
Development of subtype-selective covalent ligands for the adenosine A2B receptor by tuning the reactive group†
Bert L. H.
Beerkens
 ,
Xuesong
Wang
,
Maria
Avgeropoulou
,
Lisa N.
Adistia
,
Jacobus P. D.
van Veldhoven
,
Willem
Jespers
,
Rongfang
Liu
,
Laura H.
Heitman
,
Adriaan P.
IJzerman
and
Daan
van der Es
*
,
Xuesong
Wang
,
Maria
Avgeropoulou
,
Lisa N.
Adistia
,
Jacobus P. D.
van Veldhoven
,
Willem
Jespers
,
Rongfang
Liu
,
Laura H.
Heitman
,
Adriaan P.
IJzerman
and
Daan
van der Es
*
Division of Drug Discovery and Safety, Leiden Academic Centre for Drug Research, Leiden University, Einsteinweg 55, 2333 CC, Leiden, The Netherlands. E-mail: d.van.der.es@lacdr.leidenuniv.nl
First published on 21st June 2022
Abstract
Signalling through the adenosine receptors (ARs), in particular through the adenosine A2B receptor (A2BAR), has been shown to play a role in a variety of pathological conditions, ranging from immune disorders to cancer. Covalent ligands for the A2BAR have the potential to irreversibly block the receptor, as well as inhibit all A2BAR-induced signalling pathways. This will allow a thorough investigation of the pathophysiological role of the receptor. In this study, we synthesized and evaluated a set of potential covalent ligands for the A2BAR. The ligands all contain a core scaffold consisting of a substituted xanthine, varying in type and orientation of electrophilic group (warhead). Here, we find that the right combination of these variables is necessary for a high affinity, irreversible mode of binding and selectivity towards the A2BAR. Altogether, this is the case for sulfonyl fluoride 24 (LUF7982), a covalent ligand that allows for novel ways to interrogate the A2BAR.
Introduction
The endogenous molecule adenosine acts as a signalling molecule on the G protein-coupled receptor (GPCR) subfamily of adenosine receptors (ARs): the A1, A2A, A2B and A3 adenosine receptors (A1AR, A2AAR, A2BAR and A3AR).1 Elevated concentrations of adenosine have been observed in various pathological conditions, e.g. cancer, inflammation and hypoxia, implying an important role for AR signaling.2,3 Antagonizing ARs and blocking the adenosine-induced signalling pathways is therefore an interesting strategy to tackle a broad spectrum of pathological conditions.4A2BAR activation has been linked to hallmarks of cancer, i.e. cancer cell proliferation, tumour growth, tumour metastasis and the suppression of surrounding immune cells, among others.5–7 In fact, multiple clinical trials are currently investigating the inhibition of the A2BAR in cancers, e.g. in combination with an A2AAR antagonist or immune stimulants.5 Nevertheless, persistent high levels of extracellular adenosine in the tumour microenvironment might hinder the proper inhibition of A2BAR-induced signalling pathways.
Covalent A2BAR ligands on the other hand, cause an ‘infinite’ blockade of the A2BAR which constitutes a new strategy that may be deployed in targeting cancer progression as well as studying the inhibition of A2BAR signalling in cancerous cell lines and tissues.8 After binding reversibly, covalent ligands react with an electrophilic substituent (‘warhead’) to a nearby amino acid residue, allowing the formation of an irreversible bond with the target protein.9 This in turn leads to an ‘infinite’ occupancy of the ligand binding pocket, which in case of the A2BAR would prevent even high levels of adenosine from binding to and activating the receptor.
Besides their medicinal potential, covalent ligands have proven especially useful as tools to study GPCR functioning, as they ‘lock’ the highly dynamic GPCRs into one conformation.10 This facilitates purification, isolation and crystallization of the receptor and allows for a more thorough pharmacological characterization on a molecular level.9,11
Over the past decades, various high affinity xanthine derivatives have been developed as antagonists for the adenosine receptors.12 In case of the A2BAR these are mostly N1,N3-dipropylxanthines, developed by the lab of Jacobson,13,14 and N1-propylxanthines, developed by the lab of Müller.15–17 While both classes exhibit high affinity, the latter type of compounds generally show higher selectivity towards the A2BAR over the other adenosine receptors. This prompted us to design covalent xanthine derivatives, based on the N1,N3-dipropyl and N1-propyl series. Looking at the A2AAR, structurally the most similar to the A2BAR, multiple covalent ligands have been developed.18–20 A lysine residue on the second extracellular loop (EL2) of the A2AAR is the target of at least one these ligands.20 We therefore decided to substitute the herein synthesized xanthines with various electrophilic groups known to react with lysine residues. To increase the chances of covalent binding, we also varied the location of the warhead: either meta- or para-substituted at the C8-phenyl ring.
Altogether, we have developed a set of twelve potential xanthine-based covalent ligands. Here, we show the synthesis, affinity, selectivity and covalent mode of action of these ligands.
Results and discussion
Design of covalent A2BAR ligands
Analysing the binding mode of xanthines into the A2AAR binding pocket,21,22 as well as the amino acid sequences of the A2AAR and A2BAR, we found three interesting potential anchors for covalent binding: lysines K265EL3, K267EL3 and K2697.32.23 In this respect, sulfonyl fluoride, fluorosulfonate and isothiocyanate groups were chosen to target either one of these lysine residues. Sulfonyl fluoride groups (–SO2F) have recently emerged as warheads that have a weak intrinsic reactivity, are often stable under physiological conditions and, under the same conditions, can be directed to react selectively with lysine or tyrosine moieties on drug targets.24–26 These beneficial properties have helped to coin the term ‘SuFEx’ (sulfonyl fluoride exchange) as a type of ‘click’ chemistry.27 However, even before the use of sulfonyl fluorides in click chemistry applications, they were incorporated in ligands for the A1, A2A and A3 adenosine receptors.20,28–31 Besides sulfonyl fluorides, we also decided to synthesize ligands containing fluorosulfonate groups (–OSO2F). Fluorosulfonate groups have shown to bear a much lower intrinsic reactivity, as compared to sulfonyl fluoride groups,25,32 which might reduce off-target binding events. Lastly, we chose the isothiocyanate group (–NCS) as warhead to be incorporated in the series of ligands. Although known for its reactivity towards cysteine residues, the isothiocyanate group has shown to form a more stable product upon reacting with lysine residues.33,34 Moreover, the isothiocyanate group has been used to develop potent agonists and antagonists that irreversibly bind to the A1AR.35–37 In recent work from our lab, the isothiocyanate group was incorporated in a putative covalent ligand for the A2BAR.38 This inspired us to further investigate this electrophilic substituent as a warhead to target the A2BAR.Synthesis of covalent A2BAR ligands
Twelve potential covalent ligands were targeted for synthesis, each containing one of the abovementioned electrophilic warheads at the meta or para position on the C8-substituted phenyl ring of the xanthines. The synthesis started with 1,3-dipropyl 5,6-diamino uracil (1) (commercially obtained), or 1-propyl 5,6-diamino uracil (16), synthesized according to procedures reported by Müller et al.39–42 These building blocks were subjected to an EDC-mediated peptide coupling, using 3- or 4-fluorosulfonyl benzoic acid (2, 3, 17 and 18), 3- or 4-fluorosulfonate benzoic acid (4, 5, 19 and 20), or benzoic acid containing a protected amine group at the 3- or 4-position (6, 7, 21 and 22) (Scheme 1). Purification of Boc-protected anilines turned out to be cumbersome in case of the N1-propyl series, therefore an Fmoc-protection was chosen instead. Next, the substituted uracil derivatives were subjected to a ring closure using trimethylsilyl polyphosphate (PPSE).43,44 Gratifyingly, the electrophilic sulfonyl fluoride and fluorosulfonate groups stayed intact upon heating at 170 °C and in the presence of PSSE for several hours. In case of the Boc-protected anilines, basic conditions (reflux in 2 M NaOH) were chosen to achieve ring closure.41 The anilines were then deprotected and subsequently subjected to thiophosgene to yield the corresponding isothiocyanates. Altogether this yielded sulfonyl fluoride-containing ligands 8, 9, 23 and 24, fluorosulfonate-containing ligands 10, 11, 25 and 26, and isothiocyanate-containing ligands 13, 15, 28 and 30.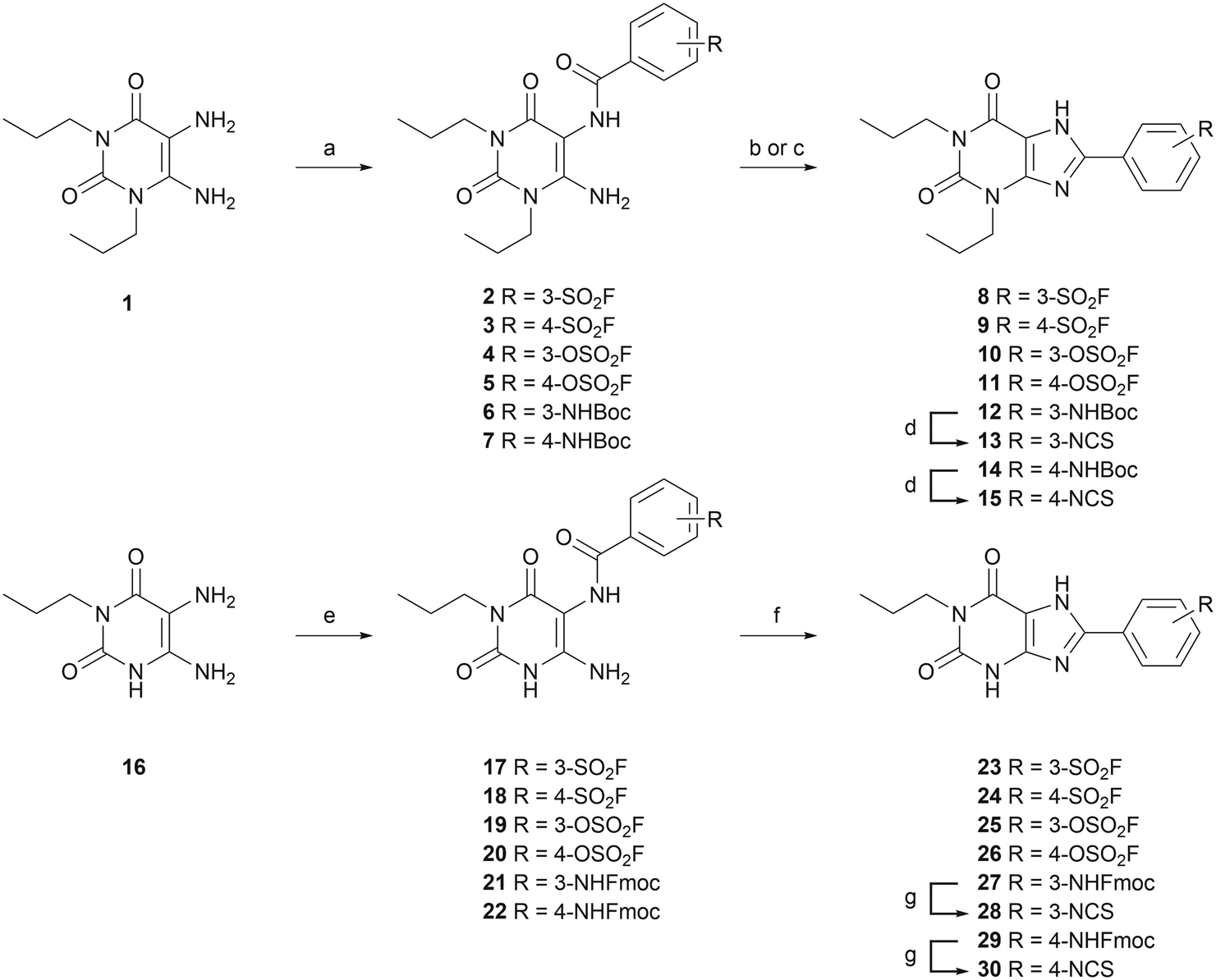 | ||
| Scheme 1 Synthesis of potential covalent ligands for the A2BAR. Reagents and conditions: (a) EDC·HCl, DIPEA, respective benzoic acid, dry DMF, rt, 2–20 h, 41–68%; (b) PPSE, 170 °C, 1–4 h, 10–53%; (c) 2 M NaOH, dioxane, 120 °C, 2–3 h, 60–84%; (d) (i) TFA, DCM, rt, 1 h; (ii) thiophosgene, 3 M HCl, rt, 2 h, 68–77%; (e) EDC·HCl, respective benzoic acid, dry DMF, rt, 1 h – 2 days, 13–54%; (f) PPSE, 150–170 °C, 2–7 h, 55–88%; (g) (i) piperidine, DMF, rt, 5 min; (ii) thiophosgene, 3 M HCl, rt, 2–4 h, 71–75%. | ||
Assessment of time-dependent affinity towards the A2BAR
To investigate the affinity of the twelve ligands and their potential to bind irreversibly to the A2BAR, radioligand displacement assays were carried out using CHO-spap membranes stably overexpressing the A2BAR. Two different conditions were chosen: no pre-incubation of receptor with ligand (pre 0 h) or a 4 h pre-incubation of receptor with ligand (pre 4 h), prior to the addition of radioligand. The pre-incubation step should allow any covalently binding ligand to irreversibly block the available receptor binding sites, thus increasing its apparent affinity for the receptor.20,30,31 The reference A2BAR antagonist PSB-1115 was taken along as a non-covalent control.Interestingly, substitution of the chosen warheads onto the xanthines mostly increased the apparent affinity towards the A2BAR (Table 1; pre 0 h), as compared to the affinity of PSB-1115 in our hands.15 Various patterns were deducted. First of all, the para-substituted xanthines all show a higher apparent affinity than their meta-substituted counterparts at 0 h of pre-incubation. Secondly, at 0 h of pre-incubation, the N1-propyl xanthines show a higher apparent affinity than the N1,N3-dipropyl xanthines. The best performing compounds are thus N1-propyl xanthines containing a para-substituted group. This is in line with the compounds presented in literature.14–17 Looking at 4 h of pre-incubation (Table 1; pre 4 h), the SO2F-substituted xanthines and NCS-substituted xanthines all show decent shifts in Ki (>3), regardless of the positioning of the warhead (meta or para) (examples depicted in Fig. 1A and C). On the other hand, the shifts observed for the OSO2F-substituted xanthines are rather small, close to the values found for PSB-1115 (example in Fig. 1B). This suggests a reversible binding mode. The SO2F-containing xanthines have a higher affinity and Ki shift when substituted at the 4-position, while the NCS-containing xanthines show a higher shift when substituted at the 3-position. The biggest shifts are observed for the 3-NCS-substituted xanthines 13 and 28 (Ki shift of 19 and 74). This is probably the result of a low apparent affinity at 0 h of pre-incubation, in combination with the relatively high reactivity of the NCS group. This Ki shift data hints towards a covalent mode of action among the majority of the xanthine-based ligands.
|
|
|||||
|---|---|---|---|---|---|
| Compound | R1 | R2 | pKia (pre 0 h) | pKib (pre 4 h) | K i shiftc |
| a Apparent affinity determined from displacement of specific [3H]PSB-603 binding on CHO-spap cell membranes stably expressing hA2BAR at 25 °C after 0.5 h co-incubation. b Apparent affinity determined from displacement of specific [3H]PSB-603 binding on CHO-spap cell membranes stably expressing hA2BAR at 25 °C with compounds pre-incubated for 4 h, followed by a 0.5 h co-incubation with [3H]PSB-603. c K i shift determined by ratio Ki(0 h)/Ki(4 h). Data represent the mean ± SEM of three individual experiments performed in duplicate. *p < 0.05, **p < 0.01 compared to the pKi values obtained from the displacement assay with 0 h pre-incubation of [3H]PSB-603, determined by a two-tailed unpaired Student's t-test. | |||||
| 8 | Propyl | 3-SO2F | 6.02 ± 0.10 | 6.80 ± 0.16* | 7.1 |
| 9 | Propyl | 4-SO2F | 7.22 ± 0.28 | 8.62 ± 0.18* | 27.3 |
| 10 | Propyl | 3-OSO2F | 6.25 ± 0.14 | 6.28 ± 0.09 | 1.2 |
| 11 | Propyl | 4-OSO2F | 7.31 ± 0.04 | 7.29 ± 0.04 | 1.0 |
| 13 | Propyl | 3-NCS | 6.21 ± 0.21 | 7.31 ± 0.15* | 19.3 |
| 15 | Propyl | 4-NCS | 7.62 ± 0.11 | 8.49 ± 0.18* | 7.7 |
| 23 | H | 3-SO2F | 6.88 ± 0.08 | 7.88 ± 0.14** | 10.2 |
| 24 (LUF7982) | H | 4-SO2F | 8.10 ± 0.06 | 9.17 ± 0.12** | 12.1 |
| 25 | H | 3-OSO2F | 6.85 ± 0.44 | 7.15 ± 0.32 | 1.6 |
| 26 (LUF7993) | H | 4-OSO2F | 7.93 ± 0.22 | 8.26 ± 0.20 | 2.2 |
| 28 | H | 3-NCS | 6.96 ± 0.27 | 8.55 ± 0.08** | 74.3 |
| 30 (LUF8002) | H | 4-NCS | 8.67 ± 0.14 | 9.18 ± 0.01* | 3.4 |
| PSB-1115 | H | 4-SO2OH | 6.71 ± 0.09 | 6.72 ± 0.18 | 1.1 |

 | ||
| Fig. 1 Displacement of [3H]PSB-603 from the A2BAR by (A) SO2F-substituted LUF7982; (B) OSO2F-substituted LUF7993 and (C) NCS-substituted LUF8002. Displacement measured after 0 or 4 h of pre-incubation of the respective ligand with CHO-spap membranes stably overexpressing the A2BAR. Data represent the mean ± SEM of three individual experiments performed in duplicate. | ||
Evaluation of binding towards the other adenosine receptors
As mentioned in the introduction, xanthine-based ligands are prone to promiscuous AR binding. To investigate the selectivity of the synthesized ligands towards the A2BAR specifically, radioligand displacement experiments were carried out using 1 μM of ligand on CHO (A1AR and A3AR) or HEK (A2AAR) membranes stably overexpressing the respective other adenosine receptor (Table 2). Similar to the experiments for the A2BAR, the compounds were tested either with or without 4 hours of pre-incubation prior to radioligand addition. In our experiments, hardly any ligand showed a strong displacement of radioligand from the structurally similar A2AAR. Only compound 11 seems to bind decently, showing a displacement that exceeds 50%. In case of the A1AR, all of the N1,N3-dipropyl xanthines (8–13 and 15) show a strong displacement (>50%) of radioligand from the receptor. This is in line with earlier reports on such substituted N1,N3-dipropyl xanthines as generally excellent A1AR antagonists.28,45,46| Compound | (%) displacement at 1 μM | |||||
|---|---|---|---|---|---|---|
| hA1ARa | hA2AARb | hA3ARc | ||||
| 0 h | 4 h | 0 h | 4 h | 0 h | 4 h | |
| a % displacement at 1 μM concentration of specific [3H]DPCPX binding on CHO cell membranes stably expressing hA1AR pre-incubated with the compounds for 4 or 0 hours at 25 °C, followed by a co-incubation with [3H]DPCPX for 0.5 h at 25 °C. b % displacement at 1 μM concentration of specific [3H]ZM241385 binding on HEK293 cell membranes stably expressing hA2AAR pre-incubated with the compounds for 4 or 0 hours at 25 °C, followed by a co-incubation with [3H]ZM241385 for 0.5 h at 25 °C. c % displacement at 1 μM concentration of specific [3H]PSB-11 binding on CHO cell membranes stably expressing hA3AR pre-incubated with the compounds for 4 or 0 hours at 25 °C, followed by a co-incubation with [3H]PSB-11 for 0.5 h at 25 °C. Data represent the mean of two individual experiments performed in duplicate. | ||||||
| 8 | 52 | 61 | 19 | 23 | 30 | 43 |
| 9 | 61 | 69 | 54 | 55 | 12 | 5 |
| 10 | 76 | 77 | 13 | 12 | 64 | 58 |
| 11 | 79 | 85 | 63 | 69 | 47 | 49 |
| 13 | 73 | 96 | 47 | 34 | 89 | 100 |
| 15 | 77 | 92 | 40 | 39 | 48 | 96 |
| 23 | 6 | 36 | 20 | 6 | 14 | 10 |
| 24 (LUF7982) | 29 | 41 | 52 | 43 | 7 | 9 |
| 25 | 6 | 1 | 2 | 0 | 14 | 5 |
| 26 (LUF7993) | 17 | 12 | 35 | 24 | 8 | 8 |
| 28 | 7 | 16 | 0 | 6 | 58 | 95 |
| 30 (LUF8002) | 51 | 97 | 25 | 27 | 15 | 66 |
Considering the isothiocyanates (13, 15, 28 and 30), a moderate to high displacement of radioligand from A1 and A3 receptors was observed. Interestingly, the 3-NCS substituted xanthines (13 and 28) seem to perform especially well at the A3AR. A notable loss of displacement at the other adenosine receptors is observed upon removal of the N3-propyl group. Also the introduction of a sulfonyl group has a beneficial effect on selectivity towards the A2BAR over the other ARs. This group might be stabilized by interactions with K2697.32, not present in any of the other ARs.17 This is especially seen for the OSO2F-containing 26 (LUF7993), showing the highest selectivity for the A2BAR. Among the compounds with the highest apparent pKi values, 24 (LUF7982) shows a good selectivity towards the A2BAR and about 50% displacement of radioligand at the A2AAR. The latter suggests 24 (LUF7982) displays a 100-fold selectivity for the A2BAR without pre-incubation (and >1000-fold after 4 h of pre-incubation). Besides, the displacement at the A2AAR is not time-dependent and therefore it is expected that 24 (LUF7982) does not bind covalently to the A2AAR. The high affinity compound 30 (LUF8002) on the other hand, also binds to the A1AR and A3AR.
Investigation of the covalent mode of action of selected compounds
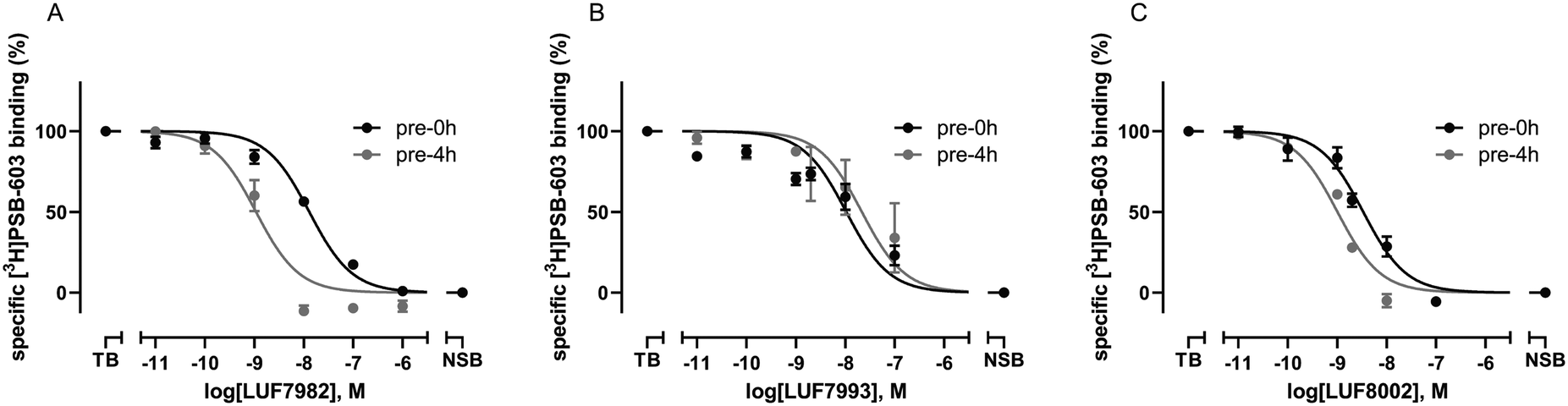
As final validation of the putative covalent mode of binding, wash-out experiments were performed using the compounds highest in affinity and selectivity: 24 (LUF7982), 26 (LUF7993) and 30 (LUF8002). PSB-1115 was taken along as reversible control compound. CHO-spap membranes stably overexpressing the A2BAR were incubated with ligand, followed by either a four-cycle wash treatment or no washing (control), before being exposed to radioligand (Fig. 2). Both PSB-1115 and 26 (LUF7993) show an almost full recovery of radioligand binding after washing, indicating that all receptor-bound ligand has been washed away. These results correspond to the previously observed Ki shifts (Table 1), in which no great shifts were observed for PSB-1115 and the OSO2F-containing xanthines. Of note: it is possible that LUF7993 forms an adduct with the receptor, which is then hydrolysed to produce a sulfonylated lysine and a reversibly bound phenol.3224 (LUF7982) and 30 (LUF8002) on the other hand, show a persistent mode of binding, with no recovery of radioligand binding after four wash treatments (Fig. 2). 24 (LUF7982) and 30 (LUF8002) thus form a stable adduct with the A2BAR, resistant to multiple washing steps and are therefore most likely covalent ligands for the A2BAR. 24 (LUF7982) is the most interesting of these two irreversible compounds due to its high selectivity towards the A2BAR. This compound was therefore further examined in docking experiments.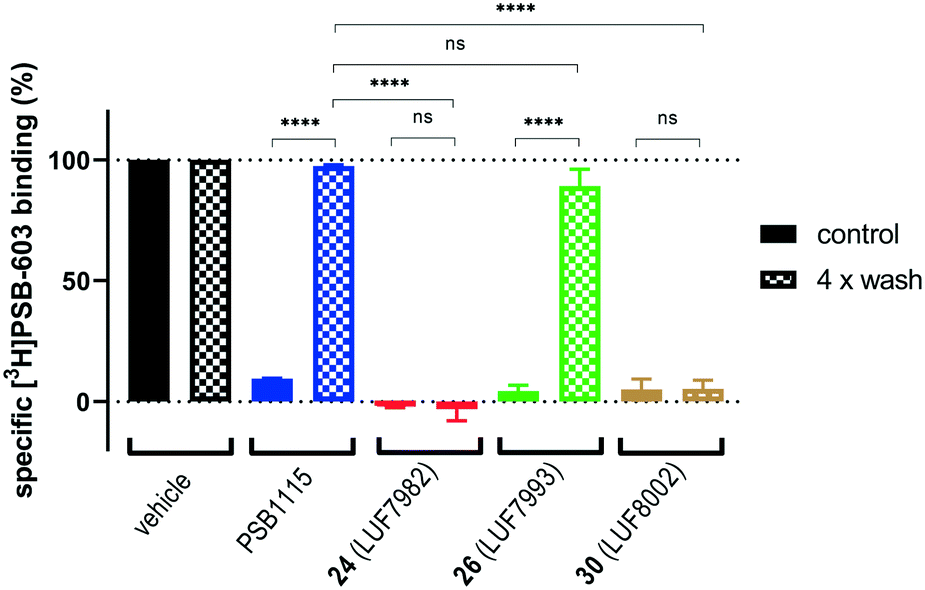 | ||
| Fig. 2 Wash-out assays on the adenosine A2B receptor using the N1-propyl xanthines with para-substituted warheads. CHO-spap cell membranes stably expressing the adenosine A2B receptor were pre-incubated with buffer (vehicle) or 1 μM of ligand (10 μM in case of PSB-1115), followed by a four-cycle washing treatment (4× wash) or no washing at all (control) before being exposed to [3H]PSB-603. Data represent the mean ± SEM of three individual experiments performed in duplicate. Statistics were determined using unpaired student's t tests. ns: no significant difference; ****P < 0.0001. | ||
Docking of LUF7982 into the A2BAR binding pocket
To predict the binding mode of LUF7982, we generated a model of the A2BAR–LUF7982 binding site based on homology modelling and docking. The first step was to identify the orientation of the xanthine core. The orientation of this chemotype in the AR family binding site is well studied and typically involves hydrogen bonding with N2546.55 and π–π stacking with F173EL2.47 This pattern was also observed for the predicted A2BAR–LUF7982 binding complex (Fig. 3A). This leaves the warhead of LUF7982 oriented towards the extracellular vestibule, pointing towards the region of the third extracellular loop (EL3). As mentioned in the Introduction, three lysine residues (K265EL3, K267EL3 and K2697.32) in and near EL3 were identified as potential attachment point for covalent binding of the compounds herein reported. In our model, two out of three lysine residues were in close vicinity to the warhead, namely K267EL3 and K2697.32 (5.7 and 3.9 Å, respectively) (Fig. 3B). K267EL3 is predicted to form a salt bridge with E174EL2, similar to the salt bridge observed between a histidine and glutamic acid in the A2AAR.47 K265EL3 on the other hand was too far away from the warhead in our model (10.6 Å) to form a plausible target for covalent attachment. Whilst K267EL3 thus is within range, it would be energetically more unfavourable to disrupt the formed salt-bridge, and we therefore expect that K2697.32 is the most likely target for covalent attachment. | ||
| Fig. 3 Predicted binding mode of LUF7982. Panel A: Overview of the key interactions of LUF7982 in the binding site, which include two hydrogen bonds (yellow dashed lines) with N2546.55 and π–π stacking with F173EL2, both are conserved interactions in adenosine receptor ligand recognition. The sulfonyl fluoride warhead points towards the extracellular vestibule. Panel B: Top view of the A2BAR–LUF7982 binding pocket, showing potential lysine residues involved in covalent binding (K267EL3 and K2697.32). | ||
Lysine residues K265EL3 and K2697.32 are not present on the other three adenosine receptors, while K267EL3 is also present on the A1AR. A covalent mode of binding involving K2697.32 might therefore further explain the selectivity of 24 (LUF7982) towards the A2BAR. Further studies using single point mutations in the receptor would need to be carried out to prove this.
Conclusion
Herein we present the development of a set of twelve novel xanthine ligands for the A2BAR, all containing electrophilic groups to covalently target lysine residue(s) on the receptor. The xanthine moiety is a well-known and promiscuous scaffold for all four of the adenosine receptors. Nevertheless, among the synthesized ligands, sulfonyl fluoride- and fluorosulfonate-substituted xanthines appear to be highly selective towards the A2BAR over the other adenosine receptors. This selectivity might be explained by the covalent (SO2F) and/or non-covalent (OSO2F) interactions with lysine residue K2697.32. Isothiocyanate (NCS)-containing ligands on the other hand, showed to be less selective towards the A2BAR. This is most likely due to a higher intrinsic reactivity of the NCS group. Furthermore, sulfonyl fluoride 24 (LUF7982) showed persistent binding to the A2BAR in radioligand displacement and wash-out assays. This points towards a covalent mode of action of the respective compound.The A2BAR is an emerging drug target that has been found to play a role in a broad spectrum of pathologies, such as cancers and immune disorders. Antagonizing adenosine signalling through inhibition of the A2BAR is therefore an interesting strategy to tackle a broad spectrum of conditions. Having covalent ligands for the A2BAR will pave the way for studies towards irreversible blockade of the receptor, e.g. in biochemical assays. LUF7982 might thus be used to study the behaviour of the A2BAR in pathological conditions, to obtain insight in the structure of the A2BAR and to pharmacologically characterize the receptor.
Author contributions
Conceptualization: BLHB, APIJ and DE. Investigation and validation: BLHB, XW, MA, LNA, JV, WJ and RL. Data curation, formal analysis and methodology: BLHB, XW, WJ, LHH, APIJ and DE. Funding acquisition, project administration, resources and supervision: LHH, APIJ and DE. Writing – original draft: BLHB and WJ. Writing – review & editing: BLHB, XW, WJ, LHH, APIJ and DE.Conflicts of interest
The authors declare no conflicts of interest.References
- B. B. Fredholm, A. P. IJzerman, K. A. Jacobson, K.-N. Klotz and J. Linden, Pharmacol. Rev., 2001, 53, 527–552 CAS.
- B. B. Fredholm, A. P. IJzerman, K. A. Jacobson, J. Linden and C. E. Müller, Pharmacol. Rev., 2011, 63, 1–34 CrossRef CAS.
- B. Allard, D. Allard, L. Buisseret and J. Stagg, Nat. Rev. Clin. Oncol., 2020, 17, 611–629 CrossRef CAS.
- J.-F. Chen, H. K. Eltzschig and B. B. Fredholm, Nat. Rev. Drug Discovery, 2013, 12, 265–286 CrossRef CAS PubMed.
- Z. Gao and K. A. Jacobson, Int. J. Mol. Sci., 2019, 20, 5139 CrossRef CAS PubMed.
- C. Cekic and J. Linden, Nat. Rev. Immunol., 2016, 16, 177–192 CrossRef CAS.
- P. A. Borea, S. Gessi, S. Merighi, F. Vincenzi and K. Varani, Physiol. Rev., 2018, 98, 1591–1625 CrossRef CAS.
- T. Zhang, J. M. Hatcher, M. Teng, N. S. Gray and M. Kostic, Cell Chem. Biol., 2019, 26, 1486–1500 CrossRef CAS.
- D. Weichert and P. Gmeiner, ACS Chem. Biol., 2015, 10, 1376–1386 CrossRef CAS PubMed.
- A. Glukhova, D. M. Thal, A. T. Nguyen, E. A. Vecchio, M. Jörg, P. J. Scammells, L. T. May, P. M. Sexton and A. Christopoulos, Cell, 2017, 168, 867–877 CrossRef CAS PubMed.
- X. Yang, L. H. Heitman, A. P. IJzerman and D. van der Es, Purinergic Signalling, 2021, 17, 85–108 CrossRef CAS.
- C. E. Müller and K. A. Jacobson, Biochim. Biophys. Acta, 2011, 1808, 1290–1308 CrossRef PubMed.
- K. A. Jacobson, A. P. IJzerman and J. Linden, Drug Dev. Res., 1999, 47, 45–53 CrossRef CAS.
- Y.-C. Kim, X. Ji, N. Melman, J. Linden and K. A. Jacobson, J. Med. Chem., 2000, 43, 1165–1172 CrossRef CAS PubMed.
- A. M. Hayallah, J. Sandoval-Ramírez, U. Reith, U. Schobert, B. Preiss, B. Schumacher, J. W. Daly and C. E. Müller, J. Med. Chem., 2002, 45, 1500–1510 CrossRef CAS PubMed.
- T. Borrmann, S. Hinz, D. C. G. Bertarelli, W. Li, N. C. Florin, A. B. Scheiff and C. E. Müller, J. Med. Chem., 2009, 52, 3994–4006 CrossRef CAS PubMed.
- J. Jiang, C. J. Seel, A. Temirak, V. Namasivayam, A. Arridu, J. Schabikowski, Y. Baqi, S. Hinz, J. Hockemeyer and C. E. Müller, J. Med. Chem., 2019, 62, 4032–4055 CrossRef CAS PubMed.
- X.-D. Ji, C. Gallo-Rodriguez and K. A. Jacobson, Drug Dev. Res., 1993, 29, 292–298 CrossRef CAS PubMed.
- J. C. Shryock, S. Snowdy, P. G. Baraldi, B. Cacciari, G. Spalluto, A. Monopoli, E. Ongini, S. P. Baker and L. Belardinelli, Circulation, 1998, 98, 711–718 CrossRef CAS.
- X. Yang, G. Dong, T. J. M. Michiels, E. B. Lenselink, L. H. Heitman, J. Louvel and A. P. IJzerman, Purinergic Signalling, 2017, 13, 191–201 CrossRef CAS.
- A. S. Doré, N. Robertson, J. C. Errey, I. Ng, K. Hollenstein, B. Tehan, E. Hurrell, K. Bennett, M. Congreve, F. Magnani, C. G. Tate, M. Weir and F. H. Marshall, Structure, 2011, 19, 1283–1293 CrossRef.
- R. K. Y. Cheng, E. Segala, N. Robertson, F. Deflorian, A. S. Doré, J. C. Errey, C. Fiez-Vandal, F. H. Marshall and R. M. Cooke, Structure, 2017, 25, 1275–1285 CrossRef CAS PubMed.
- J. A. Ballesteros and H. Weinstein, in Receptor Molecular Biology, ed. S. Sealfon, Academic Press, 1995, vol. 25, pp. 366–428 Search PubMed.
- A. Narayanan and L. H. Jones, Chem. Sci., 2015, 6, 2650–2659 RSC.
- H. Mukherjee, J. Debreczeni, J. Breed, S. Tentarelli, B. Aquila, J. E. Dowling, A. Whitty and N. P. Grimster, Org. Biomol. Chem., 2017, 15, 9685–9695 RSC.
- D. E. Mortenson, G. J. Brighty, L. Plate, G. Bare, W. Chen, S. Li, H. Wang, B. F. Cravatt, S. Forli, E. T. Powers, K. B. Sharpless, I. A. Wilson and J. W. Kelly, J. Am. Chem. Soc., 2018, 140, 200–210 CrossRef CAS PubMed.
- J. Dong, L. Krasnova, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2014, 53, 9430–9448 CrossRef CAS PubMed.
- A. R. Beauglehole, S. P. Baker and P. J. Scammells, J. Med. Chem., 2000, 43, 4973–4980 CrossRef CAS.
- M. Jörg, A. Glukhova, A. Abdul-Ridha, E. A. Vecchio, A. T. N. Nguyen, P. M. Sexton, P. J. White, L. T. May, A. Christopoulos and P. J. Scammells, J. Med. Chem., 2016, 59, 11182–11194 CrossRef.
- X. Yang, J. P. D. van Veldhoven, J. Offringa, B. J. Kuiper, E. B. Lenselink, L. H. Heitman, D. van Der Es and A. P. IJzerman, J. Med. Chem., 2019, 62, 3539–3552 CrossRef CAS.
- X. Yang, M. A. Dilweg, D. Osemwengie, L. Burggraaff, D. van der Es, L. H. Heitman and A. P. IJzerman, Biochem. Pharmacol., 2020, 180, 114144 CrossRef CAS.
- A. Baranczak, Y. Liu, S. Connelly, W.-G. H. Du, E. R. Greiner, J. C. Genereux, R. L. Wiseman, Y. S. Eisele, N. C. Bradbury, J. Dong, L. Noodleman, K. B. Sharpless, I. A. Wilson, S. E. Encalada and J. W. Kelly, J. Am. Chem. Soc., 2015, 137, 7404–7414 CrossRef CAS PubMed.
- M. Gehringer and S. A. Laufer, J. Med. Chem., 2019, 62, 5673–5724 CrossRef CAS PubMed.
- T. Nakamura, Y. Kawai, N. Kitamoto, T. Osawa and Y. Kato, Chem. Res. Toxicol., 2009, 22, 536–542 Search PubMed.
- G. L. Stiles and K. A. Jacobson, Mol. Pharmacol., 1988, 34, 724–728 CAS.
- K. A. Jacobson, S. Barone, U. Kammula and G. L. Stiles, J. Med. Chem., 1989, 32, 1043–1051 CrossRef CAS.
- J. Zhang, L. Belardinelli, K. A. Jacobson, D. H. Otero and S. P. Baker, Mol. Pharmacol., 1997, 52, 491–498 CrossRef CAS PubMed.
- A. Vlachodimou, H. de Vries, M. Pasoli, M. Goudswaard, S.-A. Kim, Y.-C. Kim, M. Scortichini, M. Marshall, J. Linden, L. H. Heitman, K. A. Jacobson and A. P. IJzerman, Biochem. Pharmacol., 2022, 200, 115027 CrossRef CAS PubMed.
- C. E. Müller, Tetrahedron Lett., 1991, 32, 6539–6540 CrossRef.
- C. E. Müller, Synthesis, 1993, 1, 125–128 CrossRef.
- C. E. Müller and J. Sandoval-Ramirez, Synthesis, 1995, 10, 1295–1299 CrossRef.
- J. Hockemeyer, J. C. Burbiel and C. E. Müller, J. Org. Chem., 2004, 69, 3308–3318 CrossRef CAS PubMed.
- Y. Imai, A. Mochizuki and M. Kakimoto, Synthesis, 1983, 851 CrossRef CAS.
- C. E. Müller, D. Shi, M. Manning and J. W. Daly, J. Med. Chem., 1993, 36, 3341–3349 CrossRef PubMed.
- J. R. Pfister, L. Belardinelli, G. Lee, R. T. Lum, P. Milner, W. C. Stanley, J. Linden, S. P. Baker and G. Schreiner, J. Med. Chem., 1997, 1773–1778 CrossRef CAS PubMed.
- S. Weyler, F. Fülle, M. Diekmann, B. Schumacher, S. Hinz, K.-N. Klotz and C. E. Müller, ChemMedChem, 2006, 1, 891–902 CrossRef CAS.
- W. Jespers, A. C. Schiedel, L. H. Heitman, R. M. Cooke, L. Kleene, G. J. P. van Westen, D. E. Gloriam, C. E. Müller, E. Sotelo and H. Gutiérrez-de-Terán, Trends Pharmacol. Sci., 2018, 39, 75–89 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2md00132b |
| This journal is © The Royal Society of Chemistry 2022 |