 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chalcogen bonding in copper(II)-mediated synthesis†
Vusala A.
Aliyeva
a,
Atash V.
Gurbanov
 *ab,
Abdallah G.
Mahmoud
ac,
Rosa M.
Gomila
d,
Antonio
Frontera
*d,
Kamran T.
Mahmudov
*ab and
Armando J. L.
Pombeiro
*a
*ab,
Abdallah G.
Mahmoud
ac,
Rosa M.
Gomila
d,
Antonio
Frontera
*d,
Kamran T.
Mahmudov
*ab and
Armando J. L.
Pombeiro
*a
aCentro de Química Estrutural, Institute of Molecular Sciences, Departamento de Engenharia Química, Instituto Superior Técnico, Universidade de Lisboa, Av. Rovisco Pais, 1049–001 Lisbon, Portugal. E-mail: organik10@hotmail.com; kamran.mahmudov@tecnico.ulisboa.pt; pombeiro@tecnico.ulisboa.pt; Tel: +351 920210423
bExcellence Center, Baku State University, Z. Xalilov Str. 23, Az 1148 Baku, Azerbaijan
cDepartment of Chemistry, Faculty of Science, Helwan University, Ain Helwan, Cairo 11795, Egypt
dDepartament de Química, Universitat de les Illes Balears, Crta. de Valldemossa km7.5, Palma, Baleares, Spain. E-mail: toni.frontera@uib.es
First published on 6th January 2023
Abstract
The chalcogen bond (ChB) is a noncovalent attraction between an electrophilic chalcogen atom and a nucleophilic (Nu) region in the same (intramolecular) or another (intermolecular) molecular entity: R–Ch⋯Nu (Ch = O, S, Se or Te; R = substituents; Nu = nucleophile). ChB is comparable to the hydrogen and halogen bonds both in terms of strengths and directionality. However, in contrast to the monovalent halogen atoms, usually the divalent or tetravalent chalcogen atoms are able to display more than one electrophilic centre (on account of the existence of two or three species bonded to the chalcogen atom), which provides an additional opportunity in the use of this type of noncovalent binding in synthetic operations. In this work, the role of ChB at the secondary coordination sphere of metal complexes through copper(II)-mediated activation of dioxygen or of one nitrile group of a 1,2,5-selenadiazole-3,4-dicarbonitrile ligand to form a carbimidate or an imino-carboxylic acid is demonstrated. DFT calculations allowed evaluation of the strength of the ChBs and proved their relevant structure directing role in the solid state architectures. The effect of metal-coordination on the σ-hole opposite to the coordinated Se![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond has been analysed using molecular electrostatic potential (MEP) surfaces and explains the greater ability of the coordinated selenoxide derivatives to form strong ChBs.
O bond has been analysed using molecular electrostatic potential (MEP) surfaces and explains the greater ability of the coordinated selenoxide derivatives to form strong ChBs.
Introduction
Selective catalysis and molecular recognition through noncovalent interactions are a central theme in synthetic chemistry.1 Inspired by the advanced function of noncovalent interactions in enzyme catalysis, a variety of artificial systems has already been developed for catalytic transformations,1,2 molecular recognition,3etc. In contrast to a mono activation/attraction mode, the use of dual noncovalent centres in organocatalysis or chemical sensors gives an additional opportunity to improve the functional properties of catalysts or receptors (Scheme 1a).1–3 Moreover, replacement of one noncovalent centre in the dual activation mode via engineering of the secondary coordination sphere of metal complexes can enhance the reaction rate and/or selectivity or binding constant in cooperative catalysis or molecular sensing, respectively (Scheme 1b).4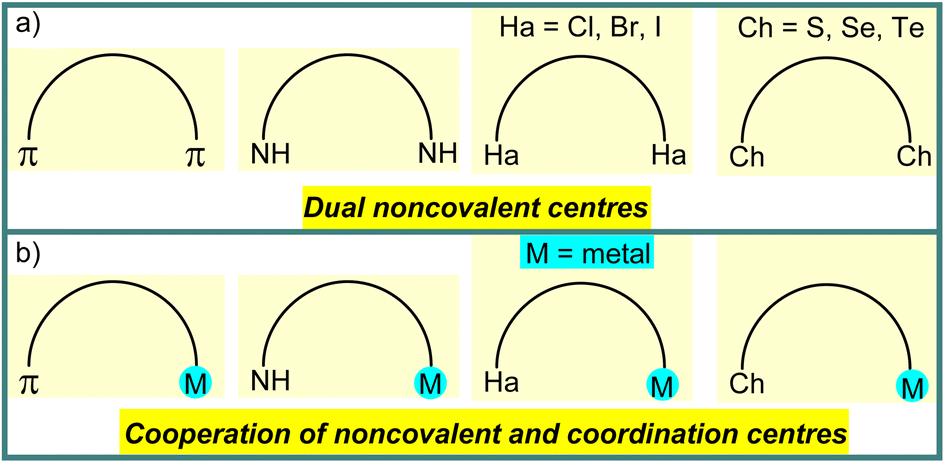 | ||
| Scheme 1 Schematic illustration of dual noncovalent centres (a) and cooperation of noncovalent and coordination centres (b). | ||
Secondary coordination sphere decoration by employing a wide range of weak forces, such as hydrogen and halogen bonds, hydrophobic, charge transfer, van der Waals, cation–π, anion–π or π–π interactions, has been well explored as a synthetic strategy for the selective functionalization of a target/desired bond among multiple bonds in a substrate molecule in metal-mediated synthesis or catalysis.1,2,5 The bond parameters (strength, directionality and tunability) as well as two electrophilic centres on the chalcogen atom (multiplicity) can make ChB a useful supramolecular tool in building of the secondary coordination sphere of metal complexes.6 Due to the only recent recognition of the chalcogen bonding by IUPAC,7 its crucial role in coordination chemistry was not necessarily highlighted in most of the original publications, and we refer the reader to our recently published review on this topic that discusses several relevant examples taken from the Cambridge Structural Database.8
On the other hand, metal-mediated synthesis allows the reactivity of the metal ion to transform a proligand, activating it through an in situ reaction, usually to produce coordination compounds with organic scaffolds that are not normally accessible by conventional organic synthesis.9 For example, the H-bond driven reaction of an arylhydrazone (A) with ethylenediamine results in the formation of diimidazoline (B),10 whereas the Cu(II)-mediated activation of one cyano group at this ligand leads to a coordination compound (C) which allows liberation of the organic part (D) through the cooperation of a resonance assisted hydrogen bond and ionic interactions (Scheme 2).11 Compound D can only be isolated by a metal-mediated pathway, and used as a starting material in the synthesis of B (Scheme 2). We anticipate that, like hydrogen bonding, the chalcogen bond can also be employed in the Cu(II)-mediated synthesis, a matter that has not yet been explored.
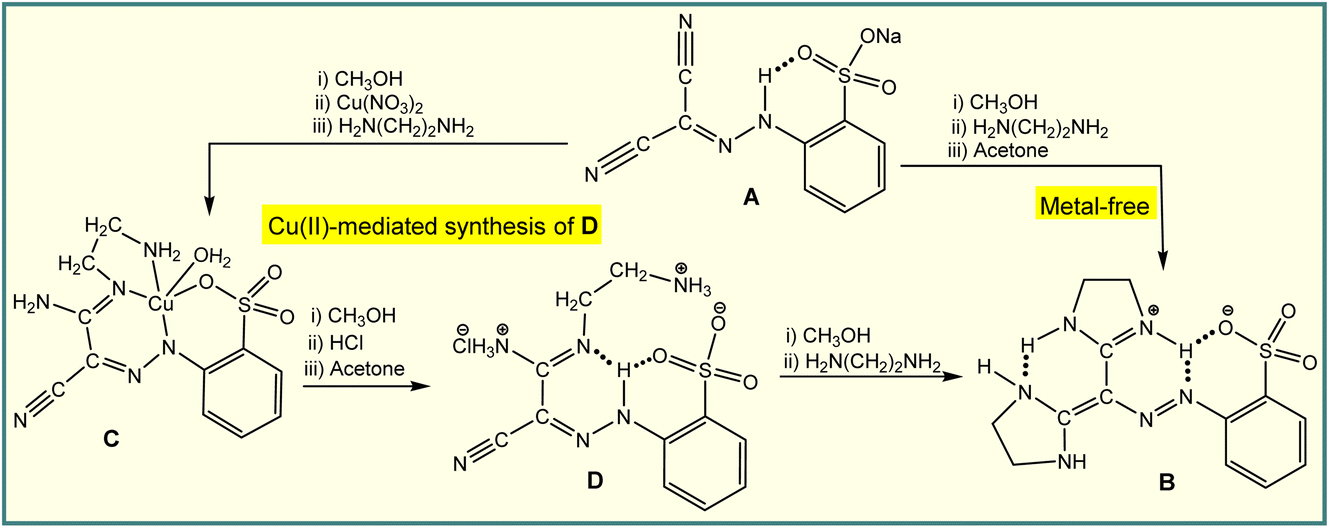 | ||
| Scheme 2 Hydrogen bond and Cu(II)-mediated transformation of cyano group(s). | ||
Taking in mind the above considerations, we focused this work on the following aims: (i) to use a chalcogen bond donor selenium centre in the design of the secondary coordination sphere of copper(II) complexes (Scheme 3); (ii) to demonstrate the crucial role of ChB in Cu(II)-mediated activation of one nitrile group of 1,2,5-selenadiazole-3,4-dicarbonitrile (Scheme 4).
Results and discussion
Synthesis and characterization of HL2 and 1–5
The synthesis and characterization of 2-((phenylselanyl)methyl)pyridine (L1) (Scheme 3) was reported earlier,12 and hence will not be discussed herein. Reaction of CuCl2·2H2O with L1 in methanol leads to [CuCl2(L1)2] (1), whereas in less volatile solvents, i.e., ethanol or dimethylformamide, the selenium atom of the ligand molecule is oxidized by air producing the dinuclear copper(II) complex [Cu2(Cl)4(μ-L1a)2] (2, L1a = 2-((phenylseleninyl)methyl)pyridine) (Scheme 3). Compound [Cu2(Br)4(μ-L1a)2] (3) was also obtained in ethanol or dimethylformamide by using CuBr2 instead of CuCl2·2H2O (Scheme 3). Similarly to the synthesis of 2 and 3, the selenium atom of HL2 undergoes oxidation in the synthesis of [Cu2(Br)2(μ-L2a)2] (5) (L2a = (E)-1-(((2-(phenylseleninyl)ethyl)imino)methyl)naphthalen-2-olate) in ethanol (with a few drops of DMF) solution, whereas no oxidation occurs in the preparation of [Cu(L2)2] (4) in the highly volatile dichloromethane (with a few drops of DMF) solution.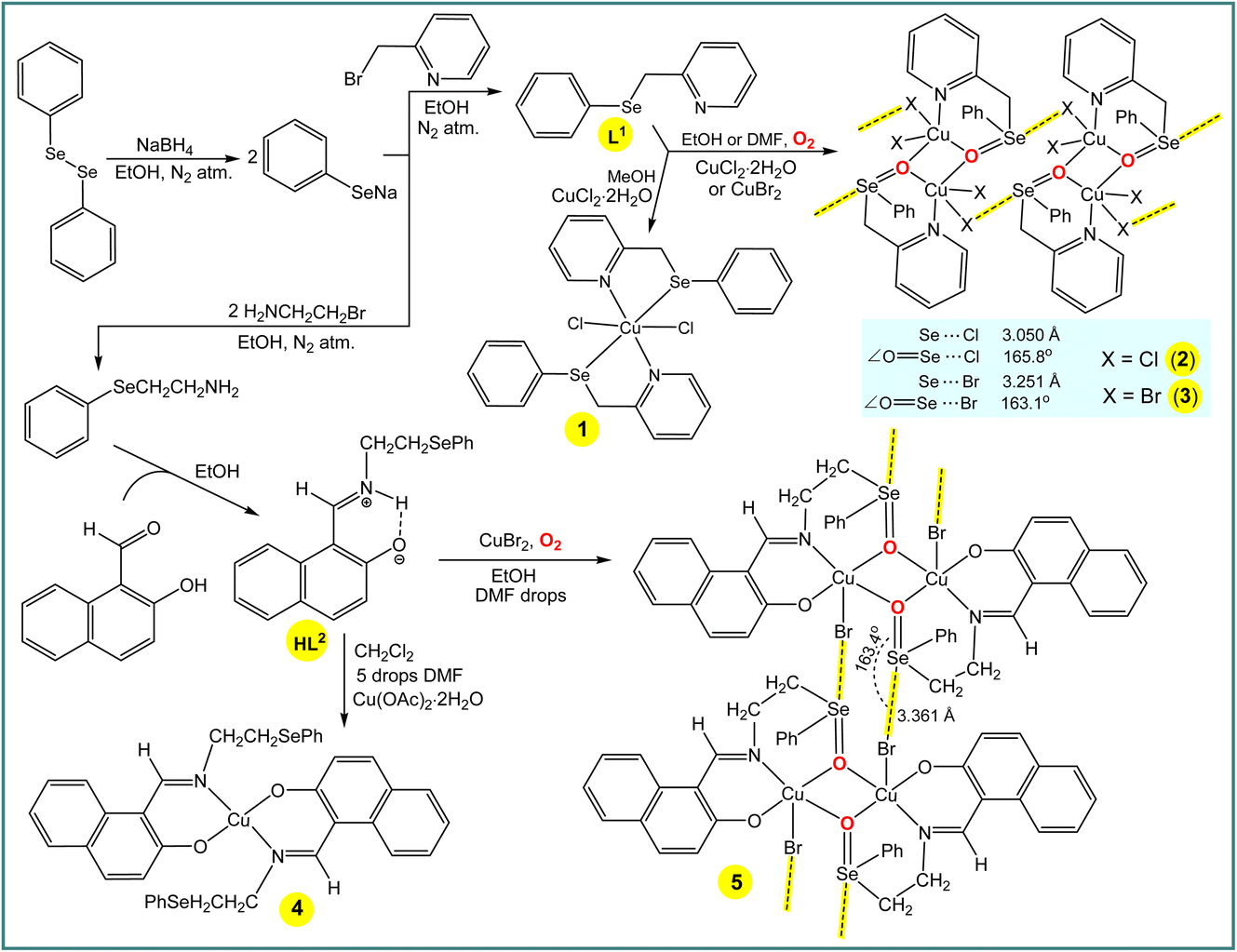 | ||
| Scheme 3 Synthesis of L1, HL2 and 1–5. | ||
The ESI-MS spectra of methanol solutions of the copper(II) complexes show relevant peaks at m/z = 631.88 [CuCl2(L1)2] + H+ (for 1), 798.73 [Cu2(Cl)4(μ-L1a)2] + H+ (for 2) and 976.53 [Cu2(Br)4(μ-L1a)2] + H+ (for 3). In the IR spectra of 1, 2 and 3, the phenyl and pyridine signals appear at 1431 and 1647, 1438 and 1640, and 1435 and 1629 cm−1, respectively, values that are significantly shifted in relation to the corresponding signals of L1 (1453 and 1651 cm−1, respectively). Both elemental analysis and X-ray crystallography (see below) are also in agreement with the proposed formulations of the copper(II) complexes in the solid state.
In the crystal structure of 1, the copper(II) ion is coordinated in a highly distorted octahedral coordination geometry formed by two selenium and two nitrogen atoms of two L1 ligands and two chlorine atoms (Fig. 1). Interestingly, the selenium atoms do not act as Ch donors in the crystal packing of 1, whereas tetravalent Se centres participate in intermolecular interactions between tectons in the structures of 2 and 3, leading to 1D supramolecular chains (Fig. 1). Both the strength and the directionality of ChB in 2 and 3 depend on the nature of the halogen atoms: Se⋯Cl 3.050 Å, ΣrvdW (Se⋯Cl) = 3.65 Å, ∠OSe⋯Cl 165.78° (in 2) and Se⋯Br 3.251 Å, ΣrvdW (Se⋯Br) = 3.75 Å, ∠OSe⋯Br 163.10° (in 3) (Fig. 1). In 2 or 3, the primary coordination sphere of the penta-coordinated copper can be described as a distorted square-pyramid (τ5 = 0.20 and τ5 = 0.27),13 with an equatorial plane containing one N and two O atoms from 2-((phenylseleninyl)methyl)pyridine ligands and one Cl or Br atom; the apical position is filled by C1(1) or Br(1) at 2.5237(6) or 2.6625(9) Å, respectively (Fig. 1).
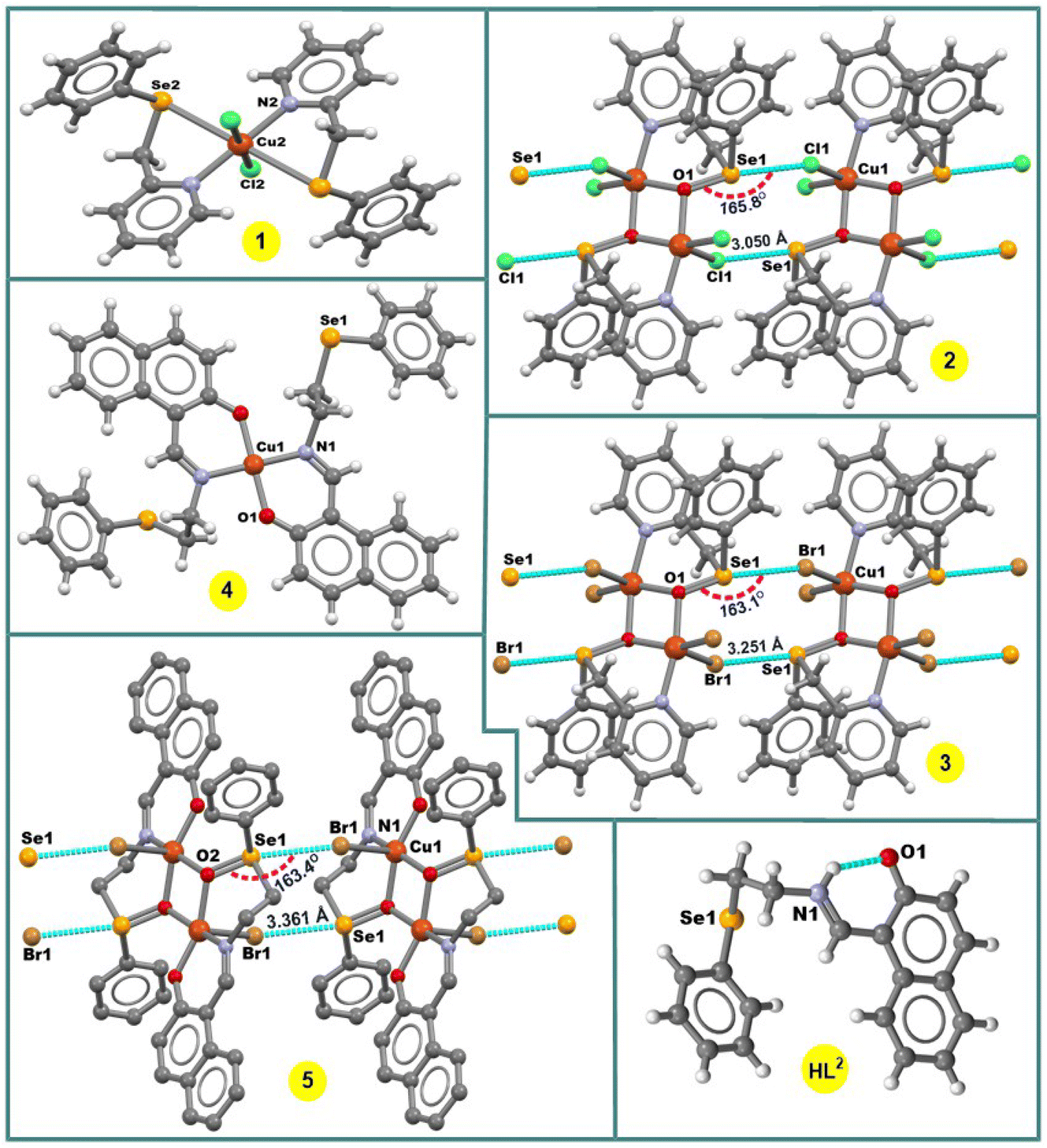 | ||
| Fig. 1 Crystal structures of HL2 and 1–5. H atoms in 5 are omitted for clarity. | ||
The new Schiff base (E)-1-(((2-(phenylselanyl)ethyl)iminio)methyl)naphthalen-2-olate (HL2) was synthesized by reaction of 2-(phenylselanyl)ethan-1-amine with 2-hydroxy-1-naphthaldehyde in ethanol (Scheme 3). In the 1H-NMR spectrum of HL2 in DMSO-d6, C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) N and N are observed at 9.07 and 14.01 ppm, respectively. In the IR spectrum, ν(CN) is observed at 1629 cm−1, which is significantly shifted due to coordination of the N atom to the metal centre in 4 (1602 cm−1) and 5 (1605 cm−1). Elemental analysis and ESI-MS data also support the structures of HL2, 4 and 5. Thus, the methanol solutions of HL2, 4 and 5 display, by ESI-MS, parent peaks at m/z = 355.05 [Mr + H]+, 771.01 [Mr + H]+ and 1026.76 [Mr + H]+, respectively.
N and N are observed at 9.07 and 14.01 ppm, respectively. In the IR spectrum, ν(CN) is observed at 1629 cm−1, which is significantly shifted due to coordination of the N atom to the metal centre in 4 (1602 cm−1) and 5 (1605 cm−1). Elemental analysis and ESI-MS data also support the structures of HL2, 4 and 5. Thus, the methanol solutions of HL2, 4 and 5 display, by ESI-MS, parent peaks at m/z = 355.05 [Mr + H]+, 771.01 [Mr + H]+ and 1026.76 [Mr + H]+, respectively.
In the crystal structure of [Cu(L2)2] (4), the Cu2+ ion, located on a center of inversion, is coordinated by two imine N atoms and two phenolate O atoms from two Schiff base ligands in trans positions leading to a square-planar arrangement around the metal centre (Fig. 1). Since the Cu(1)–O(1) and Cu(1)–N(1) bond lengths of 1.895(2) and 1.971(3) Å, respectively, are different, the coordination geometry is distorted. The –CH2CH2SePh group of the ligand does not coordinate and points away from the primary coordination sphere of the Cu(II) centre, and the selenium atom acts as hydrogen bond acceptor in the crystal packing diagram of 4.
The complex 5 crystallizes in the triclinic system, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . It contains two Cu atoms, two Schiff-base ligands and two Br atoms (Fig. 1). The seleninyl oxygen of the –CH2CH2Se(O)Ph group of the L2a ligands acts as a bridging atom, leading to a dinuclear Cu2O2 core, with a Cu⋯Cu distance of about 3.089 Å (Fig. 1). A crystallographic inversion center passes through the center of the Cu2O2 plane, i.e., the second part of the dimer is generated by symmetry. The coordination geometry around the Cu(II) centres can be described as a distorted square pyramid (τ5 = 0.12)13 with the N(1)–Cu(1)–O(2) and O(1)–Cu(1)–O(2) angles of 167.35(11)° and 160.01(10)°, respectively. In the crystal packing diagram of 5, the discrete copper(II) dimers are linked into a one-dimensional supramolecular chain via the Se⋯Br 3.361 Å [ΣrvdW (Se⋯Br) = 3.75 Å] intermolecular chalcogen bonds with the ∠OSe⋯Br directionality of 163.4° (Fig. 1).
. It contains two Cu atoms, two Schiff-base ligands and two Br atoms (Fig. 1). The seleninyl oxygen of the –CH2CH2Se(O)Ph group of the L2a ligands acts as a bridging atom, leading to a dinuclear Cu2O2 core, with a Cu⋯Cu distance of about 3.089 Å (Fig. 1). A crystallographic inversion center passes through the center of the Cu2O2 plane, i.e., the second part of the dimer is generated by symmetry. The coordination geometry around the Cu(II) centres can be described as a distorted square pyramid (τ5 = 0.12)13 with the N(1)–Cu(1)–O(2) and O(1)–Cu(1)–O(2) angles of 167.35(11)° and 160.01(10)°, respectively. In the crystal packing diagram of 5, the discrete copper(II) dimers are linked into a one-dimensional supramolecular chain via the Se⋯Br 3.361 Å [ΣrvdW (Se⋯Br) = 3.75 Å] intermolecular chalcogen bonds with the ∠OSe⋯Br directionality of 163.4° (Fig. 1).
Cu(II)-mediated activation of one nitrile group of 1,2,5-selenadiazole-3,4-dicarbonitrile
One-pot Cu(CH3COO)2·H2O (in the presence of imidazole), Cu(NO3)2·2H2O (with a few drops of DMF) or CuBr2 mediated activation of one cyano group of 1,2,5-selenadiazole-3,4-dicarbonitrile (L3)14 in methanol produces [Cu(imidazole)6](L3a)2 (6) (L3a = 4-cyano-1,2,5-selenadiazole-3-carboxylate), [4-cyano-1,2,5-selenadiazole-3-carboxylic acid·DMF] (7) or [CuBr2(L3b)2] (8) (L3b = methyl 4-cyano-1,2,5-selenadiazole-3-carbimidate), respectively (Scheme 4). The IR spectra of the compounds 6–8 show unreacted ν(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) vibrations at 2236, 2221 and 2236 cm−1, respectively, with a significant shift in relation to the corresponding signal of L3 (2243 cm−1). The ν(CO) are observed at 1684 and 1654 cm−1 for 6 and 7, correspondingly, while a broad band at 1640 cm−1 is assigned to ν(CN) for 8. Elemental analysis also supports the formulations of 6–8, and fragmentation peaks in mass spectra of the compounds can be related as follows: 235.57 [Cu(imidazole)6]2+ and 201.92 (L3a)− for 6, 73.05 (DMF) and 202.92 (4-cyano-1,2,5-selenadiazole-3-carboxylic acid) for 7 and 654.67 [Mr + H]+ for 8. In the 1H and 13C NMR spectra of co-crystal 7, the signals of the –COOH proton and CO carbon of 4-cyano-1,2,5-selenadiazole-3-carboxylic acid were detected at 9.15 and 160.3 ppm, respectively. Due to the participation in the intermolecular chalcogen bond, the acidic C–H proton and CO carbon signals of DMF in 7 were observed at lower field (δ 8.67 and 165.3) compared to the respective signals of its free form (δ 7.95 and 162.29).15
N) vibrations at 2236, 2221 and 2236 cm−1, respectively, with a significant shift in relation to the corresponding signal of L3 (2243 cm−1). The ν(CO) are observed at 1684 and 1654 cm−1 for 6 and 7, correspondingly, while a broad band at 1640 cm−1 is assigned to ν(CN) for 8. Elemental analysis also supports the formulations of 6–8, and fragmentation peaks in mass spectra of the compounds can be related as follows: 235.57 [Cu(imidazole)6]2+ and 201.92 (L3a)− for 6, 73.05 (DMF) and 202.92 (4-cyano-1,2,5-selenadiazole-3-carboxylic acid) for 7 and 654.67 [Mr + H]+ for 8. In the 1H and 13C NMR spectra of co-crystal 7, the signals of the –COOH proton and CO carbon of 4-cyano-1,2,5-selenadiazole-3-carboxylic acid were detected at 9.15 and 160.3 ppm, respectively. Due to the participation in the intermolecular chalcogen bond, the acidic C–H proton and CO carbon signals of DMF in 7 were observed at lower field (δ 8.67 and 165.3) compared to the respective signals of its free form (δ 7.95 and 162.29).15
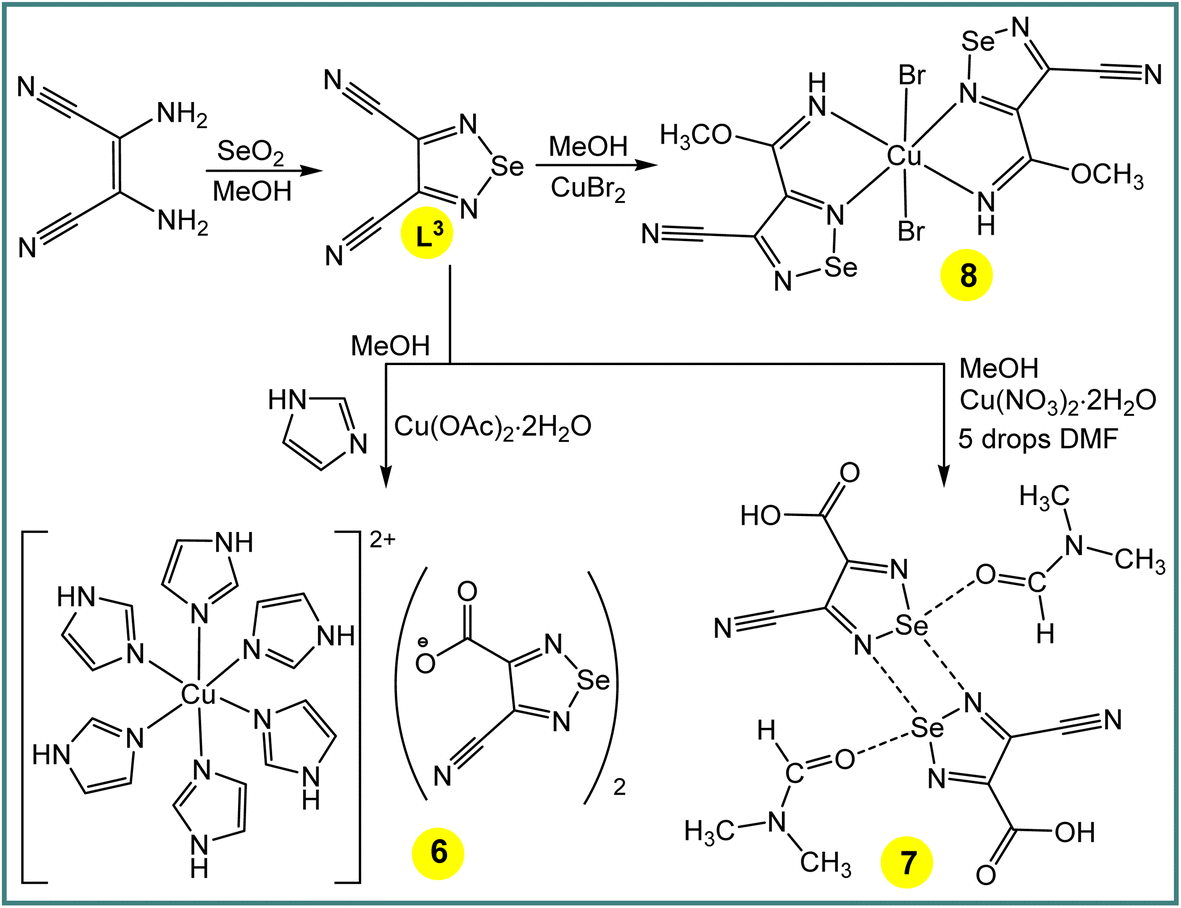 | ||
| Scheme 4 Copper(II)-mediated transformation of one nitrile group of L3. | ||
The compound [Cu(imidazole)6](L3a)2 (6) contains six imidazole ligands, one copper cation and two non-coordinated 4-cyano-1,2,5-selenadiazole-3-carboxylate anions (Fig. 2). The copper cation adopts a tetragonally elongated octahedron coordination geometry and is located on an inversion center. The four short Cu–N distances in the equatorial plane [both Cu(1)–N(6) and Cu(1)–N(8)] are 2.037 and 2.047 Å, respectively, and the two long Cu(1)–N(4) distances in the apical positions are 2.482 Å. In the crystal packing diagram of 6, the hydrogen bond donor N–H centres of the imidazole ligands of the [Cu(imidazole)6]2+ cation participate in intermolecular negative charge assisted hydrogen bonds with the carboxylate anion of L3a, with the N⋯O− distances in the range of 2.751–2.819 Å. The role of the counter-anion is not limited to its H-bond acceptor ability, but the selenium atom of 4-cyano-1,2,5-selenadiazole-3-carboxylate acts as a bifurcate ChB donor towards the oxygen and nitrogen atoms of CO and CN groups of the neighboring molecules at the distances Se(1)⋯O(2) 3.068 Å [ΣrvdW (Se⋯O) = 3.42 Å] and Se(1)⋯N(3) 3.053 Å [ΣrvdW (Se⋯N) = 3.45 Å] and the ∠N(1)–Se(1)⋯O(2) and ∠N(2)–Se(1)⋯N(3) angles of 164.50 and 174.39°, respectively (Fig. 2).
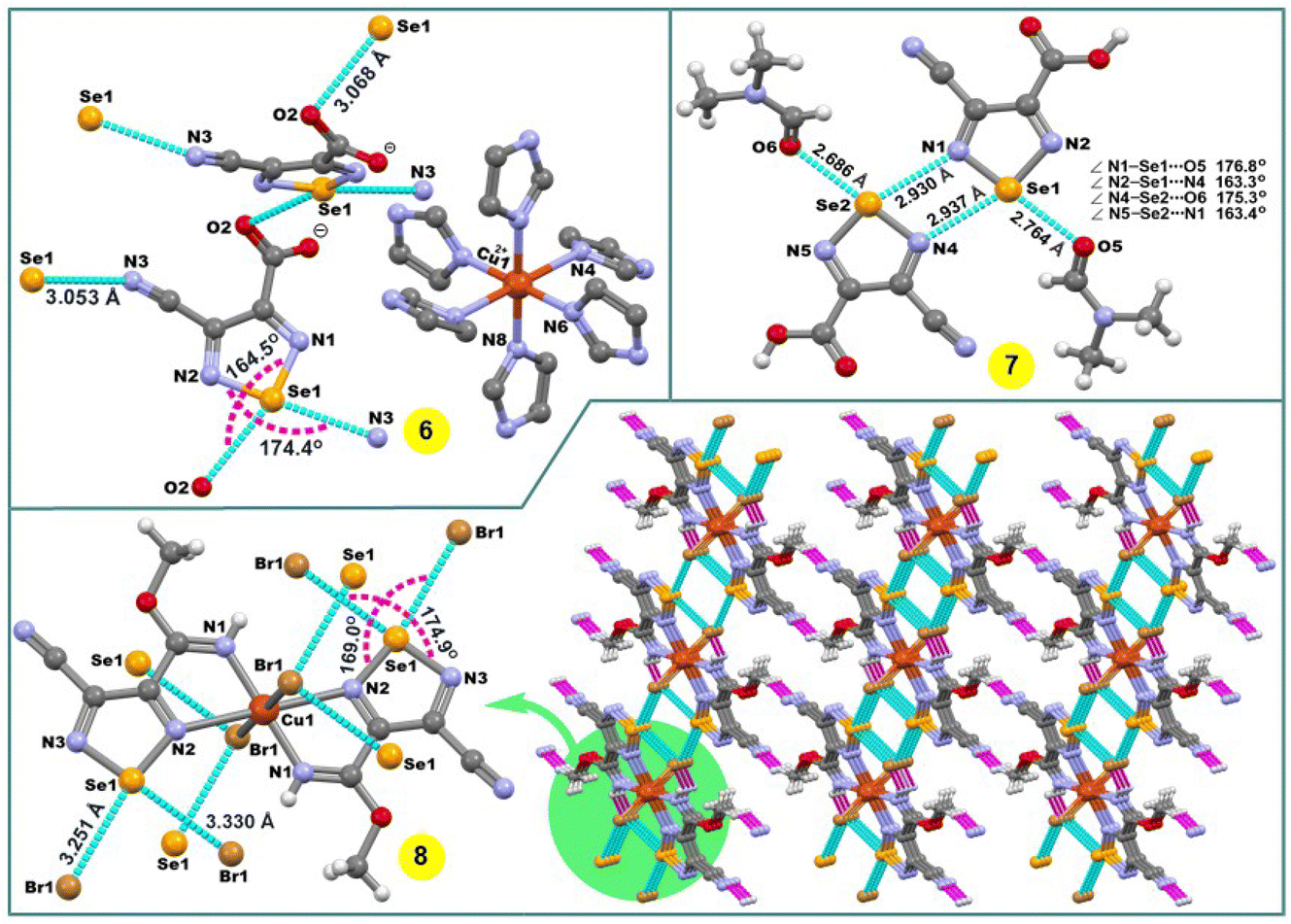 | ||
| Fig. 2 Crystal structures of 6–8. H atoms in 6 are omitted for clarity. The chalcogen and hydrogen bonds are shown in light blue and pink colour, respectively. | ||
The crystal structure of 7 shows a four-membered cyclic supramolecular synthon via head-to-head dimerization with intermolecular Se(1)⋯N(4) and Se(2)⋯N(1) chalcogen bonds with the distances of 2.937 and 2.930 Å, respectively (Fig. 2). The chalcogen bond angles ∠N(2)–Se(1)⋯N(4) and ∠N(5)–Se(2)⋯N(1) in this dimeric synthon are 163.3 and 163.4°, respectively (Fig. 2). In addition, there are short intermolecular chalcogen bond contacts between selenium and the oxygen atoms of dimethylformamide molecules, in which both the Se⋯O distance and ∠N–Se⋯O directionality are in accord with the terms of ChB7 (Fig. 2). Thus, an aggregation of two 4-cyano-1,2,5-selenadiazole-3-carboxylic acids and two dimethylformamide molecules through Se⋯N and Se⋯O types of chalcogen bonds, respectively, lead to a 0D supramolecular aggregate (Scheme 4 and Fig. 2).
In the structure of 8, the Cu(1)–N(2) 2.498 Å bond lengths are markedly longer in comparison to Cu(1)–N(1) 1.933 Å, thus, the coordination geometry around the copper atom is a distorted octahedron, and the remaining axial positions are occupied by two bromine atoms with the Cu(1)–Br(1) bond lengths of 2.524 Å (Fig. 2). Both selenium atoms act as bifurcate chalcogen bond donors towards the coordinated bromine atoms in the crystal packing diagram of 8 (Fig. 2). A cooperation of chalcogen bonds [Se(1)⋯Br(1) 3.283 and 3.330 Å [ΣrvdW (Se⋯Br) = 3.75 Å], ∠N(2)–Se(1)⋯Br(1) 169.0° and ∠N(3)–Se(1)⋯Br(1) 174.9°] with the C(5)–H(5A)⋯N(4) hydrogen bonds between the tectons lead to a 3D supramolecular network (Fig. 2).
DFT study of chalcogen bonds in 2, 3 and 5–8
As explained above, compounds 2 and 3 form infinite 1D chains in the solid state that propagate via Se⋯X ChBs (see Fig. 1 and Scheme 3). In principle, it is expected that the Cu-coordinated selenoxide group exhibits three available σ-holes opposite to the three covalent bonds. However, in compounds 2 and 3, two of them (those opposite to the carbon atoms) are blocked since they point to the coordinated halide ligands, leaving accessible only the σ-hole opposite to oxygen atom. This can be appreciated in the MEP surface plots depicted in Fig. 3a and b for compounds 2 and 3, respectively, where only one deep σ-hole per Se atom is observed. The large and positive MEP values at the σ-holes (+43.9 and +44.5 kcal mol−1 for 2 and 3, respectively), which also correspond to the MEP maximum, are due to the coordination of the O-atom of the selenoxide group to the Cu-metal centers, increasing the polarization of the SeO bond. In both compounds the MEP minimum is located at the chlorido/bromido ligand. Therefore, the chalcogen bonds observed in the solid state of compounds 2 and 3 are the most electrostatically favoured contacts.
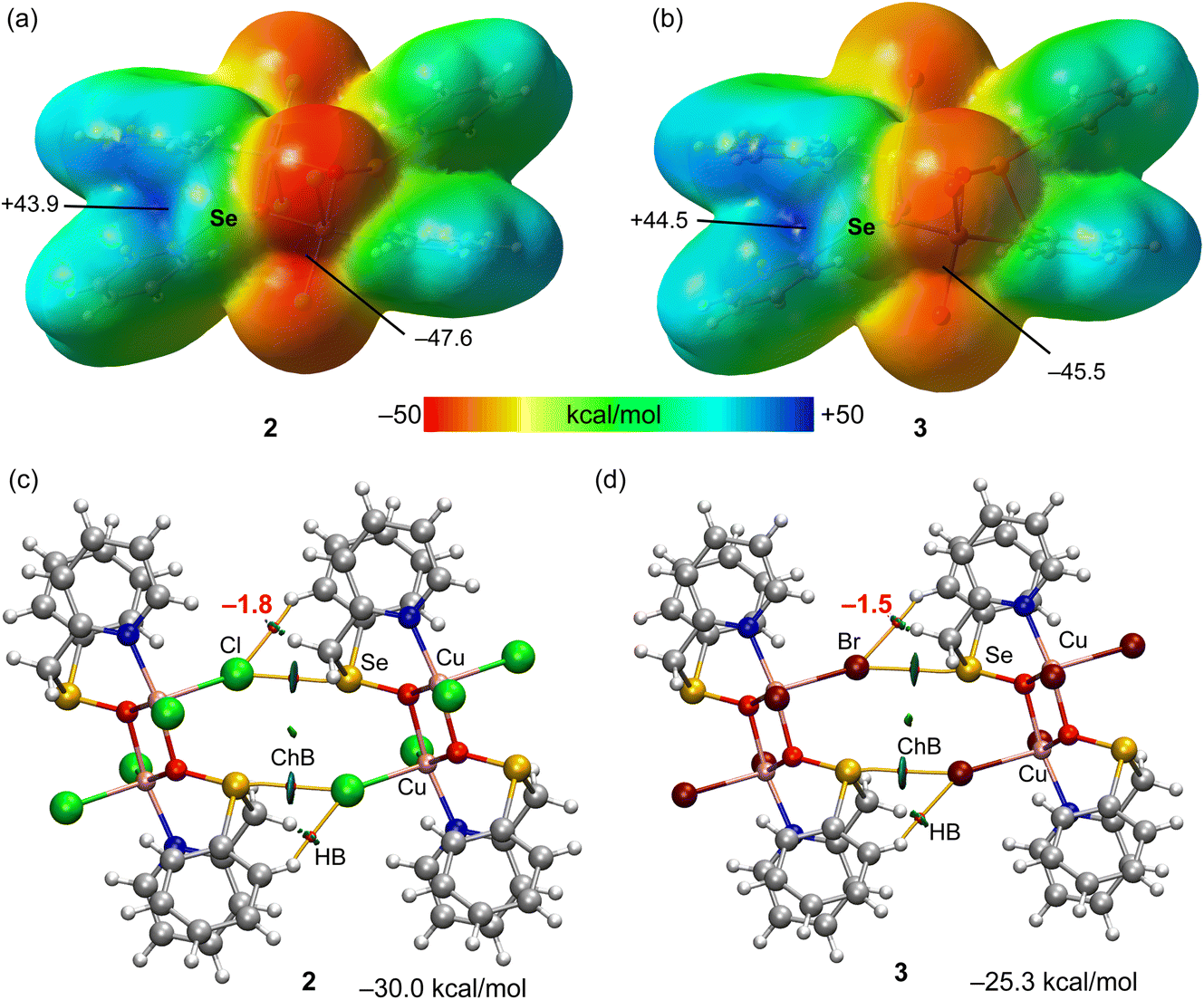 | ||
| Fig. 3 (a and b) MEP surfaces of compounds 2 and 3 with indication of the MEP maximum and minimum. Energies in kcal mol−1. (c and d) QTAIM (bond critical points in red and bond paths as orange lines) and NCIPlot (RDG = 0.4, density cut-off = 0.04 a.u., color scale ±0.04 a.u.) representation for compounds 2 and 3. Only intermolecular interactions are represented. The association energies in kcal mol−1 of the hydrogen bonds derived from Vr are given in red adjacent to the bond CP. | ||
The combined QTAIM and NCIplot analyses of two dimers of compounds 2 and 3 are represented in Fig. 3c and d, including the dimerization energies. The QTAIM/NCIplot analysis shows that the halogen atom is connected to the Se atom by a bond critical point (CP, represented as a red sphere) and bond path (represented as an orange line), thus confirming the existence of the ChBs. The interaction is also characterized by a disk-shape reduced density gradient (RDG) isosurface that is coincident with the position of the bond CP. The QTAIM/NCIPlot analysis also reveals the existence of two symmetrically equivalent CH⋯X H-bonds. Each H-bonding interaction is established between an aromatic C–H bond and the halogen atom. It is characterized by a bond CP, bond path and green RDG isosurface. The dimerization energies are large (−30.0 kcal mol−1 and −25.3 kcal mol−1 for 2 and 3, respectively), in line with the large MEP energies observed for the electron donor and acceptors atoms. The strength of the ancillary H-bonds has been estimated by using the potential energy density (Vr) energy predictor (E = 0.5 × Vr) measured at the bond CP that characterizes the H-bond. These values are indicated in red adjacent to the bond CPs in Fig. 3. The total contribution of the H-bonds is −3.6 kcal mol−1 in 2 and −3.0 kcal mol−1 in 3, thus evidencing that the formation of the supramolecular chain in the solid state is mostly governed by the ChBs.
A similar analysis has been performed for compound 5 that also forms 1D supramolecular polymers in the solid state that propagate by means of ChBs (see Fig. 1 and Scheme 3). The MEP surface plot of compound 5 is represented in Fig. 4a, showing that only one σ-hole of Se is available, opposite to the SeO bond, which corresponds to the MEP maximum (+39.5 kcal mol−1). The MEP minimum is located at the coordinated bromido ligands, as expected. These values are comparable to those observed for compounds 2 and 3 (see Fig. 3a and b).
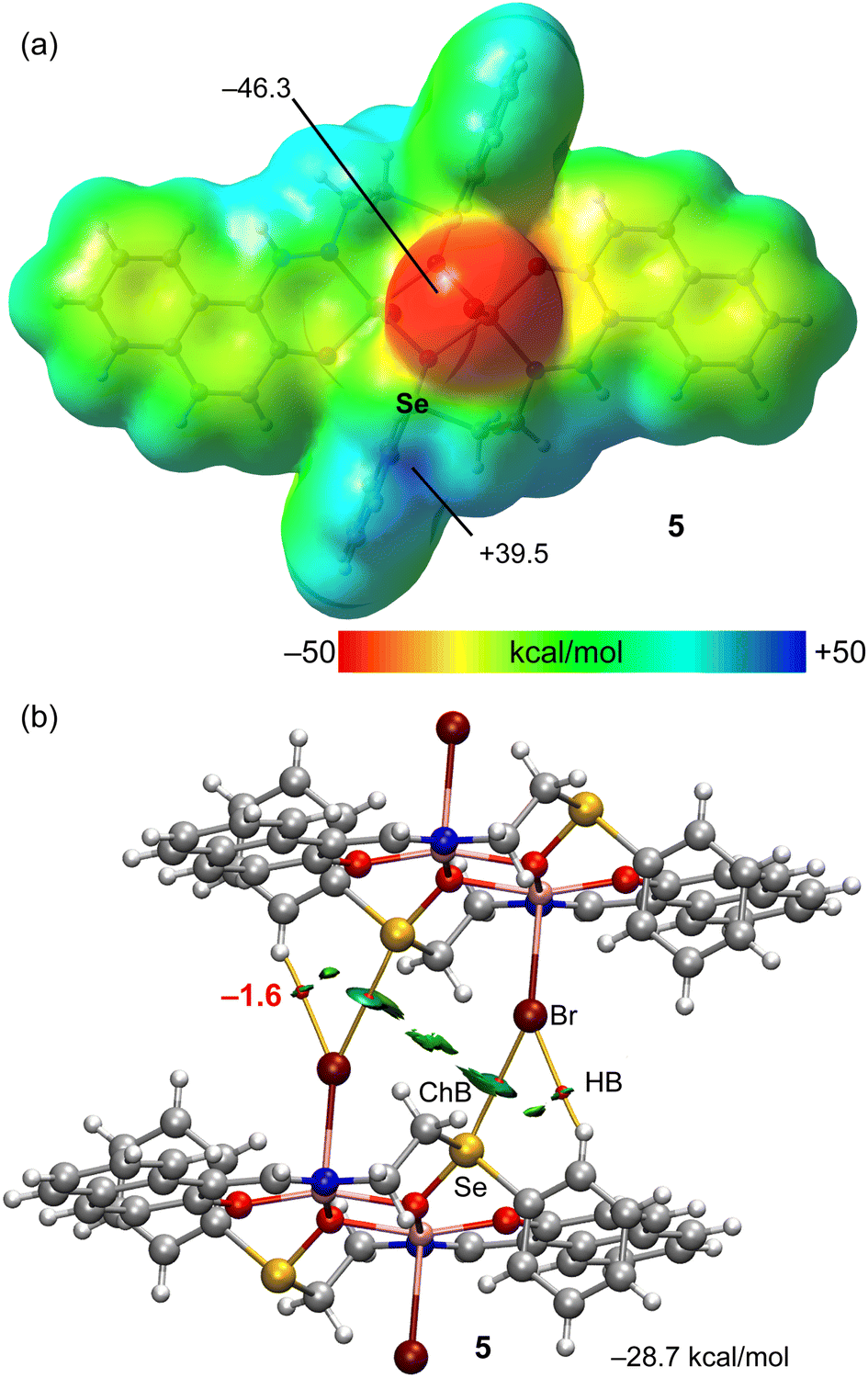 | ||
| Fig. 4 (a) MEP surface of compound 5 with indication of the MEP maximum and minimum. Energies in kcal mol−1. (b) QTAIM (bond critical points in red and bond paths as orange lines) and NCIPlot (RDG = 0.4, density cut-off = 0.04 a.u., color scale ±0.04 a.u.) representation for compound 5. Only intermolecular interactions are represented. The association energies in kcal mol−1 of the hydrogen bonds derived from Vr are given in red adjacent to the bond CP. | ||
Fig. 4b shows the combined QTAIM/NCIplot analysis of the dimer extracted from the infinite 1D chain shown in Fig. 1 (see bottom-left). The interaction energy is −28.7 kcal mol−1 which is similar to the dimerization energies of compounds 2 and 3. The QTAIM/NCIplot analysis reveals the expected bond CP, bond path and blue RDG isosurface connecting the Br and Se-atoms. The dimerization is further assisted by the formation of two symmetric H-bonds, similar to those described above for compounds 2 and 3, both geometric and energetically (total contribution of the H-bonds is −3.2 kcal mol−1). The results of compounds 2, 3 and 5 suggest that the bifurcated X⋯H,Se is a recurrent motif in these systems.
The MEP surface of compound 7 is represented in Fig. 5a, showing the presence of two deep σ-holes with similar MEP values (37.7 and 36.4 kcal mol−1) due to the electron withdrawing effect of the substituent of the five membered ring. The ChBs observed in the solid state of compounds 6 and 7 have been analysed energetically and using the QTAIM/NCIplot computational tool. Since ChBs in compound 6 are established between the anionic parts of the salt (4-cyano-1,2,5-selenadiazole-3-carboxylate), the energetic study has been performed using the Vr predictor. This is very convenient to estimate the strength associated to the ChBs in a system where the electrostatic repulsion between two anions dominates. A trimeric anion extracted from the solid state of 6 is represented in Fig. 5b, showing the formation of two ChBs, characterized by the corresponding bond CPs, bond paths and green RDG isosurfaces. The total stabilization energy due to the ChBs is modest (−3.2 kcal mol−1) likely due to the anion⋯anion nature of the interaction and the concomitant long O,N⋯Se distances (see Fig. 2 for distance values) observed in compound 6. For compound 7, we have analyzed the formation of the self-assembled dimer shown in Fig. 5c (see also Scheme 4) and the interaction with the co-crystallized solvent molecule. The dimerization energy is moderately strong (Fig. 5c) likely due to the small nucleophilicity of the selenodiazole N-atoms (MEP value is −18.2 kcal mol−1, see Fig. 5a). Each ChB is characterized by the corresponding bond CP and bond path. The RDG isosurface embraces the whole space between the interacting molecules, thus suggesting that this interaction can be also understood as an antiparallel N–Se⋯N–Se interaction. Finally, in Fig. 5d the ChB complex with the DMF molecule is shown, also exhibiting a modest interaction energy (−2.8 kcal mol−1). The weaker ChBs observed in the assemblies of compounds 6 and 7 compared to compounds 2, 3 and 5 are likely due to the better electron donor ability of the halides.
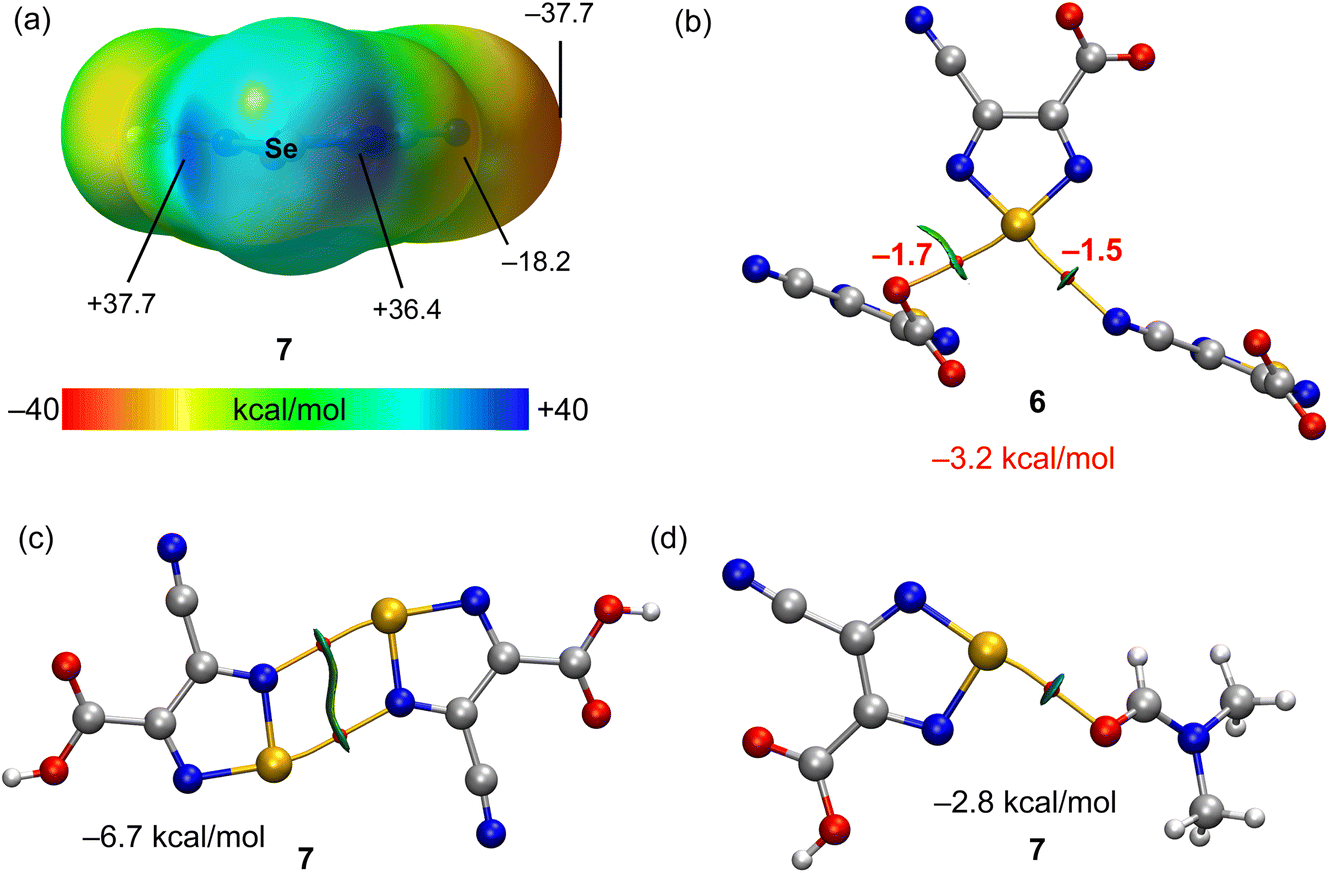 | ||
| Fig. 5 (a) MEP surface of compound 7 with indication of the MEP maximum and minimum and other selected points of the surface. Energies in kcal mol−1. (b–d) QTAIM (bond critical points in red and bond paths as orange lines) and NCIPlot (RDG = 0.4, density cut-off = 0.04 a.u., color scale ±0.04 a.u.) representation for compounds 6 and 7. Only intermolecular interactions are represented. The association energies in kcal mol−1 of the chalcogen bonds derived from Vr are given in red adjacent to the bond CP. | ||
The MEP surface of compound 8 is represented in Fig. 6a, revealing the existence of two deep σ-holes at the selenium atoms (44.6 kcal mol−1 and 38.3 kcal mol−1). However, the MEP maximum is located at the NH group (52.1 kcal mol−1). The MEP minimum is located at the N-atom of the cyano group (−31.4 kcal mol−1) followed by the value at the coordinated bromido ligand (−29.4 kcal mol−1). The QTAIM/NCIplot analysis of one of the two ChB self-assembled dimers observed in the solid state of 8 is represented, evidencing the formation of two ChBs, four H-bonds and one Se⋯Se interaction, all of them characterized by the corresponding bond CPs and bond paths. The dimerization energy is large (−23.8 kcal mol−1) due to the formation of two strong ChBs (good electron donor) and also the contribution of four H-bonds involving the methoxy and NH groups. The contribution of the H-bonds is only 2.2 kcal mol−1, likely due to the low nucleophilicity of the electron donor N-atom (MEP value −14.4 kcal mol−1, see Fig. 6a).
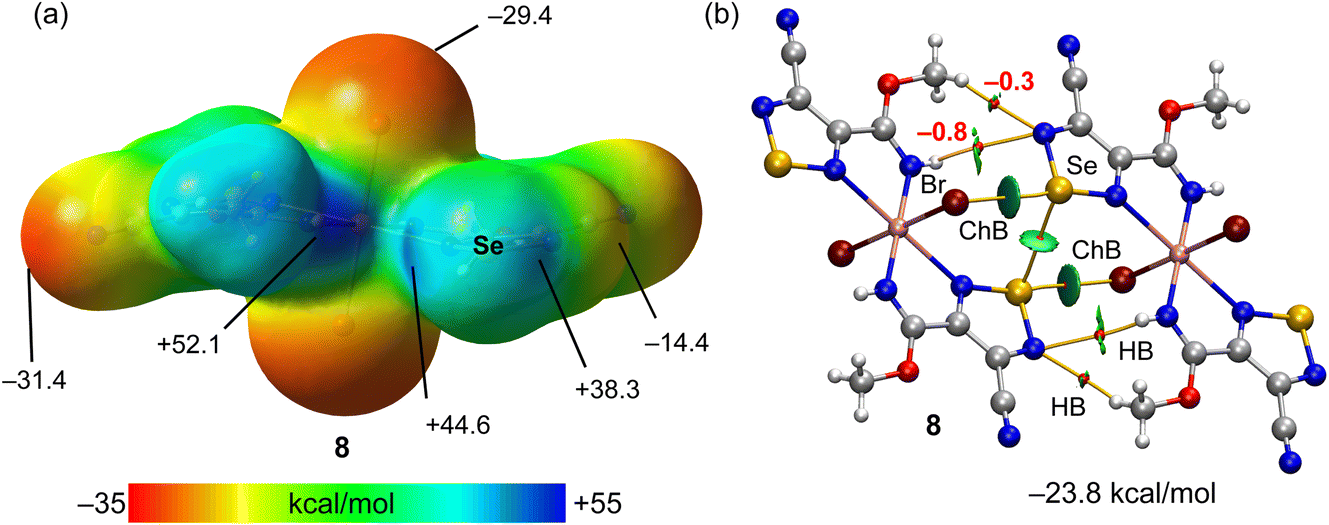 | ||
| Fig. 6 (a) MEP surface of compound 8 with indication of the MEP maximum and minimum and other selected points of the surface. Energies in kcal mol−1. (b) QTAIM (bond critical points in red and bond paths as orange lines) and NCIPlot (RDG = 0.4, density cut-off = 0.04 a.u., color scale ±0.04 a.u.) representation for compound 8. Only intermolecular interactions are represented. The association energies in kcal mol−1 of the hydrogen bonds derived from Vr are given in red adjacent to the bond CP. | ||
The other self-assembled ChB dimer analysed for compound 8 is shown in Fig. 7 where the distribution of bond CPs and bond paths reveals an intricate combination of interactions, including two symmetrically equivalent ChBs, six H-bonding contacts (2 × NH⋯Br and 2 × CH⋯Br,Se) and two Se⋯C contacts. The dimerization energy is −31.9 kcal mol−1, which is larger (in absolute value) than the other ChB self-assembled dimer of compound 8. The total contribution of the H-bonds is also larger (−6.0 kcal mol−1).
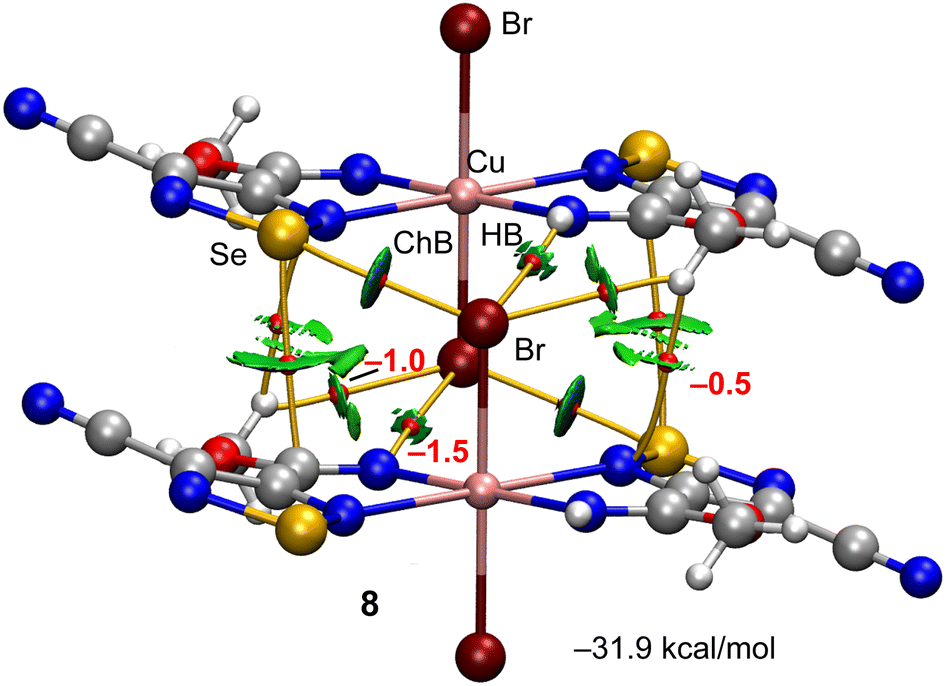 | ||
| Fig. 7 QTAIM (bond critical points in red and bond paths as orange lines) and NCIPlot (RDG = 0.4, density cut-off = 0.04 a.u., color scale ±0.04 a.u.) representation for compound 8. Only intermolecular interactions are represented. The association energies in kcal mol−1 of the hydrogen bonds derived from Vr are given in red adjacent to the bond CP. | ||
It is interesting to highlight that the strength of the ChBs is stronger in the Cu-complexes of selenoxides and selenadiazole (compounds 2, 3, 5 and 8) than in the non-complexed selenadiazole rings (compounds 6 and 7) due to the presence of the more polarized SeO bonds and better electron donor groups (halides). The fact that most compounds exhibit ChBs in the solid state that are crucial in their solid state architecture emphasize the relevance of ChBs in crystal engineering and supramolecular chemistry.
Conclusions
The chalcogen bond (ChB) has been recently exploited as a supramolecular tool for crystal growth and design involving metal complexes as tectons.8,19–21 In this work, we have demonstrated that the simple design of ligands with a ChB donor selenium centre can contribute to the molecular oxygen and one-pot nitrile activation in copper(II)-mediated synthesis. The tetravalent selenium atom acts as a ChB donor towards coordinated halogen atoms (Cl or Br) in the secondary coordination sphere of copper(II) complexes (see 2, 3 and 5 in Scheme 3 and Fig. 1), leading to a 1D supramolecular chain. The divalent selenium atom behaves as a bifurcated ChB donor centre in the crystal packing diagrams of 6–8, which associates the supramolecular aggregates and networks. In addition, theoretical calculations combined with QTAIM/NCIplot analysis identify the chalcogen bonds between tectons in the second coordination sphere of copper(II) complexes and the existence of ancillary CH⋯X interactions, which are significantly weaker. Those compounds where the selenoxide is coordinated to the copper(II) metal centre exhibit deeper σ-holes and stronger interactions. This study outlines an action of chalcogen bonding in the copper(II)-mediated synthesis and decoration of the secondary coordination sphere of metal complexes, which can be used in catalysis and in molecular recognition.Experimental section
Materials and instrumentation
All the chemicals were obtained from commercial sources and used as received. The 2-((phenylselanyl)methyl)pyridine (L1)12 and 1,2,5-selenadiazole-3,4-dicarbonitrile (L3)14 were synthesized according to reported synthetic procedures. The IR spectra (4000–400 cm−1) were recorded on a Bruker Alpha-P ATR-IR Spectrometer. The 1H and 13C NMR spectra were obtained at room temperature (r.t.) on a Bruker 400.13 MHz spectrometer using tetramethylsilane [Si(CH3)4] as the internal reference. Carbon, hydrogen and nitrogen elemental analyses were carried out by the Microanalytical Service of the Instituto Superior Técnico. Electrospray mass spectra (ESI-MS) were run with an ion-trap instrument (Varian 500-MS LC Ion Trap Mass Spectrometer) equipped with an electrospray ion source. For electrospray ionization, the drying gas and flow rate were optimized according to the particular sample with 35 p.s.i. nebulizer pressure. Scanning was performed from m/z 0 to 1500 in methanol solution. The compounds were observed in the positive or negative ion mode (capillary voltage = 80–105 V).Synthesis of HL2
A 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 equimolar ethanolic solution (50 mL) of 2-(phenylselanyl)ethan-1-amine (0.200 g, 1 mmol) and 2-hydroxy-1-naphthaldehyde (0.172 g, 1 mmol) was slightly heated (80 °C) for 2 h with stirring. The characteristic yellow precipitate obtained by the Schiff condensation was filtered off and recrystallized from methanol. Crystals suitable for X-ray analysis were obtained by slow evaporation of an ethanol solution.
:1 equimolar ethanolic solution (50 mL) of 2-(phenylselanyl)ethan-1-amine (0.200 g, 1 mmol) and 2-hydroxy-1-naphthaldehyde (0.172 g, 1 mmol) was slightly heated (80 °C) for 2 h with stirring. The characteristic yellow precipitate obtained by the Schiff condensation was filtered off and recrystallized from methanol. Crystals suitable for X-ray analysis were obtained by slow evaporation of an ethanol solution.
Yield, 77% (based on 2-(phenylselanyl)ethan-1-amine), soluble in methanol, ethanol, DMF and acetone. Anal. calcd for C19H17NOSe (M = 354.32): C, 64.41; H, 4.84; N, 3.95. Found: C, 64.39; H, 4.80; N, 3.91%. IR (ATR, 298 K): 1433 ν(CC) and 1629 ν(CN) cm−1. MS (ESI, positive ion mode), m/z: 355.05 [Mr + H]−. 1H-NMR in DMSO-d6, δ (ppm): 3.30 and 3.90 (2CH2), 6.72–8.04 (5H, Ar–H and 6H naphthyl–H), 9.07 (s, 1H, C–H) and 14.01 (s, 1H, N–H). 13C-{1H} NMR in DMSO-d6, δ (ppm): 27.2 and 51.0 (2CH2), 105.9, 118.6, 122.3, 125.3, 125.4, 126.8, 127.9, 128.9, 129.3, 129.4, 131.6, 134.3 and 137.1 (naphthyl and aromatic carbons), 159.3 (Cnaphthyl–O−) and 176.8 (CN).
Synthesis of 1
0.01 mmol (1.70 mg) of CuCl2·2H2O was dissolved in 5 mL methanol, then 0.02 mmol (4.96 mg) of L1 was added, and the system was stirred for 5 min. After ca. 1 d at room temperature, the precipitated green crystals of 1 were filtered off and dried in air.
1: Yield, 55% (based on Cu). Calcd. for C24H22Cl2CuN2Se2 (Mr = 630.88): C 45.70, H 3.52, N 4.44; found C 45.67, H 3.50, N 4.41. MS (ESI, positive ion mode), m/z: 631.80 [Mr + H]+. IR (ATR, 298 K): 1431 ν(CC) and 1647 ν(CN) cm−1.
Synthesis of 2 and 3
0.01 mmol (1.70 mg) of CuCl2·2H2O or 0.01 mmol (2.23 mg) of CuBr2 was dissolved in 5 mL ethanol (or DMF), then 0.02 mmol (4.96 mg) of L1 was added, and the system was stirred for 5 min. After ca. 2 days (5 days in DMF) at room temperature, the formed green or brown crystals of 2 or 3, respectively, were then filtered off and dried in air.
2: Yield, 48% (based on Cu). Calcd. for C24H22Cl4Cu2N2O2Se2 (Mr = 797.29): C 36.16, H 2.78, N 3.51; found C 36.11, H 2.76, N 3.48. MS (ESI, positive ion mode), m/z: 798.73 [Mr + H]+. IR (ATR, 298 K): 1438 ν(CC) and 1640 ν(CN) cm−1.
3: Yield, 41% (based on Cu). Calcd. for C24H22Br4Cu2N2O2Se2 (Mr = 975.53): C 29.56, H 2.27, N 2.87; found C 29.49, H 2.22, N 2.84. MS (ESI, positive ion mode), m/z: 976.06 [Mr + H]+. IR (ATR, 298 K): 1435 ν(CC) and 1629 ν(CN) cm−1.
Synthesis of 4
0.01 mmol (2.20 mg) of Cu(OAc)2·2H2O was dissolved in 15 mL dichloromethane, then 0.02 mmol (7.08 mg) of HL2 and 5 drops of DMF were added, and the system was stirred for 5 min. After ca. 2 days at room temperature, the precipitated green crystals of 4 were filtered off and dried in air.
4: Yield, 38% (based on Cu). Calcd. for C38H32CuN2O2Se2 (Mr = 770.17): C 59.26, H 4.19, N 3.64; found C 59.21, H 4.14, N 3.59. MS (ESI, positive ion mode), m/z: 771.01 [Mr + H]+. IR (ATR, 298 K): 1408 ν(CC) and 1602 ν(CN) cm−1.
Synthesis of 5
0.01 mmol (1.43 mg) of CuBr2 was dissolved in 15 mL ethanol, then 0.02 mmol (7.08 mg) of HL2 and 5 drops of DMF were added, and the system was stirred for 5 min. After ca. 5 days at room temperature, the precipitated green crystals of 5 were filtered off and dried in air.
5: Yield, 43% (based on Cu). Calcd. for C38H32Br2Cu2N2O4Se2 (Mr = 1025.53): C 44.51, H 3.15, N 2.73; found C 44.48, H 3.10, N 2.72. MS (ESI, positive ion mode), m/z: 1026.76 [Mr + H]+. IR (ATR, 298 K): 1411 ν(CC) and 1605 ν(CN) cm−1.
Synthesis of 6
0.01 mmol (2.20 mg) of Cu(OAc)2·2H2O was dissolved in 15 mL methanol, then 0.02 mmol (2.66 mg) of L3 and 0.06 mmol (4.08 mg) of imidazole were added. The mixture was stirred and heated to 80 °C for 24 h and then left at room temperature for slow evaporation; deep green crystals of 6 suitable for X-rays started to form after ca. 2 days.
6: Yield, 52% (based on Cu). Calcd. for C26H24CuN18O4Se2 (Mr = 874.09): C 35.73, H 2.77, N 28.84; found C 35.69, H 2.73, N 28.80. MS (ESI, positive ion mode), m/z: 235.57 [Cu(imidazole)6]2+ and 201.92 (L3a)−. IR (ATR, 298 K): 1684 ν(CO) and 2236 ν(CN), cm−1.
Synthesis of 7
0.01 mmol (2.24 mg) of Cu(NO3)2·2H2O was dissolved in 15 mL methanol, then 0.02 mmol (2.66 mg) of L3 and 5 drops of DMF were added. The mixture was stirred and heated to 80 °C for 24 h and then left at room temperature for slow evaporation. After ca. 3 days at room temperature, the precipitated green crystals of 7 were filtered off and dried in air.
7: Yield, 48% (based on L3). Calcd. for C7H8N4O3Se (Mr = 275.14): C 30.56, H 2.93, N 20.36; found C 30.52, H 2.92, N 20.29. MS (ESI, positive ion mode), m/z: 73.05 (DMF) and 202.92 (4-cyano-1,2,5-selenadiazole-3-carboxylic acid). IR (ATR, 298 K): 3265 ν(OH), 2221 ν(CN), 1654 ν(CO) and 1580 ν(CN) cm−1. 1H-NMR in DMSO-d6, δ (ppm): 2.90 (6H of –N(CH3)2), 8.67 (1H of (CH3)2NCO) and 9.15 (1H, –COOH). 13C-{1H} NMR in DMSO-d6, δ (ppm): 34.2 (–N(CH3)2), 115.3 (CN), 145.9 (![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) COOH), 155.6 (CN), 160.3 (OOH) and 165.3 (CHO).
COOH), 155.6 (CN), 160.3 (OOH) and 165.3 (CHO).
Synthesis of 8
0.01 mmol (1.43 mg) of CuBr2 was dissolved in 10 mL methanol, then 0.02 mmol (2.66 mg) of L3 was added. The mixture was stirred and heated to 80 °C for 24 h and then left at room temperature for slow evaporation. After ca. 3 days at room temperature, the precipitated brown crystals of 8 were then filtered off and dried in air.
8: Yield, 47% (based on Cu). Calcd. for C10H8Br2CuN8O2Se2 (Mr = 653.52): C 18.38, H 1.23, N 17.15; found C 18.35, H 1.80, N 17.12. MS (ESI, positive ion mode), m/z: 654.67 [Mr + H]+. IR (ATR, 298 K): 1640 ν(CN) and 2236 ν(CN) cm−1.
X-ray structure determinations
X-ray diffraction intensities of HL2 and 1–8 were collected using a Bruker SMART APEX-II CCD area detector equipped with graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å) at 296 K. Absorption correction was applied by SADABS.16,17 The structure was solved by direct methods and refined on F2 by full-matrix least-squares using Bruker’s SHELXTL-97.18 All non-hydrogen atoms were refined anisotropically. The details of the crystallographic data for HL2 and 1–8 are summarized in Table 1. Crystallographic data for the structural analysis have been deposited in the Cambridge Crystallographic Data Centre [CCDC 2213816 (for HL2), 2214683 (for 1), 2213808 (for 2), 2213810 (for 3), 2213811 (for 4), 2213812 (for 5), 2213813 (for 6), 2213814 (for 7), and 2213815 (for 8)].†| HL2 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | |
|---|---|---|---|---|---|---|---|---|---|
| a R 1 = Σ‖Fo| − |Fc‖/Σ|Fo|. b wR2 = [Σ[w(Fo2 − Fc2)2]/Σ[w(Fo2)2]]1/2. | |||||||||
| Empirical formula | C19H17NOSe | C24H22Cl2CuN2Se2 | C24H22Cl4Cu2N2O2Se2 | C24H22Br4Cu2N2O2Se2 | C38H32CuN2O2Se2 | C44H46Br2Cu2N4O6Se2 | C26H24CuN18O4Se2 | C7H8N4O3Se | C10H8Br2CuN8O2Se2 |
| fw | 354.29 | 630.79 | 797.23 | 975.07 | 770.11 | 1171.67 | 874.09 | 275.13 | 653.52 |
| Temperature (K) | 150(2) | 150(2) | 150(2) | 150(2) | 150(2) | 150(2) | 150(2) | 150(2) | 296(2) |
| Cryst. syst. | Monoclinic | Triclinic | Monoclinic | Monoclinic | Monoclinic | Triclinic | Monoclinic | Orthorhombic | Triclinic |
| Space group | C2/c |
P |
P21/n | P21/n | P21/c |
P |
P21/n | Pca21 |
P |
| a (Å) | 30.890(2) | 9.5330(3) | 7.7872(2) | 8.1270(8) | 30.1433(6) | 8.2849(5) | 8.2710(4) | 21.779(3) | 5.4742(15) |
| b (Å) | 5.5621(3) | 10.8335(3) | 13.1414(4) | 13.0444(15) | 5.71030(10) | 11.2981(9) | 20.2764(11) | 3.8696(4) | 7.767(2) |
| c (Å) | 18.6660(12) | 12.5170(4) | 14.2735(5) | 14.4480(17) | 18.3935(4) | 12.3104(9) | 10.1890(5) | 24.330(3) | 11.921(4) |
| α, ° | 90 | 77.9040(10) | 90 | 90 | 90 | 83.249(3) | 90 | 90 | 73.520(8) |
| β, ° | 96.111(2) | 73.2640(10) | 100.9450(10) | 99.666(4) | 90.4560(10) | 81.394(3) | 101.361(2) | 90 | 79.658(8) |
| γ, ° | 90 | 73.8940(10) | 90 | 90 | 90 | 81.805(3) | 90 | 90 | 70.146(7) |
| V (Å3) | 3188.9(3) | 1177.49(6) | 1434.10(8) | 1509.9(3) | 3165.92(11) | 1122.23(14) | 1675.28(15) | 2050.4(4) | 2.384 |
| Z | 8 | 2 | 2 | 2 | 4 | 1 | 2 | 8 | 1 |
| ρ calc (g cm−3) | 1.476 | 1.779 | 1.846 | 2.145 | 1.616 | 1.734 | 1.733 | 1.783 | 2.384 |
| μ(Mo Kα) (mm−1) | 2.356 | 4.261 | 4.419 | 9.136 | 3.027 | 4.401 | 2.889 | 3.656 | 9.609 |
| F(000) | 1440 | 622 | 780 | 924 | 1548 | 582 | 870 | 1088 | 307 |
| R 1 (I ≥ 2σ) | 0.0247 | 0.0404 | 0.0184 | 0.0381 | 0.0380 | 0.0313 | 0.0200 | 0.0421 | 0.0506 |
| wR2b(I ≥ 2σ) | 0.0530 | 0.0853 | 0.0391 | 0.0915 | 0.0729 | 0.0774 | 0.0489 | 0.0886 | 0.0894 |
| GOOF | 1.030 | 1.215 | 1.048 | 1.068 | 1.022 | 1.032 | 1.047 | 1.110 | 1.060 |
Theoretical methods
The calculations reported herein were performed using the Turbomole 7.2 program.22 The crystallographic coordinates were used for the calculations of the supramolecular assemblies. We used the crystallographic coordinates for the assemblies because we are interested in evaluating the interactions as they stand in the solid state, instead of finding the most favourable geometry of the dimeric assemblies in the gas phase. The level of theory used for the calculations was PBE023-D324/def2-TZVP.25,26 The MEP surface plots were generated using the wavefunction obtained at the same level of theory and the 0.001 a.u. isosurface to simulate the van der Waals envelope. The topological analysis of the electron density was carried out according to the quantum theory of atoms in molecules (QTAIM) method proposed by R. F. W. Bader27 and represented using the VMD program.28 The noncovalent interaction plot (NCIplot)29 was used to represent the interactions in real space by plotting the reduced density gradient (RDG) isosurfaces. They were represented using the VMD software.28Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been supported by the Fundação para a Ciência e a Tecnologia (FCT) (Portugal), projects UIDB/00100/2020, UIDP/00100/2020 and LA/P/0056/2020 of Centro de Química Estrutural. A. V. G. and K. T. M. thank FCT and Instituto Superior Técnico (DL 57/2016, L 57/2017 and CEEC Institutional 2018 Programs, Contracts no: IST-ID/110/2018 and IST-ID/85/2018). V. A. A. and A. G. M. are grateful to Associação do Instituto Superior Técnico para Investigação e Desenvolvimento for their research and post-doctoral fellowships through grants no: BL110/2022-IST-ID and BL133/2021-IST-ID, respectively. K. T. M. and A. V. G. acknowledge the financial support by the Baku State University (Azerbaijan). Authors thank the Portuguese NMR Network (IST-UL Centre) and the IST Node of the Portuguese Network of mass-spectrometry.References
- (a) Noncovalent Interactions in Catalysis, ed. K. T. Mahmudov, M. N. Kopylovich, M. F. C. Guedes da Silva and A. J. L. Pombeiro, Royal Society of Chemistry, 2019 Search PubMed; (b) Supramolecular Chemistry I – Directed Synthesis and Molecular Recognition, ed. E. Weber, Springer, 2022 Search PubMed.
- (a) R. R. Knowles and E. N. Jacobsen, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 20678–20685 CrossRef CAS PubMed; (b) S. E. Wheeler, T. J. Seguin, Y. Guan and A. C. Doney, Acc. Chem. Res., 2016, 49, 1061–1069 CrossRef CAS PubMed; (c) A. J. Neel, M. J. Hilton, M. S. Sigman and F. D. Toste, Nature, 2017, 543, 637–646 CrossRef CAS PubMed; (d) F. D. Toste, M. S. Sigman and S. J. Miller, Acc. Chem. Res., 2017, 50, 609–615 CrossRef CAS PubMed; (e) K. T. Mahmudov, A. V. Gurbanov, F. I. Guseinov and M. F. C. Guedes da Silva, Coord. Chem. Rev., 2019, 387, 32–46 CrossRef CAS; (f) A. Fanourakis, P. J. Docherty, P. Chuentragool and R. J. Phipps, ACS Catal., 2020, 10, 10672–10714 CrossRef CAS PubMed; (g) Y. Jiao, X.-Y. Chen and J. F. Stoddart, Chem, 2022, 8, 414–438 CrossRef CAS.
- (a) P. A. Gale, E. N. W. Howe and X. Wu, Chem, 2016, 1, 351–422 CrossRef CAS; (b) J. Y. C. Lim and P. D. Beer, Chem, 2018, 4, 731–783 CrossRef CAS; (c) M. S. Taylor, Coord. Chem. Rev., 2020, 413, 213270 CrossRef CAS; (d) N. Biot and D. Bonifazi, Coord. Chem. Rev., 2020, 413, 213243 CrossRef CAS.
- (a) Cooperative Catalysis: Designing Efficient Catalysts for Synthesis, ed. R. Peters, Wiley-VCH, 2015 Search PubMed; (b) Ligand Design in Metal Chemistry: Reactivity and Catalysis, ed. M. Stradiotto and R. L. Lundgren, Wiley, 2016 Search PubMed.
- (a) R. Bertani, P. Sgarbossa, A. Venzo, F. Lelj, M. Amati, G. Resnati, T. Pilati, P. Metrangolo and G. Terraneo, Coord. Chem. Rev., 2010, 254, 677–695 CrossRef CAS; (b) K. T. Mahmudov, M. N. Kopylovich, M. F. C. Guedes da Silva and A. J. L. Pombeiro, Coord. Chem. Rev., 2017, 345, 54–72 CrossRef CAS; (c) J. N. H. Reek, B. de Bruin, S. Pullen, T. J. Mooibroek, A. M. Kluwer and X. Caumes, Chem. Rev., 2022, 122, 12308–12369 CrossRef CAS PubMed; (d) V. Nemec, K. Lisac, N. Bedeković, L. Fotović, V. Stilinović and D. Cinčić, CrystEngComm, 2021, 23, 3063–3083 RSC.
- (a) K. T. Mahmudov, M. N. Kopylovich, M. F. C. Guedes da Silva and A. J. L. Pombeiro, Dalton Trans., 2017, 46, 10121–10138 RSC; (b) A. Bauzá, T. J. Mooibroek and A. Frontera, ChemPhysChem, 2015, 16, 2496–2517 CrossRef PubMed.
- C. B. Aakeröy, D. L. Bryce, G. R. Desiraju, A. Frontera, A. C. Legon, F. Nicotra, K. Rissanen, S. Scheiner, G. Terraneo, P. Metrangolo and G. Resnati, Pure Appl. Chem., 2019, 91, 1889–1892 CrossRef.
- K. T. Mahmudov, A. V. Gurbanov, V. A. Aliyeva, M. F. C. Guedes da Silva, G. Resnati and A. J. L. Pombeiro, Coord. Chem. Rev., 2022, 464, 214556 CrossRef CAS.
- V. Y. Kukushkin and A. J. L. Pombeiro, Chem. Rev., 2002, 102, 1771–1802 CrossRef CAS PubMed.
- M. N. Kopylovich, K. T. Mahmudov, A. Mizar and A. J. L. Pombeiro, Chem. Commun., 2011, 47, 7248–7250 RSC.
- M. N. Kopylovich, A. Mizar, M. F. C. Guedes da Silva, T. C. O. Mac Leod, K. T. Mahmudov and A. J. L. Pombeiro, Chem.–Eur. J., 2013, 19, 588–600 CrossRef CAS PubMed.
- R. C. Jones, A. J. Canty, M. G. Gardiner, B. W. Skelton, V.-A. Tolhurst and A. H. White, Inorg. Chim. Acta, 2010, 363, 77–87 CrossRef CAS.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- V. Kumar, Y. Xu and D. L. Bryce, Chem.–Eur. J., 2020, 26, 3275–3286 CrossRef CAS PubMed.
- H. E. Gottlieb, V. Kotlyar and A. Nudelman, J. Org. Chem., 1997, 62, 7512–7515 CrossRef CAS PubMed.
- SMART & SAINT Software Reference Manuals, Version 6.22, Bruker AXS Analytic X-ray Systems, Inc., Madison, WI, 2000 Search PubMed.
- G. M. Sheldrick, SADABS Software for Empirical Absorption Correction, University of Göttingen, Germany, 2000 Search PubMed.
- G. M. Sheldrick, SHELXTL V5.1, Software Reference Manual, Bruker AXS Inc., Madison, WI, 1997 Search PubMed.
- A. Frontera and A. Bauza, Int. J. Mol. Sci., 2022, 23, 4188 CrossRef CAS PubMed.
- R. M. Gomila, A. Bauza and A. Frontera, Dalton Trans., 2022, 51, 5977–5982 RSC.
- A. V. Gurbanov, M. L. Kuznetsov, G. Resnati, K. T. Mahmudov and A. J. L. Pombeiro, Cryst. Growth Des., 2022, 22, 3932–3940 CrossRef CAS.
- R. Ahlrichs, M. Bar, M. Haser, H. Horn and C. Kolmel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6169 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- E. R. Johnson, S. Keinan, P. Mori-Sanchez, J. Contreras-Garcia, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
Footnote |
| † CCDC 2213816 (HL2), 2214683 (1), 2213808 (2), 2213810 (3), 2213811 (4), 2213812 (5), 2213813 (6), 2213814 (7) and 2213815 (8). For crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2fd00160h |
| This journal is © The Royal Society of Chemistry 2023 |