
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConcurrent tandem catalysis enabled by nanomechanical motion in heteroleptic four-component dual-catalyst machinery†
Emad
Elramadi‡
 ,
Sohom
Kundu‡
,
Debabrata
Mondal
and
Michael
Schmittel
*
,
Sohom
Kundu‡
,
Debabrata
Mondal
and
Michael
Schmittel
*
Center of Micro and Nanochemistry and (Bio)Technology, Organische Chemie I, School of Science and Technology, University of Siegen, Adolf-Reichwein-Str. 2, Siegen D-57068, Germany. E-mail: schmittel@chemie.uni-siegen.de; Tel: +49(0) 2717404356
First published on 3rd April 2023
Abstract
When the basic ligand 3 was added to the heteroleptic three-component slider-on-deck [Ag3(1)(2)]3+ (sliding frequency k298 = 57 kHz), it operated as a moderate brake pad (k298 = 45 kHz). Due to motion in the resulting four-component slider-on-deck [Ag3(1)(2)(3)]3+, both ligand 3 and silver(I) were continuously exposed and became catalytically active in a concurrent tandem Michael addition/hydroalkoxylation.
Inside multicellular organisms, enzymes catalyze a gamut of reactions, ranging from simple to cascaded1 and concurrent tandem reactions.2 In many cases, enzymatic activity critically depends on protein mobility, e.g., for substrate binding or product release.3 If enzymes with multiple components are concerned, then mobility may even involve cooperative motions of distinct parts for achieving rate and efficiency increase,4,5 as convincingly demonstrated by the ATP synthase.6
Only recently, chemists have been able to mimic such spectacular enzyme capabilities by developing multicomponent catalysts,7–9 but few have succeeded to connect catalytic function with intrinsic motion.10 Herein, we illustrate the suitability of four-component machinery to act as a dual catalytic effector for concurrent tandem catalysis with both processes occurring in one solution. Dual catalytic activity of a single slider-on-deck – instigated only by its dynamics – has not yet been realized.11
In earlier work, we demonstrated that the superior action of nanorotors and slider-on-deck systems as catalysts in both base-12 or metal-catalyzed13 reactions depended crucially on their motional speed. Notably, the speed correlated with the ability of the machinery to free the catalyst12 or product13 (reducing product inhibition). Such effect is conceptually different from static liberation of a catalyst as used in switchable catalysis.14,15
In detail, the four-component slider-on-deck was constructed from deck 1,16 biped 2, silver(I) ions and the chelating base 3 as shown in Scheme 1 (for synthesis, see ESI,† Chapter 1). The resulting catalytic machinery [Ag3(1)(2)(3)]3+ exhibited a stochastic sliding motion that in parallel liberated the silver(I) sites and base 3 for catalysis. The resulting dual-catalyst machinery proved to be active in concurrent tandem base and silver(I)-catalyzed reactions, while dual catalysis in static reference structures was minor.
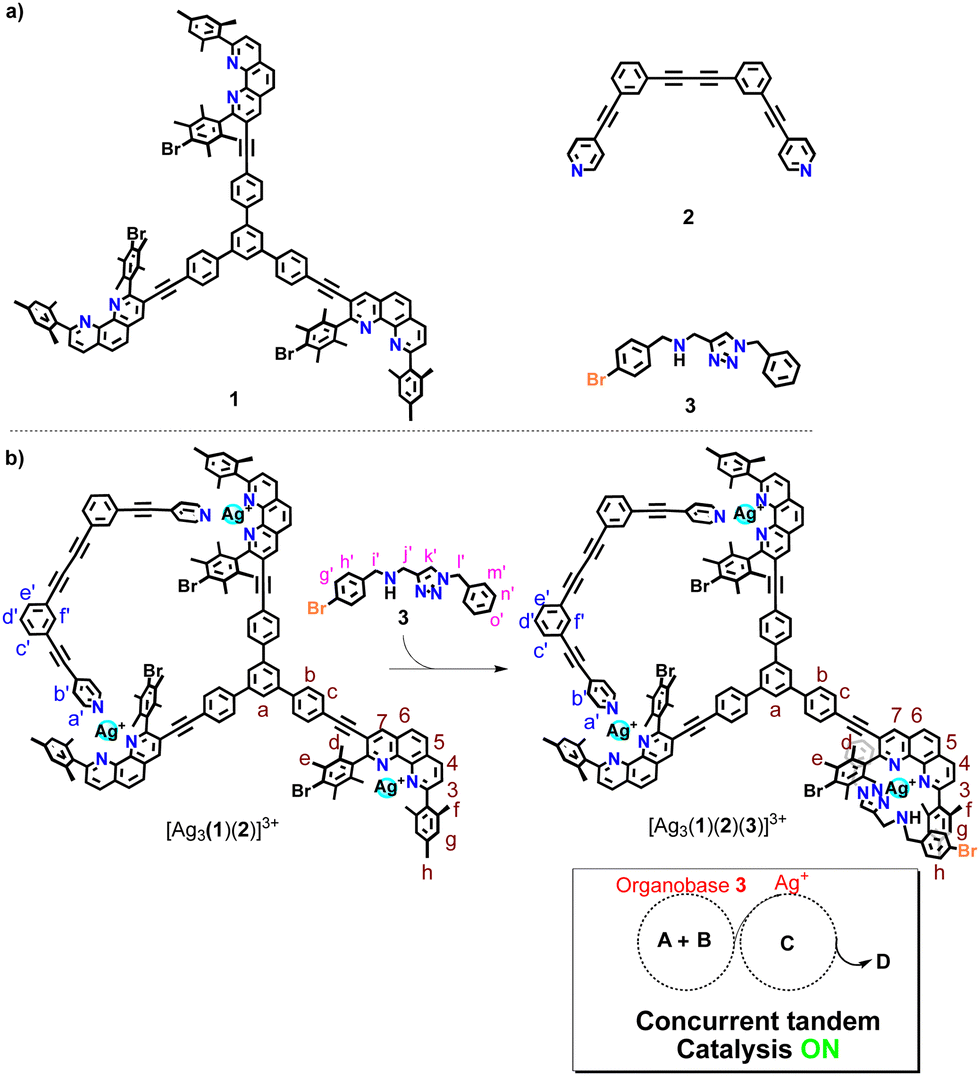 | ||
| Scheme 1 (a) Chemical structure of ligands 1–3. (b) Transformation of the three- into the four-component dual-catalyst machinery. Both organobase 3 and the silver(I) centers are liberated due to the sliding motion in [Ag3(1)(2)(3)]3+, which enables concurrent tandem catalysis. A & B represent the substrates, C denotes the intermediate product and D the final product of the concurrent tandem reaction. | ||
Before designing the main system, model studies were required to warrant the operation of the catalytic machinery. Thus, the chelating amine 3 and AgBF4 were mixed in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 2:1 ratio to investigate their combined catalytic activity. At a ratio of 1:1, 1H NMR spectroscopy showed formation of complex [Ag(3)]+. In case of a 2:1 ratio, the bishomoleptic complex [Ag(3)2]+ was furnished (ESI,† Fig. S16).
:1 and 2:1 ratio to investigate their combined catalytic activity. At a ratio of 1:1, 1H NMR spectroscopy showed formation of complex [Ag(3)]+. In case of a 2:1 ratio, the bishomoleptic complex [Ag(3)2]+ was furnished (ESI,† Fig. S16).
To check the potential of both silver complexes to concurrently perform the base-catalyzed Michael addition 4 + 5 → 6 and the silver(I)-catalyzed cyclization of 7 → 8 (Scheme 2), we mixed 4, 5 and 7 in CD2Cl2 in presence of the silver(I) complex (either [Ag(3)]+ or [Ag(3)2]+) at a ratio 10:10:10:1. At room temperature (for time, see Table 1) the Michael addition catalyzed by base 3 afforded only small amounts of 6. The yields (11% vs. 13%) were similar although in [Ag(3)2]+ the base concentration formally was 2-fold the one in [Ag(3)]+. In contrast, the yield of the silver(I)-catalyzed reaction was high with [Ag(3)]+ and ca. 8-fold less with [Ag(3)2]+ (Table 1).
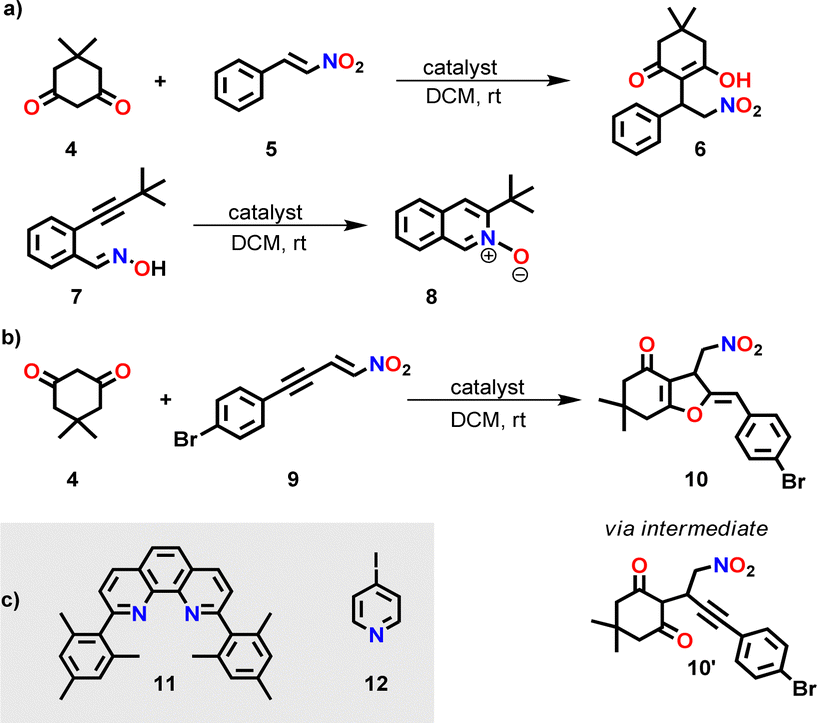 | ||
| Scheme 2 (a) Parallel one-pot base-catalyzed Michael addition and silver(I)-catalyzed cyclization. For catalyst(s), see Tables 1 and 2. (b) Sequential Michael addition/hydroalkoxylation reaction (for mechanism, see ESI,† Scheme S4).17 (c) Further ligands used in this study. | ||
| Yield with [Ag(3)]+ | Yield with [Ag(3)2]+ | |||||
|---|---|---|---|---|---|---|
| Time (h) | Of 8 (Ag+ cat.) (%) | Of 6 (base cat.) (%) | Of 10 (%) | Of 8 (Ag+ cat.) (%) | Of 6 (base cat.) (%) | Of 10 (%) |
| 2 | 6 | <1 | 1 | 0 | <1 | 1 |
| 7 | 38 | 6 | 2 | 2 | 7 | 7 |
| 14 | 68 | 11 | 4 | 9 | 13 | 11 |
Further investigation with [Ag(3)]+ or [Ag(3)2]+ as catalyst toward the concurrent tandem reaction of 4 and 9 (in a ratio 1:10:10) affording 10 (Scheme 2b and Table 1) revealed 4% and 11% yield of 10 after 14 h, respectively, and in case of [Ag(3)2]+ only a minor increase of the yield after 40 h, i.e., to 17% (ESI,† Fig. S32–S35). Thus, high dual catalytic activity cannot be realized by simply combining chelating base 3 and silver ions.
We carried out further model studies to predict the catalytic strength of the final assembly [Ag3(1)(2)(3)]3+, if it were static! Thus, benzyl amine 3, AgBF4, 2,9-dimesitylphenanthroline (11) and 4-iodopyridine (12) were mixed in a 1:3:3:2 ratio to furnish 2 × [Ag(11)(12)]+ + [Ag(3)(11)]+, reflecting the three coordination units in the assembly [Ag3(1)(2)(3)]3+. While silver catalysis was ON, base catalysis was completely OFF (ESI,† Table S1). Apparently, base 3 remains strongly bound to [Ag(11)]+ as the binding constant is logK = 5.96 (see ESI,† Fig. S45). Hence, the question emerged, whether the motion of biped 2 in the slider-on-deck [Ag3(1)(2)(3)]3+ would lead to a steady exposure of both the base and silver(I)-ions thus setting up a dual-catalyst machinery for concurrent tandem reactions.
Upon mixing 1, 2 and silver(I) in 1:1:3 ratio at room temperature in CD2Cl2, the three-component slider-on-deck immediately self-assembled in quantitative yield, as evidenced by NMR and mass spectroscopy. The 1H NMR of [Ag3(1)(2)]3+ shows a single signal set as both pyridine feet of biped 2 are sliding across all three silver(I) phenanthroline units of deck [Ag3(1)]3+. Since the pyridine feet of 2 are placed between the shielding phenanthroline's aryl groups, diagnostic upfield shifts are seen for proton signals a′-H and b′-H from 8.62 and 7.38 ppm in 2 to 6.84 and 7.32 ppm, respectively, despite their binding to the silver(I) ions. In contrast to the inner deck's proton signals, i.e., a-, b- and c-H, that remained constant, most phenanthroline signals are slightly shifted downfield when compared to those in [Ag3(1)]3+ (Fig. 1). Such finding suggests that the positive charge at the silver(I) is less neutralized by the BF4− counter anion in [Ag3(1)(2)]3+ than in [Ag3(1)]3+, because motion in the former impedes close ion pairing.
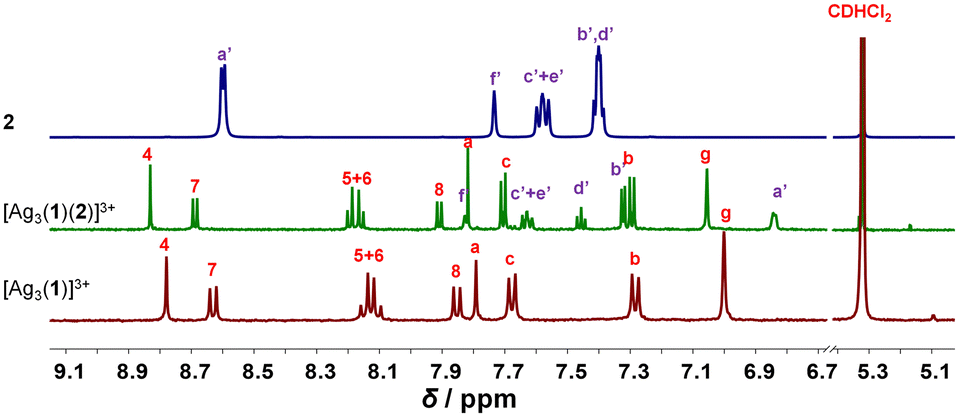 | ||
| Fig. 1 Partial 1H NMR (600 MHz, CD2Cl2, 298 K) of silver(I)-loaded deck [Ag3(1)]3+, slider-on-deck [Ag3(1)(2)]3+ and biped 2. | ||
Upon addition of 3 to [Ag3(1)(2)]3+, the four-component slider-on-deck [Ag3(1)(2)(3)]3+ was afforded as a stand-alone nano-assembly, as seen in the 1H NMR (Fig. 2) and ESI-MS (ESI,† Fig. S25). Since the proton signals of the phenanthroline core, 3-H, 4-H, 5-H, 6-H, 7-H and 8-H, exhibit identical shifts for both slider-on-deck systems, one can deduce that [Ag3(1)(2)(3)]3+ is also in motion despite the addition of ligand 3. By comparison of the shifts of proton signals i′, j′-H in 3 (δi′,j′ = 3.75, 3.83 ppm), [Ag3(1)(2)(3)]3+ (δi′,j′ = 3.33 ppm) and reference [Ag(3)(11)]+ (δi′,j′ = 3.21 ppm), similar upfield shifts were observed for both complexes attesting that 3 is situated in the shielding pocket of the silver(I) phenanthroline sites (Fig. 2). As expected from product liberation in moving rotors,13 ligand 3 in [Ag3(1)(2)(3)]3+ shows a shift that lies in between those of free 3 and [Ag(3)(11)]+, indicating that some is liberated by the motion in the four-component slider-on-deck.
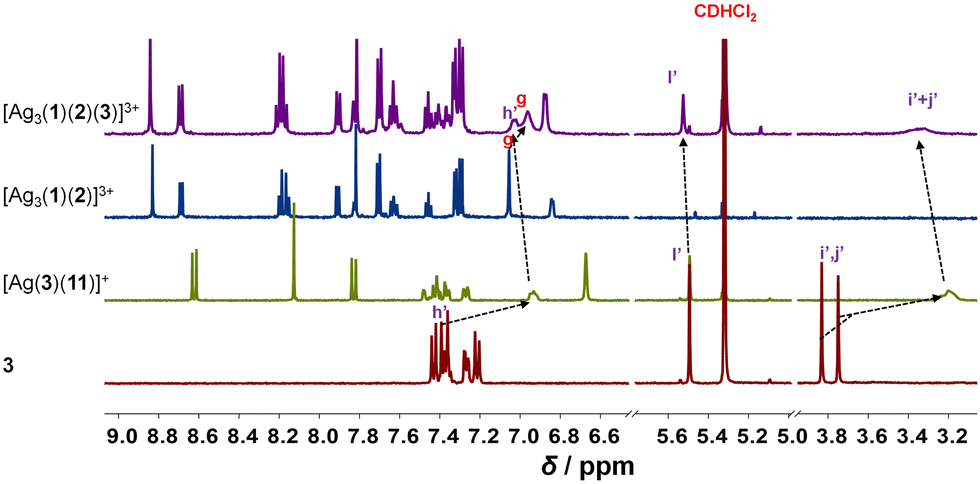 | ||
| Fig. 2 Comparison of partial 1H-NMR spectra (600 MHz, CD2Cl2, 298 K) of 3, silver(I) phenanthroline complex [Ag(3)(11)]+, three-component slider-on-deck [Ag3(1)(2)]3+, and four-component slider-on-deck [Ag3(1)(2)(3)]3+. | ||
By means of variable temperature (VT) 1H NMR spectroscopy, we determined the exchange frequency of both slider-on-deck assemblies. In case of the three-component [Ag3(1)(2)]3+, the 4-H proton signal split at −60 °C into two peaks (ratio 2:1) due to freezing the sliding of biped 2 across deck 1 on the NMR time scale (Fig. 3). Using WinDNMR,18 the 1H NMR traces were simulated over a large temperature range, providing the activation data as ΔH‡ = 45.8 kJ mol−1, ΔS‡ = 0.7 J mol−1 K−1 and ΔG‡298 = 45.6 kJ mol−1 as well as the exchange rate k298 = 57 kHz. Contrastingly, in the four-component slider-on-deck [Ag3(1)(2)(3)]3+, the proton 4-H signal split already at −40 °C. The kinetic analysis afforded k298 = 45 kHz along with the activation data ΔH‡ = 47.8 kJ mol−1, ΔS‡ = 5.4 J mol−1 K−1 and ΔG‡298 = 46.3 kJ mol−1. As a result of the addition of 3 to [Ag3(1)(2)]3+, the motion thus slowed down by 21% at room temperature and even more at lower temperature. This trend is a result of the higher positive activation entropy of [Ag3(1)(2)(3)]3+ that is increased due to the liberation of 3 into solution in the rate-determining step. As in the rate-limiting step the biped must depart from one silver(I) binding site to kick out the base 3, both the silver(I) and 3 are temporarily available for catalysis.
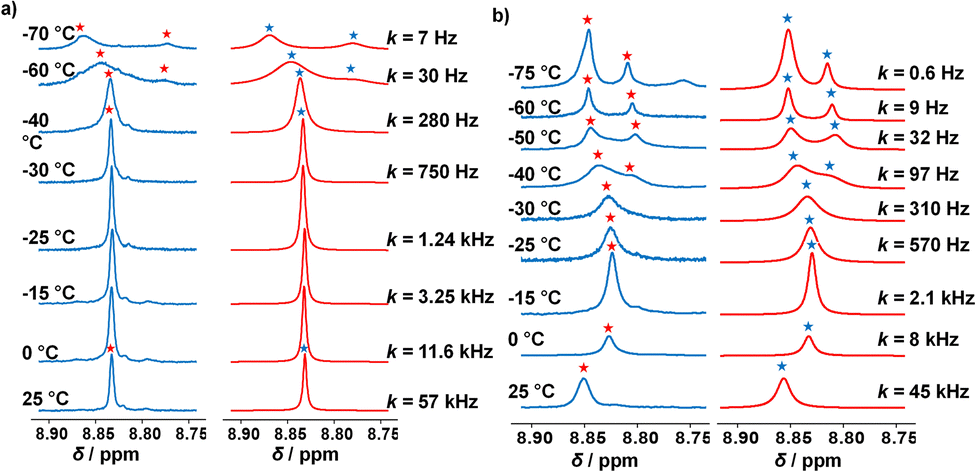 | ||
| Fig. 3 Experimental (left) and simulated (right) partial VT 1H NMR spectra (CD2Cl2, 600 MHz) of (a) [Ag3(1)(2)]3+ and (b) of [Ag3(1)(2)(3)]3+. | ||
To investigate whether both catalytic units in [Ag3(1)(2)(3)]3+ are simultaneously active we tested for concurrent base and silver(I) catalysis. Thus, the slider-on-deck [Ag3(1)(2)(3)]3+ and reactants 4, 5, and 7 were combined (1:30:30:30) in CD2Cl2 for running both transformations parallel (Scheme 2a). The reactions were monitored by 1H NMR over 14 h at room temperature, showing formation of the silver(I)-catalyzed product 8 (67%) and base-catalyzed product 6 (38%) as displayed in Table 2. The data demonstrate that the base-catalyzed formation of 6 is slower than the silver-catalyzed reaction affording 8.
| Time (h) | Yield of 6 (base cat.) (%) | Yield of 8 (Ag+ cat.) (%) | Yield of 10 (dual seq. cat.) (%) |
|---|---|---|---|
| 0 | 0 | 0 | 0 |
| 2 | 7 | 17 | 2 |
| 4 | 18 | 35 | 7 |
| 6 | 25 | 47 | 12 |
| 8 | 29 | 53 | 15 |
| 10 | 33 | 61 | 21 |
| 12 | 35 | 64 | 27 |
| 14 | 38 | 67 | 34 |
Since dual activity of this slider-on-deck is increased in comparison with the static model compounds, we probed a concurrent tandem catalysis consisting of a Michael addition followed by hydroalkoxylation (see Scheme 2b). Hence, the slider-on-deck [Ag3(1)(2)(3)]3+ and reactants 4 & 9 (1:30:30) were mixed in CD2Cl2. The kinetic profile of this reaction was monitored at room temperature by 1H NMR over 14 h at 2 h intervals. 1H NMR spectra showed 34% of 10 after 14 h (black curve in Fig. 4).
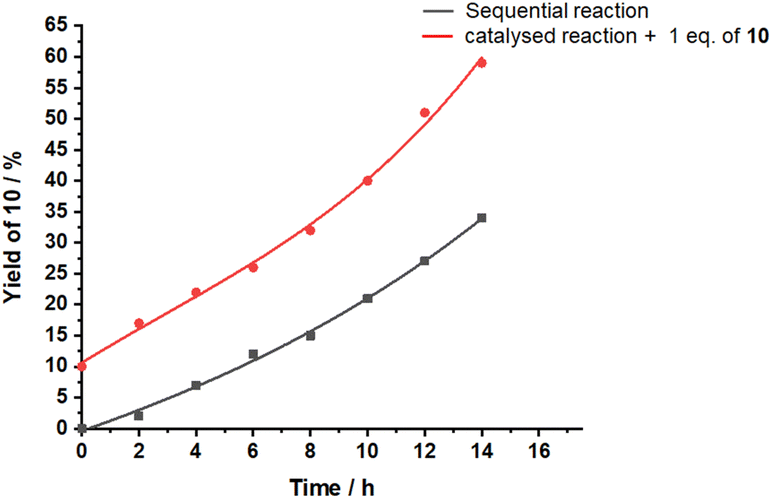 | ||
| Fig. 4 Yield of 10 with time in the tandem reaction catalysed by [Ag3(1)(2)(3)]3+. | ||
Closer inspection revealed several surprising features. Initially, the base catalyzed reaction is the rate-determining step in the tandem reaction to 10, because intermediate 10′ created by base catalysis does not show up in the 1H NMR. Only, when product 10 of the tandem base- and silver(I) catalysis had formed to more than 15%, then intermediate 10′ (Scheme 2b) became visible. Furthermore, the kinetic profile of the tandem reaction showed an increasing rate the more product 10 had formed (Fig. 4), indicative of some autocatalytic effect. To check whether it is the final product 10 that impacts on the rate, the slider-on-deck [Ag3(1)(2)(3)]3+, reactants 4, 9 and product 10 were mixed in a 1:30:30:3 ratio in CD2Cl2. Now 59% of 10 (49% after subtracting 10% of the initially added 10) was afforded after 14 h with higher rate (initial rate at t = 0: ν0 = 2.8 μM s−1) than in absence of product 10 (ν0 = 2.1 μM s−1) (Fig. 4). This effect is attributed to the competitive displacement of base 3 at the silver(I)-loaded phenanthroline by product 10 as it may act as ligand as well. Thus, in presence of sliding motion and 1 equiv. of 10, more base 3 is released.
In conclusion, we report a three-component slider-on-deck assembly [Ag3(1)(2)]3+ with a sliding speed of k298 = 57 kHz. Addition of amine 3 to the slider-on-deck converted it into a four-component dual-catalyst machinery [Ag3(1)(2)(3)]3+ with a reduced sliding speed (k298 = 45 kHz). The sliding motion in the machinery instigated two simultaneous events that enabled concurrent tandem catalysis: (a) liberation of base 3, and (b) exposure of the silver(I) sites, so that they could act synergistically19 as dual catalyst in the two-step synthesis of 10. Thus, the present system portrays a lucid example of unleashing multiple catalytic functions instigated by a single nanomechanical motion within artificial catalytic machinery.
We thank the DFG (Schm 647/22-1, no. 491092614) and the Univ. of Siegen for continued support. Moreover, we are indebted to Dr Paululat (Siegen) for the VT-1H NMR measurements.
Conflicts of interest
There are no conflicts to declare.Notes and references
- Y. Huang, A. M. Walji, C. H. Larsen and D. W. C. MacMillan, J. Am. Chem. Soc., 2005, 127, 15051–15053 CrossRef CAS PubMed.
- J.-C. Wasilke, S. J. Obrey, R. T. Baker and G. C. Bazan, Chem. Rev., 2005, 105, 1001–1020 CrossRef CAS PubMed.
- P. Ojeda-May, A. U. Mushtaq, P. Rogne, A. Verma, V. Ovchinnikov, C. Grundström, B. Dulko-Smith, U. H. Sauer, M. Wolf-Watz and K. Nam, Biochemistry, 2021, 60, 2246–2258 CrossRef CAS PubMed.
- M. J. Wiester, P. A. Ulmann and C. A. Mirkin, Angew. Chem., Int. Ed., 2011, 50, 114–137 CrossRef CAS PubMed.
- K. A. Henzler-Wildman, V. Thai, M. Lei, M. Ott, M. Wolf-Watz, T. Fenn, E. Pozharski, M. A. Wilson, G. A. Petsko, M. Karplus, C. G. Hübner and D. Kern, Nature, 2007, 450, 838–844 CrossRef CAS PubMed.
- P. Neupane, S. Bhuju, N. Thapa and H. K. Bhattarai, Biomol. Concepts, 2019, 10, 1–10 CAS.
- (a) Z. Dong, Q. Luo and J. Liu, Chem. Soc. Rev., 2012, 41, 7890–7908 RSC; (b) M. Raynal, P. Ballester, A. Vidal-Ferran and P. W. N. M. van Leeuwen, Chem. Soc. Rev., 2014, 43, 1734–1787 RSC; (c) D. Zhang, T. K. Ronson and J. R. Nitschke, Acc. Chem. Res., 2018, 51, 2423–2436 CrossRef CAS PubMed; (d) N. Mittal, M. S. Özer and M. Schmittel, Inorg. Chem., 2018, 57, 3579–3586 CrossRef CAS PubMed; (e) F. J. Rizzuto, L. K. S. von Krbek and J. R. Nitschke, Nat. Rev. Chem., 2019, 3, 204–222 CrossRef; (f) G. Olivo, G. Capocasa, D. Del Giudice, O. Lanzalunga and S. Di Stefano, Chem. Soc. Rev., 2021, 50, 7681–7724 RSC; (g) P. Howlader and M. Schmittel, Beilstein J. Org. Chem., 2022, 18, 597–630 CrossRef CAS PubMed; (h) A. W. Heard, J. M. Suárez and S. M. Goldup, Nat. Rev. Chem., 2022, 6, 182–196 CrossRef; (i) R. Saha, B. Mondal and P. S. Mukherjee, Chem. Rev., 2022, 122, 12244–12307 CrossRef CAS PubMed.
- Supramolecular Catalysis: New Directions and Developments, ed. P. W. N. M. van Leeuwen and M. Raynal, Wiley-VCH GmbH, Weinheim, 1st edn, 2022, ISBN: 9783527349029 Search PubMed.
- (a) N. Mittal, S. Pramanik, I. Paul, S. De and M. Schmittel, J. Am. Chem. Soc., 2017, 139, 4270–4273 CrossRef CAS PubMed; (b) Y. Li, X. Caumes, M. Raynal and L. Bouteiller, Chem. Commun., 2019, 55, 2162–2165 RSC; (c) E. Iniesta and A. Vidal-Ferran, Chem. Commun., 2020, 56, 6364–6367 RSC.
- L. van Dijk, M. J. Tilby, R. Szpera, O. A. Smith, H. A. P. Bunce and S. P. Fletcher, Nat. Rev. Chem., 2018, 2, 1–18 CrossRef.
- V. V. Rajasekaran, A. Ghosh, S. Kundu, D. Mondal, T. Paululat and M. Schmittel, Angew. Chem., Int. Ed., 2022, 61, e2022124 CrossRef PubMed.
- I. Paul, A. Goswami, N. Mittal and M. Schmittel, Angew. Chem., Int. Ed., 2018, 57, 354–358 CrossRef CAS PubMed.
- P. K. Biswas, S. Saha, T. Paululat and M. Schmittel, J. Am. Chem. Soc., 2018, 140(29), 9038–9041 CrossRef CAS PubMed.
- S. Gaikwad, A. Goswami, S. De and M. Schmittel, Angew. Chem., Int. Ed., 2016, 55, 10512–10517 CrossRef CAS PubMed.
- S. De, S. Pramanik and M. Schmittel, Angew. Chem., Int. Ed., 2014, 53, 14255–14259 CrossRef CAS PubMed.
- E. Elramadi, A. Ghosh, I. Valiyev, P. K. Biswas, T. Paululat and M. Schmittel, Chem. Commun., 2022, 58, 8073–8076 RSC.
- U. Kaya, P. Chauhan, K. Deckers, R. Puttreddy, K. Rissanen, G. Raabe and D. Enders, Synthesis, 2016, 3207–3216 CAS.
- H. J. Reich, NMR Spectrum Calculations: WinDNMR, Version 7.1.13, Department of Chemistry, University of Wisconsin Search PubMed.
- L. Zhou and L. Liu, Chem. Commun., 2021, 57, 5690–5693 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization, VT-NMR, spectral data. See DOI: https://doi.org/10.1039/d3cc00637a |
| ‡ Emad Elramadi and Sohom Kundu contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |