
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Achieving enhanced peroxidase-like activity in multimetallic nanorattles†
Flavia G.
da Silva
 a,
Eric V.
Formo
b and
Pedro H. C.
Camargo
*a
a,
Eric V.
Formo
b and
Pedro H. C.
Camargo
*a
aUniversity of Helsinki, Department of Chemistry, A.I. Virtasen aukio 1, Helsinki, Finland. E-mail: pedro.camargo@helsinki.fi
bUniversity of Georgia, Georgia Electron Microscopy, Athens, Georgia 30602, USA
First published on 8th September 2022
Abstract
Gold nanoparticles (Au NPs) have been extensively used as artificial enzymes, but their performance is still limited. We address this challenge by focusing on multimetallic nanorattles comprising an Au core inside a bimetallic AgAu shell, separated by a void (Au@AgAu NRs). They were prepared by a galvanic replacement approach and contained an ultrathin and porous shell comprising an AgAu alloy. By investigating the peroxide-like activity using TMB oxidation as a model transformation, we have found an increase of 152 fold in activities for the NRs relative to conventional Au NPs. Based on the kinetics results, the NRs also showed the lowest Km, indicating better interaction with the substrate and faster product formation. We also observed a linear relationship between the concentration of the product and oxTMB as a function of H2O2 concentration, which could be further applied for H2O2 sensing applications (colorimetric detection). These data suggest that the NRs enable the combined effect of an increased surface area relative to solid counterparts, the possibility of exposing highly active surface sites, and the exploitation of nanoconfinement effects due to the void regions between the core and shell components. These results provide important insights into the optimization of peroxidase-like performances beyond what can be achieved in conventional NPs and may inspire the development of better-performing artificial enzymes.
Introduction
Enzymes are the catalysts of biological processes,1 as their complex structures allow for high specificity and sensitivity to several biomolecules, which also makes them important tools for biosensing.1,2 For example, one of the most common laboratory assays, blood glucose determination, relies on the natural enzyme glucose oxidase.3,4 However, enzymes are often sensitive to changes in the reaction media, such as temperature, pH, and solvent, which restrict their storage conditions, shorten the shelf life and limit end-use applications.5 In addition, the synthesis and purification of enzymes involve complex and expensive processes, making their large-scale production not cost-effective.6 Consequently, the development of artificial enzymes comprising organic and/or inorganic materials that can reproduce an enzyme's function in a chemical reaction has received increased attention in the last few decades.7Among the several materials which mimic the function of enzymes are certain inorganic nanoparticles that have been reported to present oxidase, catalase, and peroxidase-like activities.8 Specifically, peroxidase-like activity, which involves the decomposition of peroxide and oxidation of substrates, is highly desirable for biosensing and clinical applications.9 Under the umbrella of inorganic nanoparticles presenting peroxidase-like activity (also referred to as nanozymes), gold (Au) nanoparticles (NPs) have received great attention due to their biocompatibility, easy-to-functionalize surface, stability, and straightforward synthesis.10,11–13 Nevertheless, their peroxidase-like activity is still limited relative to enzymes.14 In this context, strategies for the improvement of activities in Au-based NPs have focused on the optimization of individual NP features, such as NP size, shape, composition, surface morphology, and metal–support interactions. Although these strategies have allowed for enhanced enzyme-mimicking performances relative to conventional Au NPs, it is clear that further improvements in activity are desperately still needed.15–19
In this paper, we address this challenge by engineering NP morphologies to enable the combined optimization of several morphological features for the improved performances of Au-based nanozymes. We focus on multimetallic nanorattles (NRs) comprising an Au core inside a bimetallic AgAu shell, separated by a void (Au@AgAu NRs).20 NRs show a combination of an increased surface area relative to solid counterparts due to the hollow interiors and ultrathin walls, the possibility of exposing highly active surface sites due to the porous nature of the shell, and the exploitation of nanoconfinement effects due to the void regions between the core and shell components. Although some examples of the use of NRs as nanozymes have been described, they remain limited and the enhancement in performance has been mostly attributed to an increase in the surface area.19 A better understanding of how metallic NRs perform as nanozymes in terms of structure–performance relationships relative to solid and hollow NP counterparts is essential to unlock their full potential and develop better performing nanozymes. We confront this issue by investigating the peroxidase-like activity of NRs relative to Au NPs and AgAu nanoshells (NSs) using TMB oxidation in the presence of H2O2 as a model transformation. We have found that the NRs displayed significantly higher activities relative to Au NPs as nanozymes (an increase of 152 fold). By comparing the activity of conventional Au NPs with that of NSs and NRs, we found that nanoconfinement played a major role in the significant enhancement of peroxidase-like activity.
Experimental section
Materials and methods
Analytical grade chemicals chloroauric acid trihydrate (HAuCl4·3H2O, 99.9%, Sigma-Aldrich), sodium citrate dihydrate (C6H5Na3O7·2H2O, 99%, Sigma-Aldrich), silver nitrate (AgNO3, 99%, Sigma-Aldrich), polyvinylpyrrolidone (PVP, 99%, Sigma-Aldrich), sodium hydroxide (NaOH, 99.2%, VWR), ascorbic acid (C6H8O6, 99%, Sigma-Aldrich), 3,3′,5,5′-tetramethylbenzidine (C16H20N2, 98%, Sigma-Aldrich), hydrogen peroxide (H2O2, 33%, VWR), acetic acid (C2H2O2, Fischer Chemical), and sodium acetate trihydrate (C2H3NaO2·3H2O,99%, VWR) were used as received. Deionized water (DI) was used to prepare all the solutions.Ultraviolet–Visible (UV/Vis) spectroscopy was performed in aqueous suspensions containing NPs using a Shimadzu UV-2600 spectrophotometer. Absorbance was measured over a range of 200–1000 nm using a quartz cuvette (optical path length 1 cm). Field emission scanning electron microscopy (FESEM) was performed using a Hitachi S-4800 field emission scanning electron microscope. Samples were prepared by drop-casting a suspension containing the NPs over a Si wafer followed by drying under ambient conditions. Transmission electron microscopy (TEM) images were obtained on a Jeol JEM-1400 microscope operating at an accelerating voltage of 120 kV. To prepare TEM samples, an aqueous suspension of the catalysts was dripped onto a carbon film-supported copper grid (400 square mesh). Scanning transmission electron microscopy (STEM) bright field and high-angle annular dark-field (HAADF) images were obtained using a Hitachi SU9000 low kV STEM with an accelerating voltage of 30 kV. High-resolution STEM bright field and HAADF images were obtained with a Hitachi HD-2700 aberration-corrected STEM operating at 200 kV. Samples for STEM/HAADF were prepared using an analogous procedure as described for TEM. The composition of the NPs and the metal concentration in the catalytic experiments were analyzed by inductively coupled plasma-atomic emission spectroscopy (ICP-AES) using an Agilent Technologies 4100 MP AES. Three independent measurements were performed for each sample.
Synthesis of Au and Ag NPs
Au NPs were prepared using the Turkevich method.21 In a typical synthesis, 100 mL of 0.25 mM AuCl4− (aq) were added into a 250 mL round-bottomed flask under magnetic stirring and heated to 110 °C for 15 minutes. When the solution reached the boiling point, 3.0 mL of 1 wt% aqueous sodium citrate dihydrate was added to the solution and the reaction was allowed to proceed for another 10 min, leading to the formation of a red suspension containing the Au NPs.Ag NPs were synthesized by a homogeneous nucleation and growth method.22 To this end, 25 mL of an aqueous 5.0 mM sodium citrate and 0.25 mM tannic acid solution was heated to 100 °C for 15 minutes, followed by the addition of 250 μL of 25 mM AgNO3. After 10 minutes of magnetic stirring, the suspension was centrifuged to remove excess tannic acid and the NPs were resuspended in polyvinylpyrrolidone (PVP) 0.1 wt% aqueous solution.
Synthesis of Au@Ag core–shell NPs
Au@Ag core-shell particles (Au@Ag NPs) were obtained by a stepwise seeded-growth process using Au NPs as seeds for Ag deposition.20 In the first step, 10 mL of the as-prepared suspension containing the Au NPs was transferred to a 25 mL round-bottomed flask, followed by the quick and successive addition of 150 μL of 100 mM NaOH and 120 μL of 100 mM ascorbic acid (both solutions were aqueous). After the mixture was stirred for 5 min, 30 μL of aqueous 100 mM AgNO3 was added to the mixture and the reaction was allowed to proceed at room temperature for 30 min. This was the first step of Ag deposition and growth which was then repeated every 30 minutes until 4 deposition steps were completed. These multiple deposition steps lead to the formation of a dark orange suspension.Synthesis of Au@AgAu nanorattles and AgAu nanoshells
The synthesis of Au@AgAu nanorattles (Au@AgAu NRs) and AgAu nanoshells (AgAu NSs) was based on the galvanic replacement reaction between Au@Ag or Ag NPs (for nanorattles and nanoshells, respectively) and AuCl4− (aq).20 Initially, 5 mL of PVP 0.1 wt% (aq) and 2 mL of the suspensions containing Au@Ag or Ag NPs were stirred at 100 °C for 10 min in a 25 mL round-bottomed flask. Then, 1 mL of 0.4 mM AuCl4− (aq) was added dropwise and the reaction proceeded for another 10 min. The reaction mixture was cooled to room temperature, and the product was washed three times with ethanol and three times with hot water by successive rounds of centrifugation at 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 rpm and removal of the supernatant.
500 rpm and removal of the supernatant.
The peroxidase-like activity of the NPs, NSs, and NRs
The oxidation of 3,3′,5,5′-tetramethylbenzidine (TMB) was used to evaluate the catalytic performance (peroxidase-like activity) of the Au NPs, AgAu NSs, or Au@AgAu NRs.23 The oxidation reactions were carried out in 2 mL centrifuge tubes containing a 1 mL solution of H2O2 0.6 M, TMB 750 μM, and 37.8 μL of Au NPs, 190 μL of AgAu NSs or 50 μL of Au@AgAu NRs in HAc–NaAc buffer, with pH 4.5. The volume of each catalyst was determined to maintain the same metal loading in the reaction (8.7 μM). The reactions were carried out at room temperature with a Grant-bio PHMT Thermo Shaker at 1000 rpm. After 20 minutes, stirring was suspended and 3 drops of H2SO4 1.0 M were added to stop the reaction. The UV/Vis absorbance values were measured for each sample and the concentrations of oxTMB were obtained using the Beer-Lambert law assuming a molar absorption coefficient of 59000 M−1 cm−1 at 450 nm. The total metal loading in all catalytic reactions was kept constant to allow the comparison among the NP, NS, and NR samples. Three independent reactions were performed for each sample.
Kinetics studies
In addition to the oxidation reactions, kinetics studies were performed by measuring the absorbance at 652 nm as a function of time. Measurements were done in a 1 cm × 1 cm × 4 cm PMMA cuvette containing a 3 mL solution of H2O2 (concentrations varied from 0.0–0.600 M), TMB 750 μM, and Au NPs, AgAu NSs, or Au@AgAu NRs in HAc–NaAc buffer, pH 4.5. The UV/Vis absorption spectra were obtained using the extinction spectra of the NPs, NRs, and NSs for baseline subtraction. The concentrations of oxTMB were obtained using the Beer-Lambert law assuming a molar absorption coefficient of 39000 M−1 cm−1 at 652 nm.
The kinetics data for the NRs (concentration 1.6 × 10−10 NPs mL−1) were treated using the Michaelis–Menten model, in which the Michaelis–Menten constant (Km) and the maximal velocity at saturating substrate concentrations (Vmax) were calculated using the Lineweaver–Burk equation, in which C is the initial concentration of the substrate.24
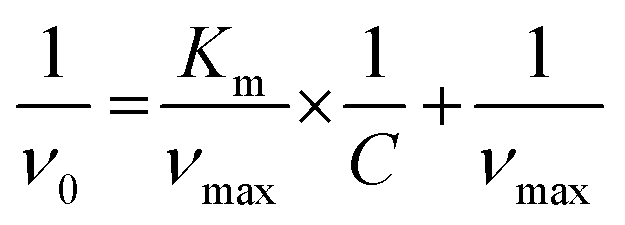
Colorimetric detection of H2O2
The colorimetric detection of H2O2 was performed in 2 mL centrifuge tubes containing a 1 mL solution of H2O2 0.0–0.6 M, TMB 750 μM, and 50 μL of Au@AgAu NRs in HAc–NaAc buffer, pH 4.5. Reactions proceeded at room temperature with a Grant-bio PHMT Thermo Shaker at 1000 rpm. After 20 minutes, stirring was suspended and 3 drops of H2SO4 1.0 M were added to stop the reaction. The UV/Vis absorbance values were measured for each sample and the concentrations of oxTMB were obtained using the Beer-Lambert law assuming a molar absorption coefficient of 59000 M−1 cm−1 at 450 nm. Three independent reactions were performed for each sample.
Results and discussion
The NRs can be prepared by using Au NPs as seeds for Ag deposition at their surface to generate Au@Ag core–shell NPs. This is followed by a galvanic replacement reaction between Ag at the shells and AuCl4− (aq) leading to the formation of an AgAu shell containing the Au NPs in their interiors (nanorattle morphology) (Fig. S1†).20 For the benchmarking of the NR performances, we also prepared AgAu nanoshells (NSs) by a similar approach, i.e., a galvanic replacement reaction between Ag NPs and AuCl4− (aq).Fig. 1A–F show the SEM images and histograms of the size distribution for the obtained Au NPs (Fig. 1A and D), AgAu NSs (Fig. 1B and E), and Au@AgAu NRs (Fig. 1C and F). It can be observed that all nanomaterials displayed a spherical shape and were relatively uniform in terms of size distribution. The Au NPs, AgAu NSs, and Au@AgAu NRs were 15, 27, and 32 nm in diameter, respectively. HRTEM and STEM images for the Au@AgAu NRs (Fig. 1G–I) indicate the formation of the nanorattle morphology, in which Au NPs are present inside a shell around <5 nm in thickness and separated by a void. The STEM-HAADF images (Fig. 1I and 2A–C) further confirm the formation of the nanorattle morphology comprising an Au core that appears brighter in the image inside a bimetallic AgAu shell, in agreement with previous reports.20,25 The higher magnification STEM-HAADF images (Fig. 2A–C and Fig. S2†) show that the shells contain polycrystalline domains, are <5 nm in thickness and are porous. To elucidate and characterize the pore sizes, we obtained higher magnification HRTEM images of different regions in the NRs (Fig. S3A–C†), STEM images using a secondary electron detector (Fig. S3D†), and additional higher magnification STEM-HAADF images of the NRs (Fig. S4†). These images clearly show the presence of various pores at the surface. It can be observed that they are <10 nm in diameter, having sizes as low as <3 nm.
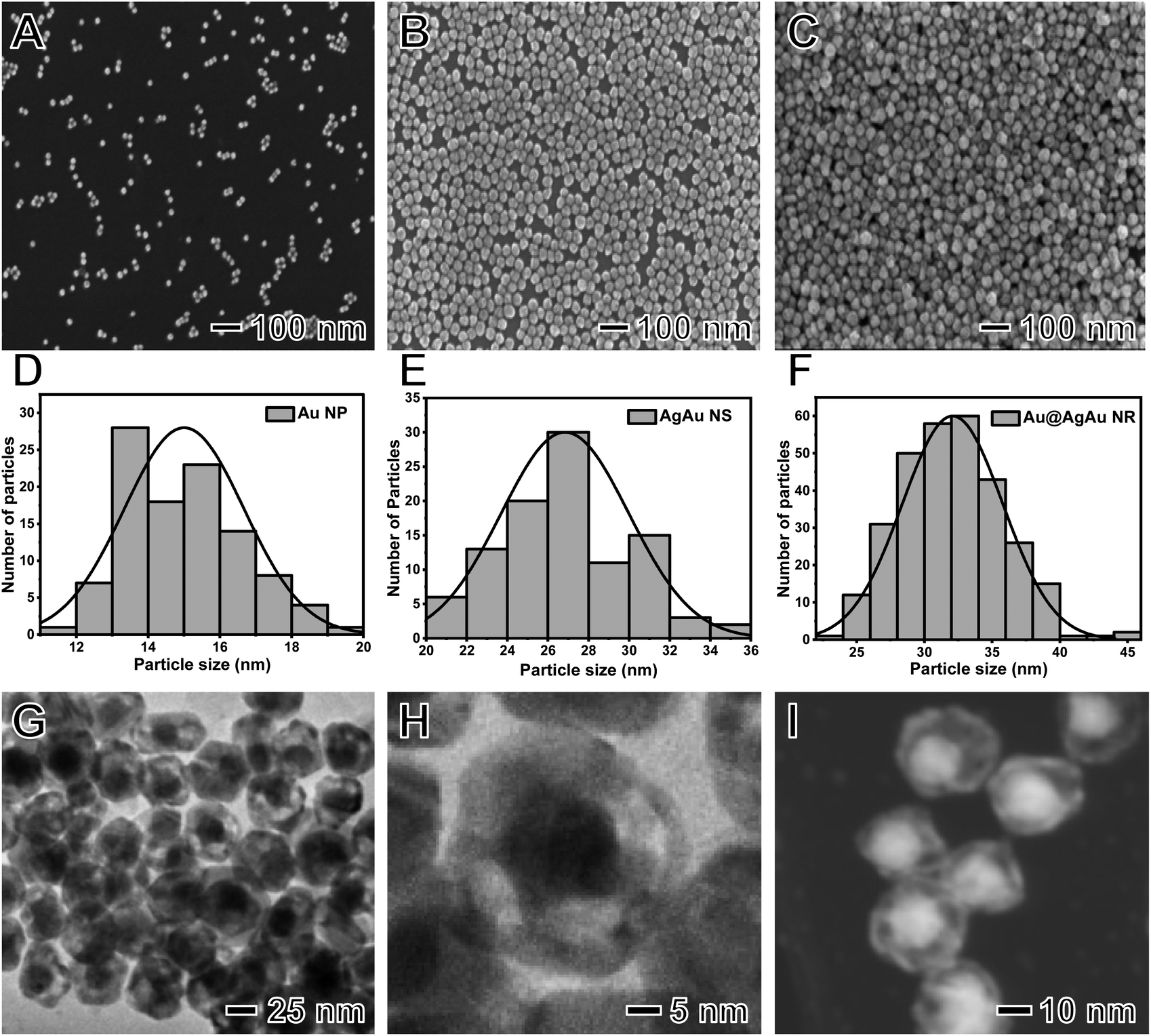 | ||
| Fig. 1 (A–C) SEM images of (A) Au NPs, (B) Ag@Au NSs, and (C) Au@AgAu NRs. (D–F) Histograms of the size distribution of the (D) Au NPs, (E) Ag@Au NSs, and (F) Au@AgAu NRs. (G and H) TEM and (I) STEM-HAADF images of the Au@AgAu NRs depict the presence of an Au nanosphere inside an AgAu shell separated by a void. | ||
 | ||
| Fig. 2 (A–D) STEM-HAADF images of the Au@AgAu NRs, in which the brightest contrast from the Au core results from its higher Z (atomic number) as ZAu > ZAg. (E and F) Corresponding EDS elemental maps of (E) Au (blue) and (F) Ag (yellow) of the region shown in (D). | ||
We used STEM-energy dispersive X-ray spectroscopy (EDS) elemental mappings to confirm the distribution of Au and Ag in the NRs (Fig. 2D–F). The elemental distribution for Au and Ag in Fig. 2D is depicted in blue (Fig. 2E) and yellow (Fig. 2F), respectively. It can be observed that the NRs indeed possessed a bimetallic composition, in which Au is present both at the core and the shells of the NRs, while Ag is also present in the shells due to its incomplete oxidation during the galvanic replacement reaction. In this case, during the synthesis of the NRs, it has been reported that the partial oxidation of Ag from the starting Au@Ag NPs is not observed and the resulting shells in the NRs are bimetallic and comprise an AgAu alloy.20,26 Thus, the STEM-EDS support the formation of bimetallic NRs. The EDS spectrum recorded from the STEM-EDS maps is shown in Fig. S5† and suggests that the Au content corresponds to 23 wt%. The XRD diffractogram (Fig. S6†) shows the presence of peaks characteristic of fcc Au and Ag, indicating that our synthesis procedure does not lead to the incorporation of any crystalline impurities in the NRs. It is important to note that as the Au cores are not stuck to the shell walls, they are expected to move freely inside the NRs in solution.27,28
The UV/Vis extinction spectra of Au NPs, Ag@Au NSs and Au@AgAu NRs shown in Fig. 3 displayed a maximum extinction band centered at 520, 440, and 466 nm, respectively. The absorbance peak at 520 nm for Au NPs is characteristic of the dipolar mode for the localized surface plasmon resonance (LSPR) and agrees with the presence of NPs 15 nm in size.29 The band at 440 nm is also in agreement with the formation of AgAu NSs as the deposition of Au, as well as the formation of hollow interiors are reported to redshift the characteristic Ag LSPR band at 420 nm.30 The broad band at 466 nm for the NRs is a result of the coupling of the core and shell LSPR effects.20 This indicates that both the Au core and AgAu shell contribute to the optical and electronic properties of the rattle structure. This band is red-shifted relative to AgAu NSs and blue-shifted relative to the Au NPs.
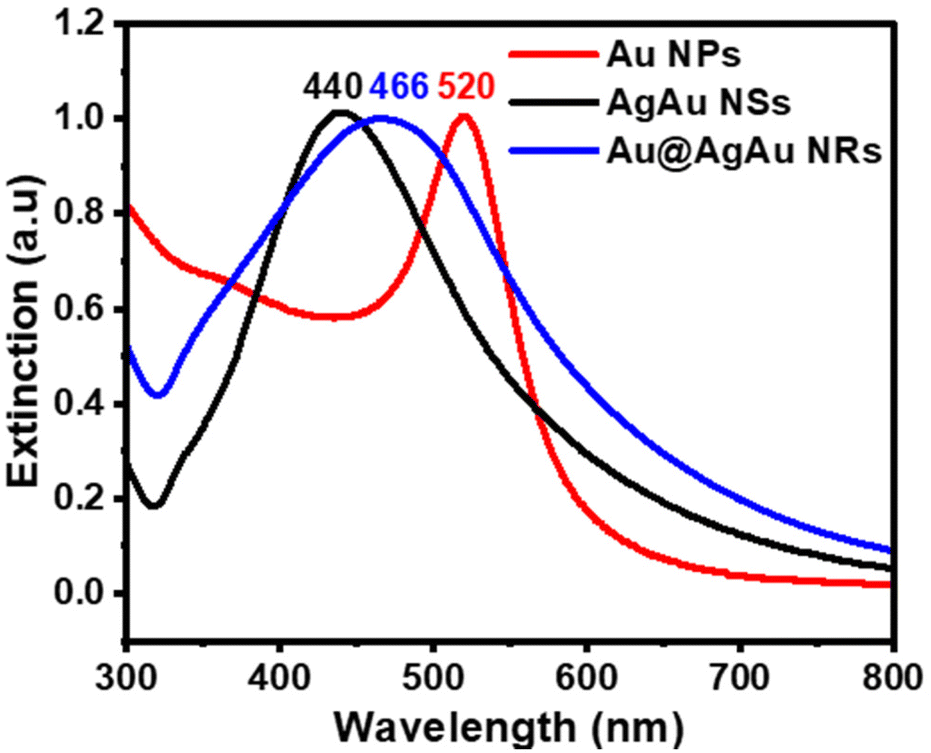 | ||
| Fig. 3 Normalized UV/Vis extinction spectra were recorded from aqueous suspensions containing Au NPs (red trace), AgAu NSs (black trace), and Au@AgAu NRs (blue trace). | ||
The NR morphology and its synthesis strategy enable the combination of the following features that are attractive for applications as nanozymes: (i) an increased surface area relative to solid counterparts;31 (ii) the possibility of exposing highly active surface sites due to the porous nature of the shell;32 and (iii) the exploitation of nanoconfinement effects due to the void regions between the core and shell components.33 All these factors are envisioned to contribute to higher nanoenzymatic performances relative to conventional Au NPs or NSs. While hollow interiors enable higher surface-to-volume ratios relative to solid NP counterparts, a nanosphere inside the void enables the combination of properties between core and shell components.34 Moreover, it has been shown that the surface charge as enabled by different surfactants in the synthesis of Au NPs affects their performances as peroxidase mimics, with citrate capped Au NPs (negatively charged) having lower activities relative to PVP (positively charged) counterparts.35 Thus, this indicates that PVP-capped NSs and NRs would have better peroxidase activity than citrate-capped Au NPs.
The application of NRs as nanozymes was evaluated focusing on the peroxidase-like activity of the NRs by using TMB oxidation in the presence of H2O2 as a model reaction as depicted in Fig. 4A. The activity was benchmarked against citrate-capped Au NPs and AgAu NSs. All catalytic experiments were performed under the same reaction conditions and metal loading. A significant change in the colour of the reaction mixture could be observed in the presence of the NRs, TMB, and H2O2 because of the formation of oxTMB (Fig. S7†). However, no change in color was detected in the absence of NRs or H2O2 (Fig. S7†). The concentration of the product (oxTMB) increased as a function of time as displayed in the UV/Vis spectra shown in Fig. 4B when the NRs were employed as nanozymes. This is visualized by the increase in the intensity of the absorption band assigned to oxTMB at 370 and 652 nm (which leads to the appearance of blue colour) as a function of time (from 0 to 20 min of the reaction).23Fig. 4C shows a comparison of the peroxidase-like activity for the NRs relative to the NPs and NSs in which the concentration of oxTMB after 20 min of reaction when the NRs were used as nanozymes corresponded to 35 μM. This was significantly higher relative to both the NSs (11 μM) and NPs (0.23 μM).
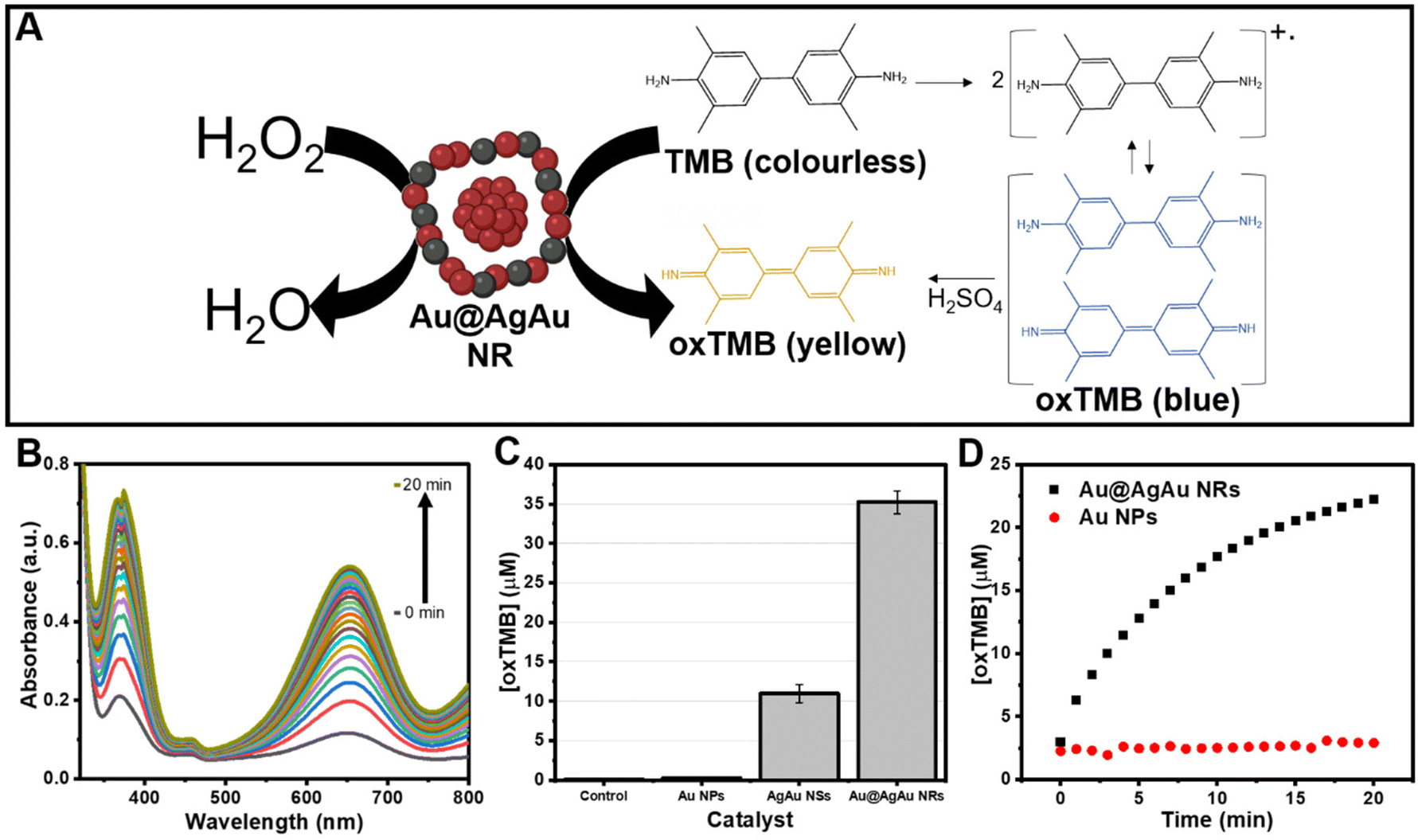 | ||
| Fig. 4 (A) Schematic illustration of the oxidation of TMB catalyzed by the NRs. (B) Absorption spectra of the TMB oxidation reaction were recorded in 1 min intervals for 20 min, using NRs as the catalyst. The spectra show an increase in the band centered at 652 nm assigned to oxTMB. (C) Comparison of the final concentration of oxTMB under blank conditions (no catalyst, control) as well as Au NPs, AgAu NSs, and Au@AgAu NRs as catalysts. Reaction conditions and metal loading were kept the same for all catalysts. The error bars represent the standard deviation of the three independent measurements. (D) The concentration of oxTMB as a function of time using the NRs (black trace) and NPs (red trace) as catalysts. The concentrations of oxTMB were obtained using the Beer-Lambert law assuming a molar absorption coefficient of 39000 M−1 cm−1 at 652 nm. | ||
The turnover frequency (TOFs) was also calculated by considering the total metal loading in each of the nanomaterials. The TOF for the particles corresponded to 337.4 × 10−5 s−1 for NRs, 104.9 × 10−5 s−1 for NSs, and 2.2 × 10−5 s−1 for NPs. Thus, it can be observed that the control over the morphology as enabled by the synthesis of NRs caused an increase in TOF by 3 and 151 fold in comparison to NSs and NPs, respectively. We also compared the performances with Ag NPs to isolate the effect of Ag over the peroxidase-like activity. Our data show that, under our employed conditions, Ag does not contribute significantly to the detected peroxidase-like activity.
It is well established that the peroxidase-like activity of Au nanozymes occurs via a surface binding mechanism, in which adsorbed H2O2 is decomposed into hydroxyl radicals, which are in turn responsible for the oxidation of the substrate TMB, resulting in the blue product oxTMB (Fig. 4A).36 The presence of hydroxyl radicals can be determined by a decrease in the concentration of the reaction product (oxTMB) upon the addition of the hydroxyl radical scavenger isopropanol (IPA) as shown in Fig. S8.† Thus, it is expected that the peroxidase-like activity of Au nanozymes will be affected by their surface area and the nature of capping agents or surface charge.37 Although the effect of particle size on the peroxidase-like properties of nanozymes has not been extensively studied, it is expected that a decrease in size38,39 and increased surface-to-volume ratios would lead to higher peroxidase-like activities.40–42 Therefore, we hypothesize that one of the reasons why NRs have better catalytic performance is their higher surface-to-volume ratios due to the nanorattle morphology, i.e., hollow interiors, ultrathin walls, and porous shells. In this case, regions comprising the pores, the interior of the nanorattles, and the Au core surface can become accessible to the H2O2 and TMB molecules and represent a catalytically active site for driving the conversion of TMB. This means that molecules can access the surface of the Au cores, inner and outer AgAu shell walls, and the regions within the pores in the NRs.19,20,43 Although these factors can contribute to the peroxidase-like activity of the NRs and NSs, it is plausible that they do not fully account for the substantial enhancement when compared to NPs.
We would like to propose herein that the hollow and porous structure can also lead to enhanced reaction rates due to the confinement effect on the interior of the NRs and NSs.44 In nanocatalysis (and therefore also in nanoenzymatic performances), activity is determined by a balance between mass transport and conversion. This is particularly important when catalytic centers (such as metallic surfaces) are inside confining materials (shells in this case). Although the understanding of nanoconfinement effects in catalysis is complex, it is noted that they can increase reaction rates relative to non-confined counterparts by altering (decrease) the adsorption strength of surface species as well as the enrichment of the concentration of intermediate or reactant species in the confining space.45 For example, in Au NPs the OH radicals that are formed from H2O2 at the NP surface could dissociate from the Au NPs and diffuse into the bulk solution before the combination with TMB. In the NSs and NRs, the OH radicals would be temporarily trapped inside the shells (NSs) or between the NP core and the shell (NRs void) as it can be formed from the Au NP surface and AgAu shell inner surface in the NRs. This can create an increase in the local concentration of OH radicals and therefore increase the reaction rate for the formation of oxTMB.23,45,46
This hypothesis would support the significant enhancement of the NSs and NRs relative to the NPs and support the results in which the NSs are compared with the NRs. In the NSs, there was an increase in activity relative to the Au NPs due to nanoconfinement effects, a higher surface-to-volume ratio relative to the Au NPs enabled by the nanoshell/porous morphology, and the absence of citrate as the stabilizer. However, when compared to the NRs, a much lower activity was observed. In the NRs, it is possible that the interior core comprising Au NPs that is present in the NRs substantially increases the magnitude of the nanoconfinement effects relative to the NSs. It is noteworthy that although Ag is also able to promote the homolytic cleavage of the HO–OH bond,47 the presence of Ag in the AgAu NSs and NRs does not significantly contribute to the increased activity since the pH of the reaction is not optimum for hydroxyl radical generation by Ag.48
The temporal evolution studies for the formation of oxTMB catalyzed by the NRs and Au NPs are shown in Fig. 4D. While no significant increase was detected for the Au NPs, an increase in the absorbance values and, consequently, in the concentration of oxTMB in the reaction media with time was detected in the presence of the Au@AgAu NRs. The rate of increase in the concentration of oxTMB produced is higher in the first 5 minutes of the reaction. Then, it starts to decrease as the reaction proceeds, reaching its maximum at 20 minutes. Therefore, in our subsequent experiments, we chose to evaluate the reaction kinetics on the effect of the H2O2 concentration in the 0–5 min interval.
Fig. 5A shows the increase in [oxTMB] as a function of time for different concentrations of H2O2. It can be observed that the produced [oxTMB] increased linearly as a function of time and H2O2 concentration between 0.1 to 0.6 M. We then performed the apparent steady-state kinetic analysis to acquire the Michaelis–Menten constant (Km) and maximal velocity at saturating substrate concentrations (Vmax) which can give further insights into the peroxidase-activity of the NRs. In the case of enzymatic reactions, Vmax/[E0], where [E0] is the initial concentration of the enzyme or catalyst (in the case of nanozymes), corresponds to kcat (catalytic constant). Typical Michaelis–Menten curves by varying the concentration of H2O2 were obtained, and the corresponding Lineweaver–Burk plot for H2O2 is shown in Fig. 5B. The Lineweaver–Burk plot reinforces that there is a well-defined linear correlation between the concentration of H2O2 and oxTMB, and it was used to obtain the Michaelis–Menten constant, Km = 1.65 mM, and the maximal velocity at saturating substrate concentrations, Vmax = 2.0 μM min−1. As previously established, the lower the Km value, the higher the affinity between the substrate (H2O2 in this case) and the catalyst surface. The higher the Vmax for a fixed concentration of catalyst, the higher the catalytic activity. These parameters were compared with the natural horseradish peroxidase (HRP, Km = 3.7 mM and Vmax 5.53 μM min−1) as reported by Gao et al.49 It can be observed that the NRs displayed a higher affinity for H2O2 than HRP, while Vmax values were higher for HRP.
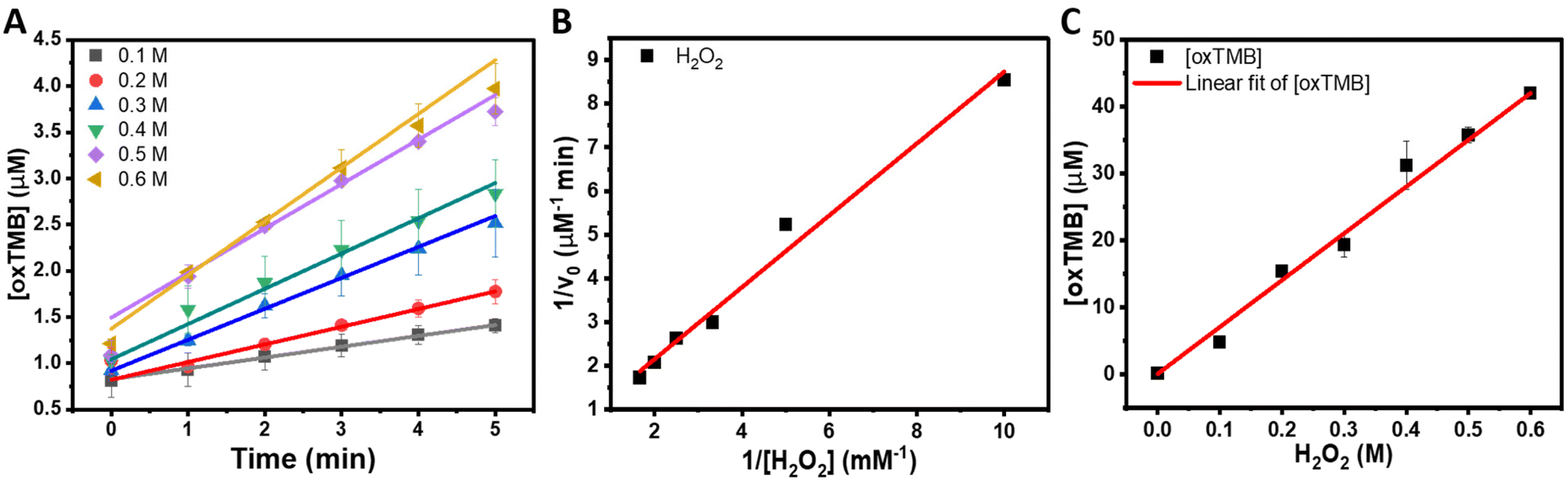 | ||
| Fig. 5 (A) Temporal evolution of oxTMB concentrations in 0–5 min of the reactions catalyzed by Au@AgAu NRs with different concentrations of H2O2 and (B) Lineweaver-Burk plots for H2O2. The pH of the reaction solutions was kept at 4.5, and the concentrations of NRs and TMB were 1.6 × 10−10 NPs mL−1 and 750 μM, respectively. The concentrations of oxTMB were obtained using the Beer-Lambert law assuming a molar absorption coefficient of 39000 M−1 cm−1 at 652 nm. The error bars represent the standard deviation of the three independent measurements. (C) Linear fitting of the variation of oxTMB concentration for different concentrations of H2O2 (0.1–0.6 M). | ||
It is noteworthy that direct comparison of Km, Vmax, and Kcat with other reported systems is challenging as they can depend on a variety of experimental parameters. In this context, as an approximation, we calculated the TOF (turnover frequencies) values as a function of metal loading for the NRs, NSS, and NPs as well as other reported systems based on Au for various reaction conditions as shown in Table 1. It can be observed that the NRs displayed improved TOFs relative to many reported systems, except for chitosan–Au NPs, which required higher temperatures during the reaction and a much higher concentration of TMB.50 Relative to systems comprising conventional Au NPs, the NRs are more versatile in terms of future optimization for tailoring the morphology and surface compositions. NRs could also serve as carriers for other biomolecules that could be encapsulated in the void of the rattle structure. Furthermore, the NRs have the potential to enable the exploitation of LSPR excitation to further increase the peroxidase-like activity. In this case, the NRs enable the plasmonic hybridization between core and shell components that can lead to electromagnetic hot spots, leading to much higher plasmonic properties relative to individual Au NPs. Finally, the synthesis of the NRs can be adapted to incorporate a different, 3rd metal at the shell enabling the design of nanoenzymes that can be employed in bifunctional or cascade molecular transformations. Our reported data illustrate that the NRs are promising to obtain improvement in nanoenzymatic activities due to the combination of higher surface areas, exposure to highly active sites, and nanoconfinement effects. Regarding stability, horseradish peroxidase is commonly used as the benchmark enzyme in the TMB oxidation reaction in the presence of H2O2. It has been reported that denaturation of this enzyme starts at temperatures as low as 45 °C, and even the buffer solution or pH can influence stability.51,52 On the other hand, the Au@AgAu NRs can withstand a large range of temperatures since they are synthesized at 100 °C. Moreover, as they are not stabilized by electrostatic interactions, variations in pH do not affect their stability.
| Nanozyme | Metal loading (μM) | [TMB] (μM) | [H2O2] (M) | T (°C) | TOF (×10−5 s−1)a | Ref. |
|---|---|---|---|---|---|---|
| a The TOF was calculated based on the total metal loading and final [oxTMB]. b This work. | ||||||
| Au@AgAu NRs | 8.7 | 750 | 0.600 | 25 | 337.4 | |
| AgAu NSs | 8.7 | 750 | 0.600 | 25 | 104.9 | |
| Au NPs | 8.7 | 750 | 0.600 | 25 | 2.2 | |
| Au SRNPs | 15.7 | 750 | 0.568 | 25 | 11 | 23 |
| Chitosan-Au NPs | 2.0 | 3495 | 0.097 | 50 | 3053 | 53 |
| Au NPs | 228.6 | 143 | 0.0001 | 37 | 8.3 | 54 |
| Ag-CDs | 36 | 1600 | 0.0016 | RT | 18.6 | 55 |
| Ag NP-N-GQD | 81.7 | 333 | 0.03 | 37 | 23 | 56 |
Based on the dependence of TMB oxidation on H2O2 concentration, the peroxidase-like activity of the NRs was used for the colorimetric H2O2 detection as shown in Fig. 5C. It can be observed that the concentration of oxTMB was linearly dependent on the concentration of H2O2 from 0.1 M to 0.6 M, indicating that this colorimetric assay could be applied to measure the amount of H2O2 in this concentration range. The detection limit of H2O2 under our experimental conditions was 13.42 mM. It is important to note that the detection limit can be further optimized with changes in the concentration of NRs employed in the experiments.
Conclusions
We have demonstrated that the nanorattle morphology, comprising a nanosphere inside a nanoshell and separated by a void, can be used to significantly enhance the peroxidase-like activity of Au-based nanozymes. The enhancement in peroxidase-like activity in these systems can be assigned to a combination of increased surface areas relative to conventional solid NPs, exposure of active surface sites within the pores of the shells, and nanoconfinement effects between nanosphere and nanoshell components. By comparing the activity of conventional Au NPs with NSs and NRs, we found that nanoconfinement played a major role in the significant enhancement of peroxidase-like activity. Specifically, by using the oxidation of TMB in the presence of H2O2 as a model transformation, we found that the NR morphology led to an increase in the peroxidase-like activity of 3.2 and 152 fold relative to the NSs and NPs, respectively, under our experimental conditions. Based on the nanozyme kinetics results, the NRs also showed the lowest Km, indicating better interaction with the substrate and faster product formation. We observed a linear relationship between the concentration of the product and oxTMB as a function of H2O2 concentration, which can be further applied for H2O2 sensing applications (colorimetric detection). We anticipate that these results provide important insights and inspire the development of nanozymes with high catalytic activity. It is envisioned that this strategy can be extended to incorporate other metals at the shells of the NRs, such as Pd and Pt, to enable the design of systems that allow for nanoenzymatic and catalytic cascade transformations. Moreover, due to the NR morphology, plasmonic coupling between the Au at the cores and the plasmonic shell can lead to the formation of electromagnetic hot spots due to the localized surface plasmon resonance excitation. This can be exploited to trigger and further enhance nanoenzymatic and catalytic activities under light excitation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Jane and Aatos Erkko Foundation, start-up funds from the University of Helsinki, Academy of Finland, the Fortum and Neste Foundation, the seed funding from the Helsinki Institute of Sustainability Science (HELSUS), and the European Union's Horizon 2020 Research and Innovation Program under Grant Agreement No 101022633.References
-
G. Cooper, in The Cell: A Molecular Approach, Sinauer Associates Inc., 2nd edn, 2000 Search PubMed
.
- H. H. Nguyen, S. H. Lee, U. J. Lee, C. D. Fermin and M. Kim, Materials, 2019, 12, 121 CrossRef CAS PubMed
-
H. Walker, W. Hall and J. Hurst, in Clinical Methods: The History, Physical, and Laboratory Examinations, Buttekrworths, Boston, 1990 Search PubMed
- S. Horton, K. A. Fleming, M. Kuti, L.-M. Looi, S. A. Pai, S. Sayed and M. L. Wilson, Am. J. Clin. Pathol., 2019, 151, 446–451 CrossRef CAS PubMed
-
H. C. Holm, P. M. Nielsen and F. Longin, in Lipid Modification by Enzymes and Engineered Microbes, ed. U. T. Bornscheuer, AOCS Press, 2018, pp. 343–373 Search PubMed
- V. Naresh and N. Lee, Sensors, 2021, 21, 1109 CrossRef CAS PubMed
-
S. Lin, J. Wu, J. Yao, W. Cao, F. Muhammad and H. Wei, Biomedical Applications of Functionalized Nanomaterials: Concepts, Development and Clinical Translation, 2018, pp. 171–209 Search PubMed
- F. Attar, M. G. Shahpar, B. Rasti, M. Sharifi, A. A. Saboury, S. M. Rezayat and M. Falahati, J. Mol. Liq., 2019, 278, 130–144 CrossRef CAS
- M. Hamid and K. U. Rehman, Food Chem., 2009, 115, 1177–1186 CrossRef CAS
- J. Lou-Franco, B. Das, C. Elliott and C. Cao, Nano-Micro Lett., 2020, 13, 1–36 Search PubMed
- Y. Jv, B. Li and R. Cao, Chem. Commun., 2010, 46, 8017 RSC
- R. Niu, C. Song, F. Gao, W. Fang, X. Jiang, S. Ren, D. Zhu, S. Su, J. Chao, S. Chen, C. Fan, L. Wang, R. J. Niu, C. Y. Song, F. Gao, X. Y. Jiang, S. K. Ren, D. Zhu, S. Su, J. Chao, S. F. Chen, L. Wang, W. N. Fang and C. H. Fan, Angew. Chem., Int. Ed., 2021, 60, 11695–11701 CrossRef CAS PubMed
- J. Yin, M. Xie, J. Wang, M. Cui, D. Zhu, S. Su, C. Fan, J. Chao, Q. Li, L. Wang, J. Yin, M. Xie, J. Wang, M. Cui, D. Zhu, S. Su, J. Chao, L. Wang, C. Fan and Q. Li, Small, 2022, 18, 2200824 CrossRef CAS PubMed
- H. Ye, Z. Xi, K. Magloire and X. Xia, ChemNanoMat, 2019, 5, 860–868 CrossRef CAS
- Y. Zhang, E. Villarreal, G. G. Li, W. Wang and H. Wang, J. Phys. Chem. Lett., 2020, 11, 9321–9328 CrossRef CAS
- Z. Huang, B. Liu and J. Liu, Nanoscale, 2020, 12, 22467–22472 RSC
- M. Sharifi, S. H. Hosseinali, P. Yousefvand, A. Salihi, M. S. Shekha, F. M. Aziz, A. JouyaTalaei, A. Hasan and M. Falahati, Mater. Sci. Eng., C, 2020, 108, 110422 CrossRef CAS PubMed
- L. Han, C. Li, T. Zhang, Q. Lang and A. Liu, ACS Appl. Mater. Interfaces, 2015, 7, 14463–14470 CrossRef CAS PubMed
- S. Ghosh, P. Singh, S. Roy, K. Bhardwaj and A. Jaiswal, ChemBioChem, 2022, 23, e202100691 CAS
- A. G. M. da Silva, T. S. Rodrigues, V. G. Correia, T. V. Alves, R. S. Alves, R. A. Ando, F. R. Ornellas, J. Wang, L. H. Andrade and P. H. C. Camargo, Angew. Chem., Int. Ed., 2016, 55, 7111–7115 CrossRef CAS PubMed
- J. Turkevich, P. C. Stevenson and J. Hillier, Discuss. Faraday Soc., 1951, 11, 55–75 RSC
- N. G. Bastús, F. Merkoçi, J. Piella and V. Puntes, Chem. Mater., 2014, 26, 2836–2846 CrossRef
- Y. Zhang, E. Villarreal, G. G. Li, W. Wang and H. Wang, J. Phys. Chem. Lett., 2020, 11, 9321–9328 CrossRef CAS
- P. J. F. HENDERSON, Biochem. J., 1973, 135, 101–107 CrossRef CAS
- T. Akita, M. Kohyama and M. Haruta, Acc. Chem. Res., 2013, 46, 1773–1782 CrossRef CAS PubMed
- Y. G. Sun, B. Wiley, Z. Y. Li and Y. N. Xia, J. Am. Chem. Soc., 2004, 126, 9399–9406 CrossRef CAS PubMed
- M. J. Schnepf, M. Mayer, C. Kuttner, M. Tebbe, D. Wolf, M. Dulle, T. Altantzis, P. Formanek, S. Förster, S. Bals, T. A. F. König and A. Fery, Nanoscale, 2017, 9, 9376–9385 RSC
- M. A. Mahmoud, J. Phys. Chem. C, 2014, 118, 10321–10328 CrossRef CAS
- N. Sarfraz and I. Khan, Chem.– Asian J., 2021, 16, 720–742 CrossRef CAS PubMed
- O. Pryshchepa, P. Pomastowski and B. Buszewski, Adv. Colloid Interface Sci., 2020, 284, 102246 CrossRef CAS PubMed
- X. Wang, J. Feng, Y. Bai, Q. Zhang and Y. Yin, Chem. Rev., 2016, 116, 10983–11060 CrossRef CAS
- J. Liu, S. Z. Qiao, J. S. Chen, X. W. (David) Lou, X. Xing and G. Q. (Max) Lu, Chem. Commun., 2011, 47, 12578 RSC
- A. B. Grommet, M. Feller and R. Klajn, Nat. Nanotechnol., 2020, 15, 256–271 CrossRef CAS PubMed
- M. Priebe and K. M. Fromm, Chem. – Eur. J., 2015, 21, 3854–3874 CrossRef CAS
- C.-P. Liu, K.-C. Chen, C.-F. Su, P.-Y. Yu and P.-W. Lee, Catalysts, 2019, 9, 517 CrossRef CAS
- M. Kumawat, H. Madhyastha, A. Umapathi, M. Singh, N. Revaprasadu and H. K. Daima, Langmuir, 2022, 38, 1877–1887 CrossRef CAS PubMed
- H. Ye, Z. Xi, K. Magloire and X. Xia, ChemNanoMat, 2019, 5, 860–868 CrossRef CAS
- R. Serra-Maia, S. Chastka, M. Bellier, T. Douglas, J. D. Rimstidt and F. M. Michel, J. Catal., 2019, 373, 58–66 CrossRef CAS
- X. Xia, J. Zhang, N. Lu, M. J. Kim, K. Ghale, Y. Xu, E. McKenzie, J. Liu and H. Ye, ACS Nano, 2015, 9, 9994–10004 CrossRef CAS PubMed
- S. Liu, F. Lu, R. Xing and J. Zhu, Chem. – Eur. J., 2011, 17, 620–625 CrossRef CAS PubMed
- A. Tripathi, K. D. Harris and A. L. Elias, PLoS One, 2021, 16, e0257777 CrossRef CAS PubMed
- C. A. S. Ballesteros, L. A. Mercante, A. D. Alvarenga, M. H. M. Facure, R. Schneider and D. S. Correa, Mater. Chem. Front., 2021, 5, 7419–7451 RSC
- M. Ahlawat, A. Sarkar, S. Roy and A. Jaiswal, ChemNanoMat, 2019, 5, 625–633 CrossRef CAS
- B. Dong, Y. Pei, F. Zhao, T. W. Goh, Z. Qi, C. Xiao, K. Chen, W. Huang and N. Fang, Nat. Catal., 2018, 1, 135–140 CrossRef
- B. Dong, Y. Pei, N. Mansour, X. Lu, K. Yang, W. Huang and N. Fang, Nat. Commun., 2019, 10, 1–6 CrossRef CAS PubMed
- J. Li, W. Liu, X. Wu and X. Gao, Biomaterials, 2015, 48, 37–44 CrossRef CAS PubMed
- H. Jiang, Z. Chen, H. Cao and Y. Huang, Analyst, 2012, 137, 5560 RSC
- W. He, Y.-T. Zhou, W. G. Wamer, M. D. Boudreau and J.-J. Yin, Biomaterials, 2012, 33, 7547–7555 CrossRef CAS PubMed
- L. Gao, J. Zhuang, L. Nie, J. Zhang, Y. Zhang, N. Gu, T. Wang, J. Feng, D. Yang, S. Perrett and X. Yan, Nat. Nanotechnol., 2007, 2, 577–583 CrossRef CAS PubMed
- C. Jiang, J. Zhu, Z. Li, J. Luo, J. Wang and Y. Sun, RSC Adv., 2017, 7, 44463–44469 RSC
- S. Asad, S. F. Torabi, M. Fathi-Roudsari, N. Ghaemi and K. Khajeh, Int. J. Biol. Macromol., 2011, 48, 566–570 CrossRef CAS PubMed
- K. Chattopadhyay and S. Mazumdar, Biochemistry, 1999, 39, 263–270 CrossRef PubMed
- C. Jiang, J. Zhu, Z. Li, J. Luo, J. Wang and Y. Sun, RSC Adv., 2017, 7, 44463–44469 RSC
- V. Kumar, D. Bano, D. K. Singh, S. Mohan, V. K. Singh and S. H. Hasan, ACS Sustainable Chem. Eng., 2018, 6, 7662–7675 CrossRef CAS
- A. Wang, C. Guan, G. Shan, Y. Chen, C. Wang and Y. Liu, Microchim. Acta, 2019, 186, 644 CrossRef
- J. Ju, R. Zhang and W. Chen, Sens. Actuators, B, 2016, 228, 66–73 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2dt02389j |
| This journal is © The Royal Society of Chemistry 2022 |