
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Controlling through-space and through-bond intramolecular charge transfer in bridged D–D′–A TADF emitters†
Hector
Miranda-Salinas
 a,
Yi-Tzu
Hung
b,
Yi-Sheng
Chen
b,
Dian
Luo
c,
Hao-Che
Kao
c,
Chih-Hao
Chang
*c,
Ken-Tsung
Wong
*bd and
Andrew
Monkman
*a
a,
Yi-Tzu
Hung
b,
Yi-Sheng
Chen
b,
Dian
Luo
c,
Hao-Che
Kao
c,
Chih-Hao
Chang
*c,
Ken-Tsung
Wong
*bd and
Andrew
Monkman
*a
aDepartment of Physics, Durham University, South Road, Durham DH1 3LE, UK. E-mail: a.p.monkman@durham.ac.uk
bDepartment of Chemistry, National Taiwan University, No. 1, Sec. 4, Roosevelt Road, Taipei, 10617, Taiwan. E-mail: kenwong@ntu.edu.tw
cDepartment of Electrical Engineering, Yuan Ze University, Taoyuan 32003, Taiwan. E-mail: chc@saturn.yzu.edu.tw
dInstitute of Atomic and Molecular Science, Academia Sinica, Taipei 10617, Taiwan
First published on 25th June 2021
Abstract
Donor–donor′–acceptor molecules where the donor′ bridges the donor and acceptor have different possible interaction pathways for charge transfer. Here we study a series of donor–donor′–acceptor molecules, having the same acceptor and donor′ but different donors, and donor′–phenyl spacer–acceptor to change the spatial separation and overlap between potential through-space donor–acceptor charge transfer (CT) in competition with donor′–acceptor through-bond CT. We determine that the charge transfer driving force plays a large role in dictating which charge transfer channel is favoured. Strong donors and acceptors with large driving force favour through-space CT. We also find that solid state host packing plays an important role, with small molecule hosts that pack tightly, distorting the guest molecules, reducing D–A separation to stabilise the through-space CT state over the through-bond state. Only the through-space CT states give fast reverse intersystem crossing and efficient TADF. These results give the first insight into the photophysics of through-space CT compared to through-bond states on the same molecule.
Introduction
Organic molecules showing thermally-activated delayed fluorescence (TADF) are considered as the third generation of materials for organic light-emitting diodes (OLEDs).1 Unlike phosphorescent emitters, TADF molecules harvest triplet excitons by a reverse intersystem crossing (rISC) mechanism between the triplet (3CT) and the singlet (1CT) charge-transfer states, mediated by vibrionic coupling between 3CT and a local excited triplet state (3LE) that enables spin orbit coupling (SOC), when the energy gap between all three states becomes small, <100 meV. Mediation by the third state is required when 3CT and 1CT are so close in energy because direct spin orbit coupling (SOC) between them is forbidden because they have effectively the same orbital and no change in orbital angular momentum occurs during the direct transition 3CT → 1CT.2,3 One of the simplest ways to achieve extremely small electron correlation energy, i.e. singlet–triplet splitting energy (ΔEST), is the use of conformational twisting between directly bridged donor (D) and acceptor (A) units in order to minimize the overlap between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO).4,5 Beyond the subtle manipulations of the degree of intramolecular through-bond charge transfer to control TADF properties, recent interest in through-space charge transfer between D and A, mediated by strong D A intramolecular π–π dipolar interactions has been motivated by the potential of increasing photoluminescence quantum yield.6,7 Such intramolecular through-space charge transfer is much like an exciplex state except that it offers far more control over the spatial seperation and orientation of D and A compared to the random orientation of D and A in the intermolecular exciplex state. In a conjugated system, the D and A can be arranged co-facially, in close spatial proximity, using a non-coplanar molecular scaffold (bridge).8 This approach has also been used in the design of non-conjugated polymers with spatially separated pendant D and A.9,10 Despite the growing interest of intramolecular through-space charge transfer states as a means to TADF, very little photophysical study of the excited state dynamics and molecular conformations of such systems has been made.11 Here we investigate the excited state dynamics of new molecules configured with asymmetrical donor–donor′–acceptor (D–D′–A) architectures, where the co-facial overlap between D and A is controlled by the introduction of a common weak (rigid) carbazole donor bridge (D′) and by the introduction of phenyl spacer units between the acceptor and bridge, shown in Fig. 1. A common diphenyltriazine (dTRZ) acceptor is used and either a weak phenyl carbazole (PhCBZ) or strong triphenylamine (TPA) donor. | ||
| Fig. 1 The chemical structures (a) and X-ray structures (b) of Ph3TRZCzTPA, Ph2TRZCzTPA, Ph2TRZCzPhCz and Ph3TRZCzPhCz, thermal ellipsoids were drawn at a 50% probability level. | ||
Results
Synthetic methods and characterization of these new D–D′–A molecules and model compounds used in this study are reported in ESI† (Scheme S1–S3). Fig. 1b shows their single crystal structures and crystallographic data is summarized in Table S1 (ESI†). As shown, the pendant (spacer) aryl rings are highly twisted from the carbazole bridge (D′) due to congested steric interactions. All dihedral angles between the D, A and bridging D′, selected closest atom-to-atom distances between the donor and the acceptor branches (3.09–3.37 Å), and distances between the donor N-atom and the centre of triazine acceptor ring (4.93–5.30 Å) are summarized in Table 1.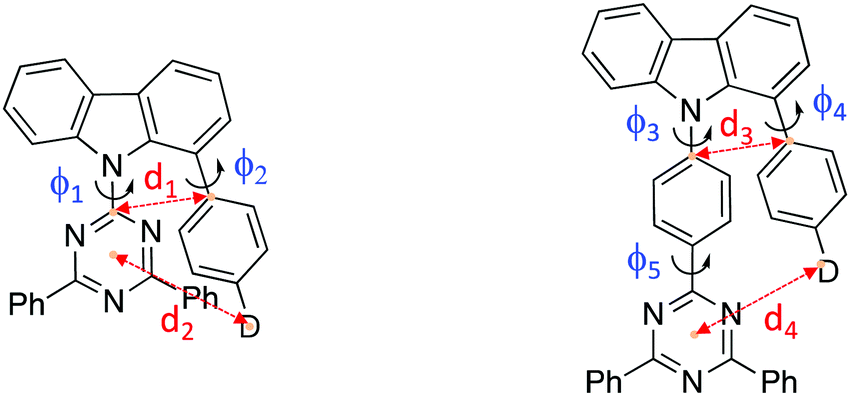
| φ 1 (°) | φ 2 (°) | φ 3 (°) | φ 4 (°) | φ 5 (°) | d 1 (Å) | d 2 (Å) | d 3 (Å) | d 4 (Å) | |
|---|---|---|---|---|---|---|---|---|---|
| Ph2TRZCzTPA | 49.3 | 55.2 | 3.093 | 4.985 | |||||
| Ph2TRZCzPhCz | 36.5 | 47.1 | 3.373 | 4.926 | |||||
| Ph3TRZCzTPA | 75.1 | 70.9 | 0.6 | 3.150 | 5.295 | ||||
| Ph3TRZCzPhCz | 65.4 | 57.8 | 2.5 | 3.201 | 4.924 |
In general, Ph3TRZCzTPA and Ph2TRZCzTPA (triphenylamine donor) show larger donor-bridge dihedral angles as compared to those of the counterpart Ph2TRZCzPhCz and Ph3TRZCzPhCz (PhCBZ donor). In addition, Ph3TRZCzTPA and Ph2TRZCzTPA show shorter closest atom-to-atom distances, but slightly longer distances between the donor N-atom and the centre of triazine ring. These distorted conformations and short distances imply that a through-space interaction between the donor and acceptor branches due to the stronger electron-donating ability of TPA may stabilize the twisted molecular structures. All of the molecules show excellent thermal stability required for stable film and device formation, and calculated HOMO/LUMO energy levels obtained from cyclic voltammetry (Fig. S1, ESI†) are given in Table 2. Ph3TRZCzTPA and Ph2TRZCzTPA show higher HOMO energy levels (−5.28/−5.27 V) as compared to Ph2TRZCzPhCz and Ph3TRZCzPhCz (−5.85/−5.57 V) in accord with donor strength. Since the acceptor of all compounds is the same, the LUMO energy levels are similar (ca. −2.75 V) in all cases.
E
oxionset
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) (V)
(V) |
E
red1/2
(V) |
HOMOc (eV) | LUMOc (eV) | E g (eV) | T d (°C) | T g (°C) | T m (°C) | |
|---|---|---|---|---|---|---|---|---|
| a Calculated from the onset potential. b E = (Ep,a + Ep,c)/2, where Ep,a and Ep,c stand for the peak potential at which anodic- and cathodic-direction scan, respectively. c HOMO and LUMO were determined from the electrochemical results in CH2Cl2 and DMF solution, respectively. d E g = LUMO–HOMO using electrochemical results. e T d was analyzed using TGA (5% weight loss). f T g and Tm were determined by DSC. g Not detected. | ||||||||
| Ph2TRZCzTPA | 0.98b | −1.45 | −5.28 | −2.73 | 2.55 | 324 | 111 | 212 |
| Ph3TRZCzTPA | 0.97b | −1.53 | −5.27 | −2.74 | 2.53 | 389 | 111 | 279 |
| Ph2TRZCzPhCz | 1.21 | −1.47 | −5.85 | −2.79 | 3.06 | 340 | 127 | n.d.g |
| Ph3TRZCzPhCz | 1.23 | −1.50 | −5.57 | −2.77 | 2.80 | 419 | 129 | 338 |
DFT calculations
The ground state structures were first optimized at a B3LYP/6-311G(d) level. The calculated HOMO and LUMO distributions as well as energy levels are shown in Fig. S2 (ESI†). The HOMOs of Ph3TRZCzTPA and Ph2TRZCzTPA are mainly located at the TPA moiety and slightly extend to the carbazole bridge. The HOMOs of Ph2TRZCzPhCz and Ph3TRZCzPhCz are mostly positioned at the pendant PhCBZ donor with limited contribution from the carbazole bridge. Not surprisingly, the LUMOs of Ph2TRZCzTPA and Ph2TRZCzPhCz are located at the dTRZ, which are well separated from their HOMOs. Whereas, the LUMOs of Ph3TRZCzTPA and Ph3TRZCzPhCz are located at the TRZ together with a small contribution on the carbazole bridge, giving weak but non zero HOMO–LUMO overlap. Obviously, the stronger donor, TPA, leads Ph3TRZCzTPA and Ph2TRZCzTPA to have higher HOMO levels as compared to those of Ph2TRZCzPhCz and Ph3TRZCzPhCz. Whereas Ph2TRZCzTPA and Ph2TRZCzPhCz without the aryl spacer between acceptor and bridge show slightly lower LUMOs. The calculated results are consistent with the observed CV data.TD-DFT calculations were then made using a hybrid meta-generalized gradient-approximation functional, m06-2x/6-311(d), to optimize the S1 and T1 structure of these four molecules and calculate the natural transition orbitals (NTOs). The HONTO and LUNTO distributions are shown in Fig. S3 (ESI†). For the S1 state of Ph3TRZCzTPA and Ph2TRZCzTPA, the HONTOs distribute on the TPA moiety, while the LUNTOs delocalises at the dTRZ and TRZ, respectively. Since there is nearly no overlap between the HONTO and LUNTO, a through-space charge transfer character for the S1 state is suggested. For the T1 state configuration, the HONTO of the Ph3TRZCzTPA is delocalised on the TPA and TRZ, exhibiting both CT and LE transition character. On the other hand, the HONTO of the Ph2TRZCzTPA only distributes on the dTRZ, indicating the 3LE state is the lowest triplet state. Notably, a distinguishable change between the S1 and T1 orbitals could be observed for Ph3TRZCzTPA and Ph2TRZCzTPA, which is a prerequisite for an allowed SOC spin–flip transition. Very interestingly, the different orbital distributions of S1 and T1 states could also be observed in the case of Ph2TRZCzPhCz. The HONTO of the S1 state distributes on the PhCBZ donor and the carbazole bridge, while the LUNTO locates on the dTRZ. On the other hand, the HONTO of the T1 state of Ph2TRZCzPhCz delocalises mainly on the dTRZ group and slightly on the phenylene of the pendant PhCBZ donor, whereas the LUNTO only locates on the dTRZ group. Thus, Ph2TRZCzPhCz is anticipated to possess both through-bond and through-space CT character due to the slight overlap of HONTO and LUNTO. For Ph3TRZCzPhCz, the HONTO of the S1 state distributes on the pendant PhCBZ, whereas the LUNTO distributes on the TRZ group, leading to a through-space CT transition character. The HONTO and LUNTO distributions of the T1 state are similarly delocalised on the PhCRZ and slightly coupled with the carbazole bridge, resulting in an apparent LE transition character. Although Ph3TRZCzPhCz shows considerable orbital deformation between S1 and T1 states, the larger ΔEST undermines the probability of reverse intersystem crossing process.
Steady state photophysics
Steady state absorption and emission of each compound was measured in methylcyclohexane (MCH) are shown in Fig. 2a and b, and in a range of different polarity solvents and solid state hosts in Fig. 3, for direct comparison.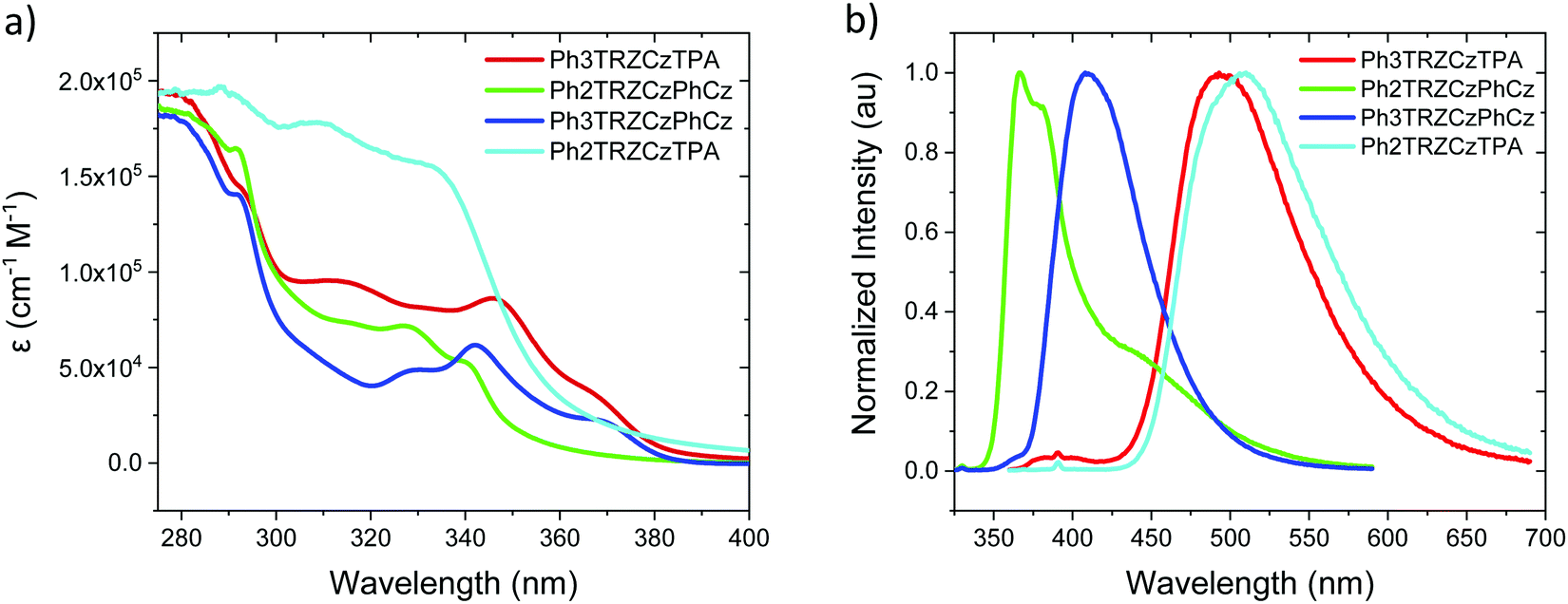 | ||
| Fig. 2 Extinction coefficients (a) and emission spectra (b) of the molecules in MCH solution (20 μM L−1). | ||
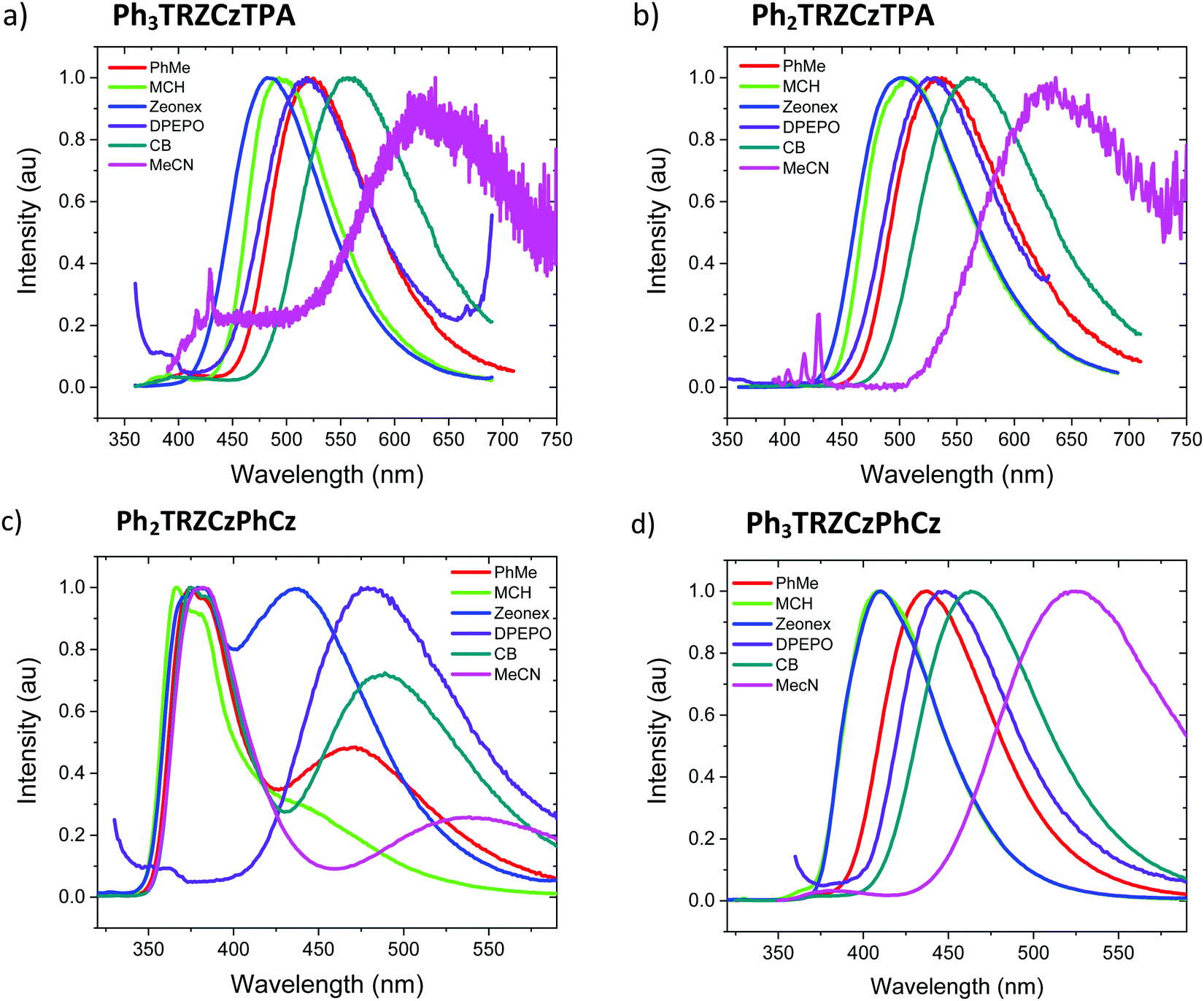 | ||
| Fig. 3 Emission spectra of the compounds (a) Ph3TRZCzTPA, (b) Ph2TRZCzTPA, (c) Ph2TRZCzPhCz and (d) Ph3TRZCzPhCz in film and different solvents, methylcyclohexane (MCH), chlorobenzene (CB) and acetonitryl (MeCN). | ||
The lowest energy absorption band, peaking at ca. 375 nm in Ph3TRZCzPhCz and Ph3TRZCzTPA is absent in Ph2TRZCzTPA and Ph2TRZCzPhCz. In Ph3TRZCzPhCz this band correlates very well to the lowest energy absorption band in the model compound, 9-(4-(4,6-diphenyl-1,3,5-triazin-2-yl)phenyl)-9N-carbazole, CzPhTrz, as observed by Sharma et al.,12 having strong ππ* character, and indicates a more delocalised acceptor unit, i.e. the phenyl spacer between A and bridge forms part of the acceptor unit itself in both cases, as suggested by our TD-DFT calculations. The magnitude of the extinction coefficients of these lowest energy bands is also >2 × 104 cm−1 M−1 indicating that they are not direct CT absorption bands as observed in typical D–A–D through bond TADF molecules.4
Emission spectra measured for each compound in MCH, Fig. 2b, show energy on-sets at; 2.89 eV, 2.79 eV, 3.59 eV and 3.32 eV for Ph3TRZCzTPA, Ph2TRZCzTPA, Ph2TRZCzPhCz and Ph3TRZCzPhCz, respectively. Ph2TRZCzPhCz gives dual emission in MCH from a local donor at 3.59 eV and a charge transfer (CT) state at lower energy, in excellent agreement with the TD-DFT calculations. Ph2TRZCzTPA emission shows some mixed local/CT character, indicated by the weak structure on the emission band. The stronger donor strength of Ph3TRZCzTPA and Ph2TRZCzTPA is clearly manifest by the large Stokes shift observed in their CT emission spectra. With increased solvent polarity, Fig. 3. Ph3TRZCzTPA, Ph2TRZCzTPA, and Ph3TRZCzPhCz all show strong solvatochromism, giving a positive Stokes shift indicative of CT emission. Emission of Ph3TRZCzTPA and Ph2TRZCzTPA in MeCN is highly red shifted and extremely weak indicating very low emission yields.
In general, the molecules with a Cz donor emit at higher energy than those having a TPA donor, in line with donor strength. This also dictates the driving force for charge transfer in each pair. The CT driving force may be estimated from the following equation;
| ΔG0 = EAA − EAD − ΔEb |
The solution measurements for Ph3TRZCzTPA and Ph2TRZCzTPA are similar as can be seen in Fig. 3a and b, the bands shift by the same amount in different polarity solvents. Comparing the film spectra, we see that the zeonex films behave just like the MCH solutions for these materials with Ph3TRZCzTPA even showing a small blue shift compare to MCH. Ph2TRZCzPhCz also shows the same 1LE and 1CT dual emission bands, but the CT state has increased relative intensity in zeonex. However, in DPEPO films, no 1LE component is observed from Ph2TRZCzPhCz, only the 1CT emission is observed, at about the same peak wavelength as the corresponding Ph2TRZCzPhCz CT emission in toluene solution.
Ph3TRZCzPhCz, Fig. 3d, is seen to have a very similar spectrum with weakly structured emission in MCH compared to the model systems CzPhTrz12 and meta-CzTRZ reported by Liu et al.13 Thus, we ascribe its excited state as a through-bond CT state between the bridge Cz and TRZ D′–A pair. The CT state shows both weak solvatochromism and in solid state the emission has very similar band shape and width as in solution, mostly unaffected by host packing effects. Only in highly polar MeCN does the band become highly red shifted and very broad, which may point to a different excited state origin such as a through-space CT state.
Ph2TRZCzPhCz on the other hand is obviously different, being the only compound showing dual CT emission and very strong local emission, consistent with the X-ray data that shows a shallow 36.5° dihedral angle between D′–A. Cz donor units tend to have shallow ground state D-A dihedral angles giving strong LE character.14 This is further emphasised by the localisation of the HOMO on the pendant Cz unit found in our DFT calculations. We therefore synthesised the model compound, 2-(carbazol-9-yl)-4,6-diphenyl-1,3,5-triazine (CzTrz), Fig. S4 (ESI†). The absorption spectrum of CzTrz indicates electronically decoupled D and A units, absorbing above 350 nm. Excitation into the A (TRZ) band yields both TRZ15 and weak CT emission, whereas excitation into the D (Cz) band yields strong CT emission, onset at 3.26 eV. The CT emission on-set shifts to 3.1 eV in toluene and dominates the emission spectra. This shift is 35 nm more to the red than for the model CzPhTrz in toluene, which we ascribe to a smaller electron hole separation in CzTrz therefore larger Coulomb attraction energy, which is relaxed more strongly by the increasing solvent polarity red shifting the energy of the CT state to a greater extent.16 This clearly corresponds with the CT state seen in Ph2TRZCzPhCz which can then be identified as a through-bond CT state between the bridge Cz (D′) and dTRZ unit. The higher intensity, narrow linewidth blue emission band with vibronic structure, also observed in Ph2TRZCzPhCz, onset ca. 3.54 eV, showing little or no solvatochromic shift, must be emission from a local excited state. Considering the individual D and A units in the molecule, PhTRZ is only very weakly emissive through excitation at 280 nm, emission onset at 3.44 eV, with slight vibronic structure.15 Emission in our model CzTrz peaking at ca. 290 nm seems highly plausible to be from the dTRZ unit. This is not what we observe in Ph2TRZCzPhCz, however, carbazole shows strong emission, onset at 3.54 eV17 and so we conclude that this local emission band in Ph2TRZCzPhCz comes from the pendent Cz D unit, consistent with the DFT calculations. This Cz must be strongly decoupled from the rest of the molecule as the emission is always present even in high polarity solvent, ruling out a through-space CT state. In highly polar MeCN, there is a small population of CT states in giving rise to the highly red shifted band which we believe to be through-space CT states, Fig. 3c.
With this in mind, we turn to Ph2TRZCzPhCz in zeonex (a branched polyolefin) and DPEPO (bis[2-(diphenylphosphino)phenyl]ether oxide) host films. In both cases the CT band is much stronger than in solution. In zeonex dual emission is still observed but the CT emission has equal intensity to the local emission, whereas in tightly packed DPEPO host, only a strong, red shifted and very broad CT emission is observed. As there can be no ‘solid state solvatochromic effect’ in a rigid solid host,18,19 this must be due to a packing effect of the host. If the host forces the D and A units closer together, improving co-facial overlap, a through-space CT state can be stabilized and the increased charge transfer rate would effectively quench donor emission from the pendent Cz unit. Thus, in DPEPO we conclude that a through-space CT state is stabilized on Ph2TRZCzPhCz, in-line with TD-DFT calculations that strongly suggest both through-space and through-bond CT states are possible. We also note that this CT emission band in DPEPO is much broader and more red shifted than the CT emission observed from Ph3TRZCzPhCz in DPEPO. This we believe indicates a very inhomogeneous process stabalising the through-space CT state, clearly in-line with random host packing effects. The stable through-bond CT state in Ph3TRZCzPhCz remains narrow in DPEPO, which potentially indicates that the extra flexibility introduced by the spacer phenyl ring enables closer co-facial separation of D and A stabalising the through-space CT state so that host packing has little extra effect on this conformation. To confirm this hypothesis we measured Ph2TRZCzPhCz in both mCP (1,3-bis(N-carbazolyl)benzene) and CBP (4,4′-bis(N-carbazolyl)-1,1′-biphenyl) hosts, both having very low dielectric and polarizability values compared in DPEPO. In both cases clear CT emission is observed from Ph2TRZCzPhCz, Fig. S5 (ESI†), confirming that host packing plays a major role in stabilising through-space CT in these materials.
To understand Ph3TRZCzTPA and Ph2TRZCzTPA we recall an important observation from our previous work. A CT complex can form between TRZ and TPA in solution.11 The resulting TRZ:TPA excited state emits at 492 nm (peak) in toluene (with solvatochromism being observed, Fig. S6, ESI†). This is at the same energy with the same solvatochromic behaviour as the band observed in both Ph3TRZCzTPA and Ph2TRZCzTPA. We therefore identify the CT emission in both as coming from a through-space TRZ⋯TPA (strong D – strong A) CT excited state. No contribution from a through-bond, strong A – weak D′ CT pair is observed. Concomitantly, emission is seen well to the red of either Ph2TRZCzPhCz and Ph3TRZCzPhCz CT emission in line with a strong-D strong-A pair CT state. We note from X-ray measurements that this behaviour does not correlated simply to D–A co-facial separation, indicating the role of CT driving force. We also previously found that the intermolecular TRZ⋯TPA state has a ground state interaction, i.e. a heterodimer complex,11 and we postulate that such a ground state interaction between D and A units may also help to drive and stabilise the through-space CT state over a through-bond interaction. Such ground state absorption is also seen in other intramolecular through-space CT systems.20
Time-resolved spectroscopy
Starting with the compounds in zeonex host matrix. Ph3TRZCzTPA at room temperature, Fig. 4a, shows initial CT like emission (Gaussian band shape) in the first few ns, onset at 3.01 eV which relaxes in energy over 100 ns to give strong prompt (PF) CT emission at 2.88 eV and delayed fluorescence (DF) at 2.87 eV (on-sets). The DF then undergoes a monotonic blue shift of some 50 meV over the first 40 μs, Fig. S7 (ESI†). Phosphorescence, onset at 2.90 eV is seen at millisecond times. Fitting the emission decay indicates a fast 11 ns and a slow 77 ns prompt CT decays along with a 1 μs and 13 μs DF components. All parameters are given in Table 3. Here we note that the DF decays are highly non exponential at longer times, turning into a power law decay. This is very typical of TADF materials in the solid state. Thus, here we fit the early time DF with one or two exponential terms to model the majority of the DF emission and calculate rISC rates from a kinetic model as detailed previously.21 The possible interpretation of the power law DF region will be discussed later.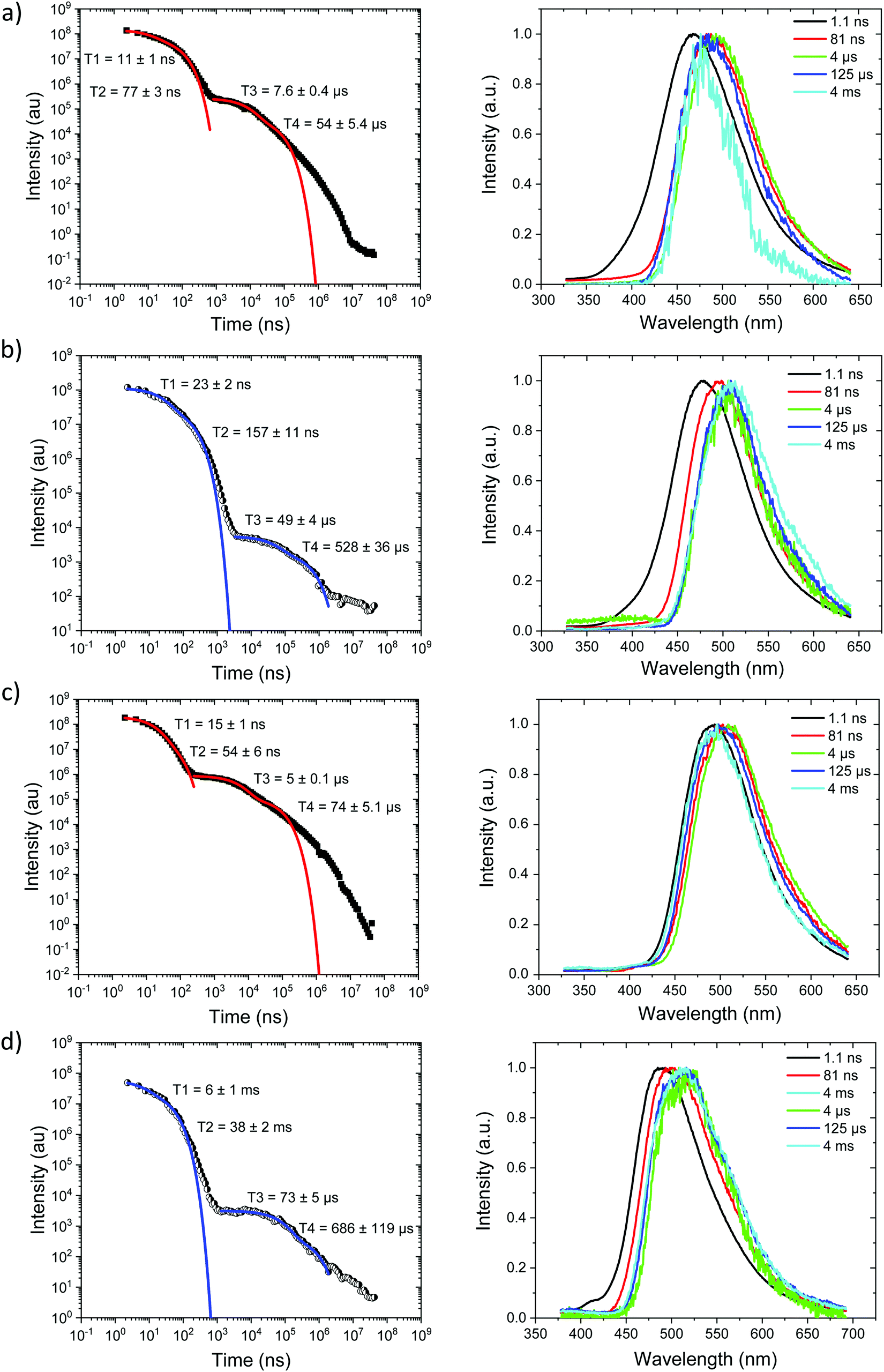 | ||
| Fig. 4 Time-resolved spectrally integrated intensity decay (left) and representative time resolved spectra (right) of Ph3TRZCzTPA (a) 300 K and (b) 80 K; Ph2TRZCzTPA (c) 300 K and, (d) 80 K in zeonex matrix. Spectral times given in ns, and decay fitting data included in the decay plots. | ||
| Molecule | 1LEa (eV) | τ LE (ns) | 1CTa (eV) | τ PF1 (ns) | τ PF2 (ns) | τ DF1 (μs) | τ DF2 (μs) | 1CT′a (eV) | 3LEc (eV) | ΔESTd (meV) |
|---|---|---|---|---|---|---|---|---|---|---|
| a Local (LE) and charge transfer (CT) emission energies calculated from the on-set of the emission band. b Prompt (τLE and τPF) and delayed fluorescence (τDF) component lifetimes calculated from fits to the measured intensity decay curves. c Local triplet energy taken from the on-set of the phosphorescence measured at 80 K. d ΔEST is the energy difference between the 1CT and 3LE energies. | ||||||||||
| Ph3TRZCzTPA | 3.01 | ∼1 | 2.88 | 11 | 77 | 7.6 | 54 | — | 2.90 | 20 |
| Ph2TRZCzTPA | — | — | 2.82 | 15 | 54 | 5 | 74 | — | 2.80 | 20 |
| Ph2TRZCzPhCz | 3.54 | 3 | 3.10 | 3 | 9 | 342 | 4900 | 2.92 | 2.76 | 340 |
| Ph3TRZCzPhCz | — | — | 3.35 | 9 | — | — | — | — | 2.88 | 470 |
At 80 K, Fig. 4b and Fig. S8 (ESI†), we see similar behaviour but the ns region prompt emission energy relaxation is slower. However, the emission band has a constant additional 60 meV red shift and exactly matches the band shape and energy position (in time) of the emission observed in Ph2TRZCzTPA (both at RT and 80 K, Fig. 4c and d). The large decrease in DF intensity shows clear thermal activation of the DF mechanism. Phosphorescence at 80 K, Fig. 4b, also has a much lower onset of 2.76 eV. This large difference in CT state energy between RT and 80 K implies that thermal motion on average leads to a higher energy CT state in Ph3TRZCzTPA. As this behaviour is not observed in Ph2TRZCzTPA we conclude that it must be an effect due to the presence of the spacer phenyl ring between A and bridge. One explanation for this is that the thermal energy drives the spacer phenyl ring more planar (conjugated). So at 80 K the phenyl ring has a more orthogonal configuration with respect to the A and bridge, changing the coupling of the phenyl ring to the dTRZ and localising the LUNTO on the dTRZ unit analogous with Ph2TRZCzTPA as shown in our TDDFT calculations. This will also greatly affect the localisation of the lowest energy triplet state as well.
Ph2TRZCzTPA, Fig. 4c and d, shows constant 1CT emission behaviour, onset at 2.82 eV both at RT and 80 K with a monotonic blue shift of 50 meV over the first 40 μs, Fig. S7 and S8 (ESI†). Phosphorescence, onset at 2.80 eV is seen at millisecond times. At RT, we see faster prompt decay, earlier time DF and faster DF decay compared to Ph3TRZCzTPA. As the ΔEST are less than 30 meV (at RT) in both cases, this could indicate stronger magnetic coupling between D and A in Ph2TRZCzTPA, so more efficient SOC, given by the different spatial overlap and orientation of D and A units in the two compounds, again an effect that can be related to the spacer phenyl unit. As seen in the DFT calculations the D–A separation in Ph2TRZCzTPA (4.985 Å) is smaller compared to Ph3TRZCzTPA (5.295 Å) fully supporting this argument. Previously, Wada et al.20 have proposed that the D A co-facial angle is critically in determining the SOC and rISC rate in a similar through-space CT system.
Ph2TRZCzPhCz at RT in zeonex, Fig. 5a, has a PF energy of 3.54 eV that decays within a few nanoseconds as expected for D emission. A simultaneous CT emission band (on our 1 ns timescale), onset at 3.10 eV, is also observed that decays very rapidly, within 100 ns, shown in detail in Fig. S18 (ESI†). At 100 ns we also see a weak transient CT emission band, onset at 2.95 eV, which also decays rapidly. This is similar to what we observe in DPEPO (see later) and may indicate a small population of weakly stabilised through-space CT states in Ph2TRZCzPhCz, as predicted by our calculations, and that are stabilised in high polarity MeCN solution, Fig. 3c. This band is better resolved at 80 K, Fig. 5b, with all prompt emission decayed by 200 ns. From 200 ns to 10 μs we observe no emission (above the noise floor of the iCCD), Fig. S7 (ESI†). At millisecond times, we observe an emission with a new band shape and onset energy of 2.92 eV, which we assume to be phosphorescence. At 80 K the prompt spectra show minor shifts in energy of ca. 10 meV only. However, a much larger shift is seen in the phosphorescence which red shifts to an onset of 2.76 eV at 80 K. This is a large change in triplet energy, and looking critically at the band shape it is different. Careful inspection of the 40 ms emission at room temperature (compared to that at 4 ms) shows a shoulder has grown in on the red side of the band which we believe is a contribution from the low energy triplet species observed at 80 K. We have observed such dual phosphorescence before in D–A–D systems and this implies two coupled local triplet states in thermal equilibrium co-existing on the molecule.22 As the lowest energy triplet state is at 2.76 eV, this yields a true ΔEST of 340 meV and so it is not surprising that we observe little or no delayed emission.
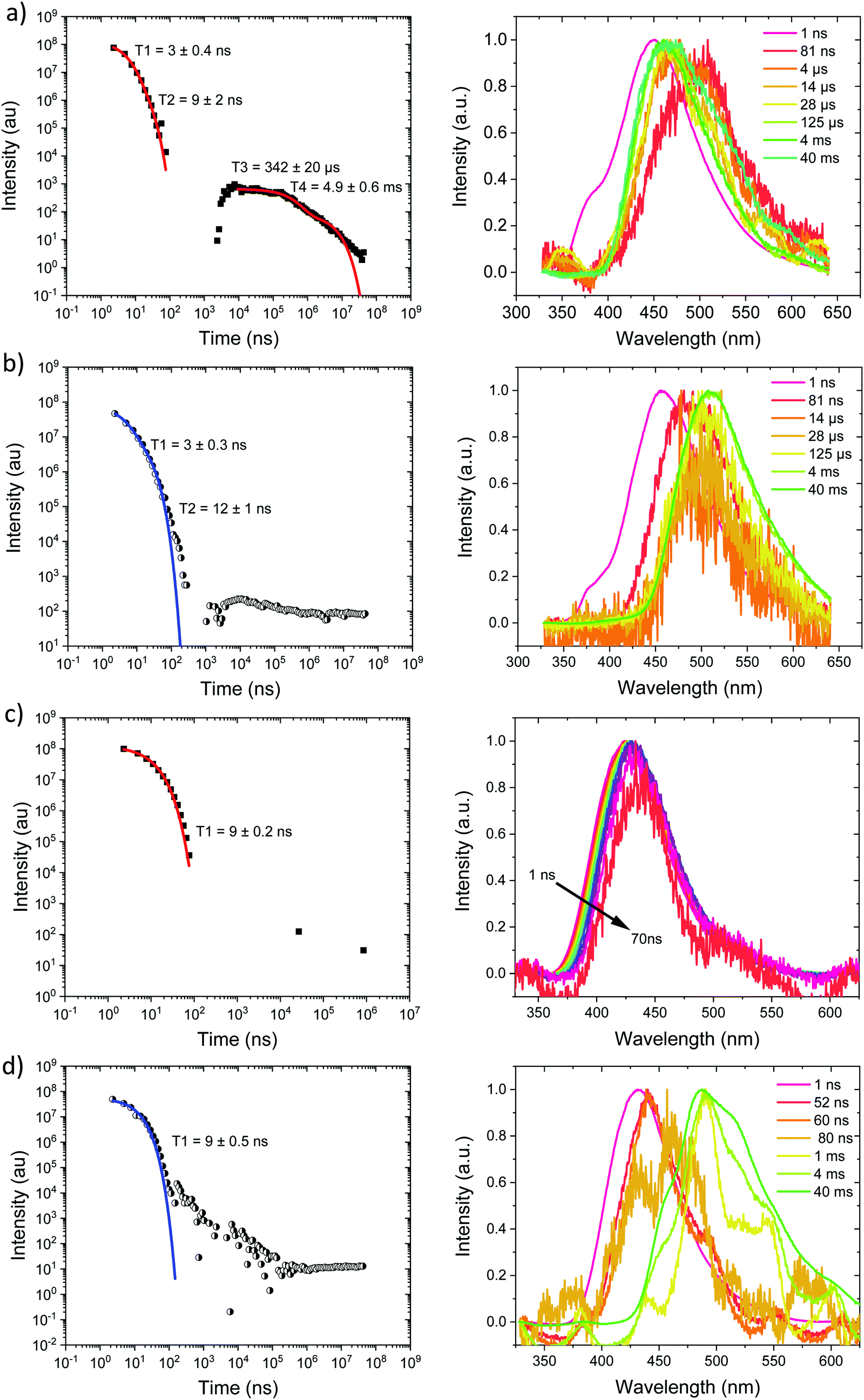 | ||
| Fig. 5 Time-resolved spectrally integrated intensity decay (left) and representative time resolved spectra (right) of (a) Ph2TRZCzPhCz, (a) 300 K and (b) 80 K; Ph3TRZCzPhCz, (c) 300 K and, (d) 80 K in zeonex matrix. Spectral times given in ns, and decay fitting data included in the decay plots. | ||
Ph3TRZCzPhCz at RT and 80 K, Fig. 5c and d, behaves like a simple fluorescence emitter, having only a single emission band, onset 3.35 eV with mono exponential lifetime of 9 ns. Over its lifetime, the blue edge of this band decays faster than the red edge, a typical red edge effect. No DF is observed from this band at either temperature. At 80 K phosphorescence is observed in the 1–10 ms range, having an extremely well resolved spectrum, onset at 2.88 eV which is a clear match to a carbazole phosphorescence spectrum.23 Further, by 40 ms the band shape has evolved, losing much of its structure, onset at 2.91 eV. The spectral band shape can be deconvolved into a linear combination of the carbazole phosphorescence and the phosphorescence band we observe in Ph2TRZCzPhCz, Fig. S9 (ESI†), indicating dual phosphorescence in Ph3TRZCzPhCz as well. This yields a ΔEST of ca. 470 meV.
Measurements were also made at 10% by weight loading in DPEPO host, which has higher dielectric coefficient and polarizability than zeonex, also the DPEPO matrix greatly restricts the movement of the molecules because of tighter packing than the zeonex polymer matrix. Concomitantly, all CT emission is seen to be red shifted. In the case of Ph3TRZCzTPA the prompt emission at RT has onset at 3.0 eV, Fig. 6a. By 80 ns the emission has relaxed to 2.85 eV and from 500 ns until 4 μs we see DF at 2.89 eV, Fig. S10 (ESI†). After 12 μs until 4 ms weak emission is observed with onset at 2.85 eV. At 80 K, Fig. 6b, bi-exponential prompt emission, with lifetimes of 7 ns and 169 ns is observed, onset at 3.0 eV with a very weak blue knee ascribed to rapid D emission along with the longer lived prompt CT emission. The prompt and delayed CT emission behave similarly to room temperature but the DF intensity is lower, again showing thermal activation, Fig. S11 (ESI†). From 120 μs to 40 ms emission is seen with slight structure and onset at 2.75 eV consistent with phosphorescence from the lowest triplet state. Ph2TRZCzTPA at RT shows dual PF, onset at 3.26 eV with lifetimes of 8.3 ns and 48 ns, Fig. 6c. From 80 ns until 4 μs DF is observed at 2.64 eV, Fig. S10 (ESI†). From 12 μs until 4 ms very weak emission onset ca. 2.79 eV is seen, ascribed to phosphorescence. At 80 K, Fig. 6d, the PF is again bi-exponential and the same delayed emission behaviour is observed, Fig. S11 (ESI†). At 40 ms we again see phosphorescence at about 2.73 eV but the signal is very weak here. In both Ph3TRZCzTPA and Ph2TRZCzTPA the 3LE energy from low temperature zeonex measurements at 2.76 and 2.80 eV respectively, is very consistent with the phosphorescence seen in DPEPO. This would then set ΔEST at around 150–200 meV in DPEPO, consistent with the observed thermally activated delayed emission.24 The calculated rISC rates of Ph3TRZCzTPA and Ph2TRZCzTPA in DPEPO films where calculated using a kinetic model. At RT, we estimated rates of 6.3 × 104 s−1 and 1 × 105 s−1 respectively, and at 80 K, 1.4 × 104 s−1 and 1 × 104 s−1 respectively, Fig. S12–S15 (ESI†).
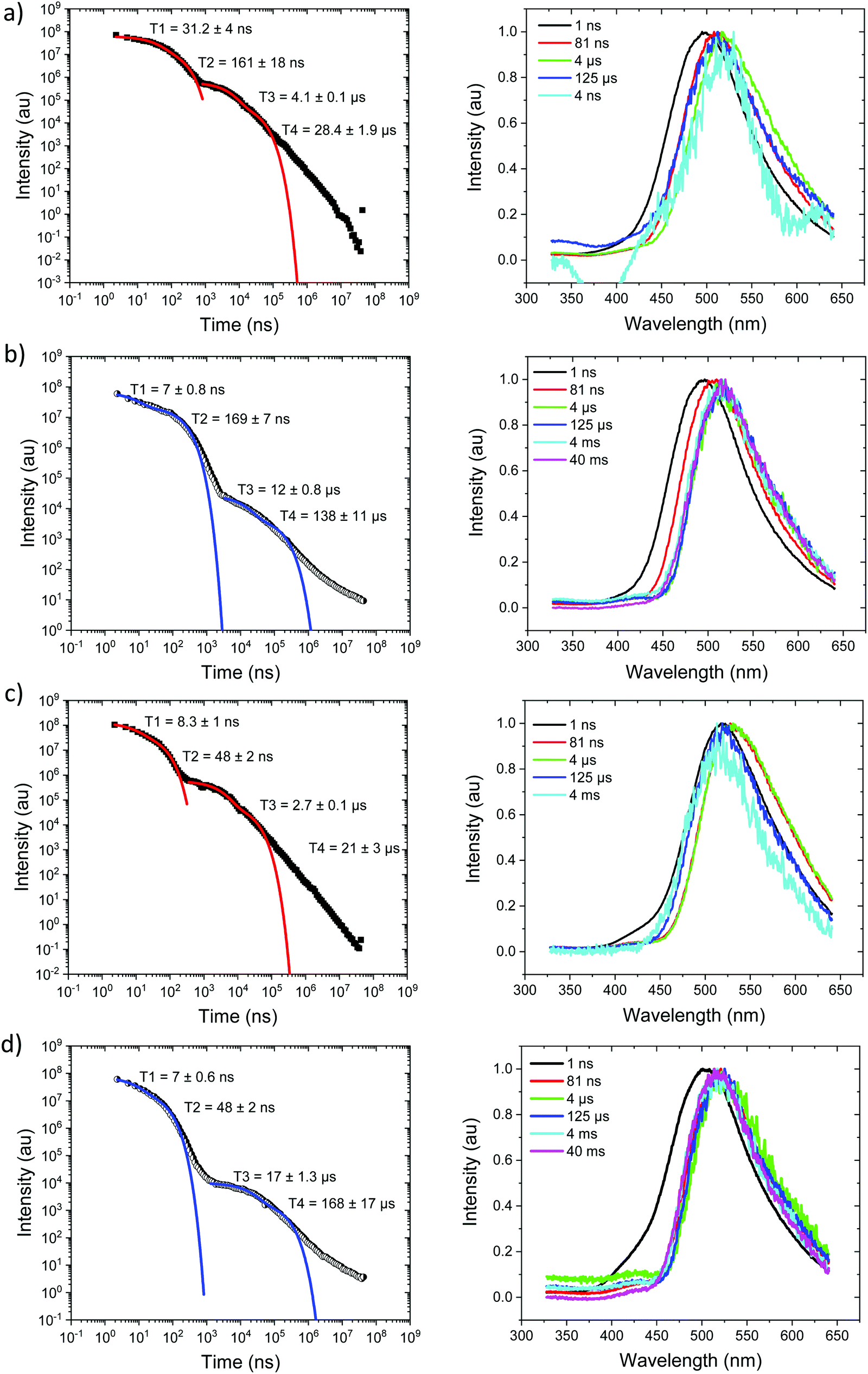 | ||
| Fig. 6 Time-resolved spectrally integrated intensity decay (left) and representative time resolved spectra (right) of Ph3TRZCzTPA (a) 300 K and (b) 80 K; Ph2TRZCzTPA (c) 300 K and (d) 80 K in DPEPO host. Spectral times given in ns, and decay fitting data included in the decay plots. | ||
For Ph2TRZCzPhCz in DPEPO at RT, Fig. 7a, emission is observed at 3.1 eV (onset) having a bi-exponential decay of 5.8 ns and 32 ns. From 500 ns to 35 μs very weak emission is seen with an onset of 2.88 eV, Fig. S10 (ESI†), and at 4 ms there may be phosphorescence with onset at 2.92 eV but the signal is very weak. At 80 K, Fig. 7b, again we observe prompt emission at 3.1 eV and then very weak emission from 500 ns to 35 μs at 2.88 eV, Fig. S11 (ESI†). At 40 ms emission is still seen at 2.83 eV which we ascribe to phosphorescence. The 3.1 eV CT band is red shifted by over 300 meV compared with that in zeonex and so we believe this shows that in DPEPO the through-space CT state is stabilised by the packing effect of the DPEPO forcing the D and A units closer together. In order to confirm this hypothesis, we measured Ph2TRZCzPhCz decays in both mCP and CBP hosts as well. In both cases, clear 3.1 eV CT emission is observed from Ph2TRZCzPhCz with long lifetime confirming DF, Fig. S5 (ESI†). These observations confirm that host packing plays a major role in the underlying physics of the CT states in Ph2TRZCzPhCz. All delayed emission in Ph2TRZCzPhCz is rather weak, and if we assume the same triplet energy as found in zeonex then ΔEST will be of order 250 meV implying inefficient rISC in line with the very long lived weak DF. The CT band is also broaden which we assume implies a high degree of disorder within the host DPEPO, again consistent with an inhomogeneous, packing induced through-space CT state. Ph3TRZCzPhCz also shows similar behaviour to Ph2TRZCzPhCz, Fig. 7c and d. At RT we observe bi-exponential decay with lifetimes of 6 ns and 20 ns, emission onset at 3.1 eV. From 100 ns to 4 μs this emission shifts to 2.91 eV, Fig. S10 (ESI†). Again the CT band is red shifted compared to zeonex pointing to DPEPO stabilising the through-space CT excited state and giving weak DF. At 80 K, prompt emission starts at 3.26 eV and shifts to 3.1 eV by 80 ns. The decay is bi-exponential, 6.3 ns and 31 ns, in line with a D emission as well as prompt through-space CT emission. There is little DF at 80 K, Fig. S11 (ESI†), in line with inefficient TADF, but at 40 ms clear phosphorescence emission with some structure is seen, onset at 2.95 eV in line with that observed in zeonex. All lifetimes are summarised in Table 4.
 | ||
| Fig. 7 Time-resolved spectrally integrated intensity decay (left) and representative time resolved spectra (right) of Ph2TRZCzPhCz, (a) 300 K and (b) 80 K; Ph3TRZCzPhCz, (c) 300 K and (d) 80 K in DPEPO host. Spectral times given in ns, and decay fitting data included in the decay plots. | ||
| Molecule | 1LEa (eV) | τ LE (ns) | 1CTa (eV) | τ PF1 (ns) | τ PF2 (ns) | τ DF1 (μs) | τ DF2 (μs) | 3LEc (eV) | ΔESTd (meV) |
|---|---|---|---|---|---|---|---|---|---|
| a Local (LE) and charge transfer (CT) emission energies calculated from the on-set of the emission band. b Prompt (τPF) and delayed fluorescence (τDF) component lifetimes calculated from fits to the measured intensity decay curves. c Local triplet energy taken from the on-set of the phosphorescence measured at 80 K. d ΔEST is the energy difference between the 1CT and 3LE energies. | |||||||||
| Ph3TRZCzTPA | 3.00 | ∼1 | 2.85 | 31 | 161 | 4 | 28 | 2.76 | 90 |
| Ph2TRZCzTPA | 2.95 | ∼1 | 2.67 | 8.3 | 48 | 2.7 | 21 | 2.80 | −130 |
| Ph2TRZCzPhCz | 3.10 | ∼1 | 2.88 | 5.8 | 32 | 2 | 59 | 2.76 | ∼250 |
| Ph3TRZCzPhCz | 3.10 | ∼1 | 2.91 | 6 | 20 | 4 | 51 | 2.95 | −40 |
Device results
Ph3TRZCzTPA and Ph2TRZCzTPA were selected as the emitters to investigate electroluminescent (EL) characteristics. Four commonly used high-triplet energy hosts including; 4,4′-bis(N-carbazolyl)-1,1′-biphenyl (CBP) (ET = 2.56 eV), 2,6-bis(3-(9H-carbazol-9-yl)phenyl)pyridine (26DCzPPy) (ET = 2.71 eV), 1,3-bis(carbazol-9-yl)benzene (mCP) (ET = 2.9 eV), and 9-(4-tert-butylphenyl)-3,6-bis(triphenylsilyl)-9H-carbazole (CzSi) (ET = 3.0 eV) were tried. To match the HOMO and the LUMO levels of the emitting layer, di-[4-(N,N-ditolyl-amino)-phenyl]cyclohexane (TAPC) and 1,3,5-tri[(3-pyridyl)-phen-3-yl]benzene (TmPyPB) were selected as the hole and electron transport layers respectively, see Fig. 8. Devices using mCP as the host gave the best performance in line with the photophysics, including relatively low operation voltages, higher maximum luminance, and improved efficiency. For doping concentration (2–20 wt%) of Ph2TRZCzTPA in the mCP further fine-tuning gave optimal performance, Fig. S16 (ESI†) depicts the EL characteristics and parameters are summarized in Table S4 (ESI†). From the EL spectra, no emission from mCP and/or carrier transport materials were observed when the doping concentration was greater than or equal to 2 wt%, implying effective charge transfer and exciton confinement on Ph2TRZCzTPA. The EL spectra of the devices were red-shifted from 505 nm to 529 nm as the doping concentration increased from 2 wt% to 20 wt%. The current density–voltage (J–V) curves shown in Fig. S16(b) (ESI†) indicate that current density increases with doping concentration whilst the turn-on voltages (at 1 cd m−2) decrease from 3.3 V to 2.6 V, in line with Ph2TRZCzTPA being a bipolar charge transporting material. Fig. S16(d) (ESI†) depicts the external quantum efficiency (EQE) as a function of Ph2TRZCzTPA concentration, with peak EQE of 16.3%, 49.7 cd A−1, and 59.3 lm W−1 found at 12 wt% Ph2TRZCzTPA. | ||
| Fig. 8 (a) Structural drawings of the materials used in OLEDs; (b) schematic structures of the fabricated OLEDs with different emitters. | ||
26DCzPPy was found to be the best host for Ph3TRZCzTPA. In addition, MoO3-doped TAPC was adopted as a hole injection layer to decrease the energy barrier between the ITO anode and the organic layer, Fig. 8. Fig. S17 (ESI†) depicts the EL characteristics while the corresponding parameters are summarized in Table S5 (ESI†). A weak emission from 26DCzPPy was observed in the EL spectra at doping concentrations of Ph3TRZCzTPA below 2 wt%, indicating the incomplete host–guest energy transfer. As the doping concentration increased from 2 wt% to 24 wt%, the devices showed the EL spectra red-shifted from 518 nm to 530 nm. The current density of the devices increases with the doping concentration, indicating the favourable carrier transport capability of Ph3TRZCzTPA. Given the inferior carrier mobility of 26DCzPPy; 2 wt% Ph3TRZCzTPA the turn-on voltage was 5.0 V but continuously decreased to 4.4 V with a higher doping concentration, to 24 wt%. The optimized concentration was found at 20 wt% Ph3TRZCzTPA, giving maximum EQE of 13.3%, 40.3 cd A−1, and 28.6 lm W−1. The corresponding EL characteristics of the optimized devices with Ph3TRZCzTPA and Ph2TRZCzTPA (i.e. devices A and B) are shown in Fig. 9 and the data are summarized in Table 5. Overall, the results demonstrate that both Ph3TRZCzTPA and Ph2TRZCzTPA give efficient TADF devices from through-space CT excited states.
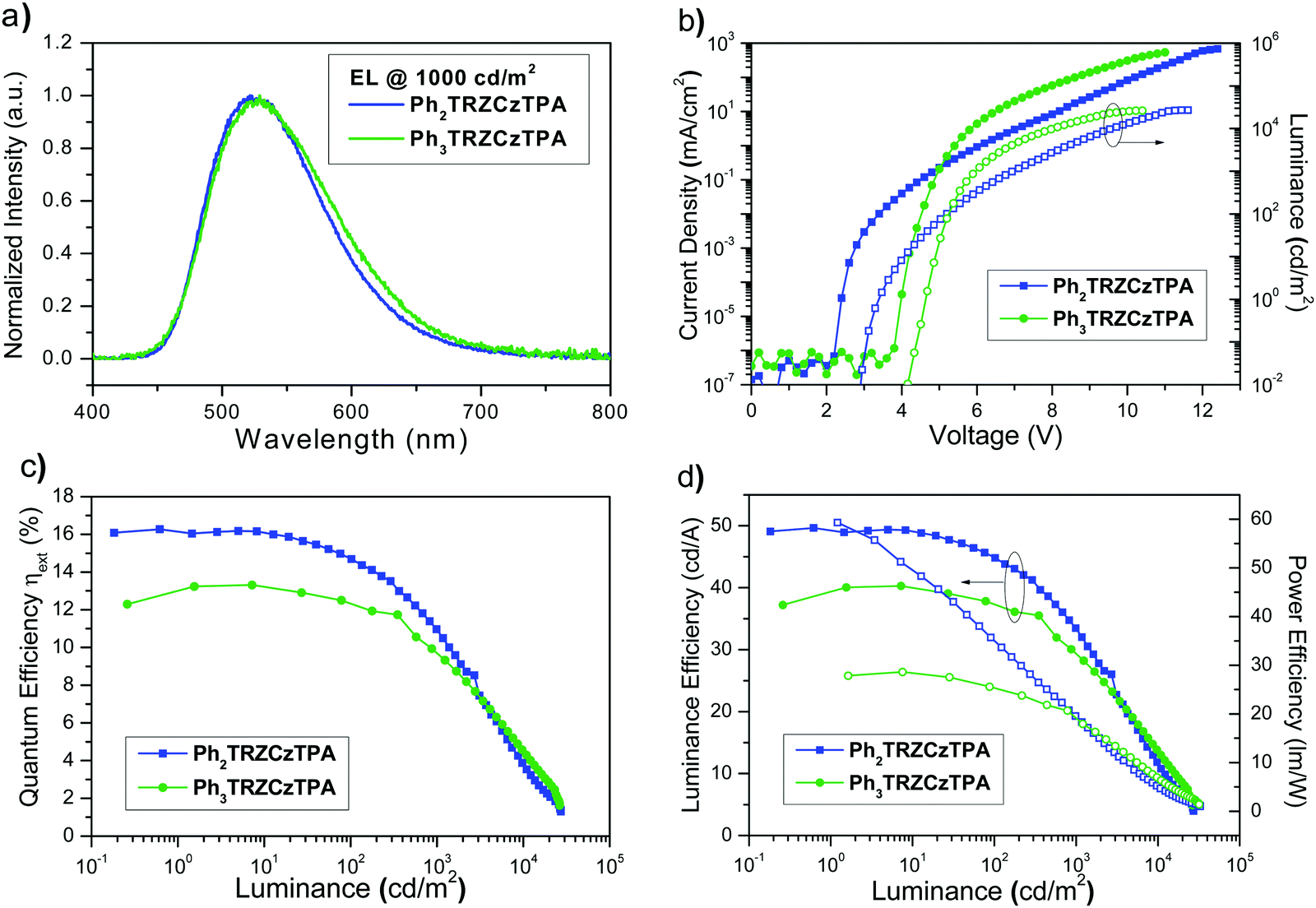 | ||
| Fig. 9 (a) Normalized EL spectra at a luminance of 103 cd m−2; (b) current density–luminance–voltage (J–V–L) characteristics; (c) external quantum efficiency versus luminance, (d) luminance/power efficiency versus luminance for devices A and B. | ||
| Device | Host | Emitter | V on [V] | L max [cd m−2] | EQE [%] | CE [cd A−1] | PE [lm W−1] | λ Peak [nm] | CIEd [x, y] |
|---|---|---|---|---|---|---|---|---|---|
| a Turn-on voltage measured at 1 cd m−2. b Maximum efficiency. c Measured at 102 cd m−2. d Measured at 103 cd m−2. | |||||||||
| A | CBP | Ph2TRZCzTPA (12 wt%) | 2.9 | 27158 (12.4 V) |
16.3,b 14.7,c 10.9d | 49.7,b 44.9,c 33.4d | 59.3,b 28.2,c 15.0d | 522 | 0.32, 0.55 |
| B | 26DCzPPy | Ph3TRZCzTPA (20 wt%) | 4.4 | 26434 (11.0 V) |
13.3,b 12.4,c 9.7d | 40.3,b 37.5,c 29.4d | 28.6,b 23.3,c 15.8d | 529 | 0.34, 0.54 |
Discussion
We find Ph2TRZCzPhCz and Ph3TRZCzPhCz to have different photophysics compared to Ph3TRZCzTPA and Ph2TRZCzTPA, primarily because of their relative weak donor strength and smaller driving force for CT. A through-bond CT state is observed to be favoured (by comparison to model systems) and in Ph2TRZCzPhCz stabilization of any CT state is hard to achieve so that D emission is always observed. In zeonex, the lowest energy triplet state is always found with onset at 2.76 eV, ascribed to the local triplet state of the pendent Cz donor, giving large 1CT–3LE energy gaps, >0.35 eV, thus we observe little or no TADF from the through-bond CT states. In Ph3TRZCzPhCz highly resolved Cz donor phosphorescence is observed indicative of a large local triplet population, remaining uncoupled from the rest of the molecule. In DPEPO films we observe a physical effect on the photophysics arising from the small molecule host packing, whereas in zeonex, which is an open polymer network having high free volume, the guest molecules behave much like in MCH solution, i.e. they are not constrained by the host. In DPEPO, Ph2TRZCzPhCz and Ph3TRZCzPhCz show no D emission but instead a highly red shifted, stable CT band. We propose that the packing forces of the DPEPO host distorts the guest molecules, forcing the D and A closer together, stabilizing a through-space CT excited state. This was confirmed measuring Ph2TRZCzPhCz in both CBP and mCP hosts which also gave the same strong CT stabalisation as DPEPO does with long lived CT delayed emission. This DF, although weak, is seen to be temperature dependent in both compounds, i.e. TADF. In the case of the through-space CT state, with D and A still have a larger co-facial separation than an A–D′ through-bond CT state (see X-ray data) and so the Coulomb attraction energy between separated electron and hole is reduced and thus the CT energy is red shifted. The 1CT through-space state being red shifted compared to the through-bond state, reduces the 1CT–3LE energy gap to ca. 120 meV and we thus observe DF in both cases.In all cases where we observe a measurable DF signal, we observe a strongly non-exponential decay at long times. We believe this comes from the inhomogeneous environment of the doped films, with the hosts imposing various degrees of restriction and packing effects on the TADF molecules. The through-space CT state systems seem to be particularly prevalent to this which is not surprising, showing a potentially high sensitivity to exact spatial separation and orientation of the D and A in a host environment. Recently, Suresh et al.25 have discussed such a long time non-exponential DF tail in terms of combined TADF and TTA emission, however here we have only used 1% loading of the TADF molecules in zeonex and 10% in DPEPO which probably rules out TTA. However in devices were much higher loadings are used a TTA contribution along with TADF cannot be ruled out.
Ph3TRZCzTPA and Ph2TRZCzTPA, by comparison to the model TRZ⋯TPA excited state CT complex (exciplex), are seen to have stable through-space CT excited states. The stabalising of the through space CT state over a A–D′ through bond CT state is ascribed to the larger CT driving force arising from their strong D, strong A, structure. From the time resolved decays and spectra, we do though see that in Ph3TRZCzTPA, an initial higher energy transient CT state which we tentatively ascribe to a short lived through-bond CT state (by comparison to model systems), potentially stabalised by the effect of the extra phenyl ring between acceptor and bridge. Relaxation of this phenyl ring configuration could then stabilise the through space CT state. Alternatively the through bond state may occur on a small population of Ph3TRZCzTPA molecules trapped in a non relaxed geometry in the film. This behaviour is observed in both zeonex and DPEPO which we believe favours the former explanation. Certainly we see no such behaviour in Ph2TRZCzTPA which lacks the spacing phenyl unit. Ph3TRZCzTPA and Ph2TRZCzTPA show intense temperature dependent delayed emission, clearly TADF. The measurable differences between rISC rates of the two molecules we believe arises from differences in relative co-facial separation and orientation of D and A due to the phenyl spacer between A and D′ (bridge) in Ph3TRZCzTPA, as seen in the X-ray data. This controls their magnetic coupling, i.e. spin orbit coupling matrix elements, that dictate rISC rate, given they have very similar, small singlet triplet energy gaps. By comparing the photophysics of these two systems, we see that strong acceptor, strong donor pairs favour through-space charge transfer whereas strong acceptor weak donor favours through-bond charge transfer, in line with both the driving force for charge transfer and the spatial separation and orientation of the non-conjugated D A pair. This clearly sets some basic design criteria for TADF intramolecular through-space charge transfer systems. The differences between the two molecules are also reflected in the device performance. We find that Ph2TRZCzTPA gives better device performance than Ph3TRZCzTPA, very much in line with the difference in rISC rates and the lack of possible residual through-bond CT states in Ph3TRZCzTPA. From these device results we again see sensitivity to D A orientation, flexibility of linkage to the bridge and also host packing effects. Much like exciplex emitters, these effects need to be carefully managed and controlled.26
In Ph2TRZCzPhCz and Ph3TRZCzPhCz, the lowest local triplet state of the molecular resides on the pendant Cz unit which quenches higher energy D′ (bridge) triplets leading to negligible mediation of rISC for the through-bond CT state. All of our observations clearly align very well with ideas recently put forward by Wada et al. from their through-space charge transfer TADF molecule.20 We also see that the effect of host packing has profound effects on the through-space CT states and these effects cannot be ignored, especially in devices.
Conclusions
From this new series of D–D′–A materials we have gained a great deal of new understanding about through-space CT states, competition with through-bond CT formation and the role a host plays in controlling this equilibrium. In the two materials having strong D and strong A, Ph3TRZCzTPA and Ph2TRZCzTPA, we unambiguously identify stable through-space CT states via comparison to model compounds and solution state D A complexes. This stabilisation we ascribe to the large driving force for charge transfer (ca. 1 eV) and subsequent larger charge separation distance. For the weak D strong A pair, Ph2TRZCzPhCz and Ph3TRZCzPhCz, a through-bond CT state between the A and the bridge D′ is stabilised as the CT driving force is much less, ca. 0.5 eV. Little DF is observed unless an external perturbation further stabilises the CT state, i.e. solvent polarity or host packing forces. Indeed, in DPEPO, CBP and mCP with large packing forces we observe highly red shifted CT formation and subsequent TADF, which we ascribe to stabilization of a through-space CT state in Ph2TRZCzPhCz and Ph3TRZCzPhCz. This shows that such external forces can distort the molecule, forcing the D and A branches closer together to fully stabilise the through-space CT state over the through-bond state. Very satisfyingly, our TDDFT calculations identified the possibility of Ph3TRZCzPhCz having both through-space and through-bond CT excited states. In Ph3TRZCzTPA having an additional ‘spacer’ phenyl group between the bridge D′ and A we observe a large temperature dependent red shift in the energies of both the 1CT and 3LE excited states. We ascribe this to thermal motion of this phenyl ring which reduces planarity between the A and the bridge, decoupling the phenyl ring from the dTRZ A moiety, making the Ph3TRZCzTPA act in a very similar fashion to Ph2TRZCzTPA. This decrease in conjugation also localises the 3LE state on the dTRZ unit, increasing the phosphorescence energy as we observe. In view of these observations, having such a spacing phenyl unit that can rotate between the D and A units, might not be the optimum design strategy for TADF materials, as we find that Ph2TRZCzTPA gives better performing devices than Ph3TRZCzTPA. We also find that Ph2TRZCzTPA has faster rISC rates than Ph3TRZCzTPA which we believe indicates more optimal co-facial orientation (not separation) of D and A forming the through-space CT state, which is critical in controlling SOC properties and thus rISC, as proposed by Wada et al.27 These differences in photophysics are also reflected in device performance with Ph2TRZCzTPA giving better performance in optimised devices. This first in-depth study of the physical properties and correlated device performance of through-space compared to through-bond CT states and TADF, gives many new pointers and ideas to design more efficient and stable TADF emitters.Methods
Full details of the synthesis and physical characterisation of the materials reported here, along with photophysical measurements and data analysis are given in the ESI† file published alongside this paper.Author contributions
Hector Miranda-Salinas performed the majority of the optical and photoluminescence measurements and contributed to the analysis and helped in the preparation of the manuscript. Yi-Tzu Hung synthesized and characterize the physical property of titled compounds. Yi-Sheng Chen made the theoretical analysis of the titled compounds. Dian Luo and Hao-Che Kao fabricate the OLED device and collect the device data. Chih-Hao Chang (analyse the device data and helped to prepare the manuscript. Ken-Tsung Wong designed the project and molecules, and helped in the preparation of the manuscript. APM conceived and the model for the analysis of the photophysical results, lead the analysis of the photophysics and helped write the manuscript.Data
All data resulting from this work is given in the paper and ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
HMS acknowledges the Mexican National Council for Science and Technology, CONACYT for his studentship (2019-000021-01EXTF-00308). APM acknowledges the EPSRC for funding under grant number EP/P012167/1. KTW acknowledges funding from the Ministry of Science and Technology, Taiwan (MOST 107-2113-M-002-019-MY3).References
- P. L. dos Santos, J. S. Ward, P. Data, A. Batsanov, M. R. Bryce, F. Dias and A. P. Monkman, J. Mater. Chem. C, 2016, 4, 3815–3824 RSC.
- M. K. Etherington, J. Gibson, H. F. Higginbotham, T. J. Penfold and A. P. Monkman, Nat. Commun., 2016, 7, 13680 CrossRef CAS.
- J. Gibson and T. J. Penfold, Phys. Chem. Chem. Phys., 2017, 19, 8428–8434 RSC.
- F. B. Dias, T. J. Penfold and A. P. Monkman, Methods Appl. Fluoresc., 2017, 5, 012001 CrossRef PubMed.
- X.-K. Chen, D. Kim and J.-L. Brédas, Acc. Chem. Res., 2018, 51, 2215–2224 CrossRef CAS PubMed.
- M.-Y. Zhang, Z.-Y. Li, B. Lu, Y. Wang, Y.-D. Ma and C.-H. Zhao, Org. Lett., 2018, 20, 6868–6871 CrossRef CAS PubMed.
- K. Kawasumi, T. Wu, T. Zhu, H. S. Chae, T. Van Voorhis, M. A. Baldo and T. M. Swager, J. Am. Chem. Soc., 2015, 137, 11908–11911 CrossRef CAS PubMed.
- H. Tsujimoto, D. G. Ha, G. Markopoulos, H. S. Chae, M. A. Baldo and T. M. Swager, J. Am. Chem. Soc., 2017, 139, 4894–4900 CrossRef CAS PubMed.
- Z. Ren, R. S. Nobuyasu, F. B. Dias, A. P. Monkman, S. Yan and M. R. Bryce, Macromolecules, 2016, 49, 5452–5460 CrossRef CAS.
- S. Y. Shao, J. Hu, X. D. Wang, L. X. Wang, X. B. Jing and F. S. Wang, J. Am. Chem. Soc., 2017, 139, 17739–17742 CrossRef CAS PubMed.
- K.-L. L. Woon, C. L. Yi, K. C. Pan, M. K. Etherington, C. C. Wu, K. T. Wong and A. P. Monkman, J. Phys. Chem. C, 2019, 123, 12400–12410 CrossRef CAS PubMed.
- N. Sharma, E. Spuling, C. M. Mattern, W. Li, O. Fuhr, Y. Tsuchiya, C. Adachi, S. Bräse, I. D. W. Samuel and E. Zysman-Colman, Chem. Sci., 2019, 10, 6689–6696 RSC.
- D. Liu, D. Li, H. Meng, Y. Wang and L. Wu, J. Mater. Chem. C, 2019, 7, 12470–12481 RSC.
- M. Aydemir, S. Xu, C. Chen, M. R. Bryce, Z. Chi and A. P. Monkman, J. Phys. Chem. C, 2017, 121, 17764–17772 CrossRef CAS.
- H. Tanaka, K. Shizu, H. Nakanotani and C. Adachi, J. Phys. Chem. C, 2014, 118, 15985–15994 CrossRef CAS.
- M. Colella, A. Danos and A. P. Monkman, J. Phys. Chem. Lett., 2019, 10, 793–798 CrossRef CAS PubMed.
- E. Stanislovaityte, J. Simokaitiene, S. Raisys, H. Al-Attar, J. V. Grazulevicius, A. P. Monkman and V. Jankus, J. Mater. Chem. C, 2013, 1, 8209 RSC.
- P. L. dos Santos, M. K. Etherington and A. P. Monkman, J. Mater. Chem. C, 2018, 6, 4842–4853 RSC.
- T. Northey, J. Stacey and T. J. Penfold, J. Mater. Chem. C, 2017, 5, 11001 RSC.
- Y. Wada, H. Nakagawa, S. Matsumoto, Y. Wakisaka and H. Kaji, Nat. Photonics, 2020, 14, 643–649 CrossRef CAS.
- N. Haase, A. Danos, C. Pflumm, A. Morherr, P. Stachelek, A. Mekic, W. Brütting and A. P. Monkman, J. Phys. Chem. C, 2018, 122, 29173–29179 CrossRef CAS.
- R. S. Nobuyasu, Z. Ren, G. C. Griffiths, A. S. Batsanov, P. Data, S. Yan, A. P. Monkman, M. R. Bryce and F. B. Dias, Adv. Opt. Mater., 2016, 4, 597–607 CrossRef CAS.
- J. Pina, J. S. de Melo, H. D. Burrows, A. P. Monkman and S. Navaratnam, Chem. Phys. Lett., 2004, 400, 441–445 CrossRef CAS.
- V. Jankus, C. J. Chiang, F. Dias and A. P. Monkman, Adv. Mater., 2013, 25, 1455–1459 CrossRef CAS PubMed.
- S. M. Suresh, E. Duda, D. Hall, Z. Yao, S. Bagnich, A. M. Z. Slawin, H. Bässler, D. Beljonne, M. Buck, Y. Olivier, A. Köhler and E. Zysman-Colman, J. Am. Chem. Soc., 2020, 142, 6588–6599 CrossRef CAS PubMed.
- M. Colella, A. Danos and A. P. Monkman, J. Phys. Chem. C, 2019, 123, 17318–17324 CrossRef CAS.
- Y. Wada, H. Nakagawa, S. Matsumoto, Y. Wakisaka and H. Kaji, ChemRix, 2019, 1–21 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1499013 and 2043941–2043943. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1tc02316k |
| This journal is © The Royal Society of Chemistry 2021 |