
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Probing the reactivity of a transient Al(I) species with substituted arenes†
Imogen
Squire
,
Michelangelo
Tritto
,
Juliana
Morell
and
Clare
Bakewell
 *
*
Department of Chemistry, King's College London, 7 Trinity Street, London, SE1 1DB, UK. E-mail: clare.bakewell@kcl.ac.uk
First published on 30th September 2024
Abstract
With only a handful of compounds known, opportunities to explore the structure and reactivity of dialumenes and related dialumene adducts have been limited. For the first time, a series of dialumene-arene adducts have been synthesised; adduct formation has been probed experimentally and through DFT, and their reactivity investigated.
Dialumenes, molecules that feature an Al
![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) Al double bond, are a notoriously challenging class of compound to isolate, owing to difficulties in stabilisation of the low oxidation state centre and the subtle difference in coordination environment required favour an Al(I) dimer over a monomer.1–3 To date, there have been only three examples of isolated dialumenes, all of which are base stabilised.3–5 In 2017, Inoue and co-workers reported the first neutral AlAl compound (I), with the Al(I) centres stabilised by N-heterocyclic carbenes.4 The integrity of this bond was borne out by DFT, and through reactivity studies with a series of unsaturated molecules which confirmed it reacts as a double bond, forming [2+2] cycloaddition products. This was followed by work from Cowley and Krämer, whose base stabilised dialumene (II) was shown to react with alkynes, both as the dialumene and as the Al(I) monomer formed through dissociation of the double bond.3 Comparison of these two classes of compound shows a significantly different bonding picture. Single crystal X-ray diffraction (SCXRD) reveals a longer Al–Al bond in IIversusI, as well as an increased trans-bent geometry. Electron localisation function (ELF), quantum theory of atoms in molecules (QTAIM) calculations, as well as analysis of the Wiberg Bond Indices all indicate a weaker, non-classical Al–Al double bond in II, with a slipped π-bond structure. This more closely resembles the structure of theoretical base-free RAlAlR dialumenes.1
Al double bond, are a notoriously challenging class of compound to isolate, owing to difficulties in stabilisation of the low oxidation state centre and the subtle difference in coordination environment required favour an Al(I) dimer over a monomer.1–3 To date, there have been only three examples of isolated dialumenes, all of which are base stabilised.3–5 In 2017, Inoue and co-workers reported the first neutral AlAl compound (I), with the Al(I) centres stabilised by N-heterocyclic carbenes.4 The integrity of this bond was borne out by DFT, and through reactivity studies with a series of unsaturated molecules which confirmed it reacts as a double bond, forming [2+2] cycloaddition products. This was followed by work from Cowley and Krämer, whose base stabilised dialumene (II) was shown to react with alkynes, both as the dialumene and as the Al(I) monomer formed through dissociation of the double bond.3 Comparison of these two classes of compound shows a significantly different bonding picture. Single crystal X-ray diffraction (SCXRD) reveals a longer Al–Al bond in IIversusI, as well as an increased trans-bent geometry. Electron localisation function (ELF), quantum theory of atoms in molecules (QTAIM) calculations, as well as analysis of the Wiberg Bond Indices all indicate a weaker, non-classical Al–Al double bond in II, with a slipped π-bond structure. This more closely resembles the structure of theoretical base-free RAlAlR dialumenes.1
Prior to the isolation of I and II, the formation of transient dialumenes had been invoked in the formation of dialumene-arene adducts (III, IV).6,7 Tokitoh further showed that IV was able to react as a masked source of the dialumene, forming addition products with alkynes and dihydrogen amongst others (Scheme 1b).8 More recent examples of transient dialumenes and dialumene-arene adducts have also been reported.9–15
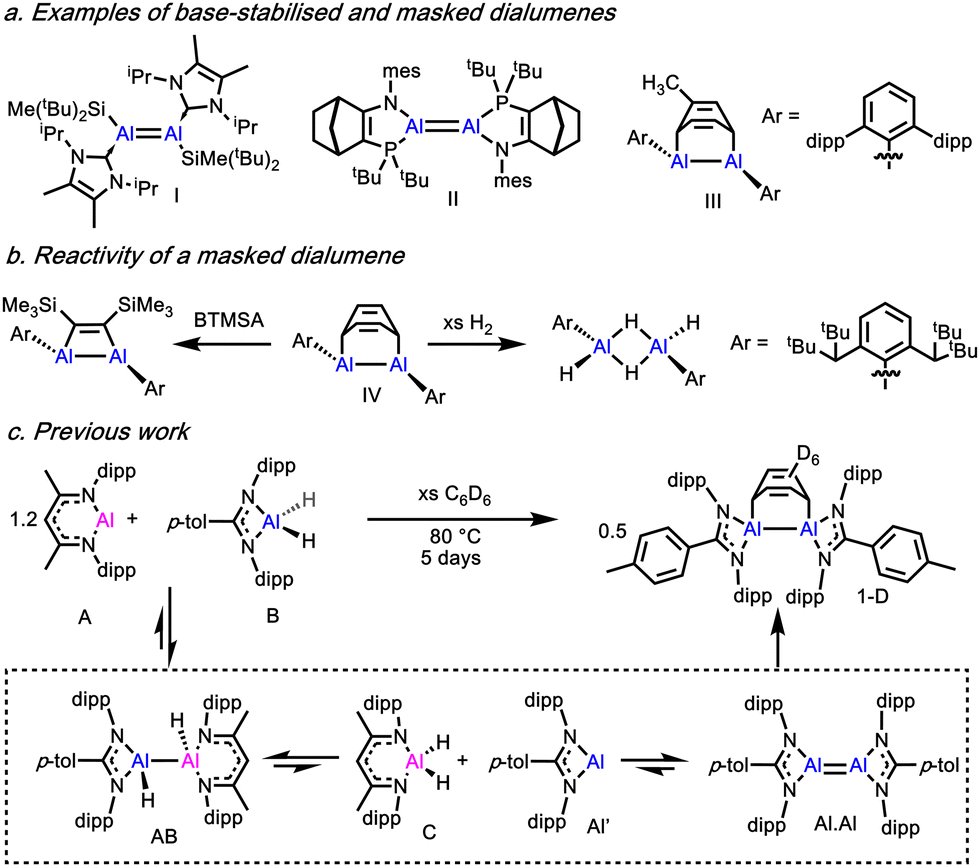 | ||
| Scheme 1 Examples of isolated and masked dialumenes. | ||
In 2022 we reported the synthesis of the deuterated dialumene benzene adduct, 1-D, which was formed through reaction between Roesky's Al(I) complex (A) and an amidinate aluminium dihydride (B), in benzene solution at elevated temperature (Scheme 1c).16 The reaction proceeds via a series of equilibria involving comproportionation and disproportionation reactions (Scheme 1c). The product, 1-D, is proposed to form directly from a transient Al(I) species (monomer (Al′) or dimer (Al.Al)) and is likely a thermodynamic sink in the complex equilibrium network. According to theoretical calculations on model dialumenes, electronegative substituents, such as N, and the strained 4-membered coordination geometry should lead to a weaker Al–Al bond that more readily dissociates to the Al monomer.3 This suggests that Al(I) amidinate complexes should be highly reactive species, capable of reacting as either the Al monomer or as the dialumene. However, despite the precedent for IV acting as a masked source of Al(I), 1 showed no reaction with a selection of arenes, alkynes or H2.16
This led us to consider the mechanism by which the dialuminum-benzene adduct is formed and if the use of more sterically encumbered or electronically divergent arenes might enable us to access masked reactivity. Herein, we report the reaction of A and B in the presence of a range of arenes with varying steric and electronic profiles, the resultant dialumene adduct reactivity, as well as computational mechanistic insight.
Heating a mixture comprised of a slight excess of A, with B in toluene-d8 at 110 °C (Scheme 2) led to the formation of a bright red solution after 1 hour, with the colour intensifying over 4 days, in line with observations using benzene solvent (1). However, analysis of the 1H NMR spectrum revealed a complex mix of products (including C), with a less distinctive pattern of upfield CH(CH3)2 doublet environments than was observed in 1 (∼0.5 ppm, Fig. S2, ESI†). Due to the promising colour change, the reaction was continued to work-up, and dark red crystals were isolated (39%).‡ SCXRD confirmed formation of a dialumene-toluene adduct (2-D), which is directly comparable with 1 (Fig. 1). The Al–Al bond length (2.5520(6) Å) is slightly longer than in 1 (2.5419(7) Å),16 and similar to that reported for a related toluene adduct.13 The six-membered ring (C1–C6) bridging the Al atoms shows a pattern of two shorter bonds (C2–C3 1.321(2) Å and C5–C6 1.322(3) Å) opposite each other, and four longer bonds (in the range 1.499(3)–1.510(3) Å), consistent with loss of aromaticity.
 | ||
| Scheme 2 The formation of dialumene-arene adducts from the reaction of A and B using a range of substrates. Isolated yields (NMR yields). | ||
 | ||
| Fig. 1 Solid state structures of 2–5. Selected bond lengths (averaged) (Å): 2 Al–Al 2.5520(6), Al1–N1 1.949(1), Al1–C1 2.016(2), C2–C3 1.321(2); 3 Al–Al 2.5485(7), Al1–N1 1.959(1), Al1–C1 2.017(1), C2–C3 1.343(3); 4 Al–Al 2.561(1), Al1–N1 1.973(2), Al1–C1 2.019(2), C2–C3 1.337(2); 5 Al–Al 2.5592(7), Al1–N1 1.953(1), Al1–C1 2.016(2), C2–C3 1.343(2) (full data Tables S1–S4, ESI†). | ||
Analysis of the 1H NMR spectra showed a complicated set of signals, however formation of the protio-toluene adduct (2-H) allowed us to obtain more information. The diene protons, that result from the [2+4] addition of toluene across the dialumene bond (5–6 ppm) indicate two distinct, persistent sets of signals in a ratio that remained consistent between samples (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.4, Fig. S3, ESI†). Exchange spectroscopy (EXSY) spectra show exchange peaks for the CH3 of the toluene fragment, as well as for CH and CH3 environments from the dipp groups (Fig. S4–S5, ESI†), suggesting that the complex exists in two distinct, but slowly interconverting conformations. The two sets of signals are not a result of two different non-interconverting products. Variable temperature (VT) NMR spectroscopy (Fig. S6–S8, ESI†) showed coalescence of the toluene fragment protons when the temperature was increased (T > 55 °C for Al–CH and >95 °C for CCH, Fig. S8, ESI†), confirming that at room temperature the system is in slow exchange.
:0.4, Fig. S3, ESI†). Exchange spectroscopy (EXSY) spectra show exchange peaks for the CH3 of the toluene fragment, as well as for CH and CH3 environments from the dipp groups (Fig. S4–S5, ESI†), suggesting that the complex exists in two distinct, but slowly interconverting conformations. The two sets of signals are not a result of two different non-interconverting products. Variable temperature (VT) NMR spectroscopy (Fig. S6–S8, ESI†) showed coalescence of the toluene fragment protons when the temperature was increased (T > 55 °C for Al–CH and >95 °C for CCH, Fig. S8, ESI†), confirming that at room temperature the system is in slow exchange.
With the toluene adduct in hand, we next targeted a series of substituted arenes with varying steric and electronic properties. Firstly, ortho-, para- and meta-xylene were combined with a slight excess of A and B and heated between 80–100 °C for 5–8 days, with the xylene acting as both substrate and reaction solvent (Scheme 2). Compounds 3–5 were isolated by fractional crystallisation (slow evaporation from hexane). In all cases, crystals suitable for SCXRD were grown, confirming the formation of the respective dialumene-xylene adducts (Fig. 1). The CH3 groups of the o-xylene adduct (3) reside on one of the CC bonds in a 1,2-fashion, in the m-xylene adduct (4) one CH3 group is bound to each CC bond, straddling the Al–C carbon. In both cases, the CH3 groups reside preferentially on the alkene carbons instead of the Al–C carbons, which is presumably due to a steric preference. Another notable feature of 3 and 5 is that the electron density of the xylene CH3 groups is localised to one position, indicating no conformational disorder in the crystal. In both 2 and 4 there is conformational disorder leading to CH3 groups being located at different positions around the diene fragment. However, it is notable that in neither case is there any residual electron density arising from a CH3 group at the Al–C carbon of the diene fragment.
Unlike with 2, analysis of the 1H NMR spectra of 3–5 reveals the presence of just one conformational isomer. In each case there are one or two signals corresponding to the Al–CH protons in the aliphatic region (3, 2.58 (1H, d), 2.75 (1H, d); 4, 2.44 (1H, s), 2.71 (1H, t); 5, 2.57 (2H, d) ppm) and the alkene CH protons between 5.2–6.4 ppm (3, 5.44 (1H, t), 6.33 (1H, determined from COSY as coincident with another resonance, so splitting unobservable); 4, 5.27 (1H, d), 6.03 (1H, d); 5, 5.91 (2H, d) ppm) (Fig. S9-S10, ESI†). The presence of two sets of alkene CH signals in 3 and 4 suggests a lack of symmetry in the ligand framework, rendering the CHs inequivalent; this is also observed in the solid state. Conversely, 5 has a centre of symmetry diagonally through the Al–Al core leading to equivalent xylene CH signals. This centre of symmetry is observed in the SCXRD structure with half the molecule in the asymmetric unit (C2/c space group).
The trend towards reduced dialumene-adduct yield with increased steric bulk (16–23% 3–5, vs. 39–50% 1–2), is in line with a less thermodynamically favoured product, which could point to an increased persistence of ‘unmasked’ Al(I) species in solution. To probe this further, more sterically demanding arenes were investigated. Increasing the number of CH3 substituents on the arene (mesitylene and durene) did not lead to adduct formation, as evidenced by the lack of a distinctive colour change indicative of the dialumene-arene adducts, and an absence of any characteristic signals in the 1H NMR spectra (Fig. S11, ESI†). However, increasing the steric bulk at the 1-position of the arene did not appear to preclude adduct formation, with both cumene and N,N-dimethylaniline forming the characteristic red colour associated with these reactions (Fig. S12 and S13, ESI†). The crude reaction mixtures contain peaks in the alkene region (5–6 ppm) of the 1H NMR spectrum, with cumene showing similar splitting to that seen in the toluene adduct (Fig. S13, ESI†). However in both cases, repeated attempts to isolate the adduct were unsuccessful.
Due to the highly reactive nature of the equilibrium-based reaction mixture, and the wide range of arene C–E bonds that A is known to activate, the scope for testing electron donating and electron withdrawing substituents was limited. For example, despite A and B immediately forming the asymmetric dihydrodialane intermediate (AB) when combined in solution,16 reaction of A and B in anisole at 80 °C led to the known C–O oxidative addition product of A and anisole (Fig. S14 and S15, ESI†), along with complex C and a previously reported dihydrodialane complex (D in Fig. S15, ESI†).17–19 The breaking of this C–O bond likely represents a more thermodynamically favoured product, and clearly confirms that the equilibrium reaction that features disproportion and comproportionation reactions between AlI/AlII/AlIII intermediates is active in both directions (Fig. S1, ESI†).
Introduction of electron withdrawing substituents was also limited, as even the C–F bonds in 1-fluorobenzene and α,α,α-trifluorotoluene are known to undergo reaction with A under forcing conditions, albeit in an undefined manner. In line with these observations, when A and B were combined in the presence of trifluorotoluene or fluorobenzene and subjected to the standard reaction conditions a complex mix of products was observed, but with no evidence of the adduct (Fig. S16–S18, ESI†). The presence of several compounds could be identified in the reaction (C, dippBDIAlF2, dippBDIAlHF), which suggests C–F bond activation precludes adduct formation, likely assisted by favourable Al–F bond formation.20,21
We next turned to DFT; investigating the [4+1] reaction between Al′ and benzene, as well the [4+2] reaction from the dialumene (Al.Al). Using the full ligand system we tested a range of functionals and basis sets, with M06L/6-31G** (C, H, N) + SDDALL (Al) found to best support our experimental data (Table S5, ESI†). Formation of the asymmetric dihydrodialane, AB, from A and B is exergonic, occurring immediately upon mixing and is thus taken as our zero point energy. AB disproportionates to Al′ and C (+17.5 kcal mol−1), and dimerisation of Al′ to Al.Al is thermodynamically favoured (−12.3 kcal mol−1), indicating that the Al(I) species is more likely to exist as a dialumene. Analysis of the dialumene intermediate (Al.Al) reveals a bond length of 2.674 Å, significantly longer than reported for Cowley and Kramer's isolated dialumene (Al–Al 2.5190(14) Å).3 The Wiberg bond index of 1.12 is also smaller, whilst QTAIM shows a bond critical point (bcp) between the two Al atoms (ρbcp = 0.05 e Å−3, ∇2ρbcp = +1.180 e Å−5, Fig. S27, ESI†). Taken together, these initial calculations suggest Al.Al would have a weaker Al–Al bond and more extreme slipped π-bond character, in line with predictions.
Approach of benzene, to Al.Al, led to the encounter complex, Int-1di, which is slightly endergonic and a transition state for the [4+2] cycloaddition reaction (TS-1di) was located, with an energy of 17.1 kcal mol−1, relative to the dialumene (22.3 kcal mol−1 relative to AB). Here, the ligands are pinched back to allow the approach of the substrate, and the transition state is highly asymmetric, with the ligand at the first Al centre nearly perpendicular to that of the second (Fig. 2). This is similar to a related transition state previously reported by Roesky and co-workers.13TS-1di leads to 1, which is the thermodynamically favoured product (−4.5 kcal mol−1). A [4+1] reaction pathway was also investigated, given recent precedent for [4+1] addition products.22,23 A transition state (TS-1mono) could be located at 42.9 kcal mol−1 relative to AB, which proceeds to a high energy intermediate (Int-2mono). From here a second equivalent of Al′ could insert, although it has not been possible to locate a transition state for this process. It therefore seems most likely that 1 is formed via a [4+2] cycloaddition from a dialumene intermediate. This [4+2] pathway was also investigated for toluene and mesitylene substrates. The transition state TS-1di (tol) was found to be slightly lower than for benzene (ΔΔG 2.7 kcal mol−1), but TS-1 (mes) was significantly higher (ΔΔG 6.9 kcal mol−1). Moreover, the mesitylene product was found to be thermodynamically unfavoured, compared to AB. A combination of both kinetic and thermodynamic effects likely explain why the dialumene adduct is not observed experimentally. Indeed 1H NMR investigations show only the presence of AB with decomposition occurring over time.
 | ||
| Fig. 2 Calculated reaction pathways for the [4+2] and [4+1] cycloaddition reactions between Al(I) species and benzene, toluene and mesitylene. Gibbs free energies (kcal mol−1). | ||
As previously reported, 1 showed no reaction with a range of substrates including H2 and diphenylacetylene. This is exemplified by the reaction of 1-H with C6D6, where even after prolonged heating the formation of 1-D was not observed. However, when 2 was heated in benzene-d6 at 80 °C exchange of the arene ring occurred, and after 2 days full conversion to 1-D was achieved (Scheme 3 and Fig. S18, ESI†). The reverse reaction of 1 (or 1-D) in toluene-d8 showed no change even after prolonged heating at 80 °C, suggesting the 2 is more labile than the equivalent benzene adduct (1), and is evidence of a true masked dialumene. This observation is supported by DFT, where TS-1di (tol) is lower in energy than TS-1di (bz) (ΔΔG‡ = 2.7 kcal mol−1).
 | ||
| Scheme 3 Reactivity of 1 and 2 with arenes. | ||
It could therefore be expected that the transient dialumene formed when 2 is heated may react with other small molecules (Fig. S19, ESI†). Despite this, no reaction was observed when 2 was exposed to 2 bar of H2 in cyclohexane-d12 or benzene-d6, although in the latter case arene exchange occurred, forming 1-D (Fig. S21, ESI†). This may be a concentration effect, where dialumene formation is too transient to encounter H2.§ This points to the proposed dialumene being highly reactive; only stable and persistent enough to react with physically and energetically accessible substrates.
The xylene adducts, 3–5, were shown to be unreactive to both benzene and toluene. This is somewhat surprising, although the transition state to the formation of 3 is slightly higher in energy than for 2 (20.6 kcal mol−1, Fig. S30, ESI†).
In conclusion, we have synthesised a series of dialumene-arene adducts with varying steric profiles. These sterics were shown to be crucial in determining both if an adduct can be isolated and whether masked reactivity is observed; we can say with certainty that with significant increase in steric hinderance adduct formation becomes less favoured. However, the factors governing the reactivity appear to be subtle and nuanced, not directly attributable to steric factors alone. Nevertheless, this represents the first time that structure activity relationships have been probed for masked dialumenes, and will no doubt inform future attempts to isolate elusive free dialumenes.
CB acknowledges the EPSRC (EP/Y000129/1), Royal Society (RGS R2 222051) and KCL for funding this research. IS acknowledges the NMES faculty (KCL) for studentship funding. We are grateful for the support of Dr Thomas Hicks with NMR experimentation.
Data availability
All data supporting this article have been included in the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- J. Moilanen, et al. , Inorg. Chem., 2010, 49, 10992–11000 CrossRef CAS.
- K. Hobson, et al. , Chem. Sci., 2020, 11, 6942–6956 RSC.
- R. L. Falconer, et al. , Angew. Chem., Int. Ed., 2021, 60, 24702–24708 CrossRef CAS.
- P. Bag, et al. , J. Am. Chem. Soc., 2017, 139, 14384–14387 CrossRef CAS.
- C. Weetman, et al. , Chem. Sci., 2020, 11, 4817–4827 RSC.
- R. J. Wright, et al. , J. Am. Chem. Soc., 2003, 125, 10784–10785 CrossRef CAS.
- T. Agou, et al. , Angew. Chem., Int. Ed., 2013, 52, 10818–10821 CrossRef CAS.
- K. Nagata, et al. , Angew. Chem., Int. Ed., 2016, 55, 12877–12880 CrossRef CAS.
- D. Dhara, et al. , Chem. Sci., 2022, 13, 5631–5638 RSC.
- D. Dhara, et al. , Chem. Sci., 2022, 13, 9693–9700 RSC.
- J. D. Queen and P. P. Power, Chem. Commun., 2022, 59, 43–46 RSC.
- D. Dhara, et al. , Chem. – Eur. J., 2023, 29, e202300483 CrossRef CAS.
- A. Kumar, et al. , Chem. – Eur. J., 2023, 29, e202300546 CrossRef CAS.
- H. Zhu, et al. , Chem. – Eur. J., 2023, 29, e202301973 CrossRef CAS.
- X. Wang, et al. , Dalton Trans., 2024, 53, 15441–15450 RSC.
- C. Bakewell, et al. , Angew. Chem., Int. Ed., 2022, 61, e202205901 CrossRef CAS.
- R. K. Brown, et al. , Chem. Commun., 2021, 57, 11673–11676 RSC.
- T. Chu, et al. , J. Am. Chem. Soc., 2014, 136, 9195–9202 CrossRef CAS.
- S. J. Bonyhady, et al. , Nat. Chem., 2010, 2, 865–869 CrossRef CAS.
- S. Yow, et al. , Angew. Chem., Int. Ed., 2012, 51, 12559–12563 CrossRef CAS.
- C. Bakewell, et al. , Angew. Chem., Int. Ed., 2018, 57, 6638–6642 CrossRef CAS.
- D. Sarkar, et al. , J. Am. Chem. Soc., 2024, 146, 11792–11800 CrossRef CAS.
- X. Zhang and L. L. Liu, Angew. Chem., Int. Ed., 2022, 61, e202116658 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2365635–2365638. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc03904a |
| ‡ Co-crystallisation limited the isolated yield of products 1–2 (∼40–45%). However 1H NMR experiments conducted using internal standards showed the % product formed in the reaction to be >80%. |
| § All reactions require an extremely large excess of substrate to form product, with attempts to conduct reactions under more dilute condition unsuccessful. This suggests that the adduct formation is dependent on a high concentration of arene. |
| This journal is © The Royal Society of Chemistry 2024 |