
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Gas phase vs. liquid phase: monitoring H2 and CO adsorption phenomena on Pt/Al2O3 by IR spectroscopy
Michele
Carosso
a,
Thibault
Fovanna
b,
Alberto
Ricchebuono
a,
Eleonora
Vottero
 a,
Maela
Manzoli
c,
Sara
Morandi
a,
Riccardo
Pellegrini
d,
Andrea
Piovano
e,
Davide
Ferri
*b and
Elena
Groppo
*a
a,
Maela
Manzoli
c,
Sara
Morandi
a,
Riccardo
Pellegrini
d,
Andrea
Piovano
e,
Davide
Ferri
*b and
Elena
Groppo
*a
aDepartment of Chemistry, INSTM and NIS Centre, University of Torino, via Quarello 15/A, I-10135 Torino, Italy. E-mail: elena.groppo@unito.it
bPaul Scherrer Institut, 5232 Villigen PSI, Switzerland. E-mail: davide.ferri@psi.ch
cDepartment of Drug Science and Technology, INSTM and NIS Centre, University of Torino, via Pietro Giuria 9, I-10125 Torino, Italy
dChimet SpA – Catalyst Division, via di Pescaiola 74, I-52041, Viciomaggio Arezzo, Italy
eInstitut Laue-Langevin (ILL), 71 avenue des Martyrs, 38000 Grenoble, France
First published on 13th January 2022
Abstract
The adsorption phenomena occurring at the surface of a highly-dispersed Pt/Al2O3 catalyst for hydrogenation reactions were thoroughly investigated in the gas-phase by transmission IR spectroscopy and in the liquid-phase by ATR-IR spectroscopy. The reduction of Pt/Al2O3 in H2 with the formation of Pt–hydrides and adsorption of CO were used as case studies for the comparison of the two different environments under otherwise similar experimental conditions. We found that compared to gas-phase, the selected solvent (cyclohexane) greatly affects the reducibility of Pt. Incomplete reduction under the adopted liquid-phase conditions was demonstrated by the formation of carbonate species at very low CO coverage. Moreover, cyclohexane competes with the adsorbates (H2 as well as CO) for the occupancy of the available surface sites, thus affecting the shape, position and relative intensity of the infrared signals corresponding to the surface Pt–H and Pt–CO species and leading to an overall lower H and/or CO coverage. Our spectroscopic data show also that the presence of the solvent is crucial in maintaining the Pt nanoparticles in partially H-solvated state even in a H2-free environment. This evidence might have important implications with respect to the catalyst reactivity and stability.
1. Introduction
Platinum based catalysts are massively employed in industrial processes involving hydrogenation reactions, such as in the synthesis of specialty chemicals and drugs.1–4 This is largely due to the superior ability of platinum in splitting molecular hydrogen into its atomic form.5,6 Understanding the nature and the behaviour of the platinum hydride (Pt–H) species under reaction conditions is crucial to design more efficient and profitable catalysts. However, characterizing the phenomena occurring at the surface of supported metal nanoparticles is one of the main challenges in surface science, due to the combination of many factors, among which are the intrinsic complexity of metal nanoparticles7–13 and their dynamic behaviour in the presence of adsorbates.14–24 In this context, the H2@Pt system was extensively investigated both theoretically15,16,21,24–28 and experimentally, especially by means of X-ray absorption spectroscopy (XAS),13,14,17,18,22,25,29–36 IR spectroscopy37–43 and inelastic neutron scattering (INS) spectroscopy.44–48 While XAS provides direct information on the structural and electronic changes experienced by nanometric Pt particles in the presence of hydrogen, vibrational spectroscopies have the capability to discern between different Pt–H surface species, since the Pt–H vibration is sensitive to even small changes in the coordination geometry.In such a scenario, some of us were involved in a systematic investigation of the hydride species formed over an industrial Pt/Al2O3 catalyst as a function of the H-coverage in the gas-phase, exploiting a multi-technique approach comprising INS, FT-IR and synchronous DRIFT/XAS/MS.49 INS, being extremely sensitive towards vibrational modes involving hydrogen motion and almost silent towards all other elements, allowed the detection of the vibrational fingerprints of multi-coordinated Pt–H species under high H-coverage conditions, which are not visible by FT-IR spectroscopy because they fall in the spectral region dominated by the vibrational modes of the alumina framework. By comparison, because the threshold of absorption of MgO is at lower energy, multi-folded hydrides were observed on Pt/MgO.50 By operando DRIFT/XAS/MS we were able to distinguish with an unprecedented level of detail at least four different species of linear Pt–hydrides characterized by different adsorption strengths and local environment, and to follow their interconversion into multi-coordinated Pt–H species upon changing the H-coverage.49 This phenomenon was correlated with a structural and electronic reconstruction of the Pt nanoparticles, in good agreement with theoretical predictions.15,16,21,24,25,27 It should be mentioned for the sake of clarity that the assignment of the IR signals in the 2100–1700 cm−1 to Pt–H species rather than to adsorbed CO is unambiguously confirmed by identical experiments with D2, which shifts them to the 1600–1200 cm−1 region.49
Industrial reactions are often carried out in a liquid solvent and the transferability of the results obtained in the gas-phase to liquid-phase cannot be assumed a priori. In order to close the gap of knowledge between gas-phase and liquid-phase conditions, similar experiments should be performed in the presence of a solvent. Unfortunately, the reports dealing with the vibrational characterization of Pt–hydrides at a solid–liquid interface are rare,43 likely because of technical limitations.51 In the liquid phase, the absorption bands related to the solvent can be orders of magnitude more intense than those associated with the adsorbed species. In this context, attenuated total reflection IR (ATR-IR) spectroscopy offers interesting opportunities.52 The catalyst can be deposited as a thin layer on the ATR crystal and contacted with the solvent. By exploiting this geometry, the amount of liquid sampled by the IR radiation is very small and most of the information is retrieved from the catalyst surface.53–59 Nevertheless only very few studies dealing with a detailed characterization of Pt-based catalysts in organic solvents and/or water are available,60 and they mainly focused on the use of CO as a molecular probe.61–63 As a further consequence of the lack of studies in liquid-phase environment a systematic comparison between gas-phase and liquid-phase experiments performed on the same catalyst is rare.94
In this work, we bridged this gap of knowledge in the characterization of surface phenomena occurring at solid–gas and solid–liquid interfaces, by thoroughly investigating a highly-dispersed Pt/Al2O3 catalyst during H2 and CO adsorption using transmission IR spectroscopy for the gas-phase and ATR-IR spectroscopy for the liquid-phase.
2. Experimental
2.1 Catalyst synthesis and preliminary characterization
The 5 wt% Pt/Al2O3 catalyst was prepared in the laboratories of the Catalyst Division of Chimet S.p.A., adopting a high-surface-area transitional alumina as a support (specific surface area = 116 m2 g−1; pore volume = 0.41 cm3 g−1) and following a proprietary deposition–precipitation method similar to the one reported by Kaprielova et al.64 After preparation, the sample was carefully washed with water and dried at 120 °C overnight. The nominal Pt dispersion (D = 63%) was determined by a H2/O2 titration method.65 As previously reported,49 HR-TEM micrographs showed very small and homogeneously distributed Pt nanoparticles, with an almost spherical shape and a regular size. A representative HR-TEM micrograph is reported in Fig. 1A, while Fig. 1B shows the particle size distribution obtained from the analysis of more than 700 particles. The average value (1.4 ± 0.4 nm) is in good agreement with the dispersion determined by H2/O2 titration, and was previously confirmed also by EXAFS data analysis.49 Temperature programmed reduction (TPR) was performed to evaluate the reducibility of the Pt phase. The main reduction peak (Fig. 1C), which is due to the Pt2+ → Pt0 reduction, starts at a temperature as low as −30 °C, reaches its maximum around 55 °C and ends around 130 °C. The integrated area below the peak delivers the overall hydrogen consumed in the process: it accounts for a Pt/H ratio of 3.6, indicating that the majority of the Pt phase was in the PtO2 form in the pristine catalyst.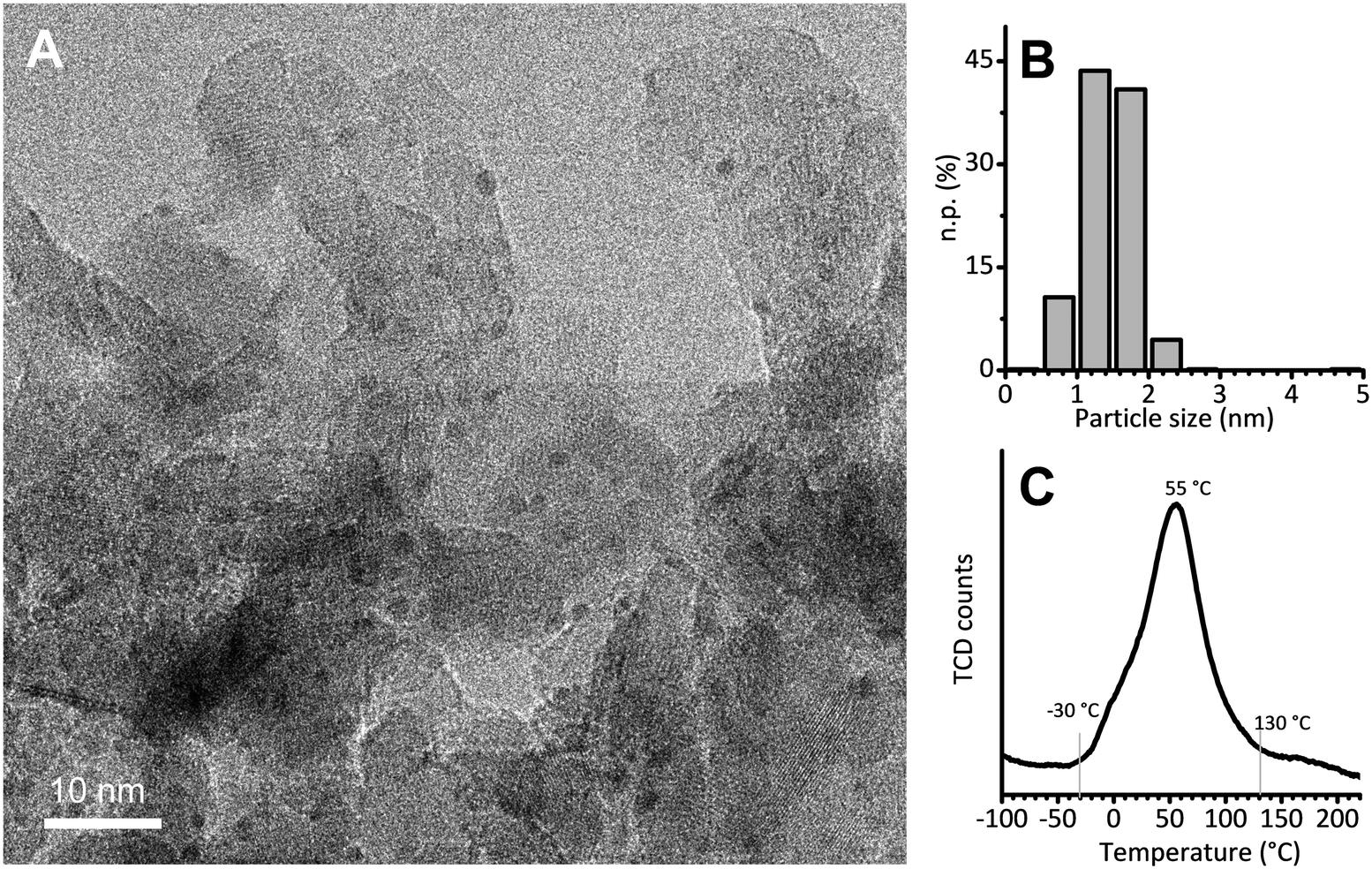 | ||
Fig. 1 (A) Representative HR-TEM micrograph of Pt/Al2O3 (instrumental magnification 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000X). (B) Corresponding particle size distribution determined by analysing more than 700 particles. (C) H2-TPR. 000X). (B) Corresponding particle size distribution determined by analysing more than 700 particles. (C) H2-TPR. | ||
2.2 Methods
The CO adsorption experiment was performed in static conditions. The self-supported pellet was inserted into a gold envelope and successively placed inside a homemade quartz cell equipped with KBr windows, allowing for the sample activation in the 25–700 °C range and successive measurements at room temperature, in the 10−4–103 mbar pressure range. The sample was heated up to 70 °C in dynamic vacuum and left at this temperature up to a final vacuum of 10−4 mbar. Successive reduction was performed by dosing an excess of H2 (equilibrium pressure of pH2 = 100 mbar, 2 times × 15 min) over the sample, followed by a final dynamic vacuum treatment down to 10−4 mbar. A FT-IR spectrum was collected at room temperature before contacting the sample at room temperature with pulses of a 10 vol% CO/He mixture up to an equilibrium pressure of pHe/CO = 30 mbar. An IR spectrum was collected at each pulse.
For the experiments in gas-phase the FT-IR spectra were collected with a Bruker Invenio IR spectrometer equipped with a MCT detector by averaging 32 scans at 2 cm−1 resolution.
For the CO adsorption experiment, the catalyst layer was reduced in H2 at 70 °C as described above, then cooled to room temperature in Ar-saturated cyclohexane and contacted with cyclohexane saturated with a 5 vol% CO/Ar mixture. ATR-IR spectra were recorded continuously every minute for 60 min.
All the measurements were performed with a Bruker Vertex70 spectrometer equipped with a MCT detector and a commercial ATR mirror unit. The ATR-IR spectra were collected by accumulating 20 scans at 4 cm−1 resolution.
3. Results and discussion
3.1 Pt–Hydrides formation and dynamics upon reducing the H-coverage
Fig. 2A shows the sequence of IR spectra collected during gas-phase reduction of Pt/Al2O3 at 70 °C in the region characteristic for the ν(Pt–H) vibrational modes of linear Pt–H species. For the sake of clarity, this experiment is equivalent to the one reported in our previous work,49 but performed at 70 °C instead of 120 °C in order to allow for a direct comparison with the experiments in the liquid phase. As soon as the catalyst was contacted by the N2/H2 flow, the growth of bands in the ν(OH) and δ(OH) region (not shown) testified the formation of H2O that was accompanied by the simultaneous appearance of a few weak bands in the ν(Pt–H) region. According to the literature37–43 and following our previous work,49 band I at 2115 cm−1 (which is detectable only in the presence of gaseous H2) is attributed to a weakly adsorbed hydride. Bands II and III at 2045 and 1990 cm−1 (the latter appearing as a shoulder of the former) are assigned to strongly adsorbed hydrides differing in terms of their local environment. Finally, the weaker bands IVa and IVb at about 1865 and 1740 cm−1 are assigned to interfacial Pt–H species, i.e. linear hydrides formed at Pt atoms in direct interaction with the alumina support. The assignments are summarized in Table 1. All the five bands gradually intensified over time almost at the same rate and reached a steady condition after about 10 min (spectrum 1GAS). At that point H2 was removed from the flow, and the desorption process was monitored (Fig. 2A′). Band I rapidly disappeared, while the other bands evolved in a counterintuitive way. The intensity of bands II and III passed through a minimum and then rose, while that of bands IVa and IVb increased constantly. These ν(Pt–H) bands reached their maximum intensity after about 1 h (spectrum 2GAS) and then they all vanished rapidly, following the observations reported in our previous work.49 | ||
| Fig. 2 (A) Evolution of the IR spectra in the region characteristic of the ν(Pt–H) vibrational modes for linear Pt–H species during the gas-phase reduction of Pt/Al2O3 in N2/H2 at 70 °C. Spectrum 1GAS was collected after 10 min in N2/H2 flow. (A′) Evolution of the IR spectra during the successive H2 desorption in N2 flow at 70 °C, up to the maximum intensities of the ν(Pt–H) bands. Spectrum 2GAS was collected after about 1 h in desorption conditions. (B) and (B′) are the same as (A) and (A′) for the ATR-IR experiment performed in liquid phase. Spectrum 1LIQ was collected after about 1 h in hydrogenation conditions, while spectrum 2LIQ after about 1 h in inert conditions. (C) Comparison of the final spectra reported in (A) and (B). (C′) Comparison of the final spectra reported in (A′) and (B′). | ||
| Linear Pt–hydrides, ν(Pt–H) | ||||||
|---|---|---|---|---|---|---|
| Species I | Species II | Species III | Species IVa and IVb | |||
| Gas-phase | 2115 | 2045 | 1990 | 1865 and 1740 | ||
| Cyclohexane | 2088 | 2020 | 1990 | 1835 and 1740 | ||
| Pt–Carbonyls, ν(Pt–CO) | ||||||
|---|---|---|---|---|---|---|
| Linear Pt–carbonyls | Multi-folded Pt–carbonyls | |||||
| Terrace sites (L1) | Edge sites (L2) | Corner sites (L3) | Bridged on terraces (B1) | Bridged on edges (B2) | 3-Fold (B3) | |
| Gas-phase | 2088 | 2070 | 2010 | 1878 | 1830 | 1790 |
| Cyclohexane | 2080 | 2060 | — | 1862 | 1818 | — |
This counterintuitive behavior is the same observed for the same sample measured in the presence of H2 at 120 °C, that was explained with a reconstruction of the hydrogenated Pt nanoparticles driven by a change in the H-coverage.49 Several theoretical models predict the occurrence of this phenomenon for Pt particles of subnanometric and nanometric size, which is also in accordance with experimental structural data obtained by XAS.15,16,21,24,25,27,34 According to theoretical models, at high H-coverage Pt13 nanoparticles adopt preferentially a cuboctahedral geometry poorly interacting with the support because they are H-solvated, mainly by multi-folded hydrides which are not visible by IR spectroscopy. Upon decreasing the H-coverage, the Pt13 nanoparticles reconstruct into a biplanar geometry strongly interacting with the support, which is accompanied by a conversion of multi-folded hydrides into linear ones, thus explaining the intensification of the ν(Pt–H) bands in the IR spectrum.
Fig. 2B and B′ show the results of the same experiment performed in liquid phase. The evident similarity to the gas-phase experiment allows us to translate the assignment of the observed signals to Pt–H species (Table 1). Focusing the attention on the hydrides formation step (Fig. 2B), from a qualitative perspective the ATR-IR spectra possess the same profile of those collected in the gas-phase (Fig. 2A), but there are important differences in the position, relative intensity and temporal evolution of the bands. Band I was barely present in cyclohexane (2088 cm−1); band II was less intense and red-shifted by ca. 25 cm−1; band IVa was more evident and red-shifted by ca. 30 cm−1, while bands III and IVb remained unaffected. Moreover, the five bands did not grow simultaneously as observed in the gas-phase experiment: band III was the first one to appear, followed by band II. In general, the kinetics of hydrides formation in the liquid-phase was slower than in the gas-phase, likely as a consequence of the low H2 concentration in cyclohexane, which is dictated by its solubility, and its higher diffusion limitation from the liquid-phase onto the catalyst surface than in the gas-phase. A direct comparison of the two final spectra collected in the presence of H2 is provided in Fig. 2C.
The differences between gas-phase and liquid-phase experiments are even larger when looking at the evolution of the IR spectra during the desorption step in cyclohexane (Fig. 2B′). Band I rapidly disappeared, while band II was quickly converted into band III within a few minutes as indicated by the formation of an isosbestic point at 1988 cm−1. At longer time, only a constant drift in the baseline was observed without further evolution in the position and/or intensity of the bands. The kinetics involved is very different from that observed in the gas-phase: the conversion of species II into species III is very fast and the spectra do not change further even after a very prolonged treatment in Ar-saturated solvent, while in the gas-phase the increase in intensity of species II and III is much slower. The final spectrum (2LIQ) is compared in Fig. 2C′ to that corresponding to the maximum intensity of the ν(Pt–H) bands obtained under gas-phase conditions (spectrum 2GAS). The complete absence of band II in the ATR-IR spectrum is the major difference between the two experiments. It is important to notice that, within the time interval investigated, the Pt–H bands did not disappear, in contrast to what was observed in the gas-phase and already reported in our previous work.49 This indicates that cyclohexane stabilizes the Pt nanoparticles in a hydrogenated form even in absence of hydrogen, preventing their reconstruction.
The set of data reported in Fig. 2 reveals that the presence of the solvent alters the relative distribution of the Pt–H species with respect to the gas-phase situation, both during the Pt oxide reduction and during the H2 desorption steps. We find at least two possible explanations for this behavior, which are not mutually exclusive: i) in the presence of the solvent, the overall H-coverage is lower than in gas-phase, either because the solvent competes with hydrogen in the occupancy of the adsorption sites and/or because the morphology of the Pt nanoparticles is different in the two cases; ii) under these conditions, in liquid-phase Pt is not completely reduced, which also leads to an overall lower H-coverage. In support of hypothesis i) it must be noticed that the evolution of the ATR-IR spectra in liquid-phase during the desorption step is very much similar to what occurs during the catalytic hydrogenation of toluene in vapour-phase,49 and is ascribed to a competitive adsorption between hydrogen and toluene. In support of hypothesis ii) there is the experimental evidence that the Pt phase is indeed not fully reduced in liquid-phase at 70 °C (as demonstrated in the following section).
Another relevant effect of the solvent is the stabilization of the Pt nanoparticles in a H-solvated form even in the absence of H2. This appears particularly relevant during reaction. In the case the reaction environment becomes poor in H2, the solvent can disfavour hydrogen desorption and decrease the rate of the morphologic reconstruction the Pt nanoparticles experience in the gas phase.16,21,22,24,25,27 We speculate that in this situation, the catalyst could sustain a reaction for longer time.
3.2 CO adsorption at room temperature
The surface properties of the Pt nanoparticles were successively investigated by means of CO adsorption at room temperature, which is one of the most utilized methods for the characterization of metal surfaces and of metal nano-particles.68–84Fig. 3 shows the sequence of transmission IR spectra collected upon progressive CO adsorption on Pt/Al2O3 in the gas-phase (Fig. 3A and B) and in the liquid-phase (Fig. 3C and D) in two different spectral regions, namely the ν(CO) region (Fig. 3A and C) and the region characteristic of surface carbonates (Fig. 3B and D).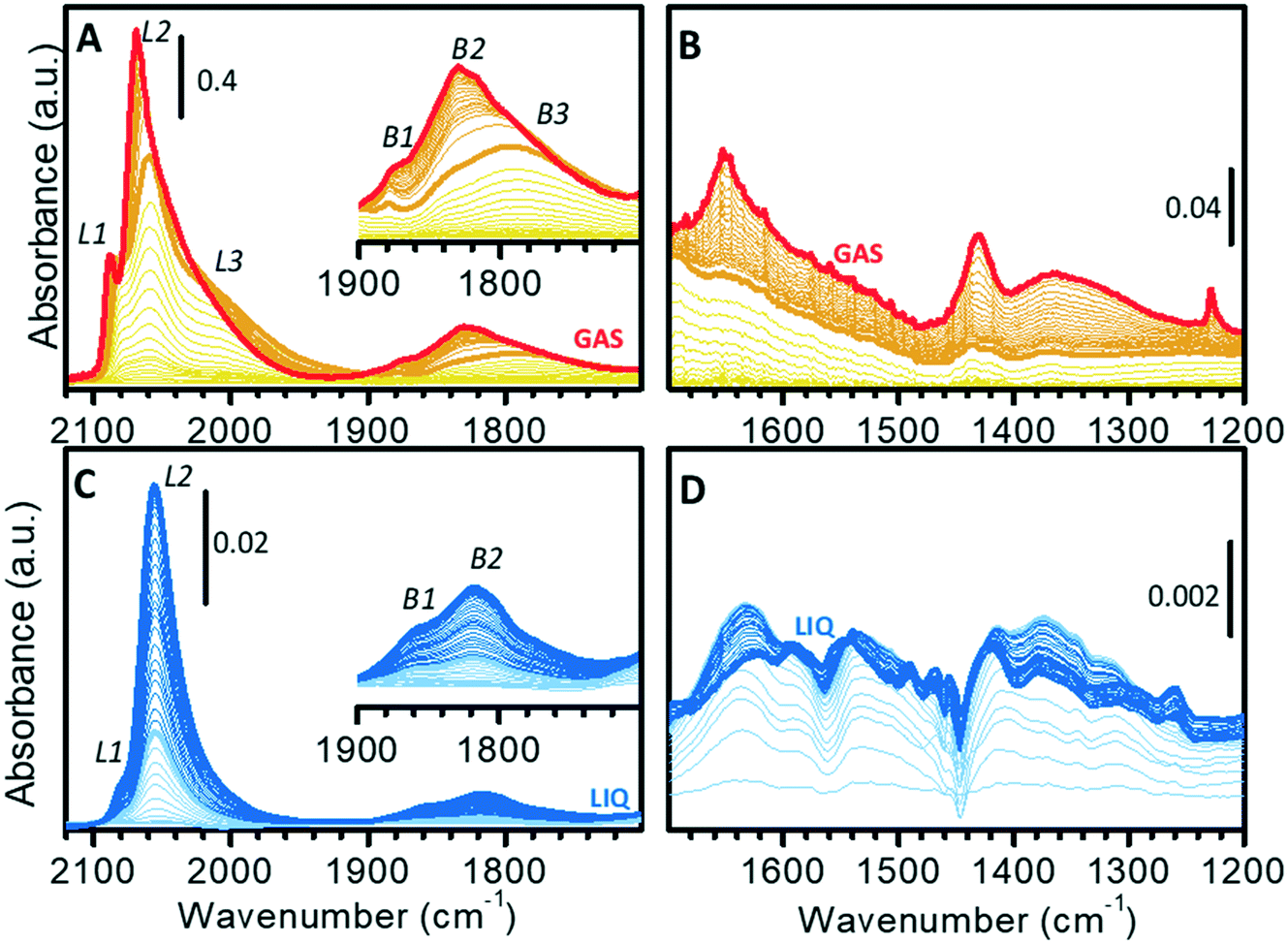 | ||
| Fig. 3 (A) FT-IR spectra of CO adsorbed at room temperature on Pt/Al2O3 (activated and reduced at 70 °C in gas-phase) in the ν(CO) region and (B) in the 1700–1200 cm−1 region, where the bands typical for surface carbonates are observed. The bold orange spectrum in (A) and (B) (PHe/CO = 2 mbar) is the last one before observing the upward shift of bands L1 and L2 (see text). (C) ATR-IR spectra of CO adsorbed at room temperature on Pt/Al2O3 (activated and reduced at 70 °C in liquid-phase) in the presence of cyclohexane in the ν(CO) region and (D) in the 1700–1200 cm−1 region. The bold light blue spectrum in (C) and (D) is that where the bands in 1700–1200 cm−1 region are maximized. The insets in (A) and (C) show a magnification of the 1900–1700 cm−1 region. | ||
The sequence of IR spectra collected upon increasing the CO coverage (θCO) in gas-phase is complex. More in detail, a total of six bands was observed in the ν(CO) region (Fig. 3A), which shift in position and grow at a different rate upon increasing the CO partial pressure. Bands in the 2100–1900 cm−1 region (L1, L2 and L3) are characteristic of linear Pt–carbonyls, while those in 1900–1700 cm−1 (B1, B2 and B3) are characteristic of multi-coordinated Pt–carbonyls. These bands can be assigned on the basis of surface science studies,68–73 as well as of the abundant literature on CO adsorption over Pt-based heterogeneous catalysts75,77–80 as follows (Table 1).
– Band L1 at 2077 cm−1 at low θCO and at 2088 cm−1 at high θCO, is attributed to CO linearly adsorbed at terrace sites.73–76,85
– Band L2 at 2060 cm−1 at low θCO and at 2070 cm−1 at high θCO, is due to CO adsorbed at the particles edges.73,85–89
– The very broad band L3 centered at ca. 2010 cm−1, which does not shift with θCO, is ascribed to CO adsorbed at particles corners/kinks.85,87
– Band B1 at 1878 cm−1 and band B2 at ca. 1830 cm−1, which are almost insensitive to θCO, are assigned to bridged carbonyls on terraces and edges, respectively.86,88–90
– The broad band B3 at ca. 1790 cm−1 is attributed to 3-fold coordinated CO at hollow sites.62,80,86,90,91
A rapid inspection of the spectra as a whole suggests a predominance of defects (edges and corners/kinks) rather than extended terraces, as expected because of the very high metal dispersion.
When analyzing the whole sequence of spectra as a function of θCO in a narrower spectral range, additional relevant information can be obtained. After the very first small pulses of CO, L1 and L2 were the first bands to appear, immediately followed by L3; bands due to bridged species appeared later. Upon increasing θCO, all the bands intensified, but did not shift until PHe/CO of about 2 mbar (bold orange spectrum in Fig. 3A). At this point, band L1 stopped growing and blue-shifted, band L2 blue-shifted while still growing and band L3 slightly attenuated. In the region of multi-folded carbonyls, bands B1 and B3 did not grow further, while band B2 became dominant. The blue-shift of bands L1 and L2 is explained in terms of a strong dipole–dipole coupling interaction between adjacent CO molecules at high θCO. The attenuation of band L3 and the simultaneous intensification of band L2 is well explained by the “transfer of intensity” concept proposed by Hollins:92 in the presence of strong dipole–dipole coupling involving more than one adsorbed species, a transfer of intensity may take place from a band at lower frequency to a band at higher frequency.
It is worth mentioning that, at the same time, in the 1700–1200 cm−1 region (Fig. 3B) a few bands started growing at ca. 1650, 1440 and 1230 cm−1. These bands are readily assigned to the ν(CO) and δ(OH) vibrational modes of a family of bicarbonates typically formed on alumina activated at low temperature.93 Interestingly, bicarbonates formed only when θCO was high enough to determine a coupling interaction among adsorbed CO (from bold orange spectra in Fig. 3B). We hypothesize that they are the result of disproportionation of two neighboring adsorbed CO molecules, which give CO2 (stabilized as bicarbonate at the partially hydrated surface of alumina) and adsorbed carbon. As a final observation, the fact that carbonate formation was not observed at low θCO confirms that the Pt phase was completely reduced, despite the low reduction temperature (70 °C). With reference to the TPR curve of Fig. 1C, this means that full reduction of the Pt–oxide phase can be achieved in the gas-phase even below 130 °C, provided that the reduction time is sufficiently long.
Fig. 3C and D show the results of a similar experiment performed in the liquid-phase, where the spectra were collected as a function of exposure time of the catalyst to a flow of CO-saturated cyclohexane. In this case, the gradual increase in θCO is also affected by the diffusion of CO from the solvent to the catalyst. In the ν(CO) region (Fig. 3C), the evolution of the spectra was much simpler than in the gas-phase. Contrary to what observed in gas-phase, the ν(CO) bands intensified at the same rate and did not shift until reaching saturation (Fig. 3C). This indicates that when the catalyst is in the liquid environment under these experimental conditions, dipole–dipole coupling between adsorbed CO molecules is not favored and thus the overall θCO is much lower than in the absence of the solvent. Nevertheless, these spectra are not the same as those collected in gas-phase at very low θCO, the most striking difference being the almost total absence of bands L3 and B3. In the carbonates region (broad signals in the 1700–1200 cm−1 region; Fig. 3D) the spectra are more complex than in gas-phase and the appearance of carbonate species already in the early stages of CO adsorption reveals the occurrence of a rich chemistry. Observation of carbonates as soon as CO reached the sample is a strong indication that the Pt phase was not fully reduced after the reduction at 70 °C in the liquid-phase. CO reduced this fraction of oxidized Pt with the consequent production of CO2, which is stabilized by the alumina support as carbonate (light blue spectra in Fig. 3D). This demonstrates that H2-reduction at 70 °C was less efficient in the presence of cyclohexane than in the gas-phase. The poor solubility of H2 in cyclohexane is a possible explanation, but we cannot exclude that the geometry of the ATR-IR cell, in which the solvent mostly by-passes the catalyst layer, could also play an important role. Formation of carbonates was completed after a few minutes, while Pt–carbonyls kept growing for longer time. Finally, it is worth noticing that in the liquid-phase experiment the simultaneous growth of carbonates and carbonyls as well as the shiftless growth of signals of carbonyls indicate that disproportionation of CO and the consequent formation of carbonates did not occur, thus confirming that disproportionation requires two vicinal adsorbed CO molecules as it was observed in the spectra collected in gas-phase.
Fig. 4 compares the IR spectra collected at the maximum θCO in the two experiments in the gas-phase and in the liquid-phase. It is evident that the spectrum of CO adsorbed on Pt/Al2O3 in cyclohexane is simpler than that of CO adsorbed in the gas-phase, with only two bands in the region of linear carbonyls, and two bands in the region of bridged carbonyls. Bands L1 and L2 are observed at 2080 and 2060 cm−1 in the liquid phase experiment, which are the values expected for linearly adsorbed CO on terraces and edges when isolated; moreover, the relative intensity of band L1 is much lower than in spectrum GAS and bands L3 and B3 are absent. All these observations suggest that the adsorption sites located at the flat domains of the nanoparticles (bands L1 and B3), as well as on corners/kinks (band L3), are less accessible to CO in the presence of the solvent, which forces CO to adsorb prevalently at the edges (bands L2 and B2). This suggests that in the presence of the solvent there are less sites available for CO adsorption, most likely because the solvent competes with CO for adsorption on the Pt surface.68
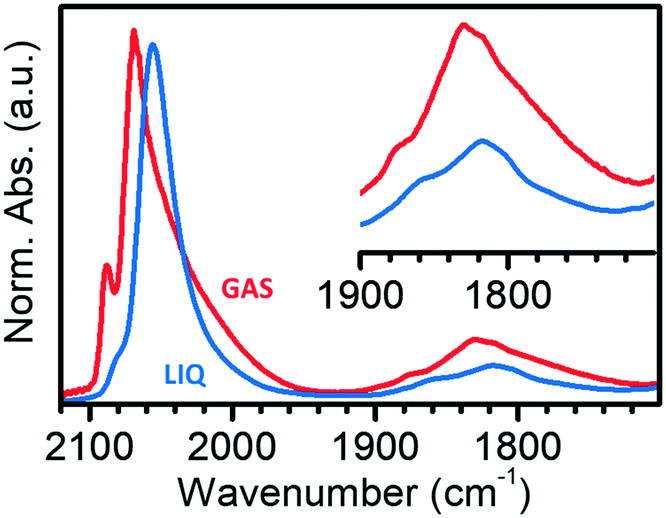 | ||
| Fig. 4 IR spectra of CO adsorbed at room temperature on Pt/Al2O3 activated and reduced at 70 °C in gas-phase (GAS) and in liquid-phase (LIQ) in the ν(CO) region. The inset reports a magnification of the 1900–1700 cm−1 region of multi-coordinated carbonyls. Spectra GAS and LIQ were arbitrarily normalized to the intensity of band L2. | ||
Finally, we should note that we have performed a purely qualitative study. We do not have yet the elements to compare quantitatively the coverages of CO and hydrides in the two different environments. We have to consider that a direct comparison of the intensities associated with adsorbed CO species in the gas and in the liquid phases cannot be performed, because of the different environment, optical geometries of the experiments and CO-coverage (likely lower in liquid-phase because of the competitive effect of the solvent). In order to proceed to a reliable quantitative prediction, the degree of the CO-coverage under the adopted experimental conditions, the metal surface area and the relative proportion of the different families of binding sites, as well as their energetics and the extinction coefficients of all the adsorbed species, should be known exactly. The data presented here allows us to qualitatively describe the surface sites available at the Pt nanoparticles and to predict which sites are favored in the liquid environment by comparing the differences in the relative intensities in the two cases.
4. Conclusions
In this work, we aimed at bridging the gap of knowledge of the adsorption phenomena occurring at the solid–gas and the solid–liquid interfaces. To this end, we investigated the adsorption of H2 and CO over an industrial Pt/Al2O3 catalyst using transmission IR spectroscopy in gas-phase and ATR-IR spectroscopy in the liquid-phase.We found that reduction by H2 at 70 °C is less efficient in the presence of the solvent (in this case, cyclohexane) than in the gas-phase, probably due to the poor H2 solubility in cyclohexane and/or to the particular geometry of the spectroscopy cell used to analyze the solid–liquid interface. Contrary to gas-phase case, the solvent stabilizes the Pt nanoparticles in a partially hydrogenated form even in the absence of H2, preventing their complete reconstruction. Not only the Pt–H species stability is affected, but also their relative distribution. This suggests that the solvent competes with H2 for the occupation of the surface sites, thus determining an overall lower H2 coverage. Competition between the solvent and the adsorbing molecule was also observed in the case of CO. A careful comparison of the transmission and ATR-IR spectra of adsorbed CO allowed us to conclude that the adsorption sites located at the terraces as well as on kinks are less accessible to CO in the presence of the solvent. Therefore, CO adsorbs prevalently at the edges of the Pt nanoparticles. This different behavior may bear important implications for the reactivity of the Pt nanoparticles in heterogeneously catalyzed reactions in the gas- and in the liquid-phase.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
T. F. and D. F. kindly acknowledge the financial support from the Paul Scherrer Institut (CROSS project).References
- R. L. Augustine, Heterogeneous Catalysis for the Synthetic Chemist, Dekker, New York, 1995 Search PubMed.
- G. M. Loudon, Organic Chemistry, Benjamin/Cummings Pub. Co., Menlo Park, 2nd edn, 1988 Search PubMed.
- S. Nishimura, Handbook of Heterogeneous Catalytic Hydrogenation for Organic Synthesis, Wiley, Chichester, 2001 Search PubMed.
- P. N. Rylander, Best Synthetic Methods: Hydrogenation Methods, Academic Press, London, 1985 Search PubMed.
- M. Che, Catal. Today, 2013, 218–219, 162–171 CrossRef CAS.
- A. Zecchina and S. Califano, Chapter 1 – From the Onset to the First Large-Scale Industrial Processes, in The Development of Catalysis: A History of Key Processes and Personas in Catalytic Science and Technology, ed. I. John, Wiley & Sons, 2017, ch. 1, pp. 1–57, DOI:10.1002/9781119181286.
- A. I. Frenkel, M. W. Small, J. G. Smith, R. G. Nuzzo, K. O. Kvashnina and M. Tromp, J. Phys. Chem. C, 2013, 117, 23286–23294 CrossRef CAS.
- A. I. Frenkel, C. W. Hills and R. G. Nuzzo, J. Phys. Chem. B, 2001, 105, 12689–12703 CrossRef CAS.
- J.-D. Grunwaldt and B. S. Clausen, Top. Catal., 2002, 18, 37–43 CrossRef CAS.
- M. A. Newton, B. Jyoti, A. J. Dent, S. G. Fiddy and J. Evans, Chem. Commun., 2004, 2382–2383, 10.1039/b405694a.
- G. Agostini, R. Pellegrini, G. Leofanti, L. Bertinetti, S. Bertarione, E. Groppo, A. Zecchina and C. Lamberti, J. Phys. Chem. C, 2009, 113, 10485–10492 CrossRef CAS.
- R. Arrigo, K. Badmus, F. Baletto, M. Boeije, M. Bowker, K. Brinkert, A. Bugaev, V. Bukhtiyarov, M. Carosso, R. Catlow, R. Chanerika, P. R. Davies, W. Dononelli, H. J. Freund, C. Friend, S. Gallarati, B. Gates, A. Genest, E. K. Gibson, J. Hargreaves, S. Helveg, H. Huang, G. Hutchings, N. Irvine, R. Johnston, S. Lai, C. Lamberti, J. Macginley, D. Marchant, T. Murayama, R. Nome, Y. Odarchenko, J. Quinson, S. Rogers, A. Russell, S. Said, P. Sermon, P. Shah, S. Simoncelli, K. Soulantica, F. Spolaore, B. Tooze, L. Torrente-Murciano, A. Trunschke, D. Willock and J. Zhang, Faraday Discuss., 2018, 208, 339–394 RSC.
- J. H. Kang, L. D. Menard, R. G. Nuzzo and A. I. Frenkel, J. Am. Chem. Soc., 2006, 128, 12068–12069 CrossRef CAS PubMed.
- M. W. Small, S. I. Sanchez, N. S. Marinkovic, A. I. Frenkel and R. G. Nuzzo, ACS Nano, 2012, 6, 5583–5595 CrossRef CAS PubMed.
- L.-L. Wang and D. D. Johnson, J. Am. Chem. Soc., 2007, 129, 3658–3664 CrossRef CAS.
- C. Mager-Maury, G. Bonnard, C. Chizallet, P. Sautet and P. Raybaud, ChemCatChem, 2011, 3, 200–207 CrossRef CAS.
- O. S. Alexeev, F. Li, M. D. Amiridis and B. C. Gates, J. Phys. Chem. B, 2005, 109, 2338–2349 CrossRef CAS PubMed.
- E. Bus and J. A. van Bokhoven, Phys. Chem. Chem. Phys., 2007, 9, 2894–2902 RSC.
- L. Li, L.-L. Wang, D. D. Johnson, Z. Zhang, S. I. Sanchez, J. H. Kang, R. G. Nuzzo, Q. Wang, A. I. Frenkel, J. Li, J. Ciston, E. A. Stach and J. C. Yang, J. Am. Chem. Soc., 2013, 135, 13062–13072 CrossRef CAS PubMed.
- S. I. Sanchez, L. D. Menard, A. Bram, J. H. Kang, M. W. Small, R. G. Nuzzo and A. I. Frenkel, J. Am. Chem. Soc., 2009, 131, 7040–7054 CrossRef CAS PubMed.
- W. Zhao, C. Chizallet, P. Sautet and P. Raybaud, J. Catal., 2019, 370, 118–129 CrossRef CAS.
- C. Dessal, A. Sangnier, C. Chizallet, C. Dujardin, F. Morfin, J. L. Rousset, M. Aouine, M. Bugnet, P. Afanasiev and L. Piccolo, Nanoscale, 2019, 11, 6897–6904 RSC.
- L. Piccolo, Catal. Today, 2021, 373, 80–97 CrossRef CAS.
- C. Chizallet and P. Raybaud, Catal. Sci. Technol., 2014, 4, 2797–2813 RSC.
- A. Gorczyca, V. Moizan, C. Chizallet, O. Proux, W. Del Net, E. Lahera, J.-L. Hazemann, P. Raybaud and Y. Joly, Angew. Chem., Int. Ed., 2014, 53, 12426–12429 CAS.
- G. Sun, A. N. Alexandrova and P. Sautet, J. Chem. Phys., 2019, 151, 1–15 Search PubMed.
- P. Raybaud, C. Chizallet, H. Toulhoat and P. Sautet, Phys. Chem. Chem. Phys., 2012, 14, 16773–16774 RSC.
- G. S. Erfani, S. Hong and T. S. Rahman, J. Phys. Chem. C, 2019, 123, 16893–16901 CrossRef CAS.
- F. W. Lytle, P. S. P. Wei, R. B. Greegor, G. H. Via and J. H. Sinfelt, J. Chem. Phys., 1979, 70, 4849–4855 CrossRef CAS.
- M. Vaarkamp, J. T. Miller, F. S. Modica and D. C. Koningsberger, J. Catal., 1996, 163, 294–305 CrossRef CAS.
- D. E. Ramaker, B. L. Mojet, M. T. Garriga Oostenbrink, J. T. Miller and D. C. Koningsberger, Phys. Chem. Chem. Phys., 1999, 1, 2293–2302 RSC.
- A. L. Ankudinov, J. J. Rehr, J. J. Low and S. R. Bare, Phys. Rev. Lett., 2002, 89, 139702-1 CrossRef.
- D. C. Koningsberger, M. K. Oudenhuijzen, J. De Graaf, J. A. Van Bokhoven and D. E. Ramaker, J. Catal., 2003, 216, 178–191 CrossRef CAS.
- M. K. Oudenhuijzen, J. A. van Bokhoven, J. T. Miller, D. E. Ramaker and D. C. Koningsberger, J. Am. Chem. Soc., 2005, 127, 1530–1540 CrossRef CAS PubMed.
- E. Bus, J. T. Miller, A. J. Kropf, R. Prins and J. A. van Bokhoven, Phys. Chem. Chem. Phys., 2006, 8, 3248–3258 RSC.
- E. Bus, J. T. Miller and J. A. van Bokhoven, J. Phys. Chem. B, 2005, 109, 14581–14587 CrossRef CAS PubMed.
- J. P. Candy, P. Fouilloux and M. Primet, Surf. Sci., 1978, 72, 167–176 CrossRef CAS.
- L. T. Dixon, R. Barth and J. W. Gryder, J. Catal., 1975, 37, 368–375 CrossRef CAS.
- D. D. Eley, D. M. Moran and C. H. Rochester, Trans. Faraday Soc., 1968, 64, 2168–2180 RSC.
- W. A. Pliskin and R. P. Eischens, Z. Phys. Chem., 1960, 24, 11–23 CrossRef CAS.
- M. Primet, J. M. Basset, M. V. Mathieu and M. Prettre, J. Catal., 1973, 28, 368–375 CrossRef CAS.
- T. Szilagyi, J. Catal., 1990, 121, 223–227 CrossRef CAS.
- D. Paleček, G. Tek, J. Lan, M. Iannuzzi and P. Hamm, J. Phys. Chem. Lett., 2018, 9, 1254–1259 CrossRef PubMed.
- P. Albers, E. Auer, K. Ruth and S. F. Parker, J. Catal., 2000, 196, 174–179 CrossRef CAS.
- P. W. Albers, M. Lopez, G. Sextl, G. Jeske and S. F. Parker, J. Catal., 2004, 223, 44–53 CrossRef CAS.
- H. Asada, T. Toya, H. Motohashi, M. Sakamoto and Y. Hamaguchi, J. Chem. Phys., 1975, 63, 4078–4079 CrossRef CAS.
- S. F. Parker, C. D. Frost, M. Telling, P. Albers, M. Lopez and K. Seitz, Catal. Today, 2006, 114, 418–421 CrossRef CAS.
- A. J. Renouprez and H. Jobic, J. Catal., 1988, 113, 509–516 CrossRef CAS.
- M. Carosso, E. Vottero, A. Lazzarini, S. Morandi, M. Manzoli, K. A. Lomachenko, M. J. Ruiz, R. Pellegrini, C. Lamberti, A. Piovano and E. Groppo, ACS Catal., 2019, 9, 7124–7136 CrossRef CAS.
- J.-P. Candy, P. Fouilloux and M. Primet, Surf. Sci., 1978, 72, 167–176 CrossRef CAS.
- D. Ferri, Toward operando infrared spectroscopy of heterogeneous catalysts, in Heterogeneous catalysts: Emerging techniques for design, characterization and applications, ed. A. U. W. Y. Teoh, Y. H. Ng and P. Sit, Wiley-VCH GmbH, 2021, pp. 311–338, DOI:10.1002/9783527813599.ch18.
- T. Bürgi and A. Baiker, Adv. Catal., 2006, 50, 227–283 Search PubMed.
- T. Bürgi, R. Wirz and A. Baiker, J. Phys. Chem. B, 2003, 107, 6774–6781 CrossRef.
- G. M. Hamminga, G. Mul and J. A. Moulijn, Chem. Eng. Sci., 2004, 59, 5479–5485 CrossRef CAS.
- A. Pintar, R. Malacea, C. Pinel, G. Fogassy and M. Besson, Appl. Catal., A, 2004, 264, 1–12 CrossRef CAS.
- J. E. Rekoske and M. A. Barteau, Ind. Eng. Chem. Res., 1995, 34, 2931–2939 CrossRef CAS.
- Z. Wang, M. L. Larsson, M. Grahn, A. Holmgren and J. Hedlund, Chem. Commun., 2004, 2888–2889, 10.1039/b410314a.
- D. Ferri and A. Baiker, Top. Catal., 2009, 52, 1323–1333 CrossRef CAS.
- C. Boulho, E. K. Gibson, M. I. McAllister, R. Moss and D. Lennon, Top. Catal., 2020, 63, 386–393 CrossRef CAS.
- R. He, R. R. Davda and J. A. Dumesic, J. Phys. Chem. B, 2005, 109, 2810–2820 CrossRef CAS PubMed.
- S. D. Ebbesen, B. L. Mojet and L. Lefferts, J. Catal., 2007, 246, 66–73 CrossRef CAS.
- D. Ferri, T. Bürgi and A. Baiker, J. Phys. Chem. B, 2001, 105, 3187–3195 CrossRef CAS.
- I. Ortiz-Hernandez, D. J. Owens, M. R. Strunk and C. T. Williams, Langmuir, 2006, 22, 2629–2639 CrossRef CAS PubMed.
- K. M. Kaprielova, O. A. Yakovina, I. I. Ovchinnikov, S. V. Koscheev and A. S. Lisitsyn, Appl. Catal., A, 2012, 449, 203–214 CrossRef CAS.
- J. E. Benson and M. Boudart, J. Catal., 1965, 4, 704–710 CrossRef CAS.
- M. Carosso, E. Vottero, S. Morandi, M. Manzoli, D. Ferri, T. Fovanna, R. Pellegrini, A. Piovano and E. Groppo, ChemCatChem, 2021, 13, 900–908 CrossRef CAS.
- J. B. Brazier, B. N. Nguyen, L. A. Adrio, E. M. Barreiro, W. P. Leong, M. A. Newton, S. J. A. Figueroa, K. Hellgardt and K. K. M. Hii, Catal. Today, 2014, 229, 95–103 CrossRef CAS.
- A. Crossley and D. A. King, Surf. Sci., 1977, 68, 528–538 CrossRef CAS.
- B. E. Heyden and A. M. Bradshaw, Surf. Sci., 1983, 125, 787–802 CrossRef.
- B. E. Hayden, K. Kretzschmar, A. M. Bradshaw and R. G. Greenler, Surf. Sci., 1985, 149, 394–406 CrossRef CAS.
- A. M. Bradshaw and F. M. Hoffmann, Surf. Sci., 1978, 72, 513–535 CrossRef CAS.
- G. Ertl, M. Neumann and K. M. Streit, Surf. Sci., 1977, 64, 393–410 CrossRef CAS.
- R. K. Brandt, M. R. Hughes, L. P. Bourget, K. Truszkowska and R. G. Greenler, Surf. Sci., 1993, 286, 15–25 CrossRef CAS.
- P. Pillonel, S. Derrouiche, A. Bourane, F. Gaillard, P. Vernoux and D. Bianchi, Appl. Catal., A, 2005, 278, 223–231 CrossRef CAS.
- S. K. Cheah, V. P. Bernardet, A. A. Franco, O. Lemaire and P. Gelin, J. Phys. Chem. C, 2013, 117, 22756–22767 CrossRef CAS.
- T. Avanesian, S. Dai, M. J. Kale, G. W. Graham, X. Pan and P. Christopher, J. Am. Chem. Soc., 2017, 139, 4551–4558 CrossRef CAS PubMed.
- J. A. Anderson, F. K. Chong and C. H. Rochester, J. Mol. Catal. A: Chem., 1999, 140, 65–80 CrossRef CAS.
- R. Barth, R. Pitchai, R. L. Anderson and X. E. Verykios, J. Catal., 1989, 116, 61–70 CrossRef CAS.
- P. Bazin, O. Saur, J. C. Lavalley, M. Daturi and G. Blanchard, Phys. Chem. Chem. Phys., 2005, 7, 187–194 RSC.
- D. M. Haaland, Surf. Sci., 1987, 185, 1–14 CrossRef CAS.
- F. Zaera, ChemCatChem, 2012, 4, 1525–1533 CrossRef CAS.
- T. Elgayyar, R. Atwi, A. Tuel and F. C. Meunier, Catal. Today, 2021, 373, 59–68 CrossRef CAS.
- C. Lentz, S. P. Jand, J. Melke, C. Roth and P. Kaghazchi, J. Mol. Catal. A: Chem., 2017, 426, 1–9 CrossRef CAS.
- I. Jbir, A. Paredes-Nunez, S. Khaddar-Zine, Z. Ksibir, F. Meunier and D. Bianchi, Appl. Catal., A, 2015, 505, 309–318 CrossRef CAS.
- F. Coloma, J. M. Coronado, C. H. Rochester and J. A. Anderson, Catal. Lett., 1998, 51, 155–162 CrossRef CAS.
- A. Bourane, O. Dulaurent and D. Bianchi, J. Catal., 2000, 196, 115–125 CrossRef CAS.
- J. Rasko, J. Catal., 2003, 217, 478–486 CrossRef CAS.
- J. Xu and J. T. Yates Jr., J. Chem. Phys., 1993, 99, 725–732 CrossRef CAS.
- J. Xu and J. T. Yates Jr., Surf. Sci., 1995, 327, 193–201 CrossRef CAS.
- B. E. Hayden and A. M. Bradshaw, The Adsorption of CO on Pt(111) Studied by Infrared-Reflection-Adsorption Spectroscopy, in Stud. Surf. Sci. Catal., ed. C. R. Brundle and H. Morawitz, Elsevier, 1983, vol. 14, p. 51 Search PubMed.
- N. P. Lebedeva, A. Rodes, J. M. Feliu, M. T. M. Koper and R. A. van Santen, J. Phys. Chem. B, 2002, 106, 9863–9872 CrossRef CAS.
- P. Hollins, Surf. Sci. Rep., 1992, 16, 51–94 CrossRef CAS.
- C. Morterra and G. Magnacca, Catal. Today, 1996, 27, 497–532 CrossRef CAS.
- S. D. Ebbesen, B. L. Mojet and L. Lefferts, Langmuir, 2006, 22, 1079–1085 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |