
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSemiflexible polymer scaffolds: an overview of conjugation strategies
Lotte
Gerrits
a,
Roel
Hammink
 *bc and
Paul H. J.
Kouwer
*a
*bc and
Paul H. J.
Kouwer
*a
aInstitute for Molecules and Materials, Radboud University, Heyendaalseweg 135, 6525 AJ Nijmegen, The Netherlands. E-mail: p.kouwer@science.ru.nl
bDepartment of Tumor Immunology, Radboud Institute for Molecular Life Sciences, Radboud University Medical Center, Geert Grooteplein 26, 6525 GA Nijmegen, The Netherlands. E-mail: roel.hammink@radboudumc.nl
cDivision of Immunotherapy, Oncode Institute, Radboud University Medical Center, Nijmegen, The Netherlands
First published on 24th February 2021
Abstract
Semiflexible polymers play an important role in nature and possess remarkable properties that render them efficient scaffolds. While flexible polymers coil up, semiflexible polymers are rigid enough to retain a straight conformation and thus facilitate access to substituents on the polymer chain. This review describes how semiflexible polymers function as scaffolds and which chemical strategies are used to conjugate substituents to this unique class of polymers. In general, biopolymeric scaffolds are functionalized through post-functionalization strategies, which generates versatility but offers low spatial control. Synthetic polymeric scaffolds can be designed and functionalized from scratch, which provides a larger freedom in design and conjugation strategies. Altogether, this review summarizes the most important methods that are available to the chemist, which have been developed to generate truly functional semiflexible scaffolds.
 From left to right: Roel Hammink, Lotte Gerrits and Paul Kouwer | Roel Hammink is a postdoctoral researcher in the Tumor Immunology group of Prof. Carl. G. Figdor. He received a Ph.D. in polymer chemistry from the Radboud University Nijmegen in 2016. His current research focusses on the development of artificial antigen presenting cells, using polymers as a scaffold for the delivery of immunolatory signals. Lotte Gerrits obtained her MSc degree in Chemistry from the Radboud University in Nijmegen in 2018. She is currently pursuing her PhD under supervision of Dr Paul H. J. Kouwer and Dr Roel Hammink in the Molecular Materials group at the same university. Her research interest is focused on developing functionalized polymers for immunotherapies. Paul Kouwer received his PhD degree from the Delft University of Technology. He currently holds an Associate Professor position in Molecular Materials at Radboud University in Nijmegen, the Netherlands, where he develops highly biomimetic polymer materials and hydrogels for diverse biomedical applications. The three authors collaborate in an interdisciplinary project to develop new (semiflexible) scaffold materials that are functionalized for immunological application and that, ultimately, can be used for in vivo immunotherapies. |
Introduction
In the last few decades, semiflexible polymers have spiked the interest of many researchers. The main rationale behind the growing interest in this class of polymers is that semiflexible polymers and their striking properties play an important role in living systems. In fact, many (structural) biopolymers, such as collagen, DNA, and the cytoskeletal components are considered semiflexible (Box 1).1–3 Due to their reduced flexibility, these polymers exhibit unique properties such as the ability to recover shape after withstanding external stress. These properties allow the cytoskeleton, a dynamic three-dimensional network that fills the cytoplasm, to give cells their overall shape and endure external stress even at relatively low polymer concentrations.3,4The properties of semiflexible polymers are not only useful in nature. The unique conformation of these polymers enables them to serve as a scaffold. Because semiflexible polymers do not collapse into a random coil, they can be used as a relatively rigid scaffold that can be decorated with all sorts of functional substituents, which will be much better accessible than on flexible polymers (Fig. 1A). In addition, semiflexible polymers are typically long, one-dimensional structures, which allows for the introduction of multiple of the same or different substituents where one can benefit optimally from, for instance, multivalent interactions, another design principle frequently used in nature.
Box 1 Unique properties of semiflexible polymersTo understand why semiflexible polymers behave differently than flexible polymers, it is important to define what semiflexibility is. In general, polymers are defined by two lengths: the total contour length (L) of the polymer, and the persistence length (lp). The contour length is the length of the fully stretched polymer chain. The persistence length quantifies the polymer stiffness (or flexibility) and is defined as the length where the correlation of angles of the tangent vectors is lost.5,6A polymer is semiflexible when its total contour length and its persistence length are of the same order of magnitude.3 Polymers (or objects) with L ≪ lp act like rigid rods, while polymer with L ≫ lp are fully flexible. For semiflexible polymers, thermal fluctuations contribute to their conformation; their behavior is well-described by the Wormlike chain model.7 Some typical persistence lengths of natural and synthetic polymers are given in Table 1, although care should be taken that direct comparison is difficult because of different measurement techniques and conditions. Table 1 Persistence lengths of some common polymersa
The difference in conformations between flexible and semi-flexible polymers is important in the context of scaffolds. In flexible polymers, thermal fluctuations dominate and the chains coil up to maximize its entropy. Functional substituents will be poorly accessible. Semiflexible polymers are stiff enough that a much more straight conformation is favored and the substituents on the polymer will be readily accessible. | ||||||||||||||||||||
 | ||
| Fig. 1 A. Schematic representation of the difference between flexible (left) and semiflexible (right) polymer scaffolds. The more stretched chain conformation of the latter provides a much better access to substituents grafted to the polymer. B. Schematic overview of the different strategies for the introduction of substituents on semiflexible polymer scaffolds as discussed in this review. | ||
In this work, we review how researchers have used semiflexible polymers as functional scaffolds and which strategies they followed to introduce (functional) substituents, ranging from small molecules to biomolecules as large as antibodies (Fig. 1B). In the first part of the review, we focus on biopolymers that form a primary source of semiflexible scaffolds and a design inspiration for synthetic equivalents. Functionalization of biopolymers primarily follows post-modification approaches, unless the biopolymers can be custom-designed, such as DNA. In the second part of the review, we discuss progress in synthetic semiflexible scaffolds. For synthetic polymers, the freedom to design is larger, which allows for different conjugation strategies.
Biopolymers
Semiflexible biopolymers are suitable scaffolds due to their abundance in nature and unique properties. We divided biopolymers in three main classes: polypeptides, polynucleotides and polysaccharides, based on the monomer units and the resulting polymer structure (Fig. 2). Generally, it is difficult to modify the monomers of a biopolymer individually. As a result, the introduction of substituents on a biopolymer scaffold is commonly achieved via post-modification reaction on the polymer chain, using accessible functional groups, such as amines and thiols on polypeptides, phosphates on polynucleotides and carboxylic acids and hydroxyls on polysaccharides. The main drawback of this approach is that the selective introduction of substituents is not possible which results in a loss of spatial control and thus gives rise to a less well-defined scaffold.8 Here, we will describe and assess the functionalization techniques that are used to introduce substituents on polypeptide, polynucleotide and polysaccharide scaffolds.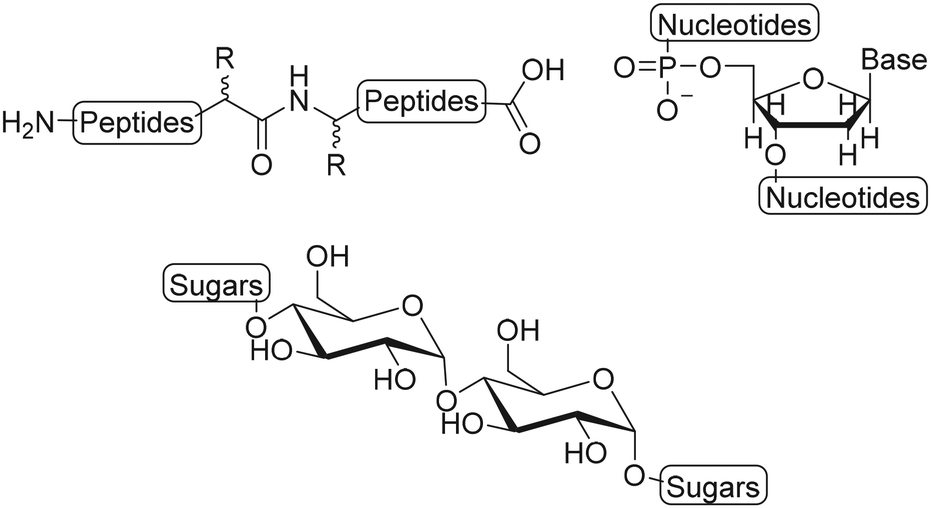 | ||
| Fig. 2 Schematic structures of biopolymers: polypeptides, polynucleotides and polysaccharides. | ||
Polypeptides
Peptide-based biopolymers gained interest as a class of biomaterials due to their unique biological, chemical and physical properties.9 They are easily degradable in the body, which makes them desirable candidates for drug delivery and tissue engineering applications.10 Furthermore, the ability of polypeptides to self-assemble into highly ordered structures provides new opportunities to develop functional biomaterials.9 Polypeptides consist of amino acid residues that are linked by peptide bonds. Natural occurring polypeptides such as collagen and fibrin consist of multiple polypeptide chains that are aggregated to form stable protein complexes.11,12 Residual functional groups of the amino acid buildings blocks provide active sites for the introduction of substituents on the polypeptide chains. In the next paragraphs we will discuss the landmark semiflexible polypeptides collagen and fibrin.Collagen
Collagen is a cross-linked fibrous protein that consists of a right handed bundle of three polypeptide chains which are aligned in a parallel fashion and coiled into a left-handed polyproline II-type helix.11 Each polypeptide chain contains around one thousand amino acid residues of which every third residue is glycine (Gly).11 The repetitive presence of Gly ensures tight packing of the three polypeptide chains into the triple helix.13 Collagen is widely distributed in connective tissues and can be formed into highly organized scaffolds which are biocompatible, -degradable and non-toxic upon exogenous application.13 Its fibers form the mechanical and structural scaffold of bone, skin, blood vessel walls and other connective tissues and is the most commonly used type of collagen in medicine.13,14 As a consequence, the material is extensively used as a scaffold for tissue engineering and wound healing applications.13,15 Various active molecules such as growth factors and antibiotics have been conjugated to collagen scaffolds.16,17Boyce and coworkers biotinylated collagen to conjugate growth factors such as epidermal growth factor (EGF) and heparin binding growth factor 2 (HBGF2) to modulate wound healing.16,18 Both growth factors were attached to collagen using a bridged avidin and biotin procedure. Biotin was covalently bound to bovine skin collagen, EGF and HBGF2 by reaction of biotinyl-N-hydroxysuccinimide (NHS) with the amine-groups of lysine residues on the protein (Fig. 3A). Biotinylated EGF and HBGF2 were subsequently attached to collagen through the pendant avidin (Fig. 3B). The authors showed both growth factors retained mitogenic activity for cultured human epidermal keratinocytes after biotinylation, thereby showing that their biotinylated collagen scaffold is suitable for wound healing applications.16
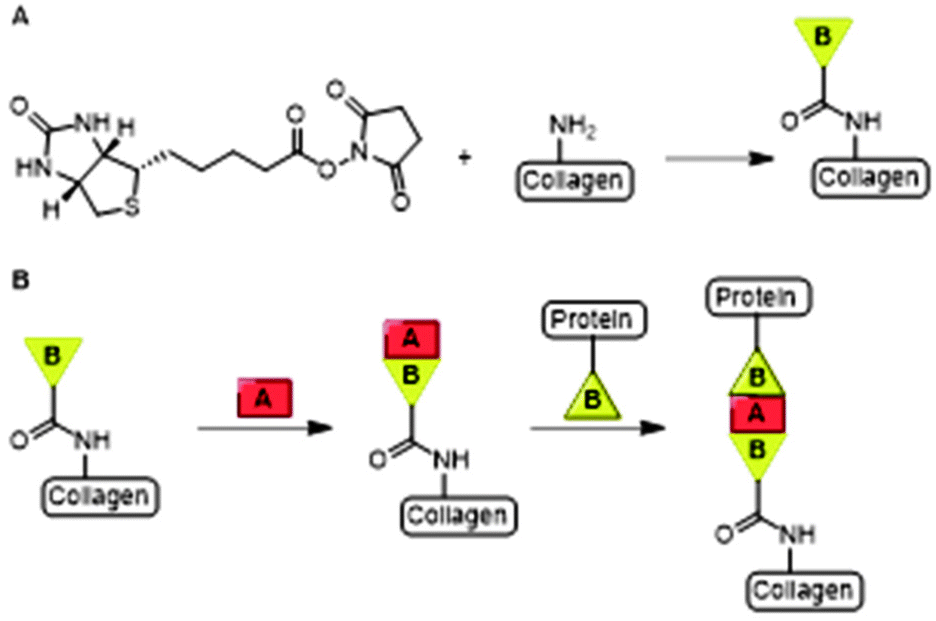 | ||
| Fig. 3 A. The bridged avidin and biotin procedure for conjugation of EGF and HBGF2 to collagen. NHS-functionalized biotin was conjugated to collagen, EGF and HBGF2 via amide bond formation. Reprinted from ref. 16 with permission from Elsevier, copyright (1989). B. Avidin (A) was used a bridge between two biotinylated (B) compounds, the scaffold and biotinylated growth factors EGF or HBGF2. Reprinted from ref. 18 with permission from John Wiley and Sons, Copyright (1969). | ||
With a similar goal, Myung and coworkers covalently conjugated EGF to a collagen scaffold. They used strain-promoted azide–alkyne cycloaddition (SPAAC) click chemistry to develop a material for epithelial wound healing applications.19 EGF was modified with an azide handle via reaction with azide-OEG5-sulfo-NHS (OEG = oligo(ethylene glycol)) ester and subsequently reacted with dibenzocyclooctyne (DBCO)-modified collagen to obtain the EGF functionalized collagen scaffold (Fig. 4A and C). The DBCO handles were introduced on collagen via reaction of the lysine residues through (sulfo)-NHS chemistry (Fig. 4B). The SPAAC anchoring method proved non-toxic and biocompatible, highlighting its broader applicability towards biomaterials. The resulting EGF-functionalized collagen scaffold showed a significant increase in adhesion and proliferation of epithelial cells compared to the soluble EGF control, which underlines the advantage of presenting substituents on a scaffold. The group of Myung also applied the SPAAC reaction to crosslink collagen fibers to develop a hydrogel with optimal mechanical properties to promote corneal keratocyte growth for corneal stromal tissue engineering applications.20
 | ||
| Fig. 4 Covalent conjugation of EGF to collagen via SPAAC ‘click’ chemistry. A. Introduction of azide handle to EGF via sulfo-NHS chemistry. B. Functionalization of collagen with DBCO via reaction via the lysine residues. C. Schematic overview of EGF coupling of collagen scaffold via SPAAC click chemistry. Reprinted from ref. 19 with permission from American Chemical Society, copyright (2017). | ||
Since collagen constitutes the majority of the extracellular matrix, scaffolds of collagen are highly attractive as a three-dimensional (3D) cell culture matrix for promotion of cell growth and differentiation, either for tissue engineering ex vivo or as an implant. Li and coworkers developed a collagen scaffold with Cetuximab to steer differentiation of neural progenitor cells (NPCs) for repair of spinal cord injuries.21 Cetuximab was covalently linked to the collagen scaffold via the use of a hetero-bi-functional crosslinker (Fig. 5). To introduce a thiol group, collagen was reacted with Traut's reagent, which conveniently reacts efficiently with primary amines at pH 7 to 9. A free amine on Cetuximab was conjugated to the commercially available sulfo-succinimidyl-4-(N-maleimidomethyl)cyclohexane-1-carboxylate (Sulfo-SMCC) crosslinker. A subsequent reaction of the thiolated collagen scaffold with the maleimide group of Sulfo-SMCC gave the Cetuximab-decorated collagen. An enzyme-linked immunosorbent assay (ELISA) showed that more Cetuximab was retained on the collagen scaffold through covalent linking compared with physical absorption, demonstrating the advantage of chemical modification of a scaffold. NPCs cultured on Cetuximab functionalized collagen scaffolds demonstrated neuronal differentiation in vitro while rats subjected to a spinal cord hemisection followed by treatment with Cetuximab functionalized collagen showed successful formation of neurons in vivo.
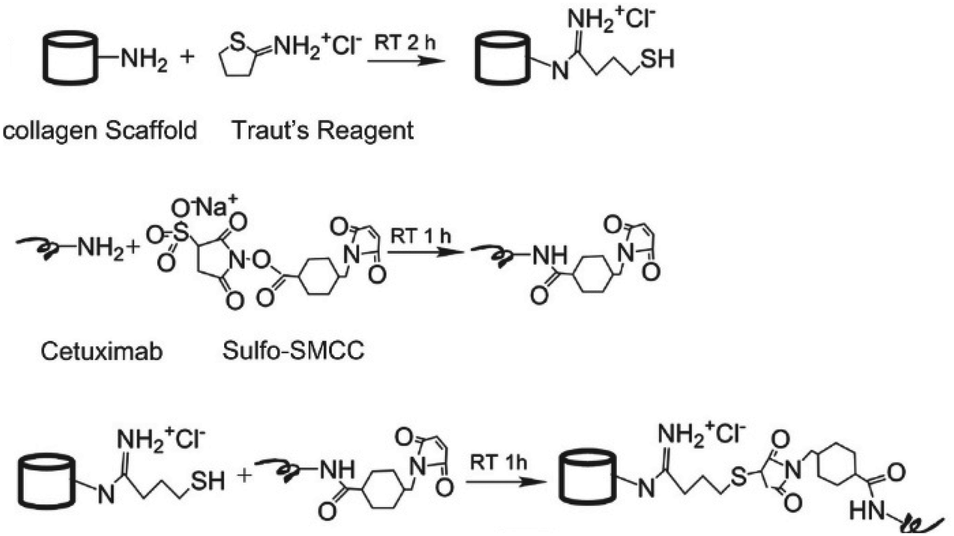 | ||
| Fig. 5 Conjugation of Cetuximab to the collagen scaffold. Reprinted from ref. 21 with permission from Elsevier, copyright (2013). | ||
Puoci et al. conjugated Ciprofloxacin (CFX), an antibiotic, to collagen.17 Collagen was activated towards radical insertion of CFX via attack of the labile residues with hydroxyl radicals, which are formed through the oxidation of ascorbic acid by H2O2, followed by insertion of CFX. The obtained material showed antimicrobial activity and stimulation of fibroblast growth, supporting its applicability as wound dressing.
Fibrin
Similar to collagen, fibrin is one of the main components of the extracellular matrix (ECM) and plays an important role as scaffold for tissue engineering applications. Fibrin biopolymers consist of protofibrils that self-assemble and bundle into semiflexible fibers (Fig. 6). The precursor of fibrin is fibrinogen, a protein that consists of three polypeptide chains, named Aα, Bβ and γ.22 Polymerization into fibrin is initiated by thrombin, which cleaves the protective fibrinopeptides FpA and FpB, initiating a two-step assembly process. In the first step, double-stranded protofibrils form after cleavage of FpA.23 Subsequently, cleavage of FpB promotes clustering of the protofibrils into fibers constituting up to hundreds of protofibrils.24 Enzyme factor XIII (FXIII) catalyzes the formation of crosslinks between Aα and γ, which causes a closer packing of the protofibrils in the fibrin fibers.25 A widely applied strategy to introduce functionalities on fibrin is via feeding of a modified substrate to FXIII, which then incorporates the modification into the fibrin fibers. Several proteins, genes and peptide–DNA conjugates have been introduced on a fibrin scaffold via this method.26–29 | ||
| Fig. 6 Schematic overview of Fibrin formation and incorporation of substituents (R) via modification of enzyme factor XIII (FXIII). | ||
Banfi and coworkers used fibrin as a scaffold for the delivery of recombinant VEGF to induce angiogenesis.30 The N-terminus of murine VEGF164 was fused to a peptide substrate for FXIII, which binds VEGF164 to fibrin during fibrinogen crosslinking (R group in Fig. 6). Bound VEGF164 is released from the scaffold via degradation of fibrin by proteases. The biodegradability of natural polymers marks a big advantage for biopolymeric scaffolds towards medical applications, as most synthetic polymers lack this feature. Implantation of the VEGF164 functionalized fibrin hydrogel in murine models of ischemic wound healing showed that sustained release of VEGF164 induced normal angiogenesis, which improved tissue perfusion and accelerated wound healing. This strategy, where the molecule of interest is fused to a peptide substrate for FXIII, was applied to functionalize fibrin scaffolds with various proteins such as β-NGF, αvβ3 integrin and DNA sequences.27,29,31
Nam et al. conjugated synthetic laminin-111 to fibrinogen monomers to prepare a fibrin hydrogel that promotes tissue regeneration in submandibular glands.32 To covalently bind the peptide, primary amines on fibrinogen were functionalized with a sulfo-LC-SPDP linker via NHS chemistry (Fig. 7A). The pyridylthiol reactive group enabled binding of the peptides via disulfide bond formation with residual thiols. The functionalized fibrinogen monomers were subsequently polymerized to form the laminin-111-functionalized fibrin hydrogel, which showed regeneration of gland tissue in vivo. This functionalization strategy was also applied to introduce other peptides and growth factors on fibrin scaffolds.33–35
 | ||
| Fig. 7 Introduction on substituents on fibrin via fibrinogen functionalization. A. Introduction of peptides on fibrinogen (Fg) via modification with sulfo-NHS-LC-SPDP linker to enable conjugation with thiol residues via disulfide formation. B. Introduction of aptamers on Fg monomers via acrylate-thiol Michael addition. C. Formation of fibrin hydrogels and subsequent binding of growth factors. Reprinted from ref. 36 with permission from American Chemical Society, copyright (2019). | ||
Another method for functionalization of fibrin was applied by Zhao et al.36,37 Here, fibrinogen was functionalized with aptamers, which can bind the growth factors VEGF and platelet-derived growth factor (PDGF) to promote angiogenesis after fibrin polymerization. The aptamers were covalently bound to fibrinogen via a Michael addition. The primary amines on native fibrinogen were acylated with NHS-acrylate. Thiolated anti-VEGF or anti-PDGF aptamers were then reduced and reacted with the acrylates on fibrinogen to form to Michael adduct (Fig. 7B). The aptamer-functionalized fibrinogen monomers were polymerized using thrombin to form fibrin hydrogels comprising VEGF and PDGF binding domains (Fig. 7C). In vitro studies of VEGF and PDGF loaded fibrin gels showed increased retention and significantly slower release of the growth factors compared to the release from native fibrin gels. The effect of the dual growth factor release on angiogenesis was examined both in vitro and in vivo and demonstrated promoted formation of stable blood vessels. A follow-up study with a VEGF-aptamer functionalized fibrin hydrogel loaded with VEGF demonstrated enhanced angiogenesis and osteogenesis in vivo compared to soluble VEGF loaded fibrin hydrogel,38 which demonstrates the advantage of scaffold functionalization. Other work showed the applicability of this approach to promote the survival of transplanted mesenchymal stem cell (MSC) spheroids in vitro, ultimately for the development of various tissue-engineering materials.39
Both collagen and fibrin are readily functionalized by using the functional groups on their backbones. Consequently, regioselectivity is difficult to realize and modifications are randomly introduced in the structure. In addition, fibrin can be modified through FXIII which is spatially controlled, albeit less dense.
Polynucleotides
Polynucleotides consist of a sequence of nucleotide monomers that are linked through a covalent bond between the phosphate group and the pentose sugar of each nucleotide, resulting in a sugar-phosphate backbone (Fig. 1). In nature, DNA exists in a double stranded form (dsDNA) comprising two polynucleotide chains that bind via the formation of hydrogen bonds between the complementary nucleobases. On the other hand, RNA primarily occurs as a single polynucleotide chain. DNA, as well as RNA are considered to be semiflexible.6,40 This fact, combined with their abundance in nature, makes them suitable scaffolds for bioengineering applications. As DNA and RNA can be assembled into highly ordered nanostructures, these biopolymers serve as excellent scaffolds to conjugate a variety of molecules which are subsequently presented on such nanostructures.DNA nanostructures
Over the past few years, a wide variety of well-defined, compact 3D DNA nanostructures has been synthesized through the DNA origami technique,41 or via the single-stranded DNA tiles technique, which uses single-stranded DNA (ssDNA) as bricks that assemble into well-defined DNA nanostructures.42 Due to their high spatial control, DNA nanostructures provide excellent scaffolds for introduction of substituents. Peptides and proteins have been conjugated to DNA nanostructures with well-controlled intermolecular distances and relative ratios.43 In the next paragraphs, we will focus on protein conjugation to DNA, which is frequently pursued for development of biosensors, drug delivery systems and multi-enzyme networks.Nucleic acids and their polymers have been chemically modified at several positions: at the nucleobase, the sugar, the phosphate backbone and the chain termini (Fig. 8A). Chemical modification at the base and sugar positions is laborious and requires the nucleobase hydrogen bond patterns to remain intact.44 Functionalization at the chain ends or at the phosphate backbone is synthetically easier and, therefore, more applied. A variety of methods for protein–DNA conjugation have been developed, which can be divided in covalent/non-covalent, and site-specific/non-site-specific methods. To enable conjugation with biomolecules, the 5′-terminus of a polynucleotide chain has been modified with functional groups such as amines, azides and thiols.43,44 Most of these functionalized DNA strands are readily synthesized by automated solid-phase synthesis or are commercially available.43
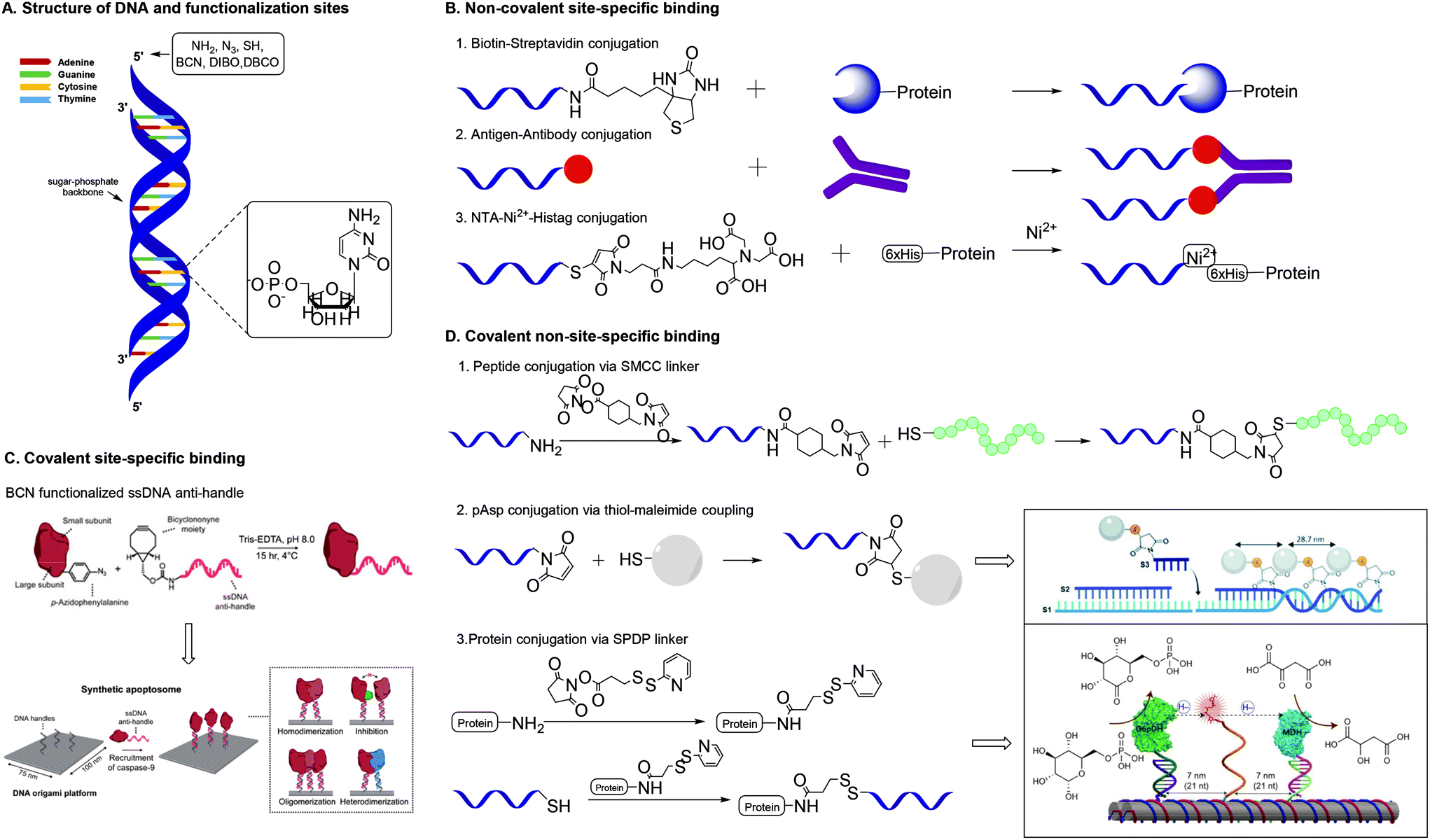 | ||
| Fig. 8 A. Structure of DNA and functionalization positions. B. Methods for non-covalent site-specific binding of proteins to a DNA scaffold. 6×His = hexahistidine tag. C. Method for covalent site-specific binding of proteins to a DNA scaffold. Reprinted from ref. 60 with permission from Nature Springer: Nature Catalysis, copyright (2020). D. Methods for covalent non-site-specific binding of proteins to a DNA scaffold. D.2 Reproduced from ref. 63 with permission from the Royal Society of Chemistry, copyright (2019). D.3 Reprinted from ref. 64 with permission from Nature Springer: Nature Nanotechnology, copyright (2014). | ||
One of the most studied methods for non-covalent site specific binding of proteins to DNA is binding through biotin–streptavidin (Sav) interactions (Fig. 8B),45 which conjugates in mild conditions and with high affinity.46 The 5′-end of the DNA strand is easily biotinylated. Proteins are functionalized with Sav via protein engineering, which is laborious and remains challenging.47 The use of monomeric Sav circumvents stoichiometry difficulties and ensures efficient conjugation.48 Other common non-covalent protein–DNA binding strategies include antigen–antibody binding49 and nitriloacetic acid (NTA)-Ni2-Histag binding.50 These methods require the 5′-end binding of an antigen, through solid phase synthesis,49 or a NTA moiety through the reaction of thiolated DNA with maleimide-C3-NTA.51
Covalent site-specific conjugation of proteins to DNA nanostructures (Fig. 8C) requires proteins to express a chemical handle that can selectively react with a functional group on the DNA scaffold. Such techniques require genetic manipulation of the protein of interest, which is challenging and needs optimization for each newly studied protein.43 Extensive reviews on the various approaches towards site specific conjugation of proteins have been published recently.43,52 A common and chemically relevant approach towards site-specific ligation is azido protein–DNA conjugation. In this strategy, the protein of interest is mutated with an azido group via incorporation of unnatural amino acids such as azido-homoalanine.53 The 5′ end of a DNA strand can be functionalized with a complementary group that enables a selective reaction with the azide. The frequently used terminal alkyne needs a Cu(I) catalyst for the cupper-catalysed azide–alkyne cycloaddition (CuAAC), which presents biological drawbacks, including cell toxicity and loss of enzyme activity.43 The SPAAC reaction forms a suitable alternative and needs DNA modification with groups such as bicyclononyne (BCN),54 dibenzoannulated cyclooctyne55 or dibenzocyclooctyne (DBCO).56
Using these strategies various proteins of interest have been conjugated on a DNA scaffold.57–59 Rosier et al. applied site-specific protein–DNA conjugation to develop a DNA-origami based synthetic apoptosome.60 Caspase-9 was conjugated to ssDNA via a SPAAC reaction between p-azidophenylalanine incorporated in the enzyme and BCN-functionalized ssDNA (Fig. 8C). Subsequent hybridization of the enzyme-ssDNA construct with complementary strands on a DNA origami platform resulted in the formation of a well-defined enzyme-DNA nanostructure, offering proximity-induced Cas9 enzyme activity.
Covalent non-site-specific protein–DNA conjugation strategies circumvent problems related with dissociation of non-covalent interactions. Generally, a protein is conjugated to DNA via the use of a heterobifunctional linker. The most common linkers contain a maleimide functionality and an NHS-ester to bind thiolated DNA strands to a protein via lysine residues.61 The heterobifunctional linker SMCC was used by Chaput and co-workers to develop a synthetic antibody.62 Chaput created a bivalent protein affinity reagent by conjugating peptide ligands on a DNA scaffold. To enable binding of peptide ligands, ssDNA was modified with a terminal amine, which was reacted with the NHS ester of the SMCC linker (Fig. 8D). Subsequent reaction of the maleimide on the linker with the C-terminal cysteine on the peptide yielded in peptide-functionalized DNA strands that self-assembled into a bivalent DNA–peptide scaffolds with tailorable peptides positions, which ultimately could bind a target protein with a 1000-fold higher affinity than the individual peptides. This result is a clear example of how a well-defined scaffold promotes a multivalent presentation of biomolecules.
Another covalent conjugation strategy was applied by the group of Carneiro, who coupled polyaspartic acid (pAsp) to ssDNA to develop pAsp functionalized nanostructures for enamel regeneration.63 The N-terminus of pAsp was functionalized with a thiol group through acetylation of the N-terminus followed by nucleophilic substitution with thioacetic acid and subsequent deprotection. The resulting terminal thiol was reacted with a maleimide functionalized DNA strand containing a sticky end. The pAsp functionalized DNA strand was then combined with two other DNA strands to form a three-stranded double helix in which the pAsp particle always faces the same side of the DNA duplex at a pre-defined distance (Fig. 8D). The results highlight that the DNA assembly technique, which resembles a block-copolymer approach by employing functionalized oligonucleotides as monomers that are incorporated on DNA strands, enables introduction of substituents on a DNA scaffold with high spatial control.
DNA assembly proves a powerful strategy to introduce different moieties (enzymes and synthetic polymers) on a scaffold with high spatial control.64–66 Fu et al. used DNA assembly to develop a multi-enzyme complex in which a synthetic swinging arm facilitates hydride transfer between two hydrogenases conjugated on a DNA scaffold.64 Lysine residues on the hydrogenases were reacted with the NHS ester of a commercially available succinimidyl 3-(2-pyridyldithio)propionate (SPDP) heterobifunctional linker (Fig. 8D). The resulting enzyme constructs were coupled to thiolated ssDNA via cleavage of the pyridylthiol reactive group of the SPDP linker. The synthetic swinging arm, consisting of NAD+-functionalized poly(thymine)20, was conjugated to ssDNA through coupling with disuccinimidyl suberate; both NHS-esters reacting with the primary amines on amine functionalized ssDNA and NAD+-functionalized poly(thymine)20. Subsequent DNA hybridization on the DNA scaffold gave rise to a multi-enzyme complex with excellent enzyme activity. The unparalleled spatial control that DNA scaffolds offer allows for manipulation and optimization of (bio)functionality, in this example enzyme activity.
RNA nanostructures
Although RNA (Fig. 9) is chemically similar to DNA, there are some key differences in their structures. In nature, DNA primarily exists as a double stranded helix, while most RNA is single stranded. The RNA helix has a smaller pitch and is shorter than that of DNA.67,68 Despite the differences, strategies for the development of DNA nanostructures can be extended to design RNA nanostructures, as long as minor modulations, such as changing the duplex length are considered.69 In this way, the DNA origami technique was extrapolated to RNA by replacing the DNA staples with RNA staples. RNA tiles, analogous to DNA tiles, were designed, which were self-assembled into uniform nanostructures.70,71 Because RNA has different tertiary folding and assembly principles than DNA, different nanostructures can also be designed, which leads to new scaffolds for substituent presentation. As this review focusses on the application of RNA nanostructures as scaffolds, only a short overview is given on the various types of RNA nanostructures. The design and structure of RNA nanostructures themselves has been reviewed elaborately elsewhere.72 | ||
| Fig. 9 RNA structure and functionalization positions. | ||
Tertiary structures of RNA consist of lower order structural motifs, which can function as building blocks for nanoassemblies. RNA structural motifs can bend, link or branch RNA molecules, which further expands the toolbox for organization at the nanometer scale.69 Due to the large variety of known structural motifs, an infinite number of RNA nanostructures can be designed.73 Similar to DNA, RNA nanostructures can function as a scaffold for the introduction of functional molecules. Aptamers, chemical ligands, fluorescent dyes and other functional molecules have been fused to RNA strands prior to their assembly into nanostructures.74 However, because RNA is chemically labile, introduction of functional molecules on RNA scaffolds via chemical modification is problematic.75 Therefore, fusion of functional molecules to RNA strands is commonly achieved through modification of the functional molecule with a phosphoramidite group followed by coupling to the 3′ or 5′ end of RNA (Fig. 10). Through this method, fluorescent dyes and folate have been conjugated to RNA nanoparticles; the latter with the goal to target tumor cells, which are known to overexpress folate receptors.76 Protein binding RNA scaffolds can be prepared through the incorporation of aptamers in RNA structural motifs. A nanostructure based on the packaging RNA three-way junction motif was functionalized with four binding aptamers to develop a multifunctional RNA–protein nanostructure (Fig. 10).77 RNA aptamers are relatively easily attached to the nucleotide chain of RNA motifs, which allows for subsequent binding of proteins of interest and further assembly into nanostructures.
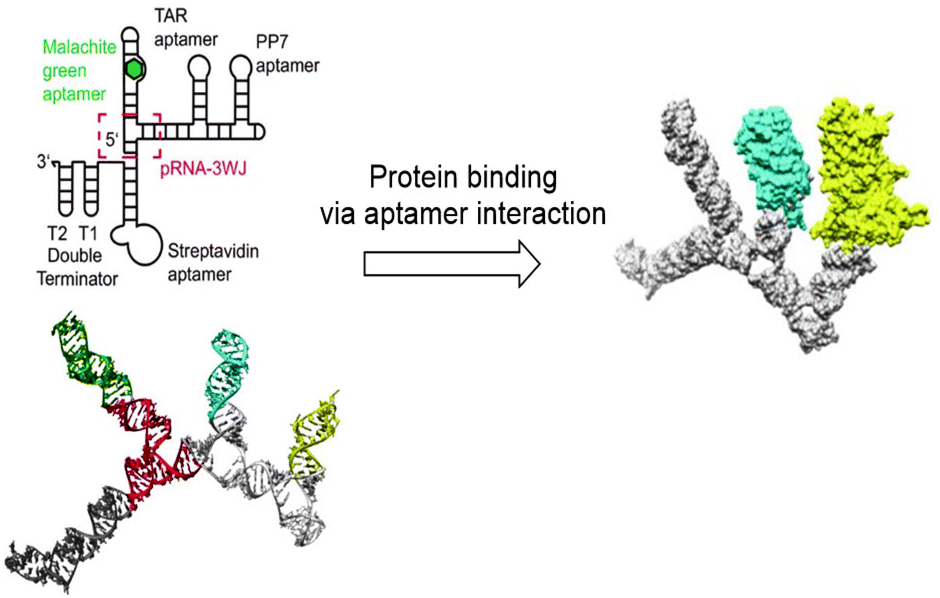 | ||
| Fig. 10 RNA nanostructure based on the three-way junction motifs that binds proteins via interaction with protein specific aptamers on the RNA motif. Reprinted from ref. 77 with permission from American Chemical Society, copyright (2018). | ||
Another method for the introduction of proteins on RNA nanostructures makes use of RNA recognition motifs, i.e. a region on the RNA strand that is recognized and bound by the protein.78 Using such RNA recognition motifs, Shibata et al. developed a RNA nanostructured device that can control cell fate via RNA–protein interaction-mediated protein assembly.79 For instance, K-turn RNA and the RNA-binding protein L7Ae function as RNA–protein interaction motif. L7Ae was fused with caspase-8 (Casp8) to develop a nanodevice that induces cell death. RNA scaffolds containing various amounts of K-turn motifs were developed. Populations with cell-death were observed for cells that were co-transfected with L7Ae-Casp8 and RNA scaffolds containing a higher number of K-turn motifs, suggesting that cell-death signals can be tuned by changing the number of assembled Casp8 proteins on the scaffold. These results show that RNA scaffolds can localize target proteins and induce strong biological effects.
Although a wide variety of DNA and RNA scaffolds with potential therapeutic application have been synthesized, their use remains restricted. Due to their phosphodiester backbone, synthetic oligonucleotides are susceptible to degradation by nucleases.80 This process makes the in vivo application of DNA and RNA scaffolds a challenge. Peptide nucleic acid (PNA) can circumvent degradation by nucleases, as its backbone is composed of peptides.81
Peptide nucleic acids (PNAs)
A PNA (Fig. 11) is a modified DNA equivalent that contains a neutral peptide backbone instead of a negatively charged sugar-phosphate backbone. Unlike DNA and RNA, PNA is chemically stable and resistant to enzymatic degradation in living cells. Functional groups are most often introduced on the backbone of PNA by modification of the α (C-2) or γ (C-5) position of the PNA monomer.82 Modification of the backbone of PNA introduces chirality and, therefore requires enantioselective synthesis. Chirality can be introduced by starting the synthesis with synthons such as D- or L-amino acids. Monomers that are functionalized at the α position may suffer from epimerization when used in solid phase synthesis, which makes it difficult to obtain optically pure PNAs.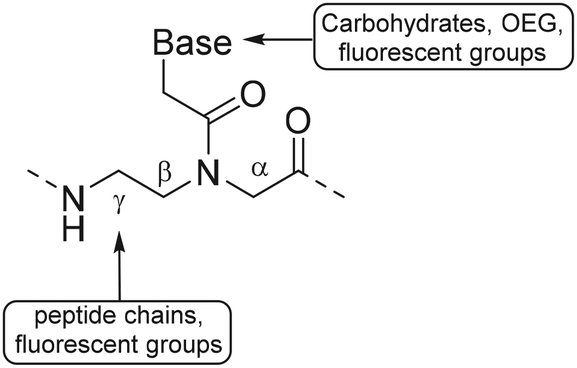 | ||
| Fig. 11 Structure of PNA and functionalization positions. | ||
Optical purity for α modified PNA is improved by a submonomeric approach, in which the nucleobase is introduced after linking the peptide to the PNA backbone.83 Modification of the α position with positively charged side chains improves cellular uptake and duplex stability but introduction of neutral or negative charged side chains on that position destabilize duplex formation.84,85 Synthetic bottlenecks and restricted options of functional groups limit the use of α modified PNA as a scaffold. Optical purity is easier obtained for PNAs comprising γ modified monomers. For example, optically pure PNA monomers derived from L-amino acids were prepared using a Mitsunobu reaction with t-butyloxycarbonyl (Boc)-protected α amine (Fig. 12A, route i) or via reductive amination with protection of the α amine with 9-phenylfluorenyl group (route ii).86
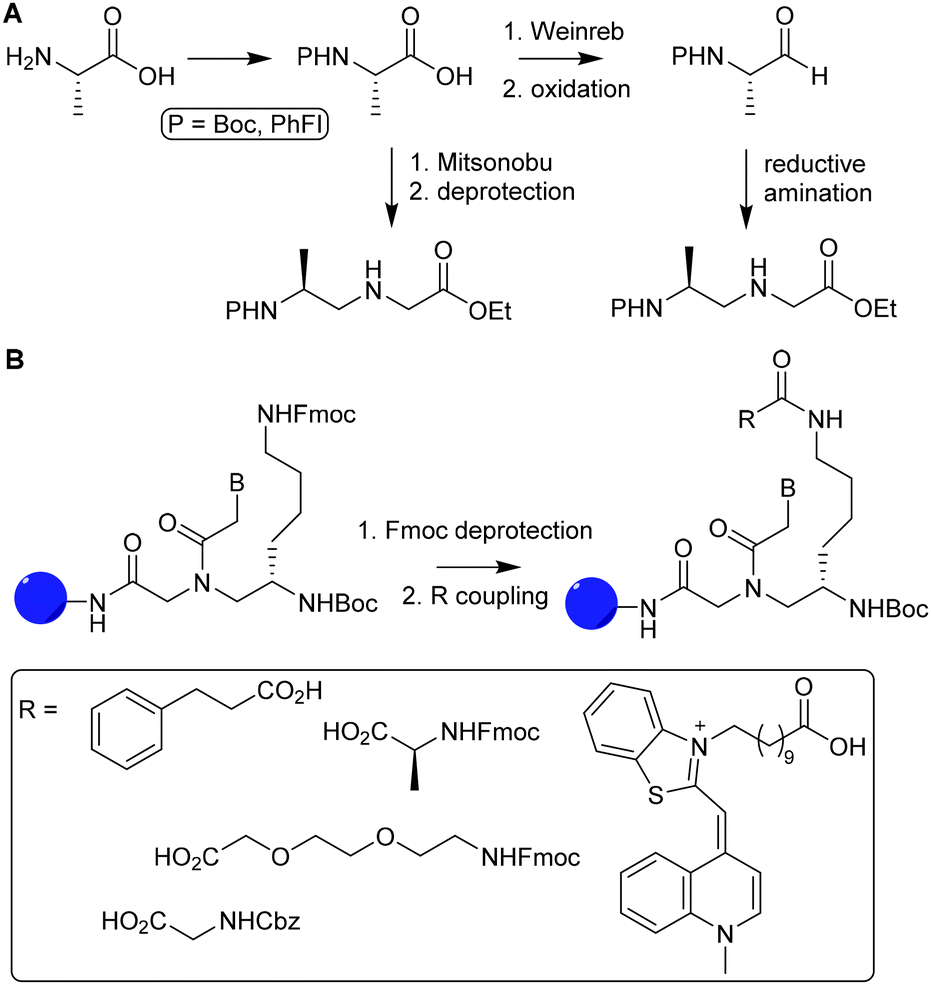 | ||
| Fig. 12 A. Synthesis of optically pure γ-PNA monomers via reductive amination (1) and Mitsunobu coupling (2). Reprinted from ref. 86 with permission from Elsevier, copyright (2015). B. Introduction of various functional groups (R) on PNA oligomers via functionalization of L-lysine γ-PNA building blocks via SPPS. Reprinted from ref. 85 with permission from John Wiley and Sons, copyright (2007). | ||
Appella and coworkers used L-lysine γ-PNA as building blocks to synthesize PNA oligomers containing various functional groups.85 The primary amine of the lysine chain was used to conjugate a variety of groups such as acetamide, phenylpropanoic acid, small peptide chains and fluorescent groups. Introduction of these functional groups was carried out using fluorenylmethyloxycarbonyl (Fmoc) protected amine residues, which were deprotected during PNA oligomer synthesis on solid support. After deprotection, carboxylic acids were coupled using hexafluorophosphate benzotriazole tetramethyl uronium as a coupling reagent (Fig. 12B). The authors found that introduction of these functional groups did not interfere with the ability of PNA to bind complementary nucleic acid sequences, showing that PNA can function as an excellent scaffold for the development of for instance nucleic acid detection systems and antisense molecules.87,88
A more straightforward method to introduce functionalities is through modification of the PNA nucleobase, which circumvents the introduction of chiral centers. To this end C-5 modified pyrimidines were developed using 5-hydroxymethyl uracil, and 5-iodocytosine as building blocks.89
5-Hydroxymethyluracil is easily obtained from uracil through reaction with formaldehyde.90 Dambenieks and coworkers conjugated a variety of functionalities such as carbohydrates, cationic groups and oligo(ethylene glycol) tails to hydroxymethylated uracil PNA monomers (Fig. 13A).89 The 5-iodonucleobase allows for introduction of alkynyl derivatives using the Sonogashira coupling. Using this method, fluorescent groups were conjugated to PNA monomers and subsequently incorporated in PNA oligomers to detect increased binding affinity with DNA.91 Substitution at the C-6 position of pyrimidines is slightly more sensitive to steric effects than the C-5 position as it is closer to the PNA backbone, making introduction of functional groups at the C-6 position less favorable.
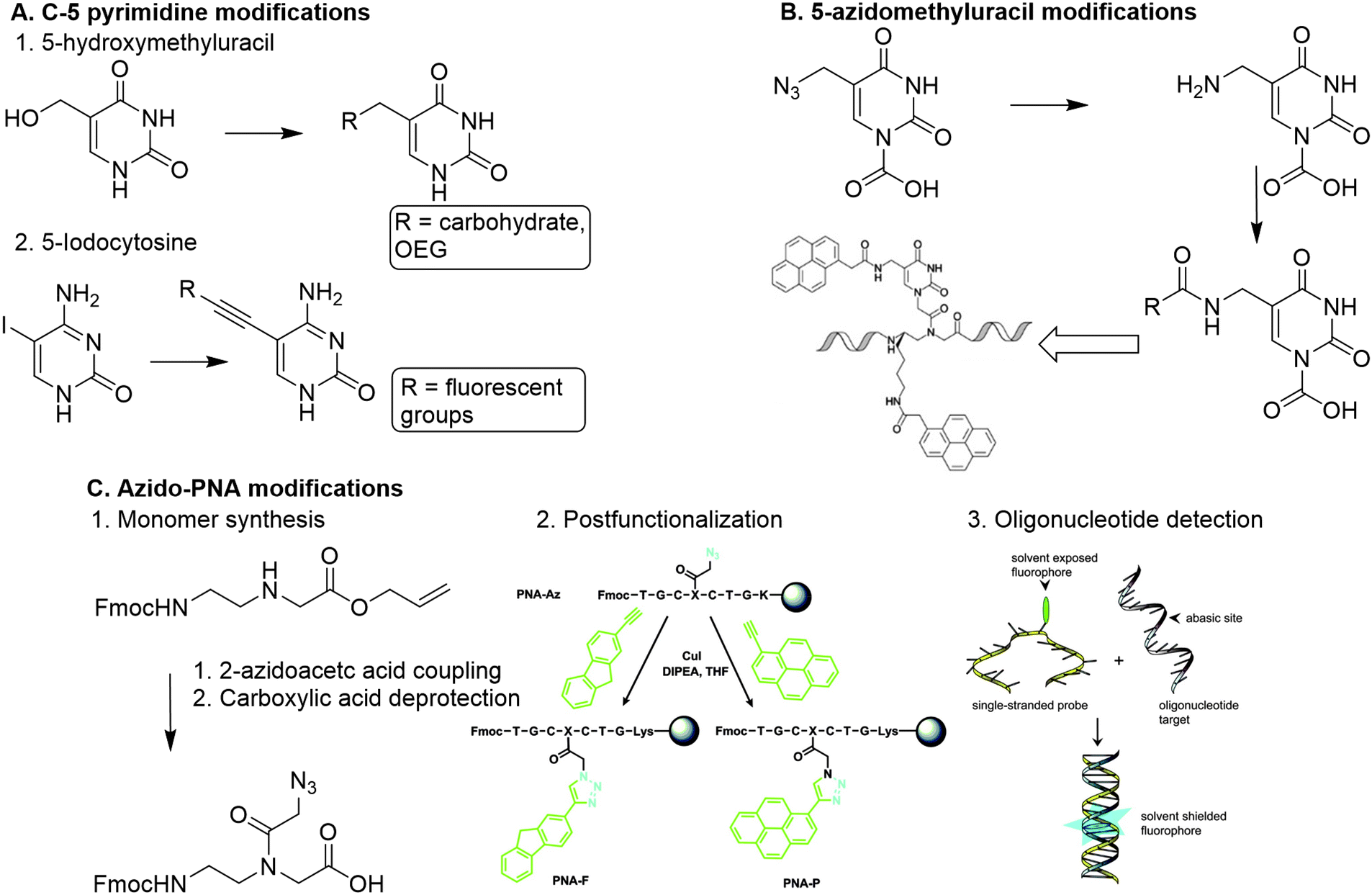 | ||
| Fig. 13 A. Functionalization of PNA via nucleobase modification. 1. Introduction of carbohydrates and oligo(ethylene glycol) on a PNA monomer via reaction with 5-hydroxylmethyluracil. Adapted from ref. 89. 2. Introduction of fluorescent groups on a PNA monomer via Sonogashira coupling with 5-iodocytosine. B. Modification of 5-azidomethyluracil towards introduction of pyrenes on a polynucleotide scaffold. Reproduced from ref. 93. https://pubs.acs.org/doi/10.1021/acs.orglett.6b02363. Reprinted with permission from ACS, copyright (2016). Further permissions related to the material excerpted should be directed to the ACS. C. 1. Direct modification of the PNA backbone with 2-azidoacetic acid. 2. Post-functionalization of a PNA sequence with fluorescent groups (PNA-F and PNA-P) via CuAAC reaction. Reprinted from ref. 94 with permission from Taylor & Francis, copyright (2013). 3. Detection of target oligonucleotides via hybridization and subsequent fluorescence. Reprinted from ref. 94 with permission from Taylor & Francis, copyright (2013). | ||
Analogously, PNA monomers bearing 5-azidomethyluracil were developed as a versatile building block for the introduction of functional groups.92 This building block is easily obtained via acid catalyzed nucleophilic substitution of the hydroxyl of hydroxymethylated uracil with chloride, followed by conversion into an azide using sodium azide. Introduction of a carboxymethylene linker at the N-1 position of uracil enables the insertion on the PNA backbone. The azido group can be converted into an amine to enable conjugation of carboxylic acid containing substituents, or it can be used to couple alkyne containing substituents via CuAAC or SPAAC click chemistry. Both conjugation strategies can also be applied on PNA oligomers which enables post-modification of the PNA scaffold. Manicardi et al. applied this strategy to introduce pyrenes on a PNA scaffold to develop a PNA-based probe that detects target oligonucleotides via a conversion in fluorescence emission upon hybridization.93
Another approach was developed by Hudson and coworkers, who synthesized an azide containing PNA monomer without nucleobases (Fig. 13C).94 Hudson and coworkers argue that conjugation of a modified base to the monomer backbone is laborious and leads to loss of material during purification, which can be circumvented by the use of a reactive monomer that can be transformed into a variety of derivatives. To this end, azidoacetic acid was directly conjugated to the PNA backbone (Fig. 13B). To demonstrate its post-modification abilities, the azido PNA monomer was incorporated into an oligonucleotide using solid-phase peptide synthesis (SPPS), and subsequently conjugated to a fluorophore while on the solid support. This on-resin approach is beneficial for the copper mediated click reaction between the azido modified oligomer and the alkyne containing fluorophore, as it enabled using an excess of reagents and simplified the purification. The resulting fluorophore functionalized PNA scaffold functioned as a probe for detecting abasic sites on oligonucleotides. The on-resin method facilitates easy derivatization of PNAs via separation of the resin and subsequent modification reactions. Furthermore, SPPS of PNA scaffolds gives the opportunity to introduce functional groups with high spatial control, comparable to the DNA assembly approaches.
Polysaccharides
Polysaccharides are linear or branched long chains of carbohydrate molecules that are linked via glycosidic bonds (Fig. 1). Linear polymers such as alginate, cellulose, chitin, chitosan and hyaluronic acid are semiflexible and have a high abundance in nature, which makes them interesting for scaffold appliations.95–98 The ample amines, primary hydroxyls and carboxylic acids on polysaccharide chains provide active sites to introduce substituents. The open-chain aldehyde form of the terminal monosaccharide can also be subjected to modifications such as reductive amination. An overview of the possibilities for chemical modifications on polysaccharides was reviewed earlier.99 Here, we focus on polysaccharides as scaffolds and describe the modification and conjugation strategies used to introduce substituents on these scaffolds.Hyaluronic acid
Hyaluronic acid (HA) is an anionic glycosaminoglycan of D-glucuronic acid (D-GlcA) and N-acetyl-D-glucosamine (D-GlcNAc) disaccharide units which are linked via β-1,3 and β-1,4 glycosidic bonds (Fig. 14). Hyaluronan synthases produce long HA chains with an average contour lengths of >15 μm (Mw > 7 MDa).100 HA is a chief component in the ECM and plays an essential role in tissue regeneration.101 Due to its physical and biochemical properties, HA is gaining interest in biomaterials science for applications such as bone regeneration, drug delivery, wound healing and tissue engineering.102 | ||
| Fig. 14 Structure of hyaluronic acid (HA) and its functionalization positions. | ||
The combination of its straightforward chemical modification, biocompatibility and degradability make HA a particularly attractive scaffold for biochemical applications.103 Particularly, the primary hydroxyl of D-GlcNac and the carboxylic acid of D-GlcA are readily modified and can function as handles for conjugation of HA with various substituents. The anticancer drug Dihydroartemisinin (DHA) was conjugated to a HA scaffold to improve its bioavailability.104 HA itself can function as a tumor targeting ligand as it can bind to CD44, a transmembrane glycoprotein and cell surface receptor for HA, and RHAMM, a receptor for HA-mediated motility. DHA was coupled to HA via ester bond formation between the hydroxyl of DHA and the carboxylic acid of HA (Fig. 15A). Because of its amphiphilic character, the HA–DHA conjugate forms nanoparticles in aqueous solution, resulting in a higher bioavailability than DHA, which is poorly soluble in aqueous solutions due to its lipophilic character. In vitro cytotoxicity tests indicated that the efficacy of DHA is enhanced when conjugated to HA, which demonstrates the targeting and solubility effect of the HA scaffold.
 | ||
| Fig. 15 A. Conjugation of DHA on a HA scaffold to form HA–DHA nanoparticles (NPs). Reprinted from ref. 104 with permission from Elsevier, copyright (2019). B. Conjugation of Cu(II)CHl on a HA scaffold to prepare Cu(II)Chl-HA NPs. Reproduced from ref. 110 with permission from the Royal Society of Chemistry, copyright (2019). | ||
Shen and co-workers also applied the targeting ability of HA for the delivery of Cu(II)-chlorophyll (Cu(II)Chl) to cancer cells105 and subsequent photodynamic/photothermal therapy.106,107 Cu(II)Chl-HA nanoparticles were generated in two steps: first PEG-diamine was covalently linked to the carboxylic acid of Cu(II)Chl after NHS activation; then the resulting PEG-Cu(II)Chl was conjugated to the NHS-activated HA to form a carbamate linkage (Fig. 15B). In vitro, the construct showed good selectivity for CD44-overexpressing cancer cells and high cell-killing efficacy upon irradiation at 650 nm. In vivo mouse experiments show decreasing tumor volumes on treatment. These results demonstrate how HA can be used as a scaffold for targeted delivery of drugs.
Hyaluronic acid is extensively studied as a scaffold for tissue engineering applications as it has the ability to induce intracellular signal transduction and can affect cell activities such as proliferation and differentiation.108,109 HA hydrogels are frequently used as a designer matrix for 3D cell culture or for wound healing purposes. The gels, however show poor mechanical properties and undergo rapid degradation in vivo.102 Crosslinking and chemical modification can circumvent these disadvantages.
Chaudhuri and coworkers developed a HA hydrogel with adjustable crosslinks to resemble the dynamic ECM microenvironment.111 To synthesize a hydrogel with tunable stress-relaxation, HA was crosslinked via hydrazone bond formation. Hydrazone formation is efficient, biocompatible and can form dynamically exchangeable crosslinks, which form the basis of the controlled stress relaxation profiles in the HA hydrogels.112 The carboxylic acid of HA was NHS-activated and coupled to propargyl amine, which was then transformed via the CuAAC reaction to HA decorated with either hydrazine or aldehyde groups (Fig. 16). Upon mixing both, hydrazone crosslinked hydrogels readily formed and their tunable stress relaxation was established with rheology. Next, dynamic HA-collagen hydrogels were developed to form matrices for 3D cell culture. Investigation of cell morphology showed that faster hydrogel relaxation promotes cell spreading, collagen fiber realignment, and focal adhesion (FA) formation. Functionalization of the HA scaffold with such dynamic covalent crosslinks enables development of synthetic 3D cell cultures in which function and fate of cells can be directed, comparable to the ECM.
 | ||
| Fig. 16 Crosslinking of HA with adjustable crosslinks. A. Propargylamine was conjugated to NHS-activated HA. Aldehyde and hydrazone functionalities were introduced via CuAAC chemistry. B. Crosslinking of HA via dynamic hydrazone bond formation. Adapted and reprinted from ref. 111 with permission from Elsevier, copyright (2018). | ||
To enhance the stability of HA hydrogels, Lee et al. crosslinked the hydroxyls of HA with poly(ethylene glycol) (PEG) diglycidyl ether.113 Additionally, tannic acid (TA) that was incorporated via hydrogen bond formation, gave the hydrogel improved mechanical properties, and, as a hyaluronidase inhibitor, also provided enzymatic stability. A similar chemical approach was taken by Lin and coworkers, who crosslinked HA with 1,4-butanediol diglycidyl ether (BDDE) followed by the introduction of chlorhexidine (CHX) to develop an antibacterial hydrogel for pacemaker pocket infection prevention.114 The pre-crosslinking with BDDE ensured formation of a stable but injectable hydrogel, while the weak physical bonding between the imines of CHX and the carboxylic acids of HA facilitated continuous release of CHX.
Bencherif and coworkers developed methacrylated HA to incorporate covalent crosslinks in gels and nanogels after free-radical photopolymerization.115–117 Methacrylate groups were introduced on a HA scaffold via reaction of the carboxylic acids or the primary hydroxyls with the epoxides of glycidyl methacrylate (Fig. 17A) with good control over the degree of methacrylation (DM).118 Subsequent photo crosslinking of HA with varying DM yielded hydrogels with tailorable material properties; the DM affects the crosslinking density, mechanical properties and swelling ratios. Methacrylated HA (MeHA) hydrogels are effective scaffolds for drug delivery as they demonstrate excellent swelling ratios and are biodegradable. In a follow-up paper, the group incorporated methacrylated nanogels that give additional control over drug release.115
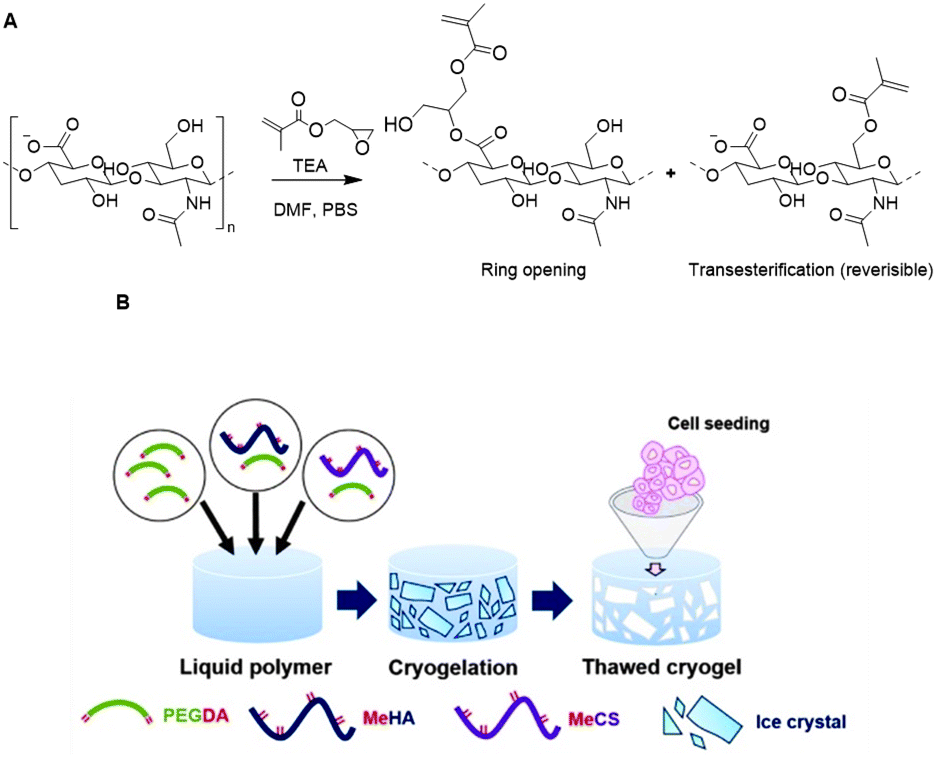 | ||
| Fig. 17 Functionalization of HA scaffolds. A. Crosslinking of HA with glycidyl methacrylate via a competition between the ring opening and transesterification mechanisms. Adapted from ref. 118. B. Preparation of HA cryogels trough free-radical photopolymerization mediated crosslinking with PEG diacrylate. Reprinted from ref. 119 with permission from Elsevier, copyright (2016). | ||
To develop ideal microenvironments for tissue engineering facilitating cellular growth in 3D, a scaffold resembling the interconnected macroporous structure of tissue is desirable. To this end, the Bencherif group developed an ECM-based cryogel from MeHA.116 PEG diacrylate was used as a crosslinker and was covalently linked to MeHA trough free radical polymerization under freezing conditions (Fig. 17B). The resulting cryogel exhibited a macroporosity of 75% and supported infiltration of cartilage cells. Culturing of rabbit chondrocytes on the cryogels gave rise to stimulated collagen type II gene expression and collagen accumulation. Another HA-based cryogel was developed through crosslinking with alginate (see next section).117 Both polysaccharides were functionalized with methacrylates, which enabled chemical crosslinking through free radical polymerization. The resulting cryogel displayed suitable mechanical properties and bioactivity, and regained their original shape and size after injection. The straightforward introduction of methacrylate groups on HA scaffolds together with the promising properties of the resulting cryogels clearly demonstrate the efficacy of polysaccharide-based cryogels for tissue engineering applications.
Alginate
Alginate is a linear copolymer derived from Phaeophyceae seaweed and consists of residues of 1,4-linked β-D-mannuronic acid (M-block) and α-L-guluronic acid (G block) residues (Fig. 18).119 Alginate chains are composed of a random sequence of alternating M-, G- and MG-blocks. Because of its biocompatibility and tailorability, alginate is a suitable candidate for a variety of biomedical applications.120 Furthermore, aqueous alginate solutions crosslink and gel almost instantly in the presence of multivalent cations, which results in the formation of hydrogels with highly tunable mechanical properties.119,121 The gels are suitable as vehicles for delivery of cells, genes and drugs.122–124 In analogy to HA, the carboxylic acid groups in alginate can be employed for crosslinking or introduction of substituents and for covalent as well as ionic crosslinking. Thus far, crosslinking has been largely limited to the formation of ionic bridges between polymer chains via the use of divalent cations such as Ca2+. Such gels are weak and lose their mechanical integrity in vitro and in vivo.125 | ||
| Fig. 18 Structures of Alginate and its functionalization position. | ||
To circumvent the bottlenecks of cationic crosslinking, Desai et al. developed alginate hydrogels that are crosslinked via click chemistry, using the biorthogonal inverse electron demand Diels–Alder (IEDDA) reaction between tetrazine and norbornene.126 To this end, alginate was functionalized with norbornene and tetrazine by reacting the carboxylic acid with norbornene methanamine and benzylamino tetrazine, respectively (Fig. 19A). The two polymer solutions were mixed to form a stable gel within 1 hour (Fig. 19B). Unreacted norbornene groups facilitated post-gelation functionalization of the alginate hydrogel with thiol-bearing molecules via a photoinitiated thiol–ene reaction (Fig. 19C), for instance to introduce cell-adhesive peptides. In vitro tests demonstrated the cytocompatibility of the click alginate hydrogels for 2D cell culture. Furthermore, subcutaneous injection of the hydrogel with embedded cells in mice demonstrated its suitability for in vivo applications.
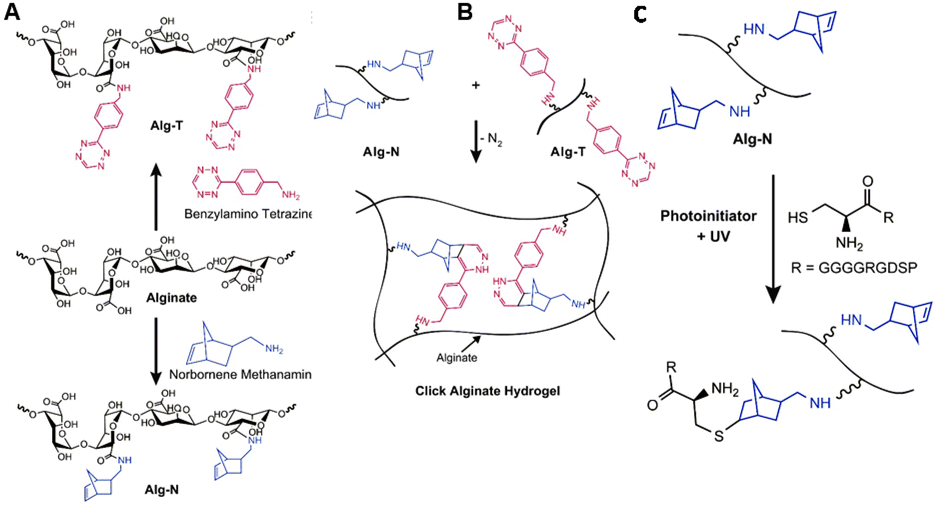 | ||
| Fig. 19 A. Functionalization of alginate with tetrazine (Alg-T) and norbornene groups (Alg-N) via reaction of the carboxylic acids with benzylamine tetrazine and norbornene methanamine. Reprinted from ref. 128 with permission from Elsevier, copyright (2015). B. Preparation of click alginate hydrogel via inverse electron demand Diels–Alder (IEDDA) reaction between tetrazine and norbornene. Reprinted from ref. 126 with permission from Elsevier, copyright (2015). C. Incorporation of cell-adhesive peptides via a photoinitiated thiol–ene reaction. Reprinted from ref. 126 with permission from Elsevier, copyright (2015). | ||
Similar to HA, alginate is also used as a scaffold for the development of cryogels. The biocompatibility of alginate together with its tunable mechanical properties make alginate a suitable scaffold for development of cryogels for delivery of biomolecules.127,128 Mooney and coworkers prepared an alginate based cryogel via a free-radical crosslinking mechanism.129 To this end, methacryloyl groups were introduced in the alginate chains via reaction of NHS-activated carboxylic acids with 2-aminoethyl methacrylate (Fig. 20A). Macroporous cryogels were obtained by crosslinking the MA-alginate at subzero temperatures via free-radical polymerization (Fig. 20B). The polymeric network of the resulting cryogel collapses when subjected to shear-stress during injection but fully recovers, which enables alginate cryogel scaffolds to be administered through needles with almost full geometric reconstruction.
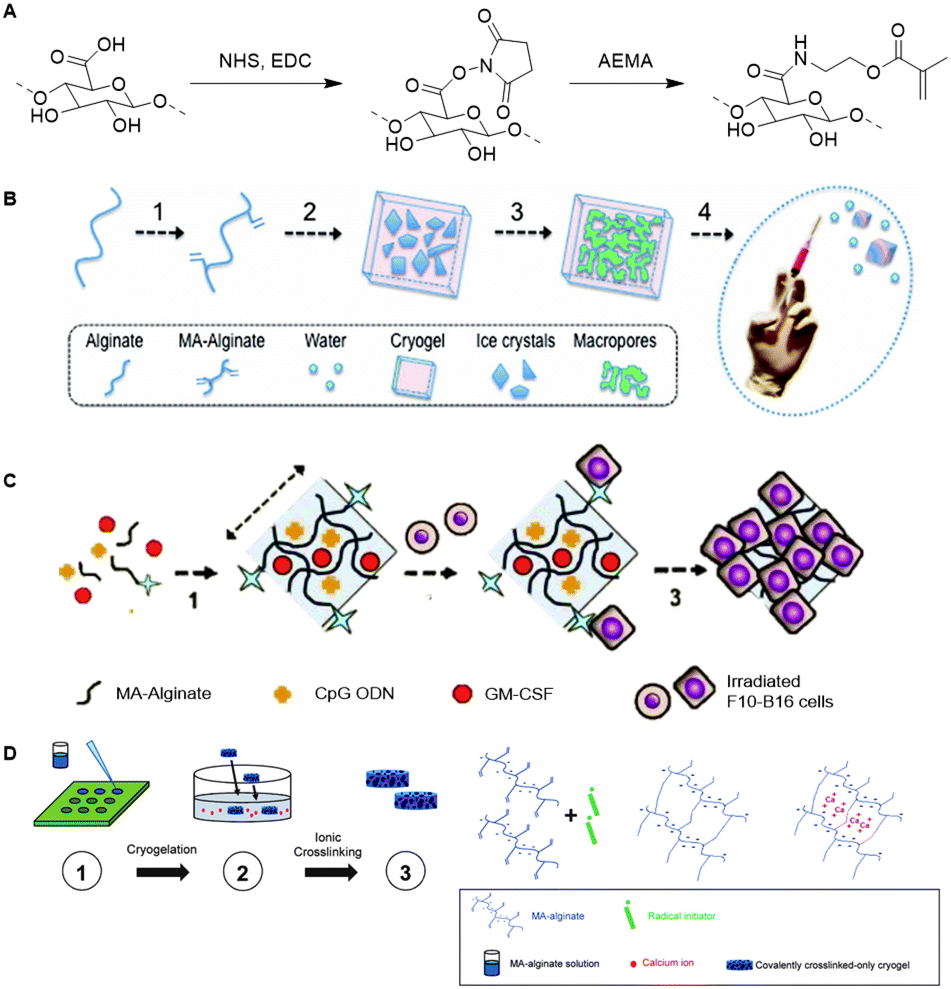 | ||
| Fig. 20 A. Introduction of methacryloyl groups on alginate via activation of the carboxylic acids with NHS followed by reaction with 2-aminoethylmethacrylate (AEMA). B Preparation of injectable alginate cryogels through free-radical polymerization mediated crosslinking of MA-Alginate at subzero temperatures. Reprinted from ref. 129 with permission from National Academy of Sciences, copyright (2012) C. Loading of the alginate cryogels with antigen carrying tumor cells, with dendritic cell enhancement factor CPG ODN and activation factor GM-CSF for the development of an injectable cancer vaccine. Reprinted from ref. 130 with permission from Nature Springer, Nature Communications, copyright (2015) D. Preparation of an injectable tough alginate cryogel via free-radical polymerization mediated crosslinking of the MA-alginate and subsequent ionic crosslinking of the carboxylic acids on the alginate chains with Ca2+ ions. Reprinted from ref. 128 with permission from John Wiley and Sons, copyright (2018). | ||
In one example, alginate cryogels were loaded with antigen carrying tumor cells, a dendritic cell (DC) enhancement factor and a DC activating factor, to function as an injectable vaccine platform (Fig. 20C).127 Subcutaneous injection of the loaded cryogel in mice demonstrated potent and specific anti-tumor T cell responses, indicating that alginate cryogels have a high potential as scaffold for (cancer cell) vaccinations. To enable injection of the alginate cryogels trough smaller needles without breaking of the gel, Mooney and coworkers crosslinked the alginate scaffold with Ca2+ (Fig. 20D).128 The combination of covalent (methacrylate-based free-radical polymerization) and ionic (Ca2+) crosslinking yielded a tough cryogel that could be injected trough a small 18G needle without sustaining damage.
Besides functioning as a carrier, alginate also serves as scaffold for targeted release of active molecules. Royzen and coworkers functionalized alginate with tetrazines for local activation of Doxorubicin (Dox), which protected by a trans-cyclooctene (TCO) moiety can be administered systemically (Fig. 21).131,132 Tetrazine methanamine was conjugated to the carboxylic acids of alginate to obtain a tetrazine-functionalized hydrogel. The high density of carboxylates on the alginate scaffold allowed for the introduction of a large number of tetrazine groups, which leads to high effective concentrations of the TCO-protected molecules injected at the location of choice. Additionally, the large number of tetrazines on the alginate scaffold permits multiple rounds of Dox administration, and its injectability provides a viable in vivo strategy for local activation of molecules as it does not rely on presence of molecular markers or local processes. In vivo efficacy studies with TCO-protected Dox demonstrated the excellent catch-and-release ability of tetrazine functionalized hydrogel, validating the use of the local activation strategy for administration of cytotoxic drugs in vivo.
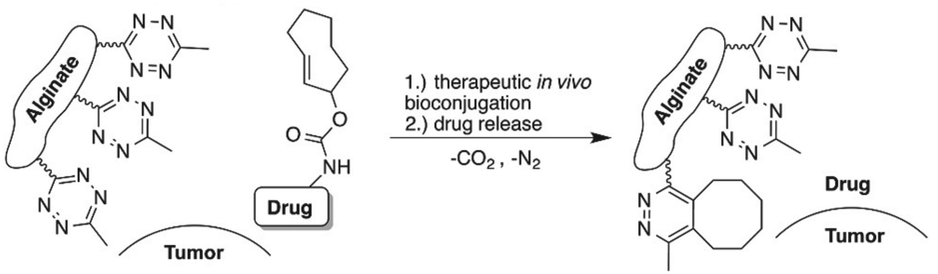 | ||
| Fig. 21 Local activation strategy for “catch and release” of drugs. Reproduced from ref. 131. https://pubs.acs.org/doi/10.1021/acscentsci.6b00150 with permission from ACS, copyright (2016). Further permissions related to the material excerpted should be directed to the ACS. | ||
Other polysaccharides
Other semiflexible polysaccharides such as cellulose, chitin, and chitosan (Fig. 22) can similarly function as scaffolds. Cellulose consists of β-D-glucopyranose units linked by 1,4-glycosidic bonds.133 The primary alcohols of cellulose have been functionalized with amine ligands to prepare environmentally-friendly regenerative catalysts for formation of C–C bonds in organic synthesis (Fig. 23A).134 Cellulose also functioned as an anion exchange resin to remove chromate ions from aqueous solutions.135 To enable interaction with chromium ions, quaternary ammonium groups were introduced on the scaffold via reaction of the primary hydroxyls with succinic anhydride, followed by conjugation of triethylenetetraamine and subsequent reaction with methyl iodide (Fig. 23B). The resulting cellulose scaffold effectively removed chromate from water. | ||
| Fig. 22 Structures of cellulose, chitin and chitosan and their primary functionalization positions. | ||
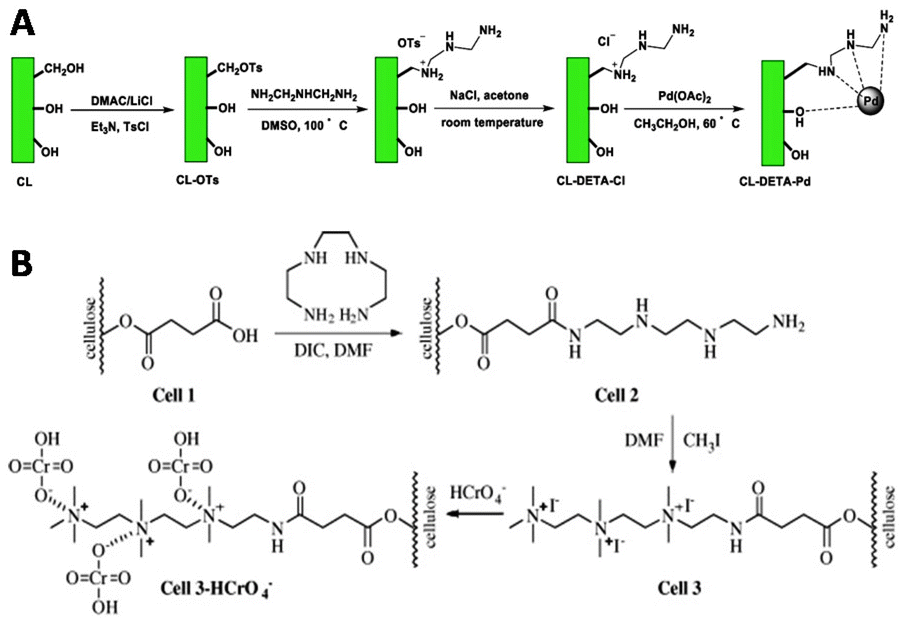 | ||
| Fig. 23 A. Introduction of amine ligands via tosylation of the primary hydroxyls on the cellulose chain followed by a substitution reaction with the amine ligand and subsequent coordination with Palladium (Pd(OAc)2) to afford a regenerative catalyst for the formation of C–C bonds. Reprinted from ref. 134 with permission from Elsevier, copyright (2019). B. Functionalization of the cellulose scaffold with quaternary ammonium groups via reaction of the primary hydroxyls with succinic anhydride, followed by conjugation of triethylenetetraamine and subsequent reaction with methyl iodide. Reprinted from ref. 135 with permission from Elsevier, copyright (2009). | ||
Chitin and chitosan biopolymers are extensively investigated for wound healing and tissue engineering applications.136 Chitin is a linear polymer of repeating N-acetyl-D-glucosamine units. Chitosan is the deacetylated form of chitin and consists of randomly distributed β(1,4)-linked D-glucosamine and N-acetyl-D-glucosamine. Yang et al. conjugated cysteine to chitin nanofibers to prepare a material for the removal of arsenic from aqueous solutions.137 Chitin was deacetylated with sodium hydroxide and cysteine moieties were conjugated on chitin nanofibers via reaction of the free amines on chitin with the NHS-activated carboxylic acids of cysteine (Fig. 24). The small size of the cysteine-functionalized nanofibers gave rise to a high surface-to-volume ratio which provided abundant sites for arsenic absorption via interaction with the cysteine thiols. As a result, the absorption capacity of the scaffold was higher than existing absorption systems, attributed to multivalency, and likely to the good availability provided by the semiflexible chitin scaffold.
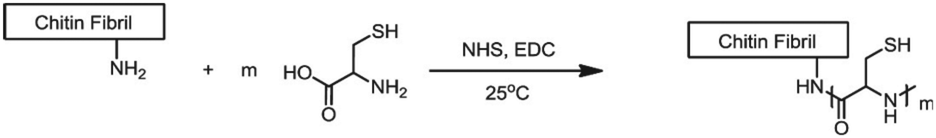 | ||
| Fig. 24 Conjugation of cysteine moieties on deacetylated chitin nanofibers. Reprinted from ref. 137 with permission from Elsevier, copyright (2015). | ||
Liu and coworkers modified chitosan with decanoic acid to develop a wound dressing material that enhances wound healing.138 Decanoic acid (DA) was conjugated to chitosan via an NHS-mediated coupling with the amine groups of D-glucosamine (Fig. 25A). In vivo wound healing studies in rats with full-thickness excisional wounds showed accelerated wound healing for gauze dressings soaked in DA functionalized chitosan.
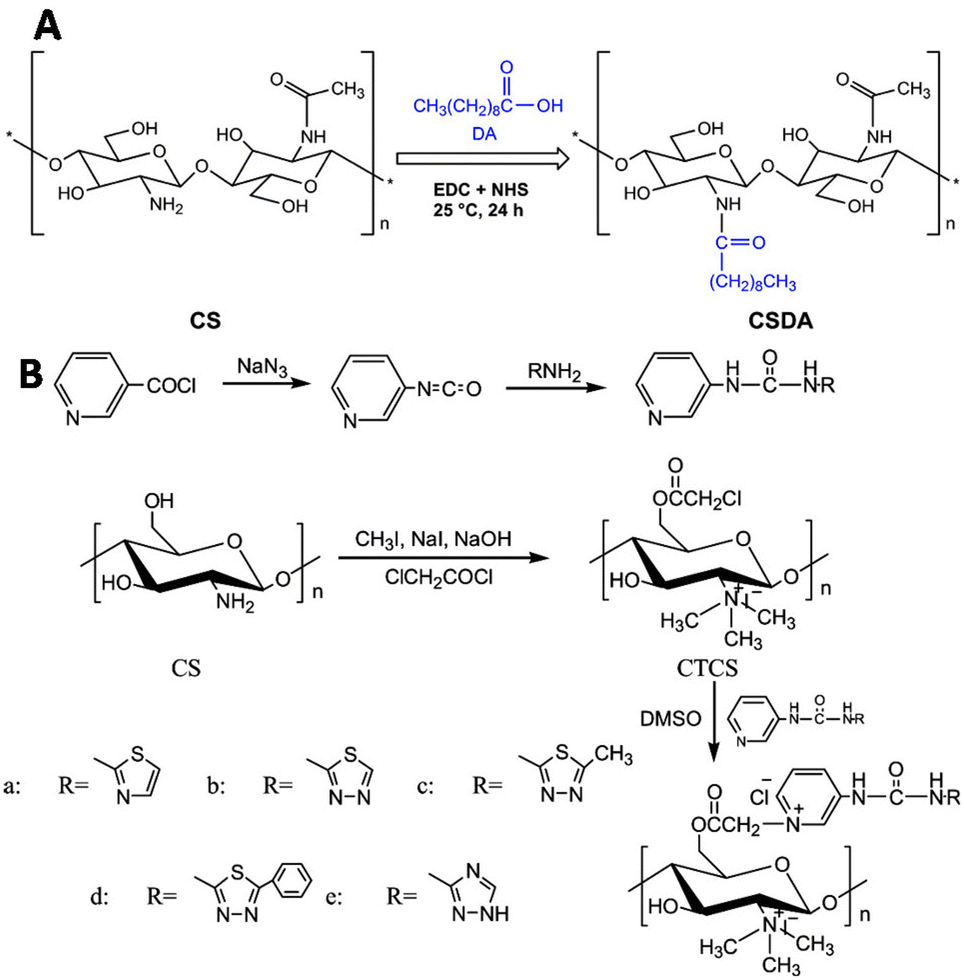 | ||
| Fig. 25 Functionalization of chitosan scaffolds. A. Introduction of decanoic acid (DA) via NHS activation of the carboxylic acid of DA followed by reaction with the amine groups of chitosan. Reprinted from ref. 138 with permission from Elsevier, copyright (2019). B. Conjugation of urea compounds to the chitosan scaffold via chloroacetylation of the primary hydroxyls on the chitosan chain with 2-choroacetyl chloride followed by conjugation of the urea compounds through reaction with the nitrogen of the heterocyclic urea compounds. Reprinted from ref. 120 with permission from Elsevier, copyright (2019). | ||
Urea groups were conjugated to a chitosan scaffold for the development of a biomaterial with antifungal and antioxidant applications.139 The primary hydroxyls of chitosan were chloroacetyled and further conjugated with nitrogen-containing heterocycles. The amine groups of chitosan were reacted with methyl iodide to form quaternary ammonium groups (Fig. 25B), which improved its water solubility. The functionalized chitosan scaffold showed increased antifungal and antioxidant activities in comparison with chitosan.
Overall, the functionalization of polysaccharides primarily occurs at the available functional groups of the backbone, quite similar to collagen derivatization. Polysaccharides, however offer the advantage that the density and variety of these groups is very high, giving access high loading capacities. The downside of this class of semi-flexible polymers is the poor spatial control, unless the chain-end is specifically targeted.
Synthetic polymers
Despite the rapidly increasing efforts in the field, semiflexibility in synthetic polymers remain a rare find. In contrast to semiflexible biopolymers, synthetic polymers provide more strategies to introduce substituents. Monomer modification and post-functionalization approaches allow grafting of substituents on polymer chains to give rise to well-defined polymer scaffolds. In the following sections, we will review common conjugation approaches of some of the major classes of synthetic semiflexible polymers, including synthetic polypeptides, conducting polymers and polyisocyanides. Moreover, we include applications of the grafts in biosensing, bioimaging, drug delivery, optoelectronics, photovoltaics and tissue engineering.140–145Synthetic polypeptides
The amino acid building blocks that comprise synthetic polypeptide scaffolds retain various functional handles that enable the introduction of virtually any substituent. Amine, carboxyl, hydroxyl and thiol functional groups on amino acids can be utilized for conjugation strategies with functional molecules.Typically, synthetic polypeptides are prepared via SPPS or ring opening polymerization (ROP) of α-amino acid N-carboxyanhydrides (NCAs). SPPS is generally used to produce oligopeptides whereas ROP enables the synthesis of polypeptides with high molecular weights.146 NCA monomers are conveniently synthesized in large scales from the corresponding α-amino acids147 and living polymerization techniques yield polypeptides with low polydispersity (PDI) and allow for the controlled incorporation of multiple amino acids.148,149 The use of (orthogonal) protection strategies allows for subsequent conjugation of substituents to amino acid side chains at specific positions along the polymer chain, resulting in a well-defined scaffold. Additionally, aminolysis-based chemoenzymatic polymerizations of peptides have been reported but reaching high molecular weights and good sequence control remains challenging.150
Many functional molecules have been introduced on the synthetic polypeptide backbone, including crosslinks, fluorescent dyes or binding sites for drug delivery.151–153 Frequently, the applied functionalization strategies follow unselective post-polymerization conjugations, analogous to what is described in the biopolymer section of this review. Here, we will zoom in on alternative strategies that employ the polymerization reaction or use monomer modification to introduce substituents.
The Pochan group used SPPS to develop small computationally designed chains that assemble into well-defined semiflexible tetrameric bundles (bundlemers).154 A Michael addition of the maleimide groups at the N-terminus with a thiol functionalized linker yields co-polymer chains (Fig. 26A). The introduction of functional handles in the peptide chain enables further conjugation with substituents in any desired pattern. Pouchan functionalized the peptide chains with an alkyne handle to conjugate azide-terminated PEG polymers to the polypeptide, producing polymer chains with a regular nanometer scale pattern (Fig. 26B), which they confirmed with atomic force microscopy studies.
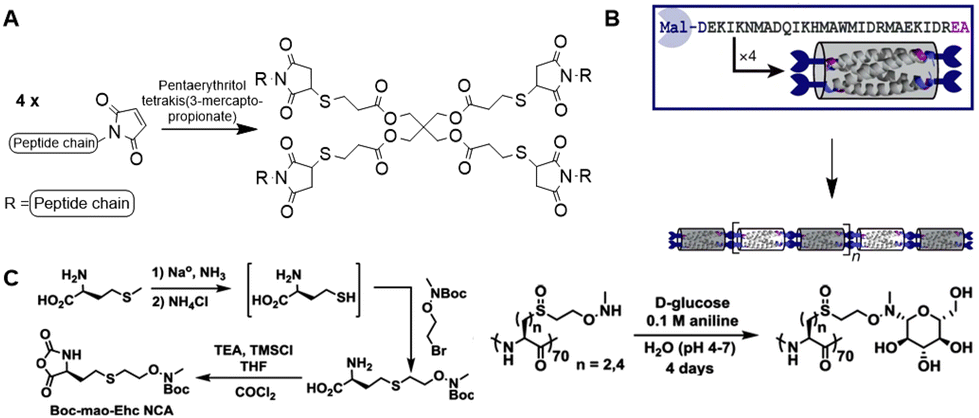 | ||
| Fig. 26 A. Michael addition of the maleimide-functionalized peptide bundlemers with a thiol functionalized linker to afford a semiflexible polymer chain. Adapted from ref. 154. B. Single letter code and schematic presentation of peptide bundlemers and their assembly. Reprinted from ref. 154 with permission from Nature Springer, Nature, copyright (2019). C. Synthesis of NCA monomer (left) and conjugation of D-glucose to polypeptide scaffold (right). Reprinted from ref. 157 with permission from American Chemical Society, copyright (2019). | ||
ROP has been applied to develop homopolypeptide scaffolds that can be functionalized post-polymerization. Anas et al. prepared alkyne functionalized poly(cysteine) via NCA polymerization, to which they grafted azide-containing polymer chains to generate self-assembling vesicles.155 A poly(glutamic acid) scaffold bearing furan side chains was developed via the same strategy, which is compatible with conjugation of maleimide-modified Dox via a Diels–Alder reaction.156 Deming and coworkers synthesized polypeptides with N-methylaminooxy groups via ROP to enable conjugation of unmodified sugars (Fig. 26C).157
The synthesis of neoglycopolypeptides that mimic glycoproteins is often laborious, as it requires multistep monomer synthesis and the use of protecting groups. Development of a polypeptide scaffold that allows post modification with unmodified sugars enables the synthesis of neoglycopolypeptides in a facile, versatile manner. To this end, NCA monomers carrying an aminooxy handle were developed from L-methionine (Fig. 26C) after deprotection of the thiol group followed by a substitution reaction with 2-(N-Boc-N-methylaminooxy)ethyl bromide and subsequent conversion into the NCA monomer using phosgene. ROP polymerization followed by Boc-deprotection yielded N-methylaminooxy functionalized homopolypeptide chains. These peptide scaffolds were conjugated with a variety of mono- and disaccharides in high yields, which makes them attractive for development of glycoprotein mimics. The straightforward synthesis and functionalization of the polypeptide chain demonstrates its versatility as a scaffold.
Copolymerization techniques broaden the potential of polypeptide scaffolds by enabling the spatially controlled introduction of substituents along the polymer backbone. The group of Lei incorporated catechol (DOPA) on a polypeptide scaffold to prepare copolymer glues as surgical adhesives.158 Various polypeptide–Pluronic–polypeptide block-copolymers were synthesized through ROP of L-DOPA-N-carboxyanhydride with L-arginine-NCA, L-cysteine-NCA, or ε-N-acryloyl lysine-NCA (Fig. 27A). The polymerization reaction was initiated by Pluronic-L31, a thermo-responsive polymer that ensures gelation of the copolymer in aqueous solution under physiological conditions. The pendant catechol groups on L-DOPA form (non)covalent bonds with various substrates. Via this mechanism, L-DOPA can form homo-crosslinks or with the alkyne or thiol groups of cysteine and lysine units via a Michael type addition (Fig. 27B). The guanidinium ions on the scaffold form salt bridges with a protein surface, which together with the catechol-substrate interactions gives attachment of the polymer to skin (Fig. 27B). In vivo application of the copolymer solutions demonstrated its efficiency in the wound healing process, highlighting that polymerization of functionalized NCAs is an excellent strategy to introduce substituents on a polypeptide scaffold.
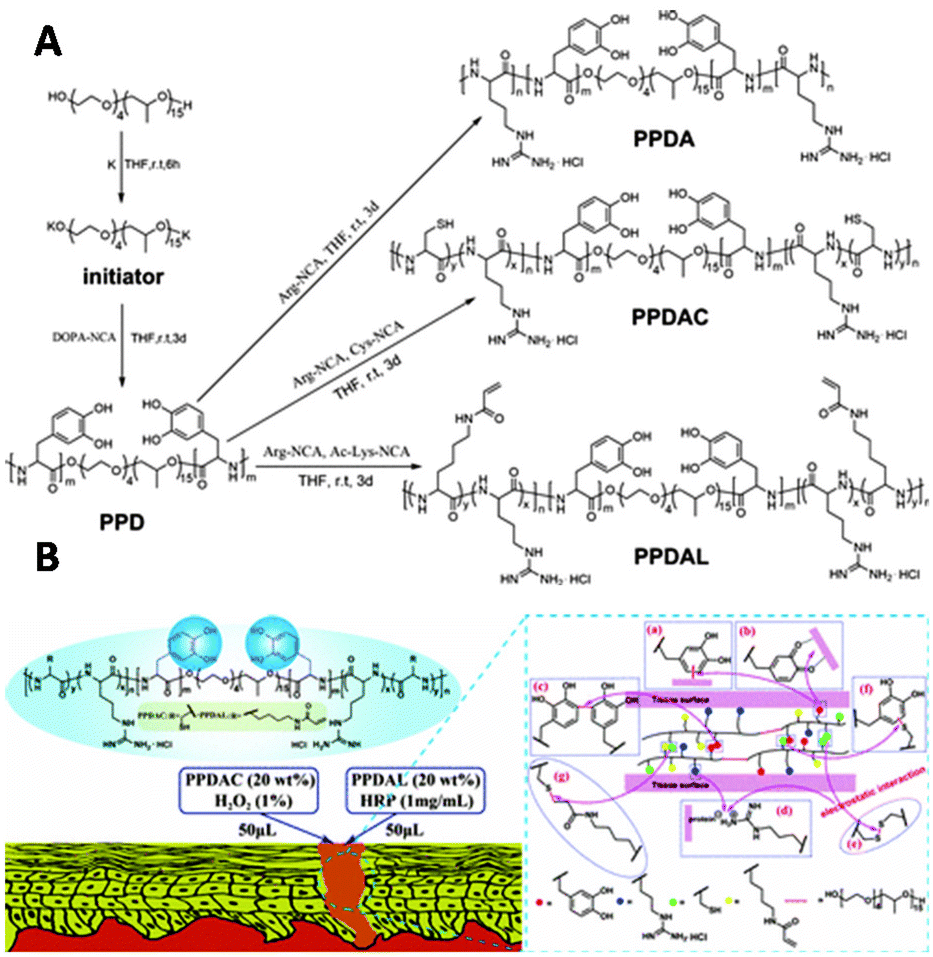 | ||
| Fig. 27 A. Synthesis of PPDA (from L-arginine NCA), PPDAC (from L-cysteine NCA) and PPDAL (from ε-N-acryloyl lysine-NCA) polypeptides. Reprinted from ref. 158 with permission from American Chemical Society, copyright (2017). B. Schematic representation of polymer interactions with skin substrate. Reprinted from ref. 158 with permission from American Chemical Society, copyright (2017). | ||
In addition, ROP-synthesized polypeptide copolymers with structure poly(lysine)x(alanine)y were crosslinked with 6-arm PEG-glutarate-NHS via amide bond formation with the amino groups of the lysine units.159 This crosslinking reaction afforded hydrogels with significant cell adhesion, proliferation and antibacterial activities that can function as scaffolds for wound healing applications. A similar method was applied to crosslink the glutamic acid units on PEG-poly(glutamine)x(phenyl alanine)y block co-polymers.160 The three blocks in the polymer chain form multi-compartment micelles that enable co-delivery of hydrophilic and hydrophobic anti-cancer drugs. Crosslinking of the glutarate carboxylic acids with 1,2-ethylenediamine stabilizes the micelle towards proteases.
Nucleophiles such as amines can initiate ROP and are, as such, readily introduced at one chain end of polypeptide scaffolds. This method is extensively applied to introduce alkyne or azide handles, fluorescent dyes, nanodiamonds and a wide variety of polymer chains.161–167 Wei and coworkers applied this strategy to develop amphiphilic poly(amino acid)s that self-assemble into luminescent polymer nanoparticles (LPNs) starting from a amine containing aggregation induced emission dye to polymerize OEG-functionalized glutamate (OEG-glu) NCA.163 The resulting dye functionalized polyglutamate scaffolds (dye-OEG-Pglu) formed LPNs in aqueous solution with the hydrophobic dye aggregated in the core while the hydrophilic OEG chains functioned as a shell. The LPNs exhibited efficient luminescence and were potent for cell imaging applications. Initiating polymerization with the dye ensured its position on the polymer chain end, which resulted in a scaffold with desired amphiphilic properties for LPN formation. Dong and coworkers exploited a similar method to prepare amphiphilic block copolymers that form micelles and can function as light responsive drug carriers.168 ROP of photo responsive S-(o-nitrobenzyl)-L-cysteine (NBC) NCA monomers was initiated by amine-functionalized poly(ethylene glycol) (PEG). The resulting micelles were loaded with Dox. Cleavage of the photosensitive nitrobenzyl moiety by UV irradiation resulted in controlled release of Dox from the micelles as the hydrophobic binding interactions between Dox and nitrobenzyl reduced after cleavage. Although functionalization of a scaffold via ROP initiation is an efficient strategy for the introduction of substituents in a controlled manner, its versatility remains limited as functional groups can only be introduced at one chain end.
Polypeptides offer many advantages, including high versatility and availability of the building blocks. While (NCA) monomer modification followed by ROP or SPPS and post functionalization are great techniques to functionalize polypeptide scaffolds with high functional group densities, ROP initiation is very useful for development of amphiphilic polypeptides. SPPS offers sequence control and with that high spatial control. Through chain extension strategies, also longer peptide polymers are within reach.
Conducting polymers
Conducting polymers combine useful properties of both organic molecules and semiconductors and have formed a mature research field over the past decades. Many polymers, however are intrinsically rigid and poorly processable.169 Introduction of substituents adds flexibility to the rigid backbone of conducting polymers, which improves their processability and provides opportunity to add more functionality to the scaffolds.170Polyacetylene (PA), polyaniline and polypyrrole are semiflexible conducting polymers that have been functionalized for the development of various electric devices and chemical sensors. Substituted polythiophenes are considered more flexible.171 Functionalization strategies for polythiophenes have been reviewed earlier.172
Polyacetylene
Research on unfunctionalized PA (Fig. 28A) stagnated due to a lack in processability and stability.173 Functionalized PAs, however remain soluble in many solvents and are stable in air for a long period of time, which makes these polymers easier to handle.173 The carbon–carbon alternating double bonds in the PA backbone contribute to a variety of properties such as conductivity, gas permeability, and photo- and electroluminescence.173 These findings induced a regained interest PAs and their applications as a scaffold. A variety of transition metal catalysts have been used to form functionalized PAs via the insertion or metathesis polymerization mechanisms. Currently, Rh-based catalysts are used most frequently as they enable living polymerization and the synthesis of well-defined PA (block co)polymers with high initiator efficiency.174 Due to the insolubility of unsubstituted PAs, post-polymerization functionalization is not feasible. Therefore, substituents are generally introduced via modification of acetylene monomers. One or two substituents can be placed on each repeating unit in the chain. In this review, we highlight a number of examples of substituted polyacetylenes, in which the polyacetylene backbone functions as a clear scaffold. We refer the reader to an extensive overview on substituted polyacetylenes for more information.174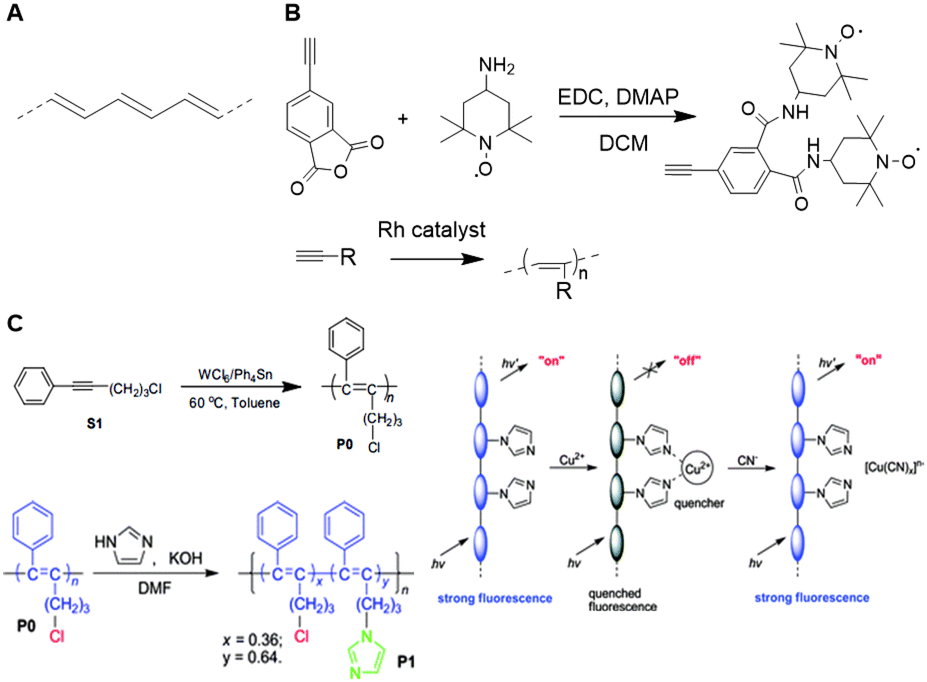 | ||
| Fig. 28 A. Structure of PA. B. Schematic overview of the synthesis of TEMPO substituted acetylene monomers and subsequent polymerization. Reprinted from ref. 183 with permission from John Wiley and Sons, copyright (2007). C. Synthesis of disubstituted PA and post functionalization of the PA scaffold with imidazole (left). Schematic overview of the probe, where interaction with Cu2+ quenches the fluorescent signal and subsequent interaction with CN− restores the fluorescence (right). Reproduced from ref. 184 with permission from the Royal Society of Chemistry, copyright (2008). | ||
Monosubstituted acetylene polymers are readily synthesized through direct polymerization,175 which tolerates a wide variety functional groups, different polarities and a substantial bulkiness.176–182 An example of such monosubstituted PA scaffold was prepared by Masuda and coworkers, who functionalized PA with 2,2,6,6-tetramethyl-1-piperidinoxy (TEMPO, Fig. 28B) radicals to develop organic radical batteries.183 Oxidation of the nitroxy group of TEMPO to form an oxoammonium cation is reversible and this process can be used to develop cathode active materials. PA is a particularly suitable scaffold due to its semiconductive properties and the formation of a rigid scaffold. The direct polymerization method ensures quantitative incorporation of the TEMPO groups on the scaffold for optimal charging and discharging.
Most disubstituted PAs cannot be synthesized via direct polymerization of their monomers due to deactivation of the catalyst by active protons or coordinative ligands in the monomers.175 Tang and coworkers developed a strategy that allows introduction of a second substituent after polymerization.184 An acetylene monomer with a phenyl substituent and a chlorine handle was polymerized and subsequently functionalized with imidazole via nucleophilic substitution of the chlorine (Fig. 28C). The resulting disubstituted scaffold could function as a chemosensor for Cu2+ and CN− through quenching or recovery of the fluorescent signal of the PA scaffold. These results show that incorporation of an active handle such as chlorine on a polyacetylene scaffold facilitates post-polymerization functionalization and provides a versatile strategy to develop multiple disubstituted polyacetylene materials.
Polyaniline
Over the past few decades, polyaniline (PANI, Fig. 29A) has materialized as a key conducting polymer for commercial applications.185 Its easy synthesis, stability and controlled electrical properties make PANI an attractive functional scaffold.186,187 Conventional methods to synthesize PANI chains include chemical or electrochemical oxidative polymerization of aniline.188 Despite the facile synthesis, both methods result in inconsistent PANI products instead of the desired nanofibrillar morphology.189 Alternatively, interfacial synthesis of PANI allows spontaneous reaction between aniline and an oxidant such as ammonium peroxydisulfate at the interface between two immiscible solvents. This method results in the formation of PANI nanofibers with uniform diameters between 30 and 50 nm and lengths up to micrometer scale.190 Although PANI synthesis is experimentally easy, its polymerization mechanism is intricate; for detailed information, we refer to a review.187 | ||
| Fig. 29 A. Structures of PANI, from top to bottom; leucoemeraldine (reduced), emeraldine (partly oxidized) and pernigraniline (fully oxidized). B. Reaction mechanism of surface modification of PANI nanostructures with thiol compounds. Adapted from ref. 194. C. Schematic overview of the preparation of an antibody sensor: (i) introduction of amine or carboxylic acid handles via surface modification; (ii) conjugation of antibodies through EDC-mediated reactions; (iii) detection of secondary antibody, labeled with horseradish peroxidase (HRP). Reprinted from ref. 186 with permission from Elsevier, copyright (2011). D. Schematic overview of the synthesis of a DNA probe: functionalization of Pt/PANI-Au-NP with β-mercaptoethylamine, followed by conjugation of oligonucleotides (dA) and detection of target nucleotides (dT). dTs were detected via cyclic voltammetry (DPV). Reprinted from ref. 140 with permission from Elsevier, copyright (2012). | ||
PANI can be functionalized via modification of aniline or via post-polymerization modification. Direct introduction of substituents on the aniline monomer can induce steric challenges during the polymerization with the possibility that the conducting properties of the scaffold are compromised.191 As such, surface modification is frequently used as a post-polymerization functionalization strategy for PANI nanofiber functionalization.192 Surface modification allows covalent attachment of functional groups via nucleophilic addition to the quinoid ring of the PANI backbone.191,193 The Hanks group applied surface modification to introduce thiol-terminated PEG on PANI nanostructures to improve PANI water solubility (Fig. 29B).194 Thiols react efficiently with PANI to change the polymers’ surface properties without affecting its conductivity. The authors found that the degree of PEG-SH substitution increases with temperature, which in turn increases solubility and promoted further access to the PANI scaffold. This effect was used to control the PEG coverage of the PANI scaffolds. The prepared PEGylated polyaniline polymers were water dispersible and maintained their original morphology and electroactivity, showcasing that this approach is a suitable technique to modify polyaniline scaffolds in a controlled manner and without disrupting the properties of polyaniline.
O'Kennedy and coworkers used a similar surface modification technique to immobilize antibodies on polyaniline nanofibers for the development of immunosensors.195 PANI nanofibers were prepared via interfacial polymerization and subsequently modified with mercaptoundecanoic acid, mercaptopropionic acid or cysteamine via employing the reactivity of the thiol nucleophiles.193,196 IgG antibodies were conjugated to both carboxyl and amino functionalized nanofibers via ethyl diaminocarbodiimide (EDC) chemistry to form amide bonds (Fig. 29C). Cyclic voltammetry experiments showed direct electrical communication between the polyaniline nanofibers and the covalently linked peroxidase-linked antibodies, which forms the working mechanism of the antibody conjugated PANI scaffolds as immunosensors. Furthermore, the amino and carboxylate functionalized polyaniline polymers provide versatile scaffolds for further derivatization.
Analogous to previous examples, Gangopadhyay et al. applied interfacial polymerization to incorporate gold nanoparticles on PANI nanowires for biosensing applications,140 where the electrical properties of PANI change upon biomolecule binding. To this end, the gold nanoparticle (AuNP) functionalized PANI nanowires were deposited on a platinum electrode and the gold was functionalized with β-mercaptoethylamine (Fig. 29D). The introduced primary amine facilitates attachment of a variety of biomolecules. An oligonucleotide (dA) was conjugated to the electrode to monitor its hybridization and thus sensing ability of a target nucleotide (dT) (Fig. 29D). Hybridization with the target nucleotide at a minimal concentration (10−18 M) led to peak current values and changes in impedance, demonstrating the extreme sensitivity of the sensing platform. The AuNP-functionalized PANI nanowires on the electrode facilitate introduction of various functional groups, which enables binding to a variety of biomolecules and thus generates a multifunctional biosensing platform.
Polypyrrole
Because of its easy synthesis, high conductivity and stability in the oxidized state, the interest in polypyrrole (PPy, Fig. 30A) remains high.197 PPy is readily obtained by electrochemical198 or oxidative199 polymerization of pyrrole. For both methods holds that controlling the oxidation potential of the reaction mixture increases the conductivity of the synthesized polymers and results in the formation of polypyrrole with a homogenous, fibril-like surface morphology.199,200 | ||
| Fig. 30 A. Structure of PPy. B. Synthetic route towards biotin functionalized pyrrole. Adapted from ref. 202. C. Synthetic route towards amine and COOH functionalized pyrrole monomers and subsequent polymerization of pristine and functionalized pyrrole. Adapted from ref. 203 and 204. D. Synthetic route towards NHS-functionalized pyrrole. Adapted from ref. 206. E. Overview of PPy functionalization with porphyrin (TTP) via metal coordination (SnTPP-PPy) and a combination of covalent and coordination (TPP-PPy-SnTPP). Reproduced from ref. 209 with permission from the Royal Society of Chemistry, copyright (2019). F. Synthetic route towards RGD capped PPy. Reprinted from ref. 209 with permission from American Chemical Society, copyright (2006). | ||
Substituents are introduced on the polypyrrole scaffold via the direct modification of the pyrrole monomer or via the introduction of a handle on pyrrole followed by post-polymerization functionalization. Bidan and coworkers synthesized homopolymer biotin-functionalized PPy sensors by polymerizing biotin-modified pyrrole (Fig. 30B).201 After avidin treatment of the deposited polymer, the layer serves as an electrode that can immobilize biotinylated enzymes or oligonucleotides.202
Lee and Schmidt prepared amine functionalized pyrrole scaffolds via a similar method.203 Aminopropyl-modified pyrrole was polymerized via electrochemical polymerization with and without pristine pyrrole to afford homo- and copolymers (Fig. 30C). In line with earlier work,204,205 the authors found that modified pyrrole is poorly incorporated, likely due to increased steric interactions. Introduction of positive charges on PPy by introducing amine groups resulted in a scaffold that improved adhesion of fibroblasts and Schwann cells and shows promise as a substrate for cell culture applications. Schmidt and coworkers used the same monomer modification strategy to develop carboxylic acid functionalized PPy scaffolds.204 The cyano group in 1-(2-cyanoethyl)pyrrole was hydrolyzed to a carboxylic acid using KOH and HCl to afford 1-(2-carboxyethyl)pyrrole which was subsequently polymerized to afford a carboxylic acid-functionalized pyrrole homopolymer (Fig. 30C). Via a similar monomer modification strategy, PPy scaffolds were synthesized from NHS-functionalized pyrrole monomers (Fig. 30D).206 Such amine-, carboxylic acid-, and NHS-functionalized polypyrroles have been used as a scaffold for post-functionalization with oligonucleotides, peptides or electrochemical probes.205–207
As steric hindrance of bulky pendants interferes with polymerization, direct monomer modification is not always advantageous. Post polymerization functionalization circumvents such limitations. Wang et al. applied this strategy to conjugate porphyrins on a polypyrrole-like scaffold.208 4-[Di(1H-pyrrol-2-yl)methyl]phenol monomers were polymerized using ammonium perchlorate as oxidant. Next, the scaffold was functionalized (Fig. 30E) with porphyrins covalently (via a straightforward substitution reaction) and/or noncovalently (through coordination to the metal inside the porphyrin ring). Charge transfer interactions between the porphyrins and the conducting polymer scaffold gives rise to unique electrooptical properties.
To facilitate spatially controlled introduction of substituents, PPy scaffolds can be capped with a functionalized pyrrole monomer via electrochemical coupling. The group of Schmidt applied this method to prepare a carboxylic acid end-capped PPy scaffold that was used for the conjugation of the cell-adhesive tripeptide arginylglycylaspartic acid (RGD in single letter code) to develop a cell culture scaffold.209 PPy was electrochemically synthesized on an electrode. The resulting films were modified with a carboxylic acid functionalized pyrrole via a final electrochemical coupling and were subsequently decorated with RGD-peptides through sulfo-NHS and EDC chemistry (Fig. 30F). The selective introduction of the carboxylic acid handle at the chain end of PPy gives rise to a well-defined scaffold structure without compromising electrical conductivity and mechanical integrity. Jang et al. applied an equivalent strategy, albeit with a pyrrole monomer already with the RGD peptide attached. Electrochemical introduction of the functional monomer was controlled by the deposition time.210
Analogous to initiator modification, electrochemical coupling facilitates scaffold functionalization with high spatial control, but its application remains limited as it only allows introduction at the polymer chain end.
Polyisocyanides
We end this review with a class of polymers named polyisocyanides, which is one of the largest classes of synthetic semiflexible polymers. Although known from the 1970s, recent developments moved these polymers to the forefront of various biomedical applications. Because of the extensive research in the field, we divided poly(isocyanides) (PICs) further into poly(isocyanopeptides) (PICPs) and water soluble PICPs.Poly(isocyanides)
Similar to many bio-macromolecules, semiflexible PICs possess a helical conformation211 as a result of the polymerization process (Fig. 31A).212,213 The most common methods to prepare PIC involve transition metal complexes of Ni(II), Pd(II)–Pt(II) or Rh(III).214 The steric demands of each carbon in the polymer backbone bearing a substituent restricts rotation of the single bonds in the backbone, which stabilizes the helical structure. This stable helical conformation makes PIC polymers suitable building blocks for development of chiral macromolecules or compounds that mimic bio-macromolecules. | ||
| Fig. 31 A. Formation of stable helix by polymerizing tert-butylisocyanide with a nickel(II) catalyst. Reproduced from ref. 214 with permission from the Royal Society of Chemistry, copyright (2011). B. Isocyanide polymerization with an alkyne-modified nickel(II) catalyst to introduce an alkyne handle on the polymer chain-end. Reprinted from ref. 225 with permission from John Wiley and Sons, copyright (1969). | ||
The preserved helical conformation of PICs provides an excellent scaffold to develop highly ordered materials with applications in optoelectronics, photonics, memory devices and cell imaging.143,215–217 Isocyanide monomers are readily obtained via dehydration of the corresponding formamides. Substituents are introduced on PIC scaffolds via direct modification of the isocyanide monomer or by introduction of an active handle targeted towards post-polymerization functionalization.
Takei et al. modified isocyanide monomers to prepare arrays of porphyrins for development of light-harvesting systems.218 Aryl-isocyanide monomers with porphyrin pendants were prepared via the introduction of a nitrite, which was reduced to an amino-group followed by formylation and dehydration into an isocyanide. The monomers were polymerized to give precisely stacked porphyrin arrays. The porphyrin bulkiness combined with π–π stacking of adjacent aryl moieties in the side-chains further stabilized the helical conformation and the well-defined arrangement of the porphyrins in the stacks. Various chiral groups, chromophores, fulvenyl moieties, metal complexes, perylenes and saccharides have been introduced on poly(aryl isocyanide) scaffolds using the same monomer modification strategy.216,219–223 Direct monomer modification has also been applied to introduce carbazoles and a variety of bulky pendants on poly(isocyanide) chains.143,224
Although polymerization of modified monomers is a successful strategy to prepare functionalized PIC scaffolds with high spatial control, it is not a versatile strategy as introduction of a new substituent requires a completely new monomer synthesis and consecutive polymerization. Decoration of the PIC scaffold with handles such as azido, alkyne or thiol groups allows for post-polymerization functionalization, which enables facile derivatization of the scaffold towards multiple purposes. The Weck group integrated a terminal alkyne handle on PIC via modification of the nickel(II) catalyst (Fig. 31B).225 This approach enables coupling of the polymer scaffold to a variety of substituents such as dyes or other polymers to generate (block) copolymers.222,226,227 While the introduction of a handle via initiator modification is an elegant functionalization strategy, its applications are restricted, analogous to electrochemical coupling and ROP initiator modification mentioned earlier.
A method to enable post-functionalization of the full PIC scaffold was develop by Wu and coworkers217 who incorporated a pentafluorophenyl (PFP) ester on the PIC side chains, which provided a handle for covalent crosslinking. Amphiphilic block-copolymers were synthesized through one-pot sequential polymerization of PFP-functionalized phenyl isocyanide monomers (Fig. 32, M1), tetraphenylethene-functionalized phenyl isocyanide monomers (M2) and hydrophilic phenyl isocyanide monomers (M3). PFP proved to be an efficient handle for post polymerization functionalization and subsequent micelle formation. In later work, it has been used to introduce a variety of molecules such as pyrene and PEG chains.228,229
 | ||
| Fig. 32 Synthetic route for the successive copolymerization of pentafluorophenyl-, tetraphenylethene-, and hydrophilic phenyl PICs (PFPPI-TPEPI-HPPI-HPPI-polymers) (left). Schematic representation of micelle formation and subsequent luminescent behavior and reducing agent triggered payload release. Reprinted from ref. 197 with permission from American Chemical Society, copyright (2017). | ||
Poly(isocyanopeptides)
Nolte and co-workers demonstrated that two or three amino acids directly substituted to the isocyanide strongly stabilize the helical conformation via the formation of a network of intramolecular hydrogen bonds between the amide groups of the nth and (n + 4)th side chains (Fig. 33A).230–233 This discovery gave rise to the formation of a subclass of poly(isocyanides) termed poly(isocyanopeptides) (PICPs) and circumvented the use of bulky substituents on the isocyanide. The stable helical structure of the polymer chain introduces an increased persistence length,234 thereby facilitating the arrangement of substituents in well-defined arrays, which is favorable for development of functional materials in the field of photonics and electronics.235 | ||
| Fig. 33 A. Schematic representation of the hydrogen bonding network between the pendant alanine units and 41 helical conformation of PICs/PICPs. Reprinted from ref. 242 with permission from John Wiley and Sons, copyright (2010). B. Post-functionalization of PICPs bearing acetylene groups via CuAAC reaction from azide-pendant monomers. C. Post-modification of PICPs through conjugation of substituents with the thiol groups in the side chains. Reprinted from ref. 242 with permission from John Wiley and Sons, copyright (2010). | ||
Substituents such as carbazoles, perylenes, porphyrins and thiophenes were introduced on a PICP scaffold via the monomer modification strategy previously described in this paragraph.215,236–238 The well-defined alignment of perylenes along the polymer backbone resulted in overlapping pathways along which excitons and electrons rapidly migrate, showing potential application in the field of optoelectronics and photovoltaics.144,215,239
While monomer modification yields full substitution of the backbone, monomer synthesis is laborious and requires optimization of polymerization conditions for each monomer. To overcome this disadvantage, monomers carrying an acetylene group were incorporated on the polymer chain to provide handles for post-functionalization via click-chemistry.240 Researchers demonstrated that this route allows the introduction of perylenes, phthalocyanines and OEG substituents (Fig. 33B).235,241
Nolte's group prepared PICPs bearing an alanine-cysteine motif in the side chain to develop a versatile scaffold for post-polymerization conjugation of various active molecules via reaction with the thiol (Fig. 33C).242 Thiol-maleimide click chemistry between the scaffold and maleimide functionalized pyrenes resulted in a well-defined arrangement of chromophores. To demonstrate the versatility of the PICP scaffold, pyrene derivatives bearing different thiol-click substrates were conjugated. Similarly, the iodoacetamide- and thioester-modified pyrenes were efficiently conjugated to the scaffold and formed well-defined arrangements. With these results, the researchers show that incorporation of cysteines on the PICP side-chains provides a versatile scaffold for the highly efficient introduction of substituents. Additionally, Nolte and coworkers decorated the side chains with OEG tails to improve polymer solubility in water while retaining the helical conformation.242
Water-soluble poly(isocyanopeptides)
For polyisocyanides to enter the biomedical field, water-soluble derivatives have been prepared. Kitto et al. conjugated azido functionalized OEG chains to acetylene bearing PICPs thrugh the efficient CuAAC click reaction.235 The post-functionalization, however led to a change in the helical conformation of the polymer. The alternative route, i.e. modification of the isocyanopeptide monomers with OEG tails followed by polymerization gave rise to water-soluble poly(isocyanopeptides) (wsPCIPs) that retained their stable helical conformation.243 This subclass of PICs have become one of the dominating examples of synthetic semiflexible polymers over the course of a few years. Aqueous solutions of the wsPICPs show a lower critical solution temperature and, reversibly, form gels upon heating.244 The gelation temperature Tgel depends on the environment245 as well as the exact molecular design and can be finetuned by the molecular weight,245 the peptides246 and, most strongly, by the length of the oligo(ethylene glycol) tail.247 In the next section, we will discuss the two workhorses of the family, the tri(ethylene glycol)PICP with Tgel ∼ 18 °C (gel at 37 °C) and the tetra(ethylene glycol)PICP with Tgel ∼ 40 °C (soluble at 37 °C) sequentially (Fig. 34A). | ||
| Fig. 34 A. Schematic structure of a (fictional) wsPICP comprising tri(ethylene glycol), tetra(ethylene glycol) and azido-tetra(ethylene glycol) side chains, respectively. B. Conjugation of BCN to GRGDS peptide and functionalization of a wsPICP scaffold with GRGDS via the SPAAC reaction. Reprinted from ref. 263 with permission from Nature Springer, Nature Materials, copyright (2015). C. Overview of the preparation of DNA-responsive wsPICP hydrogels (up, left) with the pH or thrombin dependent contraction and expansion motifs in the crosslinker. Copied from ref. 275. https://pubs.acs.org/doi/abs/10.1021/acs.biomac.7b00964. Reprinted with permission from ACS, copyright (2017). Further permissions related to the material excerpted should be directed to the ACS. D. Schematic overview of post-functionalization of a wsPICP scaffold with anti-CD3 and anti-CD28 via the SPAAC reaction between BCN-Sav and the azide handles on the scaffold followed by introduction of anti-CD3 and anti-CD28 through biotin-Sav coupling. Reprinted from ref. 278 with permission from American Chemical Society, copyright (2015). E. Schematic representation of the grafting block-copolymers (PIC brushes) containing allyl and azide handles through ‘SPAAC first’ or ‘graft first’ strategies and biotin-based grafting route to generate PICP-brushes through the NITEC reaction (below) that can be grafted to Sav surfaces. Copied from ref. 281. https://pubs.acs.org/doi/10.1021/acs.biomac.9b00385. Reprinted with permission from ACS, copyright (2019). Further permissions related to the material excerpted should be directed to the ACS. | ||
The hydrogels formed by tri(ethylene glycol)PICP display a fibrous structure,248 quite similar to gels made of biopolymers such as collagen, actin and intermediate filaments. Not only the semiflexible architecture is similar, also the mechanical properties closely mimic those of the biogels, including a strong strain-stiffening response, i.e. an increase in the stiffness (sometime 100-fold) upon small deformations of the gel244,249,250 even under complex multiaxial deformations.251 The gels have been used extensively as functional soft scaffolds in a wide range of applications. Wagener and colleagues demonstrated its use as a washable dressing for abdominal wounds.252,253 Yang and coworkers developed an injectable (at 5 °C) and in situ gel forming composite with Lipoxin A4-containing poly(lactic-co-glycolic acid) (PLGA) microparticles and found sustained release in treatment against periodontitis in animal studies.254,255 Xing and coworkers used the scaffolding capacity of the PICP backbone to induce conformational changes in water-soluble conducting polymers (polythiophenes and polyfluorenes).256–258 The stretched conformations red shifts the absorption band of the polymers, which allows the formation of reactive oxygen species (ROS) after irradiation with red light. The hydride forms an efficient photodynamic antimicrobial material with activity towards various pathogens. The efficiency further increases after the addition of nanoparticles which introduce also a thermal component.257 More in general, the addition of a second component, either particle or polymeric, is an efficient route towards responsive synergistic properties.259,260 For instance, the addition of poly(N-isopropylacrylamide) (PNIAPM) gives rise to a strong thermal stiffening response that can be optimized to a 10 fold increase in stiffness with a mere 1 °C increase in temperature.261
To benefit from the unique biomimetic properties of wsPICPs for cell culture applications, the scaffold usually requires functionalization with a cell-adhesive peptide; the most frequently used contain RGD. To this end, a monomer with a terminal azide handle is randomly copolymerized with the non-functional monomer.262 Peptides, equipped with a DBCO or BCN group are then cleanly introduced via a SPAAC reaction, resulting in a biofunctional and biomimetic scaffold (Fig. 34B).262,263 The materials have been used in a number of in vitro 3D cell culture studies, where the readily controlled (mechanical) properties were used to tailor cell behavior, for instance towards stem cell differentiation263,264 and secretome modification,265 cancer therapy,266 morphogenesis,267 organoid formation,268,269 vascularization270 and T-cell expansion.271
Using the same SPAAC approach other functional groups, such as dyes248 or PET probes253 have been introduced. To tune the mechanical behavior and the thermal response of the wsPICPs, crosslinks were introduced via the same SPAAC approach. Whereas simple bifunctional covalent crosslinks were shown to merely stabilize the gel,272 the use of multi-functional capsid-based crosslinkers introduces a healing mechanism after mechanical damage as well as a mechanical-induced payload release system.273
Tetra(ethylene glycol)-decorated wsPICP also forms gels when crosslinkers are introduced at the azide handles of the polymers. Deshpande et al. used DNA-based linkers to generate highly stimulus-responsive hydrogels.274 DNA strands A and B were functionalized with DBCO and clicked to the azide-decorated polymer to give wsPICPs-DNA A and wsPCIP-DNA-B (Fig. 34C). DNA strand C that is complementary to A and B forms the crosslinks, which generates a hydrogel below Tgel, which (nonlinear mechanical) properties are customizable by varying the crosslinker C to A/B ratio. A pH-responsive element or thrombin binding DNA sequence into crosslinker C introduces further response motifs to gel.275 The advantages of this approach has been demonstrated in DNA functionalized PNIPAM nanogels in the context of controlled Dox release.276 Furthermore, Grad et al. recently developed a wsPCIP hydrogel with tunable mechanical properties via a similar crosslinking method as described above, using coiled coils as crosslinks instead of DNA,277 clearly demonstrating the potential of the use of functional crosslinks.
Beyond functional hydrogels, tetra(ethylene glycol)PICPs with azide handles have been developed into an efficient artificial dendritic cell scaffold.262,278 For antibody conjugation to the wsPICP backbone, BCN-functionalized Sav was linked to the polymer by the SPAAC reaction (Fig. 34D).278 Biotinylated αCD3 and αCD28 antibodies were then coupled to the polymer through biotin–Sav interactions. In vitro T cell experiments demonstrated that the antibody functionalized polymers were able to activate T cells at much lower concentrations than soluble antibodies or antibodies immobilized on rigid PGLA spheres. These results show that using the Sav approach also relatively large biomolecules can be immobilized on a polymer scaffold. Further work underlined the advantageous effect of the semiflexible polymer scaffold in terms of multivalent binding interactions.279 Increasing the polymer length and antibody density enhanced the multivalent character of the polymer and lowered the effective concentration required for T cell activation. Furthermore, prolonged activation of the stimulated T cells was observed, indicating that the synthetic dendritic cells support T cell signaling. Eggermont et al. applied a covalent coupling strategy with DBCO functionalized proteins interleukin-2 and interferon-α and showed that binding of cytokines in combination with αCD3 on a semiflexible scaffold further optimizes T cell activation.280
While sequence control in synthetic polymers remains challenging, sequential addition of monomers during PIC polymerization yields block copolymers. Voerman et al. prepared blocks with two orthogonal functional groups; a small block containing allyl groups for surface grafting and a larger azide-containing block that enables a SPAAC-based introduction of bioactive molecules (Fig. 34E).281 The allyl handles were conjugated with tetrazole-functionalized biotin linkers via a nitrile-imine-mediated tetrazole-ene cycloaddition reaction that allows binding to Sav-coated substrates. The authors showed that the sequence of polymer grafting and functionalization influenced the polymer density on the substrate as well as the degree of substitution of the azide handles. First substrate grafting, then bioconjugation with DBCO-functionalized bovine serum albumin (BSA) resulted in surfaces with a high density of polymer brushes but with a limited amount of coupled BSA proteins. The reverse order gave more BSA conjugation per chain but fewer brushes on the substrate. These experiments show how block copolymers facilitate introduction of substituents in a moderate spatially controlled manner and simultaneously shows the challenges associated with more complex orthogonal conjugation strategies.
Conclusion and perspective
Over the past years semiflexible polymers have emerged as attractive scaffolds that are able to present many different functional groups, ranging from small molecules to large proteins. Their relatively linear conformation and ability to withstand deformations ensures an efficient presentation of these groups, sometimes even with high spatially control. In this review, we focused on different methods to introduce functional groups through conjugation strategies that are available to introduce these substituents, either through covalent or noncovalent bonding.In summary, we consider three ways to conjugate substituents to a polymer (see Table 2): (1) via modification of the monomers that comprise the polymer; (2) via modification of the catalyst that initiates polymerization; and (3) via post-polymerization functionalization. Typically, natural polymeric scaffolds are functionalized through post-modification approaches, while substituents can be introduced on synthetic polymers via any method. Post functionalization strategies offer the advantage of versatility –virtually any molecule can be introduced– but offers limited control on the molecules’ position on the scaffold. Introduction of a substituent via catalyst modification ensures its position on the polymer chain-end and is a functionalization strategy with high spatial control, but low functional group density. Direct monomer modification enables introduction of substituents with some control over their position on the polymer chain by making use of polymerization reactions such as block-copolymerization or SPSS. The downside, however is that monomer modification is laborious and demands that the introduced functional group is tolerated by the polymerization technique, which may severely limit the scope of this approach, particularly for the field of semiflexible polymers. Introduction of functional groups on a polymer chain that facilitate conjugation of substituents post-polymerization provides a versatile route and allows for insertion of substituents with more spatial control. Although this combination of monomer modification and post polymerization functionalization has already been applied and generated well-defined functionalized scaffolds, it is only applicable for synthetic polymers and still requires laborious monomer modification. A functionalization method that facilitates conjugation of substituents on polymer chains in a spatially controlled manner would circumvent these bottlenecks but, to the best of our knowledge, is yet to be revealed.
| Semiflexible scaffold | 1: Monomer modification | 2: Initiator modification | 3: Post-functionalization | Most frequent applied chemical functionalization strategiesa,b |
|---|---|---|---|---|
| a For references we refer to the corresponding paragraphs in this review. b Numbers refer to classes of modifications 1–3. c Monomer modification refers in this case to modification of oligonucleotides which assemble into larger structures. d Monomers are prepared through a wide variety of synthetic routes instead of one or two often applied chemical strategies. e For disubstituted polyacetylene, post-polymerization functionalization is possible (ref. 179). | ||||
| Biopolymers | ||||
| Polypeptides | X | NHS-chemistry (3), bifunctional linkers (3) | ||
| Polynucleotides: | ||||
| - DNA | Xc | X | NHS-chemistry (3), bifunctional linkers (3) | |
| - RNA | Xc | X | No chemical modification | |
| - PNA | X | X | Nucleobase modification (1,3), SPPS (1,3) | |
| Polysaccharides | X | Amine coupling (3), NHS-chemistry (3) | ||
| Synthetic polymers | ||||
| Polypeptides | X | X | X | NCA modification (1,3), SPPS (1,3), ROP initiation (2) |
| Conducting polymers: | ||||
| - Polyacetylenes | Xd | Xe | Chlorine substitution (3) | |
| - Polyanilines | Xd | X | (Thiol) surface modification (3) | |
| - Polypyrroles | X | |||
| Polyisocyanides | Xd | X | X | PFP substitution (3), SPAAC (3) |
Finally, it is interesting to reflect where functional semiflexible polymers may have the biggest impact. When we consider the broad spectrum of applications discussed in this review, it may be hard to predict. The field that currently jumps out most clearly is chemical biology, which regularly requires scaffolds to present one or multiple (similar or different) chemical cues simultaneously. It may also be this particular field, where the intrinsic benefits of semiflexible scaffolds, i.e. an intermediate flexibility that offers excellent cue presentation compared to flexible polymers and a more flexible conformation for multivalent binding compared to functional nanoparticles, are optimally exploited.
Conflicts of interest
The authors declare no conflict of interests.Acknowledgements
This work received funding from the Ministry of Education, Culture and Science through the Institute of Chemical Immunology (NWO Gravitation programme 024.002.009) and the Research Center for Functional Molecular Systems (NWO Gravitation program 024.001.035).Notes and references
- K. Kubota, Y. Tominaga and S. Fujime, Biopolymers, 1987, 26, 1717–1729 CrossRef CAS.
- J. F. Marko and E. D. Siggia, Macromolecules, 1995, 28, 8759–8770 CrossRef CAS.
- P. Benetatos and E. Frey, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2003, 67, 051108 CrossRef CAS.
- H. Herrmann, H. Bär, L. Kreplak, S. V. Strelkov and U. Aebi, Nat. Rev. Mol. Cell Biol., 2007, 8, 562–573 CrossRef CAS.
- P. Gutjahr, R. Lipowsky and J. Kierfeld, Eur. Phys. Lett., 2006, 76, 994 CrossRef CAS.
- C. P. Broedersz and F. C. MacKintosh, Rev. Mod. Phys., 2014, 86, 995 CrossRef CAS.
- O. Kratky and G. Porod, Recl. Trav. Chim. Pays-Bas, 1949, 68, 1106–1122 CrossRef CAS.
- A. S. Carlini, L. Adamiak and N. C. Gianneschi, Macromolecules, 2016, 49, 4379–4394 CrossRef CAS.
- D. Chow, M. L. Nunalee, D. W. Lim, A. J. Simnick and A. Chilkoti, Mater. Sci. Eng., R, 2008, 62, 125–155 CrossRef.
- H. Lu, J. Wang, Z. Song, L. Yin, Y. Zhang, H. Tang, C. Tu, Y. Lin and J. Cheng, Chem. Commun., 2014, 50, 139–155 RSC.
- M. D. Shoulders and R. T. Raines, Annu. Rev. Biochem., 2009, 78, 929–958 CrossRef CAS.
- J. W. Weisel and R. I. Litvinov, in Fibrous proteins: structures and mechanisms, Springer, 2017, pp. 405–456 Search PubMed.
- S. Chattopadhyay and R. T. Raines, Biopolymers, 2014, 101, 821–833 CrossRef CAS.
- E. Makareeva and S. Leikin, in Osteogenesis Imperfecta, Elsevier, 2014, pp. 71–84 Search PubMed.
- L. Cen, W. Liu, L. Cui, W. Zhang and Y. Cao, Pediatr. Res., 2008, 63, 492–496 CrossRef CAS.
- B. E. Stompro, J. F. Hansbrough and S. T. Boyce, J. Surg. Res., 1989, 46, 413–421 CrossRef CAS.
- F. Puoci, C. Piangiolino, F. Givigliano, O. I. Parisi, R. Cassano, S. Trombino, M. Curcio, F. Iemma, G. Cirillo, U. G. Spizzirri, D. Restuccia, R. Muzzalupo and N. Picci, J. Funct. Biomater., 2012, 3, 361–371 CrossRef CAS.
- S. T. Boyce, B. E. Stompro and J. F. Hansbrough, J. Biomed. Mater. Res., 1992, 26, 547–553 CrossRef CAS.
- H. J. Lee, G. M. Fernandes-Cunha, I. Putra, W.-G. Koh and D. Myung, ACS Appl. Mater. Interfaces, 2017, 9, 23389–23399 CrossRef CAS.
- H. J. Lee, G. M. Fernandes-Cunha, K. S. Na, S. M. Hull and D. Myung, Adv. Healthcare Mater., 2018, 7, e1800560 CrossRef.
- X. Li, Z. Xiao, J. Han, L. Chen, H. Xiao, F. Ma, X. Hou, X. Li, J. Sun and W. Ding, Biomaterials, 2013, 34, 5107–5116 CrossRef CAS.
- J. M. Kollman, L. Pandi, M. R. Sawaya, M. Riley and R. F. Doolittle, Biochemistry, 2009, 48, 3877–3886 CrossRef CAS.
- J. W. Weisel and R. I. Litvinov, Blood, 2013, 121, 1712–1719 CrossRef CAS.
- A. L. C. La Corte, H. Philippou and R. A. Ariens, in Advances in protein chemistry and structural biology, Elsevier, 2011, vol. 83, pp. 75–127 Search PubMed.
- N. A. Kurniawan, J. Grimbergen, J. Koopman and G. H. Koenderink, J. Thromb. Haemostasis, 2014, 12, 1687–1696 CrossRef CAS.
- S. E. Sakiyama-Elbert and J. A. Hubbell, J. Controlled Release, 2000, 69, 149–158 CrossRef CAS.
- S. E. Sakiyama-Elbert, A. Panitch and J. A. Hubbell, FASEB J., 2001, 15, 1300–1302 CrossRef CAS.
- V. Sacchi, R. Mittermayr, J. Hartinger, M. M. Martino, K. M. Lorentz, S. Wolbank, A. Hofmann, R. A. Largo, J. S. Marschall and E. Groppa, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 6952–6957 CrossRef CAS.
- D. Trentin, J. Hubbell and H. Hall, J. Controlled Release, 2005, 102, 263–275 CrossRef CAS.
- V. Sacchi, R. Mittermayr, J. Hartinger, M. M. Martino, K. M. Lorentz, S. Wolbank, A. Hofmann, R. A. Largo, J. S. Marschall, E. Groppa, R. Gianni-Barrera, M. Ehrbar, J. A. Hubbell, H. Redl and A. Banfi, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 6952–6957 CrossRef CAS.
- H. Hall, Curr. Pharm. Des., 2007, 13, 3597–3607 CrossRef CAS.
- K. Nam, C. L. Maruyama, C. S. Wang, B. G. Trump, P. Lei, S. T. Andreadis and O. J. Baker, PLoS One, 2017, 12, e0187069 CrossRef.
- K. Nam, S. M. Dean, C. T. Brown, R. J. Smith Jr., P. Lei, S. T. Andreadis and O. J. Baker, Acta Biomater., 2019, 91, 186–194 CrossRef CAS.
- H. T. Dos Santos, K. Nam, C. T. Brown, S. M. Dean, S. Lewis, C. S. Pfeifer, P. Lei, M. J. Petris, S. T. Andreadis and O. J. Baker, J. Dent. Res., 2020, 22034520964784 Search PubMed.
- C. T. Brown, K. Nam, Y. Zhang, Y. Qiu, S. M. Dean, H. T. Dos Santos, P. Lei, S. T. Andreadis and O. J. Baker, J. Histochem. Cytochem., 2020, 68, 305–318 CrossRef CAS.
- N. Zhao, A. Suzuki, X. Zhang, P. Shi, L. Abune, J. Coyne, H. Jia, N. Xiong, G. Zhang and Y. Wang, ACS Appl. Mater. Interfaces, 2019, 11, 18123–18132 CrossRef CAS.
- N. Zhao, J. Coyne, M. Xu, X. Zhang, A. Suzuki, P. Shi, J. Lai, G. H. Fong, N. Xiong and Y. Wang, Chem. Mater., 2019, 31, 1006–1015 CrossRef CAS.
- O. Juhl IV, N. Zhao, A.-B. Merife, D. Cohen, M. Friedman, Y. Zhang, Z. Schwartz, Y. Wang and H. Donahue, ACS Biomater. Sci. Eng., 2019, 5, 6152–6160 CrossRef.
- N. Zhao, J. Coyne, L. Abune, P. Shi, X. L. Lian, G. Zhang and Y. Wang, ACS Appl. Mater. Interfaces, 2020, 12, 24599–24610 CrossRef CAS.
- J. N. Milstein and J.-C. Meiners, Worm-like chain (wlc) model, 2013 Search PubMed.
- S. M. Douglas, H. Dietz, T. Liedl, B. Högberg, F. Graf and W. M. Shih, Nature, 2009, 459, 414–418 CrossRef CAS.
- Y. Ke, L. L. Ong, W. M. Shih and P. Yin, Science, 2012, 338, 1177–1183 CrossRef CAS.
- Y. R. Yang, Y. Liu and H. Yan, Bioconjugate Chem., 2015, 26, 1381–1395 CrossRef CAS.
- J. Goodchild, Bioconjugate Chem., 1990, 1, 165–187 CrossRef CAS.
- A. Kuzuya, M. Kimura, K. Numajiri, N. Koshi, T. Ohnishi, F. Okada and M. Komiyama, ChemBioChem, 2009, 10, 1811–1815 CrossRef CAS.
- M. Ozawa, T. Ozawa, M. Nishio and K. Ueda, J. Mol. Graphics Modell., 2017, 75, 117–124 CrossRef CAS.
- M. Leisola and O. Turunen, Appl. Microbiol. Biotechnol., 2007, 75, 1225–1232 CrossRef CAS.
- M. Howarth, D. J. Chinnapen, K. Gerrow, P. C. Dorrestein, M. R. Grandy, N. L. Kelleher, A. El-Husseini and A. Y. Ting, Nat. Methods, 2006, 3, 267–273 CrossRef CAS.
- Y. He, Y. Tian, A. E. Ribbe and C. Mao, J. Am. Chem. Soc., 2006, 128, 12664–12665 CrossRef CAS.
- E. Hochuli, H. Dobeli and A. Schacher, J. Chromatogr., 1987, 411, 177–184 CrossRef CAS.
- J. Shimada, T. Maruyama, T. Hosogi, J. Tominaga, N. Kamiya and M. Goto, Biotechnol. Lett., 2008, 30, 2001–2006 CrossRef CAS.
- M. Meldal and S. Schoffelen, F1000Research, 2016, 5, 2303 Search PubMed.
- K. L. Kiick, E. Saxon, D. A. Tirrell and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 19–24 CrossRef CAS.
- J. Dommerholt, S. Schmidt, R. Temming, L. J. Hendriks, F. P. Rutjes, J. C. van Hest, D. J. Lefeber, P. Friedl and F. L. van Delft, Angew. Chem., Int. Ed., 2010, 49, 9422–9425 CrossRef CAS.
- X. Ning, J. Guo, M. A. Wolfert and G. J. Boons, Angew. Chem., Int. Ed., 2008, 47, 2253–2255 CrossRef CAS.
- M. F. Debets, S. S. van Berkel, S. Schoffelen, F. P. Rutjes, J. C. van Hest and F. L. van Delft, Chem. Commun., 2010, 46, 97–99 RSC.
- S. L. Khatwani, J. S. Kang, D. G. Mullen, M. A. Hast, L. S. Beese, M. D. Distefano and T. A. Taton, Bioorg. Med. Chem., 2012, 20, 4532–4539 CrossRef CAS.
- D. Kostrz, H. K. Wayment-Steele, J. L. Wang, M. Follenfant, V. S. Pande, T. R. Strick and C. Gosse, Nat. Nanotechnol., 2019, 14, 988–993 CrossRef CAS.
- G. Marth, A. M. Hartley, S. C. Reddington, L. L. Sargisson, M. N. Parcollet, K. E. Dunn, D. D. Jones and E. Stulz, ACS Nano, 2017, 11, 5003–5010 CrossRef CAS.
- B. Rosier, A. J. Markvoort, B. Gumi Audenis, J. A. L. Roodhuizen, A. den Hamer, L. Brunsveld and T. F. A. de Greef, Nat. Catal., 2020, 3, 295–306 CrossRef CAS.
- B. A. Williams and J. C. Chaput, Curr. Protoc. Nucleic Acid Chem., 2010, 42, 4.41.41–44.41.20 Search PubMed.
- B. A. Williams, C. W. Diehnelt, P. Belcher, M. Greving, N. W. Woodbury, S. A. Johnston and J. C. Chaput, J. Am. Chem. Soc., 2009, 131, 17233–17241 CrossRef CAS.
- F. Kim, T. Chen, T. Burgess, P. Rasie, T. L. Selinger, A. Greschner, G. Rizis and K. Carneiro, Chem. Sci., 2019, 10, 10537–10542 RSC.
- J. Fu, Y. R. Yang, A. Johnson-Buck, M. Liu, Y. Liu, N. G. Walter, N. W. Woodbury and H. Yan, Nat. Nanotechnol., 2014, 9, 531–536 CrossRef CAS.
- S. I. Liang, J. M. McFarland, D. Rabuka and Z. J. Gartner, J. Am. Chem. Soc., 2014, 136, 10850–10853 CrossRef CAS.
- D. de Rochambeau, M. Barłóg, T. G. Edwardson, J. J. Fakhoury, R. S. Stein, H. S. Bazzi and H. F. Sleiman, Polym. Chem., 2016, 7, 4998–5003 RSC.
- P. Guo, Nat. Nanotechnol., 2010, 5, 833–842 CrossRef CAS.
- B. Alberts, A. Johnson, J. Lewis, M. Raff, K. Roberts and P. Walter, in Molecular Biology of the Cell, Garland Science, 4th edn, 2002 Search PubMed.
- H. Ohno, S. Akamine and H. Saito, Curr. Opin. Biotechnol., 2019, 58, 53–61 CrossRef CAS.
- M. Endo, Y. Takeuchi, T. Emura, K. Hidaka and H. Sugiyama, Chemistry, 2014, 20, 15330–15333 CrossRef CAS.
- J. Yu, Z. Liu, W. Jiang, G. Wang and C. Mao, Nat. Commun., 2015, 6, 1–6 Search PubMed.
- L. Jaeger and A. Chworos, Curr. Opin. Struct. Biol., 2006, 16, 531–543 CrossRef CAS.
- N. B. Leontis, A. Lescoute and E. Westhof, Curr. Opin. Struct. Biol., 2006, 16, 279–287 CrossRef CAS.
- Y. Shu, D. Shu, F. Haque and P. Guo, Nat. Protoc., 2013, 8, 1635 CrossRef CAS.
- A. K. Rana and S. Ankri, Front. Genet., 2016, 7, 99 Search PubMed.
- S. Abdelmawla, S. Guo, L. Zhang, S. M. Pulukuri, P. Patankar, P. Conley, J. Trebley, P. Guo and Q. X. Li, Mol. Ther., 2011, 19, 1312–1322 CrossRef CAS.
- M. Schwarz-Schilling, A. Dupin, F. Chizzolini, S. Krishnan, S. S. Mansy and F. C. Simmel, Nano Lett., 2018, 18, 2650–2657 CrossRef CAS.
- A. Cléry, M. Blatter and F. H. Allain, Curr. Opin. Struct. Biol., 2008, 18, 290–298 CrossRef.
- T. Shibata, Y. Fujita, H. Ohno, Y. Suzuki, K. Hayashi, K. R. Komatsu, S. Kawasaki, K. Hidaka, S. Yonehara and H. Sugiyama, Nat. Commun., 2017, 8, 1–13 CrossRef CAS.
- W. Yang, Q. Rev. Biophys., 2011, 44, 1–93 CrossRef CAS.
- P. E. Nielsen, M. Egholm and O. Buchardt, Bioconjugate Chem., 1994, 5, 3–7 CrossRef CAS.
- T. Sugiyama and A. Kittaka, Molecules, 2013, 18, 287–310 CrossRef CAS.
- S. Sforza, T. Tedeschi, R. Corradini, D. Ciavardelli, A. Dossena and R. Marchelli, Eur. J. Org. Chem., 2003, 1056–1063 CrossRef CAS.
- P. Zhou, M. Wang, L. Du, G. W. Fisher, A. Waggoner and D. H. Ly, J. Am. Chem. Soc., 2003, 125, 6878–6879 CrossRef CAS.
- E. A. Englund and D. H. Appella, Angew. Chem., Int. Ed., 2007, 46, 1414–1418 CrossRef CAS.
- A. Manna, S. Rapireddy, G. Sureshkumar and D. H. Ly, Tetrahedron, 2015, 71, 3507–3514 CrossRef CAS.
- S. Ghosh, S. Saini and I. Saraogi, Chem. Commun., 2018, 54, 8257–8260 RSC.
- M. Ali, R. Neumann and W. Ensinger, ACS Nano, 2010, 4, 7267–7274 CrossRef CAS.
- R. Hudson, R. Viirre, Y. Liu, F. Wojciechowski and A. Dambenieks, Pure Appl. Chem., 2004, 76, 1591–1598 CAS.
- R. E. Cline, R. M. Fink and K. Fink, J. Am. Chem. Soc., 1959, 81, 2521–2527 CrossRef CAS.
- F. Wojciechowski and R. H. Hudson, Org. Lett., 2009, 11, 4878–4881 CrossRef CAS.
- A. Manicardi, A. Accetta, T. Tedeschi, S. Sforza, R. Marchelli and R. Corradini, Artif. DNA PNA XNA, 2012, 3, 53–62 CrossRef.
- A. Manicardi, A. Bertucci, A. Rozzi and R. Corradini, Org. Lett., 2016, 18, 5452–5455 CrossRef CAS.
- A. H. St Amant, C. Engbers and R. H. Hudson, Artif. DNA PNA XNA, 2013, 4, 4–10 CrossRef.
- I. M. N. Vold, K. A. Kristiansen and B. E. Christensen, Biomacromolecules, 2006, 7, 2136–2146 CrossRef.
- P.-J. Flory, Proc. R. Soc. London, Ser. A, 1956, 234, 60–73 CAS.
- K. Mazeau, S. Pérez and M. Rinaudo, J. Carbohydr. Chem., 2008, 19, 1269–1284 CrossRef.
- M. K. Cowman and S. Matsuoka, Carbohydr. Res., 2005, 340, 791–809 CrossRef CAS.
- I. Cumpstey, ISRN Org. Chem., 2013, 2013, 417672 Search PubMed.
- J. R. Fraser, T. C. Laurent and U. B. Laurent, J. Intern. Med., 1997, 242, 27–33 CrossRef CAS.
- M. Litwiniuk, A. Krejner, M. S. Speyrer, A. R. Gauto and T. Grzela, Wounds, 2016, 28, 78–88 Search PubMed.
- M. N. Collins and C. Birkinshaw, Carbohydr. Polym., 2013, 92, 1262–1279 CrossRef CAS.
- C. Chircov, A. M. Grumezescu and L. E. Bejenaru, Rom. J. Morphol. Embryol., 2018, 59, 71–76 Search PubMed.
- R. Kumar, M. Singh, J. Meena, P. Singhvi, D. Thiyagarajan, A. Saneja and A. K. Panda, Int. J. Biol. Macromol., 2019, 133, 495–502 CrossRef CAS.
- Z. Guo, X. Zhou, C. Hou, Z. Ding, C. Wen, L. J. Zhang, B. P. Jiang and X. C. Shen, Biomater. Sci., 2019, 7, 3886–3897 RSC.
- M. Chu, H. Li, Q. Wu, F. Wo and D. Shi, Biomaterials, 2014, 35, 8357–8373 CrossRef CAS.
- D. Pemmaraju, T. Appidi, G. Minhas, S. P. Singh, N. Khan, M. Pal, R. Srivastava and A. K. Rengan, Int. J. Biol. Macromol., 2018, 110, 383–391 CrossRef CAS.
- E. A. Turley, P. W. Noble and L. Y. Bourguignon, J. Biol. Chem., 2002, 277, 4589–4592 CrossRef CAS.
- P. W. Noble, Matrix Biol., 2002, 21, 25–29 CrossRef CAS.
- Z. Guo, X. Zhou, C. Hou, Z. Ding, C. Wen, L.-J. Zhang, B.-P. Jiang and X.-C. Shen, Biomater. Sci., 2019, 7, 3886–3897 RSC.
- J. Lou, R. Stowers, S. Nam, Y. Xia and O. Chaudhuri, Biomaterials, 2018, 154, 213–222 CrossRef CAS.
- A. Dirksen, S. Yegneswaran and P. E. Dawson, Angew. Chem., Int. Ed., 2010, 49, 2023–2027 CrossRef CAS.
- H. Y. Lee, C. H. Hwang, H. E. Kim and S. H. Jeong, Carbohydr. Polym., 2018, 186, 290–298 CrossRef CAS.
- Q. Dong, X. Zhong, Y. Zhang, B. Bao, L. Liu, H. Bao, C. Bao, X. Cheng, L. Zhu and Q. Lin, Carbohydr. Polym., 2020, 245, 116525 CrossRef CAS.
- S. A. Bencherif, D. J. Siegwart, A. Srinivasan, F. Horkay, J. O. Hollinger, N. R. Washburn and K. Matyjaszewski, Biomaterials, 2009, 30, 5270–5278 CrossRef CAS.
- M. E. Han, S. H. Kim, H. D. Kim, H. G. Yim, S. A. Bencherif, T. I. Kim and N. S. Hwang, Int. J. Biol. Macromol., 2016, 93, 1410–1419 CrossRef CAS.
- M. Rezaeeyazdi, T. Colombani, A. Memic and S. A. Bencherif, Materials, 2018, 11, 1374 CrossRef.
- S. A. Bencherif, A. Srinivasan, F. Horkay, J. O. Hollinger, K. Matyjaszewski and N. R. Washburn, Biomaterials, 2008, 29, 1739–1749 CrossRef CAS.
- H. Hecht and S. Srebnik, Biomacromolecules, 2016, 17, 2160–2167 CrossRef CAS.
- J. Sun and H. Tan, Materials, 2013, 6, 1285–1309 CrossRef CAS.
- I. Donati, Y. A. Morch, B. L. Strand, G. Skjak-Braek and S. Paoletti, J. Phys. Chem. B, 2009, 113, 12916–12922 CrossRef CAS.
- S. J. Bidarra, C. C. Barrias and P. L. Granja, Acta Biomater., 2014, 10, 1646–1662 CrossRef CAS.
- M. D. Krebs, E. Salter, E. Chen, K. A. Sutter and E. Alsberg, J. Biomed. Mater. Res., Part A, 2010, 92, 1131–1138 Search PubMed.
- E. Josef, M. Zilberman and H. Bianco-Peled, Acta Biomater., 2010, 6, 4642–4649 CrossRef CAS.
- M. S. Shoichet, R. H. Li, M. L. White and S. R. Winn, Biotechnol. Bioeng., 1996, 50, 374–381 CrossRef CAS.
- R. M. Desai, S. T. Koshy, S. A. Hilderbrand, D. J. Mooney and N. S. Joshi, Biomaterials, 2015, 50, 30–37 CrossRef CAS.
- S. A. Bencherif, R. Warren Sands, O. A. Ali, W. A. Li, S. A. Lewin, T. M. Braschler, T. Y. Shih, C. S. Verbeke, D. Bhatta, G. Dranoff and D. J. Mooney, Nat. Commun., 2015, 6, 7556 CrossRef CAS.
- T. Y. Shih, S. O. Blacklow, A. W. Li, B. R. Freedman, S. Bencherif, S. T. Koshy, M. C. Darnell and D. J. Mooney, Adv. Healthcare Mater., 2018, 7, 1701469 CrossRef.
- S. A. Bencherif, R. W. Sands, D. Bhatta, P. Arany, C. S. Verbeke, D. A. Edwards and D. J. Mooney, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 19590–19595 CrossRef CAS.
- S. A. Bencherif, R. W. Sands, O. A. Ali, W. A. Li, S. A. Lewin, T. M. Braschler, T.-Y. Shih, C. S. Verbeke, D. Bhatta and G. Dranoff, Nat. Commun., 2015, 6, 1–13 Search PubMed.
- J. M. Mejia Oneto, I. Khan, L. Seebald and M. Royzen, ACS Cent. Sci., 2016, 2, 476–482 CrossRef CAS.
- R. M. Versteegen, R. Rossin, W. ten Hoeve, H. M. Janssen and M. S. Robillard, Angew. Chem., 2013, 125, 14362–14366 CrossRef.
- A. C. O'sullivan, Cellulose, 1997, 4, 173–207 CrossRef.
- Y. Dong, Y. Lai, X. Wang, M. Gao, F. Xue, X. Chen, Y. Ma and Y. Wei, Int. J. Biol. Macromol., 2019, 130, 778–785 CrossRef CAS.
- L. V. Gurgel, J. C. Perin de Melo, J. C. de Lena and L. F. Gil, Bioresour. Technol., 2009, 100, 3214–3220 CrossRef CAS.
- A. Francesko and T. Tzanov, in Biofunctionalization of Polymers and their Applications, Springer, 2010, pp. 1–27 Search PubMed.
- R. Yang, Y. Su, K. B. Aubrecht, X. Wang, H. Ma, R. B. Grubbs, B. S. Hsiao and B. Chu, Polymer, 2015, 60, 9–17 CrossRef CAS.
- Q. Dang, Q. Zhang, C. Liu, J. Yan, G. Chang, Y. Xin, X. Cheng, Y. Cao, H. Gao and Y. Liu, Int. J. Biol. Macromol., 2019, 139, 1046–1053 CrossRef CAS.
- J. Zhang, W. Tan, L. Wei, Y. Chen, Y. Mi, X. Sun, Q. Li, F. Dong and Z. Guo, Carbohydr. Polym., 2019, 215, 108–118 CrossRef CAS.
- R. Gangopadhyay, A. D. Chowdhury and A. De, Sens. Actuators, B, 2012, 171, 777–785 CrossRef.
- E. R. Thapaliya, Y. Zhang, P. Dhakal, A. S. Brown, J. N. Wilson, K. M. Collins and F. M. Raymo, Bioconjugate Chem., 2017, 28, 1519–1528 CrossRef CAS.
- L. Chambre, A. Degirmenci, R. Sanyal and A. Sanyal, Bioconjugate Chem., 2018, 29, 1885–1896 CrossRef CAS.
- X. Tian, Y. Cao, B. Zhang, S. Huang and Y. Chen, Eur. Polym. J., 2018, 106, 196–201 CrossRef CAS.
- C. E. Finlayson, R. H. Friend, M. B. Otten, E. Schwartz, J. J. Cornelissen, R. J. Nolte, A. E. Rowan, P. Samori, V. Palermo and A. Liscio, Adv. Funct. Mater., 2008, 18, 3947–3955 CrossRef CAS.
- S. M. Bruekers, M. Jaspers, J. M. Hendriks, N. A. Kurniawan, G. H. Koenderink, P. H. J. Kouwer, A. E. Rowan and W. T. S. Huck, Cell Adhes. Migr., 2016, 10, 495–504 CrossRef CAS.
- H. R. Kricheldorf, Angew. Chem., Int. Ed., 2006, 45, 5752–5784 CrossRef CAS.
- J. E. Semple, B. Sullivan and K. N. Sill, Synth. Commun., 2017, 47, 53–61 CrossRef CAS.
- T. J. Deming, Macromolecules, 1999, 32, 4500–4502 CrossRef CAS.
- T. J. Deming, Nature, 1997, 390, 386–389 CrossRef CAS.
- K. Tsuchiya and K. Numata, Macromol. Biosci., 2017, 17, 1700177 CrossRef.
- J. Ding, C. Xiao, L. Yan, Z. Tang, X. Zhuang, X. Chen and X. Jing, J. Controlled Release, 2011, 152, e11–e13 CrossRef CAS.
- D. Perego, N. Masciocchi, A. Guagliardi, J. Manuel Dominguez-Vera and N. Galvez, Nanotechnology, 2013, 24, 075102 CrossRef.
- C. Apfelthaler, M. Anzengruber, F. Gabor and M. Wirth, Eur. J. Pharm. Biopharm., 2017, 115, 131–139 CrossRef CAS.
- D. Wu, N. Sinha, J. Lee, B. P. Sutherland, N. I. Halaszynski, Y. Tian, J. Caplan, H. V. Zhang, J. G. Saven and C. J. Kloxin, Nature, 2019, 574, 658–662 CrossRef CAS.
- M. Anas, S. Jana and T. K. Mandal, Polym. Chem., 2020, 11, 2889–2903 RSC.
- H. Liu, Y. Quan, X. Jiang, X. Zhao, Y. Zhou, J. Fu, L. Du, X. Zhao, J. Zhao and L. Liang, Colloids Surf., B, 2020, 111165 CrossRef CAS.
- A. L. Wollenberg, P. Perlin and T. J. Deming, Biomacromolecules, 2019, 20, 1756–1764 CrossRef CAS.
- D. Lu, H. Wang, T. Li, Y. Li, F. Dou, S. Sun, H. Guo, S. Liao, Z. Yang, Q. Wei and Z. Lei, ACS Appl. Mater. Interfaces, 2017, 9, 16756–16766 CrossRef CAS.
- A. Song, A. A. Rane and K. L. Christman, Acta Biomater., 2012, 8, 41–50 CrossRef CAS.
- S. S. Desale, S. M. Cohen, Y. Zhao, A. V. Kabanov and T. K. Bronich, J. Controlled Release, 2013, 171, 339–348 CrossRef CAS.
- J. Rao, Y. Zhang, J. Zhang and S. Liu, Biomacromolecules, 2008, 9, 2586–2593 CrossRef CAS.
- L. Fu, C. Sun and L. Yan, ACS Appl. Mater. Interfaces, 2015, 7, 2104–2115 CrossRef CAS.
- J. Tian, R. Jiang, P. Gao, D. Xu, L. Mao, G. Zeng, M. Liu, F. Deng, X. Zhang and Y. Wei, Mater. Sci. Eng., C, 2017, 79, 563–569 CrossRef CAS.
- D. Xu, M. Liu, Q. Zhang, Q. Huang, H. Huang, J. Tian, R. Jiang, Y. Wen, X. Zhang and Y. Wei, Mater. Sci. Eng., C, 2018, 91, 496–501 CrossRef CAS.
- H. Y. Tian, C. Deng, H. Lin, J. Sun, M. Deng, X. Chen and X. Jing, Biomaterials, 2005, 26, 4209–4217 CrossRef CAS.
- J. Jacobs, N. Gathergood and A. Heise, Macromol. Rapid Commun., 2013, 34, 1325–1329 CrossRef CAS.
- H. Song, G. Yang, P. Huang, D. Kong and W. Wang, J. Mater. Chem. B, 2017, 5, 1724–1733 RSC.
- G. Liu and C. M. Dong, Biomacromolecules, 2012, 13, 1573–1583 CrossRef CAS.
- M. Aldissl, Polym.-Plast. Technol. Eng., 1987, 26, 45–70 CrossRef.
- W. Feast, J. Tsibouklis, K. Pouwer, L. Groenendaal and E. Meijer, Polymer, 1996, 37, 5017–5047 CrossRef CAS.
- B. Kuei and E. D. Gomez, Soft Matter, 2017, 13, 49–67 RSC.
- P. Sista, K. Ghosh, J. S. Martinez and R. C. Rocha, J. Nanosci. Nanotechnol., 2014, 14, 250–272 CrossRef CAS.
- T. Masuda, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 165–180 CrossRef CAS.
- T. Masuda, Polym. Rev., 2017, 57, 1–14 CrossRef CAS.
- J. W. Lam and B. Z. Tang, Acc. Chem. Res., 2005, 38, 745–754 CrossRef CAS.
- H. Zhao, F. Sanda and T. Masuda, Polymer, 2006, 47, 1584–1589 CrossRef CAS.
- H. P. Xu, B. Y. Xie, W. Z. Yuan, J. Z. Sun, F. Yang, Y. Q. Dong, A. Qin, S. Zhang, M. Wang and B. Z. Tang, Chem. Commun., 2007, 1322–1324 RSC.
- W. Z. Yuan, J. Z. Sun, Y. Dong, M. Häussler, F. Yang, H. P. Xu, A. Qin, J. W. Lam, Q. Zheng and B. Z. Tang, Macromolecules, 2006, 39, 8011–8020 CrossRef CAS.
- T. Fulghum, S. A. Karim, A. Baba, P. Taranekar, T. Nakai, T. Masuda and R. C. Advincula, Macromolecules, 2006, 39, 1467–1473 CrossRef CAS.
- V. Percec, J. G. Rudick, M. Peterca, M. Wagner, M. Obata, C. M. Mitchell, W. D. Cho, V. S. Balagurusamy and P. A. Heiney, J. Am. Chem. Soc., 2005, 127, 15257–15264 CrossRef CAS.
- E. Anger, H. Iida, T. Yamaguchi, K. Hayashi, D. Kumano, J. Crassous, N. Vanthuyne, C. Roussel and E. Yashima, Polym. Chem., 2014, 5, 4909–4914 RSC.
- R. Sakai, S. Okade, E. B. Barasa, R. Kakuchi, M. Ziabka, S. Umeda, K. Tsuda, T. Satoh and T. Kakuchi, Macromolecules, 2010, 43, 7406–7411 CrossRef CAS.
- J. Qu, T. Katsumata, M. Satoh, J. Wada, J. Igarashi, K. Mizoguchi and T. Masuda, Chemistry, 2007, 13, 7965–7973 CrossRef CAS.
- Q. Zeng, P. Cai, Z. Li, J. Qin and B. Z. Tang, Chem. Commun., 2008, 1094–1096, 10.1039/b717764j.
- W. Salaneck, Science and applications of conducting polymers: papers from the 6th European Physical Society Industrial Workshop held in Lofthus, Norway, 28–31 May 1990, Hilger, 1991 Search PubMed.
- W. Salaneck, I. Lundström, W.-S. Huang and A. MacDiarmid, Synth. Met., 1986, 13, 291–297 CrossRef CAS.
- I. Sapurina and J. Stejskal, Polym. Int., 2008, 57, 1295–1325 CrossRef CAS.
- Y. Wei, X. Tang, Y. Sun and W. W. Focke, J. Polym. Sci., Part A: Polym. Chem., 1989, 27, 2385–2396 CrossRef CAS.
- D. Li, J. Huang and R. B. Kaner, Acc. Chem. Res., 2009, 42, 135–145 CrossRef CAS.
- J. Huang, S. Virji, B. H. Weiller and R. B. Kaner, J. Am. Chem. Soc., 2003, 125, 314–315 CrossRef CAS.
- D. F. Acevedo, H. J. Salavagione, M. C. Miras and C. A. Barbero, J. Braz. Chem. Soc., 2005, 16, 259–269 CrossRef CAS.
- P. Fabbri and M. Messori, in Modification of Polymer Properties, Elsevier, 2017, pp. 109–130 Search PubMed.
- E. Lahiff, S. Scarmagnani, B. Schazmann, A. Cafolla and D. Diamond, Int. J. Nanomanuf., 2010, 5, 88–99 CrossRef CAS.
- B. T. DiTullio, C. J. Wright, P. Hayes, P. J. Molino and T. W. Hanks, Colloid Polym. Sci., 2018, 296, 637–645 CrossRef CAS.
- C. Crean, E. Lahiff, N. Gilmartin, D. Diamond and R. O'Kennedy, Synth. Met., 2011, 161, 285–292 CrossRef CAS.
- E. Lahiff, T. Woods, W. Blau, G. G. Wallace and D. Diamond, Synth. Met., 2009, 159, 741–748 CrossRef CAS.
- V. V. Tat'yana and O. N. Efimov, Russ. Chem. Rev., 1997, 66, 443 CrossRef.
- A. Diaz, K. K. Kanazawa and G. P. Gardini, J. Chem. Soc., Chem. Commun., 1979, 635–636 RSC.
- S. Machida, S. Miyata and A. Techagumpuch, Synth. Met., 1989, 31, 311–318 CrossRef CAS.
- Y. Whang, J. Han, H. Nalwa, T. Watanabe and S. Miyata, Synth. Met., 1991, 43, 3043–3048 CrossRef CAS.
- A. Dupont-Filliard, M. Billon, S. Guillerez and G. Bidan, Talanta, 2001, 55, 981–992 CrossRef CAS.
- A. Dupont-Filliard, M. Billon, T. Livache and S. Guillerez, Anal. Chim. Acta, 2004, 515, 271–277 CrossRef CAS.
- J. Y. Lee and C. E. Schmidt, J. Biomed. Mater. Res., Part A, 2015, 103, 2126–2132 CrossRef CAS.
- J.-W. Lee, F. Serna, J. Nickels and C. E. Schmidt, Biomacromolecules, 2006, 7, 1692–1695 CrossRef CAS.
- J. Y. Lee, J. W. Lee and C. E. Schmidt, J. R. Soc., Interface, 2009, 6, 801–810 CrossRef CAS.
- P. Godillot, H. Korri-Youssoufi, P. Srivastava, A. El Kassmi and F. Garnier, Synth. Met., 1996, 83, 117–123 CrossRef CAS.
- H. Korri-Youssoufi, F. Garnier, P. Srivastava, P. Godillot and A. Yassar, J. Am. Chem. Soc., 1997, 119, 7388–7389 CrossRef CAS.
- A. Wang, L. Cheng, X. Chen, C. Li, J. Zhang and W. Zhu, Dalton Trans., 2019, 48, 14467–14477 RSC.
- J. W. Lee, F. Serna and C. E. Schmidt, Langmuir, 2006, 22, 9816–9819 CrossRef CAS.
- L. K. Jang, S. Kim, J. Seo and J. Y. Lee, Biofabrication, 2017, 9, 045007 CrossRef.
- R. J. Nolte, Chem. Soc. Rev., 1994, 23, 11–19 RSC.
- R. Nolte, R. Stephany and W. Drenth, Recl. Trav. Chim. Pays-Bas, 1973, 92, 83–91 CrossRef CAS.
- W. Drenth and R. J. Nolte, Acc. Chem. Res., 1979, 12, 30–35 CrossRef CAS.
- E. Schwartz, M. Koepf, H. J. Kitto, R. J. Nolte and A. E. Rowan, Polym. Chem., 2011, 2, 33–47 RSC.
- E. Schwartz, V. Palermo, C. E. Finlayson, Y. S. Huang, M. B. Otten, A. Liscio, S. Trapani, I. Gonzalez-Valls, P. Brocorens, J. J. Cornelissen, K. Peneva, K. Mullen, F. C. Spano, A. Yartsev, S. Westenhoff, R. H. Friend, D. Beljonne, R. J. Nolte, P. Samori and A. E. Rowan, Chemistry, 2009, 15, 2536–2547 CrossRef CAS.
- N. Hida, F. Takei, K. Onitsuka, K. Shiga, S. Asaoka, T. Iyoda and S. Takahashi, Angew. Chem., 2003, 115, 4485–4488 CrossRef.
- X. Han, J. Zhang, C.-Y. Qiao, W.-M. Zhang, J. Yin and Z.-Q. Wu, Macromolecules, 2017, 50, 4114–4125 CrossRef CAS.
- F. Takei, D. Kodama, S. Nakamura, K. Onitsuka and S. Takahashi, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 585–595 CrossRef CAS.
- T. Kajitani, K. Okoshi and E. Yashima, Macromolecules, 2008, 41, 1601–1611 CrossRef CAS.
- X. Yan, S. Zhang, P. Zhang, X. Wu, A. Liu, G. Guo, Y. Dong and X. Li, Angew. Chem., Int. Ed., 2018, 57, 8947–8952 CrossRef CAS.
- S. Schraff, Y. Sun and F. Pammer, Macromolecules, 2018, 51, 5323–5335 CrossRef CAS.
- R. J. Ono, A. D. Todd, Z. Hu, D. A. Vanden Bout and C. W. Bielawski, Macromol. Rapid Commun., 2014, 35, 204–209 CrossRef CAS.
- T. Hasegawa, S. Kondoh, K. Matsuura and K. Kobayashi, Macromolecules, 1999, 32, 6595–6603 CrossRef CAS.
- T. Kajitani, H. Lin, K. Nagai, K. Okoshi, H. Onouchi and E. Yashima, Macromolecules, 2009, 42, 560–567 CrossRef CAS.
- S. K. Pomarico, C. Wang and M. Weck, Macromol. Rapid Commun., 2020, 41, e1900324 CrossRef.
- Z.-Q. Jiang, Y.-X. Xue, J.-L. Chen, Z.-P. Yu, N. Liu, J. Yin, Y.-Y. Zhu and Z.-Q. Wu, Macromolecules, 2015, 48, 81–89 CrossRef CAS.
- Z. H. Zhang, C. Y. Qiao, J. Zhang, W. M. Zhang, J. Yin and Z. Q. Wu, Macromol. Rapid Commun., 2017, 38, 1700315 CrossRef.
- J. Yin, L. Xu, X. Han, L. Zhou, C. Li and Z.-Q. Wu, Polym. Chem., 2017, 8, 545–556 RSC.
- L. Xu, X.-H. Xu, N. Liu, H. Zou and Z.-Q. Wu, Macromolecules, 2018, 51, 7546–7555 CrossRef CAS.
- J. J. Cornelissen, J. J. Donners, R. de Gelder, W. S. Graswinckel, G. A. Metselaar, A. E. Rowan, N. A. Sommerdijk and R. J. Nolte, Science, 2001, 293, 676–680 CrossRef CAS.
- J. J. Cornelissen, W. S. Graswinckel, P. Adams, G. H. Nachtegaal, A. P. Kentgens, N. A. Sommerdijk and R. J. Nolte, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 4255–4264 CrossRef CAS.
- G. A. Metselaar, P. J. Adams, R. J. Nolte, J. J. Cornelissen and A. E. Rowan, Chemistry, 2007, 13, 950–960 CrossRef CAS.
- J. J. Cornelissen, W. S. Graswinckel, A. E. Rowan, N. A. Sommerdijk and R. J. Nolte, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 1725–1736 CrossRef CAS.
- A. M. van Buul, E. Schwartz, P. Brocorens, M. Koepf, D. Beljonne, J. C. Maan, P. C. M. Christianen, P. H. J. Kouwer, R. J. M. Nolte and H. Engelkamp, Chem. Sci., 2013, 4, 2357–2363 RSC.
- H. J. Kitto, E. Schwartz, M. Nijemeisland, M. Koepf, J. J. Cornelissen, A. E. Rowan and R. J. Nolte, J. Mater. Chem., 2008, 18, 5615–5624 RSC.
- E. Schwartz, E. Lim, C. M. Gowda, A. Liscio, O. Fenwick, G. Tu, V. Palermo, R. de Gelder, J. J. Cornelissen and E. R. Van Eck, Chem. Mater., 2010, 22, 2597–2607 CrossRef CAS.
- P. A. de Witte, M. Castriciano, J. J. Cornelissen, L. Monsu Scolaro, R. J. Nolte and A. E. Rowan, Chemistry, 2003, 9, 1775–1781 CrossRef CAS.
- D. M. Vriezema, J. Hoogboom, K. Velonia, K. Takazawa, P. C. Christianen, J. C. Maan, A. E. Rowan and R. J. Nolte, Angew. Chem., Int. Ed., 2003, 42, 772–776 CrossRef CAS.
- S. Foster, C. E. Finlayson, P. E. Keivanidis, Y.-S. Huang, I. Hwang, R. H. Friend, M. B. Otten, L.-P. Lu, E. Schwartz and R. J. Nolte, Macromolecules, 2009, 42, 2023–2030 CrossRef CAS.
- E. Schwartz, H. J. Kitto, R. De Gelder, R. J. Nolte, A. E. Rowan and J. J. Cornelissen, J. Mater. Chem., 2007, 17, 1876–1884 RSC.
- I. López-Duarte, M. V. Martínez-Díaz, E. Schwartz, M. Koepf, P. H. J. Kouwer, A. E. Rowan, R. J. Nolte and T. Torres, ChemPlusChem, 2012, 77, 700–706 CrossRef.
- S. Le Gac, E. Schwartz, M. Koepf, J. J. Cornelissen, A. E. Rowan and R. J. Nolte, Chem. – Eur. J., 2010, 16, 6176–6186 CrossRef CAS.
- M. Koepf, H. J. Kitto, E. Schwartz, P. H. J. Kouwer, R. J. Nolte and A. E. Rowan, Eur. Polym. J., 2013, 49, 1510–1522 CrossRef CAS.
- P. H. J. Kouwer, M. Koepf, V. A. Le Sage, M. Jaspers, A. M. van Buul, Z. H. Eksteen-Akeroyd, T. Woltinge, E. Schwartz, H. J. Kitto, R. Hoogenboom, S. J. Picken, R. J. M. Nolte, E. Mendes and A. E. Rowan, Nature, 2013, 493, 651–655 CrossRef CAS.
- M. Jaspers, A. C. H. Pape, I. K. Voets, A. E. Rowan, G. Portale and P. H. J. Kouwer, Biomacromolecules, 2016, 17, 2642–2649 CrossRef CAS.
- H. Yuan, J. Xu, E. P. van Dam, G. Giubertoni, Y. L. A. Rezus, R. Hammink, H. J. Bakker, Y. Zhan, A. E. Rowan, C. Xing and P. H. J. Kouwer, Macromolecules, 2017, 50, 9058–9065 CrossRef CAS.
- P. H. J. Kouwer, P. de Almeida, O. ven den Boomen, Z. H. Eksteen-Akeroyd, R. Hammink, M. Jaspers, S. Kragt, M. F. J. Mabesoone, R. J. M. Nolte, A. E. Rowan, M. G. T. A. Rutten, V. A. A. Le Sage, D. C. Schoenmakers, C. Xing and J. Xu, Chin. Chem. Lett., 2018, 29, 281–284 CrossRef CAS.
- J. Vandaele, B. Louis, K. Liu, R. Camacho, P. H. J. Kouwer and S. Rocha, Soft Matter, 2020, 16, 4210–4219 RSC.
- M. Jaspers, M. Dennison, M. F. J. Mabesoone, F. C. MacKintosh, A. E. Rowan and P. H. J. Kouwer, Nat. Commun., 2014, 5, 5808 CrossRef.
- C. Storm, J. J. Pastore, F. C. MacKintosh, T. C. Lubensky and P. A. Janmey, Nature, 2005, 435, 191–194 CrossRef CAS.
- P. de Almeida, P. A. Janmey and P. H. J. Kouwer, Adv. Funct. Mater., 2021 DOI:10.1002/adfm.202010527.
- R. C. op ‘t Veld, O. I. van den Boomen, D. M. S. Lundvig, E. M. Bronkhorst, P. H. J. Kouwer, J. A. Jansen, E. Middelkoop, J. W. Von den Hoff, A. E. Rowan and F. A. D. T. G. Wagener, Biomaterials, 2018, 181, 392–401 CrossRef.
- R. C. Op't Veld, L. Joosten, O. van den Boomen, O. Boerman, P. H. J. Kouwer, E. Middelkoop, A. Rowan, J. A. Jansen, F. Walboomers and F. Wagener, Biomater. Sci., 2019, 7, 3041–3050 RSC.
- B. Wang, J. Wang, J. Shao, P. H. J. Kouwer, E. M. Bronkhorst, J. A. Jansen, X. F. Walboomers and F. Yang, J. Controlled Release, 2020, 324, 134–145 CrossRef CAS.
- B. Wang, H. E. Booij-Vrieling, E. M. Bronkhorst, J. Shao, P. H. J. Kouwer, J. A. Jansen, X. F. Walboomers and F. Yang, Acta Biomater., 2020, 116, 259–267 CrossRef CAS.
- H. Yuan, Y. Zhan, A. E. Rowan, C. Xing and P. H. J. Kouwer, Angew. Chem., 2020, 59, 2720–2724 CrossRef CAS.
- Q. Cui, H. Yuan, X. Bao, G. Ma, M. Wu and C. Xing, ACS Appl. Bio Mater., 2020, 3, 4436–4443 CrossRef CAS.
- J. Guo, C. Xing, H. Yuan, R. Chai and Y. Zhan, ACS Appl. Bio Mater., 2019, 2, 2520–2527 CrossRef.
- A. Alvarez Fernandez, R. Hammink, S. Kragt, L. Cattaneo, M. Savoini, J. van der Velden, T. Rasing, A. E. Rowan, P. J. Collings and P. H. J. Kouwer, Adv. Funct. Mater., 2016, 26, 9009–9016 CrossRef CAS.
- M. Jaspers, S. L. Vaessen, P. van Schayik, D. Voerman, A. E. Rowan and P. H. J. Kouwer, Nat. Commun., 2017, 8, 15478 CrossRef CAS.
- P. de Almeida, M. Jaspers, S. Vaessen, O. Tagit, G. Portale, A. E. Rowan and P. H. J. Kouwer, Nat. Commun., 2019, 10, 1–8 CrossRef.
- S. Mandal, Z. H. Eksteen-Akeroyd, M. J. Jacobs, R. Hammink, M. Koepf, A. J. Lambeck, J. C. van Hest, C. J. Wilson, K. Blank and C. G. Figdor, Chem. Sci., 2013, 4, 4168–4174 RSC.
- R. K. Das, V. Gocheva, R. Hammink, O. F. Zouani and A. E. Rowan, Nat. Mater., 2016, 15, 318–325 CrossRef CAS.
- K. Liu, S. M. Mihaila, A. Rowan, E. Oosterwijk and P. H. J. Kouwer, Biomacromolecules, 2019, 20, 826–834 CrossRef CAS.
- K. Liu, T. Veenendaal, M. Wiendels, A. M. Ruiz-Zapata, J. van Laar, R. Kyranas, H. Enting, B. van Cranenbroek, H. J. P. M. Koenen, S. M. Mihaila, E. Oosterwijk and P. H. J. Kouwer, ACS Appl. Mater. Interfaces, 2020, 12, 56723–56730 CrossRef CAS.
- Z. Zhang, C. Tang, R. Hammink, F. Nelissen, H. Heus and P. H. J. Kouwer, Chem. Commun., 2021 10.1039/D0CC08205H.
- Y. Zhang, M. M. Zegers, A. E. Rowan, P. N. Span and P. H. J. Kouwer, Adv. Sci., 2021, 8, 2003380 CrossRef.
- Y. Zhang, C. Tang, P. N. Span, A. E. Rowan, T. W. Aalders, J. A. Schalken, G. J. Adema, P. H. J. Kouwer, M. M. P. Zegers and M. Ansems, Adv. Sci., 2020, 7, 202001797 Search PubMed.
- S. Ye, J. W. B. Boeter, M. Mihajlovic, F. G. van Steenbeek, M. E. van Wolferen, L. A. Oosterhoff, A. Marsee, M. Caiazzo, L. J. W. van der Laan, L. C. Penning, T. Vermonden, B. Spee and K. Schneeberger, Adv. Funct. Mater., 2020, 30, 2000893 CrossRef CAS.
- J. Zimoch, J. S. Padial, A. S. Klar, Q. Vallmajo-Martin, M. Meuli, T. Biedermann, C. J. Wilson, A. Rowan and E. Reichmann, Acta Biomater., 2018, 70, 129–139 CrossRef CAS.
- J. Weiden, D. Voerman, Y. Dölen, R. K. Das, A. Van Duffelen, R. Hammink, L. J. Eggermont, A. E. Rowan, J. Tel and C. G. Figdor, Front. Immunol., 2018, 9, 2798 CrossRef.
- D. C. Schoenmakers, A. E. Rowan and P. H. J. Kouwer, Nat. Commun., 2018, 9, 1–8 CrossRef CAS.
- D. C. Schoenmakers, L. Schoonen, M. G. Rutten, R. J. Nolte, A. E. Rowan, J. C. van Hest and P. H. J. Kouwer, Soft Matter, 2018, 14, 1442–1448 RSC.
- S. R. Deshpande, R. Hammink, R. K. Das, F. H. Nelissen, K. G. Blank, A. E. Rowan and H. A. Heus, Adv. Funct. Mater., 2016, 26, 9075–9082 CrossRef CAS.
- S. R. Deshpande, R. Hammink, F. H. Nelissen, A. E. Rowan and H. A. Heus, Biomacromolecules, 2017, 18, 3310–3317 CrossRef CAS.
- W. Liwinska, I. Stanislawska, M. Lyp, Z. Stojek and E. Zabost, RSC Adv., 2019, 9, 13736–13748 RSC.
- E. M. Grad, I. Tunn, D. Voerman, A. Sanz de León, R. Hammink and K. G. Blank, ChemRxiv, 2020.
- S. Mandal, R. Hammink, J. Tel, Z. H. Eksteen-Akeroyd, A. E. Rowan, K. Blank and C. G. Figdor, ACS Chem. Biol., 2015, 10, 485–492 CrossRef CAS.
- R. Hammink, S. Mandal, L. J. Eggermont, M. Nooteboom, P. H. Willems, J. Tel, A. E. Rowan, C. G. Figdor and K. G. Blank, ACS Omega, 2017, 2, 937–945 CrossRef CAS.
- L. J. Eggermont, R. Hammink, K. G. Blank, A. E. Rowan, J. Tel and C. G. Figdor, Adv. Ther., 2018, 1, 1800021 CrossRef.
- D. Voerman, M. Schluck, J. Weiden, B. Joosten, L. J. Eggermont, T. van den Eijnde, B. Ignacio, A. Cambi, C. G. Figdor and P. H. J. Kouwer, Biomacromolecules, 2019, 20, 2587–2597 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |