
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced hydrogen bonding via epoxide-functionalization restricts mobility in poly(ethylenimine) for CO2 capture†‡
Sichi
Li
 *a,
Marcos F.
Calegari Andrade
a,
Anthony J.
Varni
a,
Glory A.
Russell-Parks
bc,
Wade A.
Braunecker
bc,
Elwin
Hunter-Sellars
a,
Maxwell A. T.
Marple
a and
Simon H.
Pang
*a
*a,
Marcos F.
Calegari Andrade
a,
Anthony J.
Varni
a,
Glory A.
Russell-Parks
bc,
Wade A.
Braunecker
bc,
Elwin
Hunter-Sellars
a,
Maxwell A. T.
Marple
a and
Simon H.
Pang
*a
aMaterials Science Division, Lawrence Livermore National Laboratory, Livermore, CA 94550, USA. E-mail: li77@llnl.gov; pang6@llnl.gov
bNational Renewable Energy Laboratory, 15013 Denver West Parkway, Golden, Co 80401, USA
cDepartment of Chemistry, Colorado School of Mines, 1500 Illinois St., Golden, CO 80401, USA
First published on 2nd August 2023
Abstract
Free energy sampling, deep potential molecular dynamics, and characterizations provide insights into the impact of epoxide-functionalization on the hydrogen bonding and mobility of poly(ethylenimine), a promising CO2 sorbent. These findings rationalize the anti-degradation effects of epoxide functionalization and open up new avenues for designing more durable CO2 sorbents.
Effective technologies to reduce CO2 emissions and atmospheric CO2 levels are crucial in the fight against global warming and climate change. Carbon capture methods that use chemisorbents containing poly(ethylenimine) (PEI) show great promise as they are efficient in capturing CO2 from flue-gas point sources or the atmosphere.1,2 However, PEI-based materials are known to undergo oxidative degradation, which causes a loss in CO2 capacity after cycling.3,4 Overcoming this degradation will enable more rapid adoption of PEI-based sorbents for carbon capture and direct air capture.
Several recent studies have focused on improving the durability of PEI-based sorbents by exploring the potential benefits of epoxide-functionalization. For instance, Choi and Min et al. reported that functionalization of PEI supported in porous silica with 1,2-epoxybutane and epoxides with other side chains significantly improves its resistance to degradation under simulated wet flue gas conditions.5–7 Notably, this improvement is especially pronounced when used in conjunction with chelators, effectively inhibiting the catalytic impact of ppm-level metal impurities. Taking a different approach, Hamdy et al. employed epoxy resin as the cross-linker to synthesize support-free cross-linked PEI, which demonstrated maintained CO2 capacity throughout extensive aggressive adsorption/desorption cycles.8 Interestingly, epoxide-functionalization also enhances the oxidative stability of amine oligomers, such as tetraethylenepentamine.9,10 Min et al.11,12 and Guo et al.9 attribute the enhanced oxidative stability to changes in amine composition resulting from the conversion of primary amines to secondary amines through alkylation with hydroxyalkyl groups. However, contrasting findings have been reported, suggesting that amine stability cannot be solely attributed to amine identity.13,14 Another hypothesis suggests that abundant hydroxyl groups generated by epoxide-functionalization play a role in stabilizing amines through hydrogen bonding.11,12 However, the precise mechanism by which hydrogen bonding slows down amine oxidation remains largely unexplored.
The basic autoxidation scheme (BAS)15 in polymer degradation, involving radical reactions, may help explain the impact of hydrogen bonding on amine oxidation. As demonstrated by Rapoport et al., the oxidative degradation rate of poly(propylene) is influenced by the mobility of amorphous chain segments, which impacts the rate of hydrogen abstraction responsible for radical propagation across polymer chains.16 Similarly, Denisov's review of literature reports on the oxidation kinetics of polymers concludes a strong correlation between polymer segmental mobility and the kinetics of radical reactions.17 Recent work by Colin et al. show that the rate constants of peroxy radical termination and propagation in epoxy-diamine networks can be controlled by modifying molecular mobility.18 Hydrogen bonding has been found to impact chain mobility in various polymer systems.19–21 Our previous studies on PEI and aminooligomers underscore the importance of radical reactions in degradation.14,22 These findings hint at potential connections between epoxide-functionalization, hydrogen bonding, polymer mobility, and degradation kinetics of amine sorbents.
In BPEI and epoxide-functionalized BPEI, primary (1°), secondary (2°), and tertiary (3°) amines, as well as hydroxyl groups (OH) resulting from funtionalization, have the potential to engage in hydrogen bonding networks. To quantitatively assess the strengths of these potential hydrogen bonds (H bonds), we use small molecules containing hydroxyl and/or amino groups as proxies (Fig. S1, ESI‡) and compute the free energy profiles of intermolecular H bond acceptor–donor interactions using constrained ab initio molecular dynamics (AIMD) in conjunction with the blue-moon ensemble method. For further computational details, please refer to ESI,‡ Section S1.1 and Fig. S2, S3.
The computed free energy profiles and bond free energies (ΔF) at 298 K for various H bonds are presented in Fig. 1. Among the amine–amine H bonds, only those involving a 2° amine as the donor exhibit shallow and elongated free energy wells, indicating weaker interactions. The remaining amine–amine H bonds have ΔF values greater than or equal to 0 kJ mol−1, suggesting thermodynamically unfavorable interactions. In contrast, the H bonds involving OH as the donor display stronger interactions, with well-defined free energy wells and ΔF values ranging from −7 to −13 kJ mol−1. Further simulations show that the H bond between a 1° amine and OH remains stable even at elevated temperatures (Fig. S4, ESI‡), indicating its extended stability under typical CO2 desorption and amine regeneration conditions. This stability is attributed to the small ΔS derived from the linear relationship between ΔF and temperature.
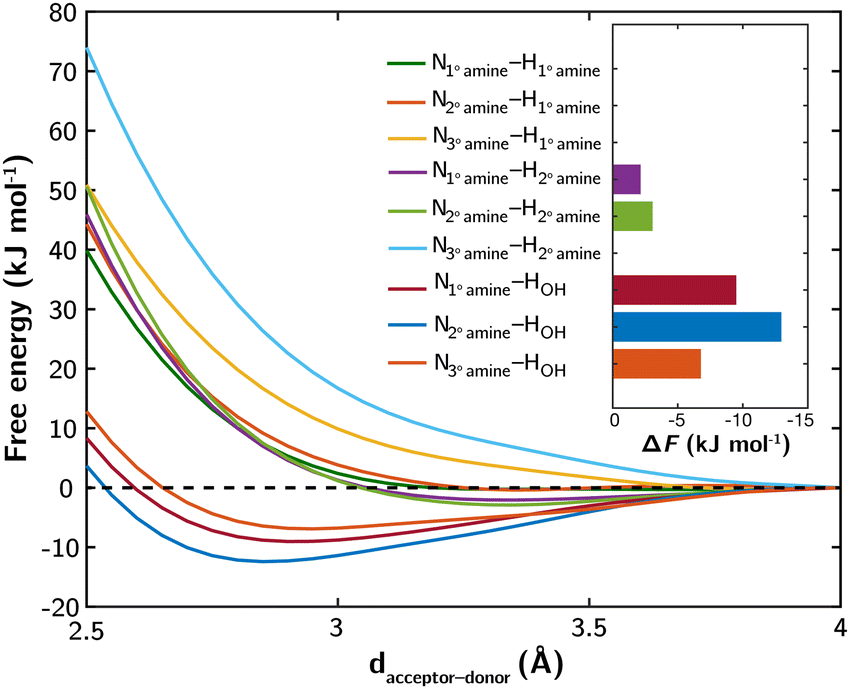 | ||
| Fig. 1 Free energy profiles of nine examined hydrogen bonds involving amines and hydroxyl groups at 298 K. Bond free energies (ΔF), characterized as the energy of local minima at the bound states relative to the unbound states, are superimposed. | ||
To investigate the hydrogen bonding environment in condensed-phase polymeric systems, we employ SCAN-trained deep potential molecular dynamics (DPMD) simulations23 on liquid TETA, representing the oligomeric form of PEI. Within these simulations, a fraction of TETA molecules are replaced with functionalized TETA, where propylene oxide (PO) is attached to one of the 1° amine sites. Details regarding the simulation cell containing thousands of atoms, training of deep neural potentials, and DPMD parameters can be found in ESI,‡ Section S1.2.
Fig. 2a presents the partial radial distribution functions between N and H (gN–H(r)) of amines and OH groups based on 1.5 ns of DPMD trajectory. The range between 2 and 3 Å corresponds to the characteristic bond distance of N–H hydrogen bonds. The intensity and shape of the peaks vary across different N–H interactions. Notably, the peak associated with N2° amine–HOH is the strongest and sharpest, consistent with its highest bond strength (as shown in Fig. 1). The weak peak associated with N1° amine–HOH is due to the abundance of 2° amines and the prevalence of N2° amine–HOH hydrogen bonds, which utilize most of the available OH groups. Among N–H interactions involving only amines, those with N2° amine exhibit more prominent peaks than N1° amine. Multiple peaks beyond 3 Å in the amine–amine gN–H(r) represent neighboring amines on the same TETA molecule.
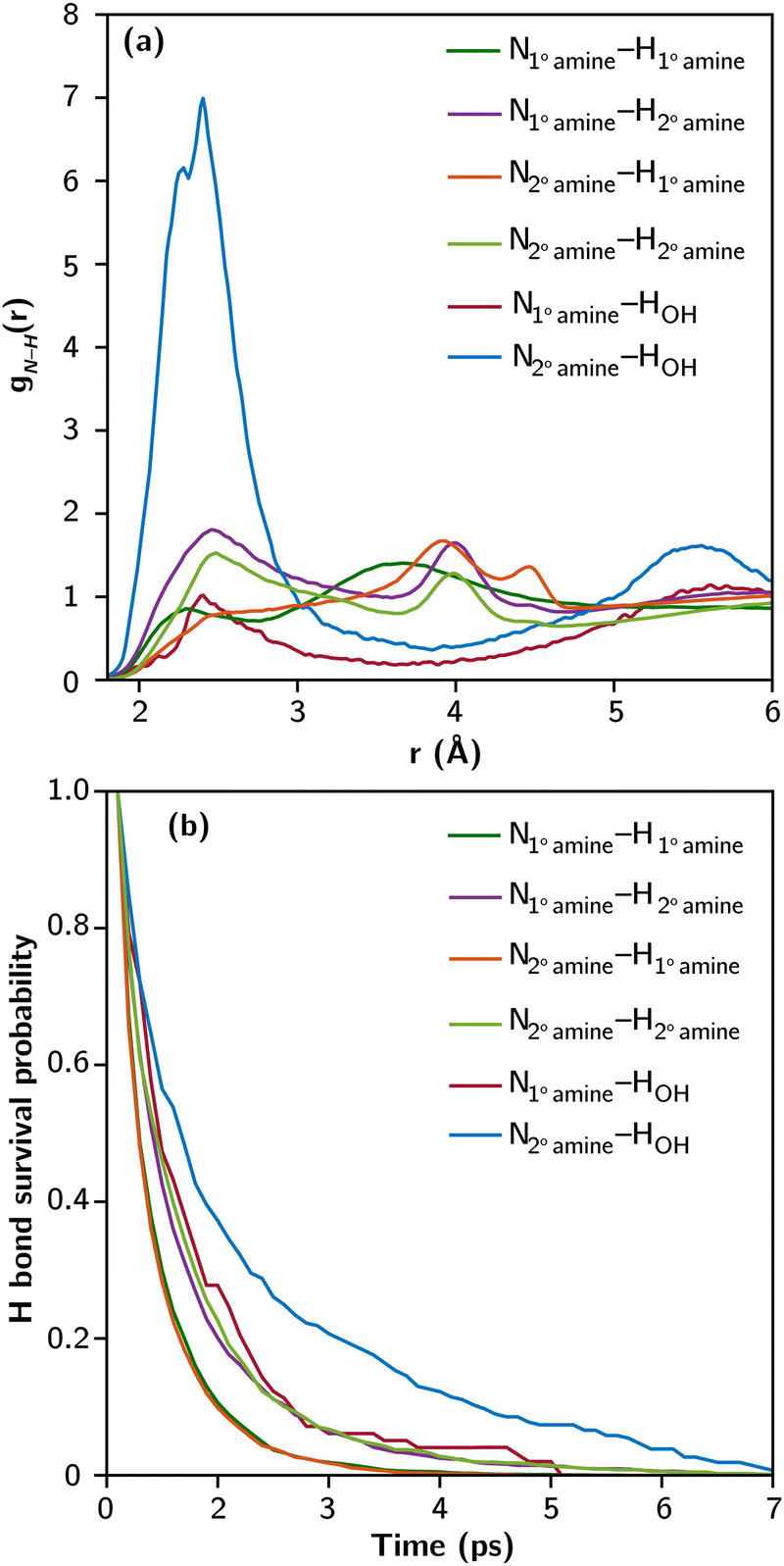 | ||
| Fig. 2 (a) Partial radial distribution functions between N and H involving each type of N–H hydrogen bond potentially present in the partially PO-functionalized liquid TETA, and (b) their survival probability averaged over all hydrogen bonds of the same type from the DPMD trajectory. | ||
The dynamics of hydrogen bonds were investigated by analyzing their survival probability, representing the history-dependent distribution of bond lifetimes. Fig. 2b shows the survival probability of N–H hydrogen bonds in the partially functionalized liquid TETA at room temperature. These bonds typically have picosecond lifetimes, slightly shorter than O–H hydrogen bonds in liquid water. The rates of decay in the survival probability inversely correlate with the ΔF values from Fig. 1, indicating that bond strength primarily influences the lifetime of N–H hydrogen bonds. Notably, hydrogen bonds involving OH groups exhibit longer lifetimes and greater stability, experiencing less fluctuation in bond breaking and reforming at equilibrium.
To further evaluate the impact of hydroxyl functionalities in aminopolymer systems, two samples of branched poly(ethylenimine) (BPEI, Mw = 800 g mol−1) were treated with different amounts of propylene oxide to yield oxygen to nitrogen molar ratios of 0.2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (0.2 O:N PO-BPEI) and 0.4:1 (0.4 O:N PO-BPEI), respectively (Fig. 3 and see ESI,‡ Section S2.1 for synthetic details and NMR spectra). The physical properties of these two materials were characterized by a variety of techniques and compared to unmodified BPEI, as described in the following sections.
:1 (0.2 O:N PO-BPEI) and 0.4:1 (0.4 O:N PO-BPEI), respectively (Fig. 3 and see ESI,‡ Section S2.1 for synthetic details and NMR spectra). The physical properties of these two materials were characterized by a variety of techniques and compared to unmodified BPEI, as described in the following sections.
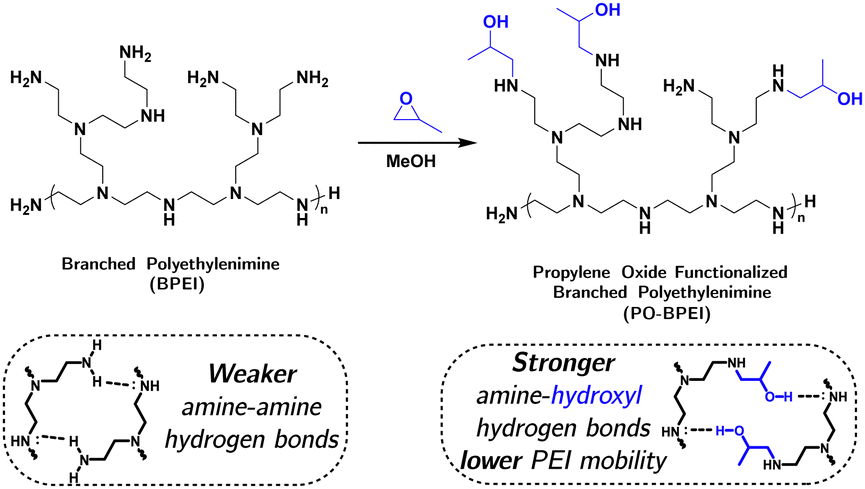 | ||
| Fig. 3 Synthesis of propylene oxide functionalized branched poly(ethylenimine) (PO-BPEI) and the consequence of resulting hydroxyl groups on hydrogen bonding. | ||
DSC experiments were conducted on BPEI, 0.2 O:N PO-BPEI, and 0.4 O:N PO-BPEI using a temperature range of −120 °C to 100 °C and a ramp rate of 10 °C min−1. Glass transition temperatures (Tg) were determine based on their respective half-widths calculated using estimated Tons and Tend values shown in Fig. S9–S11 (ESI‡). Fig. 4a illustrates that unmodified BPEI had a Tg of −70 °C, while 0.2 O:N PO-BPEI and 0.4 O:N PO-BPEI exhibited higher Tg values of −50 °C and −36 °C, respectively. The 0.2 O:N PO-BPEI (with a molecular weight of approximately 1000 g mol−1) displayed a similar Tg to a higher molecular weight (25000 g mol−1) unmodified BPEI.24 This suggests that the increased strength of hydrogen bonding between polymer chains in the PO-BPEI materials is the primary factor contributing to their higher Tg values, rather than the increase in molecular weight.
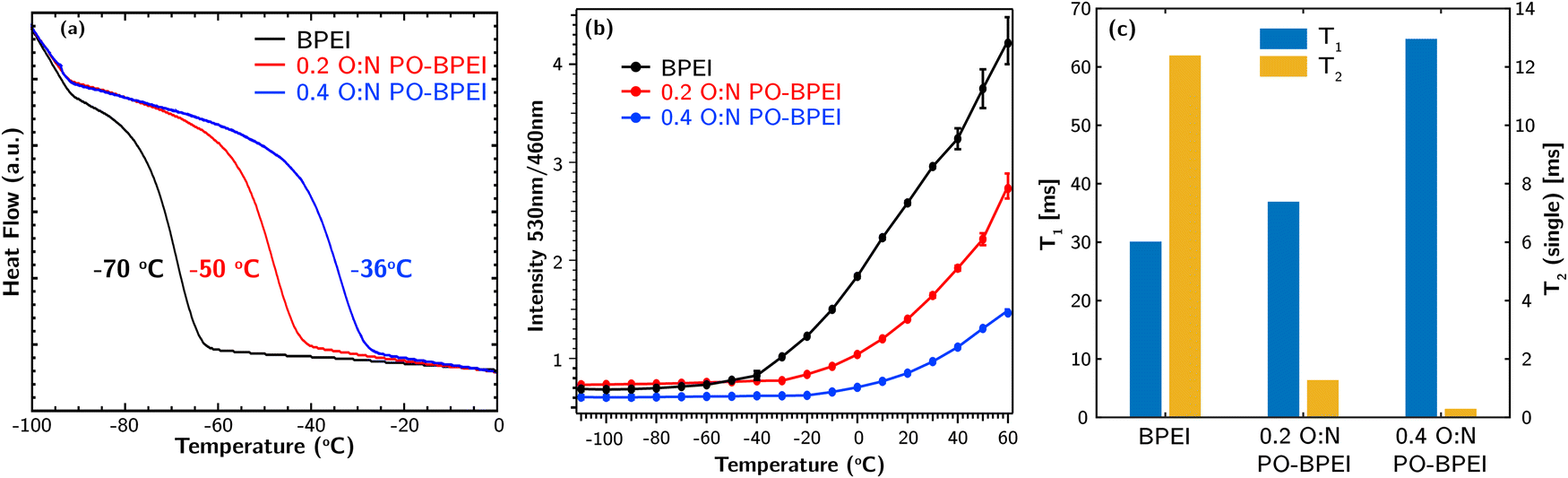 | ||
| Fig. 4 (a) DSC traces and estimated Tg values, (b) temperature dependence of the ratiometric fluorescence intensity (530 nm/460 nm) with 0.5 wt% THPE dissolved in, and (c) NMR relaxometry (T1 and T2) for BPEI, 0.2 O:N PO-BPEI, and 0.4 O:N PO-BPEI. | ||
We used ratiometric fluorescence to provide additional complementary mobility data for the three polymer samples across a wide range of temperatures, following our previous work.24,25 Tetrakis(4-hydroxyphenyl)ethylene (THPE) was employed as the fluorescent probe molecule in this work. THPE emits near 460 nm in a glassy matrix and 530 nm in a viscous solution (representative spectra are illustrated in Fig. S12 of the ESI‡). We employ the ratio of emission at these two wavelengths as a measure of the relative mobility of each polymer matrix. Further details are described in ESI,‡ Section S2.3.
As shown in Fig. 4b, between −110 °C and −60 °C, there is essentially no change in the ratiometric response of the probe in any of the polymers. In BPEI, the emission spectrum begins shifting as the sample is heated above −60 °C, consistent with literature data for this system and our DSC data that indicate the matrix is now in a rubbery state. A similar response is observed above −30 °C from the 0.2 O:N PO-BPEI sample and above −20 °C from 0.4 O:N PO-BPEI, also consistent with DSC. We can further compare the relative mobility of each sample at a given temperature. For instance, the data indicate that at room temperature, the mobility of these polymers follows the trend of BPEI > 0.2 O:N PO-BPEI > 0.4 O:N PO-BPEI. The lower mobility of PO-BPEI and its dependence on O:N molar ratio are fully consistent with the conclusions drawn from DSC measurements.
Finally, we turned to NMR relaxometry experiments to rapidly determine spin relaxation properties of the three samples at room temperature. The experimental details can be found in ESI,‡ Section S2.4. These experiments allowed us to extract both the spin–lattice relaxation time (T1) and spin–spin relaxation time (T2) for each sample.26T1 reflects motion occurring on large length scales, such as translational diffusion. Lower T1 indicates higher molecular mobility. On the other hand, T2 serves as an indicator of chain stiffness, with higher values indicating greater chain mobility.
Fig. 4c presents the measured T1 and T2 values for the synthesized samples, with additional statistics provided in Table S1 (ESI‡). The results indicate that upon PO-functionalization, T1 increases while T2 decreases. A marginal increase in T1 is observed at a 0.2 O:N functionalization level, but a more significant increase occurs at an O:N ratio of 0.4. In contrast, even at a 0.2 O:N functionalization level, T2 shows a significant decrease, which further diminishes five-fold at an O:N ratio of 0.4. These observations suggest that small amounts of functionalization primarily affect chain stiffness or mobility, while extensive functionalization reduces both translational and chain mobility of BPEI. As reported by Choi et al.,5 the stabilizing effect of 1,2-epoxybutane-functionalization kicks in only when O:N is higher than 0.37. These findings along with our T1 and T2 measurements suggest that enhancing the oxidative stability of PEI necessitates a substantial extent of hydrogen bonding, resulting in a distinct reduction in molecular mobility at larger length scales. Alternatively, it could imply that as polymer mobility continues to decrease, the rate-determining step of oxidative degradation transitions from mobility-independent to strongly mobility-dependent radical propagation reactions.
In summary, controlled simulations and characterizations provide strong evidence for the impact of epoxide-functionalization on reducing mobility in BPEI. The decreased mobility is attributed to robust hydrogen bonding interactions between hydroxyl functional groups and amines, supported by molecular simulations. Illustrated in Fig. 3, epoxide-functionalization transforms primary amines into secondary amines with anchored alkyl tails containing hydroxyl groups, enabling more stable hydrogen bonding. The presence of stable hydrogen bonding restricts polymer chain mobility, slowing down the detrimental radical propagation reactions. However, polymer mobility is unlikely the sole factor influencing degradation kinetics, but becomes relevant in kinetic regimes where mobility-dependent reactions, like radical propagation, play a determining role.
The insights gained from these findings shed light on future sorbent design. To enhance stability, future research can explore methods to promote a more uniform distribution of hydroxyl groups in the polymer structure. Incorporating functional monomers during polymerization could facilitate this. Furthermore, incorporating functional groups with stronger hydrogen bond properties than hydroxyl groups, as well as exploring weak acid–base interactions with amines for increased stability without affecting CO2 adsorption, are potential avenues for further investigation.
This work was supported by the U.S. Department of Energy (DOE), Office of Science, Basic Energy Sciences, Materials Sciences and Engineering Division. Work at Lawrence Livermore National Laboratory and National Renewable Energy Laboratory was performed under the auspices of the DOE under Contract DE-AC52-07NA27344 and Contract No. DE-AC36-08GO28308, respectively. National Energy Research Scientific Computing Center (BES-ERCAP0023097) and the LLNL Grand Challenge Program provided the computational resources.
Conflicts of interest
There are no conflicts to declare.References
- A. M. Varghese and G. N. Karanikolos, Int. J. Greenhouse Gas Control, 2020, 96, 103005 CrossRef CAS.
- X. Shen, H. Du, R. H. Mullins and R. R. Kommalapati, Energy Technol., 2017, 5, 822–833 CrossRef CAS.
- M. Jahandar Lashaki, S. Khiavi and A. Sayari, Chem. Soc. Rev., 2019, 48, 3320–3405 RSC.
- A. Heydari-Gorji and A. Sayari, Ind. Eng. Chem. Res., 2012, 51, 6887–6894 CrossRef CAS.
- W. Choi, K. Min, C. Kim, Y. S. Ko, J. W. Jeon, H. Seo, Y.-K. Park and M. Choi, Nat. Commun., 2016, 7, 12640 CrossRef CAS PubMed.
- K. Min, W. Choi, C. Kim and M. Choi, ACS Appl. Mater. Interfaces, 2018, 10, 23825–23833 CrossRef CAS PubMed.
- W. Choi, J. Park, C. Kim and M. Choi, Chem. Eng. J., 2021, 408, 127289 CrossRef CAS.
- L. B. Hamdy, R. J. Wakeham, M. Taddei, A. R. Barron and E. Andreoli, Chem. Mater., 2019, 31, 4673–4684 CrossRef CAS.
- M. Guo, S. Liang, J. Liu, J. Jin and J. Mi, ACS Sustainable Chem. Eng., 2020, 8, 3853–3864 CrossRef CAS.
- A. Goeppert, H. Zhang, R. Sen, H. Dang and G. K. S. Prakash, ChemSusChem, 2019, 12, 1712–1723 CrossRef CAS PubMed.
- S. Park, K. Choi, H. J. Yu, Y.-J. Won, C. Kim, M. Choi, S.-H. Cho, J.-H. Lee, S. Y. Lee and J. S. Lee, Ind. Eng. Chem. Res., 2018, 57, 4632–4639 CrossRef CAS.
- K. Min, W. Choi, C. Kim and M. Choi, Nat. Commun., 2018, 9, 726 CrossRef PubMed.
- C.-J. Yoo, S. J. Park and C. W. Jones, Ind. Eng. Chem. Res., 2020, 59, 7061–7071 CrossRef CAS.
- S. Li, M. R. Cerón, H. V. Eshelman, A. J. Varni, A. Maiti, S. Akhade and S. H. Pang, ChemSusChem, 2023, 16, e202201908 CAS.
- L. Bateman, G. Gee and E. K. Rideal, Proc. R. Soc. London, Ser. A, 1948, 195, 376–391 CAS.
- N. Rapoport, S. Berulava, A. Kovarskii, I. Musayelyan, Y. Yershov and V. Miller, Polym. Sci. U.S.S.R., 1975, 17, 2901–2909 CrossRef.
- Y. Denisov, Polym. Sci. U.S.S.R., 1977, 19, 2893–2905 CrossRef.
- X. Colin, F. Essatbi, J. Delozanne and G. Moreau, Polym. Degrad. Stab., 2020, 181, 109314 CrossRef CAS.
- N. S. Murthy, J. Polym. Sci., Part B: Polym. Phys., 2006, 44, 1763–1782 CrossRef CAS.
- Q. Liu, C. Wang, Y. Guo, C. Peng, A. Narayanan, S. Kaur, Y. Xu, R. A. Weiss and A. Joy, Macromolecules, 2018, 51, 9294–9305 CrossRef CAS.
- B. Wu, W. Chassé, A. Heise, A. P. M. Kentgens, D. F. Brougham and V. M. Litvinov, Mater. Chem. Front., 2022, 6, 990–1004 RSC.
- J. Racicot, S. Li, A. Clabaugh, C. Hertz, S. A. Akhade, E. W. Ping, S. H. Pang and M. A. Sakwa-Novak, J. Phys. Chem. C, 2022, 126, 8807–8816 CrossRef CAS.
- L. Zhang, J. Han, H. Wang, R. Car and E. Weinan, Phys. Rev. Lett., 2018, 120, 143001 CrossRef CAS PubMed.
- H. Correll, N. Leick, R. E. Mow, G. A. Russell-Parks, S. H. Pang, T. Gennett and W. A. Braunecker, J. Phys. Chem. C, 2022, 126, 10419–10428 CrossRef CAS.
- G. A. Russell-Parks, N. Leick, M. A. T. Marple, N. A. Strange, B. G. Trewyn, S. H. Pang and W. A. Braunecker, J. Phys. Chem. C, 2023 DOI:10.1021/acs.jpcc.3c03653.
- B. Blümich, J. Perlo and F. Casanova, Prog. Nucl. Magn. Reson. Spectrosc., 2008, 52, 197–269 CrossRef.
Footnotes |
| † The views and opinions of the authors expressed herein do not necessarily state or reflect those of the United States Government or any agency thereof. Neither the United States Government nor any agency thereof, nor any of their employees, makes any warranty, expressed or implied, or assumes any legal liability or responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product, or process disclosed, or represents that its use would not infringe privately owned rights. |
| ‡ Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cc02702c |
| This journal is © The Royal Society of Chemistry 2023 |