
Synthesis and characterization of polysulfides formed by the inverse vulcanisation of cyclosiloxanes with sulfur†
Kun Woo
Park
 ab,
Elizabeth A.
Tafili
ab,
Flora
Fan
ac,
Zoran
Zujovic
a and
Erin M.
Leitao
*ab
ab,
Elizabeth A.
Tafili
ab,
Flora
Fan
ac,
Zoran
Zujovic
a and
Erin M.
Leitao
*ab
aSchool of Chemical Sciences, University of Auckland, Private Bag, 92019, Auckland, 1142, New Zealand. E-mail: erin.leitao@auckland.ac.nz
bThe MacDiarmid Institute for Advanced Materials and Nanotechnology, Victoria University of Wellington, Wellington 6140, New Zealand
cDepartment of Chemistry, University of California, Berkeley, Berkeley, California 94720, USA
First published on 30th July 2022
Abstract
Inverse vulcanisation stabilizes polysulfide chains through cross-linking. This research focuses on the incorporation of cyclosiloxane cross-linkers containing multiple alkene moieties, namely tetravinyl-tetramethyl-cyclotetrasiloxane (TVTSi) and pentavinyl-pentamethyl-cyclopentasiloxane (PVPSi). Both siloxanes underwent successful inverse vulcanisation and two series of polysulfides were synthesized using different weight percentages of sulfur. Poly(S-r-TVTSi) required at least 20 wt% S in order to generate a solid product whereas poly(S-r-PVPSi) required only 10 wt% S. Interestingly, heating the polysulfide materials caused further cross-linking through the unreacted alkenes. The novel polysulfides were characterized using FTIR and NMR spectroscopies, TGA, DSC and SEM, which revealed that the products formed were inhomogeneous due to the presence of unreacted sulfur. Washing the products with CS2 removed the excess elemental sulfur. All polyslufide products were insoluble in common solvents, but were found to swell to various degrees in non-polar solvents such as hexanes, toluene and benzene, as well as dichloromethane. This led to a preliminary study into their potential use as selective solvent absorption polymers, specifically for the selective removal of hydrocarbons from water. The findings from this research will stimulate further studies towards potential applications as well into other polysulfides synthesized using inorganic cross-linkers.
Introduction
Polysulfides were first discovered in 1937 and have been synthesized and studied ever since.1 The discovery of a new synthetic method in 2013, by Pyun et al., has led to a resurgence in this field.2 The method was aptly named inverse vulcanisation due to the use of the inverse, alkene cross-linked polysulfide, to traditional rubber vulcanisation, sulfur cross-linked polyisoprene. This discovery showed promise with respect to the efficiency, simplicity, scalability and economic feasibility of the synthesis of sulfur based materials, including an expansion into novel polysulfides which were not synthetically achievable by other methods.2,3A wide variety of cross-linked polysulfides have been synthesized since 2013 due to significant interest in the properties of the materials formed.4 However, one of the drawbacks to inverse vulcanisation is the exclusion of certain cross-linkers due to the high reaction temperature required during the synthesis. This limitation was overcome by Wu et al. in 2019, with the discovery of the use of a catalyst for inverse vulcanisation. The researchers reported that metal diethyldithiocarbamate accelerators (e.g. Zn, Fe, Co, Cu, Ni, and Na) with one or two diethyldithiocarbamate ligands enabled the reaction to occur at significantly lower temperatures (135–160 instead of 180 °C).5 These new reaction conditions allowed for a substantial increase in the range of cross-linkers used and reduced the formation of H2S, a by-product that can form under certain reaction conditions. In a subsequent study, 32 different catalysts were investigated and catalogued to allow comparison and selection of catalyst depending on what variable outcome was required. It was found that stronger nucleophiles gave shorter reaction times at the cost of promoting H2S formation.6
The polysulfides synthesized by these methods show properties such as, self-healing, high resistance to swelling in solvents, oils and fuels, high stability from degradation by UV and ozone, minimal structural degradation under stress, high refractive indecies,7 the ability to transmit mid-wave infrared light,8 high affinity for metals,9 and enhanced capacity retention.10 The combination of these properties have given rise to numerous applications,11–14 including in IR sensing and imaging,7 sorption of heavy and/or valuable metals,5,15 batteries as Li–S cathode materials,16 self-healing polymers,8 functional coatings,17–19 fertiliser delivery,20,21 antibacterial surfaces,22 insulation23,24 and oil spill remediation.3
Inorganic cross-linkers are relatively unexplored in the field of inverse vulcanisation. Most polysulfide research focuses on cross-linkers that are readily accessible and rich in carbon, such as cooking oils. However, the presence of other elements in the cross-linker, such as silicon, can provide an opportunity to incorporate different functionality.
For example, silanes, containing a maximum of three chloro or alkoxy substituents attached to a Si atom can be used in surface modification via the formation of silanol intermediates.17,25 This technology has shown great promise in applications such as membranes,26 dentistry,27 road construction,28 metal corrosion protection,25 and other areas requiring strict control over surface properties. The use of silane cross-linkers for inverse vulcnisastion was explored in 2020 by Scheiger et al., where styrylethyltrimethoxysilane reacted with sulfur at the alkene followed by a polycondensation reaction of the methoxy groups in order to coat a surface.17
Siloxanes, molecules which contain Si–O–Si linkages and organic substituents (e.g. Me, Ph) attached to the Si atoms can also be used as cross-linkers. Modification of the substituents, shape, chain length and degree of cross-linking of siloxanes allow them to exist in a variety of states.29 Other notable properties of siloxanes include, high oxidative stability, conformational flexibility, bio-durability, biocompatibility, chemical inertness, hydrophobicity and low thermal conductivity.30 These properties have led to their wide-spread use, ranging from implants,31 to lubricants,32 to bakeware.33 By merging the beneficial chemical and physical properties of both polysulfides and siloxanes, we aim to increase the breadth of potential applications of polysulfide materials, such as their use as selective sorbents for contaminants and solvents, in addition to improving the self-healing nature of these materials.
Using siloxanes as cross-linkers in inverse vulcanisation reactions has been limited to three prior studies. These studies were more general and focused on the synthesis of the actual polysuflides in comparison to silane cross-linker study. The first was in recorded in 2019, by Wu et al. who was able to synthesize a polysulfide with tetravinyl-tetramethyl-cyclotetrasiloxane and 50 wt% S using a catalyst.5 The second by Anyszka et al., involved the reaction of polyhedral oligomeric silsesquioxanes with 90 wt% S.34 The resulting polysulfide was made in moderate 24.4 wt% yield, with a porous morphology after treatment with CS2 to remove unreacted sulfur. It was also observed from the characterization that unreacted C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds were noticeably present. The polysulfide produced showed high visual and morphological similarity to sulfur, with the porosity showing promise for applications for Li–S batteries and heavy metal sorption. The final study was highlighted in 2022 by Park et al. and involved cross-linking polysulfide with the simplest siloxane containing diene functionalities, 1,3-diallyl-tetramethyldisiloxane and 1,1,3,3-tetramethyl-1,3-divinyldisiloxane.35 It was observed, in this case, that the difference in cross-linker chain length and synthetic method used for the different cross-linkers, resulted to very different materials after inverse vulcanisation. For example, at a higher wt% S for the poly(S-r-vinyldisiloxane) series (e.g. 80 wt% S), a significant proportion of unreacted CC bonds were present whilst the poly(S-r-allyldisiloxane) series did not show unreacted CC bonds, even at 45 wt% S. The physical properties of the poly(S-r-vinyldisiloxane) series ranged from a soft rubbery solid (30–40 wt% S), to a hard rubbery solid (50–60 wt% S) to a brittle hard solid (65–90 wt% S). In contrast, the physical properties of the poly-(S-r-allyldisiloxane) series ranged from an orange liquid (15–30 wt% S), to a soft orange rubber (33–40 wt% S), to a hard orange rubber (45–50 wt% S) to a brittle hard orange solid (70–90 wt% S). Both series showed high stability towards commonly used laboratory solvents.
C bonds were noticeably present. The polysulfide produced showed high visual and morphological similarity to sulfur, with the porosity showing promise for applications for Li–S batteries and heavy metal sorption. The final study was highlighted in 2022 by Park et al. and involved cross-linking polysulfide with the simplest siloxane containing diene functionalities, 1,3-diallyl-tetramethyldisiloxane and 1,1,3,3-tetramethyl-1,3-divinyldisiloxane.35 It was observed, in this case, that the difference in cross-linker chain length and synthetic method used for the different cross-linkers, resulted to very different materials after inverse vulcanisation. For example, at a higher wt% S for the poly(S-r-vinyldisiloxane) series (e.g. 80 wt% S), a significant proportion of unreacted CC bonds were present whilst the poly(S-r-allyldisiloxane) series did not show unreacted CC bonds, even at 45 wt% S. The physical properties of the poly(S-r-vinyldisiloxane) series ranged from a soft rubbery solid (30–40 wt% S), to a hard rubbery solid (50–60 wt% S) to a brittle hard solid (65–90 wt% S). In contrast, the physical properties of the poly-(S-r-allyldisiloxane) series ranged from an orange liquid (15–30 wt% S), to a soft orange rubber (33–40 wt% S), to a hard orange rubber (45–50 wt% S) to a brittle hard orange solid (70–90 wt% S). Both series showed high stability towards commonly used laboratory solvents.
To expand the research of siloxane cross-linkers, we report two series of polysulfides with varied wt% S (X) containing cyclic siloxane cross-linkers, pentavinyl-pentamethyl-cyclopentasiloxane, X-poly(S-r-PVPSi), and tetravinyl-tetramethyl-cyclotetrasiloxane, X-poly(S-r-TVTSi), through traditional and catalytic inverse vulcanisation, respectively (Scheme 1). We rationalised that these two series would sit at a midway point between the complex polyhedral oligomeric silsesquioxanes (POSS) and the simple and vinyl and allyl disiloxane cross-linked in terms of materials properties.
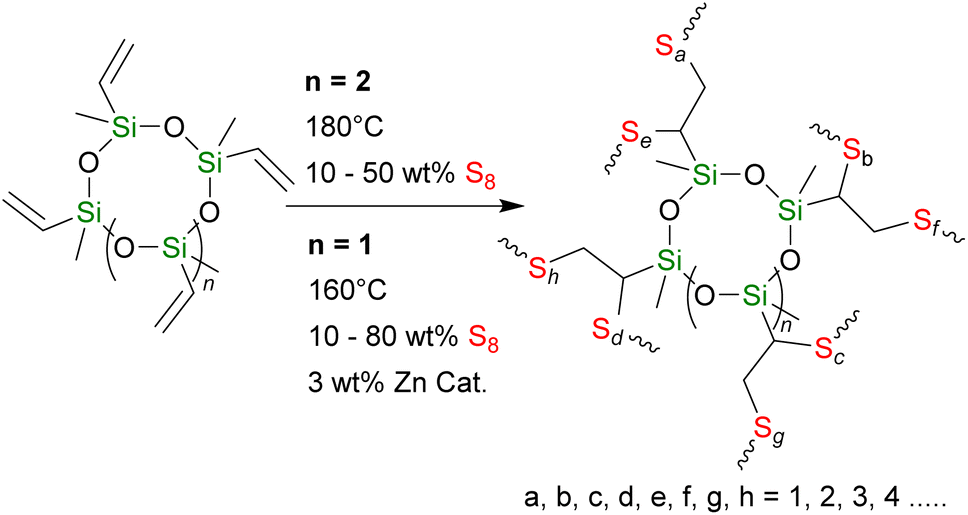 | ||
| Scheme 1 Synthesis of cyclosiloxane cross-linked polysulfides through traditional inverse vulcanisation (n = 2, top) and catalytic inverse vulcanisation (n = 1, bottom). | ||
It was previously observed with the cyclic carbon based cross-linker dicyclopentadiene, when used in inverse vulcanisation as a monomer or co-monomer, that rigidity, hardness, and solvent insolubility all increased in comparison to its noncyclic counterpart.36,37 The complex cyclosiloxane polyhedral oligomeric silsesquioxane containing 10 methacrylate units (POSS-MA) was used as a cross-linker, it formed products containing pores.38 It was hypothesized that a combination of these properties would be observed for the cyclosiloxanes.
Another intended use of these cyclic cross-linkers was to determine the source of the unreacted CC bonds in the previous studies, such as whether the catalyst or the length of the diene chain was more important in the reactivity of the CC bonds. Both series were analysed using a suite of characterization techniques such as solid state (SS) NMR spectroscopy, differential scanning calorimetry (DSC), thermogravimetric analysis (TGA), fourier-transform infrared spectroscopy (FT-IR) and scanning electron microscopy (SEM). Furthermore, solvent swelling studies were performed in an attempt to identify potential solvent sorption selectivity with these materials.
Experimental
Materials
1,3,5,7-Tetravinyl-1,3,5,7-tetramethylcyclotetrasiloxane (Gelest), carbon disulphide (Sigma-Aldrich), chloroform-D (CDCl3, Cambridge Isotope Laboratories Inc.), hexane (Scharlau), pentavinyl-pentamethyl-cyclopentasiloxane (Gelest), sodium hydroxide (ECP limited), sodium sulfate (ECP limited), sulfur (S8 powder, Riedel-de Haen), zinc diethyldithiocarbamate (97%, Sigma-Aldrich). All materials were used as received.Instrumentation
For 1H, 13C and 29Si NMR spectroscopy, solution based samples were dissolved in CDCl3 and recorded using a Bruker Avance 400 MHz spectrometer and all spectra were recorded in parts per million (ppm). Tetramethylsilane (TMS) was used to calibrate and 1H NMR and residual amounts of protium found in NMT solvents were used for reference (e.g. 7.26 ppm for CDCl3). CDCl3 (77.3 ppm) was used to reference 13C{1H} NMR spectra and tetramethylsilane (TMS at 0.0 ppm) was used as reference for 29Si{1H} NMR spectra. The resonances for 29Si{1H} NMR were obtained through a DEPT pulse sequence. Peak multiplicity is represented as follows: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, coupling constants in Hertz (Hz). Solid State NMR measurements were carried out on a Bruker AVANCE 300 (B0 = 7.05 T) spectrometer operating at 300.13 and 75.46 MHz for 1H and 13C, respectively. A double-tuning cross-polarisation magic angle spinning (CP/MAS) probe was used with 7 mm zirconia rotors and Kel-F caps. CP measurements were performed with a 5 s recycle delay for 20-poly(S-r-TVTSi) (15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 transients), 6.5 s for 40-poly(S-r-TVTSi (966 transients), 7.5 s for 80-poly(S-r-TVTSi (10000 transients), 5 s for 20-poly(S-r-TVTSi) heated (8000 transients), 5 s 20-poly(S-r-PVPSi) (15000 transients), 6 s 40-poly(S-r-PVPSi) (5000 transients) and 6 s 20-poly(S-r-PVPSi) heated (8000 transients). For the CP/MAS experiments, the 90° proton preparation pulses were 4.9 and 6 μs, followed by a 1000 μs CP contact time, 25.6 ms of data acquisition, spectral width of 40 kHz, and spinning speed of 7 kHz. Recycle delays were used to assure complete relaxation of the components present, and they were determined using 1H saturation-recovery experiments. The 13C chemical shift scale for all 13C experiments was referenced using the 13C signal of the TMS (0 ppm). The data processing was carried out using the Bruker TopSpin® software. Differential Scanning Calorimetry (DSC) was obtained using a TA Instrument Q1000 DSC, under nitrogen flow, and with heating from −80 °C to 150 °C and cooling rates of 10 °C min−1. Thermogravimetric analysis (TGA) was obtained using a TA Instrument Q500 with samples being heated to 800 °C under nitrogen at a heating rate of 10 °C min−1. Fourier-transform infrared spectroscopy (FT-IR) results were obtained using a Bruker Vertex70, between 400 cm−1 to 4000 cm−1. Scanning Electron Microscopy (SEM) images and the associated EDS spectra were obtained using Hitachi tabletop scanning electron microscope TM3030Plus.
000 transients), 6.5 s for 40-poly(S-r-TVTSi (966 transients), 7.5 s for 80-poly(S-r-TVTSi (10000 transients), 5 s for 20-poly(S-r-TVTSi) heated (8000 transients), 5 s 20-poly(S-r-PVPSi) (15000 transients), 6 s 40-poly(S-r-PVPSi) (5000 transients) and 6 s 20-poly(S-r-PVPSi) heated (8000 transients). For the CP/MAS experiments, the 90° proton preparation pulses were 4.9 and 6 μs, followed by a 1000 μs CP contact time, 25.6 ms of data acquisition, spectral width of 40 kHz, and spinning speed of 7 kHz. Recycle delays were used to assure complete relaxation of the components present, and they were determined using 1H saturation-recovery experiments. The 13C chemical shift scale for all 13C experiments was referenced using the 13C signal of the TMS (0 ppm). The data processing was carried out using the Bruker TopSpin® software. Differential Scanning Calorimetry (DSC) was obtained using a TA Instrument Q1000 DSC, under nitrogen flow, and with heating from −80 °C to 150 °C and cooling rates of 10 °C min−1. Thermogravimetric analysis (TGA) was obtained using a TA Instrument Q500 with samples being heated to 800 °C under nitrogen at a heating rate of 10 °C min−1. Fourier-transform infrared spectroscopy (FT-IR) results were obtained using a Bruker Vertex70, between 400 cm−1 to 4000 cm−1. Scanning Electron Microscopy (SEM) images and the associated EDS spectra were obtained using Hitachi tabletop scanning electron microscope TM3030Plus.
General synthesis of siloxane cross-linked polysulfides
Results and discussion
Synthesis of polysulfides through inverse vulcanization
Tetravinyl-tetramethyl-cyclotetrasiloxane (TVTSi) is unable to undergo traditional inverse vulcanisation due to its boiling point (111–112 °C) being lower than that required for the process (180 °C). It was however, able to undergo catalytic inverse vulcanisation using Zn(S2CNEt2)2, discovered by Wu et al. This catalyst allows organic compounds to undergo inverse vulcanisation at 135 °C whilst the inorganic siloxane requires 160 °C.5 Elemental sulfur and 3 wt% of the Zn catalyst was heated at 160 °C while being covered with a septum. Once the sulfur forms its radical polymeric state and turns orange, TVTSi was added (Scheme 1), resulting to the formation of two layers that merged after 30 minutes, leading to polysulfides with a range of physical properties. The synthesized series of polysulfides, X-poly(S-r-TVTSi) (X = 10–80 wt% S, increasing by increments of 10), are all black in colour due to the presence of the catalyst in the final product. The physical properties varied depending on the wt% S used. For example, 10-poly-(S-r-TVTSi) is a thin black liquid, 20-poly(S-r-TVTSi) is a soft rubbery solid, 30-poly-(S-r-TVTSi) is a harder rubbery solid and 40–80 wt% S poly(S-r-TVTSi) products are all observed to be brittle, hard solids.3 wt% of the Zn diethyldithiocarbmate catalyst was used for this reaction for a number of reasons. The higher catalyst loading enabled a shorter reaction time, with completion after 2 h, much faster than the established time frame of 3.5 h for 1 wt%. Another advantage to a high catalyst loading was the increased likelihood of activating more polysulfide chains. The activated polysulfide chains could then react with more of the vinyl groups present leading to a highly cross-linked material.35
A general trend was observed where an increase in the wt% S correlated to the brittleness of the material formed. All products were black and opaque, similar to other materials created using catalytic inverse vulcanisation (Fig. S1†).8,39 All products in this series were insoluble and stable in commonly used solvents.
Pentavinyl-pentamethyl-cyclopentasiloxane (PVPSi) has a boiling point of 261–262 °C, high enough to undergo traditional inverse vulcanisation. Using this method, a series of polysulfides, X-poly(S-r-PVPSi), with increasing amounts of sulfur (X = 10–50 wt% S, increasing in increments of 10) was generated. This method melted and heated elemental sulfur past its floor temperature (159 °C) to create and maintain radical polymeric sulfur that was reacted by adding PVPSi dropwise which formed two layers that merged after 30 minutes (Scheme 1). Similar to the previous series, a wide range of physical properties were observed in the X-poly(S-r-PVPSi) series due to the variation in quantity of sulfur used (Fig. S2†). 10-poly(S-r-PVPSi) is a soft rubbery red solid, 20-poly(S-r-PVPSi) is a slightly more brittle rubbery red solid, and 30–50 wt% S poly(S-r-PVPSi)s are slightly opaque, brittle and glass like red solids. The 30–50 wt% S poly(S-r-PVPSi) products show promise for optical ability, and all polysulfides in the series were found to be insoluble and stable in many commonly used solvents.
Characterization of siloxane cross-linked polysulfides
The FTIR spectra of the cyclosiloxane cross-linkers, TVTSi (orange, Fig. 1) and PVPSi (yellow, Fig. 1), gave peaks at 1596, 1007 and 959 cm−1. These peaks indicate the presence of Si–CHCH2 bonds.40 Peaks that correspond to C–H stretching were also observed at 3056 cm−1. After inverse vulcanisation the Si–CHCH2 peaks reduced in intensity as the wt% S increased for both the X-poly(S-r-TVTSi) series (X = 20–80 wt% S) (blue, Fig. 1) and X-poly(S-r-PVPSi) series (X = 10–50 wt% S) (grey, Fig. 1). Moreover, the presence of the sulfur chains diluted the signals as the weight percent of sulfur increased across the series (Fig. S3 and S4†).
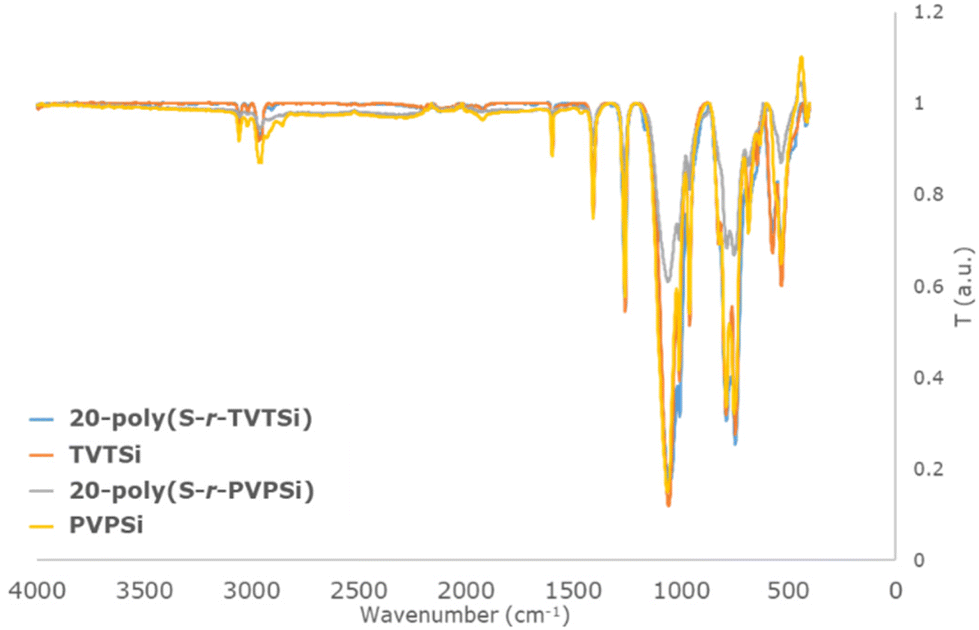 | ||
| Fig. 1 FTIR spectrum comparison of TVTSi (orange) and 20-poly(S-r-TVTSi) (blue), 20-poly(S-r-PVPSi) (grey) and PVPSi (yellow). | ||
When attempting to melt a polysulfide material with either cyclosiloxane cross-linker, to form into different shapes, an unexpected change in morphology occurred. For the rubbery polysulfides of both series, when heated to around 250 °C to melt and reform, the rubbery solids hardened and became darker and more crystalline instead of melting (Fig. S5†). As seen with acrylamide,41 this was due to presence of unreacted vinyl groups present in the polysulfides, which, once heated to a sufficiently high temperature, cross-link with other vinyl groups to form a more rigid material. This theory was tested by heating each of the cross-linkers, PVPSi and TVTSi, to 230 °C in a closed system. Both cyclosiloxanes started off as a colourless solutions and began to form yellow crystalline solids after 1–3 hours these solids were found to be insoluble in commonly used laboratory solvents. The FTIR after heating shows a reduction in the CC bonding (Fig. S6 and S7†). FTIR was also performed on the hardened polysulfides and compared to pre-heated form. For 20-poly(S-r-PVPSi) (blue, Fig. 2), after heating the peaks at 3056 cm−1, 1569 cm−1, 1007 cm−1 and 959 cm−1 were no longer observed, indicating the disappearance of the unreacted vinyl groups as they begin to cross-link (orange, Fig. 2). No changes were observed for both siloxanes and polysulfides when heated at 180 °C, but at 190 °C cross-linking/hardening was observed after 4 hours.
 | ||
| Fig. 2 FTIR spectrum comparison of 20-poly(S-r-PVPSi) before (blue) and after (orange) heating at 230 °C. | ||
Solution 13C{1H} NMR spectroscopy of the starting cyclosiloxane cross-linkers show the presence of CHCH2 bonds at similar chemical shifts (Fig. S8 and S9†). For TVTSi, these were observed at 136.9–137.2 ppm (CH2) and 134.2–134.3 ppm (CH) and for PVPSi they were observed at 137.3–137.5 ppm (CH2) and 133.8–134.0 ppm (CH). Both siloxane cross-linkers resulted to polysulfides that did not dissolve in conventional solvents, requiring the use of solid state (SS) NMR spectroscopy for characterization.
Fig. 3 and 4 show the 13C cross-polarisation spectra for X-poly(S-r-TVTSi) (X = 20, 40, 80, Fig. 3) and for X-poly(S-r-PVPSi) (X = 20 and 40). The spectra showed the presence of CC double bond (δC = ca. 136–137 ppm), along with the peaks between 18 and 35 ppm that indicate the presence of CH2–S and CH–S and a peak at ca. 0 ppm which corresponds to the methyl group attached to the Si. All five spectra show the presence of unreacted CC bonds even at a the highest sulfur weight percentages used. Nonetheless, the relative ratio of peak intensities of CH2–S, CH–S: CH3 increased as the wt% S increases, indicating that more C–S bonds were formed, as expected.
 | ||
| Fig. 3 SS 13C cross polarisation NMR spectra of X-poly(S-r-TVTSi), X = 80 (top, green), 40 (middle, red) and 20 (bottom, blue). | ||
 | ||
| Fig. 4 SS 13C cross polarisation NMR spectra of X-poly(S-r-PVPSi), X = 40 (top, red) and 20 (bottom, blue). | ||
Both spectra showed very similar results to previous work performed by Park et al. using 1,1,3,3-tetramethyl-1,3-divinyldisiloxane as a cross-linker.35 Even with increased weight percentages of sulfur, the CC were observed in the final product. This occurs as the vinyl groups that have been attacked by the polysulfide radical form a polysulfide chain that prevents the other unreacted vinyl groups from coming into contact with another polysulfide radical.35
SS 13C cross-polarisation NMR spectroscopy was also performed on the heated polymers, 20-poly(S-r-TVTSi) (Fig. 5) and 20-poly(S-r-PVPSi) (Fig. 6). Both show the presence of CC double bonds (δC = ca. 136–137 ppm) but at a significantly reduced ratio compared to the unheated materials. Furthermore, the ratio of the peaks that correspond to CH2–S, CH–S, CH2 and CH at 18–30 ppm are noticeably larger compared to the unheated versions. This indicates an increase in the formation of CH2 and CH due to CC cross-linking. Both spectra also show broadening of the peaks and an increase in the number of peaks after heating (Fig. 5 and 6, top). The broadening of peaks observed can be due to the increase in the density of cross-linking of the CC bonds,42 whilst the increase in the number of peaks alludes to a change in chemical environment.
 | ||
| Fig. 5 SS 13C cross polarisation NMR spectra of 20-poly(S-r-TVTSi) before (bottom, blue) and after (top, red) heating. | ||
 | ||
| Fig. 6 SS 13C cross polarisation NMR spectra of 20-poly(S-r-PVPSi) before (bottom, blue) and after (top, red) heating. | ||
Analyzing the SEM results in conjunction with SS NMR spectroscopy, suggests that in both cyclosiloxane cross-linked polysulfide series, most if not all the sulfur at 20 wt% S is consumed, even though there are CC bonds remaining (Fig. 7a and b). However, increasing the wt% S not only left unreacted CC bonds, but also resulted to sulfur crystallites on the surface of the material (Fig. 7c–e and Fig. S10–S11†). This is similar to what was observed previously with other siloxanes containing vinyl functionalities and matches with what we observe during SS NMR spectroscopy.35 After the vinyl groups have been attacked by the polysulfide radical, the polysulfide chain that forms prevents the other unreacted vinyl groups from being accessed. These results indicate that, under the reaction conditions, there is a maximum wt% S able to undergo cross-linking with cyclosiloxanes leaving unreacted sulfur and vinyl groups above the maximum.
 | ||
| Fig. 7 Surface analysis using SEM of selected X-poly(S-r-PVPSi) (X = wt% S) (a) X = 20 (c) 40 and X-poly(S-r-TVTSi) (b) X = 20, (d) 40, (e) 80. | ||
Thermal stability profiles of both series of cyclosiloxane cross-linked polysulfides, were measured using TGA. It was predicted that the cross-linked polysulfides would undergo one major loss corresponding to the sulfur domains (200–280 °C), and that the weight percentage lost should be similar to the wt% S used in the reaction, followed by another minor loss corresponding to C–S bond cleavage (ca. 325 °C).9,15
In the TGA spectra of the X-poly(S-r-TVTSi) series, X = 30–80 wt% S, the sulfur domains resulted to a wt% loss between 200–300 °C at an amount which corresponded to the wt% S used in the reaction, for most of the materials. Furthermore, at around 300–325 °C all spectra show another minor loss indicating the cleavage of C–S bonds (Fig. 8). However, for 20, 30, and 70 wt% S samples there was major loss starting around 72 °C and continuing to 255 °C. The lower starting degradation temperature could be caused by short chain cross-linked oligomers which would evaporate at lower temperatures, while the rest of the weight loss can be attributed to the sulfur domains of the longer chain polysulfides. This shows that the reaction was not very homogenous. The inhomogeneity could also be observed when running the samples multiple times (e.g. Fig. S12†).
 | ||
| Fig. 8 Thermogram of X-poly(S-r-TVTSi)s, X = 20 (blue), 30 (purple), 40 (black), 50 (red), 60 (green), 70 (olive) and 80 (pink). | ||
As observed with the X-poly(S-r-TVTSi) series the inhomogeneity shows TGA results that are unpredicted and hard to reproduce for the X-poly(S-r-PVPSi) series. 10-poly(S-r-PVPSi) and 20-poly(S-r-PVPSi) gave a similar trend to 20-poly(S-r-TVTSi), 30-poly(S-r-TVTSi) and 70-poly(S-r-TVTSi). Both 10-poly(S-r-PVPSi) and 20-poly(S-r-PVPSi), show a first loss occurring at around 160–250 °C and a second at 300–325 °C (Fig. 9). TGA of the rest of the X-poly(S-r-PVPSi) series, 30-poly(S-r-PVPSi), 40-poly(S-r-PVPSi) and 50-poly(S-r-PVPSi) seemed to follow the predicted trend, with the sulfur domains being lost at 200–300 °C (Fig. 9). However, similar to the X-poly(S-r-TVTSi) series, the inhomogeneity of the final products results to unexpected wt% material remaining after the loss of the sulfur domains. For example, 55 wt% remained for 30-poly(S-r-PVPSi), 65 wt% for 40-poly(S-r-PVPSi) and 45 wt% for 50-poly(S-r-PVPSi) (Fig. 9).
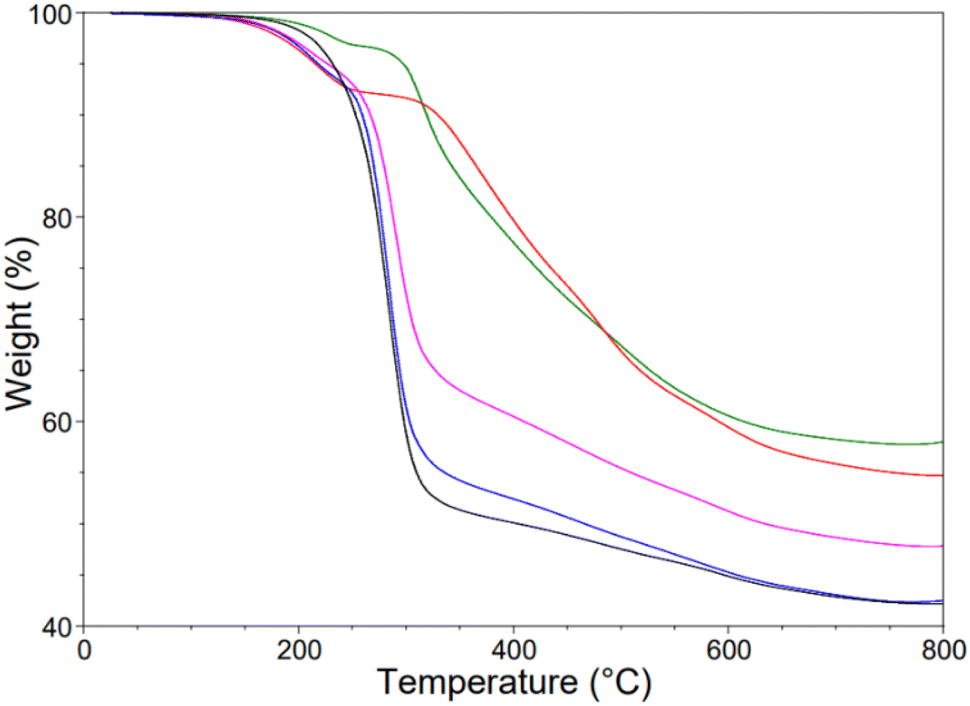 | ||
| Fig. 9 Thermogram of X-poly(S-r-PVPSi), X = 10 (green), 20 (red), 30 (blue), 40 (pink), 50 (black). | ||
From the SEM and SS NMR spectroscopy results, it was hypothesized that a maximum wt% S could be cross-linked into a given material for both TVTSi and PVPSi cross-linkers and any sulfur added above this remained on the surface after synthesis. To determine the maximum wt% S that can undergo cross-linking, higher wt% S polysulfides were chosen from each series. These were then washed in CS2 as it has been reported in literature that CS2 can be used to remove excess elemental sulfur from the surfaces of polysulfide materials.34,35 A comparison between 60-poly(S-r-TVTSi) (Fig. 10a) and 80-poly(S-r-TVTSi) (Fig. S13†) and their CS2 washed counterparts it appeared that 32–37 wt% S was the maximum wt% S range for the TVTSi cross-linker as this is the weight lost after the CS2 wash for the sulfur domains. Furthermore 30-poly(S-r-PVPSi) (Fig. 10b) and 50-poly(S-r-PVPSi) (Fig. S14†) were compared with their CS2 washed counterparts and it was observed that 24–35 wt% S was the maximum wt% S range for PVPSi cross-linking. The observation of a range for the maximum wt% S could be due to the earlier observed in inhomogeneity of the final products observed in earlier TGA results.
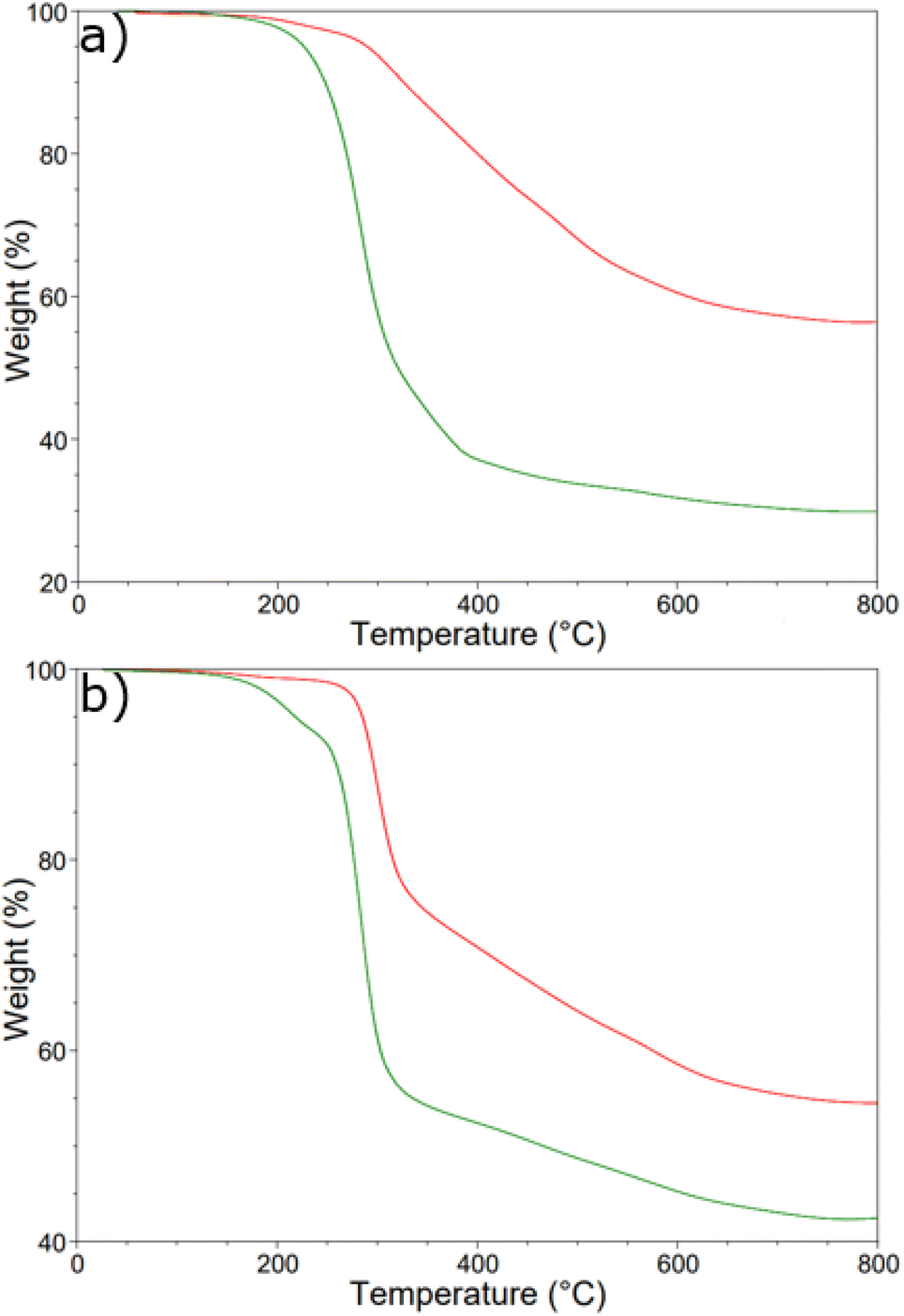 | ||
| Fig. 10 Thermogram of (a) 60-poly(S-r-TVTSi) (b) 30-poly(S-r-PVPSi) before (green) and after (red) washing with CS2. | ||
Glass transition temperatures (Tg) were determined using DSC for both series. In the X-poly(S-r-TVTSi) series, starting from 20-poly(S-r-TVTSi) to 80-poly(S-r-TVTSi), gave Tg values between −26.2 °C to 23.7 °C (Fig. S15–S21†). In the X-poly(S-r-PVPSi) series, starting from 10-poly(S-r-PVPSi) to 50-poly(S-r-PVPSi) gave Tg values which were lower, between −24.2 °C to −5.4 °C (Fig. S22–S26†). Overall the lower Tg values for the X-poly(S-r-PVPSi) series in comparison to the X-poly(S-r-TVTSi) series, could be explained by the one additional vinyl group. The additional vinyl group, as shown by the SS NMR spectra, would not undergo inverse vulcanisation and so would help the overall polysulfide become less rigid. As increasing rigidity increases the Tg, the more flexible X-poly(S-r-TVTSi) series therefore have the lower Tg values.43 The lack of a trend observed could be attributed to the inhomogeneous nature of the final product preventing a proper analysis for a series by DSC. Importantly, it was observed that there was an increase in the intensity of the sulfur melting peaks at 120 °C, as the wt% S used in the reaction increased.
While washing the polysulfides that formed from inverse vulcanisation with hexanes, the polysulfide appeared to swell and the volume extracted out after washing was noticeably less. To investigate this further, a very preliminary study of solvent sorption with the two different cyclosiloxane cross-linked polysulfides was performed. 20-poly(S-r-TVTSi) and 20-poly(S-r-PVPSi) were chosen as they were the least brittle polysulfides in their respective series. Polysulfide swelling (reported as % and mL g−1) was calculated and used to determine which polysulfide had better potential for use in selective solvent capture. The solvents tested were dichloromethane (DCM), diethyl ether (Et2O), hexane, toluene, benzene and octane. Dichloromethane was chosen as it has shown decent swelling with different polymer resins for peptide synthesis,44 while diethyl ether is volatile and the others are hydrocarbons, chosen as they are found in crude oil and petroleum. 20-poly(S-r-TVTSi) was found to hold more solvent than 20-poly(S-r-PVPSi) for all the solvents tested except hexane, toluene and octane (Fig. 11). It was observed that polar solvents such as water, methanol and ethanol were not absorbed by the polysulfides.
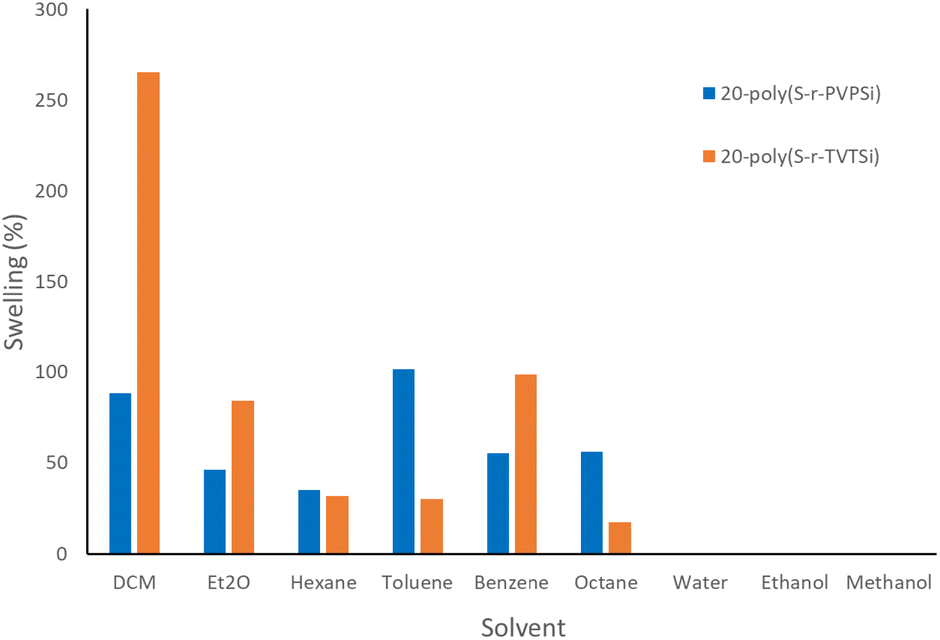 | ||
| Fig. 11 Swelling (%) of the two different cyclosiloxane cross-linked polysulfides in various solvents after 24 hours. The swelling (%) was calculated as an average of triplicates. | ||
Swelling calculations in mL g−1, provide a means to determine which solvent had the best quantitative sorption (Tables S1 and S2†). The results show that 20-poly(S-r-TVTSi) has the best sorption of dichloromethane, diethyl ether and benzene with swelling values of 1.87, 1.17 and 1.12 mL g−1 respectively, whilst the same solvents show values of 0.66, 0.65 and 0.63 mL g−1 for 20-poly(S-r-PVPSi). The remaining solvents hexane, toluene and octane show values of 0.53, 1.17 and 0.80 mL g−1 for 20-poly(S-r-PVPSi) respectively, whilst 20-poly(S-r-TVTSi) gave values of 0.48, 0.35 and 0.25 mL g−1, respectively.
Utilizing these results, future studies can be performed looking at selective solvent sorption. One such study could be the selective removal of hydrocarbon contaminants from stormwater runoff. Rain can interact with pollutants in the air from vehicle combustion processes,45 or come into contact with roads, highways and parking lots where spillage has occurred, or after improper disposal of used industrial or domestic hydrocarbons.46 This contaminated water then makes its way to either larger water ways, ground water or soil where it causes damage to the surrounding area.47 A brief preliminary study was performed to see the viability of this application. A 5000 ppm solution of hexane in water was made and analyzed using solution 1H NMR spectroscopy before and after the addition of the polysulfides for 24 hours. The water before the addition showed the presence of hexane at ca. δH 1.26 and 0.88 ppm. These peaks are reduced significantly after 24 hours for both 20-poly(S-r-TVTSi) and 20-poly(S-r-PVPSi) (Fig. S27 and S28†). Using a known amount of trimethoxybenzene as an internal standard 20-poly(S-r-TVTSi) showed ca. 69% removal of hexane and 20-poly(S-r-PVPSi) showed ca. 70% removal.
To see if the synthesized polysulfides have the potential to be recycled, 20-poly(S-r-PVPSi) was placed in a 5000 ppm solution of hexane in water, left for 24 hours, and then was analyzed using solution NMR spectroscopy. The polysulfide was then dried under reduced pressure for 2 hours before placing it into a fresh solution of 5000 ppm hexanes in water. Notably, there was no change in the appearance or mechanical properties of the polymer during regeneration. This process was repeated three times (Table S3†). It was observed that the percentage hexanes removed from the water was consistent for all three trials, demonstrating the recyclability of the polysulfide material for this application (Fig. 12).
 | ||
| Fig. 12 Hexanes removed (%) from water for a series of identical batches (1–4) using the same 20-poly(S-r-PVPSi) material. The percentage was calculated by analysing solution NMR spectra containing an internal standard. | ||
As it was observed that the polysulfides could be recycled, the next test was to see the upper limit of hexanes removal made possible by the polysulfide (Fig. 13 and Table S4†). Four cycles were tested on the same hexanes in water solution, with filtering and drying the polysulfide in between. 1H NMR spectroscopy was used to determine the % hexanes removed each cycle. In this case, it is observed that the polysulfide could only remove up to ca. 70% total from the solution which was similar to the solvation study with neat hexanes (Fig. 13).
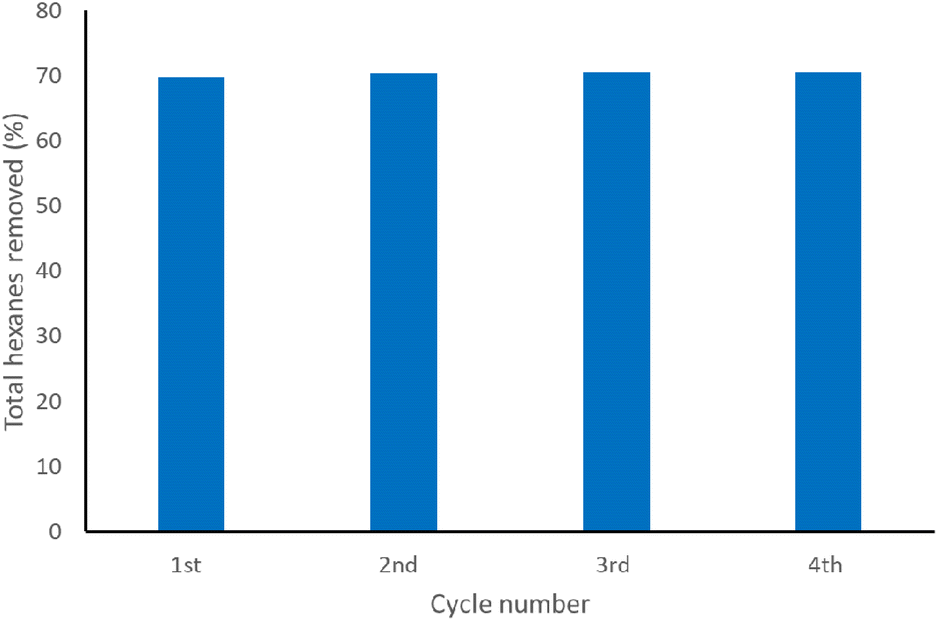 | ||
| Fig. 13 Total hexanes removed (%) from the same water solution for a number of cycles (1–4) using the same 20-poly(S-r-PVPSi) material. Percentage was calculated by analysing solution NMR spectra containing an internal standard. | ||
Based on these promising preliminary results, future studies will focus on optimizing a method to maximize the removal of the solvents, for example, utilizing the polysulfides as a sorbent filter/mesh in a flow system. In addition, relevant industrial samples of contaminated solutions will be used.
Conclusions
Two cyclosiloxanes with multiple pendant alkenes and elemental sulfur were used to synthesize cross-linked polysulfides through inverse vulcanisation. These polysulfides have shown thermal stability above 200 °C, have low glass transition temperatures and are insoluble in common laboratory solvents.They have also shown promise towards selective solvent sorption. The polysulfides synthesized were found to be inhomogeneous as determined by TGA and SEM analysis. The methods used in this paper will be optimized to improve reproducibility, increase the scale of reaction, and test further solvent selection for other applications. Other potential applications of the cyclosiloxane cross-linked polysulfides will also be investigated, along with the use of other siloxane cross-linkers to increase functionality. In addition, these follow-up studies will illuminate further understanding of the synthetic mechanisms involved in creating siloxane linked polysulfides.
Author contributions
K. W. P. performed most of the synthesis, characterization and solvent testing of the polysulfides. E. A. T. synthesized some polymers in the series and F. F. helped with initial solvent testing. Z. Z. measured and interpreted the SS NMR spectra of the cyclosiloxane cross-linker polysulfides. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the support provided by the School of Chemical Sciences at the University of Auckland. We would also like to thank Albina Avzalova and Stephen Crawley for helping with DSC and TGA measurements. We acknowledge the support of the University of Auckland's BIRU (Biomedical Imaging Research Unit) and Ratish Kurian for capturing images and EDX using the Scanning electron Microscope.Notes and references
-
T. Lee, Properties and applications of elastomeric polysulfides, iSmithers Rapra Publishing, 1999 Search PubMed
.
- W. J. Chung, J. J. Griebel, E. T. Kim, H. Yoon, A. G. Simmonds, H. J. Ji, P. T. Dirlam, R. S. Glass, J. J. Wie, N. A. Nguyen, B. W. Guralnick, J. Park, Á. Somogyi, P. Theato, M. E. Mackay, Y.-E. Sung, K. Char and J. Pyun, Nat. Chem., 2013, 5, 518–524 CrossRef CAS PubMed
- M. J. H. Worthington, C. J. Shearer, L. J. Esdaile, J. A. Campbell, C. T. Gibson, S. K. Legg, Y. Yin, N. A. Lundquist, J. R. Gascooke, I. S. Albuquerque, J. G. Shapter, G. G. Andersson, D. A. Lewis, G. J. L. Bernardes and J. M. Chalker, Adv. Sustainable Syst., 2018, 2, 1800024 CrossRef
- K. W. Park and E. M. Leitao, Chem. Commun., 2021, 57, 3190–3202 RSC
- X. Wu, J. A. Smith, S. Petcher, B. Zhang, D. J. Parker, J. M. Griffin and T. Hasell, Nat. Commun., 2019, 10, 647 CrossRef PubMed
- L. J. Dodd, Ö. Omar, X. Wu and T. Hasell, ACS Catal., 2021, 11, 4441–4455 CrossRef CAS
- J. J. Griebel, S. Namnabat, E. T. Kim, R. Himmelhuber, D. H. Moronta, W. J. Chung, A. G. Simmonds, K.-J. Kim, J. van der Laan, N. A. Nguyen, E. L. Dereniak, M. E. Mackay, K. Char, R. S. Glass, R. A. Norwood and J. Pyun, Adv. Mater., 2014, 26, 3014–3018 CrossRef CAS PubMed
- J. J. Griebel, N. A. Nguyen, S. Namnabat, L. E. Anderson, R. S. Glass, R. A. Norwood, M. E. Mackay, K. Char and J. Pyun, ACS Macro Lett., 2015, 4, 862–866 CrossRef CAS PubMed
- A. D. Tikoalu, N. A. Lundquist and J. M. Chalker, Adv. Sustainable Syst., 2020, 4, 1900111 CrossRef CAS
- P. T. Dirlam, A. G. Simmonds, T. S. Kleine, N. A. Nguyen, L. E. Anderson, A. O. Klever, A. Florian, P. J. Costanzo, P. Theato, M. E. Mackay, R. S. Glass, K. Char and J. Pyun, RSC Adv., 2015, 5, 24718–24722 RSC
- M. J. H. Worthington, R. L. Kucera and J. M. Chalker, Green Chem., 2017, 19, 2748–2761 RSC
- Y. Zhang, R. S. Glass, K. Char and J. Pyun, Polym. Chem., 2019, 10, 4078–4105 RSC
- J. M. Chalker, M. Mann, M. J. Worthington and L. J. Esdaile, Org. Mater., 2021, 3, 362–373 CrossRef CAS
- T. Lee, P. T. Dirlam, J. T. Njardarson, R. S. Glass and J. Pyun, J. Am. Chem. Soc., 2022, 144, 5–22 CrossRef CAS PubMed
- M. J. H. Worthington, R. L. Kucera, I. S. Albuquerque, C. T. Gibson, A. Sibley, A. D. Slattery, J. A. Campbell, S. F. K. Alboaiji, K. A. Muller, J. Young, N. Adamson, J. R. Gascooke, D. Jampaiah, Y. M. Sabri, S. K. Bhargava, S. J. Ippolito, D. A. Lewis, J. S. Quinton, A. V. Ellis, A. Johs, G. J. L. Bernardes and J. M. Chalker, Chem. – Eur. J., 2017, 23, 16219–16230 CrossRef CAS PubMed
- A. G. Simmonds, J. J. Griebel, J. Park, K. R. Kim, W. J. Chung, V. P. Oleshko, J. Kim, E. T. Kim, R. S. Glass, C. L. Soles, Y.-E. Sung, K. Char and J. Pyun, ACS Macro Lett., 2014, 3, 229–232 CrossRef CAS PubMed
- J. M. Scheiger, C. Direksilp, P. Falkenstein, A. Welle, M. Koenig, S. Heissler, J. Matysik, P. A. Levkin and P. Theato, Angew. Chem., Int. Ed., 2020, 59, 18639–18645 CrossRef CAS PubMed
- M. Mann, B. Zhang, S. J. Tonkin, C. T. Gibson, Z. Jia, T. Hasell and J. M. Chalker, Polym. Chem., 2022, 13, 1320–1327 RSC
- C. Miao, X. Xun, L. J. Dodd, S. Niu, H. Wang, P. Yan, X.-C. Wang, J. Li, X. Wu, T. Hasell and Z.-J. Quan, ACS Appl. Polym. Mater., 2022, 4, 4901–4911 CrossRef CAS
- S. F. do Valle, A. S. Giroto, H. P. G. Reis, G. G. F. Guimarães and C. Ribeiro, J. Agric. Food Chem., 2021, 69, 2392–2402 CrossRef CAS PubMed
- M. Mann, J. E. Kruger, F. Andari, J. McErlean, J. R. Gascooke, J. A. Smith, M. J. H. Worthington, C. C. C. McKinley, J. A. Campbell, D. A. Lewis, T. Hasell, M. V. Perkins and J. M. Chalker, Org. Biomol. Chem., 2019, 17, 1929–1936 RSC
- J. A. Smith, R. Mulhall, S. Goodman, G. Fleming, H. Allison, R. Raval and T. Hasell, ACS Omega, 2020, 5, 5229–5234 CrossRef CAS PubMed
- C. V. Lopez, M. S. Karunarathna, M. K. Lauer, C. P. Maladeniya, T. Thiounn, E. D. Ackley and R. C. Smith, J. Polym. Sci., 2020, 58, 2259–2266 CrossRef CAS
- I. Bu Najmah, N. A. Lundquist, M. K. Stanfield, F. Stojcevski, J. A. Campbell, L. J. Esdaile, C. T. Gibson, D. A. Lewis, L. C. Henderson, T. Hasell and J. M. Chalker, ChemSusChem, 2021, 14, 2352–2359 CrossRef CAS PubMed
-
D. Zhu, N. Hu and D. W. Schaefer, in Handbook of Waterborne Coatings, ed. P. Zarras, M. D. Soucek and A. Tiwari, Elsevier, 2020, pp. 1–27, DOI:10.1016/B978-0-12-814201-1.00001-9
- C. Atallah, A. Y. Tremblay and S. Mortazavi, J. Pet. Sci., 2017, 157, 349–358 CrossRef CAS
- J. P. Matinlinna, C. Y. K. Lung and J. K. H. Tsoi, Dent. Mater., 2018, 34, 13–28 CrossRef CAS PubMed
-
H. Zhang, C. Zhu, C. Wei, H. Duan and J. Yu, in Handbook of Functionalized Nanomaterials for Industrial Applications, ed. C. Mustansar Hussain, Elsevier, 2020, pp. 865–907, DOI:10.1016/B978-0-12-816787-8.00027-2
-
T. C. Kendrick, B. M. Parbhoo and J. W. White, in Comprehensive Polymer Science and Supplements, ed. G. Allen and J. C. Bevington, Pergamon, Amsterdam, 1989, pp. 459–523, DOI:10.1016/B978-0-08-096701-1.00140-3
-
S. Kobayashi and K. Müllen, Encyclopedia of polymeric nanomaterials, Springer, Berlin, Heidelberg, 2015 Search PubMed
-
S. K. Tilley and R. C. Fry, in Systems Biology in Toxicology and Environmental Health, ed. R. C. Fry, Academic Press, Boston, 2015, pp. 117–169, DOI:10.1016/B978-0-12-801564-3.00006-7
-
H. Fromme, in Encyclopedia of Environmental Health (Second Edition), ed. J. Nriagu, Elsevier, Oxford, 2019, pp. 805–812, DOI:10.1016/B978-0-12-409548-9.11241-2
-
F. Müller and S. Silber, in Polymer Science: A Comprehensive Reference, ed. K. Matyjaszewski and M. Möller, Elsevier, Amsterdam, 2012, pp. 443–451, DOI:10.1016/B978-0-444-53349-4.00276-4
- R. Anyszka, M. Kozanecki, A. Czaderna, M. Olejniczak, J. Sielski, M. Siciński, M. Imiela, J. Wręczycki, D. Pietrzak, T. Gozdek, M. Okraska, M. I. Szynkowska, P. Malinowski and D. M. Bieliński, J. Sulphur Chem., 2019, 40, 587–597 CrossRef CAS
- K. W. Park, Z. Zujovic and E. M. Leitao, Macromolecules, 2022, 55, 2280–2289 CrossRef CAS
- B. Zhang, L. J. Dodd, P. Yan and T. Hasell, React. Funct. Polym., 2021, 161, 104865 CrossRef CAS
- J. A. Smith, X. Wu, N. G. Berry and T. Hasell, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 1777–1781 CrossRef CAS PubMed
- Ç. Kip, C. Demir and A. Tuncel, J. Chromatogr. A, 2017, 1502, 14–23 CrossRef PubMed
- J. M. Chalker, M. J. H. Worthington, N. A. Lundquist and L. J. Esdaile, Top. Curr. Chem., 2019, 377, 16 CrossRef PubMed
-
B. Arkles and G. Larson, Silicon Compounds: Silanes & Silicones, 2013 Search PubMed
- K. Kishore and K. N. Santhanalakshmi, J. Polym. Sci., Part A: Polym. Chem., 1981, 19, 2367–2375 CAS
-
A. bin Ahmad and A. bin Amu, in Blends of Natural Rubber: Novel Techniques for Blending with Speciality Polymers, ed. A. J. Tinker and K. P. Jones, Springer, Netherlands, Dordrecht, 1998, pp. 40–52, DOI:10.1007/978-94-011-4922-8_4
-
R.-M. Wang, S.-R. Zheng and Y.-P. Zheng, in Polymer Matrix Composites and Technology, ed. R.-M. Wang, S.-R. Zheng and Y.-P. Zheng, Woodhead Publishing, 2011, pp. 101–548, DOI:10.1533/9780857092229.1.101
- R. Santini, M. C. Griffith and M. Qi, Tetrahedron Lett., 1998, 39, 8951–8954 CrossRef CAS
- C. J. DiBlasi, H. Li, A. P. Davis and U. Ghosh, Environ. Sci. Technol., 2009, 43, 494–502 CrossRef CAS PubMed
- V. A. Tsihrintzis and R. Hamid, Water Resour. Res., 1997, 11, 136–164 Search PubMed
- M. Dechesne, S. Barraud and J.-P. Bardin, J. Contam. Hydrol., 2004, 72, 189–205 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2py00581f |
| This journal is © The Royal Society of Chemistry 2022 |