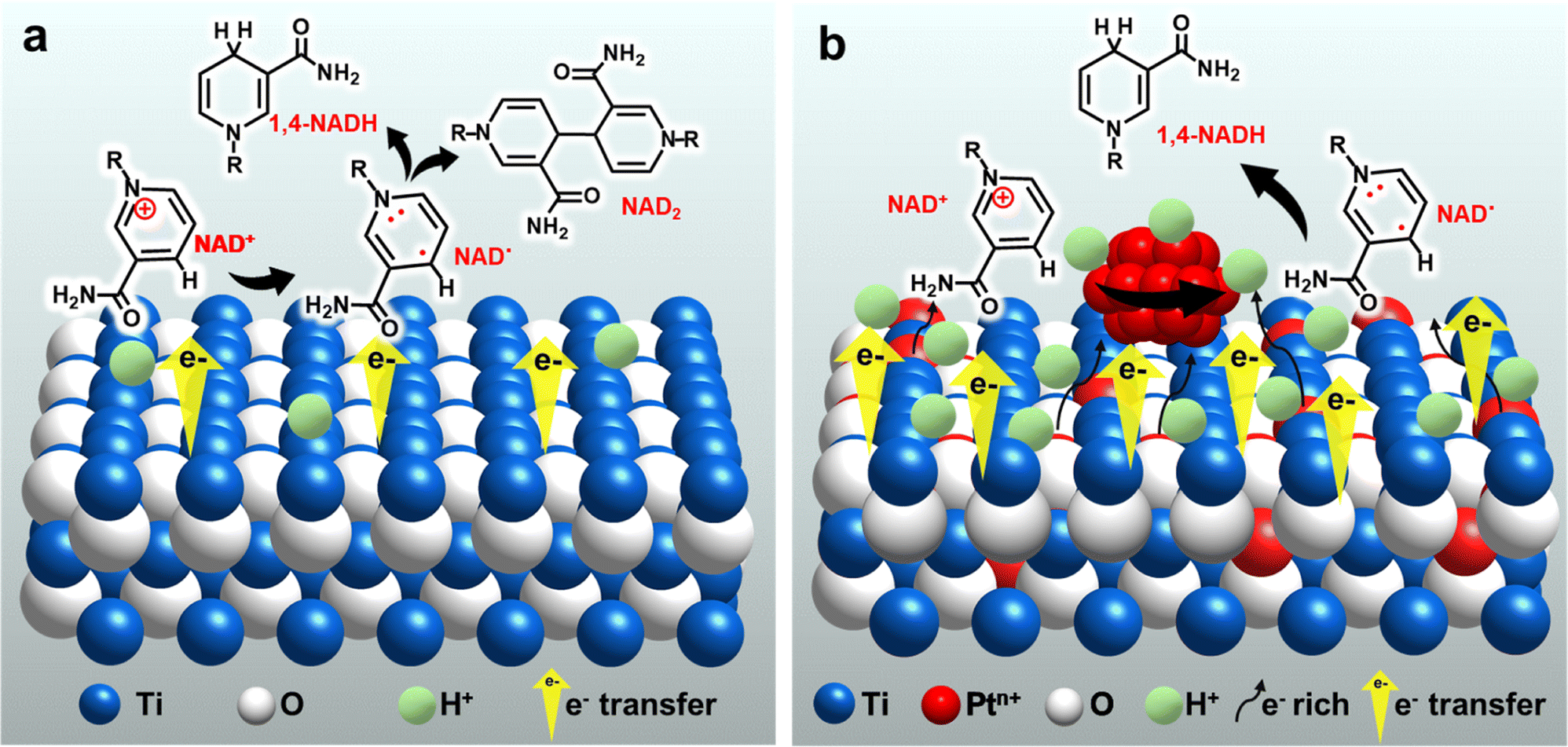 | ||
| Scheme 2 Suggested strategy and mechanism for the electrochemical reduction of NAD+ on (a) TOT and (b) Pt-TOTs. | ||
Here, we apply TiO2 grains grown on a Ti mesh cathode (TOT), and further introduce Pt into TOT by including a Pt source in a hydrothermal preparation process of TOT. The resultant Pt-modified TiO2/Ti cathode (Pt-TOT) was found to exhibit superior NADH-reg performance in terms of selectivity for 1,4-NADH, surpassing that of TOT and Pt electrodes. In-depth analyses of Pt-TOT revealed that the presence of Pt in various valence states on TiO2/Ti and defects plays a crucial role in enhancing the yield of 1,4-NADH (Scheme 2b) and lowering the overpotential for NADH-reg. Furthermore, the usability of the electrochemically generated 1,4-NADH in enzymatic lactic acid formation from pyruvic acid was then verified using lactate dehydrogenase (LDH), as shown in Scheme 1.
Results and discussion
Structural characterization of electrodes
Four Pt-modified TOT electrodes with different Pt contents were prepared using a hydrothermal process (see the ESI† for detailed methodology). The Pt concentration in the starting solution was adjusted to control the amount of Pt species in the electrodes. The resulting electrodes are denoted as Pt-TOT.X (X = 1–4), indicating increasing Pt content (Table S1†). To establish Pt contents in Pt-TOTs, inductively coupled plasma optical emission spectroscopy (ICP) was employed, and the obtained values are summarized in Table S1.†The morphology of prepared TOT and Pt-TOTs was investigated by transmission electron microscopy (TEM) and scanning electron microscopy (SEM) combined with energy dispersive X-ray spectroscopy (EDS, where Pt content was represented as Pt atom% according to EDS elemental analysis in this work) (Fig. 1a, S1–S3, and Table S2†). High contrast particles were observed on the surface of Pt-TOTs, the number of which increased proportionally with the Pt content (Fig. 1a and S1†). In TOT, undefined large grains measuring several tens of nanometers were observed (Fig. S2a†). In contrast, the grain size of Pt-TOTs decreased with increasing Pt content, as depicted in Fig. S2b–f.† Furthermore, EDS mapping analysis revealed the homogeneous distribution of Pt species throughout the grains of Pt-TOTs, with their density increasing with increasing Pt content (Fig. 1a and S3†). On the other hand, X-ray diffraction (XRD) patterns of TOT and Pt-TOTs primarily displayed weak and strong diffractions of tetragonal and hcp phases, respectively, indicating the formation of rutile-type (tetragonal) TiO2 grains on the hcp-type Ti mesh (Fig. 1b). Notably, no diffraction peaks corresponding to PtO or Pt metal phases were observed in Pt-TOTs. Rietveld analysis provided lattice constants for TiO2 grown on TOT and Pt-TOTs (Fig. S4 and Table S3†). Compared to TOT, the lattice constants a and b of Pt-TOTs slightly decreased with increasing Pt content, causing a subtle shift of diffraction peaks from the rutile phase toward the higher angle side (Fig. 1c). This observation suggests the substitution of 6-coordinated Ti3+ ions with a radius of 0.67 Å by Pt4+ ions with a smaller radius of 0.625 Å.31 Consequently, Pt species not only exist as nanoparticles on the surface but are also incorporated within the TiO2 lattice of Pt-TOTs.
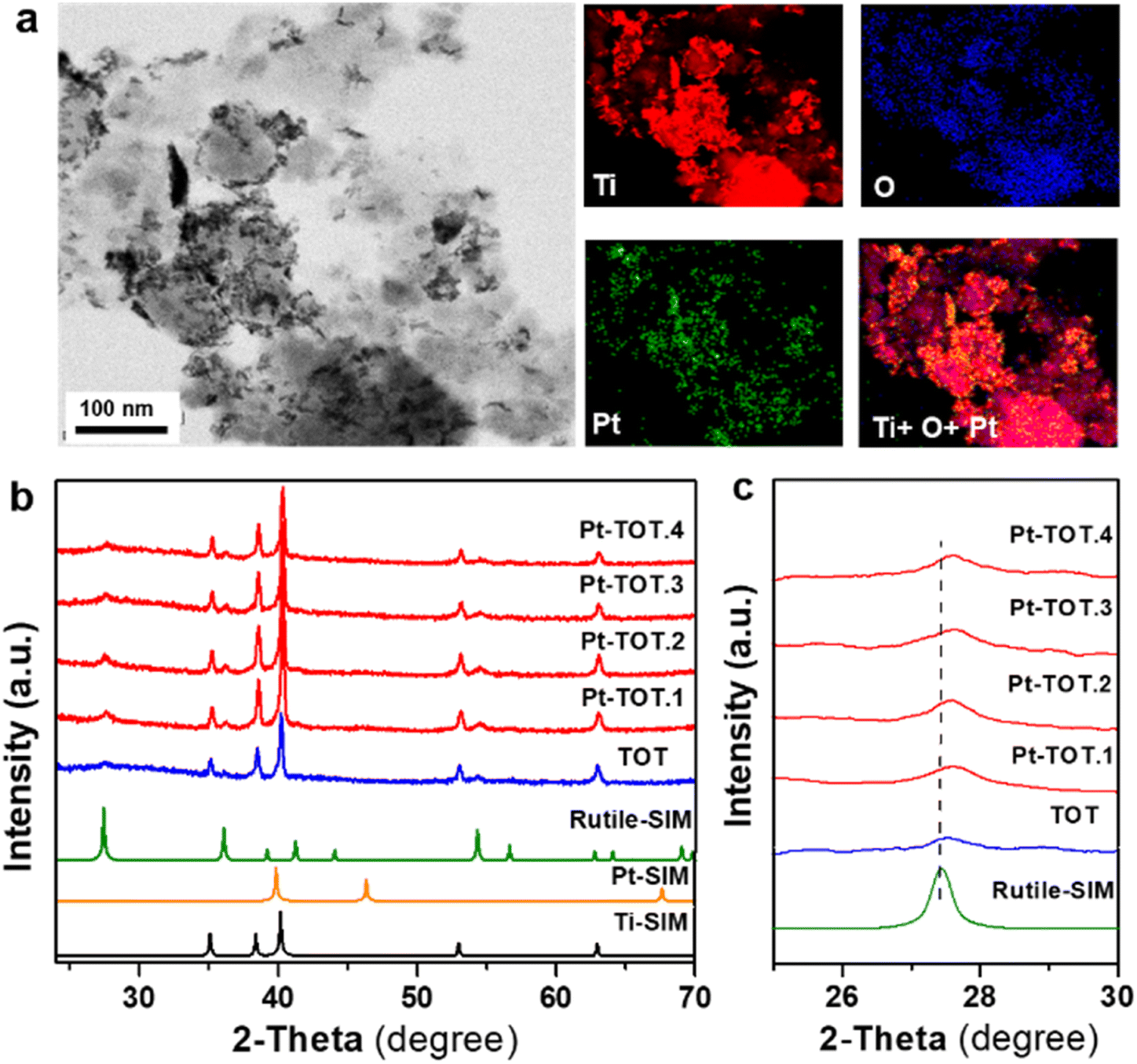 | ||
| Fig. 1 (a) STEM and EDS elemental mapping of Pt-TOT.2, (b) XRD pattern, and (c) expanded XRD pattern from 2θ = 24 to 30° of TOT and Pt-TOTs with different Pt content (the dashed line shows the diffraction peak position of rutile TiO2). | ||
Electrochemical studies
To investigate onset potentials for the NAD+ reduction, we conducted cyclic voltammetry (CV) for the prepared electrodes over a potential range from 0 to −1.8 V vs. Ag/AgCl (−0.06 to −1.2 V vs. RHE) with a scan rate of 10 mV s−1 (Fig. 2a–d). In the absence of NAD+ (blank), the Ti mesh exhibited the most negative onset potential at approximately −1.1 V vs. Ag/AgCl (Fig. 2a). The onset potential on TOT was slightly shifted to approximately −1 V vs. Ag/AgCl (Fig. 2b), whereas Pt-TOT.2 and Pt showed even lesser negative onset potentials at −0.5 (Fig. 2c) and −0.6 V vs. Ag/AgCl (Fig. 2d), respectively, suggesting that the presence of TiO2 and Pt promotes both the HER and NAD+ reduction reaction. On the other hand, a larger negative current density (j) was observed in the presence of NAD+ on bare Ti mesh (Fig. 2a) and TOT (Fig. 2b) compared to the blank, suggesting the emergence of their catalytic activity for NAD+ reduction. Interestingly, Pt-TOT.2 showed the largest j in the absence of NAD+ (Fig. 2c), suggesting that inclusion of Pt with TiO2 enhances the HER. Notably, the j on Pt-TOT.2 in the presence of NAD+ was smaller than that in the blank, but larger than those on Ti and TOT. In addition, Pt exhibited a slightly larger j on the blank than in the presence of NAD+. Given that Pt is expected to show NADH-reg activity, the decrease in j on Pt-TOT.2 with NAD+ possibly implies the suppression of the HER due to NAD+ adsorption on the surface. Additionally, the current density in the presence of NAD+ followed the order of 20.2 mA cm−2 on Pt, 16.5 mA cm−2 on Pt-TOT.2, 12 mA cm−2 on TOT, and 10 mA cm−2 on Ti at −1.8 V vs. Ag/AgCl, possibly indicating that the coexistence of both Pt and TiO2 efficiently activates both the HER and NAD+ reduction.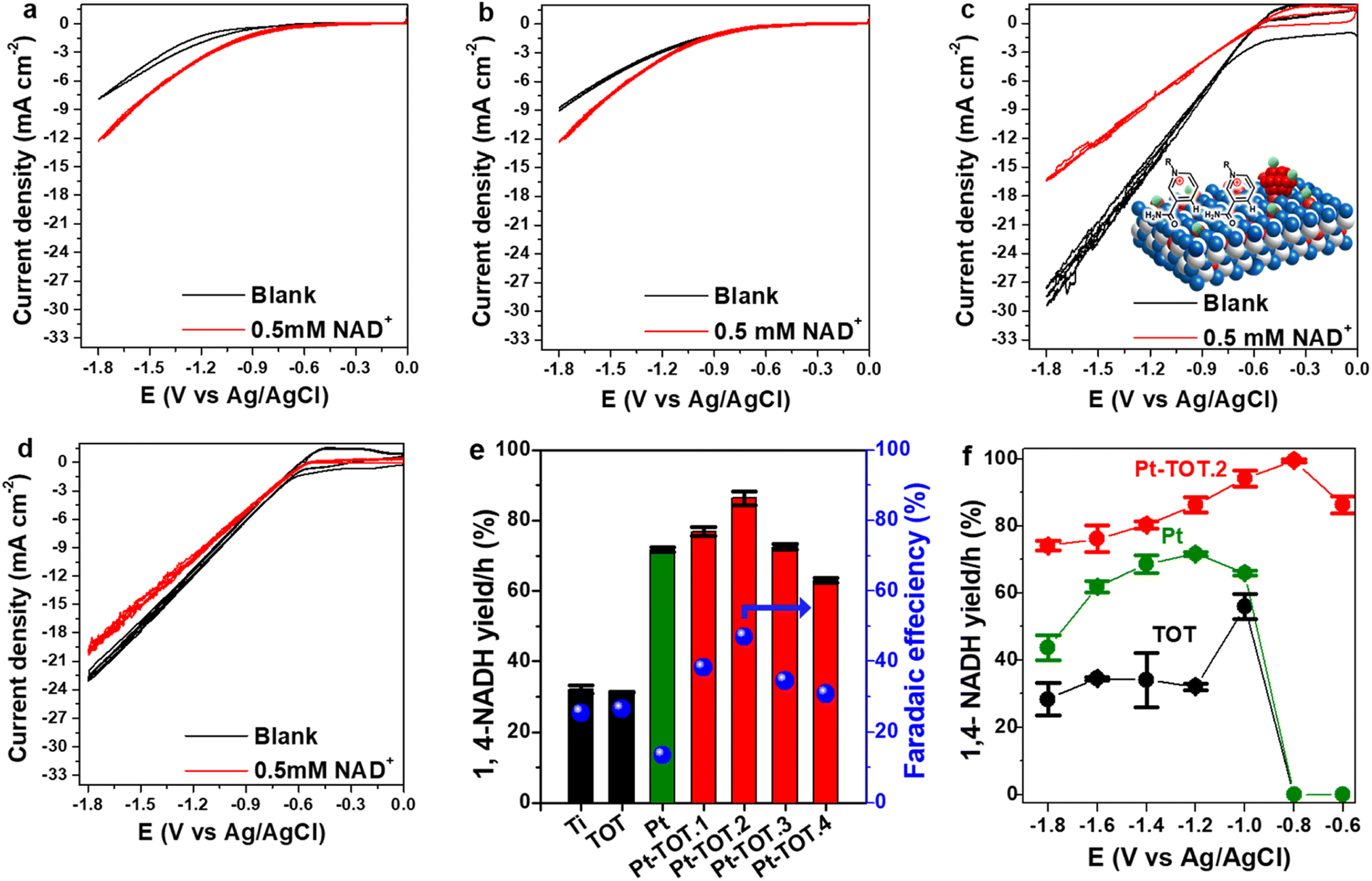 | ||
| Fig. 2 Cyclic voltammograms (CV) of (a) Ti, (b) TOT (c) Pt-TOT.2, the inset of which is a schematic representation of adsorption of NAD+, and (d) Pt in both blank (black) and 0.5 mM NAD+ solution (red), (e) yield and faradaic efficiency (FE) for NADH-reg at −1.2 V vs. Ag/AgCl, and (f) yield for NADH-reg on TOT, Pt, Pt-TOT.2 in a wide potential range. | ||
We then conducted the electrochemical NAD+ reduction on the prepared electrodes using chronoamperometry (CA) at −1.2 V vs. Ag/AgCl (−0.6 V vs. RHE) where all electrodes showed the reduction current in the CV experiments with NAD+ using an H-type cell (Fig. S5†). As presented in Scheme 1, 1,4-NADH exhibits a UV absorption peak around 340 nm, whereas the peaks for possible side products such as (NAD)2 dimer, 1,6-NADH, and 1,2-NADH are observable at similar positions of 340, 345, and 395 nm, respectively. Therefore, we evaluated the yield for NADH-reg by enzyme activity assay (Scheme S1b†), in addition to calculating faradaic efficiency (FE) and the production rate for NADHs (CNADH and C1,4-NADH, refer to the ESI for more details, Fig. S6†) to compare the selectivity on different electrodes. Fig. 2e shows that Pt and Pt-TOTs exhibit larger yields for NADH-reg than Ti and TOT, suggesting that the Pt loading significantly enhances NADH-reg. Interestingly, Pt-TOT.1, Pt-TOT.2 and Pt-TOT.3 demonstrated higher NADH-reg yields than Pt, implying that the presence of TOT is crucial for achieving higher selectivity for NADH-reg. As a result, Pt-TOT.2 showed the largest yield for NADH-reg of 86 ± 3% with the highest FE of 47% (Fig. 2e). Further increase in Pt content led to a slight decrease in catalytic activity. We then presume that NADH-reg preferentially occurs around Pt sites interacting with TiO2, whereas the HER surpasses NADH-reg once the number of Pt nanoparticles exceeds a level to form HER sites isolated from influence from the TiO2 surface.
Fig. 2f compares NADH-reg yields on Pt, TOT and Pt-TOT.2 over a wide range of potentials. Below −1.2 V vs. Ag/AgCl, the yields for NADH-reg on TOT and Ti electrodes were approximately 30% (Fig. 2f and S7a†). In contrast, at a less negative potential of −1.0 V vs. Ag/AgCl, the NADH-reg performance on TOT and Ti was enhanced, resulting in yields of 56 ± 4 and 68 ± 1%, respectively. However, at potentials less negative than −1 V vs. Ag/AgCl, no NADH-reg was observed on either Ti or TOT. Similarly, NADH-reg on Pt started at −1 V and showed the maximum with yield of 72 ± 1.5% at −1.2 V vs. Ag/AgCl. In contrast, Pt-TOT.2 demonstrated a significantly high yield of 86 ± 3% at only −0.6 V vs. Ag/AgCl (−0.06 V vs. RHE), which is the lowest overpotential among all those reported so far (Table S4†). At a slightly more negative potential of −0.8 V vs. Ag/AgCl (−0.2 V vs. RHE), the highest yield of 99.5 ± 0.4% for NADH-reg was achieved on Pt-TOT.2, combined with high stability and reusability over the number of cycles (Fig. S7b†). In addition, it showed the highest current density at −0.8 V vs. Ag/AgCl confirming the high activity towards NAD+ reduction (Fig. S7c†).
To further confirm the product selectivity, 1H NMR measurements were performed for products generated in CA at −0.8 V vs. Ag/AgCl with 4 mM NAD+ solution, which is 8 times higher than the concentration used in previous CA measurements to obtain sufficient NMR intensity. 1H NMR spectra of pure commercial NAD+ and 1,4-NADH as well as the products of our electrochemical NADH-reg on Pt, TOT, and Pt-TOT.2 are shown in Fig. 3. A singlet signal located at 9.28 ppm is characteristic of the H-2 hydrogen on the NAD+ structure of the nicotinamide moiety (Fig. 3I). The two singlets observed between 8 and 8.5 ppm are related to the hydrogen atoms on the adenine ring contained in the R group. In contrast, freshly prepared 1,4-NADH solution showed a singlet located at 6.8 ppm which is characteristic for the H-2′ of the nicotinamide ring of NADH and two singlets related to the adenine ring (Fig. 3II). 1H NMR spectra of the products on Pt and TOT presented signals assignable to NAD+ only, confirming the inability of Pt and TOT to produce active NADH at −0.8 V vs. Ag/AgCl (Fig. 3III and IV). On the other hand, the characteristic singlet at 6.8 ppm, assignable to the production of 1,4-NADH, was observed at more negative potential than −1.2 V vs. Ag/AgCl on Pt although accompanying a singlet at 7 ppm suggesting the formation of 1,6-NADH34 (Fig. S7d†). Surprisingly, Fig. 3V shows an obvious singlet signal at 6.8 ppm, confirming the 1,4-NADH formation in addition to signals of the starting NAD+. The results suggest that only pure 1,4-NADH is produced on Pt-TOT.2 without formation of other side products. To completely convert the starting NAD+, longer-time reaction (4 h) was conducted. Although signals for starting NAD+ almost disappeared and signals for 1,4-NADH grew, a small singlet was detected at 6.93 ppm (Fig. 3VI) with the integral ratio to that for 1,4-NADH of 0.086![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. According to the previous report,34 the 6.93 ppm peak is assignable to the formation of 1,6-NADH. This phenomenon can be understood by considering considerably high concentration of starting NAD+ compared to that in CA, which led to higher coverage of intermediates and induces formation of the side products.35 Thus, application of appropriate reaction conditions is critical to improve selectivity to produce 1,4-NADH.
:1. According to the previous report,34 the 6.93 ppm peak is assignable to the formation of 1,6-NADH. This phenomenon can be understood by considering considerably high concentration of starting NAD+ compared to that in CA, which led to higher coverage of intermediates and induces formation of the side products.35 Thus, application of appropriate reaction conditions is critical to improve selectivity to produce 1,4-NADH.
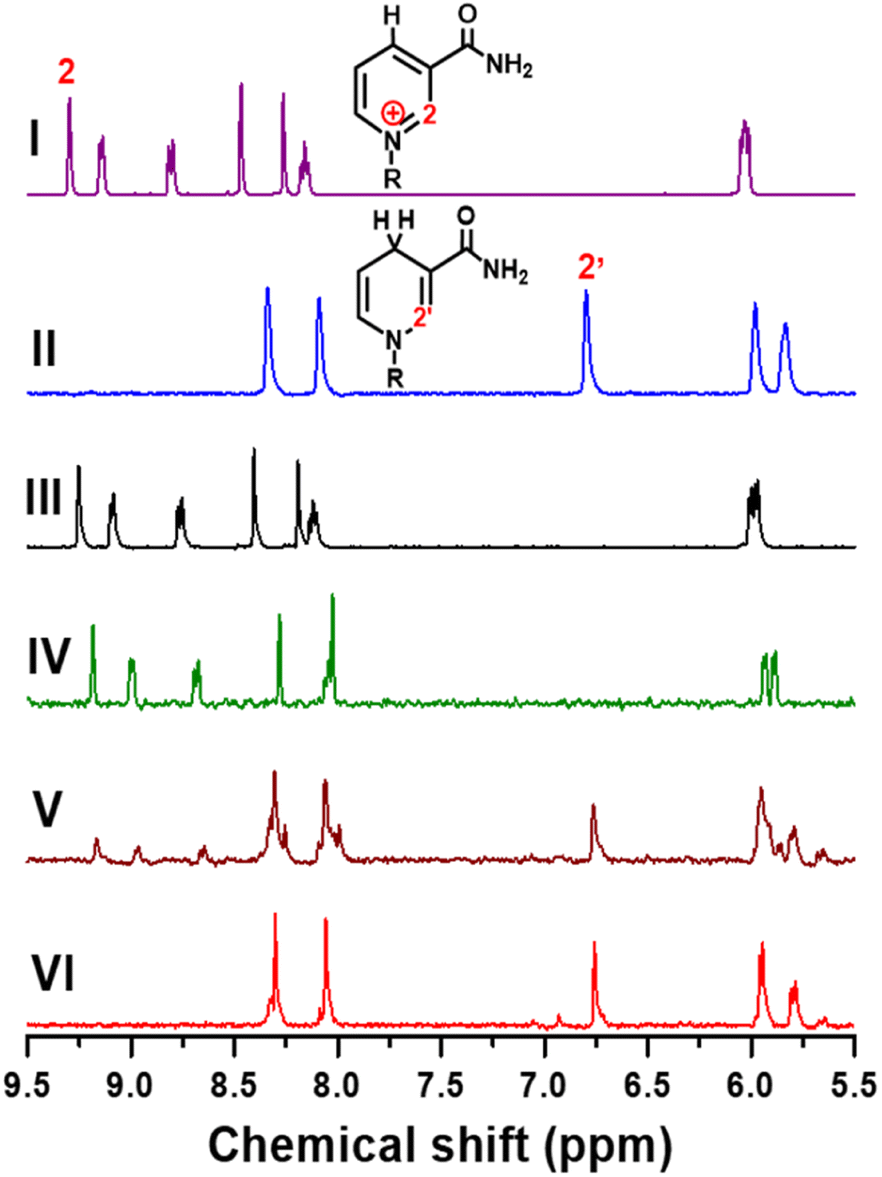 | ||
| Fig. 3 1H NMR spectra of NAD+ (4 mM in 0.1 M D2O phosphate buffer, pH 6) (I), 1,4-NADH (4 mM in 0.1 M D2O phosphate buffer, pH 6) (II), products generated from 4 mM NAD+ in 0.1 M D2O phosphate buffer with pH 5.8 at −0.8 V vs. Ag/AgCl on TOT (III) for 2 h, Pt (IV) for 2 h, and Pt-TOT.2 (V) for 2 h, and on Pt-TOT.2 (VI) for 4 h. | ||
Mechanistic understanding
To gain insight into the catalytic performance of our electrodes, we conducted XPS measurements to investigate the electronic properties of the prepared TOT and Pt-TOTs (Fig. 4, S8, and Table S5†). Interestingly, Fig. 4a and S8† revealed that increasing Pt content in Pt-TOTs resulted in a slight positive shift of the peaks of both Ti 2p and O 1 s signals compared to those on TOT.31 This shift probably indicates the interaction of Pt species with Ti and O, forming Ti–O–Pt bonds and suggesting an increase in defects (Ti3+ and oxygen vacancies, Vo) as presented in Table S5.† The XPS Pt 4f spectra of Pt-TOT.2 suggested the presence of Ptn+ species with different valence states in Pt-TOT.2 (Fig. 4b, ESI†).31 Notably, the incorporation of Pt resulted in a red shift of the Raman peak from 446.1 cm−1 to a lower wavenumber (Fig. 4c and S9†). This red shift can be attributed to the introduction of defects and an increase in Vo, likely due to the replacement of Ti by Pt. The Raman and XPS analyses collectively suggest that Pt species occupy certain Ti3+ positions and promote Ti–O–Pt interactions.31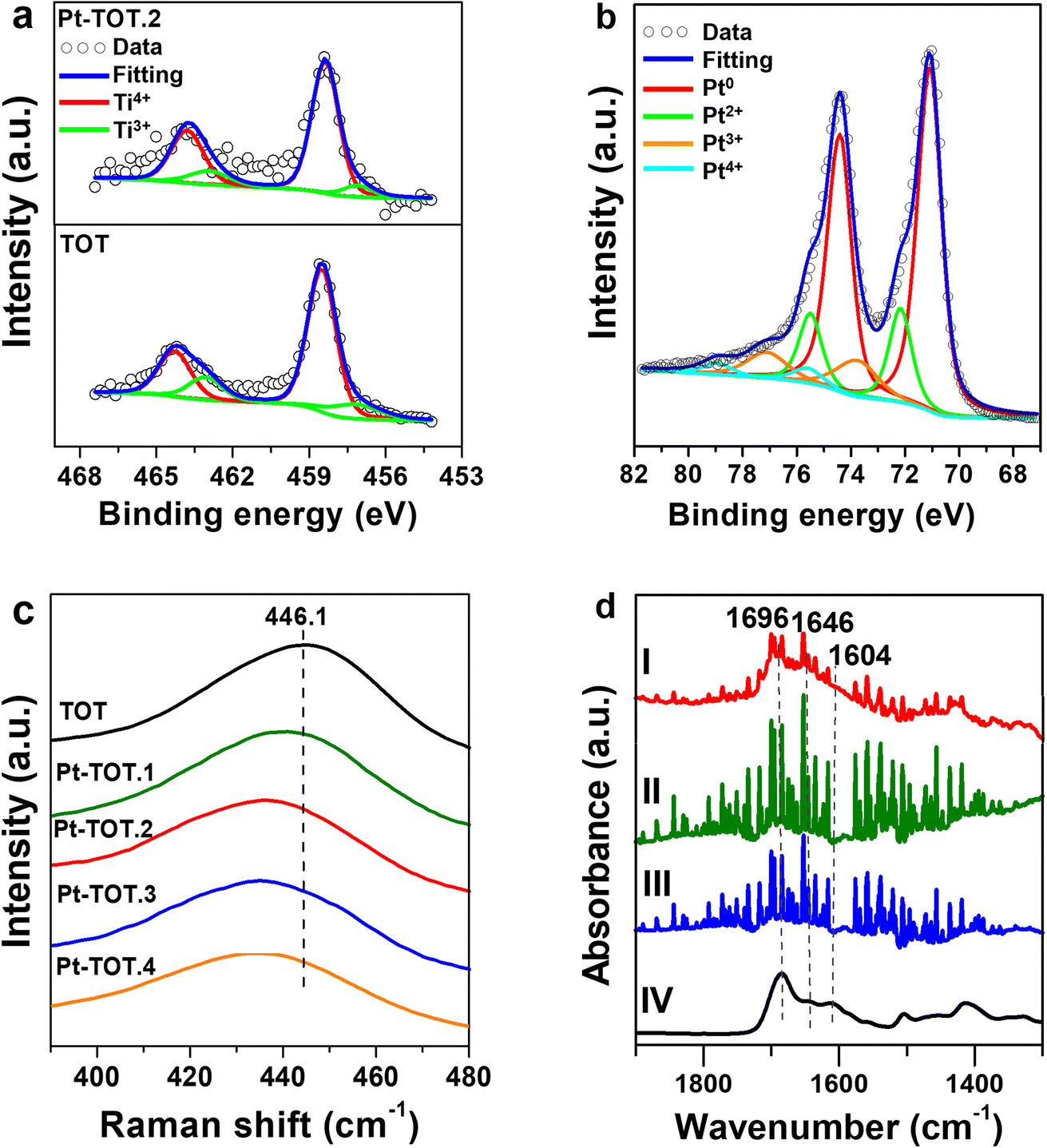 | ||
| Fig. 4 (a) Ti 2p XPS spectra of TOT and Pt-TOT.2, (b) Pt 4f XPS spectra of Pt-TOT.2, (c) Raman spectra and of TOT and Pt-TOTs in the range from 390 to 480 cm−1, and (d) FT-IR of adsorbed NAD+ on the surface of (I) Pt-TOT.2, (II) TOT (III) Ti, and (IV) ATR-IR spectrum of the NAD+ crystal (95%). | ||
To further examine the role of TOT in NAD+ adsorption, we conducted Fourier transform infrared spectroscopy using an attenuated total reflectance configuration (ATR-IR) for analysis of adsorbed NAD+ on Ti, TOT, and Pt-TOT.2 surfaces after immersing into 0.5 mM NAD+ solution, washing, and drying (Fig. 4d). We observed broad signals on all electrodes at wavenumbers similar to the reported values, with sharp signals originating from the electrolytes (water vapor). We also measured the IR spectrum of the NAD+ crystal (95%) and observed broad peaks around 1690, 1646, and 1608 cm−1, corresponding to signals observed at 1696 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch, nicotinamide), 1646 (NH2 bend, adenine), and 1604 cm−1 (C5–C6 stretch, adenine) for NAD+ powder.15b By comparing these results, all spectra seem to contain spectral components of NAD+ and Pt-TOT.2 has the largest signals assignable to NAD+, possibly indicating that favorable environments for NAD+ adsorption are created on Pt-TOT.2, probably due to cooperative interactions between TOT and Pt. In particular, this may be attributed to the presence of oxygen vacancies formed through Ti–O–Pt interactions, which facilitate the adsorption of the amide CO.15c The existence of various valence states on Pt species has been previously correlated with the favorable electron transfer,31 and the formation of linking Ti–O–Pt bonding networks in the oxide and the boundary around Pt nanoparticles on the surface can mediate the electron transfer to the adsorbed NAD+ (Scheme 2b).36 These synergistic effects contribute to the remarkable catalytic performance of Pt-TOTs in NADH-reg and provide valuable insights for the design of efficient electrochemical processes.
O stretch, nicotinamide), 1646 (NH2 bend, adenine), and 1604 cm−1 (C5–C6 stretch, adenine) for NAD+ powder.15b By comparing these results, all spectra seem to contain spectral components of NAD+ and Pt-TOT.2 has the largest signals assignable to NAD+, possibly indicating that favorable environments for NAD+ adsorption are created on Pt-TOT.2, probably due to cooperative interactions between TOT and Pt. In particular, this may be attributed to the presence of oxygen vacancies formed through Ti–O–Pt interactions, which facilitate the adsorption of the amide CO.15c The existence of various valence states on Pt species has been previously correlated with the favorable electron transfer,31 and the formation of linking Ti–O–Pt bonding networks in the oxide and the boundary around Pt nanoparticles on the surface can mediate the electron transfer to the adsorbed NAD+ (Scheme 2b).36 These synergistic effects contribute to the remarkable catalytic performance of Pt-TOTs in NADH-reg and provide valuable insights for the design of efficient electrochemical processes.
We further investigated the electrochemical double layer capacitance (Cdl) on Pt and Pt-TOT.2 to gain deeper insights into the electrode surfaces. The Cdl was evaluated based on the slope of the charging current, as shown in Fig. S10.† Subsequently, the corresponding electrochemical surface areas (ECSAs) were calculated from the Cdls and are presented in Fig. S11d.† Among the Pt-containing electrodes, it is evident that Pt-TOT.2 exhibits the highest values for both electrochemical double layer capacitance (Cdl) and electrochemical surface area (ECSA). The performance of the hydrogen evolution reaction (HER) on these electrodes at −0.8 V vs. Ag/AgCl was also assessed as shown in Fig. S10d.† Remarkably, Pt-TOT.2 demonstrated the highest partial current density for the HER among the three electrodes, whereas Pt exhibited the lowest HER. We think that the formation of different valence states of Pt (Ptn+, n = 0, 2, 3, or 4) plays an important role in enhancing the NADH-reg.
This observed trend provides a straightforward explanation for the behavior in NADH-reg; Pt-TOTs likely possess the largest number of hydrogens adsorbed on their surface (Hads), a favorable condition for enhancing the kinetics of Step 2a in Scheme 1 (for more detailed explanation of the HER, refer to the ESI†). Furthermore, Pt-TOTs predominantly adsorb NAD+, making them an ideal platform for both NAD adsorption and efficient hydrogen supply. The interplay of TiO2 in NAD+ adsorption and the presence of Ptn+ species in Pt-TOTs, responsible for enhancing Hads coverage and promoting electron transfer to the adsorbed NAD molecules, collectively contribute to accelerating the 1,4-NADH production. Notably, the rapid hydrogenation of NAD molecules on Pt-TOTs effectively limits the occurrence of dimerization processes and side products, ensuring superior selectivity.
In light of these results, we have presented a plausible mechanism for NADH-reg on TOT and Pt-TOTs, which is illustrated in Scheme 2. The findings presented in this study shed light on the mechanism underlying NADH-reg on Pt-TOTs, and we propose the mechanism based on our results in the ESI† (eqn (S14)–(S19). Therefore, Pt-TOTs emerge as a highly promising electrode for efficient electrochemical regeneration of enzymatically active 1,4-NADH at significantly low overpotentials.
Furthermore, we evaluated the enzymatic activity of the 1,4-NADH produced using Pt-TOT.2 for the electrochemical generation of lactic acid from pyruvic acid. For this purpose, we utilized purified lactate dehydrogenase (LDH) from Lactobacillus acidophilus as the enzyme in our system (refer to ESI, Fig. S11 and S12†), along with the addition of pyruvic acid to the NAD+ solution. To monitor the production of lactic acid, we employed UV spectrophotometry and an H-type electrochemical cell, analyzing the product at 15-minute intervals (Fig. 5). In addition, we employed in situ UV spectroscopy (Fig. S13†) to confirm the consumption of NADH produced by our system during the lactic acid formation over time (Fig. S14†). Our observations revealed a progressive increase in the concentration of generated 1,4-NADH over time during the electrochemical reaction in the absence of pyruvic acid and LDH, as shown in Fig. 5, the insets (a) and (b), and S14.† Conversely, when we introduced both pyruvic acid and LDH, the concentration of 1,4-NADH became negligible, indicating that the produced 1,4-NADH was immediately utilized by LDH for the reduction of pyruvic acid (Fig. 5 and S14†). Furthermore, we performed HPLC analysis (Fig. S15†) to verify the product generated from the enzymatic electrochemical formation of lactic acid on Pt-TOT.2, which conclusively confirmed the production of lactic acid. These results unequivocally demonstrate that the generated 1,4-NADH using the Pt-TOT.2 electrode effectively serves as an electron donor in the enzymatic reaction catalyzed by LDH.
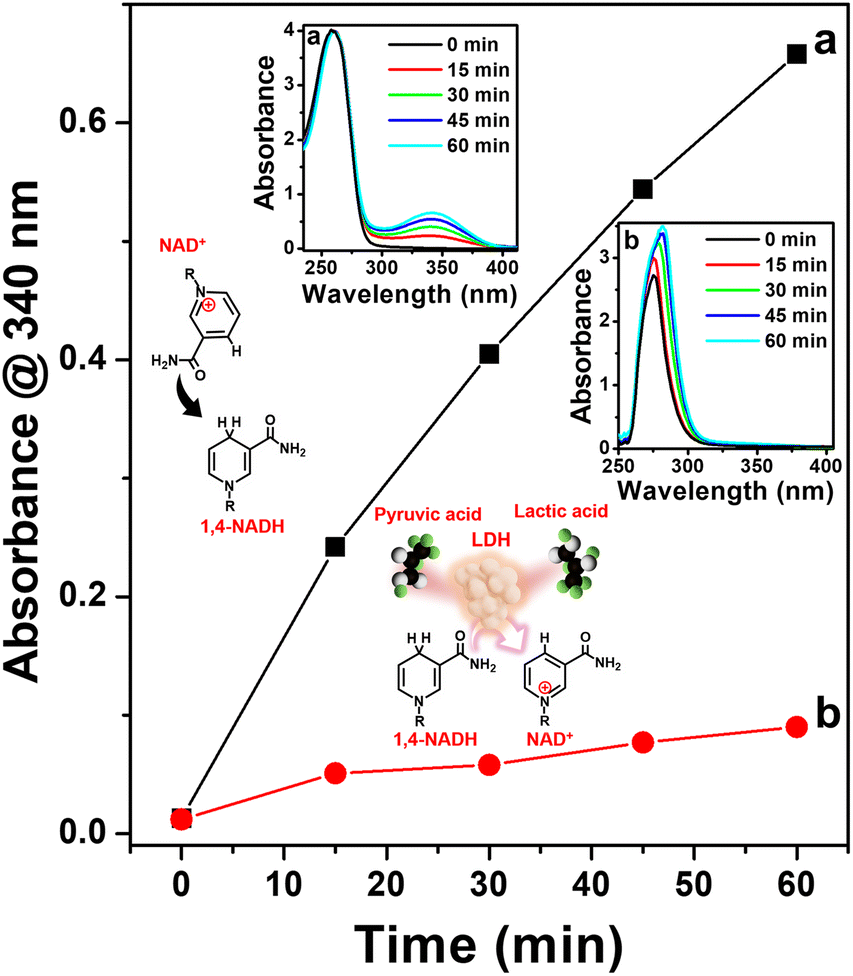 | ||
| Fig. 5 UV absorbance at 340 nm plotted against time for the electrochemical reduction of (a) 0.25 mM NAD+, and (b) 0.25 mM NAD+ with 20 mM pyruvic acid and 8 U ml−1 LDH from 0 min to 60 min. Insets show the corresponding UV absorbance measured for samples taken every 15 min during the electrochemical reduction of (a) and (b) and schematic representation of the reactions involved. | ||
Conclusion
Pt-TOTs demonstrated exceptional performance in NADH-reg. Notably, Pt-TOT.2 exhibited the highest yield of 99.5 ± 0.4% at the lowest reported overpotential of only −0.8 V vs. Ag/AgCl (−0.2 V vs. RHE), and 86 ± 3% at an even lower overpotential of −0.6 V vs. Ag/AgCl (−0.06 V vs. RHE). This outstanding performance arises from the synergistic effects of the predominant NAD adsorption on TiO2 and the rapid kinetics of hydrogenation achieved on Pt-TOT.2, with the presence of Ptn+ species in different valence states. The integration of such synergistic surface functions at the electrode interface emerges as a pivotal factor in achieving efficient electrochemical processes. Moreover, we validated the enzymatic activity of the 1,4-NADH produced using our designed electrode by converting pyruvic acid to lactic acid using LDH. The highly efficient NADH-reg demonstrated in this study holds great promise for advancing enzymatic systems and facilitating their widespread application in energy and materials transformations.Data availability
We provided the related data in the ESI.†Author contributions
Nada H. A. Besisa: conceptualization, investigation, experimental, writing – original draft, validation, analytical interpretation. Ki-SeokYoon: review and editing. M. Yamauchi: conceptualization, review and editing, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan (Grant No. 197321, JP18H05517 “Hydrogenomics” and 22K19088), and the Moonshot Research and Development Program (JPNP18016).References
- U. T. Bornscheuer, G. W. Huisman, R. J. Kazlauskas, S. Lutz, J. C. Moore and K. Robins, Engineering the third wave of biocatalysis, Nature, 2012, 485, 185–194 CrossRef CAS PubMed.
- J. Kim, H. Jia and P. Wang, Challenges in biocatalysis for enzyme-based biofuel cells, Biotechnol. Adv., 2006, 24, 296–308 CrossRef CAS PubMed.
- A. Liese, K. Seelbach and C. Wandrey, Industrial biotransformations, WILEY-VCH, Weinheim, 2nd edn, 2006 Search PubMed.
- A. Walcarius, R. Nasraoui and Z. Wang, et al., Factors affecting the electrochemical regeneration of NADH by (2,2’-bipyridyl) (pentamethylcyclopentadienyl)-rhodium complexes: Impact on their immobilization onto electrode surfaces, Biochemistry, 2011, 82, 46–54 CAS.
- I. Ali, T. Khan and S. Omanovic, Direct electrochemical regeneration of the cofactor NADH on bare Ti, Ni, Co and Cd electrodes: The influence of electrode potential and electrode material, J. Mol. Catal. A: Chem., 2014, 387, 86–91 CrossRef.
- I. Ali, A. Gill and S. Omanovic, Direct electrochemical regeneration of the enzymatic cofactor 1,4-NADH employing nano-patterned glassy carbon/Pt and glassy carbon/Ni electrodes, Chem. Eng. J., 2012, 188, 173–180 CrossRef CAS.
- S. Immanuel and R. Sivasubramanian, Electrochemical reduction of NAD+ on graphene oxide and chemically reduced graphene oxide nanosheets, Mater. Sci. Eng., B, 2020, 114705 CrossRef CAS.
- S. Immanuel, R. Sivasubramanian, R. Gul and M. A. Dar, Recent Progress and Perspectives on Electro-chemical Regeneration of Reduced Nicotinamide Adenine Dinucleotide (NADH), Chem.–Asian J., 2020, 15, 4256–4270 CrossRef CAS PubMed.
- R. Barin, S. Rashid-Nadimi, D. Biria and M. A. Asadollahi, Direct electrochemical regeneration of 1,4-NADH at the copper foam and bimetallic copper foam, Electrochim. Acta, 2017, 247, 1095–1102 CrossRef CAS.
- N. Ullah, I. Ali and S. Omanovic, Direct electrocatalytic reduction of coenzyme NAD+ to enzymatically- active 1,4- NADH employing an iridium/ruthenium-oxide electrode, Mater. Chem. Phys., 2015, 149–150, 413–417 CrossRef CAS.
- K. Warriner, S. Higson and P. Vadgama, A lactate de-hydrogenase amperometric pyruvate electrode exploiting direct detection of NAD+ at a poly(3-methylthiophene): poly (phenol red) modified platinum surface, Mater. Sci. Eng., C, 1997, 5, 91–99 CrossRef.
- Y. Shimizu, A. Kitani, S. Ito and K. Sasaki, Electro-chemical Reduction of Nicotinamide Coenzyme (NAD+) Mediated with Rhodium Ion Immobilized in Polymer Modified Electrodes, Denki Kagaku, 1993, 61, 872–873 CrossRef CAS.
- A. Damian, K. Maloo and S. Omanovic, Direct electrochemical regeneration of NADH on Au, Cu and Pt-Au electrodes, Chem. Biochem. Eng. Q., 2007, 21, 21–32 CAS.
- A. Azem, F. Man and S. Omanovic, Direct regeneration of NADH on a ruthenium modified glassy carbon electrode, J. Mol. Catal. A: Chem., 2004, 219, 283 CrossRef CAS.
- (a) A. Damian and S. Omanovic, Electrochemical reduction of NAD+ on a polycrystalline gold electrode, J. Mol. Catal. A: Chem., 2006, 253, 222–233 CrossRef CAS; (b) A. Damian and S. Omanovic, Interactive adsorption behaviour of NAD+ at a gold electrode surface, Langmuir, 2006, 23, 3162–3171 CrossRef PubMed; (c) R. Zhang, L. Pan and B. Guo, et al., Tracking the Role of Defect Types in Co3O4 Structural Evolution and Active Motifs during Oxygen Evolution Reaction, J. Am. Chem. Soc., 2023, 145, 2271–2281 CrossRef CAS PubMed.
- F. Man and S. Omanovic, A kinetic study of NAD+ reduction on a ruthenium modified glassy carbon electrode, J. Electroanal. Chem., 2004, 568, 301–313 CrossRef CAS.
- T. Saba, J. W. H. Burnett, J. Li, P. N. Kechagiopoulos and X. Wang, A facile analytical method for reliable selectivity examination in cofactor NADH regeneration, Chem. Commun., 2020, 56, 1231 RSC.
- B. M. Reddy and A. Khan, Recent Advances on TiO2-ZrO2 Mixed Oxides as Catalysts and Catalyst Supports, Cal. Rev. Sci. Eng., 2005, 47, 257–296 CrossRef CAS.
- M. Morozova, P. Kluson, J. Krysa and M. Vesely, et al., Electrochemical Properties of TiO2 Electrode Prepared by Various Methods, Procedia Eng., 2012, 42, 573–580 CrossRef.
- D. Reinhardt, S. Krieck and S. Meyer, Special titanium dioxide layers and their electrochemical behavior, Elecrochem. Acta, 2006, 52, 825–830 CrossRef CAS.
- T. Fröschl, U. Hörmann, P. Kubiak and G. Kučerová, et al., High surface area crystalline titanium dioxide: potential and limits in electrochemical energy storage and catalysis, Chem. Soc. Rev., 2012, 41, 5313–5360 RSC.
- A. Curulli, F. Valentini, G. Padeletti, M. Viticoli and D. Caschera, et al., Smart (Nano) materials: TiO2 nanostructured films to modify electrodes for assembling of new electrochemical probes, Sens. Actuators, B, 2005, 111–112, 441–449 CrossRef CAS.
- M. Yamauchi, Inorganic nano catalysts for hydrogenation reactions contributable to a sustainable material supply, Chem. Lett., 2021, 50, 1901–1908 CrossRef CAS.
- T. Fukushima and M. Yamauchi, Electrosynthesis of glycine from bio-derivable oxalic acid, J. Appl. Electrochem., 2020, 51, 99–106 CrossRef.
- H. Li, S. Wu, Z. D. Hood, J. Sun, B. Hu, C. Liang, S. Yang, Y. Xu and B. Jiang, Atomic defects in ultra-thin mesoporous TiO2 enhance photocatalytic hydrogen evolution from water splitting, Appl. Surf. Sci., 2020, 513, 145723 CrossRef CAS.
- R. Watanabe, M. Yamauchi, M. Sadakiyo, R. Abe and T. Takeguchi, CO2-free electric power circulation via direct charge and discharge using the glycolic acid/oxalic acid redox couple, Energy Environ. Sci., 2015, 8, 1456 RSC.
- M. Yamauchi, H. Saito, T. Sugimoto, S. Mori and S. Saito, Sustainable Organic Synthesis Promoted on Titanium Dioxide using Coordinated Water and Renewable Energies/Resources, Coord. Chem. Rev., 2022, 472, 214773 CrossRef CAS.
- G. S. Li, D. Q. Zhang and J. S. Yu, Thermally stable ordered mesoporous CeO2/TiO2 visible-light photocatalysts, Phys. Chem. Chem. Phys., 2009, 11, 3775–3782 RSC.
- C. Li, Z. Jiang and Z. Yao, Fabrication and characterization of multi-metal co-doped titania films for a water-splitting reaction, Dalton Trans., 2010, 39, 10692–10696 RSC.
- C. Wang, Q. Hu, J. Huang, C. Zhu and Z. Deng, et al., Electrochemical assisted photocatalytic degradation of salicylic acid with highly ordered TiO2 nanotube electrodes, Appl. Surf. Sci., 2014, 292, 161–169 CrossRef CAS.
- Z. Lian, W. Wang, G. Li, F. Tian, K. S. Schanze and H. Li, Pt-Enhanced Mesoporous Ti3+/TiO2 with Rapid Bulk to Surface Electron Transfer for Photocatalytic Hydrogen Evolution, ACS Appl. Mater. Interfaces, 2017, 9, 16959–16966 CrossRef CAS PubMed.
- (a) K. Fukutani, J. Yoshinobu, M. Yamauchi, T. Shima and S. Orimo, Hydrogenomics: Efficient and selective hydrogenation of stable molecules utilizing three aspects of hydrogen, Cat, Lett, 2022, 152, 1583–1597 CAS; (b) M. Sadakiyo, S. Hata, T. Fukushima, G. Juhász and M. Yamauchi, Electrochemical hydrogenation of non-aromatic carboxylic acid derivatives as a sustainable synthesis process: from catalyst design to device construction, Phys. Chem. Chem. Phys., 2019, 21, 5882–5889 RSC.
- (a) H. Eguchi, K. Kato, G. Juhasz and M. Yamauchi, Selectivity Enhancement in Electrochemical Reduction of Oxalic Acid on Titanium Dioxide Nanoparticles Achieved by Shape and Energy-states Controls, Catal. Sci. Technol., 2021, 11, 7592–7597 RSC; (b) M. Sadakiyo, S. Hata, X. Cui and M. Yamauchi, Sci. Rep., 2017, 7(1), 17032 CrossRef PubMed.
- T. Saba, J. Li and J. W. H. Burnett, et al., NADH regeneration: a case study of Pt-catalyzed NAD+ reduction with H2, ACS Catal., 2021, 11, 283–289 CrossRef CAS.
- E. Aamer, J. Thöming, M. Baune and N. Reimer, et al., Influence of electrode potential, pH and NAD+ concentration on the electrochemical NADH regeneration, Sci. Rep., 2022, 12(1), 16380 CrossRef CAS PubMed.
- S. Kitano, T. G. Noguchi, M. Nishihara, K. Kamitani, T. Sugiyama, S. Yoshioka, T. Miwa, K. Yoshizawa, A. Sadykov and M. Yamauchi, Heterointerface Created on Au-Cluster-Loaded Unilamellar Hydroxide Electrocatalysts as a Highly Active Site for the Oxygen Evolution Reaction, Adv. Mater., 2022, 34, 16 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc04104b |
| This journal is © The Royal Society of Chemistry 2024 |