DOI:
10.1039/D1DT04204A
(Paper)
Dalton Trans., 2022,
51, 4814-4828
Oxidation of europium with ammonium perfluorocarboxylates in liquid ammonia: pathways to europium(II) carboxylates and hexanuclear europium(III) fluoridocarboxylate complexes†
Received
14th December 2021
, Accepted 25th February 2022
First published on 1st March 2022
Introduction
Inorganic trifluoroacetates define a well-known class of compounds continuously investigated up to the present time since the first syntheses by Swarts et al. in the year 1922.1 More recently, metal trifluoroacetates gained attention as precursors for metal fluorides, the trifluoroacetato ligand being the fluoride source.2 Functional fluoridic thin-films or nanomaterials may be accessed this way.3 Furthermore, the trifluoroacetate ion proves to be an efficient ligand to stabilize low oxidation states of the d-block metals4 as well as subvalent oxidation states of heavy main group metals as in the case of the dibismuth(II) tetrakis(trifluoroacetate) molecule.5 The reason for this behavior can be assumed to be the electron-withdrawing along with the chelating or bridging character of the ligand. We assumed a similar stabilizing effect on the oxidation state + II of the lanthanoids (Ln) by trifluoroacetato and higher perfluorocarboxylato ligands, especially for europium and ytterbium, due to their particular electron configurations among the f-block metals (Eu: [Xe] 4f7 6s2; Yb: [Xe] 4f14 6s2).6 Although materials containing Ln2+ ions have been known for almost a century7 and are still the subject of current research (especially phosphors),8 only a very small number of simple europium(II) carboxylates has been reported up to date.9 Usually, in the course of crystallization these compounds form chain and network structures to satisfy the lanthanoids’ tendency to have high coordination numbers.10 The Ln3+ cations tend to give dimeric building units in their solid-state structures, with O,O′-bridging carboxylato ligands such as CH3COO,11 CF3COO12 and C2F5COO.13 However, under certain conditions, the formation of polyhedral cages is also observed. Here, in most cases, hydroxide ions cap the polyhedral faces of the [Ln]n cages and ensure maximum cohesion of the Ln3+ centers, while further anions occupy the polyhedral edges and bridge the Ln3+ centers, e.g. polyselenides,14 [PdCl4]2−,15 β-diketonates16 and trifluoromethanesulfonate.17 To date, only a few lanthanoid complexes can be found in the literature in which perfluorocarboxylate ions serve as bridging ligands.18
Herein we report the preparation of europium carboxylates with a high content of Eu2+ by the oxidation of europium with ammonium perfluorocarboxylates in liquid ammonia as solvent. Due to the oxidation sensitivity19 of Ln2+ and the Lewis acidity20 of Ln3+ ions, we assumed interesting subsequent chemistry for this class of substances, with special regard to perfluorocarboxylate ions as a potential source of fluoride ions. Based on the products of oxidation in liquid ammonia we succeeded in the synthesis and characterization of a solvated europium(II) trifluoroacetate 1, and in the further oxidation by perfluorocarboxylic acids to hexanuclear europium complexes, (NH4)2[Eu6F8(O2CR)12(RCOOH)6], with R = CF3 (2) and R = C2F5 (3). To the best of our knowledge, compounds 1, 2 and 3 are the first EuII perfluorocarboxylate and the first EuIII derivatives of hexanuclear LnIII fluoridocarboxylates,21 respectively.
Results and discussion
Oxidation of europium with ammonium perfluorocarboxylates in liquid ammonia
The stabilizing effect of liquid ammonia as a reaction medium on Eu2+ ions has been known for decades22 and has been used, for example, in the synthesis of Eu(C5H5)2,23 Eu(PH2)2,24 Eu(NH2)2,25 and of unsolvated europium(II) halides.26
It was reasonable to assume that liquid ammonia might also be a suitable solvent for the synthesis of unsolvated europium(II) trifluoroacetate, which is not yet known. In fact, the addition of ammonium trifluoroacetate to the blue solutions of europium in liquid ammonia results in the precipitation of a yellow solid. By evaporation of the ammonia an air-sensitive powder can be obtained, that did not diffract in PXRD experiments. In addition to some content of NH4F (detected via ion chromatography, elemental analysis, and IR spectroscopy), a ratio of 77% Eu2+ to 23% Eu3+ was detected via151Eu Mössbauer spectroscopy. Under similar reaction conditions, also a yellow powder with a ratio of 50% Eu2+ to 50% Eu3+ was obtained from ammonium pentafluoropropionate, generated in situ by dissolving anhydrous pentafluoropropionic acid in liquid ammonia. It can be concluded from the analytical results that the substances contain high amounts of EuII compounds, including europium(II) trifluoroacetate and europium(II) pentafluoropropionate, and that the decomposition of the perfluorocarboxylate ions, with simultaneous oxidation of the europium, plays an important role as a side reaction.
catena-Poly[europium(II)bis(μ2-N,N-dimethylformamide)bis(μ2-trifluoroacetato)], [Eu(O2CCF3)2(dmf)2]∞ (1)
Synthesis and characterization.
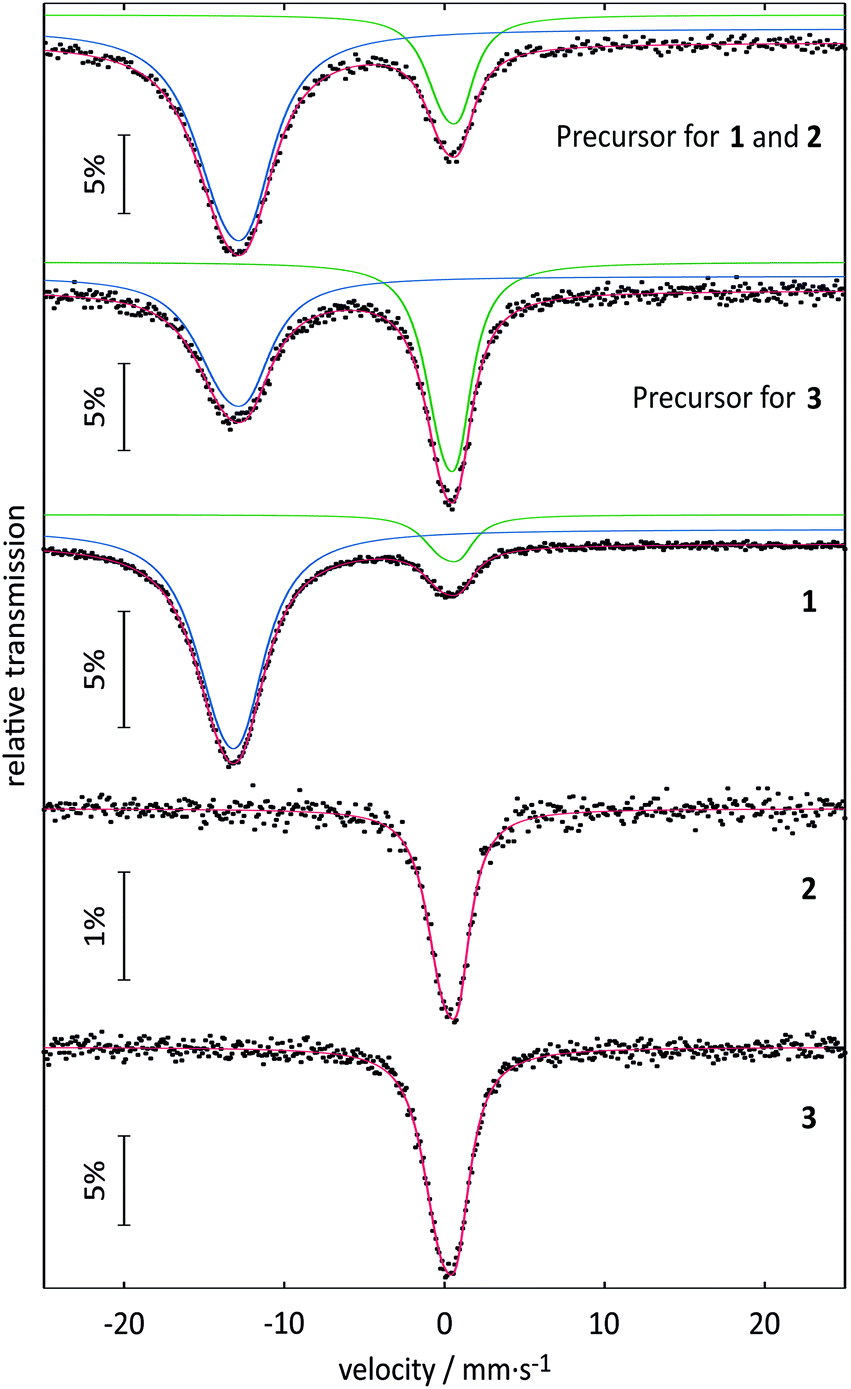
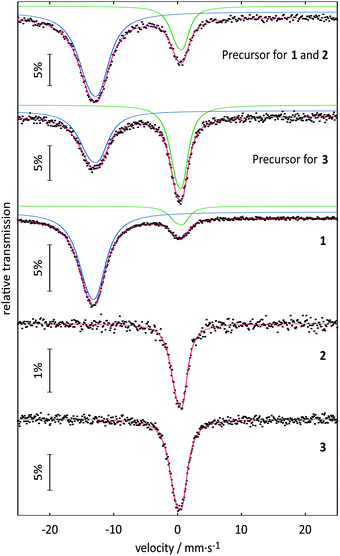
In the task of purification of the substances described above, the solvent selection is limited to aprotic solvents due to the inherent protolysis sensitivity. In trifluoroacetic anhydride, the Eu(O2CCF3)2-containing substance proved to be insoluble. With N,N-dimethylformamide (DMF) a yellow suspension results that can be separated to give a white solid and a clear, yellow solution. After this solution had been concentrated to half of its initial volume, it was stored in a closed ampoule at 8 °C. After 1–2 weeks very fine yellow crystal needles of 1 could be harvested. A single crystal of 1 suitable for crystal structure analysis could be selected directly from the solution. Subsequently, the mother liquor was removed, and the remaining yellow crystals dried in vacuo. A ratio of 88% Eu2+ to 12% Eu3+ was detected via151Eu Mössbauer spectroscopy. However, the signal intensity in a Mössbauer spectrum depends on the strength of the bonding of the europium atom in the crystal structure, which could not be considered here. Thus, the atomic ratio of Eu2+ to Eu3+ can only be approximated from the area ratio. The small amount of oxidation product is probably due to the isolation of the extremely oxidation-sensitive substance from the mother liquor and the preparation for the 151Eu Mössbauer spectroscopic measurements. The results of the 151Eu Mössbauer spectroscopic studies of 1 and of the two precursors are summarized in Table 1 along with the results for 2 and 3 that will be discussed later. The spectra are shown in Fig. 1. ATR-IR spectroscopy of freshly harvested crystals of 1 showed the typical bands of trifluoroacetates and of DMF. The compound turned out to be a labile solvate that loses some DMF even at room temperature in the absence of mother liquor. Not unexpectedly, CHN analysis in several attempts always indicated partial loss of DMF for the ‘dried’ substance. If ‘dried’ in vacuum (5 × 10−3 hPa, 1 h) about one equivalent of DMF is lost and the remaining yellow powder can be stored for at least three months in the absence of moisture. Samples were investigated via TGA/DSC-thermal analysis and PXRD. However, the latter method failed due to the lability of the substance that decomposed in the X-ray beam. The thermal analysis of the product indicated a mass loss of about one equivalent DMF (∼13%; calc. 13.95% for one eq. DMF) in connection with a broad endothermic event between room temperature and 180 °C (see Fig. S1 and S2, ESI†). A further endothermic feature indicated melting of the substance (Tonset = 221 °C), directly followed up by strong exothermic features (Tonset = 255 °C) that are related to a mass loss compatible to the complete decomposition of the substance in a narrow range of temperature. The remaining amorphous substance (∼45%) is assumed to be EuF3 mainly (calc. 46.32%, based on 1). In the course of a single crystal structure analysis 1 was finally proved to be the bis(DMF) adduct of Eu(O2CCF3)2.
 |
| | Fig. 1
151Eu Mössbauer spectra of 1, 2 and 3 (at 78 K), and their corresponding precursors (at 6 K). Signals with isomer shifts around 0 mm s−1 correspond to Eu3+ and signals around −13 mm s−1 correspond to Eu2+. The experimental data are represented by the dotted lines and the fitted data are represented by the colored lines. | |
Table 1 Fitting parameters of the 151Eu Mössbauer spectroscopic measurements of 1, 2 and 3, and their corresponding precursors
| Compound |
Temperature |
δ [mm s−1] |
ΔEQ [mm s−1] |
Γ [mm s−1] |
Area [%] |
|
δ = isomer shift, ΔEQ = quadrupole splitting, Γ = experimental line width. Parameters marked with an asterisk were kept fixed during the fitting procedure. |
| Precursor for 1 and 2 |
6 K |
–13.11(2) |
4.4(2) |
4.10(8) |
77(1) |
|
|
|
0.35(2) |
2.9(2) |
2.32(11) |
23(1) |
| Precursor for 3 |
6 K |
–13.11(2) |
4.3(2) |
3.78(13) |
50(1) |
|
|
|
0.29(1) |
2.74(11) |
2.31(6) |
50(1) |
| Compound 1 |
78 K |
–13.33(1) |
3.68(7) |
3.85(3) |
88(1) |
|
|
|
0.31(2) |
3.2(2) |
2.30(10) |
12(1) |
| Compound 2 |
78 K |
0.18(1) |
2.62(7) |
2.3* |
100 |
| Compound 3 |
78 K |
0.31(2) |
2.09(13) |
2.3* |
100 |
Crystal structure analysis.
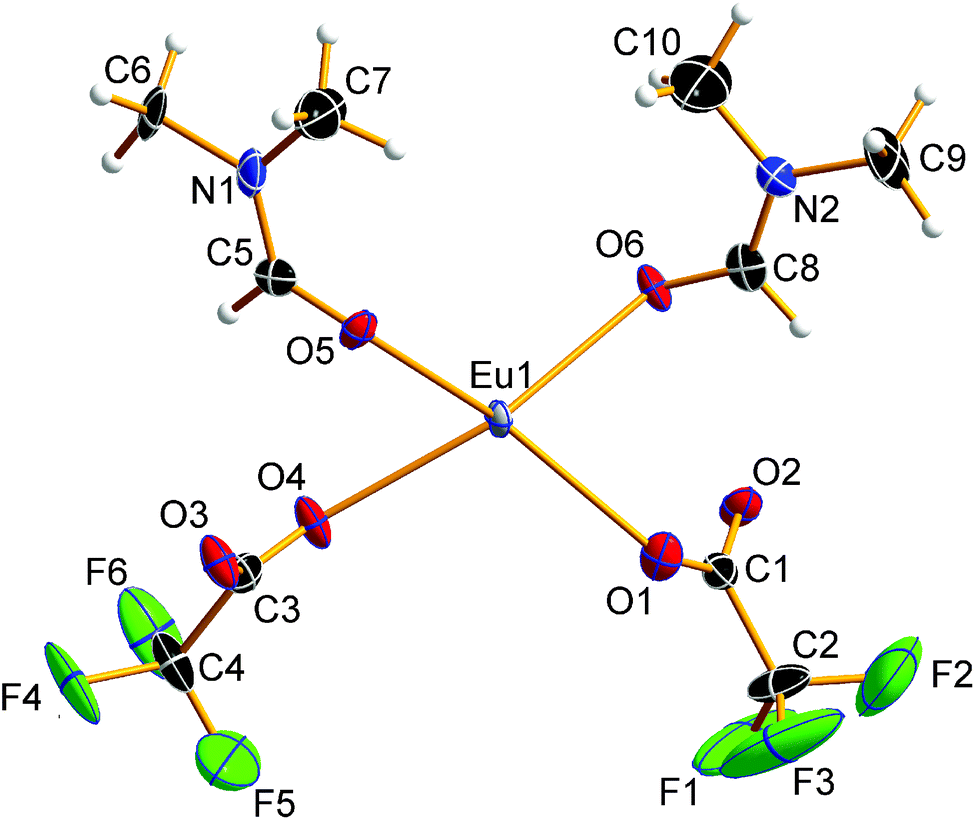
Compound 1 crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (Z = 2). The asymmetric unit of the crystal structure is shown in Fig. 2 and contains one Eu2+ ion, two trifluoroacetate ions, and two DMF molecules (all in general position). The central Eu2+ ion is coordinated by a total of eight oxygen atoms, four in the asymmetric unit and four symmetry-equivalent ones. The bond valences si and the valence sum S of the europium atoms were calculated from the Eu1–Oi bond lengths di according to the empirical bond valence method27 (see Table S1, ESI†). The valence sum of 1.92 shows good agreement with the expected value of 2.00. This ensures that the structure determination on a solvated EuII trifluoroacetate has been achieved.
(Z = 2). The asymmetric unit of the crystal structure is shown in Fig. 2 and contains one Eu2+ ion, two trifluoroacetate ions, and two DMF molecules (all in general position). The central Eu2+ ion is coordinated by a total of eight oxygen atoms, four in the asymmetric unit and four symmetry-equivalent ones. The bond valences si and the valence sum S of the europium atoms were calculated from the Eu1–Oi bond lengths di according to the empirical bond valence method27 (see Table S1, ESI†). The valence sum of 1.92 shows good agreement with the expected value of 2.00. This ensures that the structure determination on a solvated EuII trifluoroacetate has been achieved.
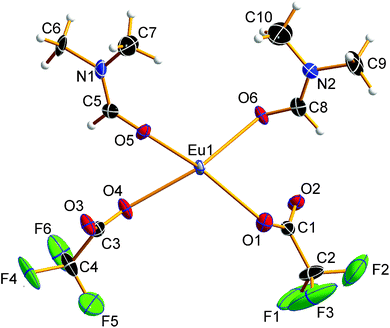 |
| | Fig. 2 Asymmetric unit of 1. Displacement ellipsoids are drawn at the 50% probability level, hydrogen atoms are drawn with an arbitrary radius. The trifluoromethyl group at carbon atom C3 is disordered. For the sake of clarity, only one of the partial occupation sites is shown. | |
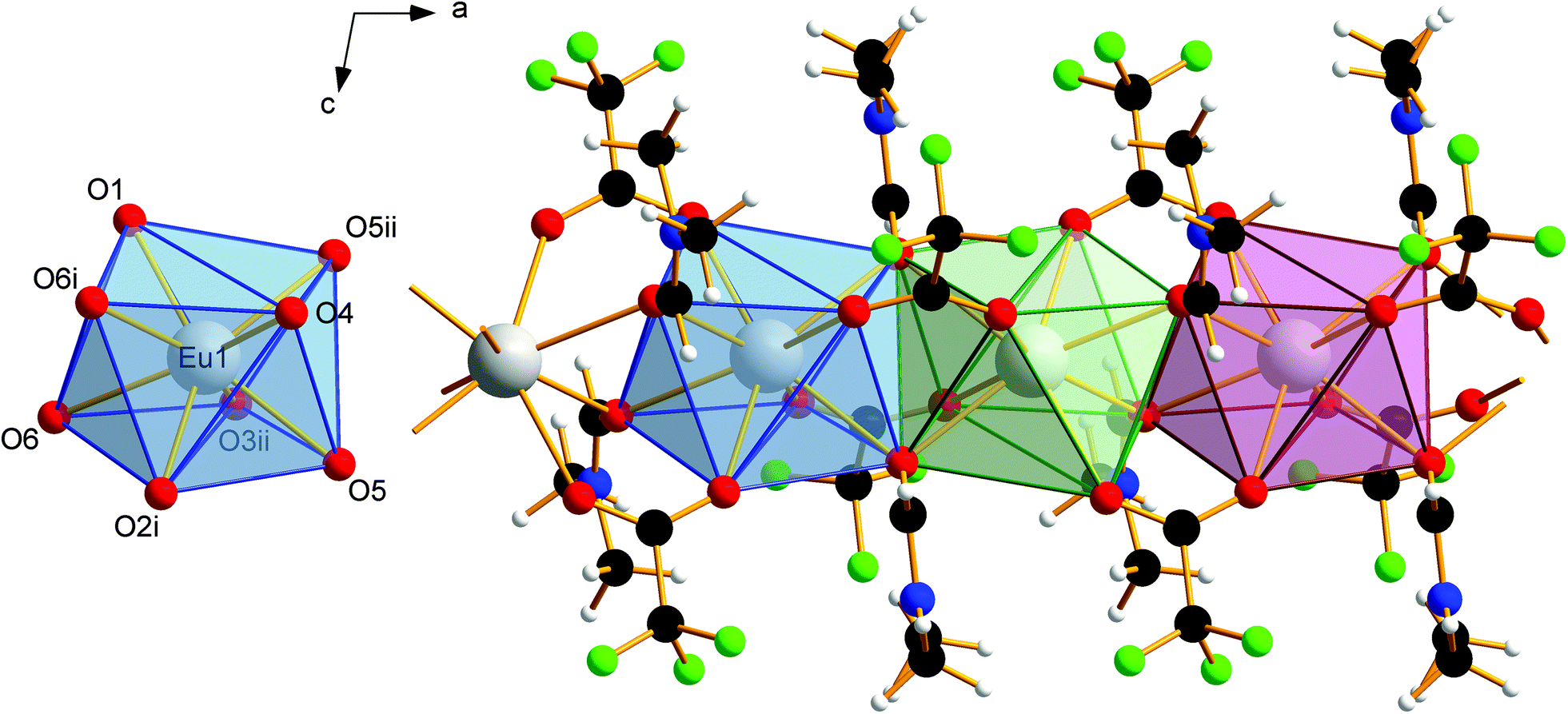
The EuII–O bond lengths in 1 are 2.542(6)–2.623(6) Å, which is within the expected range for europium(II) carboxylates, of 2.39–2.87 Å (mean 2.63 Å).9b–f The Eu2+ ions are connected to polymeric strands by two bridging trifluoroacetato ligands and two bridging molecules of DMF. The orientation of these bridging ligands is shown in Fig. 3. Note the alternating position of the ligands along the chain growth direction, to avoid the collision of adjacent trifluoroacetate groups. A coordination polymer with Eu⋯Eu distances of 4.0838(13)–4.1049(13) Å and inversion centers between two Eu2+ ions is formed in which the Eu2+ ions are in square-antiprismatic coordination. As shown in Fig. 3 the individual coordination polyhedra are edge-linked to each other. The square antiprismatic coordination polyhedron is consistent with the tendency of the lanthanoid ions to prefer high coordination numbers in solution and in the solid state.10 Lanthanoid ions prefer (capped) trigonal-prismatic coordination polyhedra with monodentate ligands, e.g., in [Ln(H2O)n]3+ ions, and square-antiprismatic coordination polyhedra with bridging ligands.10a The linkage pattern of the Eu2+ ions in 1 is closely related to that of [Eu2[C6H3(NH2)(SO3)2]2(dmf)4]∞ mentioned in a personal communication to CSD.28 The Eu⋯Eu distances are in the same range of 4.0964(5)–4.1251(5) Å. Since these distances heavily depend on the nature of the bridging ligands and their coordination mode, comparisons to other europium(II) carboxylates are less instructive (4.24–4.30 Å).9b–f Usually, Ln⋯Ln distances in exclusively perfluorocarboxylate bridged complexes are about 4.5 Å.13 The involvement of DMF or other monodentate bridging ligands seems to give shorter Eu⋯Eu distances. In contrast to the two catena-compounds mentioned above with linear arrangements of Eu2+, the Eu2+ ions in [Eu(OAc)2(HAc)2(H2O)2]∞ are arranged in a zig-zag pattern.9c The O–C–O′ bond angles of the trifluoroacetate ions in 1 are 130.8(10)–131.7(10)°, which is within the expected range for bidentate carboxylates.11g,13,18a
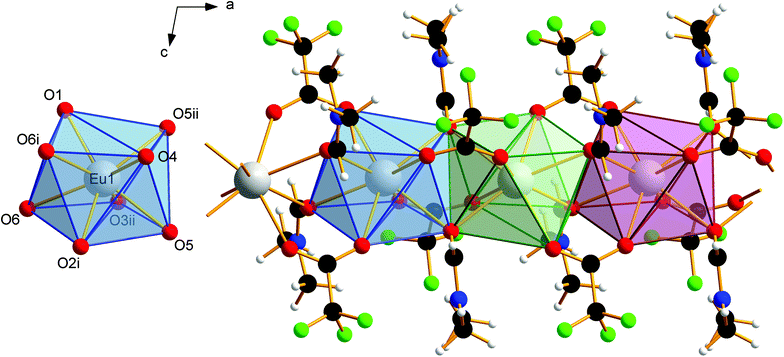 |
| | Fig. 3 Coordination polyhedron of Eu1 in 1 (left). Symmetry codes: (i) 1 − x, 1 − y, 1 − z; (ii) −x, 1 − y, 1 − z. Note the eightfold coordination of the Eu2+ ions by oxygen atoms in square antiprismatic coordination polyhedra. Section of the coordination polymer 1, with four repeating units (right). Direction of view against the crystallographic b-axis. The Eu2+ ions are linked by two bridging trifluoroacetato ligands and two bridging molecules of N,N-dimethylformamide. The individual coordination polyhedra are edge-linked to each other. Color code: C (black), H (white), N (blue), O (red), F (green), Eu (gray). | |
van der Waals interactions between methyl and trifluoromethyl groups of adjacent strands in 1 cause the solid-state association. The absence of stronger interactions, such as hydrogen bonds, perpendicular to the chain growth direction explains the crystal habit and the mechanical lability of these hair-fine needles.
Hexanuclear europium(III) fluoridocarboxylates by slow oxidation of europium(II) perfluorocarboxylates
Synthesis and characterization.
In aqueous solutions Eu2+ ions are thermodynamically unstable due to the negative standard potential of −0.38 V.29 However, in the absence of oxygen solutions containing Eu2+ ions are practically stable due to the kinetic barrier of the hydrolysis.30 We observed a similar stability of Eu2+ ions in solutions of europium(II) perfluorocarboxylates in anhydrous perfluorocarboxylic acids. In a closed tube, the resulting yellow solutions can be stored at least two years without any decolorization, that easily would allow to recognize an oxidation process. However, in PTFE ring sealed ampoules at 8 °C colorless crystals of 2 and 3 grew within 2–3 weeks. Coulometric Karl Fischer titrations of the mother liquors from the crystallizations of 2 and 3 gave water contents of 2% and 1%, respectively. The crystals of 2 and 3 were identified via single crystal structure analysis as complexes of the type (NH4)2[Eu6F8(O2CR)12(RCOOH)6], R = CF3 (2) and R = C2F5 (3), respectively. Therefore, it is likely that atmospheric oxygen and moisture penetrating the PTFE seals are responsible for a slow oxidation of the yellow solutions. However, it is known that, for example, Eu(OH)2·H2O is subject to a slow oxidative hydrolysis process in the absence of oxygen.31 In further experiments a much faster decolorization of the yellow solutions can be achieved even in the absence of oxygen by using equimolar mixtures of perfluorocarboxylic acid and water. In preliminary experiments crystals grown from such solutions are not 2 or 3 but hydrated substances.
Crystals of 2 are stable at room temperature in the absence of moisture. Crystals of 3 readily decompose in the absence of mother liquor releasing acid to give a white solid. After removing the released acid in vacuum, this solid, like 2, is stable at room temperature. Assuming the general formula given above, for both, 2 and the white solid derived from 3, TGA/DSC-thermal analysis indicated a stepwise endothermic release of about six equivalents of acid in the temperature range of room temperature to 185 °C (∼20%, calc. 21.78% for 6 eq. acid) and room temperature to 205 °C (∼22%, calc. 24.36% for 6 eq. acid), respectively (see Fig. S3–S6, ESI†). Without previous melting the step of final decomposition in the TGA curve is observed at about 20 °C higher temperature in the case of 3 (Tonset = ∼280 °C) as compared to 2 (Tonset = ∼255 °C). The remaining amorphous substances (∼36% and ∼34%) are assumed to be EuF3 mainly (calc. 39.92%, based on 2 and 31.03%, based on 3–8 C2F5COOH).
Although the isomer shifts in the 151Eu Mössbauer spectra of 2 and 3 both are compatible with trivalent europium, the isomer shifts are different (also with respect to their standard deviations), indicating a ligand-driven change in the electron density at the europium nuclei (see Table 1). Such isomer shift changes are usually a consequence of electronegativity changes, as exemplarily listed for the series: (i) EuF3 (−0.59) → EuCl3 (−0.42) → EuBr3 (−0.06); (ii) EuOF (−0.72) → EuOCl (−0.43) → EuOBr (−0.29) → EuOI (+0.02) or (iii) EuPO4 (0.16) → EuAsO4 (0.49) → EuSbO4 (0.86) (all values in mm s−1).32 Thus, within these series the highest electron density occurs at the europium nuclei of EuBr3, EuOI and EuSbO4. This trend also holds for 2 and 3 described herein. 3, with an isomer shift of 0.31(2) mm s−1 and the less electron withdrawing moiety –CF2–CF3 shows higher electron density at the europium nuclei as compared to 0.18(1) mm s−1 for 2 with –CF2–F. Thus, the 151Eu spectra nicely reflect the small electronic differences induced by the substituent in the ligand.
On the question: fluoride or hydroxide?.
There are reasons to question the suggested composition of the complexes, since fluoride and hydroxide are hardly different from each other in XRD experiments due to similar electron numbers of F and O. μ3-Capping hydroxido ligands are well known in complexes of this type that are typical hydrolysis products.33 Therefore, careful study is necessary to exclude this ion for occupation of the octahedral faces. Striking evidence for μ3-capping fluoride ligands: (i) the physical meaningful refinement of the crystal structure. The alternative structure refinement with atomic form factors for O instead of F resulted in physically absurd anisotropic displacement parameters; (ii) the calculation of the empirical valence sums gives a significantly better agreement assuming Eu–F bonds (mean S = 3.14 in 2 and 3.22 in 3) than for the approach as Eu–O bonds (mean S = 3.60 in 2 and 3.68 in 3) and confirms that these are complexes of the type [Eu6F8] and not [Eu6(OH)8]; (iii) by hydrolysis the fluoride ions bound to europium were released and could be detected via19NMR spectroscopy. That these fluoride ions do not originate from a hydrolysis of the perfluorocarboxylate ions is ensured, since Eu(O2CCF3)3 did not show this behavior (see Fig. S17, ESI†); (iv) there is a very recent example of a complex of the type [Tb6F8] in literature, with pivalic acid as a carboxylate ligand.21
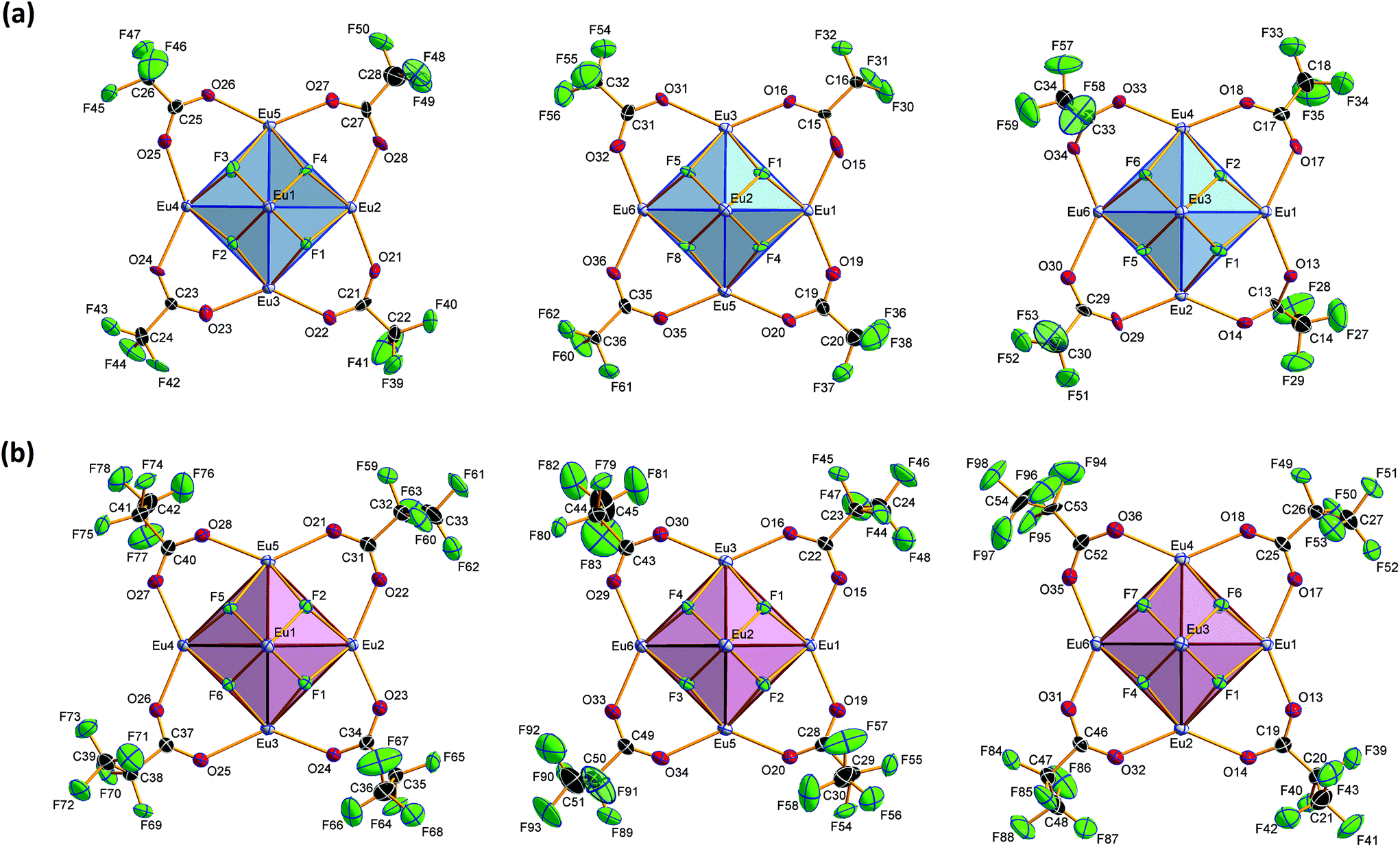
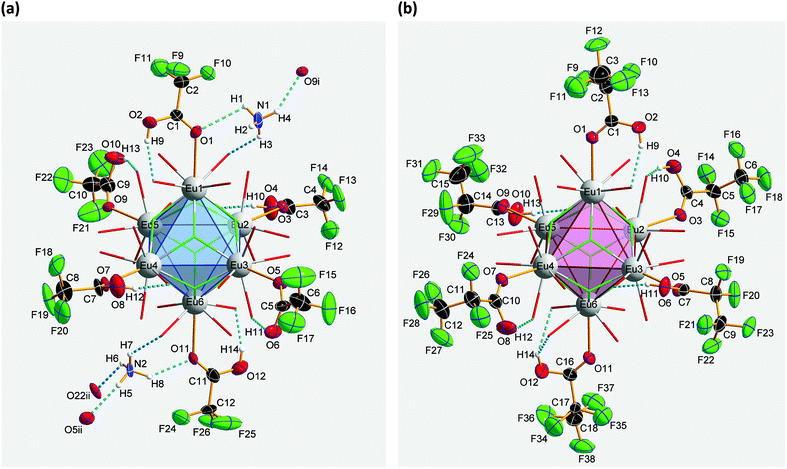
Crystal structure of bis(ammonium)[dodecakis(μ2-trifluoro-acetato)-hexakis(trifluoroacetic acid)octa-μ3-fluorido-octahedro-hexaeuropiate(III)], (NH4)2[Eu6F8(O2CCF3)12(CF3COOH)6] (2)
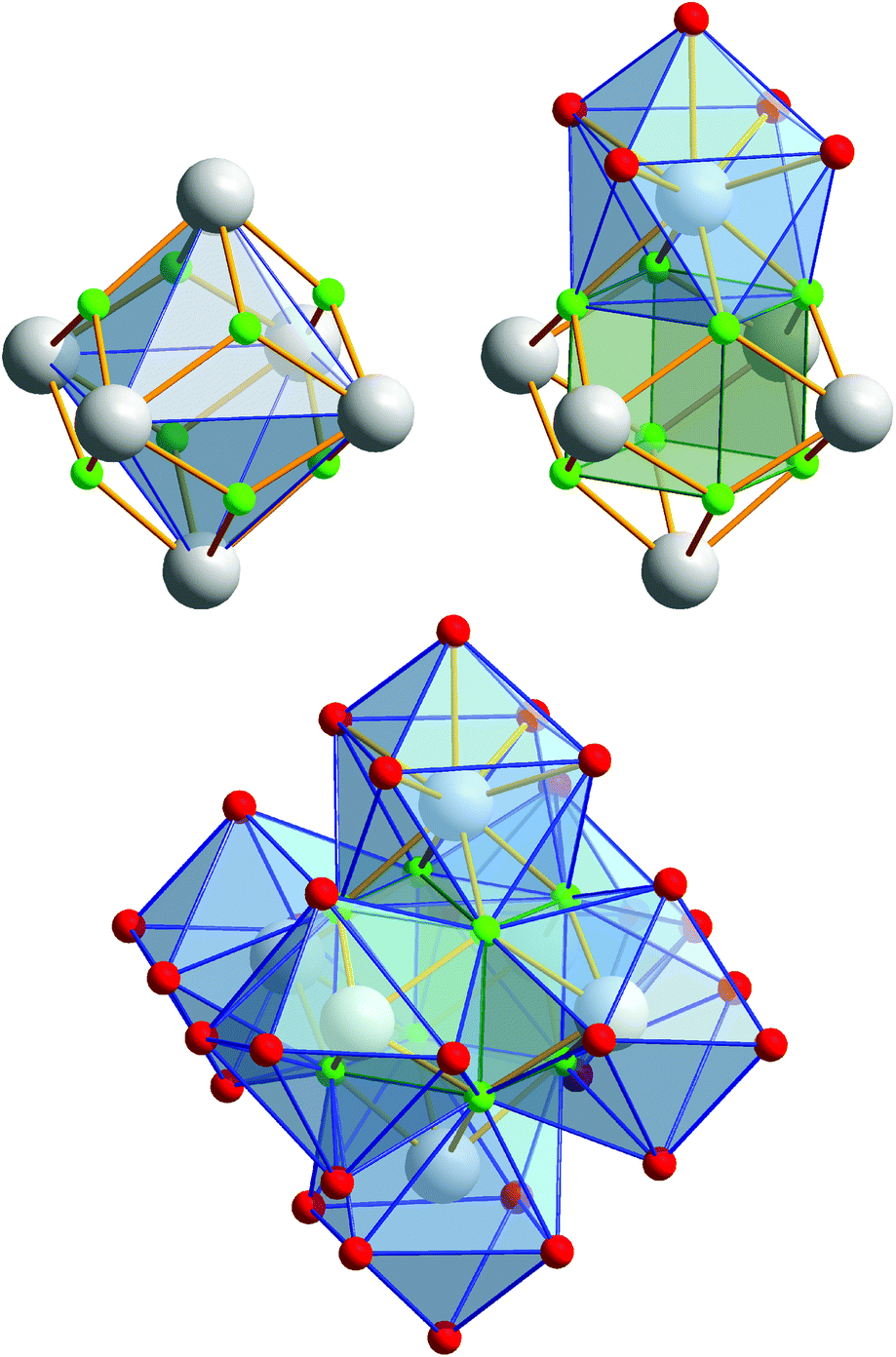
Compound 2 crystallizes in the monoclinic space group P21/n (Z = 4). The colorless crystals of 2 are notoriously twinned. For the course of X-ray crystal structure determination, twinning had to be considered. Details are given in the Experimental section. The asymmetric unit‡ of the crystal structure contains six Eu3+ ions, two NH4+ ions (including N1, N2), eight fluoride ions (F1–F8), twelve trifluoroacetate ions and six molecules of trifluoroacetic acid (all in general position). The general structural feature is an octahedral framework of six Eu3+ ions. The eight octahedral faces are capped by fluoride ions to form a [Eu6F8] unit. Each of the twelve edges and six vertices of the [Eu6] octahedron, is coordinated by oxygen atoms of peripheral ligands as shown in Fig. 4. The Eu–F and Eu–O bond lengths along with the corresponding empirical bond valences si are given in Table S2 (ESI†). The valence sums of the europium atoms have been discussed above and indicate the presence of eight μ3-fluorido ligands that form a [F8] cube interpenetrating the [Eu6] octahedron in the central [Eu6F8] unit. Each of the cube's six faces is the basis for one of the six square antiprismatic coordination polyhedra of Eu1–Eu6. The top face of each of these coordination polyhedra is capped by another O atom, giving ninefold coordinated Eu3+ ions. The Eu–F bond lengths and Eu⋯Eu distances are 2.328(13)–2.426(13) Å (mean 2.39 Å) and 3.9827(15)–4.0093(15) Å (mean 4.00 Å), respectively. The twelve octahedral edges of the central [Eu6F8] unit are coordinated by trifluoroacetato ligands in a bridging coordination mode (see Fig. 5a). Their O–C–O′ angles seem to be inhomogeneously ranging from 123(3)–132(2)° (mean 127°) but are nevertheless typical for the bidentate-bridging (μ2-O:O′) coordination mode. 130° is usually found in lanthanoid(III) perfluorocarboxylates.12,13 Smaller O–C–O′ angles of about 120° would be typical for the tridentate-chelating and -bridging (μ2-O:O′,O) mode often found in lanthanoid(III) acetates.11 The six octahedral vertices of the central [Eu6F8] unit are coordinated by trifluoroacetic acid molecules, which form classical intramolecular O–H⋯O hydrogen bonds with adjacent μ2-O2CCF3 ligands (see Fig. 6a). The hydrogen bond motifs are self-contained and can be assigned to the graph set descriptor S11(6).34 The geometrical parameters of these hydrogen bonds are given in Table S3 (ESI†). The very sharp band for the O–H stretching mode in the IR spectrum of 2 at 3661 cm−1 strongly suggests that these hydrogen bonds are rather weak than strong, what would result in band broadening and a shift to smaller frequencies.35 The Eu–O distances involving the O atoms of the acid molecules at the octahedral vertices are slightly longer than those of the trifluoroacetato ligands at the octahedral edges, in the ranges of 2.528(17)–2.766(19) Å (mean 2.67 Å) and 2.337(18)–2.502(17) Å (mean 2.40 Å), respectively. Six Eu3+ ions, eight fluoride ions, and twelve trifluoroacetate ions result in a double negatively charged complex ion. Charge compensation in the structure is ensured by two symmetry independent ammonium ions connecting the complex ions along the crystallographic a-axis.
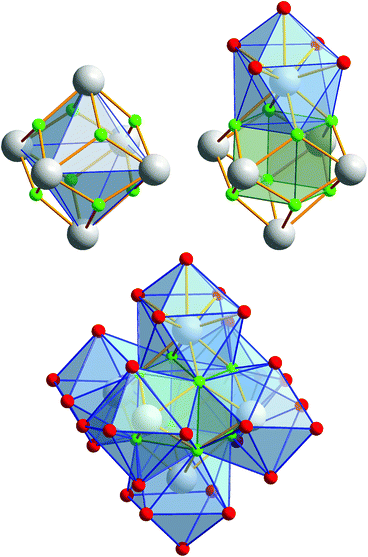 |
| | Fig. 4 Different polyhedra of the central unit of 2. The Eu3+ ions span an octahedron whose eight octahedral faces are capped by fluoride ions. These fluoride ions form a [F8] cube interpenetrating the [Eu6] octahedron. Each of the six cube faces forms the bottom face for a single-capped square antiprismatic coordination polyhedron in which the Eu3+ ions are ninefold coordinated; note the almost identical construction of the central unit of 3, discussed later. Color code: O (red), F (green), Eu (gray). | |
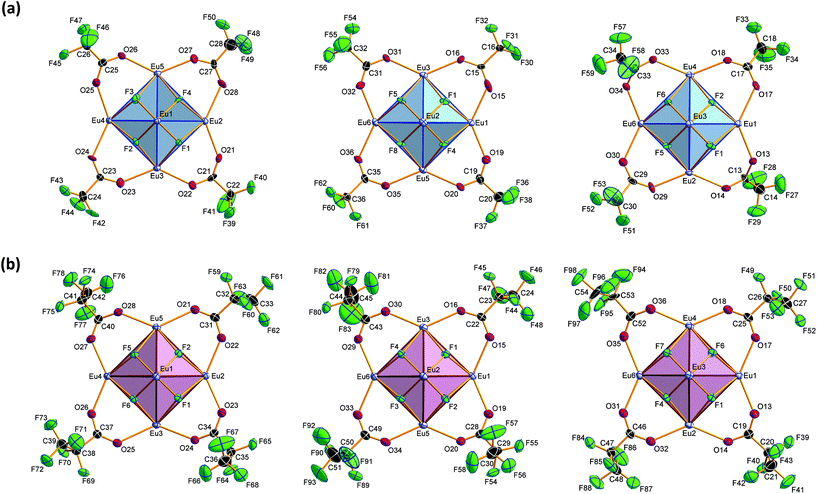 |
| | Fig. 5 Coordination of the three ‘equatorial planes’ in the central [Eu6F8] unit in 2 (part a) and 3 (part b). Direction of view along Eu1⋯Eu6 (left), Eu2⋯Eu4 (middle) and Eu3⋯Eu5 (right). Displacement ellipsoids are drawn at the 50% probability level. The CF3 (C2F5) groups at the C atoms C19, C23, C27 and C31 (C31 and C52) are disordered. For the sake of clarity, only one of the partial occupation sites is shown. The trifluoroacetato (pentafluoropropionato) ligands at the edges of the ‘meridional planes’ and the trifluoroacetic (pentafluoropropionic) acid molecules at the octahedral vertices are omitted. | |
 |
| | Fig. 6 Coordination of the octahedral vertices of the central [Eu6F8] units in 2 (part a) and 3 (part b). Displacement ellipsoids are drawn at the 50% probability level for the NH4+ ions in 2, trifluoroacetic (pentafluoropropionic) acid, and the acceptors of hydrogen bonds (segmented sticks) as well. For the sake of clarity, the remaining parts of the structure are shown partially, in a combined wire and ball-and-stick (for Eu1–Eu6) model representation. | |
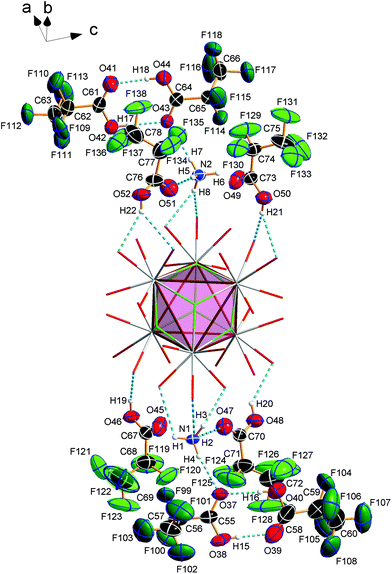
Crystal structure of bis(ammonium)[dodecakis(μ2-pentafluoro-propionato)hexakis(pentafluoropropionic acid)octa-μ3-fluorido-octahedro-hexaeuropiate(III)]—pentafluoropropionic acid (1/8), (NH4)2[Eu6F8(O2CC2F5)12(C2F5COOH)6]·8C2F5COOH (3)
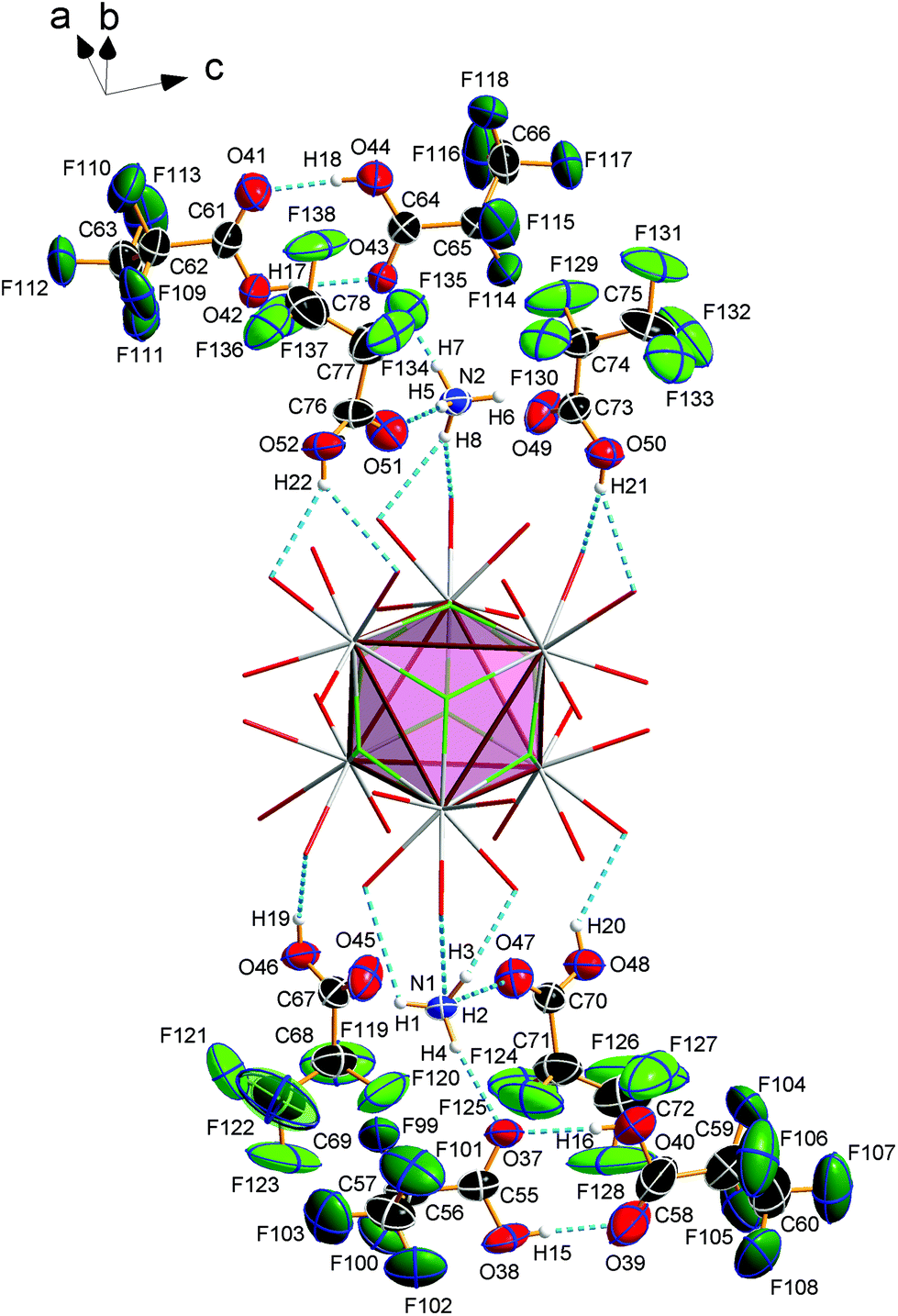
Compound 3 crystallizes in the monoclinic space group P21/n (Z = 4) and shows the same structural feature as 2 (see Fig. 4), particularly the same coordination pattern of the octahedral edges and vertices of the central [Eu6F8] unit as described for 2, with C2F5 instead of CF3 as perfluoroalkyl groups (see Fig. 5b and 6b). The asymmetric unit‡ of the crystal structure contains six Eu3+ ions, two NH4+ ions (including N1, N2), eight fluoride ions (F1–F8), twelve pentafluoropropionate ions and fourteen molecules of pentafluoropropionic acid (all in general positions). The Eu⋯Eu distances of 3.9537(4)–3.9978(5) Å (mean 3.98 Å) are in the same narrow range as those in 2. The Eu–F bond lengths of 2.343(3)–2.417(3) Å (mean 2.37 Å) are of the same magnitude, too. The Eu–O bond lengths to the pentafluoropropionato ligands and to the vertex substituting acid molecules are in the ranges of 2.358(4)–2.494(4) Å (mean 2.40 Å) and 2.644(4)–2.701(4) Å (mean 2.66 Å), respectively. The Eu–F and Eu–O bond lengths are given in Table S4 (ESI†). The O–C–O′ angles are within the range of 126.6(8)–129.6(5)°. The fourteen molecules of pentafluoropropionic acid in the asymmetric unit of 3 can be divided into six internal ones that coordinate the octahedral vertices of the central [Eu6F8] unit and eight external molecules located in layer-like regions, that extend parallel to the a,c-plane at the heights 0 and ½ of the unit cell. All of them, internal and external, are engaged in O–H⋯O hydrogen bonds. Some of these are bifurcated, the great majority are simple ones. The geometrical parameters of all these and the hydrogen bonds of the two ammonium ions are given in Table S5 (ESI†). The internal acid molecules at Eu1 to Eu5 form self-contained hydrogen bond patterns with adjacent μ2-O2CC2F5 ligands in a manner already known from 2 (see Fig. 6b) [graph set descriptor S11(6)]. The internal acid molecule at Eu6 forms a bifurcated hydrogen bond from O12 to O33 and O35 [R12(4)]. The external acid molecules can be subdivided into four acid molecules that form single (O46, O48) or bifurcated (O50, O52) hydrogen bonds with peripheral ligands of the central [Eu6F8] unit [D11(2) or R12(4)], and four acid molecules that are associated to carboxylic acid dimers [R22(8)] (see Fig. 7). The presence of these dimers as a particular structural feature is assumed to be related to the enlarged gaps between adjacent clusters, due to the enhanced perfluoroalkyl chain lengths of the peripheral ligands. The position of the hydrogen atoms can be clearly identified by aids of the C–O bond lengths, which are about 0.1 Å shorter for the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds (mean 1.19 Å) than for the C–O bonds (mean 1.29 Å). This is an expected feature of carboxylic acid dimers in the solid state except for aromatic carboxylic acids.36 The O⋯O distances of the dimers in 3 are in the range of 2.577(8)–2.752(9) Å (mean 2.66 Å), which indicates medium-strength hydrogen bonds that are rather electrostatic than related to orbital interactions.37
O bonds (mean 1.19 Å) than for the C–O bonds (mean 1.29 Å). This is an expected feature of carboxylic acid dimers in the solid state except for aromatic carboxylic acids.36 The O⋯O distances of the dimers in 3 are in the range of 2.577(8)–2.752(9) Å (mean 2.66 Å), which indicates medium-strength hydrogen bonds that are rather electrostatic than related to orbital interactions.37
 |
| | Fig. 7 Pentafluoropropionic acid molecules and dimers, located in layer-like regions, that extend parallel to the a,c-plane at the heights 0 and ½ of the unit cell, NH4+ ions and their hydrogen bond pattern (segmented sticks) in 3. Displacement ellipsoids are drawn at the 50% probability level for the NH4+ ions and pentafluoropropionic acid molecules. For the sake of clarity, the remaining parts of the structure are shown partially, in a wire model representation and F atoms related to acid dimers are given in dark green. | |
The hydrogen bonds of the other external acid molecules are much weaker, with O⋯O distances of 2.902(6)–3.081(7) Å (mean 2.99 Å). The O⋯O distances of the hydrogen bonds of the internal acid molecules are in the range of 2.669(6)–2.904(7) Å (mean 2.76 Å) and tend to be medium strong to weak. Among the small number of acid adducts of lanthanoid carboxylates,9c,11f,12e,f substances without bonds between the carboxylic acid molecules to lanthanoid cations are rare and known only from unpublished results.38 The presence of carboxylic acid dimers has not yet been reported in the context of lanthanoid carboxylates. The NH4+ ion including N1 (N2) forms three (two) single and one bifurcated hydrogen bonds with external acid molecules and peripheral ligands of the central [Eu6F8] unit [D11(2) or R12(4)] (see Fig. 7). Since these acid molecules donate further hydrogen bonds to peripheral ligands at the central [Eu6F8] unit, the ammonium ions are part of an extended hydrogen bond network with maximum ring sizes according to graph set descriptors R43(14) for N1 and R33(12) for N2.
Photoluminescence properties of 1, 2 and 3
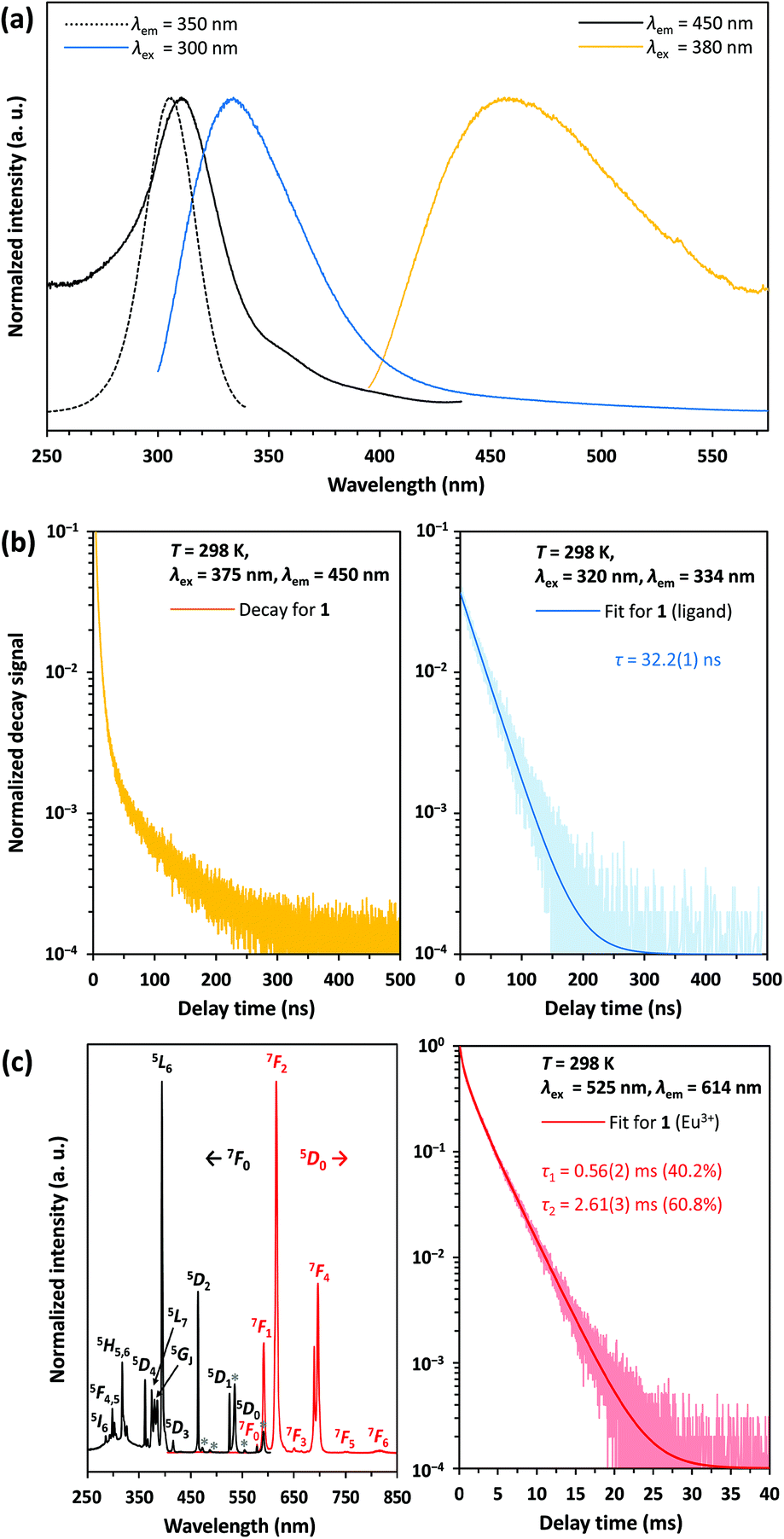
According to 151Eu Mössbauer spectroscopy, the yellow crystals of 1 isolated from DMF contain a small amount of oxidation product (see Table 1). This is confirmed by luminescence spectroscopic characterization. The barely visible luminescence of 1 under an UV lamp indicates that EuIII is present only as an impurity. The luminescence spectra confirm the findings from the Mössbauer spectroscopic measurements and reveal the typical 4f6 ↔ 4f6 transitions of EuIII (see Fig. 8c, left).39 The decay curve of 1 was fitted by a multiexponential function (see Fig. 8c, right) and luminescence decay times of 0.56(2) ms (40.2%) and 2.61(3) ms (60.8%) were determined (see Table 2). These are typical for EuIII compounds with organic ligands and slightly shorter than those of inorganic EuIII doped fluorides, which have reported decay times of the 5D0 level as high as 10 ms.40 At an excitation wavelength of 300 nm, the broadband fluorescence of the organic ligands is prominent, showing an emission maximum at 334 nm (see Fig. 8a). This assignment is confirmed by the luminescence decay in the nanosecond range indicating fluorescence of the organic ligands (see Fig. 8b, right). Upon excitation at 380 nm a weak emission band with a maximum at 456 nm is detected, which could be assigned to the 4f65d1 → 4f7 transition of EuII. However, the shortened luminescence decay time of less than 0.1 μs (see Fig. 8b, left) is untypical for this transition, which is consistently reported in the order of 1 μs.41 Together with the low intensity of the emission band this suggests an effective quenching mechanism for the luminescence of EuII at room temperature. Possible explanations are: (i) a small band gap of the coordination polymer, that induces thermal ionization of the excited 5d electrons of EuII into the conduction band.42 This may be supported by the bulk yellow color of 1 that indicates strong absorption of blue light; (ii) the presence of EuII next to EuIII allows energy transfer due to intervalence charge transfer (IVCT) processes;43 (iii) quenching of the luminescence due to non-radiative relaxation of the excited states by the vibration of DMF ligands. Future detailed studies of the photoluminescence at temperatures below 100 K could help provide additional insights into the quenching mechanism of the preliminarily assigned EuII-related blue luminescence at 458 nm.
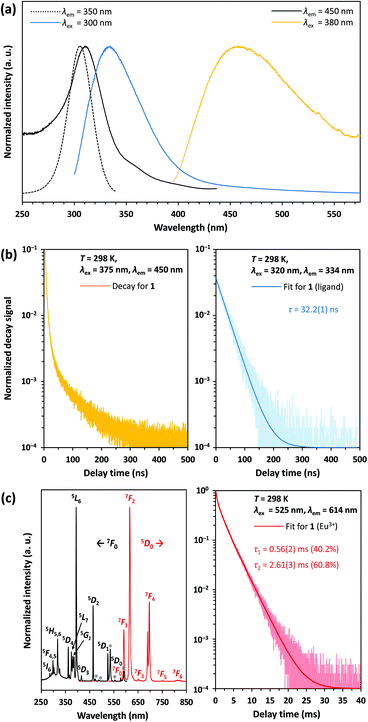 |
| | Fig. 8 (a) Absorption and emission bands of the coordination polymer 1; (b) luminescence decay of 1 at λem = 450 nm (left) and 334 nm (right); (c) excitation and emission spectra of EuIII impurities in 1 and luminescence decay of the level 5D0 at λem = 614 nm and 298 K; excitation lines marked with an asterisk indicate transitions from the thermally populated 7FJ (J = 1, 2) levels to the correspondingly labelled levels in the excitation spectrum. | |
Table 2 Decay times of the EuIII luminescence of 1, 2 and 3, and reported lifetimes of polynuclear EuIII hydroxido complexes
| Compound |
Temperature |
Species |
τ [ms] |
Percentage |
Ref. |
|
τ = luminescence decay time, L1 = (modified) 2-hydroxybenzophenone ligands; L2 = 9-hydroxyphenalen-1-one ligands. |
| Compound 1 |
298 K |
EuIII impurity |
0.56(2) |
0.402 |
Here |
|
|
|
|
2.61(3) |
0.608 |
|
| Compound 2 |
298 K |
EuIII |
0.39(1) |
0.195 |
Here |
|
|
|
|
1.15(1) |
0.805 |
|
| Compound 3 |
298 K |
EuIII |
1.48(1) |
1.000 |
Here |
| [Eu9(L1)16(μ3-OH)8(μ4-OH)(μ4-O)] |
298 K |
EuIII |
0.30–0.51 |
|
44a
|
| [Eu9(L2)16(μ3-OH)9(μ4-OH)] |
78 K |
EuIII |
0.31(2) |
|
44b
|
Compounds 2 and 3 show a very bright red luminescence under UV light (to get a better visual impression see Fig. 9d), typical for the 4f6 ↔ 4f6 transitions of EuIII.39 The photoluminescence spectra of 2 (see Fig. 9a) and 3 (see Fig. 9b) are consistent with this observation and show narrow lines in excitation and emission.
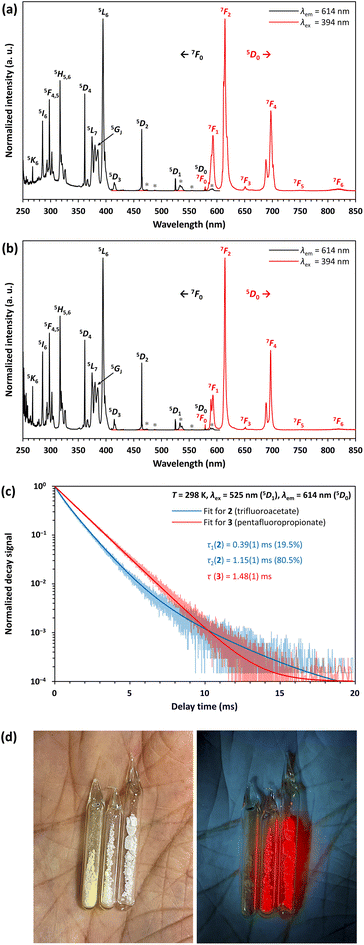 |
| | Fig. 9 Excitation (black) and emission (red) spectra of the compounds 2 (a) and 3 (b); excitation lines marked with an asterisk indicate transitions from the thermally populated 7FJ (J = 1, 2) levels to the correspondingly labelled levels in the excitation spectrum; (c) luminescence decay of the level 5D0 and fits for 2 and 3 at λem = 614 nm and 298 K; (d) compounds 1, 2 and 3 in daylight (left) and under an UV lamp with an emission maximum of 265 nm (right). The samples were placed in closed tubes after drying in vacuum. | |
As expected, the emission originates from the 5D0 level, since despite the low vibrational energies of the fluorinated organic ligands, emission from the 5D1 or even 5D2 level is unlikely.39 The most intense line in the excitation spectrum is related to the 7F0 → 5L6 transition at 394 nm, while the emission spectrum is dominated by the 5D0 → 7F2 transition at 614 nm. Especially the much higher intensity of the strongly electric dipolar 5D0 → 7F2 transition compared to the magnetic dipolar 5D0 → 7F1 transition at 591 nm indicates that the Eu3+ ions are located in non-centrosymmetric positions.39 This is in agreement with the structural models of 2 and 3. There is also no evidence for ligand-to-metal charge transfer (LMCT) transitions in the excitation spectrum. The decay curves of 2 and 3 were fitted by multiexponential functions (see Fig. 9c), giving luminescence decay times of 0.39(1) ms (19.5%) and 1.15(1) ms (80.5%) for 2 and 1.48(1) ms for 3 (see Table 3). These are typical for EuIII compounds with organic ligands and slightly shorter than those of inorganic EuIII doped fluorides, which have reported decay times of the 5D0 level as high as 10 ms.40 Interestingly, however, the lifetimes are longer than those of complexes with similar constructed [Eu9(OH)n] cores, ranging from 0.31(2) ms ([Eu9(OH)10] core) to 0.30–0.51 ms ([Eu9(OH)9(O)] core).44 Ling et al. observed that fluoride ions hardly contribute to non-radiative relaxation of the excited states of TbIII in a complex with a [Tb6F8] core due to their low vibrational energy and expanding the luminescence decay time compared to polynuclear terbium(III) hydroxido complexes.21 This is consistently confirmed for complexes with a [Eu6F8] core within this work.
Table 3 Crystal data and structure refinement parameters for 1, 2 and 3
| Parameters |
1
|
2
|
3
|
|
R
1 = ∑||Fo| − |Fc||–∑|Fo|; wR2 = [∑w(Fo2 − Fc2)2–∑w(Fo2)2]−1; S = [∑w(Fo2 − Fc2)2–(m − n)]−1; w = 1–σ2(Fo2) + (0.0215P)2 + 3.8152P; P = (Fo2 + 2Fc2)–3. |
| CCDC depository |
2125846
|
2125847
|
2125848
|
| Crystal data |
|
|
|
| Color/shape |
Yellow/needle |
Colorless/prism |
Colorless/plate |
| Chemical formula |
C10H14N2O6F6Eu6 |
C36H14N2O36F62Eu6 |
C78H22N2O52F138Eu6 |
|
M
r (g mol−1) |
524.20 |
3140.31 |
5352.80 |
| Temperature (K) |
120(2) |
223(2) |
120(2) |
| Wavelength (Å) |
0.71073 Mo Kα |
0.71073 Mo Kα |
0.71073 Mo Kα |
| Crystal system, space group |
Triclinic, P |
Monoclinic, P21/n |
Monoclinic, P21/n |
|
a, b, c (Å) |
8.1855(7), 9.5209(8), 11.2944(10) |
12.0562(4), 25.5991(8), 27.1945(9) |
17.6214(13), 33.280(2), 26.1987(19) |
|
α, β, γ (°) |
98.367(5), 99.729(5), 101.090(6) |
90, 95.521(3), 90 |
90, 91.721(4), 90 |
| Volume (Å3) |
836.81(17) |
8354.1(5) |
15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 357.2(18) 357.2(18) |
|
Z
|
2 |
4 |
4 |
|
ρ
calc (g cm−3) |
2.08 |
2.50 |
2.32 |
|
μ (mm−1) |
3.8 |
4.7 |
2.7 |
| Crystal size (mm3) |
0.084 × 0.069 × 0.022 |
0.163 × 0.113 × 0.110 |
0.386 × 0.326 × 0.120 |
| Data collection |
|
|
|
| Diffractometer |
Bruker APEX II CCD |
STOE IPDS 2T |
Bruker APEX II CCD |
| Absorption correction |
Multi-scan (SADABS) |
Gaussian (SHELXTL) |
Multi-scan (SADABS) |
|
T
min, Tmax |
0.631, 0.928 |
0.633, 0.742 |
0.309, 0.430 |
|
F
000
|
506 |
5872 |
10160 |
|
θ range for data collection (°) |
1.861 ≤ θ ≤ 25.112 |
2.157 ≤ θ ≤ 24.999 |
1.504 ≤ θ ≤ 25.000 |
| Completeness |
0.992 |
0.999 |
0.993 |
| No. of collected reflections |
10288 |
50839 |
229309 |
| No. of independent reflections |
2968 |
14759 |
26839 |
| No. of observed reflections |
1958 |
12450 |
21715 |
|
R
int
|
0.0939 |
0.1348 |
0.0553 |
| Refinement |
|
|
|
|
R values [F2 > 2σ(F2)] |
R
1 = 0.0453, wR2 = 0.0902 |
R
1 = 0.0892, wR2 = 0.2293 |
R
1 = 0.0394, wR2 = 0.0777 |
|
R values [all data] |
R
1 = 0.0868, wR2 = 0.1032 |
R
1 = 0.1114, wR2 = 0.2621 |
R
1 = 0.0555, wR2 = 0.0874 |
|
S (Goodness-of-fit) |
1.038 |
1.038 |
1.065 |
| No. of data (m) |
2968 |
14759 |
26839 |
| No. of parameters (n) |
261 |
1420 |
2567 |
| No. of restraints |
50 |
549 |
110 |
| Δρmax, Δρmin |
1.43, −1.45 |
3.98, −2.01 |
1.89, −1.00 |
Experimental
All experiments were performed on a vacuum line equipped with J. Young high-vacuum PTFE valves under an inert argon atmosphere, unless otherwise stated. Liquid ammonia for the syntheses was freshly condensed and dried over sodium before use. Perfluorocarboxylic acids were distilled with 10 vol% of the corresponding anhydride over a 20 cm Vigreux column and kept under argon. The hygroscopic salts ammonium trifluoroacetate and pentafluoropropionate were handled in an argon filled glovebox. All other chemicals were obtained from commercial sources and used as purchased.
Synthesis of catena-poly[europium(II)bis(μ2-N,N-dimethylformamide)bis(μ2-trifluoroacetato)], [Eu(O2CCF3)2(dmf)2]∞ (1)
0.684 g (4.50 mmol) of europium was weighed in a reaction vessel containing a glass-coated stir bar and dissolved in 50 ml of dried liquid ammonia. 1.179 g (9.90 mmol) of ammonium trifluoroacetate was added to the dark blue solution. The resulting yellow suspension was stirred for 1 h at approximately −30 °C. The ammonia was evaporated, and the residue dried in vacuum for 8h until a pressure of 10−3 hPa was reached. The residue was ground to a fine yellow powder (1.30 g). 151Eu Mössbauer spectrum, isomer shift [mm s−1]: EuIII: 0.35 (23%), EuII: −13.11 (77%). 400 mg of this crude product were stirred in an ampoule with 4 ml of DMF and the insoluble part was allowed to settle overnight. The supernatant yellow solution was transferred into another ampoule and DMF was distilled off in vacuo until the solution was concentrated to half of its initial volume. The ampoule, sealed with a PTFE ring seal, was stored at 8 °C. Within 1–2 weeks hair-fine yellow crystal needles of 1 grew, which rapidly redissolve at room temperature. The crystalline material was dried in vacuum to a pressure of 5 × 10−3 hPa. Yield: 0.230 g, 43% based on Eu. Elemental analysis [%]: Found: C, 20.8; H, 2.3; N, 5.0; Eu, 31.6. Calc. for C10H14N2O6F6Eu: C, 22.91; H, 2.69; N, 5.34; Eu, 28.99. IR spectrum [cm−1]: νas(COO): 1638 (s); νs(COO): 1482 (m); ν(C–F): 1200, 1160 (s); ν(C–C): 854 (w); δ(CF3): 800 (m); δ(O–C–O): 723 (m); δ(CF3): 610, 520, 450 (w). Bands caused by DMF: ν(C–H): 2942 (w); ν(CO): 1713 (m); ν(C–N) 1500 (w); δ(N–CH3): 1438, 1427 (w); δ(CH3): 1379 (m); 1351 (vw); ν(N–CH3): 1256 (vw); γ(N–CH3): 1110 (m); δ(CH3): 1061 (w); δ(OC–N): 678 (w). Raman spectrum [cm−1]: 2947 (s); νas(COO): 1660 (w); νs(COO): 1462 (s); δ(CF3): 1205 (w); 933 (w); ν(C–C): 850 (s), δ(O–C–O): 733 (m); δ(CF3): 600 (m), 520 (w), 417 (s); 270 (m); 85 (s). 19F NMR spectrum (282.38 MHz, D2O/NaOH) [ppm]: −75.2 (s, CF3). 151Eu Mössbauer spectrum, isomer shift [mm s−1]: EuIII: 0.31 (12%), EuII: −13.33 (88%).
Synthesis of bis (ammonium)[dodecakis(μ2-trifluoroacetato)-hexakis(trifluoroacetic acid)octa-μ3-fluorido-octahedro-hexaeuropiate(III)], (NH4)2[Eu6F8(O2CCF3)12(CF3COOH)6] (2)
Preparation and crystallization of 2 was as described for 1 using 150 mg of the crude product obtained from liquid ammonia and 3 ml of anhydrous trifluoroacetic acid instead of DMF. The mother liquor was removed, and the crystalline material dried in vacuo until a pressure of 10−2 hPa was reached. Yield: 81 mg, 31% based on Eu. A coulometric Karl Fischer titration of the mother liquor gave a water content of 1.9%. Elemental analysis [%]: Found: C, 13.9; H, 0.6; N, 1.1; Eu, 29.4. Calc. for C36H14N2O36F62Eu6: C, 13.76; H, 0.45; N, 0.89; Eu, 29.04. IR spectrum [cm−1]: ν(O–H): 3661 (m), 3500 (w); ν(N–H, NH4+): 3190, 3072, 2963 (w); ν(CO): 1755, 1725 (m); νas(COO): 1670 (s); 1600 (s); νs(COO): 1477 (m); δ(O–H): 1424 (w, sh); ν(C–F): 1199, 1155 (s); γ(O–H): 877 (w); ν(C–C): 844 (w); δ(CF3): 800 (m); 737 (w); δ(O–C–O): 719 (m); 701 (w); δ(CF3): 612, 521, 443 (w). Raman spectrum [cm−1]: ν(CO): 1716 (m); νas(COO): 1660 (m); 1600 (w); νs(COO): 1473 (s); ν(C–F): 1205 (m); ν(C–C): 850 (s); 835 (s); 775 (m); 741 (s); δ(CF3): 613 (m), 548 (w); 440 (m); 363 (w); 285 (m); 93 (s). 19F NMR spectrum (282.38 MHz, D2O/NaOH) [ppm]: −75.5 (s, CF3); −122.5 (s, F−). 151Eu Mössbauer spectrum, isomer shift [mm s−1]: EuIII: 0.18.
Synthesis of bis(ammonium)[dodecakis(μ2-pentafluoro-propionato)hexakis(pentafluoropropionic acid)octa-μ3-fluorido-octahedro-hexaeuropiate(III)]—pentafluoropropionic acid (1/8), (NH4)2[Eu6F8(O2CC2F5)12(C2F5COOH)6]·8C2F5COOH (3)
10 ml (50.7 mmol, ρ25 = 1.571 g cm−3) of pentafluoropropionic anhydride was slowly mixed with 95% of the 1:1 molar ratio of water (0.87 ml, 48.2 mmol) in a microdistillation device under ice cooling. The liquid was distilled over a 10 cm Vigreux column, and the main fraction collected at 96–97 °C. 0.89 ml (8.4 mmol, ρ25 = 1.561 g cm−3) of the pentafluoropropionic acid was slowly dropped into a reaction vessel containing a glass-coated stir bar to 50 ml of dried liquid ammonia. 0.638 g (4.2 mmol) of europium was added. The resulting yellow suspension was stirred for 1 h at approximately −30 °C. The ammonia was evaporated, and the residue dried in vacuum for 8 h until a pressure of 10−3 hPa was reached. The residue was ground to a fine yellow powder (1.12 g). 151Eu Mössbauer spectrum, isomer shift [mm s−1]: EuIII: 0.29 (50%); EuII: −13.11 (50%). Crystals of 3 were received as described for 1 using 230 mg of this crude product and 3 ml of anhydrous pentafluoropropionic acid instead of DMF. The mother liquor was removed, and the crystalline material dried in vacuo until a pressure of 10−2 hPa was reached. Yield: 163 mg, 28% based on Eu. A coulometric Karl Fischer titration gave a water content of the mother liquor of 0.95%. Elemental analysis [%]: Found: C, 16.4; H, 0.4; N, 0.7; Eu, 21.1. Calc. for 3–8 C2F5COOH, C54H14N2O36F98Eu6: C, 16.05; H, 0.35 N, 0.69; Eu, 22.57. IR spectrum [cm−1]: ν(O–H): 3602 (w), 3538 (vw); ν(CO): 1775 (w); νas(COO): 1693 (s); 1625 (w); νs(COO): 1439 (m); ν(CF3–CF2): 1328 (m); ν(C–F): 1213 (m), 1159 (s); ν(CF2–COO): 1032 (s); δ(C–C): 821 (w); δ(O–C–O): 729 (m); δ(CF3): 584, 539 (w); 407 (w). Raman spectrum [cm−1]: ν(CO): 1776 (w); νas(COO): 1709 (s); νs(COO): 1442 (s); ν(CF3–CF2): 1332 (w); ν(C–F): 1218 (w), ν(CF2–COO): 1039 (w); δ(C–C): 826 (s); 779 (s); δ(O–C–O): 739 (m); 624 (w); δ(CF3): 584, 542 (w); 426 (s); 394 (s); 361 (s); 295 (m); 143, 118, 78 (s). 19F NMR spectrum (282.38 MHz, CD3CN/NaOH) [ppm]: −76.9 (s, F−); −84.1 (s, CF3); −123.1 (s, CF2). 151Eu Mössbauer spectrum, isomer shift [mm s−1]: EuIII: 0.31.
Crystallography
Crystals of 1, 2 and 3 suitable for crystal structure analysis were selected in perfluorinated oil and mounted in the nitrogen-cold gas stream on a STOE IPDS 2T diffractometer in the case of 2 and on a Bruker APEX II CCD diffractometer in the cases of 1 and 3. Indexing, unit cell refinement, data collection and data processing were performed using X-AREA45 or APEX2.46 A Gaussian absorption correction was applied for 2 with the XPREP routine of SHELXTL and a multi-scan absorption correction was applied for 1 and 3 with SADABS. The structures were solved by Direct Methods and refined by full-matrix least-squares calculations on F2 with programs of the SHELX system.47 All non-hydrogen atoms were located and all H atoms were included in idealized positions. For those H atoms bound to C and O a riding model was applied, with bond lengths constrained to 0.98, 0.95 and 0.83 Å for CH3, CH and OH groups, respectively. In addition, the CH3 groups and the OH groups were allowed to rotate around the adjacent N–C and O–C bond, respectively. The NH4+ ions in 2 and 3 were included in the refinement as rigid groups with idealized tetrahedral geometry and N–H bond lengths constrained to 0.91 Å. The displacement parameters Uiso(H) were set to 1.5Ueq, 1.5Ueq, 1.5Ueq and 1.2Ueq of the parent atoms for OH, NH, CH3 and CH, respectively. For trifluoromethyl groups in 1 and 2, clearly identified as disordered, partial occupation site models were introduced. In the final stages of converging refinements, the corresponding site occupation factors refined to 0.556(14):0.444(14) and 0.58(5):0.42(5)–0.66(5):0.34(5) for 1 and 2, respectively. Partial occupation site models were introduced for disordered pentafluoroethyl groups in 3, giving site occupation factors of 0.548(8):0.452(8)–0.821(5):179(5) in course of the refinement. Crystals of 2 notoriously suffer from twinning and the plane law (1 0 0) was detected by closer inspection. Based on the entire set of intensity data from both individuals and assuming the additivity of intensities for overlapped reflections a refinement with the HKLF5 option of SHELXL was performed. However, several shortcomings of the refinement including convergence problems forced us to discard this attempt. Tolerating some feature in the residual electron density map, definitely related to the twinning, a refinement based on intensities collected with the orientation matrix of the main individual and applying the index-transforming twin matrix −1 0 0 0 −1 0 0.431 0 1 (written by rows) for an approximate treatment of the intensity contamination resulting from the twinning and applying symmetry merging as well gave the best result in terms of plausibility of geometric parameters and anisotropic atom displacement. The fraction of second individual contribution was refined to 0.252(2). Molecular graphics were generated with DIAMOND.48 Crystal data, data collection and structure refinement details for 1, 2 and 3 are given in Table 3.
Elemental analysis
The elemental analyses were performed using a vario MICRO cube (Elementar Analysensysteme GmbH).49 They were supplemented by the determination of the europium content using a titrimetric method.19,50 50–150 mg of the sample was weighed to the nearest 0.1 mg and dissolved in 10 ml of hydrochloric acid, c(HCl) = 6 mol l−1. This solution was diluted to 60 ml with water and dropped into a chromatography column of 2 cm diameter, filled with a 20 cm high layer of zinc amalgam containing 1% of mercury.51 The solution was collected in a flask containing 15 ml of FeCl3–H2SO4 reagent [dissolve 2.70 g of FeCl3·6H2O in 100 ml sulfuric acid, c(H2SO4) = 1 mol l−1] and two drops of ferroin solution (1/40 mol l−1). To exclude atmospheric oxygen, a steady stream of dry nitrogen was passed through the flask. After the solution completely passed the reducer, it was washed with 40 ml of hydrochloric acid, c(HCl) = 0.1 mol l−1, and the content of the flask (no longer under inert gas) was acidified with 2 ml conc. phosphoric acid and titrated with a solution of cerium(IV) sulfate, c[1/1 Ce(SO4)2] = 0.05 mol l−1, until the colour changed from orange to blue. Since the end point is indicated with some delay, the titration must be carried out very slowly towards the end. To take into account remaining dissolved oxygen, negatively influencing the results, the standard solution was adjusted against portions of europium(III) oxide.
151Eu Mössbauer spectroscopy
For 151Eu Mössbauer spectroscopic studies on the compounds 1, 2 and 3, and their corresponding precursors the 21.53 keV transition of a 151Sm:EuF3 source with an activity of 55 MBq (1% of the total source activity) was used in normal transmission geometry. The samples were investigated with a commercial liquid nitrogen bath cryostat (78 ± 0.5 K, for 1, 2 and 3) or with a Janis Research continuous flow helium cryostat (6 ± 0.5 K, for the precursors). The samples were cooled to the respective temperature while the source was kept at room temperature. The temperatures were controlled with a resistance thermometer. The samples were prepared by mixing them with α-quartz in an argon-filled glovebox and placing them in a thin-walled PMMA container, 2 cm in diameter. These were then rapidly glued airtight outside the glovebox. The absorber thickness was calculated so that it corresponded to 10 mg of europium per cm2 of the sample holder. The results were fitted and plotted with the WinNormos for Igor6 program package52 and graphical editing was done with CorelDraw 2017.53
Photoluminescence spectroscopy
Steady-state and time-resolved photoluminescence studies on the powdered compounds 1, 2 and 3 were performed on an FLS1000 photoluminescence spectrometer from Edinburgh Instruments equipped with a 450 W Xe arc lamp as an excitation source, double excitation and emission monochromators (focal length 2 × 325 mm) in Czerny Turner configuration and a thermoelectrically cooled (−20 °C) photomultiplier tube PMT-980 as a detector. All spectra and decay traces were measured at room temperature. Emission spectra were corrected with respect to the grating efficiency and PMT sensitivity, while excitation spectra were additionally corrected with respect to the lamp intensity. Fluorescence decay traces (delay time ranges <1 μs) were measured with pulsed laser diodes from Edinburgh Instruments (EPL-320 for excitation at 320 nm, EPL-375 for excitation at 375 nm, temporal pulse widths: ∼75 ps) and time-correlated single photon detection, while longer decay traces (delay time ranges >1 μs) were acquired using a μF2 Xe flash lamp (150 W) with an average pulse width of 2 μs and single-photon multi-channel scaling detection.
IR and Raman spectroscopy
Compounds 1, 2 and 3, and their corresponding precursors were characterized via vibrational spectroscopy. IR spectra were recorded with a PerkinElmer Spectrum Two FT-IR spectrometer equipped with a LiTaO3 detector (4000–350 cm−1) and an universal ATR unit.54 Raman spectra were recorded with a Bruker MultiRAM spectrometer equipped with a Nd:YAG laser (1064 nm) and an InGaAs detector (4000–70 cm−1).55 Band assignments for 1 were made using the IR spectra of DMF56 and metal trifluoroacetates,57 for 2 additionally of trifluoroacetic acid35 and trifluoroacetic acid esters58 and for 3 of pentafluoropropionic acid59 and some of its esters.60
19F NMR spectroscopy
Compounds 2 and 3 were characterized via19F NMR spectroscopy. 10–20 mg each were dissolved in 0.7 ml D2O (for 2) or CD3CN (for 3) and the 19F NMR spectra were recorded using a Bruker Avance III spectrometer at 282.38 MHz. The fluoride ions bound to europium were released by adding a drop of conc. NaOH solution to the NMR tube and a second spectrum was recorded. Due to the poor solubility of some EuF3 precipitated a qualitative detection of the fluoride ions is possible, only.
Karl Fischer titration
The water content in the mother liquors from the crystallization of 2 and 3 was determined by coulometric Karl Fischer titrations. 0.1 ml of the solutions were injected into a Metrohm 831 KF coulometer filled with Hydranal Coulomat AG reagent. The results were corrected against a freshly opened 100 ppm water standard.
Thermal analysis
Compounds 1, 2 and 3 were characterized via TGA/DSC-thermal analysis. 10–15 mg each were weighed into an aluminum crucible and heated under N2 atmosphere. TGA curves were recorded in the temperature range of 25–600 °C using a Netzsch TG 209 F3 system. DSC curves were recorded in the temperature range of 25–450 °C using a Mettler-Toledo DSC 1 STARe system. A baseline correction was applied.
Conclusions
The reaction of NH4(O2CCF3) or NH4(O2CC2F5) with europium metal in liquid ammonia gives yellow Eu(O2CCF3)2 or Eu(O2CC2F5)2, respectively. However, to some extent this oxidation process is accompanied by C–F bond activation resulting in a recognizable amount of NH4F in the amorphous samples that are extremely sensitive to further oxidation. Bis(N,N-dimethylformamide) complexes of the EuII compounds have been prepared and the crystal structure determination of the trifluoroacetate derivative 1 shows the solid of this first lanthanoid(II) perfluorocarboxylate coordination compound to be chain-polymeric with bridging trifluoroacetato and N,N-dimethylformamide ligands. Reactions of the NH4F-containing samples with the corresponding anhydrous perfluorocarboxylic acid yields ammonium salts of two new dodecakis(μ2-perfluorocarboxylato)octa-μ3-fluorido-octahedro-hexaeuropiate(III) complexes with additional vertex-coordinating acid molecules, (NH4)2[Eu6F8(O2CR)12(RCOOH)6], with R = CF3 (2) and R = C2F5 (3). Interestingly, in the case of the pentafluoropropionate an octakis(acid) solvate is obtained. The oxidation state of europium in the unsolvated precursors and in complexes 1, 2 and 3 derived thereof were monitored by 151Eu Mössbauer and photoluminescence spectroscopy. In a subsequent article we will report on the stepwise hydrolysis of the europium(III) complexes presented here, yielding a series of partially and fully vertex-hydrated octahedro-hexanuclear fluoridocarboxylates.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We would like to thank Jun.-Prof. Dr. M. Suta, Dr. P. Barthen, Dr. G. J. Reiss, Dr. G. Kreiner, E. Hammes, and T. Herrmann for technical support and discussions. Support for this research by the Jürgen Manchot Stiftung (scholarship to F. Morsbach) is gratefully acknowledged. Thanks to the CeMSA@HHU (Center for Molecular and Structural Analytics @ Heinrich Heine University) for recording the NMR-spectroscopic data.
Notes and references
-
(a) F. Swarts, Acad. R. Belg., 1922, 8, 343–370 CAS;
(b) R. N. Haszeldine, J. Chem. Soc., 1951, 584–587 RSC;
(c) R. Hara and G. H. Cady, J. Am. Chem. Soc., 1954, 76, 4285–4287 CrossRef CAS;
(d) G. S. Fujioka and G. H. Cady, J. Am. Chem. Soc., 1957, 79, 2451–2454 CrossRef CAS;
(e) P. Sartori, J. Fazekas and J. Schnackers, J. Fluorine Chem., 1972, 1, 463–471 CrossRef CAS;
(f) C. D. Garner and B. Hughes, Inorg. Chem., 1974, 14, 1722–1724 CrossRef;
(g) C. D. Garner and B. Hughes, Inorg. Chem., 1974, 14, 463–471 Search PubMed;
(h) C. D. Garner and B. Hughes, Adv. Inorg. Chem., 1975, 17, 1–47 CrossRef CAS;
(i) G. J. Reiss, W. Frank and J. Schneider, Main Group Met. Chem., 1995, 18, 287–294 CAS;
(j) B. F. T. Cooper and C. L. B. Macdonald, New J. Chem., 2010, 34, 1551–1555 RSC;
(k) A. Llordés, K. Zalamova, S. Ricart, A. Palau, A. Pomar, T. Puig, A. Hardy, M. K. v. Bael and X. Obradors, Chem. Mater., 2010, 22, 1686–1694 CrossRef;
(l) K. T. Dissanayake, L. M. Mendoza, P. D. Martin, L. Suescun and F. A. Rabuffetti, Inorg. Chem., 2016, 55, 170–176 CrossRef CAS PubMed.
-
(a) R. Dallenbach and P. Tissot, J. Therm. Anal., 1977, 11, 61–69 CrossRef CAS;
(b) R. Dallenbach and P. Tissot, J. Therm. Anal., 1981, 20, 409–417 CrossRef CAS;
(c) S. Fujihara, M. Tada and T. Kimura, Thin Solid Films, 1997, 304, 252–255 CrossRef CAS;
(d) S. Fujihara, S. Ono, Y. Kishiki, M. Tada and T. Kimura, J. Fluorine Chem., 2000, 105, 65–70 CrossRef CAS;
(e) D. Czajkowski, I. Simon and W. Frank, Z. Anorg. Allg. Chem., 2019, 645, 402–408 CrossRef CAS.
-
(a) S. Mishra, S. Daniele, G. Ledoux, E. Jeanneau and M.-F. Joubert, Chem. Commun., 2010, 46, 3756–3758 RSC;
(b) S. Mishra, G. Ledoux, E. Jeanneau, S. Daniele and M.-F. Joubert, Dalton Trans., 2012, 41, 1490–1502 RSC;
(c) S. Mishra and S. Daniele, Chem. Rev., 2015, 115, 8379–8448 CrossRef CAS PubMed.
-
(a) F. A. Cotton and J. G. Norman, J. Coord. Chem., 1971, 1, 161–172 CrossRef;
(b) M. Sikirica and D. Gardenic, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1974, 30, 144–146 CrossRef CAS;
(c) D. J. Santure, K. W. McLaughlin, J. C. Huffman and A. P. Sattelberger, Inorg. Chem., 1983, 22, 1877–1883 CrossRef CAS;
(d) F. A. Cotton, E. V. Dikarev and X. Feng, Inorg. Chim. Acta, 1995, 237, 19–26 CrossRef CAS;
(e) F. A. Cotton, E. V. Dikarev and M. A. Petrukhina, Inorg. Chem., 2000, 39, 6072–6079 CrossRef CAS PubMed;
(f) E. V. Dikarev, A. S. Filatov, R. Clérac and M. A. Petrukhina, Inorg. Chem., 2006, 45, 744–751 CrossRef CAS PubMed;
(g) B. Li, H. Zhang, L. Huynh, M. Shatruk and E. V. Dikarev, Inorg. Chem., 2007, 46, 9155–9159 CrossRef CAS PubMed;
(h) E. V. Dikarev, T. G. Gray and B. Li, Angew. Chem., Int. Ed., 2005, 44, 1721–1724 CrossRef CAS PubMed.
-
(a) E. V. Dikarev and B. Li, Inorg. Chem., 2004, 43, 3461–3466 CrossRef CAS PubMed;
(b) W. Frank, V. Reiland and G. J. Reiss, Angew. Chem., Int. Ed., 1998, 110, 3153–3155 CrossRef;
(c) W. Frank, V. Reiland and G. J. Reiss, Angew. Chem., Int. Ed., 1998, 37, 2983–2985 CrossRef.
-
A. F. Holleman, E. Wiberg and N. Wiberg, Anorganische Chemie, Walter de Gruyter, Berlin, 103th edn, 2017, pp. 2288–2311 Search PubMed.
-
(a) H. N. McCoy, J. Am. Chem. Soc., 1935, 57, 1756 CrossRef CAS;
(b)
G. Brauer, Handbuch der Präparativen Anorganischen Chemie, Ferdinand Enke Verlag, Stuttgart, 1st edn, 1975, pp. 1066–1116 Search PubMed.
- F. Ruegenberg, A. García-Fuente, M. Seibald, D. Baumann, S. Peschke, W. Urland, A. Meijerink, H. Huppertz and M. Suta, Adv. Opt. Mater., 2021, 9, 2101643 CrossRef CAS.
-
(a) A. Lossin and G. Meyer, Z. Anorg. Allg. Chem., 1992, 614, 12–16 CrossRef CAS;
(b) P. Starynowicz, J. Alloys Compd., 1995, 224, 217–219 CrossRef CAS;
(c) P. Starynowicz, Polyhedron, 1995, 14, 3573–3577 CrossRef CAS;
(d) P. Starynowicz, J. Alloys Compd., 1995, 225, 406–408 CrossRef CAS;
(e) P. Starynowicz, J. Alloys Compd., 1998, 268, 47–49 CrossRef CAS;
(f) P. Starynowicz, J. Alloys Compd., 1998, 275–277, 815–817 CrossRef CAS.
-
(a) D. G. Karraker, J. Chem. Educ., 1970, 47, 424–430 CrossRef CAS;
(b) F. H. Spedding, M. J. Pikal and B. O. Ayers, J. Phys. Chem., 1966, 70, 2440–2449 CrossRef CAS;
(c) K. Micskei, D. H. Powell, L. Helm, E. Brücher and A. E. Merbach, Magn. Reson. Chem., 1993, 31, 1011–1020 CrossRef CAS;
(d) D. H. Powell and A. E. Merbach, Magn. Reson. Chem., 1994, 32, 739–745 CrossRef CAS.
-
(a) J. W. Bats, R. Kalus and H. Fuess, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1979, 35, 1225–1227 CrossRef;
(b) M. C. Favas, D. L. Kepert, B. W. Skelton and A. H. White, Dalton Trans., 1980, 454–458 RSC;
(c) S. Ganapathy, V. P. Chacko, R. G. Bryant and M. C. Etter, J. Am. Chem. Soc., 1986, 108, 3159–3165 CrossRef CAS;
(d) A. Lossin and G. Meyer, Z. Naturforsch., B: J. Chem. Sci., 1992, 47, 1602–1608 CrossRef CAS;
(e) J. L. Arias, A. Cabrera, P. Sharma, N. Rosas, J. L. Garcia and S. Hernandez, Inorg. Chim. Acta, 2000, 310, 261–264 CrossRef CAS;
(f) S. Gomez-Torres, I. Pantenburg and G. Meyer, Z. Anorg. Allg. Chem., 2006, 632, 1989–1994 CrossRef CAS;
(g) L. Cañadillas-Delgado, O. Fabelo, J. Cano, J. Pasán, F. S. Delgado, F. Lloret, M. Julveb and C. Ruiz-Pérez, CrystEngComm, 2009, 11, 2131–2142 RSC;
(h) M. Evangelisti, O. Roubeau, E. Palacios, A. Camón, T. N. Hooper, E. K. Brechin and J. J. Alonso, Angew. Chem., Int. Ed., 2011, 50, 6606–6609 CrossRef CAS PubMed;
(i) G. Lorusso, O. Roubeau and M. Evangelisti, Angew. Chem., Int. Ed., 2016, 55, 3360–3363 CrossRef CAS PubMed;
(j) A. De, S. S. Pradhan and B. Biswas, J. Indian Chem. Soc., 2017, 94, 1063–1071 CAS.
-
(a) S. P. Bone, D. B. Sowerby and R. D. Verma, Dalton Trans., 1978, 1544–1548 RSC;
(b) K. V. Katti, P. R. Singh and C. L. Barnes, Synth. React. Inorg. Met.-Org. Chem., 1996, 26, 349–355 CrossRef CAS;
(c) G. V. Romanenko, N. P. Sokolova and S. V. Larionov, J. Struct. Chem., 1999, 40, 325–329 CrossRef CAS;
(d) A. A. Rastorguev, A. A. Remova, G. V. Romanenko, N. P. Sokolova, V. I. Belyi and S. V. Larionov, J. Struct. Chem., 2001, 42, 759–766 CrossRef CAS;
(e) V. I. Belyi, A. A. Rastorguev, A. A. Remova, G. V. Romanenko and N. P. Sokolova, J. Struct. Chem., 2002, 43, 587–594 CrossRef CAS;
(f) N. P. Sokolova, V. L. Varand, G. V. Romanenko, V. I. Lisoivan, V. P. Fadeeva and L. A. Sheludyakova, Russ. J. Coord. Chem., 2002, 29, 362–368 CrossRef;
(g) S. I. Gutnikov, E. V. Karpova, M. A. Zakharov and A. I. Boltalin, Russ. J. Inorg. Chem., 2006, 51, 541–548 CrossRef.
- A. Rohde and W. Urland, Z. Anorg. Allg. Chem., 2006, 632, 1141–1144 CrossRef CAS.
-
(a) C. G. Pernin and J. A. Ibers, Inorg. Chem., 1997, 36, 3802–3803 CrossRef CAS;
(b) C. G. Pernin and J. A. Ibers, J. Cluster Sci., 1999, 10, 71–90 CrossRef CAS.
- J. Liu, E. A. Meyers and S. G. Shore, Inorg. Chem., 1998, 37, 5410–5411 CrossRef CAS PubMed.
-
(a) L. G. Hubert-Pfalzgraf, N. Miele-Pajot, R. Papiernik and J. Vaissermann, Dalton Trans., 1999, 4127–4130 RSC;
(b) R.-G. Xiong, J.-L. Zuo, Z. Yu, X.-Z. You and W. Chen, Inorg. Chem. Commun., 1999, 2, 490–494 CrossRef CAS;
(c) G. Xu, Z.-M. Wang, Z. He, Z. Lu, C.-S. Liao and C.-H. Yan, Inorg. Chem., 2002, 41, 6802–6807 CrossRef CAS PubMed;
(d) P. W. Roesky, G. Canseco-Melchor and A. Zulys, Chem. Commun., 2004, 738–739 RSC;
(e) V. Baskar and P. W. Roesky, Dalton Trans., 2006, 676–679 RSC;
(f) M. T. Gamer, Y. Lan, P. W. Roesky, A. K. Powell and R. Clérac, Inorg. Chem., 2008, 47, 6581–6583 CrossRef CAS PubMed;
(g) S. Petit, F. Baril-Robert, G. Pilet, C. Reber and D. Luneau, Dalton Trans., 2009, 6809–6815 RSC.
- A. Babai and A.-V. Mudring, Z. Anorg. Allg. Chem., 2006, 632, 1956–1958 CrossRef CAS.
-
(a) A. Rohde and W. Urland, Dalton Trans., 2006, 2974–2978 RSC;
(b) D. John and W. Urland, Z. Anorg. Allg. Chem., 2007, 633, 2587–2590 CrossRef CAS.
- H. N. McCoy, J. Am. Chem. Soc., 1936, 58, 1577–1580 CrossRef CAS.
- J. K. M. Sanders and D. H. Williams, Nature, 1972, 240, 385–390 CrossRef CAS.
- B.-K. Ling, J. Li, Y.-Q. Zhai, H.-K. Hsu, Y.-T. Chan, W.-P. Chen, T. Han and Y.-Z. Zheng, Chem. Commun., 2020, 56, 9130–9133 RSC.
-
(a) J. C. Warf and W. L. Korst, J. Phys. Chem., 1956, 60, 1590–1591 CrossRef CAS;
(b) J. C. Warf, Angew. Chem., Int. Ed. Engl., 1970, 9, 383 CrossRef.
- E. O. Fischer and H. Fischer, J. Organomet. Chem., 1965, 3, 181–187 CrossRef CAS.
- L. L. Pytlewsky and J. K. Howell, Chem. Commun., 1967, 1280 RSC.
- R. Juza and C. Hadenfeldt, Naturwissenschaften, 1968, 55, 229 CrossRef CAS.
- J. K. Howell and L. L. Pytlewsky, J. Less-Common Met., 1969, 18, 437–439 CrossRef CAS.
- I. D. Brown and D. Altermatt, Acta Crystallogr., Sect. B: Struct. Sci., 1985, 41, 244–247 CrossRef.
-
M. Gudenschwager and M. S. Wickleder, CCDC 1045819: Experimental Crystal Structure Determination, 2015 Search PubMed.
- G. Biedermann and G. S. Terjosin, Acta Chem. Scand., 1969, 23, 1896–1902 CrossRef CAS.
-
(a) Y. Cai and J. H. Espenson, Inorg. Chem., 2005, 44, 489–495 CrossRef CAS PubMed;
(b) M. V. d. Voorde, B. Geboes, T. V. Hoogerstraete, K. V. Hecke, T. Cardinaels and K. Binnemans, Dalton Trans., 2019, 48, 14758–14768 RSC.
- H. Bärnighausen, Z. Anorg. Allg. Chem., 1966, 342, 233–239 CrossRef.
-
(a)
J. G. Stevens, V. E. Stevens, P. T. Deason, A. H. Muir, H. M. Coogan and R. W. Grant, Mössbauer Effect Data Index, IFI/Plenum Data Company, New York, 1975 CrossRef;
(b) S. Golbs, F. M. Schappacher, R. Pöttgen, R. Cardoso-Gil, A. Ormeci, U. Schwarz, W. Schnelle, Y. Grin and M. Schmidt, Z. Anorg. Allg. Chem., 2013, 639, 2139–2148 CrossRef CAS.
-
D. T. Richens, The Chemistry of Aqua Ions: Synthesis, Structure and Reactivity: A Tour Through the Periodic Table of the Elements, John Wiley & Sons Ltd, Chichester, West Sussex, UK, 1997 Search PubMed.
- M. C. Etter, J. C. MacDonald and J. Bernstein, Acta Crystallogr., Sect. B: Struct. Sci., 1990, 46, 256–262 CrossRef PubMed.
-
(a) N. Fuson, M.-L. Josien, E. A. Jones and J. R. Lawson, J. Chem. Phys., 1952, 20, 1627–1634 CrossRef CAS;
(b) R. E. Kagarise, J. Chem. Phys., 1957, 27, 519–522 CrossRef CAS.
- A. Gavezzotti, Acta Crystallogr., Sect. B: Struct. Sci., 2008, 64, 401–403 CrossRef CAS PubMed.
- T. K. Harris and A. S. Mildvan, Proteins: Struct., Funct., Genet., 1999, 35, 275–282 CrossRef CAS.
-
S. Gomez-Torres, Dissertation, Universität zu Köln, Köln, Germany, 2007 Search PubMed.
- K. Binnemans, Coord. Chem. Rev., 2015, 295, 1–45 CrossRef CAS.
-
(a) D. Tu, Y. Liu, H. Zhu, R. Li, L. Liu and X. Chen, Angew. Chem., 2013, 125, 1166–1171 CrossRef;
(b) D. Tu, Y. Liu, H. Zhu, R. Li, L. Liu and X. Chen, Angew. Chem., Int. Ed., 2013, 52, 1128–1133 CrossRef CAS PubMed.
- S. H. M. Poort, A. Meyerink and G. Blasse, J. Phys. Chem. Solids, 1997, 58, 1451–1456 CrossRef CAS.
- V. K. Gramm, D. Smets, I. Grzesiak, T. Block, R. Pöttgen, M. Suta, C. Wickleder, T. Lorenz and U. Ruschewitz, Chem. – Eur. J., 2020, 26, 2726–2734 CrossRef CAS PubMed.
- J. J. Joos, L. Seijo and Z. Barandiarán, J. Phys. Chem. Lett., 2019, 10, 1581–1586 CrossRef CAS PubMed.
-
(a) B. Zhang, T. Xiao, C. Liu, Q. Li, Y. Zhu, M. Tang, C. Du and M. Song, Inorg. Chem., 2013, 52, 13332–13340 CrossRef CAS PubMed;
(b) J.-F. Greisch, M. E. Harding, B. Schäfer, M. Ruben, W. Klopper, M. M. Kappes and D. Schooss, J. Phys. Chem. Lett., 2014, 5, 1727–1731 CrossRef CAS PubMed.
-
X-Area, STOE & Cie GmbH, Darmstadt, Germany, 2009 Search PubMed.
-
APEX2, v2014.11–0, Bruker AXS Inc., Madison, WI, USA, 2014 Search PubMed.
-
(a) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed;
(b) G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
-
K. Brandenburg, DIAMOND, 4.5.1, Crystal Impact GbR, Bonn, Germany, 2018 Search PubMed.
-
vario MICRO, 3.1.13, Elementar Analysensysteme GmbH, Langenselbold, Germany, 2015 Search PubMed.
-
(a) H. Pink, Z. Anorg. Allg. Chem., 1968, 356, 319–320 CrossRef CAS;
(b) M. Sato, S. Kodama and N. Mori, Bunseki Kagaku, 1971, 20, 557–561 CrossRef CAS.
- H. W. Stone and D. N. Hume, Ind. Eng. Chem., Anal. Ed., 1939, 11, 598–602 CrossRef CAS.
-
R. A. Brand, WinNormos for Igor6, version for Igor6.2 or above: 22.02.2017, Universität Duisburg-Essen, Duisburg, Germany, 2017 Search PubMed.
-
CorelDRAW Graphics Suite 2017, 19.0.0.328, Corel Corp., Ottawa, Canada, 2017 Search PubMed.
-
SpectrumTM, 10, PerkinElmer Inc., Waltham, MA, USA, 2008 Search PubMed.
-
OPUS, 6.5, Bruker Corp., Billerica, MA, USA, 2009 Search PubMed.
-
(a) V. V. Chalapathi and K. V. Ramiah, Proc. – Indian Acad. Sci., Sect. A, 1968, 68, 109–122 CrossRef CAS;
(b) A. Sharma, S. Kaur, C. G. Mahajan, S. K. Tripathi and G. S. S. Saini, Mol. Phys., 2007, 105, 117–123 CrossRef CAS.
-
(a) M. J. Baillie, D. H. Brown, K. C. Moss and D. W. A. Sharp, J. Chem. Soc., 1968, 3110–3114 RSC;
(b) J. A. Faniran and K. S. Patel, Spectrochim. Acta, Part A, 1976, 32, 1351–1354 CrossRef.
-
(a) G. A. Crowder, Spectrochim. Acta, Part A, 1971, 28, 1625–1629 CrossRef;
(b) G. A. Crowder, Spectrochim. Acta, Part A, 1971, 27, 1873–1877 CrossRef CAS;
(c) G. A. Crowder, J. Fluorine Chem., 1972, 1, 219–225 CrossRef.
-
(a) G. A. Crowder, J. Fluorine Chem., 1972, 1, 385–389 CrossRef CAS;
(b) N. Rontu and V. Vaida, J. Mol. Struct., 2006, 237, 19–26 CAS.
-
(a) G. A. Crowder, J. Fluorine Chem., 1972, 2, 217–224 CrossRef;
(b) G. A. Crowder, J. Fluorine Chem., 1973, 3, 133–140 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Selected structural parameters and empirical bond valences, TGA and DSC curves, IR and NMR spectra, packing diagrams. CCDC 2125846–2125848. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1dt04204a |
| ‡ Because of the large number of atoms in the asymmetric units of 2 and 3, these are not shown in a single figure, contrary to the usual conventions, for reasons of clarity; instead, the designation of the atoms and the illustration of their anisotropic displacement parameters is divided among several figures in the discussion of these structures. |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Steffen
Klenner
b,
Rainer
Pöttgen
a,
Steffen
Klenner
b,
Rainer
Pöttgen