
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Colloidal III–V quantum dots: a synthetic perspective
Theodore A.
Gazis
 ,
Ashleigh J.
Cartlidge
and
Peter D.
Matthews
*
,
Ashleigh J.
Cartlidge
and
Peter D.
Matthews
*
School of Chemical & Physical Sciences, Keele University, Newcastle-under-Lyme, ST5 5BG, UK. E-mail: p.d.matthews@keele.ac.uk; Tel: +44 (0)1782 733188
First published on 27th February 2023
Abstract
With the advent of stricter environmental regulation, quantum dots based on cadmium, lead and other heavy metals have become anathema. III–V semiconductors constitute a promising alternative because they not only match but indeed surpass the optoelectronic properties of classical quantum dot systems. Despite this fact, III–V semiconductors are no panacea. Their exacting synthesis is a major hurdle hampering widespread adoption. Several groups have risen to this synthetic challenge, resulting in a plethora of publications. In this perspective, we compile the disparate routes to III–V quantum dots and concisely present them, with pertinent examples for each synthetic methodology. This has allowed us to identify gaps in the field, which we highlight as perspectives in the conclusion.
1. Introduction
Albeit almost half a century has passed since quantum dots (QD) were first formulated and characterised, they continue to garner significant attention.1 This is attributable to their highly tunable optoelectronic properties, where a change in shape, size, composition, or surface state fundamentally alters the emission profile of these nanomaterials. This characteristic, alongside their high colour purity and outstanding photostability, has seen them applied in numerous fields from electricity production and lighting to biomedical imaging and display screens.2II−VI semiconductors dominate the field of QDs due to their ease of synthesis and high-performance characteristics. However, the inherent toxicity of these materials has prompted an exhaustive search for alternatives.3 III–V semiconductors have captured the imagination of chemists the world over thanks to their large excitonic Bohr radius and direct band gap (Eg) (Table 1).4 Two rapidly growing areas of research are transition metal dichalcogenides and perovskite quantum dots, but these have yet to achieve the high photoluminescent quantum yields (PLQY) of their III–V counterparts.5
The large Bohr radius and direct band gap enable size quantisation effects to theoretically manifest over the whole visible and near-infrared range. With careful fine-tuning of reaction conditions during synthesis, greater control can be established over the size-dependent properties of these materials in comparison to their II–VI congeners.12
Several excellent reviews and book chapters have extensively covered the properties of these nanomaterials.13–15 Others are devoted to the conversion of these materials into devices, and detail the challenges of converting the high PLQY of III–V QDs (>90% is attainable) to a correspondingly high external quantum efficiency (EQE).16 Consequently, these lie beyond the purview of this review, which exclusively focuses on their synthesis.
Unfortunately, III–V QDs are not without their Achilles heel. They are notorious for their arduous synthesis, a challenge discussed in exhaustive detail by Heath and Shiang.17 In brief, the highly reactive, air-sensitive precursors combined with the strongly covalent nature of the resultant products hinder the separation of nucleation and growth. As a result, the QDs exhibit a broad particle size distribution with poorly controlled photoluminescence and emission properties.
These challenges have served as the impetus for extensive research into in situ optimising of III–V QD formation, in particular, InP (Fig. 1). This has regrettably resulted in a vast array of isolated research results with little cohesion between publications. Navigating this synthetic landscape can be a daunting task for the uninitiated. This review aims to chart the considerable progress accomplished, paying particular attention to developments within the last decade.
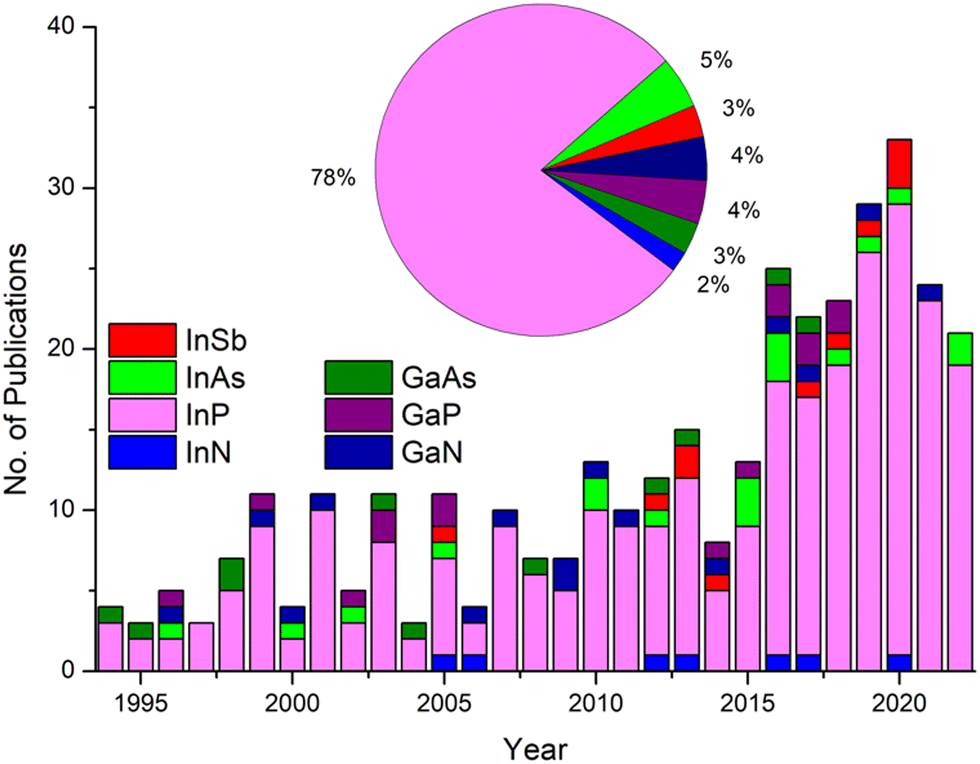 | ||
| Fig. 1 Publications on the syntheses of colloidally stable III–V QDs as a function of time, with a breakdown of publications by III–V type over the last 29 years inset. A representative total of 354 papers were analysed. | ||
For the purpose of this review, synthetic efforts are broadly divided into three categories: (a) binary reactions, where the cationic and anionic moieties are provided by separate precursors; (b) single source precursors (SSP), where the desired metal–pnictide bond is preformed and contained in a single constituent; and (c) magic-sized clusters (MSC), atomically precise nanoclusters which serve as stable intermediates of QDs. Pertinent examples are given for each category with a strong focus on InP, the most researched III–V QD (Fig. 1).18 Finally, a perspective on the future direction of the field is presented. Thus, we aim to provide the reader with a holistic, rather than exhaustive, overview of the field and equip them with the necessary tools to explore it further.
2. Binary methods
2.1 Miscellaneous reactions
Initial forays into the colloidal formation of III–V QDs were met with little success. Conventional aqueous colloidal routes proved particularly ill-suited to the hygroscopic group III salts.4 In addition, unlike II–VI QDs, group V elemental precursors do not readily lend themselves to semiconductor synthesis.19,20 Limited reports of the use of elemental phosphorus do exist, most notably a recent report utilising red phosphorus powder.21–24 However, these examples constitute the exception, not the rule, and commonly suffer from broad size distribution and poor PLQY.Moreover, simple hydride pnictogen precursors, such as PH3 and AsH3, are unfortunately gases at ambient conditions, adding an unwarranted layer of complexity to handling these exceptionally hazardous substances. Nevertheless, Buhro et al. established a solution-liquid–solid (SLS) method whereupon thermolysis of metal alkyls in the presence of arsine/phosphine gas in an organic solvent encourages nanoparticle formation (Fig. 2). As supersaturation of the liquid phase occurs, growth initiates at the solid–liquid boundary.25 Expanding this protocol, the Tilley group utilised solid hydrogen phosphide (PH)x formed from the decomposition of PBr3 with LiAlH4. The in situ formed, air-stable, solid phosphorus precursor was exposed to indium seed particles with trioctylphosphine (TOP) as the solvent to form InP wires.26
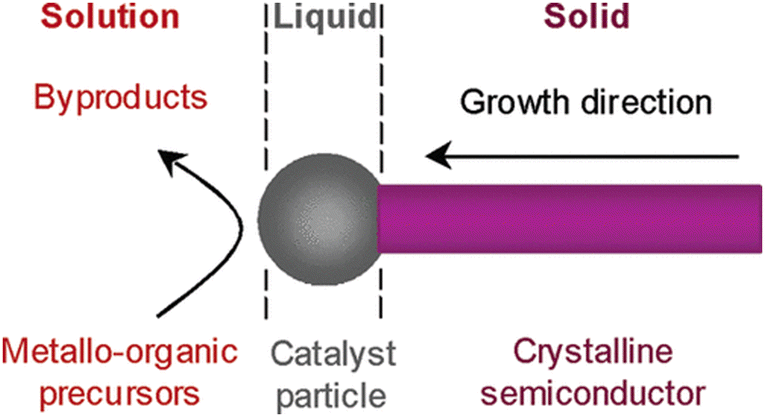 | ||
| Fig. 2 The reaction mechanism of the SLS method. Reproduced with permission from ref. 27. Copyright 2006 American Chemical Society. | ||
Furthermore, in situ generated phosphine gas (PH3), formed by adding acid (e.g., HCl, H2SO4) to M3P2 (M = Ca, Zn) under argon, can serve as a group V source. The resultant continuously produced gas is bubbled through a mixture of InCl3 and myristic acid dissolved in octadecene at 250 °C. Of note were the excellent optoelectronic properties and the high monodispersity of the resultant QDs.28 In a similar vein, in situ generated AsH3 and SbH3 have yielded InAs and InSb, respectively.29,30 Albeit both publications claimed high-quality nanoparticle formation, the high toxicity of the precursors cannot be overlooked.
As the field has continued to mature, binary reactions have come to crystallise into two main categories: (a) the dehalosilylation protocol, and (b) the zero oxidation state protocol. Dehalosilylation is by far the most widely implemented technique and constitutes the focus of the following section.
2.2 Dehalosilylation protocol
As highlighted in the previous section, it quickly became apparent that a drastically different synthetic approach to that of II–VI QDs was required. Significant inroads were made by taking advantage of silicon's natural inclination to form strong covalent bonds with halogens. The Barron and Wells groups adapted previous Chemical Vapor Deposition (CVD) methodologies to a thermolysis protocol for the growth of GaAs, InAs and InP.31,32 By mixing the appropriate group III halide (GaCl3 or InCl3) with a silylated pnictogen [As(SiMe3)3 or P(SiMe3)3] in solution at low temperatures a single precursor was formulated. Flame annealing of this intermediate at 650 °C under vacuum yielded high-purity bulk III–V semiconductors. The reactions were shown to proceed via adduct formation and subsequent elimination of Me3SiCl (Scheme 1).32,33 Small procedural changes allowed for alternative solvents to be utilised.34–37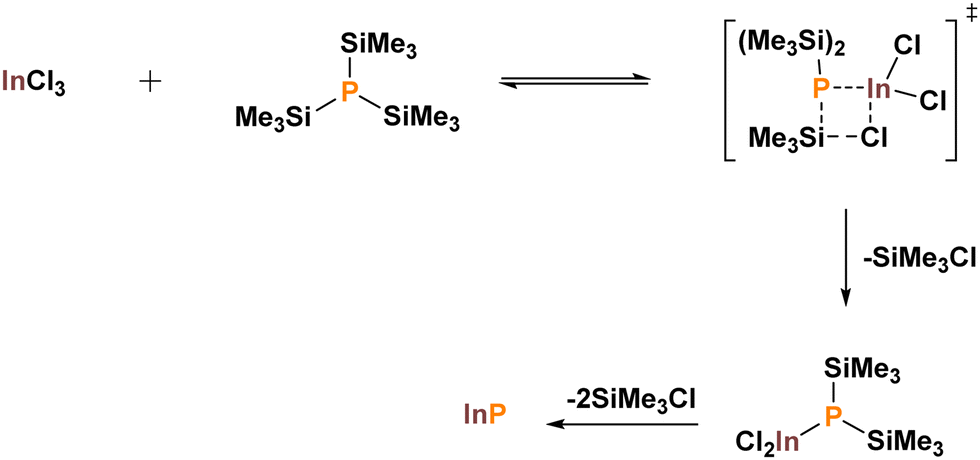 | ||
| Scheme 1 Stipulated mechanism of the dehalosilylation protocol highlighting the oligomeric intermediate. Adapted with permission from ref. 33. Copyright 1995 American Chemical Society. | ||
Following on from these seminal discoveries, the protocol has been extensively tweaked and optimised. Importantly flame annealing is no longer a prerequisite, as suitably crystalline nanoparticles can be obtained without it. Instead, steadily raising the solvent temperature is sufficient. In its current form, this protocol has been labelled as the heat-up method and alongside the hot-injection protocol (discussed in more detail in Section 3) represents over 80% of the reported literature.38
Coordinating solvents, such as the ubiquitous trioctylphosphine (TOP) and trioctylphosphine oxide (TOPO), have been probed as both reaction mediums and stabilising/solvating agents. Notable examples include the synthesis of InP QDs in either a mixture of TOP/TOPO or pure TOP using chloroindium oxalate and P(SiMe3)3. Control over particle size could be achieved by adjusting the ratio of In:P, a principle that has been extensively employed ever since.39 This methodology was later expanded to GaP and GaAs.40,41 These coordinating solvents quickly fell out of favour due to the long reaction times required (3–7 days) as well as the low size selectivity observed. As a side note, TOP itself was considered as a phosphorus precursor by employing indium nanoparticles to catalytically cleave the P–C bond. Unfortunately, the final product contained a mixture of metallic indium species and InP.42
Pushing past this frontier, the groups of Battaglia and Peng replaced the coordinating phosphino solvents/capping agents with non-coordinating alternatives containing fatty acid stabilising ligands. This resulted in markedly reduced reaction times and stimulated monodisperse QD formation.43 To date, this protocol remains the gold standard for monodisperse III–V QD synthesis and has been adapted to accommodate amines,44 fatty acids43,45 and/or alkylphosphines46,47 as stabilising ligands. In addition, numerous silylated pnictogens are amenable to this transformation. Particularly noteworthy is Sb(SiMe3)3 which yielded InSb QDs, albeit polydisperse, despite its extreme instability.48
The most crucial feature of this methodology is its adaptability. Judicious and careful fine-tuning of reaction parameters (temperature, precursor ratio etc.) allows for precise control over optoelectronic properties.
These landmark breakthroughs have established silylated pnictogens, especially P(SiMe3)3, as the foundation upon which the vast majority of research on III–V QDs is erected. However, P(SiMe3)3 is by no means a panacea. High pyrophoricity and unbridled reactivity interfere with size distribution and lead to colour saturation. II–VI QD systems respond well to a decrease in precursor consumption rate, with positive effects on size distribution and sample homogeneity.49–51 It was envisaged that additional steric crowding around the silicon atom would be sufficient to suppress the unbridled behaviour of P(SiMe3)3.
Triarylsilylphosphines of the type P[Si(C6H4–X)3]3 (X = H, Me, CF3, or Cl) were examined as precursors for InP synthesis by the Cossairt group.52 In all cases, the decreased polarisation of the P–Si bond, coupled with the added phenyl steric bulk, dramatically decreased the rate of nucleation. Unfortunately, this proved deleterious to size control. To try to address this issue, co-injection of P(SiMe3)3 and P(SiPh3)3 was performed. Nonetheless, monodispersity proved elusive once more.52 Additional reports on bulky R-groups on the silyl group do exist with modest improvements in size distribution.
When employing P(SiEt)3 and P(SitBu3)3, the quantum yields and colour saturation were improved with the resultant QDs being slightly more homogeneous in size.53 In addition, Joung et al. were able to grow larger InP QDs by substituting P(SiMe3)3 with P(SiMe2tBu)3 or P(SiMe2Ph)3. However, no improvements in size distribution were noted in this instance.54
A more radical approach involves replacing the labile –SiR3 moiety altogether with a less reactive –GeMe3 species. Although precursor depletion dropped by a factor of 4 during the initial stages of the reaction, little change was observed in particle size distribution. A similar experimental picture emerged when employing As(GeMe3)3 for InAs synthesis.55
The above reports proved successful in limiting group V precursor consumption by several orders of magnitude in some instances. Nevertheless, the resultant nanoparticles remained polydisperse casting doubt on the importance of conversion rates and precursor chemistry on size control.56 Instead, thermodynamically stable intermediates, commonly referred to as Magic Size Clusters (MSCs), are currently believed to be the major contributing factor to polydispersity. MSCs are discussed in more detail in Section 4.
2.3 Zero oxidation state reactions
Given the limitations of the dehalosilylation method, focus has shifted away from silylated precursors. The use of reductants allows for starting materials to be in the wrong oxidation state. For example, instead of using P(SiMe3)3 as a P3− synthon, which matches the oxidation state of phosphorus in InP, sources of P(+III) are used. To this end, sequential addition of PCl3 and LiEtBH3 to indium stearate at 40 °C, followed by heating to 250 °C, afforded monodisperse InP, as evidenced by powder diffraction.57 The same superhydride was employed for the co-reduction of Sb[N(SiMe3)2]3 and InCl3 resulting in InSb3.58 A later report adapted this methodology to chlorinated pnictogen precursors (AsCl3, SbCl3) yielding InAs and InSb, respectively.28The analogous reductant NaEtBH3 provides access to GaAs in squalene at 290 °C using hexadecylamine as the surfactant.59 This protocol was amenable to a range of arsine precursors [AsH3, As(SiMe3)3, and As(NMe2)3] but did require surface treatment with aqueous HCl to remove boron contaminants. Furthermore, excitonic features were observable only after molten salt flame annealing in the presence of GaI3.59
As an important side note, the above convoluted synthetic protocol stands as a testament to the challenges of synthesising colloidally stable GaAs QDs, which represent a fraction of total publications in the area (Fig. 1). The limited protocols reported in the literature based on single-source precursors,60 cation exchange61 or transmetallation62 either fail to exhibit excitonic features or appear to directly contradict each other where optoelectronic properties are concerned.63–65 However, recent concerted efforts have resulted in GaAs QDs displaying weak band edge emission upon outer shell doping with ZnSe.66
Deamination reactions represent another iteration of the wrong oxidation concept. QDs derived from aminopnictogen precursors like P(NR2)3 have been found to mirror the photoluminescence properties of their silylphosphine counterparts with little to no synthetic adjustments required.
Early reports utilising P(NMe2)3 and InCl3 relied on TOPO as a surface ligand, resulting in a broad size distribution.67 By adapting previous solvothermal protocols, Yang utilised InCl3 and P(NMe2)3 in oleylamine to generate highly monodisperse size-tunable QDs. The addition of ZnCl2 during synthesis proved vital for ensuring narrow size distributions, and it is worth noting the almost certain transamination of the P-precursor.68
As these species are in the wrong oxidation state, the mechanistic profile of these systems is markedly more complex than for dehalosilylation reactions (Scheme 2). The first step (α) involves adduct formation between InCl3 and the aminophosphine. Dissipation of the positive charge onto one of the nitrogen groups can occur through resonance. This leaves the amino group prone to nucleophilic attack by a second P(NR2)3 (β). The resultant InP intermediate has changed oxidation state from +III to +I. The phosphonium by-product, [P(NR2)4]+, which also forms, has oxidized from +III to +V. Finally, a reduction facilitated by two equivalents of P(NR2)3 yields InP and one more equivalent of [P(NR2)4]+ (γ).
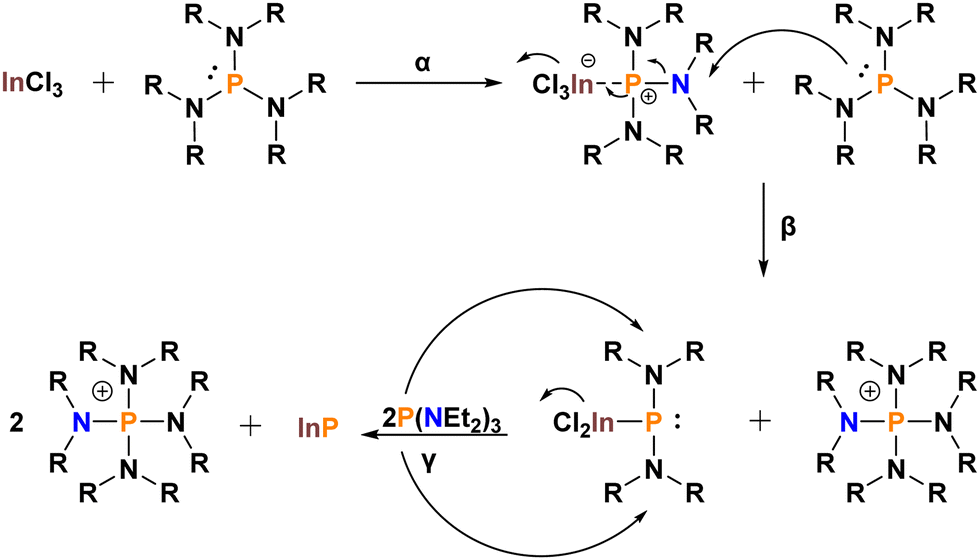 | ||
| Scheme 2 Proposed mechanism for InP formation from P[N(Et)2]3. | ||
The Tessier group, whose reported synthesis utilised P(NEt3)2, is credited with providing a facile method to tune QD size and hence their emissive properties.69 This was achieved by altering the halogen on the indium precursor (InX3, X = Cl, Br, I). Accordingly, when moving down the series of Cl, Br and I, larger nanoparticles were attained. Hence the emission colour changed respectively from red to yellow to green. Once more, doping with ZnCl2 proved essential in the formation of monodisperse QDs, with the authors attesting that the Zn atoms, which are postulated to passivate the surface by acting as Z-type ligands, are surface bound.70 However, deamination reactions can proceed in the absence of ZnCl2. A pertinent example is tetrahedron InP nanocrystals formed from InCl3 and P(NMe2)3 in oleylamine.71
The deamination protocol is not confined to InP with recent studies forming InAs from As(NR2)3 (R = Me, Et).72 However, the addition of a reductant is crucial to encourage the As0 → As3− conversion and as such it doesn’t follow the same mechanism as Scheme 2.72–74 A systematic study examined amino-P, diisobutylaluminium hydride (DIBAL-H), LiEt3BH, and alane N,N-dimethylethylamine (DMEA-AlH3) as potential reductants. The latter reagent led to the narrowest size distribution.75
Incidentally, zinc-based additives, such as zinc undecylenate44,76 and zinc stearate,77 amongst others, are known to provide a size-focusing effect to InP QDs for both silylated and amine group III-precursors. Several plausible explanations have been put forward including: (a) the formation of a Zn–P encounter complex which is less reactive than P(SiMe3)3, (b) the passivation of surface dangling bonds, or (c) increased solubility of the QDs.78 A similar effect is observed for InAs whilst utilising ZnCl2.70
In conclusion, aminopnictides represent an exciting development which could revolutionise the field. However, improvements are still required to achieve the size distribution and photoluminescence yield of the more established dehalosilylation route.70
3. Single-source precursors
Colloidal nanostructure formation is governed by two processes, nucleation and growth (Fig. 3). During nucleation, the anionic and cationic monomers combine into a new thermodynamically stable molecule or structure. Growth occurs by incorporating additional monomers on the surface of this core layer in one of two methods. First, any unreacted monomers from the nucleation are consumed. This is known as La Mer-type growth and is considered essential for narrow size distribution. Unfortunately, III–V QDs do not typically follow this pathway.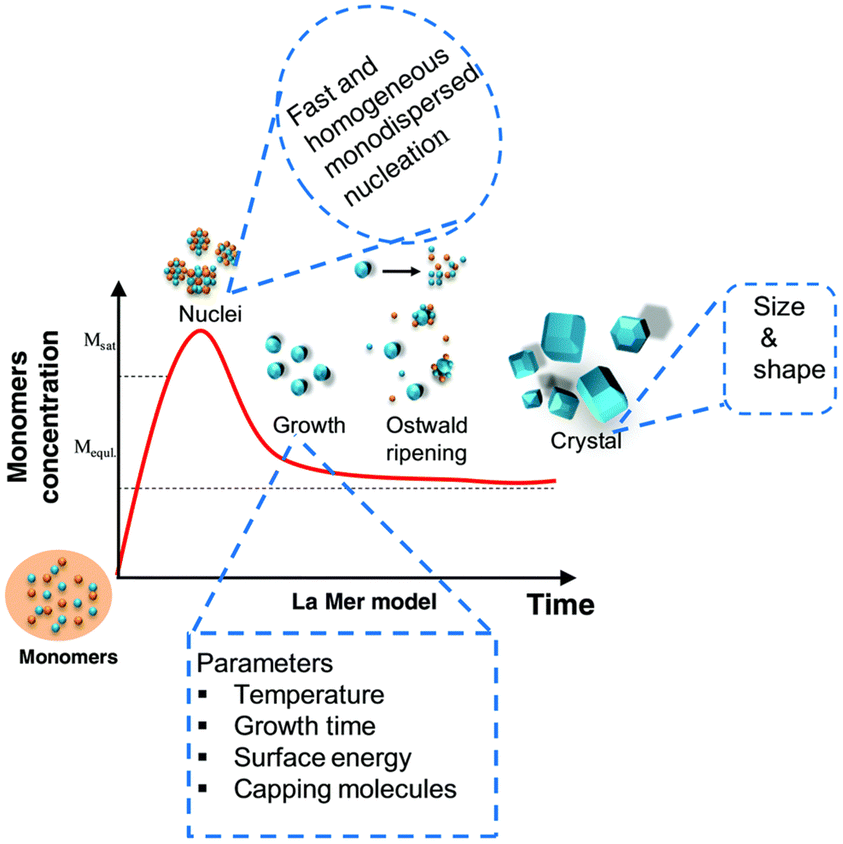 | ||
| Fig. 3 A graphical representation of the QD formation mechanism. Figure adapted with from ref. 79 with permission from the Royal Society of Chemistry. | ||
If the system has not reached thermodynamic equilibrium and the monomer reservoir is depleted, dissolution of the smallest nanoparticles occurs to produce monomers of variable composition and size. Recombination of these is known as Ostwald ripening, a poorly controlled process. For monodispersity to be achieved Ostwald ripening must be suppressed. In other words, clear temporal segregation between these two processes is necessary.80
This represents an exacting task for the binary systems discussed above. This is attributed to the highly reactive and strongly coordinated precursors normally used (Section 2.2), in addition to the covalent nature of the resultant III–V QDs.56,81–83 For instance, when synthesising InP, it is widely accepted the P(SiR3)3 precursor is all but depleted in the nucleation phase. Consequently, the growth stage is driven by an Ostwald ripening process.84 This results in a non-optimal, polydisperse range of QD sizes, which absorb and emit light over a broad range of wavelengths, with limited control. It is also common for these unstable precursors to form weakly coordinating complexes with the solvents, thus necessitating increased heat input. This further disrupts the delicate equilibrium between nucleation and growth.
A practical solution involves switching from one-pot, heat-up methodologies to hot-injection protocols.85 Herein, a room-temperature solution of the precursors is rapidly injected into the hot reaction solvent resulting in a nucleation burst as the monomers aggregate to form clusters. Within seconds, a steep temperature drop followed by a decline in monomer concentration terminates the nucleation stage with the growth process taking over. This approach represents the most common method for the synthesis of size-selective QDs regardless of elemental composition. However, studies have cast doubt on its ability to separate nucleation and growth in III–V systems.86
More recently, slow continuous injection of pnictide precursors to preheated solutions of the appropriate Group V metals has shown particular promise.87–89 This process ensures monomer depletion does not occur and significantly decreases growth rate. Judicious choice of injection rate allows for a range of sizes to be obtained.
Another elegant approach to disentangle these two processes is by utilising single source precursors (SSPs). In these inorganic compounds, the desired metal pnictide bond is already present and framed by several leaving groups. Decomposition, triggered either thermally or chemically, exposes the core M–E layer upon which growth can occur. In Section 3.1, we present a range of SSPs to InP and InAs QDs whose composition and exact stoichiometry have been carefully tailored. SSPs have been extensively employed for the generation of pnictide nitrides and Section 3.2 is devoted to this area. Finally, a comparison of the binary (Section 1) and SSP methods are presented in Section 3.3.
3.1 SSPs for group III phosphines and arsines
One of the earliest examples of a III–V SSP that was reported is [H2GaE(SiMe3)2]3 (E = P, As).90 Pyrolysis of these species leads to nanocrystalline GaP and GaAs, respectively. Other reports examined the alcoholysis of {R(Cl)In[μ-P(SiMe3)2]}2, prepared from R2InCl and P(SiMe3)3, giving a mixture of InP and In2O3.60 Likewise, the reaction of metal alkyls with silylated phosphines produced adduct compounds. Thermolysis of these precursors led to metal phosphides, although some metal impurities were also observed.91 In an analogous fashion, Wells et al. reacted GaCl3 with As(SiMe3)3 to prepare (AsCl3Ga2)n. A subsequent wash in hot hydrocarbons and heating up to 410 °C under an inert atmosphere led to GaAs nanoparticles.92Thermolysis has also been extensively employed with diorganopnictides. Consequently, GaAs and GaP 7 nm nanoparticles were obtained by the decomposition of In(PtBu2)3 and Ga(PtBu2)3 in 4-ethylpyridine.93 Experiments utilising more conventional solvents, such as TOPO, were unsuccessful. However, the use of trioctylamine as the stabilising ligand was able to generate QDs of ca. 8 nm diameter.94 Analogously, the thermal decomposition of (tBu2AsGaMe2)2 in hot hexadecylamine allowed for the preparation of GaAs nanoparticles.95 In all the above examples, the presence of elemental metal or metal oxide impurities was evident by powder XRD. In contrast, thermolysis of (tBu2AsInEt2)2 in hot hexadecylamine gave InAs particles ca. 9 nm in diameter with an XRD pattern devoid of impurities.94
3.2 SSPs in the synthesis of group III nitrides
Colloidally stable pnictonitrides are an underrepresented class of III–V QDs despite their potential applications in optoelectronic devices, solid-state lighting and laser systems96 The issues of covalent bonding and limited precursors which plague III–V QDs are exacerbated by the metastability of pnictide nitrides, with InN degrading at 500–550 °C.97 In addition, the strong N–Si bond precludes the dehalosilylation method as a viable synthesis of GaN,98 although this has been reported for InN.99 Despite the challenges solution-based routes pose, the 21st century has witnessed a resurgence of interest in the field with several solvothermal100–103 and pyrolysis104,105 protocols established for nanocrystal formation. Indium nitride has commonly been formed by the nitridation of In2O3 nanoparticles by treatment with NH3 at 500 °C,106 or through a complex system of alkali metal amides.107More recently, azide SSPs have come to the fore as nitride precursors. Pioneering work by Dingman et al. utilised an alkyl polymeric structure [(R2InN3)n, (R = alkyl)] in Lewis basic solvents. Thermolysis of this precursor in the presence of a reducing agent (H2NNMe2) yielded InN nanoparticles via an SLS mechanism (Section 2.1).108 Similarly, thermolysis of InN3(CH2CH2CH2NMe2)2 in TOPO generates InN with particle sizes approximately 2–10 nm in diameter.109
InN has also been generated by high-pressure or thermolytic decomposition of an azide intermediate derived from InBr3 and NaN3. For the thermolytic protocol trioctylamine was required as a capping agent which negatively affected the synthesis, as indium metal was detectable in the XRD patterns. On the other hand, the high-pressure route produced markedly smaller and more crystalline nanoparticles.110
Group III nitrides can also be prepared with non-azide SSPs. The purported syntheses of GaN, InN and AlN have been achieved by thermal degradation of M(H2NCONH2)3Cl3, (M = Al, In, Ga) using trioctylamine. However, the resultant nanoparticles were poorly characterised.104
Finally, gallium azides, GaN, are particularly amenable to this protocol. Hence, [Et2Ga(N3)]3, (N3)2Ga[(CH2)3NMe2] and (Et3N)Ga(N3)3 all yielded exceptionally large QDs, circa 200 nm in diameter. Their crystallinity however was not deduced despite exhibiting blue band emission patterns.111
3.3 Outlook on binary approaches versus SSPs
Other examples of SSPs do exist for III–V QDs.19 However, upon casting a cursory glance through these accounts, the disjointed nature of available precursors is revealed. To the best of our knowledge, no systematic approach to generating them has been developed unlike for binary methods.Nonetheless, combining the In and P source into a single molecule often results in a stable solid under ambient conditions, a desirable trait for industrial applications.38 In addition, the preformed bonds between the metal and chalcogenide ensure fewer defects are present within the crystal matrix. Finally, single-source precursors simplify the nucleation process by slowing down the conversion rate of the group V precursor, perhaps the greatest limitation of binary reactions.
Despite these notable advantages over binary approaches, SSPs present their own unique set of limitations. The size-dependent optical properties of QDs are influenced by the stoichiometric ratios of the precursors. In single-source precursors the stoichiometry is predetermined and cannot be influenced easily. In addition, SSPs fall victim to the very issue they purport to tackle. Albeit quantum dot preparation is streamlined, single-source precursor synthesis is markedly more complex and time consuming than binary systems.
Deciding between the two options is truly a delicate balancing act with advantages and disadvantages on both sides. The onus falls on the researcher to choose the method which best fits their needs.
4. Magic size clusters
As previously mentioned in Section 2, recent reports have cast doubt on the importance of precursor conversion kinetics.56 Instead, thermodynamically stable intermediate clusters are postulated as the rate-limiting step (Fig. 4).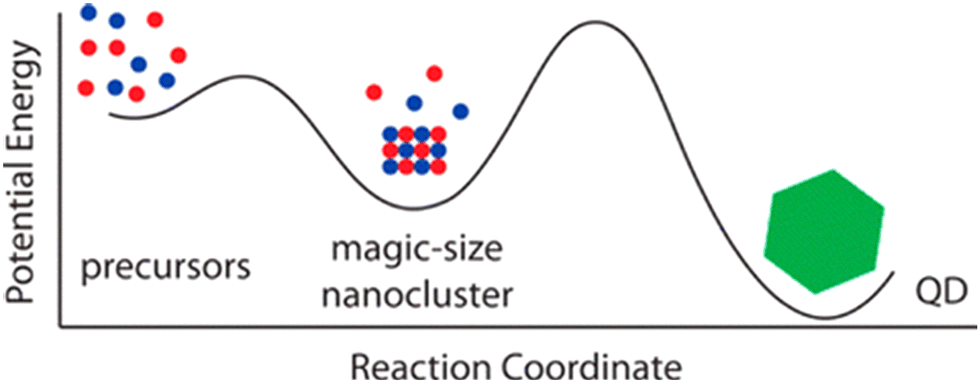 | ||
| Fig. 4 Graphical representation illustrating MSCs as reaction intermediates in the synthesis of QDs. Figure adapted with permission from ref. 84. Copyright 2015 American Chemical Society. | ||
These so-called Magic Size Clusters (MSCs) consist of a few to hundreds of atoms and are defined as small-sized nanocrystals (<2 nm).112 Their rate of formation and dissolution is hard to control under the typical high-temperature regimes used for QDs (hot-injection). As they are kinetically persistent up to 300 °C they can directly interfere with QD formation.113 Thus, the isolation and characterization of these nanomaterials are essential to understanding the growth mechanisms of III–V semiconductors.
4.1 Mechanistic insights into MSCs
An early study into these systems was conducted in 2001 by Nozik et al. by employing a chloroindium oxalate complex and P(SiMe3)3 at low temperatures (100–220 °C) to impede QD growth. The resultant nanoparticles showed a narrow excitonic absorption peak at 400 nm.114 Using these observations as a springboard Xie et al. synthesised the most studied MSC with an excitonic absorption of 386 nm utilising In(OAc)3, myristic acid and P(SiMe3)3.115 Subsequently, they performed an exhaustive kinetic study using P(SiMe3)3 and In(OAc)3 as the precursors. Variables assessed included reaction temperature (100 < T < 208 °C), surfactant type and precursor ratio. Interestingly, regardless of surface ligand, the UV-vis peak signature was always identical signifying a single species.In a seminal publication, the exact core structure and surface of the InP-386 were unambiguously deduced by single crystal X-ray diffraction by the Cossairt group.116 Thus, the cluster is composed of a [In21P20]3+ core with average In–P bond lengths of 2.528 Å. Encapsulating the core are 16 additional indium atoms coordinating with surface-positioned phosphorus atoms. Completing the structure are the 51 carboxylate ligands in a variety of binding modes.
Various modifications on the InP-386 MSC have been reported. The addition of oleylamine to In(OAc)3 and P(SiMe3)3 allowed for the isolation of two MSCs located at 365 nm (T = 45–70 °C) and 395 nm (T = 170 °C).117 On a related note, even larger MSCs could be generated upon the introduction of 3–6 equivalents of octylamine to In(My)3 and P(SiMe3)3 (λ = 450 nm) at low temperatures (158–208 °C).114 Further increasing the equivalents of octylamine relative to In(My)3 led to continuous growth of QDs.82 Furthermore, the post-synthetic addition of benzylamines to the In37P20(O2CR)51 previously crystallographically analysed, led to the red-shifting of the 386 nm cluster to 404 nm. Additional equivalents of primary amine at 160 °C failed to influence the structure any further.118
Finally, efforts have been undertaken by the Kwon group to integrate heterogeneous atoms into MSCs.117 Consequently, Zn and Cl atoms were introduced either directly during MSC synthesis or by post-synthetic modification through ion exchange or doping. As with QDs, notable differences in the optical properties of these MSCs can be seen in comparison to the heteroatom-free MSCs. Studies into III–V MSCs have been largely restricted to InP but limited reports on other III–V MSCs do exist.119
4.2 MSCs as precursors to QDs
With a comprehensive mechanistic understanding at hand, the focus has shifted to employing MSCs as precursors for QD synthesis with efforts mainly focused on the InP-386 MSC. Preliminary data points to superior monodispersity and sharper excitonic peaks when employing MSCs rather than molecular precursors.120 However, additional research is required to corroborate this hypothesis.In a typical synthesis, purified MSCs are injected into a hot solution of an appropriate solvent. Interestingly, the size of the resultant QDs can be increased simply by increasing the concentration of MSC precursors.67 Two competing growth mechanisms have been proposed: (a) a dissolution process whereupon the MSCs decompose to monomers and either deposit onto MSC seeds or recombine into larger clusters, or (b) an agglomeration process, where MSCs coalesce to form QDs.121
Concrete evidence has been presented in support of the first growth mechanism. For example, carboxylate additives promote the dissolution of InP-386 to monomer reserves. Recombination follows to form Quantum Dots.122
Where temperature is concerned, a detailed study was conducted on InP nanoparticles using HMy or In(My)3 as additives. MSC dissolution was observed within the entire temperature range examined. At temperatures below or equal to 150 °C this decomposition competed with InP QD growth leading to broad particle sizes. On the other hand, higher temperatures promoted the formation of stable and soluble monomers which form the nucleus for QD growth.122
In our group, we have recently employed Ph2PSiMe3 as a dopant for the acid-free growth of InP MSCs. The use of this species has allowed for the isolation of MSCs with markedly red-shifted UV signatures (430–490 nm). Notably, growth occurred at 100 °C in toluene, extremely mild reaction conditions for such high UV signatures.123
5. Conclusion and perspectives
In this short perspective, we have highlighted the difficulties associated with III–V quantum dot synthesis. A plethora of synthetic protocols have been conceived and implemented over the past 30 years to respond to these challenges. Thus, binary and single-source methods have matured and been refined to a point where monodispersity and respectable quantum yields are par for the course. Indeed, one may be excused for assuming the field has reached saturation point. However, progress is far from being exhausted with recent innovations utilising MSCs. Albeit at an infantile stage these techniques promise to revolutionise both the synthesis and our mechanistic understanding of QD formation. It is evident many exciting developments are yet to come. In the segment below we present the potential form these may take:1. As highlighted in Fig. 1, a staggering 80–90% of reported literature on wet chemistry QDs focuses on indium pnictides. The research is further skewed towards InP which has amassed 3/4 of total publications. We strongly believe concerted efforts towards developing other III–V QD systems are bound to be fruitful.
2. The importance of controlled precursor consumption cannot be overstated. High monomer reservoirs during the growth phase of QDs are essential to achieve La Mer-like growth and hence narrow size dispersions. The current group III precursors of choice, silylated pnictides, are not fit for purpose. Attempts to suppress this reactivity by utilising bulky silyl groups or germylphosphines have not been successful. An emerging solution is the use of dialkylaminophosphines P(NR2)3 (R = Me, Et) discussed in Section 2.3. Further exploring this area will undoubtedly prove fruitful in the years to come.
3. The major challenge of current synthetic strategies is maintaining a high enough monomer reservoir for growth to occur. Two elegant solutions to this include a continuous external supply of monomers through flow chemistry for example or weak complexation to external agents which allows for slow release of monomers during growth.
4. In these authors’ viewpoint, MSCs represent the greatest development of this decade in the field. They present a mechanistic window into QD growth, their structural properties, and any surface coordination modes, and they combine the ease of precursor ratio manipulation (hence different sizes) of binary methods with the preformed metal–pnictogen bond and clear divide between nucleation and growth which is easier to achieve with SSPs. Undoubtedly many exciting discoveries are yet to be uncovered. Mind this space.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the support of EPSRC grant EP/V043412/1 (TAG, PDM), Keele University (AJC) and the UK Government and European Union as contributors to the Smart Energy Network Demonstrator, ERDF project number 32R16P00706 (PDM) for funding.References
- A. I. Ekimov and A. A. Onushchenko, J. Exp. Theor. Phys., 1981, 34, 345 Search PubMed.
- P. V. Joglekar, D. J. Mandalkar, M. A. Nikam, N. S. Pande and A. Dubal, Int. J. Res. Advent Technol., 2019, 7, 510 CrossRef.
- L. Hu, H. Zhong and Z. He, Colloids Surf., B, 2021, 200, 111609 CrossRef CAS.
- M. M. Green, Curr. Opin. Solid State Mater. Sci., 2002, 6, 355–363 CrossRef CAS.
- For selected publications, see: (a) H. He, S. Mei, Z. Wen, D. Yang, B. Yang, W. Zhang, F. Xie, G. Xing and R. Guo, Small, 2021, 18, 2103527 CrossRef PubMed; (b) J. H. Han, M. Kwak, Y. Kim and J. Cheon, Chem. Rev., 2018, 118, 6151–6188 CrossRef CAS; (c) K. Huang, J. Liu, J. Yuan, W. Zhao, K. Zhao and Z. Zhou, J. Mater. Chem. A, 2023, 11, 4487–4509 RSC; (d) P. D. Matthews, D. J. Lewis and P. O’Brien, J. Mater. Chem. A, 2017, 5, 17135–17150 RSC; (e) P. D. Matthews, P. D. McNaughter, D. J. Lewis and P. O'Brien, Chem. Sci., 2017, 8, 4177–4187 RSC; (f) S. V. Kershaw, A. S. Susha and A. L. Rogach, Chem. Soc. Rev., 2013, 42, 3033 RSC; (g) Y. Bai, M. Hao, S. Ding, P. Chen and L. Wang, Adv. Mater., 2021, 34, 2105958 CrossRef.
- Z. Liu, L. M. Janes, M. Saniepay and R. Beaulac, Chem. Mater., 2018, 30, 5435–5443 CrossRef CAS.
- H. J. Byun, W. S. Song and H. Yang, Nanotechnology, 2011, 22, 235605 CrossRef PubMed.
- T. Puangmali, M. Califano and P. Harrison, J. Phys. Chem. C, 2010, 114, 6901–6908 CrossRef CAS.
- K. Zhu, J. Shi and L. Zhang, Solid State Commun., 1998, 107, 79–84 CrossRef CAS.
- B. Chen, D. Li and F. Wang, Small, 2020, 16, 2002454 CrossRef CAS PubMed.
- M. Califano and P. Rodosthenous, ACS Appl. Mater. Interfaces, 2019, 11, 640–646 CrossRef CAS.
- M. Green, Semicond. Quantum Dots, 2014, 53–76 Search PubMed.
- U. W. Pohl, S. Rodt and A. Hoffmann, Semicond. Nanostruct., 2008, 269–299 Search PubMed.
- A. Schliwa, G. Hönig and D. Bimberg, Multi-Band Effective Mass Approximations, 2014, pp.57–85 Search PubMed.
- A. Mittelstädt, A. Schliwa and P. Klenovský, Light: Sci. Appl., 2022, 11, 1–14 CrossRef.
- For selected publications, see: (a) H. Moon, C. Lee, W. Lee, J. Kim and H. Chae, Adv. Mater., 2019, 31, 1804294 CrossRef; (b) X. Dai, Y. Deng, X. Peng and Y. Jin, Adv. Mater., 2017, 29, 1607022 CrossRef; (c) Y. Shu, X. Lin, H. Qin, Z. Hu, Y. Jin and X. Peng, Angew. Chem., Int. Ed., 2020, 59, 22312–22323 CrossRef CAS PubMed; (d) Z. Cui, D. Yang, S. Qin, Z. Wen, H. He, S. Mei, W. Zhang, G. Xing, C. Liang and R. Guo, Adv. Opt. Mater., 2022, 2202036 Search PubMed.
- J. R. Heath and J. J. Shiang, Chem. Soc. Rev., 1998, 27, 65–71 RSC.
- B. M. May, M. F. Bambo, S. S. Hosseini, U. Sidwaba, E. N. Nxumalo and A. K. Mishra, RSC Adv., 2022, 12, 11216–11232 RSC.
- M. Green, Semicond. Quantum Dots, 2014, 224–251 RSC.
- D. Battaglia and X. Peng, Nano Lett., 2002, 2, 1027–1030 CrossRef CAS.
- P. Yan, Y. Xie, W. Wang, F. Liu and Y. Qian, J. Mater. Chem., 1999, 9, 1831–1833 RSC.
- S. Carenco, M. Demange, J. Shi, C. Boissière, C. Sanchez, P. Le Floch and N. Mézailles, Chem. Commun., 2010, 46, 5578–5580 RSC.
- U. T. D. Thuy, T. T. T. Huyen, N. Q. Liem and P. Reiss, Mater. Chem. Phys., 2008, 112, 1120–1123 CrossRef CAS.
- E. Bang, Y. Choi, J. Cho, Y. H. Suh, H. W. Ban, J. S. Son and J. Park, Chem. Mater., 2017, 29, 4236–4243 CrossRef CAS.
- T. J. Trentler, K. M. Hickman, S. C. Goel, A. M. Viano, P. C. Gibbons and W. E. Buhro, Science, 1995, 270, 1791 CrossRef CAS.
- T. H. Lim, S. Ravi, C. W. Bumby, P. G. Etchegoin and R. D. Tilley, J. Mater. Chem., 2009, 19, 4852–4856 RSC.
- F. Wang, A. Dong, J. Sun, R. Tang, H. Yu and W. E. Buhro, Inorg. Chem., 2006, 45, 7511–7521 CrossRef CAS.
- T. Zhao, N. Oh, D. Jishkariani, M. Zhang, H. Wang, N. Li, J. D. Lee, C. Zeng, M. Muduli, H. J. Choi, D. Su, C. B. Murray and C. R. Kagan, J. Am. Chem. Soc., 2019, 141, 15145–15152 CrossRef CAS PubMed.
- J. Zhang and D. Zhang, CrystEngComm, 2010, 12, 591–594 RSC.
- A. Maurice, M. L. Haro, B. Hyot and P. Reiss, Part. Part. Syst. Charact., 2013, 30, 821 CrossRef.
- R. L. Wells, C. G. Pitt, A. T. Mcphail, A. P. Purdy, S. Shafieezad and R. B. Hallock, Chem. Mater., 1989, 1, 4–6 CrossRef CAS.
- M. D. Healy, P. E. Laibinis, P. D. Stupik and A. R. Barron, J. Chem. Soc., Chem. Commun., 1989, 359–360 RSC.
- R. L. Wells, S. R. Aubuchon, S. S. Kher, M. S. Lube and P. S. White, Chem. Mater., 1995, 7, 793–800 CrossRef CAS.
- M. A. Olshavsky, A. N. Goldstein and A. P. Alivisatos, J. Am. Chem. Soc., 1990, 112, 9438–9439 CrossRef CAS.
- H. Uchida, C. J. Curtis and A. J. Nozik, J. Phys. Chem., 1991, 95, 5382–5384 CrossRef CAS.
- L. D. Potter, A. A. Guzelian, A. P. Alivisatos and Y. Wu, J. Chem. Phys., 1995, 103, 4834–4840 CrossRef CAS.
- H. Uchida, T. Matsunaga, H. Yoneyama, T. Sakata, H. Mori and T. Sasaki, Chem. Mater., 1993, 5, 716–719 CrossRef CAS.
- H. A. Nguyen, F. Y. Dou, N. Park, S. Wu, H. Sarsito, B. Diakubama, H. Larson, E. Nishiwaki, M. Homer, M. Cash and B. M. Cossairt, Chem. Mater., 2022, 34, 6296–6311 CrossRef CAS.
- M. Tomaselli, J. L. Yarger, M. Bruchez, R. H. Havlin, D. De Graw, A. Pines and A. P. Alivisatos, J. Chem. Phys., 1999, 110, 8861–8864 CrossRef CAS.
- O. I. Mićić and A. J. Nozik, J. Lumin., 1996, 70, 95–107 CrossRef.
- O. I. Mićić, J. Sprague, Z. Lu and A. J. Nozik, Appl. Phys. Lett., 1996, 68, 3150–3152 CrossRef.
- P. K. Khanna, K. W. Jun, K. B. Hong, J. O. Baeg and G. K. Mehrotra, Mater. Chem. Phys., 2005, 92, 54–58 CrossRef CAS.
- D. Battaglia and X. Peng, Nano Lett., 2002, 2, 1027–1030 CrossRef CAS.
- S. Xu, J. Ziegler and T. Nann, J. Mater. Chem., 2008, 18, 2653–2656 RSC.
- L. Li and P. Reiss, J. Am. Chem. Soc., 2008, 130, 11588–11589 CrossRef CAS PubMed.
- O. I. Mićić, C. J. Curtis, K. M. Jones, J. R. Sprague and A. J. Nozik, J. Phys. Chem., 1994, 98, 4966–4969 CrossRef.
- A. A. Guzelian, J. E. B. Katari, A. V. Kadavanich, U. Banin, K. Hamad, E. Juban, A. P. Alivisatos, R. H. Wolters, C. C. Arnold and J. R. Heath, J. Phys. Chem., 1996, 100, 7212–7219 CrossRef CAS.
- C. M. Evans, S. L. Castro, J. J. Worman and R. P. Raffaelle, Chem. Mater., 2008, 20, 5727–5730 CrossRef CAS.
- S. Abe, R. K. Čapek, B. De Geyter and Z. Hens, ACS Nano, 2012, 6, 42–53 CrossRef CAS PubMed.
- J. S. Owen, E. M. Chan, H. Liu and A. P. Alivisatos, J. Am. Chem. Soc., 2010, 132, 18206–18213 CrossRef CAS PubMed.
- J. Van Embden and P. Mulvaney, Langmuir, 2005, 21, 10226–10233 CrossRef CAS PubMed.
- D. C. Gary, B. A. Glassy and B. M. Cossairt, Chem. Mater., 2014, 26, 1734–1744 CrossRef CAS.
- H. B. Chandrasiri, E. B. Kim and P. T. Snee, Inorg. Chem., 2020, 59, 15928–15935 CrossRef CAS PubMed.
- S. Joung, S. Yoon, C. S. Han, Y. Kim and S. Jeong, Nanoscale Res. Lett., 2012, 7, 93 CrossRef.
- D. K. Harris and M. G. Bawendi, J. Am. Chem. Soc., 2012, 134, 20211–20213 CrossRef CAS.
- D. Franke, D. K. Harris, L. Xie, K. F. Jensen and M. G. Bawendi, Angew. Chem., Int. Ed., 2015, 127, 14507–14511 CrossRef.
- Z. Liu, A. Kumbhar, D. Xu, J. Zhang, Z. Sun and J. Fang, Angew. Chem., Int. Ed., 2008, 47, 3540–3542 CrossRef CAS.
- W. Liu, A. Y. Chang, R. D. Schaller and D. V. Talapin, J. Am. Chem. Soc., 2012, 134, 20258–20261 CrossRef CAS PubMed.
- V. Srivastava, W. Liu, E. M. Janke, V. Kamysbayev, A. S. Filatov, C. J. Sun, B. Lee, T. Rajh, R. D. Schaller and D. V. Talapin, Nano Lett., 2017, 17, 2094–2101 CrossRef CAS PubMed.
- T. Douglas and K. H. Theopold, Inorg. Chem., 1991, 30, 594–596 CrossRef CAS.
- B. J. Beberwyck and A. P. Alivisatos, J. Am. Chem. Soc., 2012, 134, 19977–19980 CrossRef CAS.
- J. Lauth, T. Strupeit, A. Kornowski and H. Weller, Chem. Mater., 2013, 25, 1377–1383 CrossRef CAS.
- L. Butler, G. Redmond and D. Fitzmaurice, J. Phys. Chem., 1993, 97, 10750–10755 CrossRef CAS.
- M. A. Malik, P. O’Brien, S. Norager and J. Smith, J. Mater. Chem., 2003, 13, 2591–2595 RSC.
- H. Uchida, C. J. Curtis, P. V. Kamat, K. M. Jones and A. J. Nozik, J. Phys. Chem., 1992, 96, 1156–1160 CrossRef CAS.
- J. P. Park, J.-J. Lee and S.-W. Kim, J. Am. Chem. Soc., 2016, 138, 16568–16571 CrossRef CAS PubMed.
- T. Matsumoto, S. Maenosono and Y. Yamaguchi, Chem. Lett., 2004, 33, 1492–1493 CrossRef CAS.
- W. S. Song, H. S. Lee, J. C. Lee, D. S. Jang, Y. Choi, M. Choi and H. Yang, J. Nanoparticle Res., 2013, 15, 1750 CrossRef.
- M. D. Tessier, D. Dupont, K. De Nolf, J. De Roo and Z. Hens, Chem. Mater., 2015, 27, 4893–4898 CrossRef CAS.
- D. Zhu, F. Bellato, H. Bahmani Jalali, F. Di Stasio, M. Prato, Y. P. Ivanov, G. Divitini, I. Infante, L. De Trizio and L. Manna, J. Am. Chem. Soc., 2022, 144, 1 CrossRef.
- K. Kim, D. Yoo, H. Choi, S. Tamang, J. H. Ko, S. Kim, Y. H. Kim and S. Jeong, Angew. Chem., Int. Ed., 2016, 55, 3714–3718 CrossRef CAS.
- V. Grigel, D. Dupont, K. De Nolf, Z. Hens and M. D. Tessier, J. Am. Chem. Soc., 2016, 138, 13485–13488 CrossRef CAS.
- R. Tietze, R. Panzer, T. Starzynski, C. Guhrenz, F. Frenzel, C. Würth, U. Resch-Genger, J. J. Weigand and A. Eychmüller, Part. Part. Syst. Charact., 2018, 35, 1800175 CrossRef.
- V. Srivastava, E. M. Janke, B. T. Diroll, R. D. Schaller and D. V. Talapin, Chem. Mater., 2016, 28, 6797–6802 CrossRef CAS.
- V. Srivastava, E. Dunietz, V. Kamysbayev, J. S. Anderson and D. V. Talapin, Chem. Mater., 2018, 30, 3623–3627 CrossRef CAS.
- A. M. Nightingale and J. C. Demello, J. Mater. Chem. C, 2016, 4, 8454–8458 RSC.
- S. Koh, T. Eom, W. D. Kim, K. Lee, D. Lee, Y. K. Lee, H. Kim, W. K. Bae and D. C. Lee, Chem. Mater., 2017, 29, 6346–6355 CrossRef CAS.
- N. Kirkwood, A. De Backer, T. Altantzis, N. Winckelmans, A. Longo, F. V. Antolinez, F. T. Rabouw, L. De Trizio, J. J. Geuchies, J. T. Mulder, N. Renaud, S. Bals, L. Manna and A. J. Houtepen, Chem. Mater., 2020, 32, 557–565 CrossRef CAS.
- W. M. Girma, M. Z. Fahmi, A. Permadi, M. A. Abate and J. Y. Chang, J. Mater. Chem. B, 2017, 5, 6193–6216 RSC.
- N. T. K. Thanh, N. Maclean and S. Mahiddine, Chem. Rev., 2014, 114, 7610–7630 CrossRef CAS.
- H. Bahmani Jalali, L. De Trizio, L. Manna and F. Di Stasio, Chem. Soc. Rev., 2022, 51, 9861–9881 RSC.
- Y. Kim, J. H. Chang, H. Choi, Y.-H. Kim, W. K. Bae and S. Jeong, Chem. Sci., 2020, 11, 913–922 RSC.
- T. Kim, D. Shin, M. Kim, H. Kim, E. Cho, M. Choi, J. Kim, E. Jang and S. Jeong, ACS Energy Lett., 2022, 8, 447–456 CrossRef.
- D. C. Gary, M. W. Terban, S. J. L. Billinge and B. M. Cossairt, Chem. Mater., 2015, 27, 42 CrossRef.
- O. M. B. Lutfan Sinatra and Jun Pan, Mater. Matters, 2017, 12, 3–7 Search PubMed.
- D. C. Gary, M. W. Terban, S. J. L. Billinge and B. M. Cossairt, Chem. Mater., 2015, 27, 1432–1441 CrossRef CAS.
- D. Franke, D. K. Harris, O. Chen, O. T. Bruns, J. A. Carr, M. W. Wilson and M. G. Bawendi, Nat. Commun., 2016, 7(1), 12749 CrossRef CAS PubMed.
- S. Tamang, S. Lee, H. Choi and S. Jeong, Chem. Mater., 2016, 28, 8119–8122 CrossRef CAS.
- T. Kim, S. Park and S. Jeong, Nat. Commun., 2021, 12, 3013 CrossRef PubMed.
- J. F. Janik, J. Am. Chem. Soc., 1998, 120, 532–537 CrossRef CAS.
- S. T. Barry, S. Belhumeur and D. S. Richeson, Organometallics, 1997, 16, 3588–3592 CrossRef CAS.
- R. L. Wells, R. B. Hallock, A. T. McPhail, C. G. Pitt and J. D. Johansen, Chem. Mater., 1991, 3, 381–382 CrossRef CAS.
- M. Green and P. O’Brien, Chem. Commun., 1998, 2459–2460 RSC.
- Y. H. Kim, Y. Wook Jun, B. H. Jun, S. M. Lee and J. Cheon, J. Am. Chem. Soc., 2002, 124, 13656–13657 CrossRef CAS PubMed.
- M. A. Malik, M. Afzaal, P. O’Brien, U. Bangert and B. Hamilton, Mater. Sci. Technol., 2004, 20, 959–963 CrossRef CAS.
- Z. Chen, C. Sun, W. Guo and Z. Chen, in Nonmagnetic and Magnetic Quantum Dots, ed. V. N. Stavrou, IntechOpen, London, 2018, ch. 4 Search PubMed.
- Q. Guo, O. Kato and A. Yoshida, J. Appl. Phys., 1993, 73, 7969–7971 CrossRef CAS.
- K. Sardar and C. N. R. Rao, Adv. Mater., 2004, 16, 425–429 CrossRef CAS.
- M. R. Greenberg, G. A. Smolyakov, Y.-B. Jiang, T. J. Boyle and M. Osiński, SPIE Proc., 2006, 6096, 60960D CrossRef.
- J. C. Hsieh, D. S. Yun, E. Hu and A. M. Belcher, J. Mater. Chem., 2010, 20, 1435–1437 RSC.
- R. W. Cumberland, R. G. Blair, C. H. Wallace, T. K. Reynolds and R. B. Kaner, J. Phys. Chem. B, 2001, 105, 11922–11927 CrossRef CAS.
- J. Xiao, Y. Xie, R. Tang and W. Luo, Inorg. Chem., 2003, 42, 107–111 CrossRef CAS PubMed.
- M. D. Clark, S. K. Kumar, J. S. Owen and E. M. Chan, Nano Lett., 2011, 11, 1976–1980 CrossRef CAS PubMed.
- A. C. Frank, F. Stowasser, H. Sussek, H. Pritzkow, C. R. Miskys, O. Ambracher, M. Giersig and R. A. Fischer, ChemInform, 2010, 29, 3512–3513 Search PubMed.
- K. Sardar, M. Dan, B. Schwenzer and C. N. R. Rao, J. Mater. Chem., 2005, 15, 2175–2177 RSC.
- Z. Chen, Y. Li, C. Cao, S. Zhao, S. Fathololoumi, Z. Mi and X. Xu, J. Am. Chem. Soc., 2012, 134, 780–783 CrossRef CAS PubMed.
- N. S. Karan, Y. Chen, Z. Liu and R. Beaulac, Chem. Mater., 2016, 28, 5601–5605 CrossRef CAS.
- S. D. Dingman, N. P. Rath, P. D. Markowitz, P. C. Gibbons and W. E. Buhro, Angew. Chem., Int. Ed., 2000, 39, 1470–1472 CrossRef CAS.
- P. S. Schofield, W. Zhou, P. Wood, I. D. W. Samuel and D. J. Cole-Hamilton, J. Mater. Chem., 2004, 14, 3124–3126 RSC.
- J. Choi and E. G. Gillan, J. Mater. Chem., 2006, 16, 3774–3784 RSC.
- R. A. Fischer, A. Manz, A. Birkner and M. Kolbe, Adv. Mater., 2000, 12, 569–573 CrossRef.
- Y. Kwon and S. Kim, NPG Asia Mater., 2021, 13, 1–16 CrossRef.
- A. S. Mule, S. Mazzotti, A. A. Rossinelli, M. Aellen, P. T. Prins, J. C. Van Der Bok, S. F. Solari, Y. M. Glauser, P. V. Kumar, A. Riedinger and D. J. Norris, J. Am. Chem. Soc., 2021, 143, 2037–2048 CrossRef CAS.
- O. I. Mićić, S. P. Ahrenkiel and A. J. Nozik, Appl. Phys. Lett., 2001, 78, 4022–4024 CrossRef.
- R. Xie, Z. Li and X. Peng, J. Am. Chem. Soc., 2009, 131, 15457–15466 CrossRef CAS PubMed.
- D. C. Gary, S. E. Flowers, W. Kaminsky, A. Petrone, X. Li and B. M. Cossairt, J. Am. Chem. Soc., 2016, 138, 1510–1513 CrossRef CAS.
- J. Ning and U. Banin, Chem. Commun., 2017, 53, 2626–2629 RSC.
- D. C. Gary, A. Petrone, X. Li and B. M. Cossairt, Chem. Commun., 2017, 53, 161–164 RSC.
- Y. Kwon, J. Oh, E. Lee, S. H. Lee, A. Agnes, G. Bang, J. Kim, D. Kim and S. Kim, Nat. Commun., 2020, 11, 1–15 CrossRef PubMed.
- J. L. Stein, W. M. Holden, A. Venkatesh, M. E. Mundy, A. J. Rossini, G. T. Seidler and B. M. Cossairt, Chem. Mater., 2018, 30, 6377–6388 CrossRef CAS.
- A. A. Guzelian, U. Banin, A. V. Kadavanich, X. Peng and A. P. Alivisatos, Appl. Phys. Lett., 1996, 69, 1432–1434 CrossRef CAS.
- M. R. Friedfeld, D. A. Johnson and B. M. Cossairt, Inorg. Chem., 2019, 58, 803–810 CrossRef CAS.
- T. A. Gazis and P. D. Matthews, Chem. Commun., 2022, 58, 13799–13802 RSC.
| This journal is © The Royal Society of Chemistry 2023 |