
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The chemistry of phosphines in constrained, well-defined microenvironments
Georgia R. F.
Orton
 ab,
Ben S.
Pilgrim
*a and
Neil R.
Champness
*ab
ab,
Ben S.
Pilgrim
*a and
Neil R.
Champness
*ab
aSchool of Chemistry, University of Nottingham, University Park, Nottingham, NG7 2RD, UK. E-mail: ben.pilgrim@nottingham.ac.uk
bSchool of Chemistry, University of Birmingham, Edgbaston, Birmingham, B15 2TT, UK. E-mail: n.champness@bham.ac.uk
First published on 19th February 2021
Abstract
Developments in the confinement of phosphines within micro- or nano-environments are explored. Phosphines are ubiquitous across metal coordination chemistry and underpin some of the most famous homogeneous transition metal catalysts. Constraining phosphines within confined environments influences not only their behaviour but also that of their metal complexes. Notable examples include the use of metal–organic frameworks (MOFs) or metal–organic cages (MOCs) to support phosphines which demonstrate how the microenvironment within such constructs leads to reactivity modification. The development of phosphine confinement is explored and parallels are drawn with related constrained macrocyclic systems and mechanically interlocked molecules. The review concludes by identifying areas that remain a challenge and those that will provide new avenues for research.
 Georgia R. F. Orton | Georgia R. F. Orton is a Postdoctoral Research Fellow at the University of Birmingham, UK. She studied for her MChem at the University of Sheffield from 2011–2015, with study in Bordeaux, France. Georgia received her PhD from King's College London in 2019 for work on biomimetic catalysts for hydrogen oxidation before moving to a postdoctoral fellowship at the University of Nottingham in 2019 and the University of Birmingham in 2021. Her current research focuses on mechanistic studies of reactions within MOFs. |
 Ben S. Pilgrim | Ben S. Pilgrim leads a group studying supramolecular organic chemistry in the Green Chemicals Beacon of Excellence at the University of Nottingham. He studied for his MChem at St John's College, Oxford from 2005–2009 and completed his doctoral studies with Timothy Donohoe. In 2013 he moved to the University of Cambridge, on a Herchel Smith Research Fellowship, studying metal–organic cages with Jonathan Nitschke and was subsequently awarded a Royal Commission for the Exhibition of 1851 Research Fellowship. His current research interests include supramolecular catalysis, developing responsive supramolecular systems, and designing new synthetic routes to interlocked molecules. |
 Neil R. Champness | Neil R. Champness is the Norman Haworth Professor of Chemistry at the University of Birmingham having moved from the University of Nottingham, UK in 2021. After completing his PhD at the University of Southampton, UK, with Bill Levason he moved to Nottingham in 1995, being appointed Chair of Chemical Nanoscience in 2004. His research spans chemical nanoscience and all aspects of molecular organization, including surface supramolecular assembly, the synthesis of mechanically interlocked molecules and supramolecular chemistry in the solid-state using crystal engineering approaches. |
1. Introduction
Phosphines are ubiquitous ligands, with a wealth of history and rich metal coordination chemistry. Much of modern day homogeneous transition metal catalysis is underpinned by phosphine ligands and metal–phosphine coordination complexes such as Wilkinson's,1 Crabtree's,2 and Grubbs's3 catalysts, and Vaska's complex.4 So common are phosphine ligands in transition metal catalysts that their use has become second nature to many researchers in the field. Chemists have long known that the reactivity and behaviour of phosphines is governed by their substituents. For good catalytic activity in cross-coupling reactions, phosphines typically need to be both basic (electron-rich) and sufficiently sterically encumbered (as defined by a cone angle of the substituents around the phosphorus atom).5,6 The ability to tune both the electronic and steric properties of phosphine ligands lies behind their importance in the development of effective homogeneous catalysts.However, phosphines also have some drawbacks, notably many phosphines that are used in highly active catalysts are also highly reactive in their own right, particularly with respect to oxidation, and therefore require handling under inert atmospheres. PH3 itself and many trialkylphosphines (such as PMe3 and PtBu3) are highly flammable and (like elemental white phosphorus itself) often reported as pyrophoric. Hence, they must be handled with extreme care, released in situ from their phosphonium salts, or the metal–phosphine complexes used directly. Although there have been some notable new approaches to synthesising phosphines,7 accessing novel phosphines via traditional covalent chemistry techniques is fraught with challenges of handling toxic reagents and highly reactive species. As an interesting contrast, most academic labs have an ancient tub of PPh3 which has sat in the solid state for many years largely unreactive towards oxygen.
Constraining the phosphine within a larger 3D environment offers a complementary approach to controlling the reactivity and behaviour of phosphorus, and in many cases is easier to manipulate than controlling the three covalently bonded substituents around the P centre. Confinement of reactive species in constrained microenvironments provides opportunities to influence reactivity, for example through steric control, transition state stabilisation or rate acceleration by increasing the effective concentration of encapsulated guests relative to their bulk concentrations. The phosphorus lone pair provides scope for functionalisation pre- or post- synthetically, for example via arylation, chalcogenisation or metalation. The opportunities to employ confinement of phosphines within microenvironments, therefore, represents a powerful approach to both contain and potentially enhance the rich chemistry of phosphines with molecular-level precision.
Due to the synthetic challenges of incorporating phosphines within ligands, they are rarely seen in a ligand for no purpose (or just as a linker atom) – if you see a phosphorus atom you can assume it has a crucial structural or functional role. In this review we will focus on phosphorus in well-defined artificial microenvironments and survey the structural and functional roles it plays and how these roles are influenced by the nature of the environment itself. This includes macromolecular structures such as metal–organic frameworks (MOFs) and covalent organic frameworks (COFs), as well as discrete structures such as metal–organic cages (MOCs), organic macrocycles, and mechanically interlocked architectures such as rotaxanes and catenanes. We are not including examples of phosphines in materials where the environment around phosphorus varies from one atom to the next, such as polymers and nanoparticles, which are typically polydisperse. Additionally, we will only focus on artificial systems, excluding discussions of confinement of phosphines within the 3D environment of proteins either via use of phosphine-containing amino acids,8 or by post-translational modification. The term phosphine is nowadays used more loosely than its strictest definition.9 In this review we will focus our discussion on phosphorus with three bonds to carbon or hydrogen, although will consider some derivatives or closely related compounds where particularly relevant.
2. Phosphine–metal coordination to hold structures together
2.1 MOFs
Metal–organic frameworks (MOFs) are a class of crystalline materials consisting of metal-based nodes (ions or clusters) connected through multidentate organic linkers.10 MOFs typically exhibit high permanent porosity and high surface areas leading to extensive research into potential applications in gas adsorption/release and species separation.11 In addition to this, MOFs find application as solid-state matrices for the isolation and stabilisation of reactive species, and as platforms for the characterisation of these species and their reaction mechanisms.12 Since MOFs are constructed from well-defined molecular building blocks (linkers and nodes), their macromolecular structures can be controlled at the molecular level and adapted using the reticular chemistry approach.13 As such, MOFs can offer an amalgamation of the benefits offered by standard homogeneous (high specificity) and heterogeneous catalysts (enhanced stability, and improved recyclability).Carboxylates are the most extensively used linkers in MOF synthesis.14 Since their bonding to metal centres is often reversible under solvothermal conditions, defects may be redissolved, resulting in a more uniform structure. Nevertheless, framework stability and reactivity are largely dependent on the metal-linker interactions and use of linkers with alternative coordinating groups potentially gives access to frameworks with a wider range of topologies and reactivity. The development of multidentate linkers featuring both a phosphine and other coordinating groups potentially gives access to phosphine-functionalised MOFs (P-MOFs). The phosphine group may either play an integral role in structure formation through coordination to a node (henceforth referred to as a structural phosphine),15,16 or remain uncoordinated and poised for further reactivity (open phosphine). Given the difficulties in their synthesis and handling, and the harsh conditions often required for MOF synthesis, incorporation of phosphines into MOFs has thus far been focused primarily on structures in which the phosphine plays an important role in the reactivity of the resulting structure. Consequently, examples of structural phosphines in MOFs are limited.
The first phosphine-functionalised MOF (P-MOF) was reported in 2002 by James and co-workers and used the bulky triphosphine 1,3,5-tris(diphenylphosphanyl)benzene (L1) to generate a 2D polymer network, [Ag4L13(O3SCF3)4] (Ag-PMOF) (Fig. 1).15Ag-PMOF consists of large 72-atom hexagonal rings containing 18 silver atoms and 12 L1 units which form wide channels (1.60 ± 1.84 nm). Although network interpenetration is commonly observed in coordination polymers, this is not observed for Ag-PMOF, as a consequence of the bulky nature of the phenyl groups of the linker.
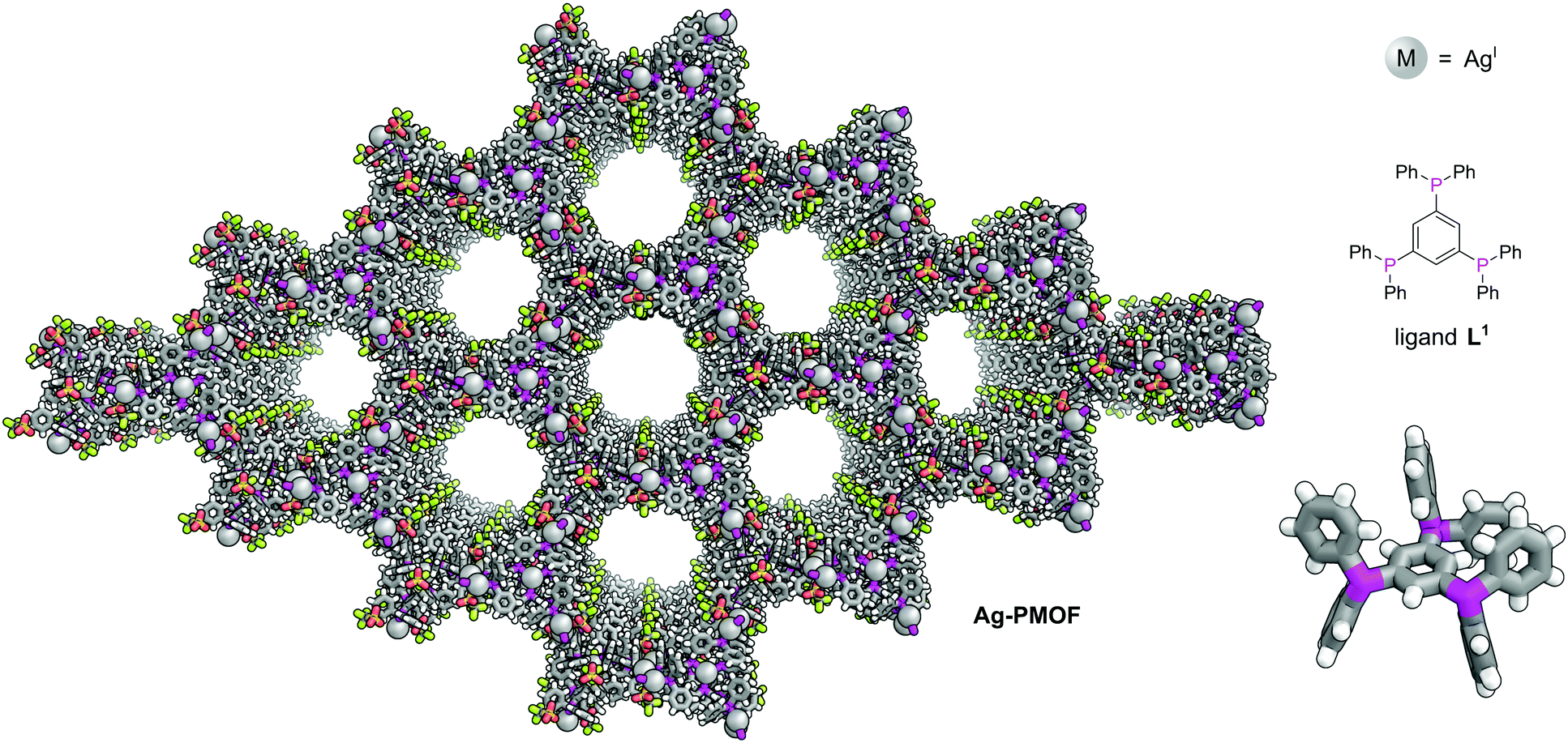 | ||
| Fig. 1 1,3,5-Tris(diphenylphosphanyl)benzene (L1) plays a structural role in Ag-PMOF. The 2D MOF is formed of sheets of hexagonal rings each consisting of 12 ligands and 18 silver atoms.15 Colours: C grey, H white, Ag light grey (shown as spheres), F lime green, O red, P magenta, S yellow. | ||
The phosphorus atom provided a useful NMR handle to study solution state precursors to Ag-PMOF and indicated the presence of two highly symmetrical coordination cage structures with one 31P chemical environment and well-resolved one-bond couplings to the spin ½ 107Ag and 109Ag isotopes. Interestingly when the anion was exchanged for the non-coordinating SbF6−, this metal and ligand combination formed either discrete M3L12 coordination cages or more ‘wormlike’ viscous coordination polymers depending on the metal:ligand ratio used.17
The first three-dimensional (3D) P-MOF, 1, was assembled from CuPF6 and the pyridyl diphosphine ligand 4-(3,5-bis(diphenylphosphino)phenyl)pyridine (L2) and the resulting structure could be solved using single crystal X-ray diffraction (SCXRD) (Fig. 2a).16 Each Cu(I) centre occupies a distorted tetrahedral coordination geometry by binding one N-donor and two P-donors from three separate ligands to form homochiral 1D channels. The crystallographically characterised nodes show heterogeneous Lewis acid catalytic activity for a ketalization reaction, and size-selectivity of reactants is provided by the pore geometry. The MOF catalyst could be isolated from the reaction solution by filtration and recycled for three successive runs with no loss of activity. Use of the pyridylphosphine linker introduces steric and electronic asymmetry at the node which, whilst not exploited here, is important in asymmetric catalysis.18 It is apparent that, despite synthetic challenges, the use of phosphines as structural components in MOFs could be further exploited to produce a range of topologically diverse functional structures.
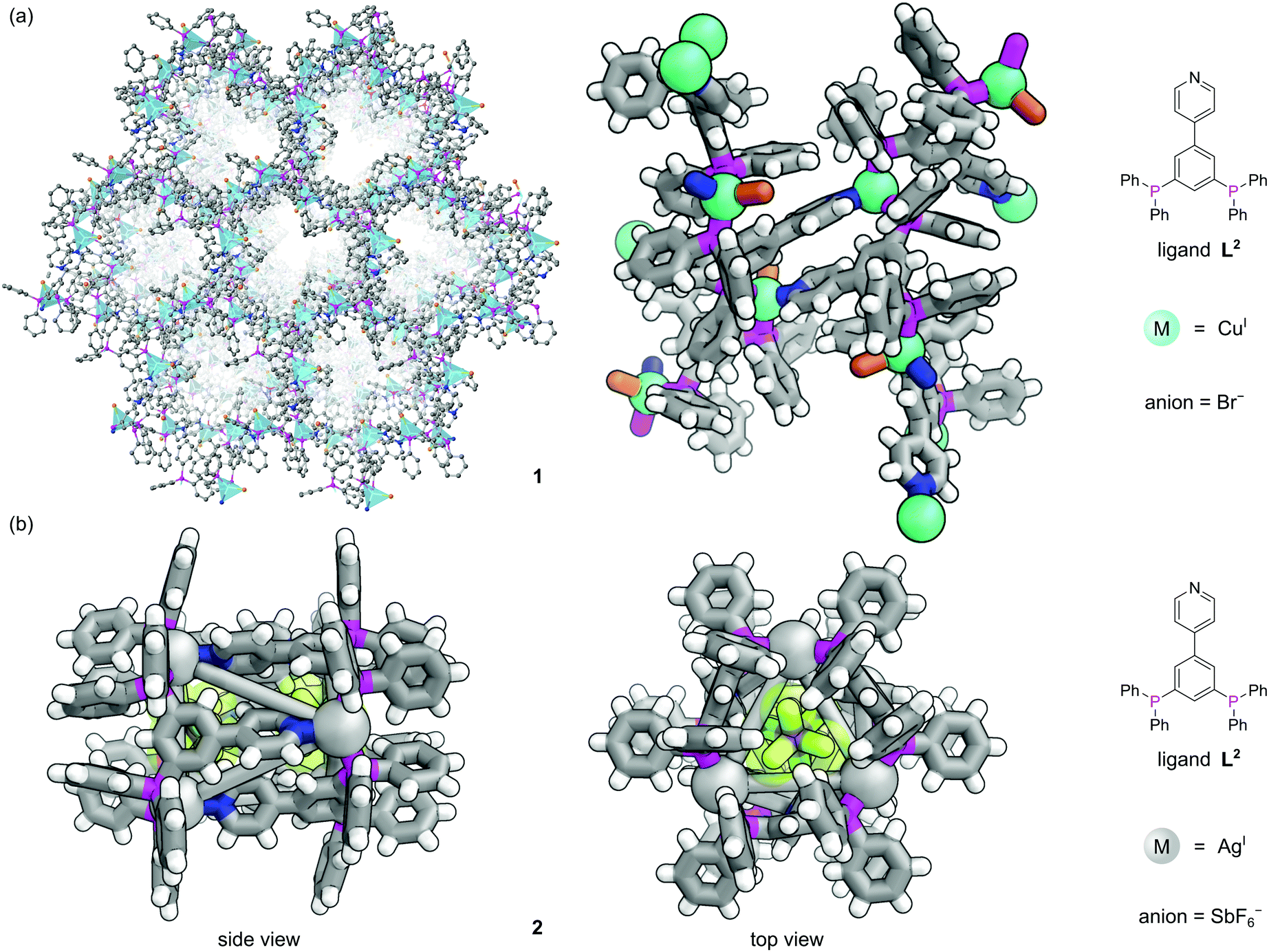 | ||
| Fig. 2 The versatile nature of metal coordination chemistry enables both extended and discrete structures to be constructed from the same 4-(3,5-bis(diphenylphosphino)phenyl)pyridine ligand L2.16 (a) Two different representations of extended MOF 1, where three different ligands meet at each tetrahedral copper centre (with bromide as the fourth ligand). Hydrogen atoms have been omitted for clarity in the left representation; (b) two views of discrete nanotubular cage 2, where three different ligands meet at each tri-coordinate silver vertex. This structure was synthesised with the non-coordinating SbF6− anions, two of which are encapsulated in the central cavity. Colours: C grey, H white, Ag light grey (shown as spheres), Br dark orange, Cu blue green (shown as spheres), F lime green, N blue, P magenta, Sb purple. | ||
2.2 Metal–organic cages
In contrast to MOFs, phosphines have been employed far more extensively as ligands for the construction of discrete metal–organic architectures, including metal organic cages,19,20 metallacycles,21 and mixed metalloporphyrin assemblies. Readers are directed towards a more detailed review of the area22 and we focus discussion here on some more recent examples. Due to the softer nature of the phosphine donors, they are often partnered with coinage metals (Cu, Ag and Au). For example, a tritopic triangular face capping phosphine ligand, L3, formed tetrahedral M4L4 metal–organic cages with Cu(I), Ag(I) or Au(I), 3 (Fig. 3a).23 Three panels meet at each vertex, with each vertex also capped by a terminal iodide ligand. Metallophilic interactions can also assist assembly. Imido μ3-NtBu2− units can hold together Au3 triangular motifs (aided by aurophilic interactions) to form multi metal nodes which can serve as the corner units for tetrahedral metal–organic cages (Fig. 3b).24 Each gold atom has one remaining coordination site for a phosphine ligand, meaning the combination of these Au3 units with triangular tritopic phosphine panels, L4, results in dodecagold metal organic cage 4. Interestingly, the same 4-(3,5-bis(diphenylphosphino)phenyl)pyridine ligand L2 that forms a MOF with Cu(I) and bromide, forms discrete M6L6 nanotubular structures 2 with Ag(I) and non-coordinating anions (Fig. 2b).25 The Ag(I) ions are tricoordinate (as opposed to the pseudo-tetrahedral Cu(I)), and crucially the anions do not coordinate to the metal centres. SCXRD however, was able to identify the presence of two SbF6− anions within the central cavity, held in place by only non-covalent interactions. More recently it has been shown that a luminescent molecular box can be constructed from four triphos ligands and six gold atoms around a central templating chloride anion. Interestingly, mechanical grinding of these blue luminescent crystals results in conversion of the cationic part into a bridged helicate cation which shows green emission.26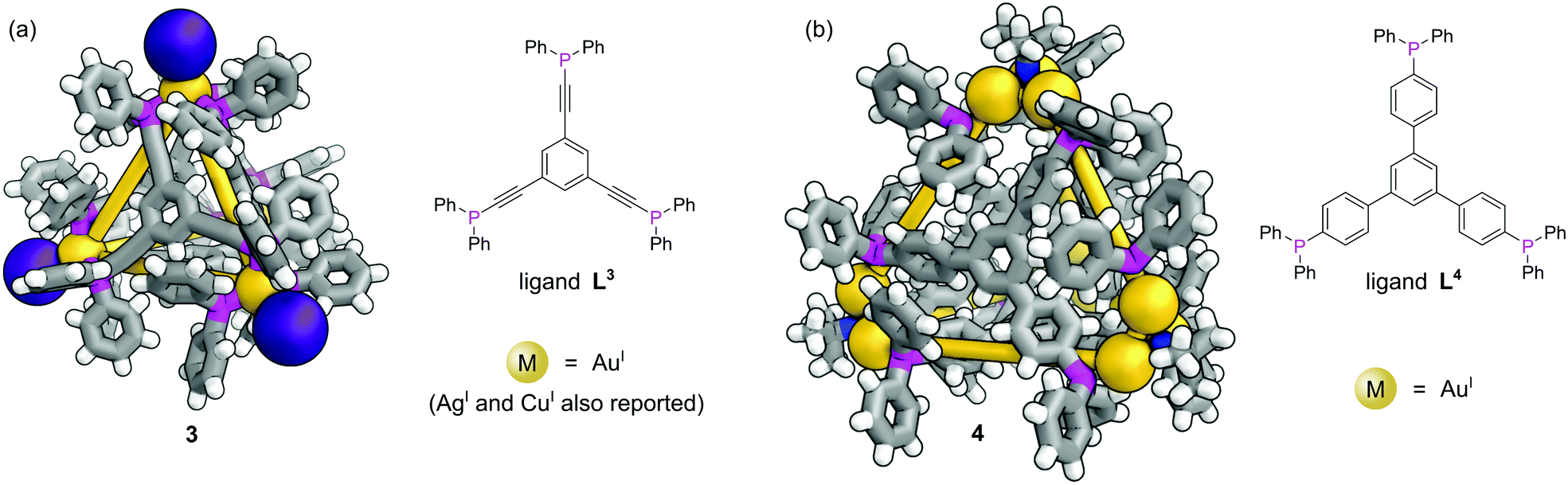 | ||
| Fig. 3 Structural metal–phosphine coordination to hold together metal–organic cages: (a) M4L4 tetrahedral metal organic cage 3 formed with AuI. AgI and CuI congeners also reported; (b) tetrahedral metal–organic cage 4, where each vertex consists of a Au3 triangle held together by a μ3-NtBu2− unit and aurophilic interactions. Colours: C grey, H white, Au gold (shown as spheres), I dark purple (shown as spheres), N blue, P magenta. | ||
When targeting the finite structure of a MOC over an extended MOF,27 certain ligands do not occupy all coordination sites on the metal nodes. These remaining coordination sites often must be blocked to prevent coordination of extra ligands. Phosphines have found particular use in this respect. It is often the case that two adjacent sites on a metal must be occupied by these ‘blocking’ ligands, and a number of the readily available bidentate phosphines such as 1,1′-bis(diphenylphosphino)ferrocene (DPPF, L5) are excellently suited for this.28 For example, four L5 ligands block extra coordination sites around the M4L2 metal–organic cage 5 (Fig. 4).29
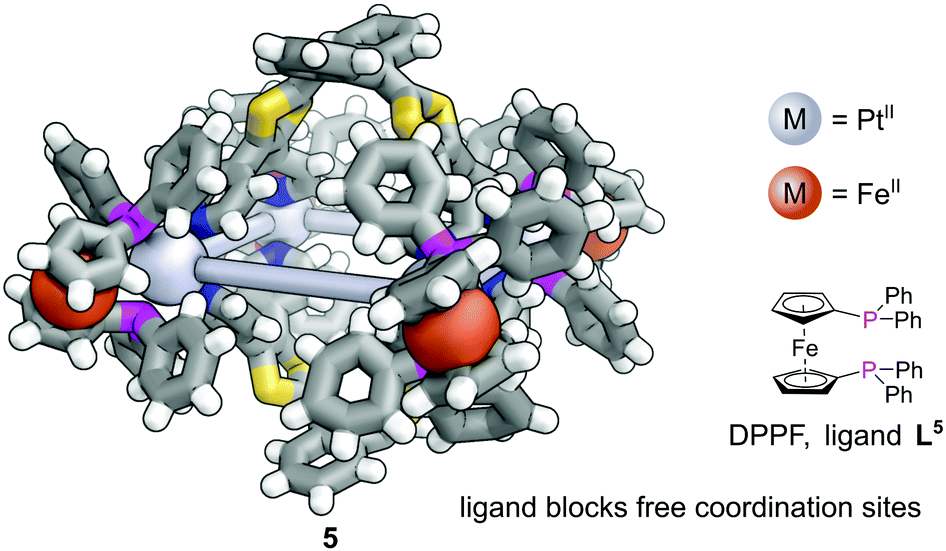 | ||
| Fig. 4 The two phosphorus atoms of each 1,1′-bis(diphenylphosphino)ferrocene (DPPF, L5) ligand each block a coordination site on the Pt4L2 metal–organic cage 5, ensuring its finite nature. Colours: C grey, H white, Fe orange (shown as spheres), N blue, P magenta, Pt light grey (shown as spheres), S yellow. | ||
3. Catalysis by non-covalently encapsulated metal–phosphine guests
The non-covalent encapsulation of phosphines and their complexes within well-defined porous structures allows for functionality to be introduced to these structures without the need for synthetic modifications. Given the synthetic difficulties of manipulating phosphines, the non-covalent approach often saves considerable time and effort.3.1 MOFs
Encapsulation of guests within the pores of MOFs is well documented within the literature.30 Encapsulation may be achieved in a variety of ways: (i) insertion of preformed guest into pores through non-covalent interactions (cartridge approach),31 (ii) assembly of the guest within the pore (‘ship-in-bottle’ approach),32 and (iii) assembly of the MOF around the guest (‘bottle-around-ship’ approach).12 Immobilisation of reactive species through encapsulation allows for a diverse array of additional functionality to be conferred to a MOF structure without the requirement for highly functionalised linkers. Not only are the matrix-isolated guests often more stable, but their reactivities may be modified, tuned and studied.12 Encapsulation has been used as a method to circumvent the challenges posed by the instability of several of homogeneous metal–phosphine complexes used in catalysis.30,33,34 Exchange of the endogenous H2NMe2+ cations within the anionic In-based MOF ZJU-28 with [Rh(dppe)(COD)]+ permitted heterogenization of the Rh catalyst by encapsulation within the MOF pores.35 As a catalyst for the hydrogenation of 1-octene to n-octane, the supported complex initially exhibits similar reactivity to its homogeneous counterpart. However, in reactions using recycled catalyst, the heterogeneous system considerably outperforms the homogeneous catalyst, maintaining its activity and crystallinity even after being recycled four times. This suggests that matrix isolation of the catalyst prevents decomposition during the reaction.The nature of the MOF pores has the potential to strongly influence the reactivity of encapsulated catalysts/substrates through extended coordination sphere interactions between guest species and their well-defined microenvironments. This is evident in the case of the heterogenization of Crabtree's catalyst, [Ir(cod)(PCy3)(py)][PF6], via encapsulation of the cationic component into the pores of sulfonated MIL-101(Cr). The resulting material outperformed its homogeneous counterpart in the hydrogenation of olefinic alcohols both in terms of overall conversion and selectivity.30 The enhanced activity of the encapsulated catalyst is attributed to (i) spatial isolation of metal centres which prevents formation of inactive polymetallic hydride clusters observed in solution, and (ii) suppression of the competing isomerisation pathway by the chemical microenvironment of the pore.30 The pores feature H-bond accepting sulfonate groups, Lewis acidic Cr(III) nodes and Na+ cations providing a range of possible guest-cavity interactions. In this case the sulfonates play a non-spectator role, forming hydrogen bonds with the olefinic hydroxyl group, disfavouring coordination to the Ir centre and thus supressing formation of the undesired aldehyde (Fig. 5).
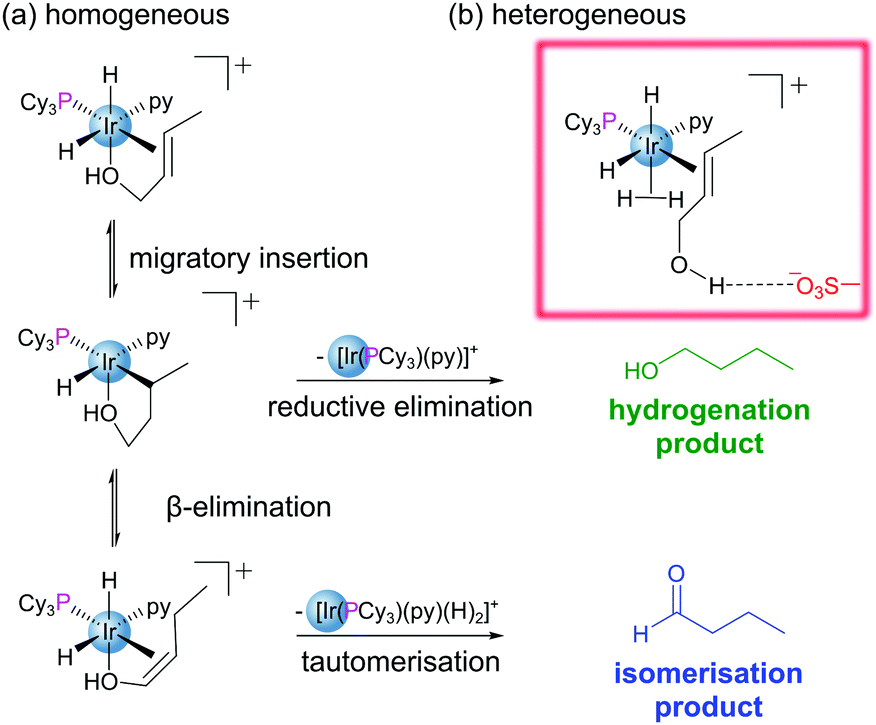 | ||
| Fig. 5 (a) Mechanism for hydrogenation of olefinic alcohols by Crabtree's catalyst in a homogeneous system via reductive elimination from the migratory insertion product, and the competing pathway involving β-elimination from the from the migratory insertion product to yield an isomerisation product; (b) when the catalyst is encapsulated in sulfonated MIL-101(Cr) the competing isomerisation pathway is suppressed by a guest–cavity interaction between the olefinic hydroxyl group and a sulfonate group present in the pore. A bound dihydrogen is shown coordinating in place of the oxygen. | ||
Nevertheless, the prerequisites for the encapsulated guest and the pore networks (size, positioning effects, stability etc.) places limits on the widespread applicability of encapsulation. Instead, species of interest may be incorporated into the MOF scaffold itself (at nodes, linkers or grafted into pores) where they occupy a defined chemical environment (see below) which facilitates their characterisation, perhaps most notably by SCXRD.5
3.2 Metal–organic cages
Fujita and co-workers have constructed a family of MnL2n nanospheres via the coordination of dipyridyl ligands to Pd or Pt metal centres.36 The cages contain both a large cavity and sizeable apertures to allow guests to move in and out easily, with the bend angle in the ligand controlling which polyhedral shape forms. Endo-functionalisation of the ligands allows large numbers of the same functional groups to be expressed projecting into the cavity. An M12L24 Pt(II) cage 6 was developed with 24 guanidinium moieties expressed in the interior (Fig. 6a).37 The target mono-phosphine gold(I) catalyst was modified with a sulfonate group to aid binding in the cage via hydrogen bonding. The 4-pentynoic acid substrate 7a is deprotonated to the carboxylate anion under the basic reaction conditions, which was also bound via similar hydrogen bonding interactions. The preorganisation of both substrate and catalyst enabled increases in the turnover frequency (TOF) and turnover number (TON) of up to 50-fold over the background reaction in the cyclisation to lactone 7b (Fig. 6b). Encapsulation also enabled additional selectivity. For example, allenol substrate 7c which normally cyclises to THF 7d under Au(I) catalysis, is unreactive, as the alcohol is insufficiently acidic to be deprotonated to the alkoxide, and so there is no driving force for encapsulation.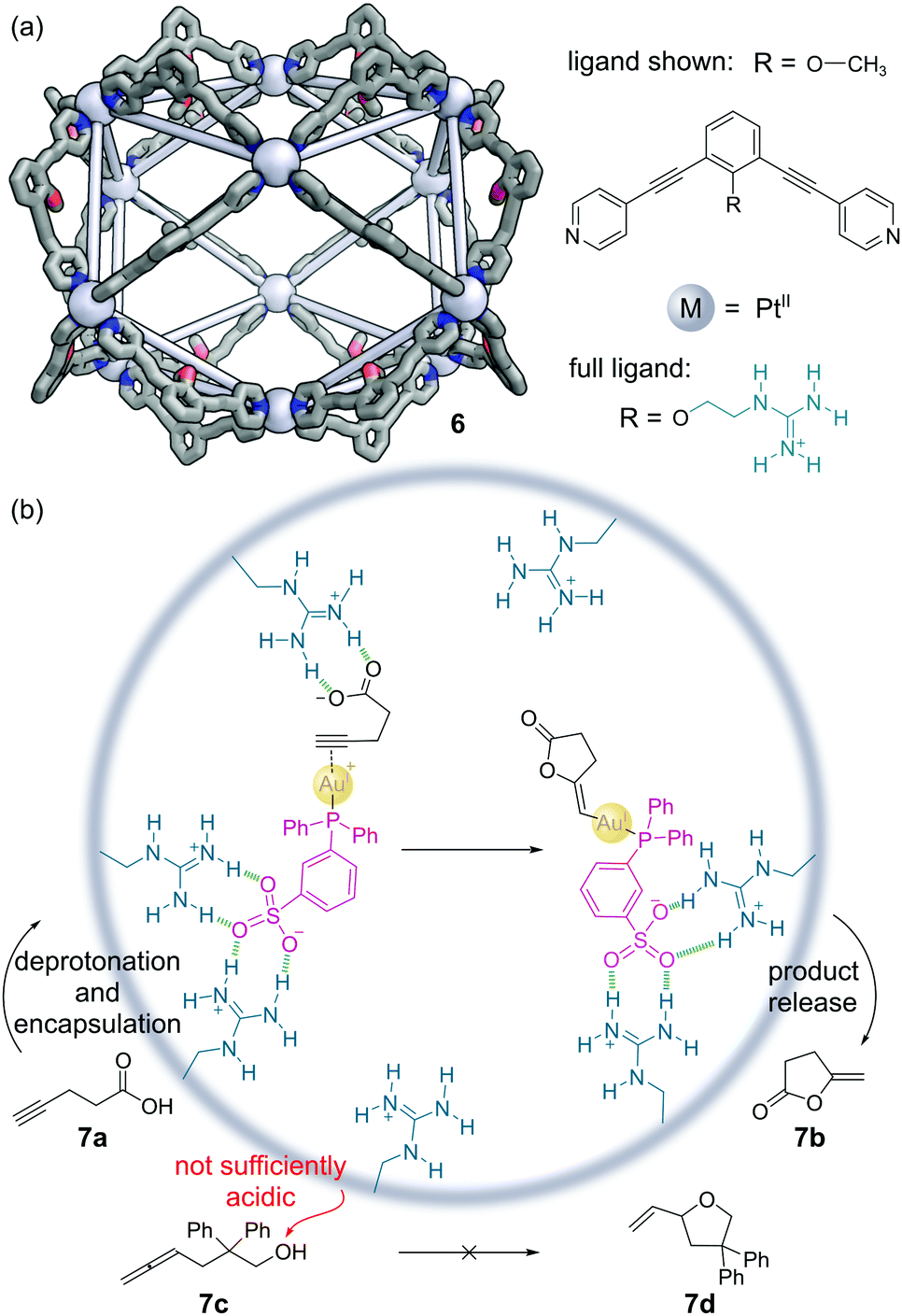 | ||
| Fig. 6 Reek and co-workers’ non-covalently bound phosphine–gold complex catalyses the cyclisation of 4-pentynoic acid 7a. (a) The outer framework of the M12L24 nanosphere 6 solved by SCXRD, with the inner groups and hydrogen atoms omitted for clarity. Colours: C grey, N blue, O red, Pt light grey (shown as spheres); (b) key steps in the cyclisation mechanism, which requires the encapsulation of both catalyst and substrate via hydrogen bonding interactions.37 | ||
The cationic component of bisphosphinorhodium(I) catalyst, [Rh(dppe)(COD)]+, can be encapsulated in the cavity of the anionic gallium(III) M4L6 cage 8 to generate a selective hydrogenation catalyst 8a (Fig. 7).38 Under an atmosphere of hydrogen gas, this host–guest complex catalyses the hydrogenation of alkene substrates that can penetrate the interior of the cage. This enables regioselective hydrogenation of large polyenes. For example, only one C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond (highlighted in green) in polyenol 9a is reduced to give 9b. The two more central CC double bonds (highlighted in red) are not touched, as they are not able to penetrate sufficiently far into the cavity to encounter the active catalyst. Hence, this system shows remarkable selectivity between three alkenes with almost identical electronic and steric properties – unsurprisingly performing the hydrogenation without the cage leads to intractable mixtures of singly and multiply hydrogenated products.
C double bond (highlighted in green) in polyenol 9a is reduced to give 9b. The two more central CC double bonds (highlighted in red) are not touched, as they are not able to penetrate sufficiently far into the cavity to encounter the active catalyst. Hence, this system shows remarkable selectivity between three alkenes with almost identical electronic and steric properties – unsurprisingly performing the hydrogenation without the cage leads to intractable mixtures of singly and multiply hydrogenated products.
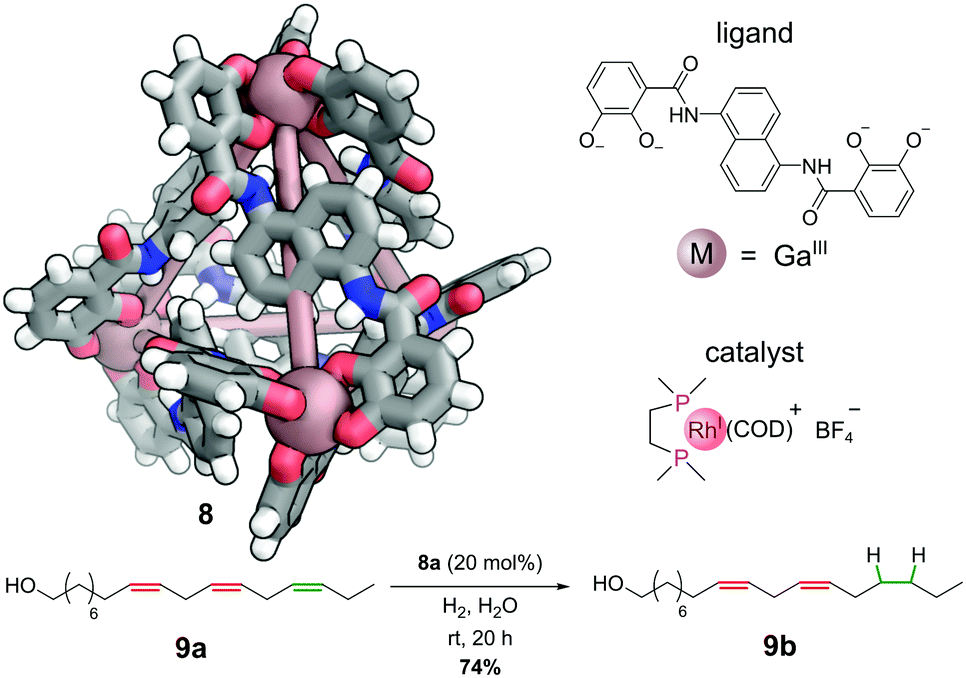 | ||
| Fig. 7 Raymond and co-workers’ regioselective hydrogenation catalysed by a non-covalently bound rhodium-phosphine complex. Colours: C grey, H white, Ga dark salmon (shown as spheres), N blue, O red. | ||
3.3 Other structures
β-Cyclodextrins and their derivatives have been shown to encapsulate sulfonated variants of triphenylphosphine (containing between one to three sulfonate groups) to form inclusion complexes. The binding of these phosphines to metal centres in the presence of these hosts alters the equilibria between species with differing numbers of phosphine ligands. The binding within the β-cyclodextrins was shown to affect reactions such as a rhodium-catalysed hydroformylation reaction (altering the linear to branched ratio) and a palladium-catalysed Tsuji–Trost reaction. The host–guest chemistry of these complexes has already been reviewed in detail, and readers are directed here for more information.39,404. Incorporation and modification of free phosphines for catalysis
4.1 Incorporation and modification of phosphine-based MOF linkers
Whilst the solution phase reactivity of phosphines and their coordination complexes has been extensively studied for catalysis, their reactivity in the solid state has yet to receive such attention. This is largely due to the challenges presented by performing and characterising reactions in the solid state, for example limitations of characterisation techniques, altered reaction kinetics compared to the solution phase, and impeded diffusion of reactants in the solid state. Nevertheless, solid-state chemistry can offer a platform for the investigation of highly reactive intermediates and reaction mechanisms, and may be used to enhance the stability or modify the selectivity of a catalyst. Thus, phosphines have been incorporated into MOFs through the use of phosphine functionalised linkers and by modification of nodes (see Section 4.2).41Differences in the binding between hard and soft coordinating groups can be harnessed to produce structures with non-coordinated phosphines pointing into the MOF pore.42,43 Post-synthetic modification strategies may be employed subsequently to perform reactions at the phosphine appendages, for example to tether metal–phosphine complexes.12 To illustrate this, we take the example of Zr-PMOF. Owing to coordination incompatibility between hard Zr4+ ions and soft phosphine groups, the reaction of ZrCl4 with a triarylphosphine linker functionalised with three carboxylic acids (e.g.L6) yields a MOF (Zr-PMOF) with Zr–oxo secondary building units (SBUs) and uncoordinated, free phosphines which point into the pores, poised for further reactivity.44,45 Post-synthetic modification of these non-coordinated phosphines with catalytic metal centres allows for the stabilisation of monophosphine complexes within the framework.46
Humphrey and co-workers have demonstrated that concomitant P–M complexation and MOF formation can be achieved in a one-pot reaction via metal–carboxylate network bonding.47 Using mixtures of higher- and lower-valent metal precursors allowed bimetallic MOFs functionalised with crystallographically defined, low-valent metal sites to be formed in-situ, removing the requirement for post-synthetic modification. Solvothermal reaction of tris(p-carboxylato)triphenylphosphine (L6) with one equivalent of 1,4-dipyridylbenzene (1,4-dpb), three equivalents of Co(BF4)2, and one equivalent of XAu(SMe2) (X = Cl, Br) gave purple crystals of PCM-107 (Fig. 8). SCXRD reveals that the structure of PCM-107 features [Co3(OCO)6(1,4-dpb)2(OH2)2] nodes where a central Co is bound only to bridging carboxylates whilst the two equivalent, peripheral Co sites are bound to four carboxylate oxygen atoms, held in infinite layers coordinated through the carboxylates to the octahedrally coordinated terminal Co(II) centres. These layers are linked to adjacent layers by 1,4-dipyridylbenzene, forming pillars. The phosphine groups, which do not coordinate Co(II) cations, remain available for tethering of linear Ar3P–AuX species (Fig. 8c).
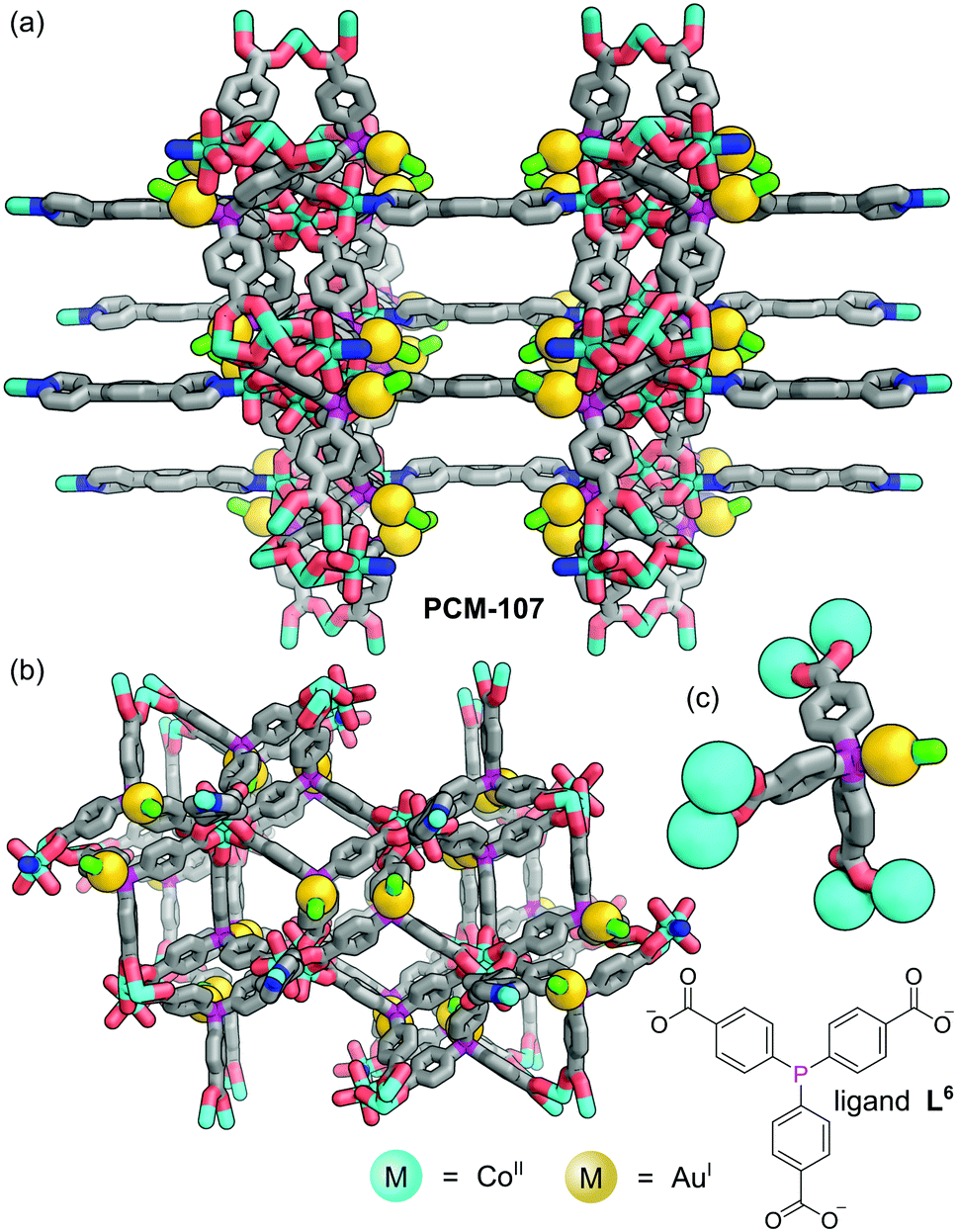 | ||
| Fig. 8 (a and b) PCM-107 features layers of [Co3(OCO)6(1,4-dpb)2(OH2)2] nodes connected through the carboxylate groups of L6 and linked to adjacent layers by 1,4-dipyridylbenzene to form pillars; (c) the nodes are linked through L6 which binds AuX species through the formation of a linear Ar3P–AuX complexes. Hydrogen atoms omitted for clarity. Colours: C grey, Au gold (shown as spheres), Cl bright green, Co light blue (shown as spheres in part (c)), N blue, O red, P magenta. | ||
Metal–phosphine complexes can also be incorporated into MOFs via pre-metalation.12 This approach requires the use of a metal–phosphine complex metalloligand with strategically positioned binding moieties in the secondary coordination sphere to take part in divergent coordination as required for MOF formation. Pre-metalation enforces high occupancy of metal complexes throughout the structure, facilitating structure determination from SCXRD data. Additionally, pre-metalation strategies also allow ligands that would otherwise not tolerate the MOF synthesis conditions to be incorporated into the framework with the metal acting to protect the ligand group.12 Transition metal complexes bearing diphosphine pincer ligands (PXEXP) (Fig. 9) find application as homogeneous catalysts for a broad range of reactions.48
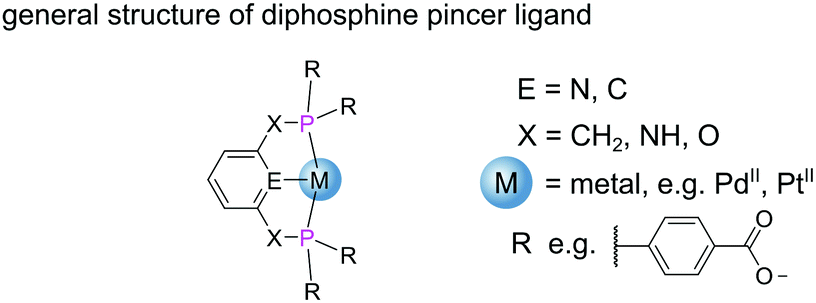 | ||
| Fig. 9 Diphosphine pincer ligands which bind to metal centres through three coplanar sites. The phosphine R groups can be chosen to incorporate additional binding sites permitting growth of extended structures. | ||
The tridentate nature of pincer ligands and the strength of the phosphine donor groups result in complexes which typically exhibit good stability to harsh reaction conditions. Consequently, a number of MOFs have been synthesised directly from pincer complexes with free carboxylates as a way to produce solid state catalysts with broad catalytic scope.48 Solvothermal reaction of the pseudo-octahedral Co(III) pincer complex [Co(PNNNP)Cl3] (where PNNNP = L7) (Fig. 10) and ZrCl4 results in the formation of a 3D MOF structure (Co-PNNNP-MOF).49 Rietveld refinement of Synchrotron Powder X-ray Diffraction (SPXRD) data reveals this structure to have csq topology. The Zr6 cluster's C4 axis is perpendicular to the crystallographic c axis and the pyridyl ring of the PNNNP ligand sits perpendicular to the ab plane, however significant missing linker defects are present throughout the structure.
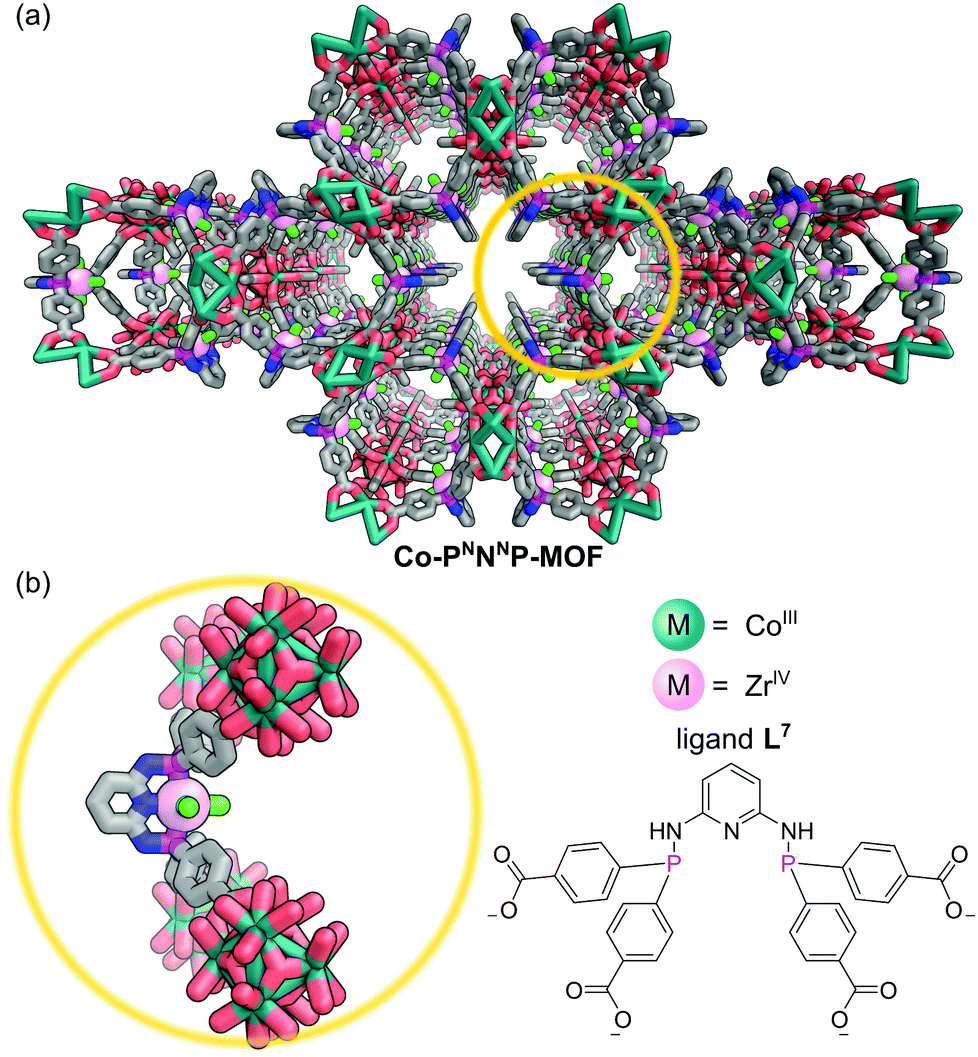 | ||
| Fig. 10 (a) The 3D MOF formed by reaction of [Co(L7)Cl3] with ZrCl4 exhibits an unusual csq topology in which Zr6 clusters are linked through the PNNNP pincer ligand; (b) the [Co(PNNNP)Cl3] complex is retained in the synthesis protruding into the MOF cavities. Hydrogen atoms omitted for clarity. Colours: C grey, Cl bright green, Co teal, N blue, O red, P magenta, Zr light pink (shown as spheres). | ||
Unsurprisingly, the geometry of the metalloligand influences the resulting MOF topology. In contrast to [Co(PNNNP)Cl3], the square planar complexes [MI(PNNNP)] (M = Pd, Pt), which feature the same pincer ligand, react with ZrCl4 under solvothermal conditions to afford cubic Zr-frameworks. Post-synthetic metal exchange (PSME) at a metalated linker is a valuable method for exploiting the geometry differences observed for different metals to give access to structures not accessible via direct solvothermal synthesis. Treating Co-PNNNP-MOF with PtCl2(SMe2)2 resulted in Pt/Co metal exchange allowing access to a different P-MOF polymorph than that obtained via the direct solvothermal route.49
The presence of chiral cavities or chiral active sites within MOFs may be used to achieve enantioselective catalysis.50,51 Chiral phosphines, such as 2,2′-bis(diphenylphosphino)-1,1′-binaphthyl (BINAP), are used extensively to achieve steroselectivity in homogeneous catalysis.52 Despite the clear benefits of incorporating chiral phosphines into MOFs, the synthesis of multidentate chiral phosphine ligands for use as linkers is synthetically challenging and their application to date is limited. Lin et al. reported the multistep synthesis of a BINAP-derived dicarboxylate linker, L8, from 4,4′-I2-BINAP which reacted with ZrCl4 with TFA as an additive to give a chiral Zr metal–organic framework (BINAP-MOF) (Fig. 11).50 Chiral metal complexes could be introduced via post-synthetic metalation of the phosphine sites to provide catalytic centres in the large open channels. For example, reaction of BINAP-MOF with one equivalent of [Rh(nbd)2](BF4) yielded BINAP-MOF-Rh, which exhibits excellent behaviour as a single-site solid asymmetric catalyst for a variety of organic addition reactions. BINAP-MOF-Rh catalyses the conjugate addition of arylboronic acids to 2-cyclohexanone at just 1 mol% catalyst loading, demonstrating activity of ca. three times higher than the homogeneous control with similar selectivity (>99% ee). Furthermore, excellent activity is observed for asymmetric addition of AlMe3 to α,β-unsaturated ketones, where it again outperformed the homogeneous control in terms of activity, with comparable selectivity, probably due to site isolation of the catalytic centres. The recyclability of the catalyst could be demonstrated as the powder X-ray diffraction (PXRD) of BINAP-MOF-Rh recovered from an AlMe3 addition reaction was the same as the freshly prepared sample, and its reuse in consecutive reactions resulted in only small decreases in activity and enantioselectivity.
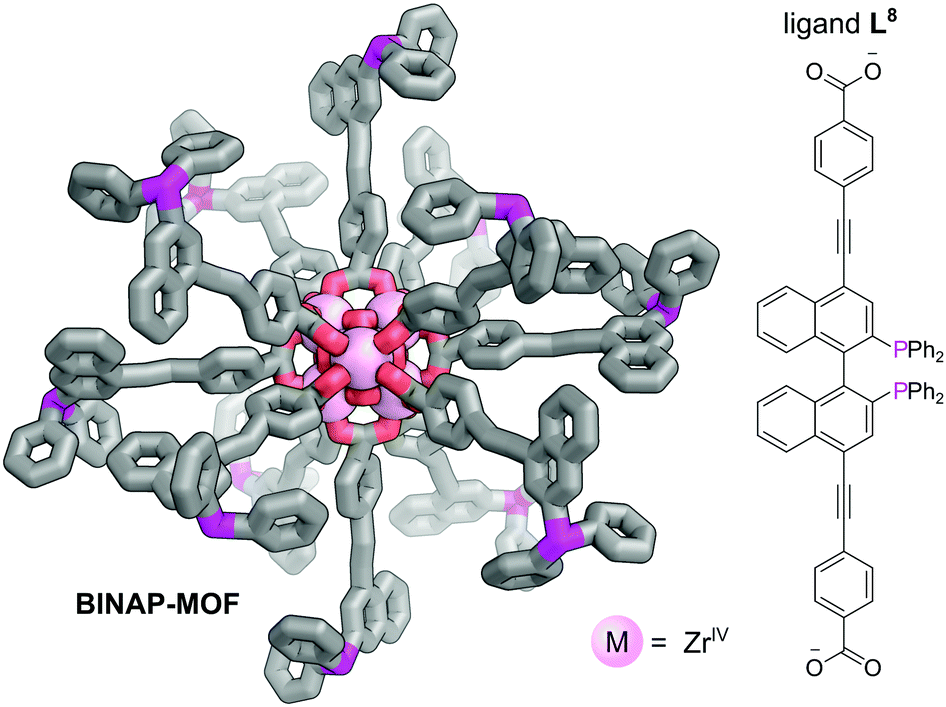 | ||
| Fig. 11 The Zr6O4(OH)4(O2CR)12 SBUs bound by 12 L8 bridging ligands which bridge adjacent clusters to form BINAP-MOF. Post-metalation with [Rh(nbd)2](BF4) yields BINAP-MOF-Rh, which acts as a catalyst for the conjugate addition of arylboronic acids to 2-cyclohexanone.50 Hydrogen atoms omitted for clarity. Colours: C grey, O red, P magenta, Zr light pink (shown as spheres). | ||
4.2 Post-synthetic modification at MOF nodes
Phosphines may also be introduced into MOFs via post-synthetic modification of the node. This approach offers the advantage that a library of isostructural P-MOFs can be produced with a range of phosphines. This allows comparisons between different ligands to be made whilst leaving other parameters unchanged and could allow for optimisation of P-MOF catalysts. Sulfonated triarylphosphine moieties could be incorporated into the large pore of MOF-808(Hf), through anion exchange of formate groups on the SBUs.53 Exchanging formate with the anionic sulfonated triarylphosphines, [Ph2P(m-SO3C6H4)]− or [PhP(p-SO3C6H4)2]2−, afforded the phosphine functionalised structures SulP1-MOF-808(Hf) or SulP2-MOF-808(Hf) respectively, with PXRD confirming retention of crystallinity following modification. Energy-dispersive X-ray spectroscopy (EDX) and inductively coupled plasma (ICP) analysis reveal a P/Hf ratio of 0.04, consistent with a single triarylphosphine sulfonate moiety bound for each pore. Such structures can be considered as porous, solid state phosphine ligands which can be used as crystalline catalyst supports. Indeed, treating SulP1-MOF-808(Hf) with [IrCl(COD)]2 results in metalation of the phosphine sites, to yield the crystalline structure SulP1-MOF-808(Hf)-Ir which has immobilised Ir sites throughout its structure. The presence of the phosphorus atom as a spectroscopic handle allows coordination to be confirmed by 31P magic angle spinning NMR since coordination results in a downfield shift of the phosphine signal from −4 ppm to 25 ppm. The presence of two distinct active sites within SulP1-MOF-808(Hf)-Ir (the SBU and the phosphine bound Ir-centre) allow the system to be exploited as a high-performance bifunctional catalyst for the reductive amination of a range of ketones, since imine formation is catalysed by the framework metal cations, and hydrogenation is catalysed at the tethered Ir centre.53 By applying the anion exchange step, introducing the phosphine tether with a variety of different phosphines and MOFs or metalating with other metal centres, a wide range of structures with different catalytic activities could potentially be accessed. Thus, this strategy offers great potential for wider development.4.3 Metal–phosphine complexes in metal–organic cages
The aforementioned M12L24 nanosphere, reported by the Fujita group,36 (see Section 3.2) can also be constructed from dipyridyl ligands with a tethered endohedral triarylphosphine gold(I) chloride complex (Fig. 12a). Self-assembly with a source of PdII gave M12L24 cuboctahedral cage 10a.54 Each cavity contained 24 gold atoms, allowing a local concentration of gold of 1.07 M to be achieved in a solution where the overall gold concentration was only 5 mM. Gold(I) chloride complexes often require chloride abstraction before they become active in catalysis, however in this system no such activation was required. The authors found evidence of d10–d10 aurophilic interactions and attributed the activity to the formation of multinuclear gold complexes inside the metal–organic cage. The cage catalysed the cyclisation of γ-allenol 7c to furan 7d in 88% yield but with a TON of 1.86. The isomeric dihydropyran 11a (which can be formed under non cage-catalysed processes) was not observed. Similarly, cyclisation of 1,6-enyne 12 proceeded with full selectivity for cyclopentene 12a over cyclohexene 12b – selectivity seldom observed in ‘normal’ gold catalysis (Fig. 12b).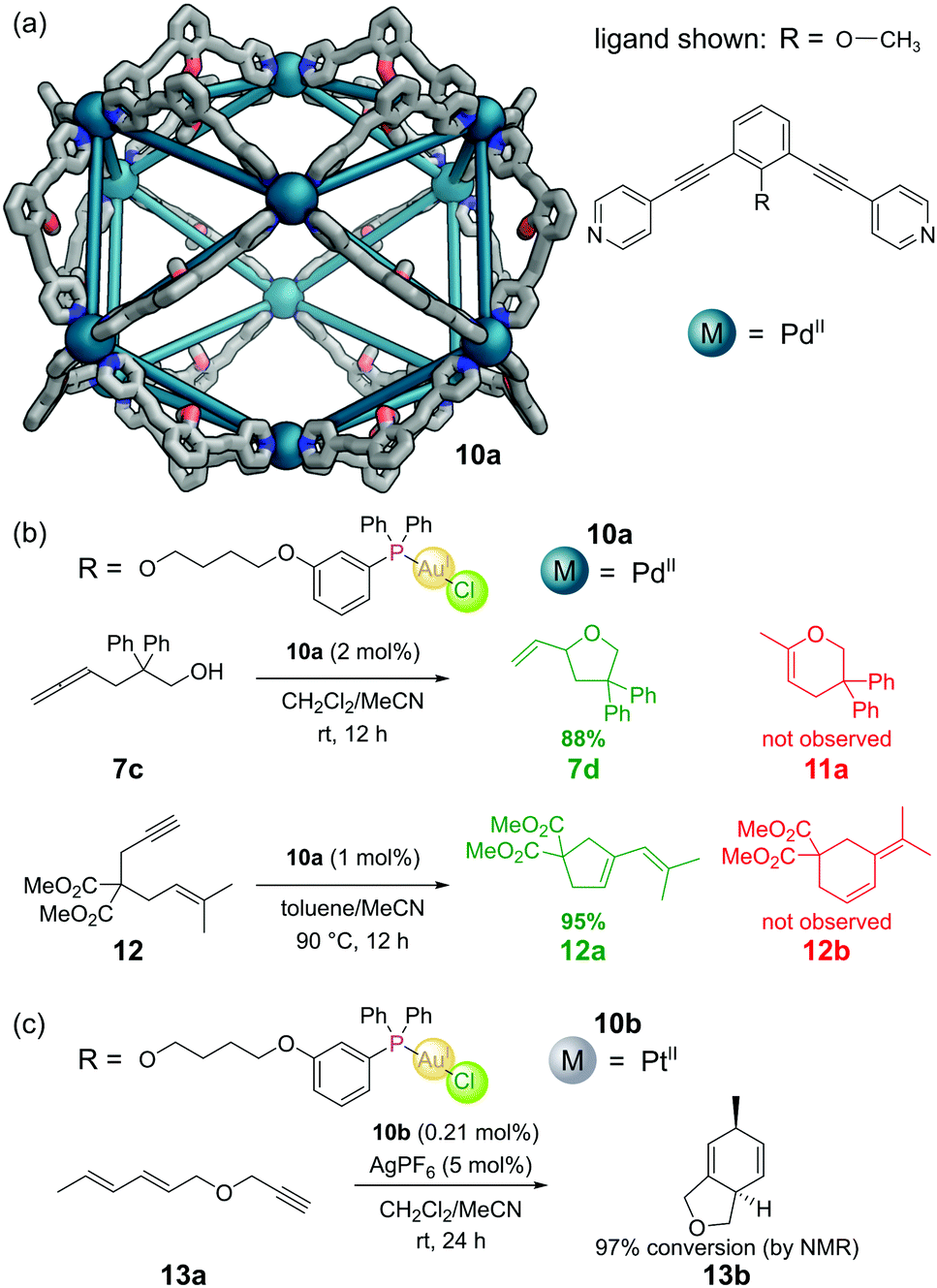 | ||
| Fig. 12 Reek and co-workers’ covalently-tethered phosphine–gold complexes for catalysis. (a) The outer framework of the M12L24 nanosphere 6 solved by SCXRD, with the inner groups and hydrogen atoms omitted for clarity. Colours: C grey, N blue, O red, Pd dark teal (shown as spheres); (b) selectivity in cyclisations catalysed by Pd nanospheres 10a; (c) the use of Pt nanospheres 10b to accomplish more challenging cyclisations. | ||
Other more challenging cyclisations required a switch from neutral to cationic Au(I) species. These could be generated by the addition of AgPF6 to extract the chloride, however under these harsher reaction conditions it was necessary to use Pt(II) nanospheres 10b as the Pd(II) nanospheres did not survive.55 In one such example, Pt nanosphere 10b catalysed the efficient conversion of diene-yne 13a into bicyclic 13bvia a [4+2] cycloaddition (Fig. 12c).
4.4 Covalently-bound phosphines in cyclodextrins
Covalent incorporation of phosphines into organic macrocycles is synthetically challenging. The most extensively studied systems have been the cyclodextrins,40 which offer several advantages. The cyclodextrins are commercially available in high purity and low cost. The primary alcohol groups which protrude from one face of the cyclodextrin macrocycle are relatively straightforward to activate into good leaving groups for substitution reactions. They can then be displaced with phosphorus(III)-based nucleophiles (which tend to be highly nucleophilic). The systems are also inherently chiral (and exist as single enantiomers) meaning that there is the possibility for enantioselective catalysis. There are many examples which have been covered in detail a recent review and so we will only mention selected examples here.56A rigid monophosphine L9 was constructed by incorporating a short phenyl phosphinidene cap on the primary face of α-cyclodextrin and β-cyclodextrin derivatives (Fig. 13a). These systems could bind a variety of different metal ions to make metal–phosphine complexes, with metals such as palladium (14), platinum, and rhodium (Fig. 13b). In many metal-catalysed reactions, the formation of an intermediate metal species bearing a single phosphine is proposed to be the most highly active species for certain steps. Even with the use of bulky phosphines, such mono-phosphine metal species are rarely observed. Typically, this is because the phosphine to metal ratio is usually kept >1 to prevent catalyst decomposition by the formation of multi-metallic species. However, with the rigid cyclodextrin monophosphines, the phosphorus lone pair is oriented towards the cavity interior, meaning only single metal single phosphine complexes can form. A robust, air stable Rh(I) complex 15 catalysed the hydroformylation of styrene (Fig. 13c).57 The reaction proceeded with both high regioselectivity for the branched isomer and high enantioselectivity.
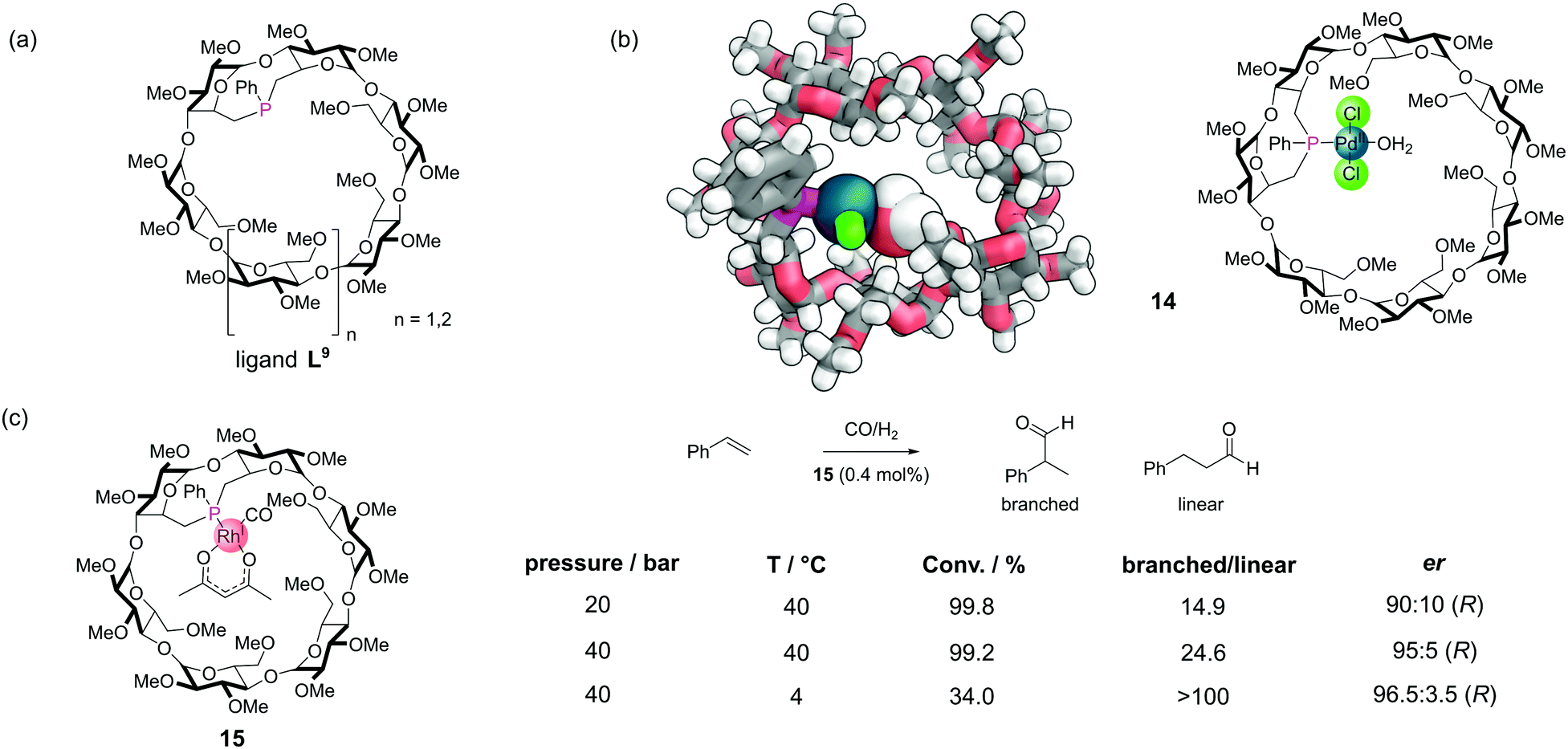 | ||
| Fig. 13 (a) Rigid monophosphine-modified α and β-cyclodextrin ligands L9; (b) SCXRD and chemical structure of a palladium–phosphine complex from a β-cyclodextrin, 14. Colours: C grey, H white, Cl bright green, O red, P magenta, Pd dark teal (shown as a sphere). The bound water molecule is also shown as spheres; (c) control of a rhodium-catalysed hydroformylation reaction with rhodium–phosphine complex 15. | ||
The same phosphines have also been employed as ligands for palladium in polymerisation reactions. The combination of tertiary phosphines with palladium is seldom used for olefin polymerisation as the resultant polymers are usually only of low molecular weight due to the tertiary phosphines increasing the rate of β-hydride elimination.58,59 Complexes formed from these α-cyclodextrin and β-cyclodextrin ligands with [Pd(allyl)Cl]2 were able to catalyse the homopolymerisation of styrene if a positive atmosphere of CO was employed.60 It was postulated this CO helped suppress β-hydride elimination in this case, and unlike other examples,61 no carbonyl insertion was detected. The resultant polymers had Mn of up to 9100, and although largely atatic, displayed optical activity imparted from the cyclodextrin scaffold.
The tethering of a dicyclohexylphosphine into a β-cyclodextrin complex gave a ligand, L10, capable of binding Ru(II) to furnish a ring-opening metathesis polymerisation (ROMP) catalyst 16 (Fig. 14a). The cyclodextrin cavity binds the 7-oxanorbornene dimethanol substrate for the reaction in water via non-covalent interactions (Fig. 14b). As the ROMP proceeds, the additional monomers insert between the metal centre and growing polymer chain passing through the molecular recognition site within the cyclodextrin on their way to the catalytic metal centre, allowing high levels of control. High conversions and molecular weights (Mn) of 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 were achieved and the polymerisation could also be inhibited by introduction of competing guests for the cavity.62
000 were achieved and the polymerisation could also be inhibited by introduction of competing guests for the cavity.62
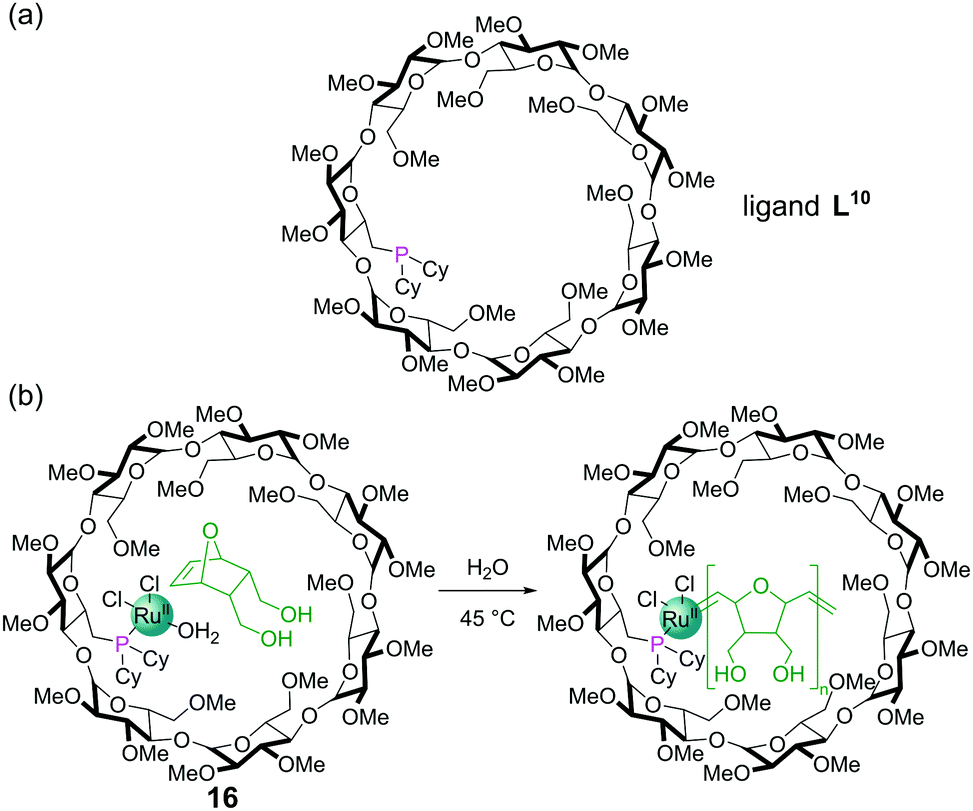 | ||
| Fig. 14 (a) Chemical structure of dicyclohexylphosphine ligand L10; (b) ring opening metathesis polymerisation catalysed by RuII complex 16. | ||
Rigid diphosphine L11 was constructed by incorporating two dialkylarylphosphine caps on the primary face of an α-cyclodextrin derivative (Fig. 15a). The two phosphine lone pairs point towards each other and into the cavity, which significantly affects their reactivity. Unlike other dialkylaryl phosphines, they are much less prone to oxidation as the lone pairs are shielded from the outside environment. As expected, diphosphine L11 was found to act as an excellent trans-spanning diphosphine, forming only trans chelate complexes with a variety of metals. The trans complex 17 with the d8 Pd(II) and two chlorides (Fig. 15b) has P–M–P angle of about 173°. This trans preference is clearly driven by the rigid backbone of the cyclodextrin and a near ideal separation between the two phosphorus atoms for complex formation. With the d10 Au(I) a distorted linear complex 18 is observed with a P–M–P angle of 164°, indicating perhaps the ‘natural’ bite angle of this ligand (Fig. 15c).63 With the d10 Ag(I) ion, a distorted trigonal planar complex 19 is formed with a P–M–P angle of 144° (Fig. 15d). The angles are influenced by the extent of the non-covalent interactions between the C–H bonds pointing into the cavity and the single other ligand that can sit in the cavity.
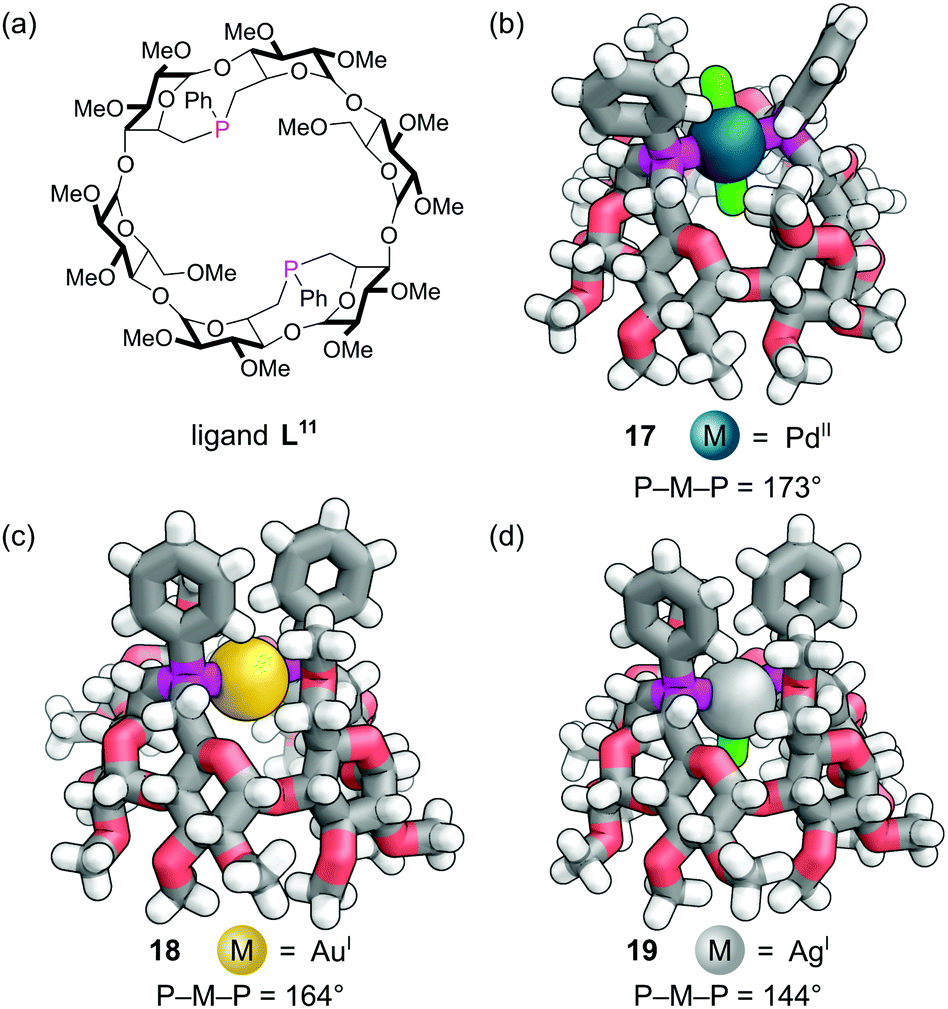 | ||
| Fig. 15 (a) Chemical structure of rigid diphosphine ligand L11; (b) the trans orientation in a square planar PdII complex 17; (c) the trans arrangement in a ‘linear’ AuI complex 18; (d) the distorted trigonal planar AgI complex 19. Colours: C grey, H white, Ag light grey (shown as a sphere), Au gold (shown as a sphere), Cl bright green, O red, P magenta, Pd dark teal (shown as a sphere). | ||
4.5 Covalently-bound phosphines in calix[n]arenes
Calix[n]arenes form another useful class of organic macrocycle for controlling the confined environment around a phosphorus atom and a variety of phosphine-modified calixarenes have been developed.64 Phosphines can be introduced to the wider upper rim by first brominating para to the phenol groups, with the bromines serving as a functional group to directly graft the phosphines to the aromatic rings. Installation of two diphenyl phosphine groups in a 1,3-relationship on a calix[4]arene gives a ligand disposed to the formation of trans complexes with metals.65 Successful formation of such complexes also relies on one of the ligands being small enough to fit into the cavity (which is substantially smaller in a calix[4]arene than in a cyclodextrin). For example the trans,trans,trans-[RuCl2(CO)2(PPh3)2] complex 20 (Fig. 16a) contains a CO within the cavity that possess a particularly low stretching frequency of 1924 cm−1, indicative of a weak bonding interaction between it and the aromatic rings. In a square planar platinum hydride complex from the same ligand, the cavity bound hydride was also observed significantly upfield in the 1H NMR due to shielding from the nearby aromatic panels. It is also possible to functionalise the phenol groups on the narrower bottom ring of calix[4]arenes. In similar fashion these ligands also ensure trans coordination around a metal, such as platinum hydride complex 21 which also has its hydrogen oriented within the cavity (Fig. 16b).66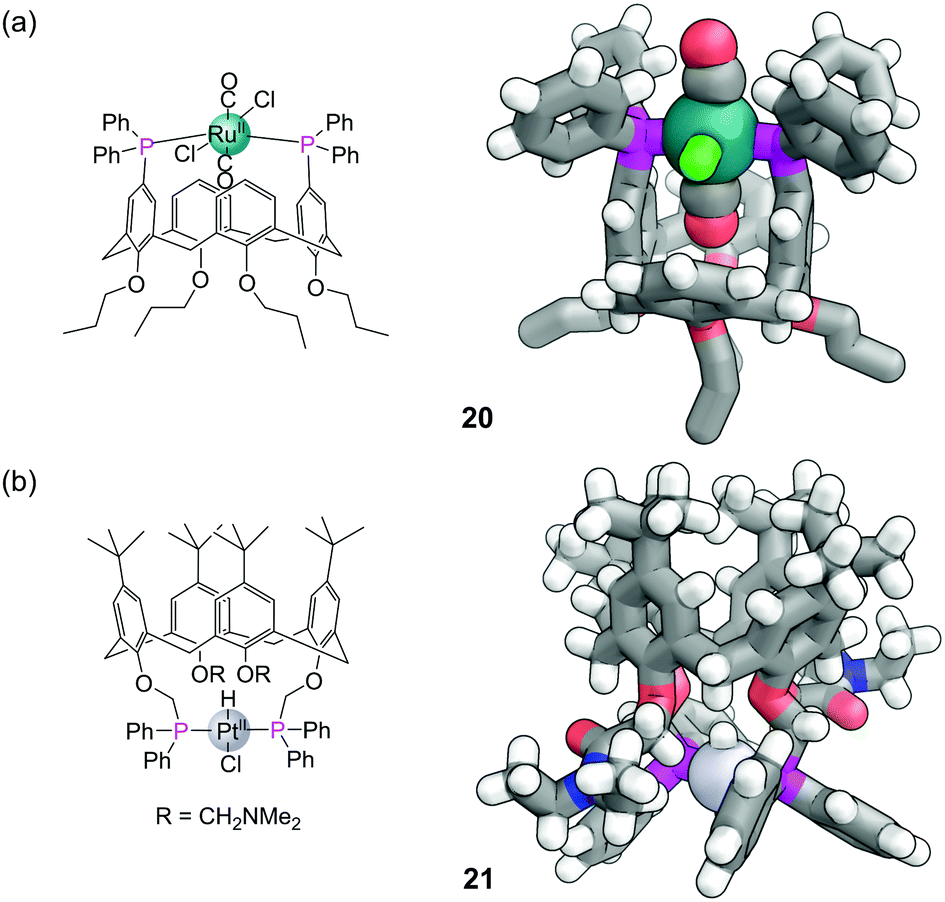 | ||
| Fig. 16 (a) trans,trans,trans-Diphosphineruthenium complex 20 with one CO ligand within cavity. Hydrogen atoms on the propyl groups have been omitted due to disorder; (b) the trans-diphosphine platinum hydride complex 21 with the hydride protruding into the cavity. Colours: C grey, H white, Cl bright green, N blue, O red, P magenta, Ru dark teal (shown as a sphere), Pt light grey (shown as a sphere). Bound CO ligands also shown as spheres. | ||
4.6 Other structures
The combination of trispyridylphosphine ligand L12 with three equivalents of zinc(II) tetraphenylporphyrin 22 (Fig. 17a) gives self-assembled complex 23, held together both by coordination of the pyridine moieties to the zinc, but also by C–H⋯π interactions between neighbouring porphyrin units (Fig. 17b). The coordination of the three porphyrin units places the central phosphorus atom in a highly constrained environment, which makes it an interesting scaffold for metal catalyst binding, as typically only a single phosphine ligand bonds to the metal centre at once. This system has been used extensively for rhodium-catalysed hydroformylation. As long as the temperature is kept sufficiently low (to avoid dissociation of the zinc porphyrins and hence loss of the confined environment around phosphorus) the monophosphine complex 23-RhH(CO)3 is formed exclusively.67 The hydroformylation of 1-octene showed high selectivity for the branched product,68 and the hydroformylation of 2-octene high selectivity for the formyl group on the inner position (Fig. 17c).69,70 Traditional hydroformylation catalysts are often unselective for these transformations and also lead to alkene isomerisation and hydrogenation. The binding of the porphyrins could also be enhanced by exchanging tetraphenylporphyrin 22 for the more electron-deficient porpholactone 24 (Fig. 17d). The higher association constant of this assembly allowed hydroformylation to be performed in more polar solvents, and the branch-selective hydroformylation of the challenging substrate propene.71 The hydroformylation could also be performed asymmetrically via the synthesis of chiral porphyrin derivatives such as 25 (Fig. 17e), although only modest enantioselectivities were obtained.72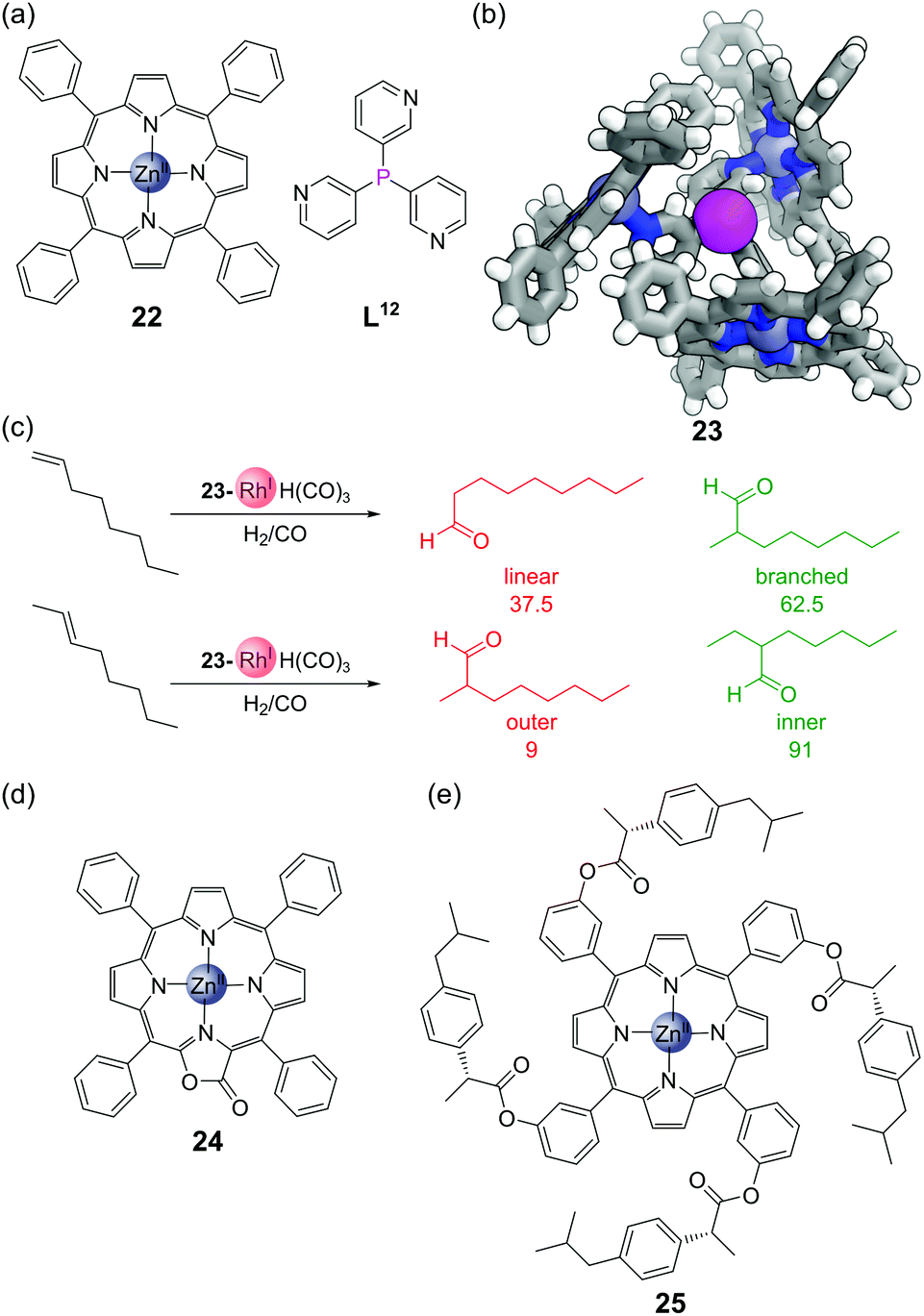 | ||
| Fig. 17 Encapsulated hydroformylation catalysts. (a) Zinc porphyrin 22 and trispyridylphosphine L12 assemble to place the phosphine in a constrained environment; (b) SCXRD of phosphine complex 23. Colours: C grey, H white, N blue, P magenta (shown as a sphere), Zn blue grey (shown as spheres); (c) selectivity for the inner product in rhodium-catalysed hydroformylation reactions; (d) electron-deficient porpholactone 24 enhances stability of the complex; (e) enantioselective hydroformylation can be promoted by using chiral porphyrins such as 25. | ||
5. Post-assembly modification of phosphines in confined environments
5.1 Post-synthetic modification of metal–organic cages to control guest binding
Post-assembly modification (PAM) of discrete supramolecular structures is ubiquitous in nature, with processes such as the post-translational modification of proteins and the epigenetic modification of DNA being crucial to control both structure and function. Recently, the covalent PAM of finite metal–organic structures has received growing attention in efforts to design novel adaptive systems on the molecular level73 and must proceed under mild conditions to not disrupt the metal–organic linkages.74 The redox and metal-coordination chemistry available to phosphines make them excellent functional handles in this regard. Nitschke and co-workers incorporated a triphenylphosphine moiety into face-capping panels for a Zn4L4 tetrahedral metal–organic cage 26 (Fig. 18a).75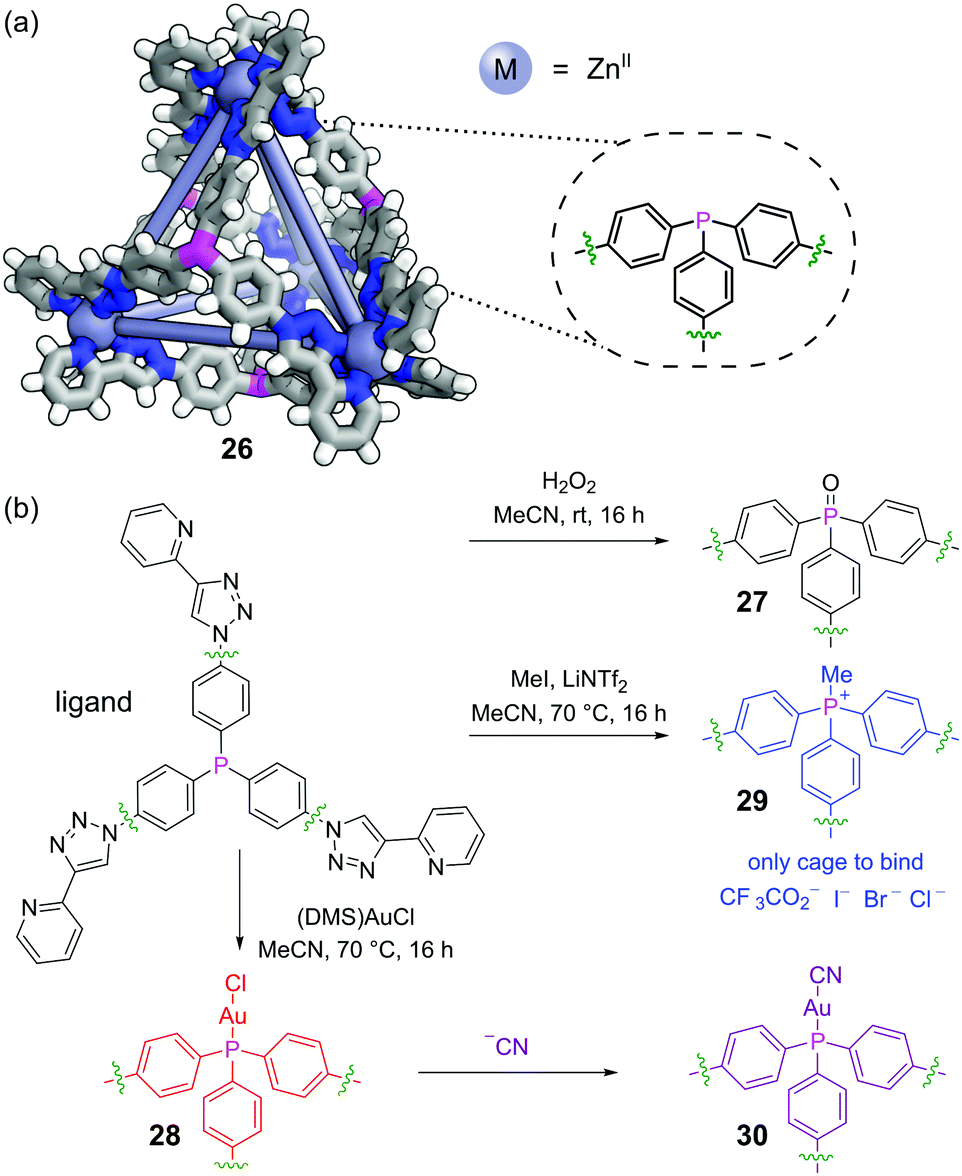 | ||
| Fig. 18 Nitschke and co-workers’ post-assembly modification (PAM) of phosphine cage 26. (a) Structure of phosphine cage 26, adapted from SCXRD of phosphonium salt derivative 29 with Me groups omitted. Colours: C grey, H white, N blue, P magenta, Zn blue grey (shown as spheres); (b) a range of post-assembly modification reactions could be performed, with oxidation giving 27, auration giving 28, and methylation giving 29. Further PAM of the bound chloride on 29 gave cyano derivative 30. The PAM altered the host–guest properties of the cage. | ||
The triaryl phosphine units cleanly underwent several different classes of PAM reactions. The phosphines could be oxidised to phosphine oxides 27 by treatment with hydrogen peroxide (Fig. 18b). Alternatively, the phosphines reacted with chloro(dimethylsulfide)gold(I) to effect auration to give 28 (although metalation with other metals was not successful). Thirdly, the free phosphines could be methylated with methyl iodide to furnish phosphonium salt cage 29. PAM had a pronounced effect on the host–guest chemistry. Whereas all three modified cages bound a range of non-coordinating anions, perhaps surprisingly it was phosphine oxide cage 27 (formal charge +8) that bound these most strongly as opposed to phosphonium cage 29 (formal charge +12). The PO groups are likely oriented into bulk solution exposing areas of high electrostatic potential to the cage interior. Aurated cage 28 could undergo further PAM by displacement of the bound chloride ions with cyanide to furnish 30.
5.2 Post-synthetic modification to control structural transformations
Mirkin and co-workers have pioneered a strategy for constructing metal–ligand coordination complexes using flexible bidentate ligands. In what they term the “Weak Link Approach”, the ligands typically consist of a ‘stronger’ diphenyl phosphine donor group separated by an ethylene space from a ‘weaker’ thioether/ether donor.76 The weaker ligand can be displaced from the metal centre by competing ligands which are more strongly coordinating, allowing complexes to be toggled between more rigid closed forms and more flexible open forms. For example, the closed form of calix[4]arene-based capsule 31 (with both sulfurs bound) has no space for guest binding (Fig. 19). The binding of guests within the capsule can be allosterically regulated by other ligands for the metal. Addition of chloride is sufficient to displace one of the sulfur ligands to form half open capsule 32 which binds pyridine N-oxide, a transformation that can be reversed with the addition of NaBArF. Addition of two equivalents of the stronger ligand cyanide leads to fully open complex 33, which binds methyl pyridinium, although this transformation cannot be reversed.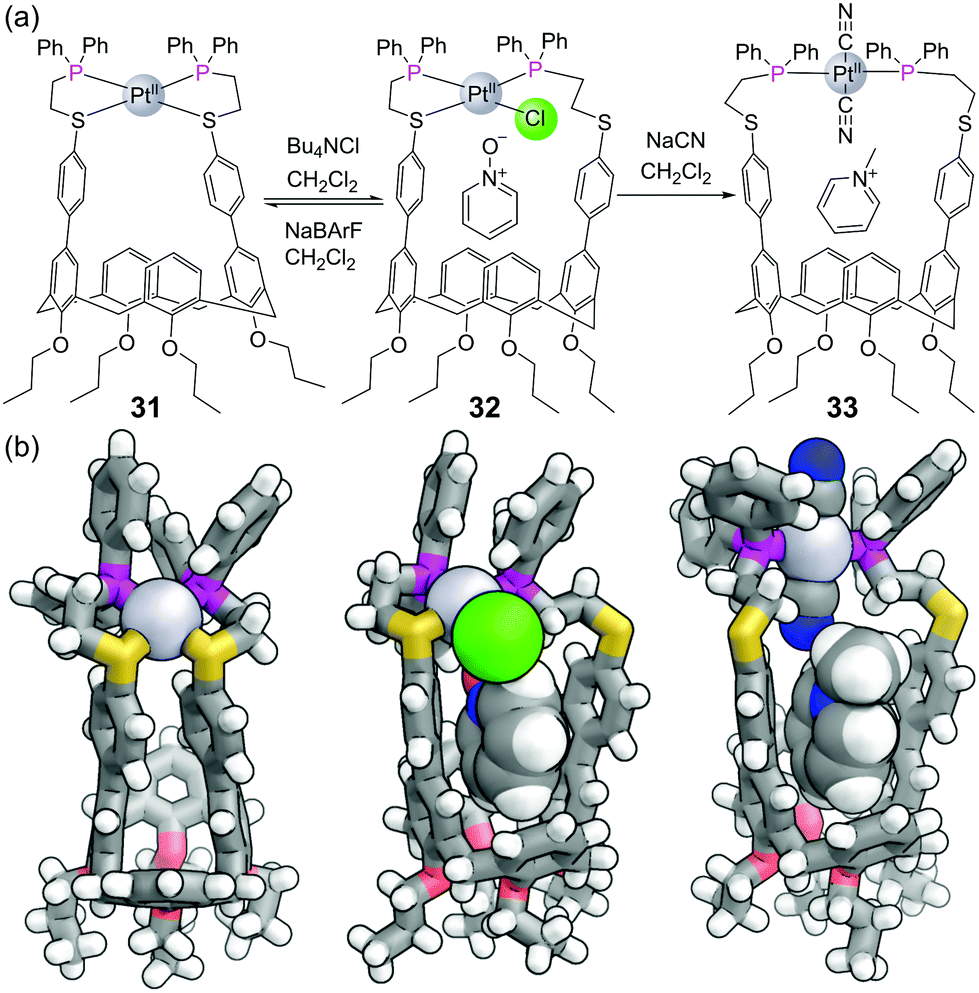 | ||
| Fig. 19 (a) Mirkin and co-workers’ ‘weak-link’ approach to achieve structural transformations between closed 31, half open 32 and fully open 33 phosphine-containing complexes; (b) SCXRD of the three complexes highlighting the binding of chloride to the metal and pyridine N-oxide within the cavity of the half open complex 32 and the binding of cyanide to the metal and methyl pyridinium in the cavity of the fully open complex 33. Colours: C grey, H white, Cl bright green, N blue, O red, P magenta, S yellow, Pt light grey (shown as spheres). Chloride and cyanide ligands, and bound guests also shown as spheres. | ||
A larger asymmetric capsule was later constructed from one calix[4]arene and one resorcin[4]arene held together by two Pt(II) metal centres.77 The binding of the pharmaceuticals dextromethorphan and β-estradiol could likewise be allosterically regulated by the addition/removal of chloride ions in this larger cavity. The same group were able to construct a macrocyclic system consisting of both N-heterocyclic carbene thioether and phosphino thioether ligands which could be allosterically regulated between four different structural states by the addition or removal of chloride ions and/or methanol.76 Signal transduction is crucial for biological systems to respond to their changing environment and synthetic chemists have looked to develop artificial systems mimicking this behaviour.74 In this regard, a macrocycle containing phosphino thioether ligands was shown to be an allosteric supramolecular catalyst.78 This macrocycle contained two Zn(II)–salen moieties, which catalyse an acyl transfer reaction from acetic anhydride to pyridyl carbinol most efficiently when held in the ‘open’ position.
6. Further microenvironments for constraining phosphines
6.1 Phosphine-containing macrocycles
The crown ethers are possibly the most well-known ligands in supramolecular chemistry. In addition to thio (sulfur) and aza (nitrogen) based variants, phosphorus variants of these macrocycles have also been known for decades. The chemistry of these phosphacrowns and other macrocyclic phosphines has previously been reviewed in detail.79 A large number have been synthesised, with varying ring sizes, number of donor atoms and rigidity of ligands. They are attractive due to their high binding constants for metals (thermodynamic macrocyclic effect) and their slow rate of ligand dissociation (kinetic macrocyclic effect), in addition to their ability to pre-organise the P donor atoms in a well-defined geometry around a central cavity. Besides the typical difficulties of macrocycle synthesis, the ready oxidation of phosphorus makes these macrocycles more challenging to access than their other heteroatom counterparts. Many synthetic strategies thus mask the phosphines as a phosphine oxides, phosphonium salts, or Lewis acid adducts (e.g. with BH3), during the synthesis, with the free phosphines only being revealed at the end. Another important factor to consider in macrocyclic phosphines is that unlike with amines, pyramidal inversion is typically slow at room temperature, giving rise to the possibility of P-stereocentres and the macrocyclic ligands existing in cis/trans or syn/anti isomeric forms.We will focus our discussion on a couple of recent examples. For medium ring-sized diphosphines to bind to a single metal centre, they must be the cis isomer. The cis-diphosphine ligand L13 (Fig. 20a) could be stereoselectively accessed from a precursor with a P–P bond, which itself is made from the combination of a diene with P4.80 Nickelalactones such as 34 (Fig. 20a and b) are known to form from the combination of Ni(0), ethene and CO2, in the presence of suitable ligands. However, to develop a system capable of the catalytic production of acrylate from ethene and CO2 requires both a base and alkali metal Lewis acid, and only a small number of electron-rich diphosphines have shown such activity.81 Characterisation of nickelalactone 34 by SCXRD indicated some interesting features (Fig. 20b). Whilst the bite angle of 91° was similar to other nickelalactones, the P–Ni–P–Cy torsion angles averaged 174°. This means the Cy substituents are rotated by ∼60° compared with typical acyclic diphosphines such as L14. The complex also has a large proportion of buried volume (55%) for the size of its substituents. The TONs in the nickel-catalysed production of acrylate from ethene and CO2 with ligand L13 were comparable to some of the best in the literature to date (Fig. 20c), attributable to the well-defined coordination environment provided by the macrocycle.
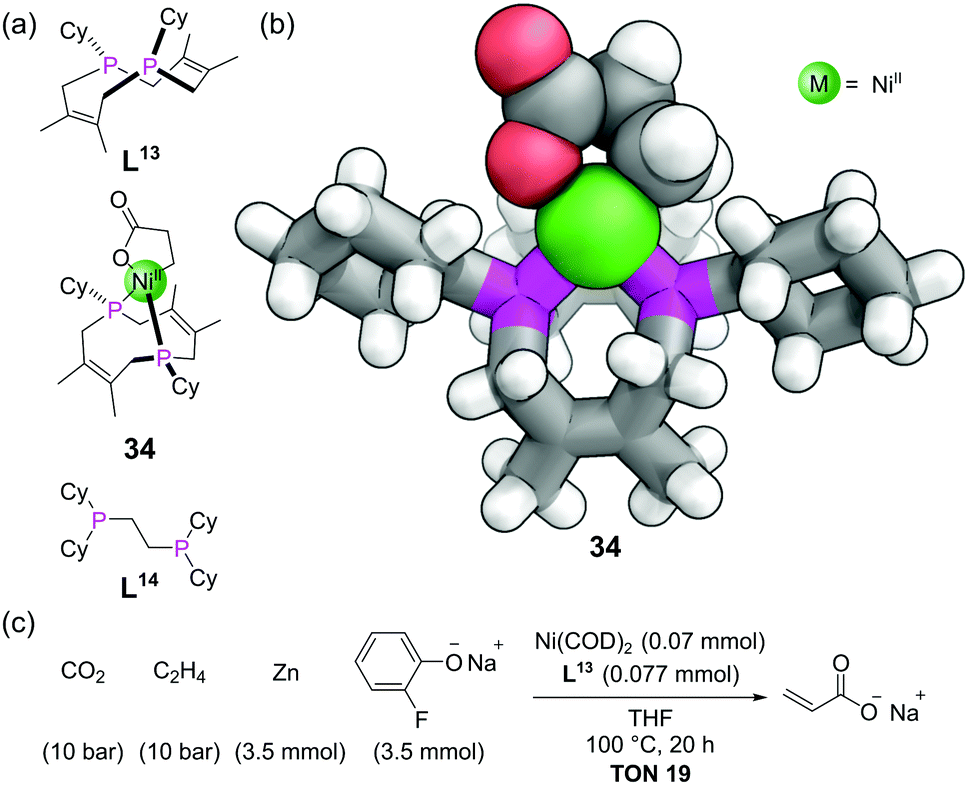 | ||
| Fig. 20 (a) Chemical structures of the macrocyclic diphosphine ligand L13, its nickelalactone complex 34, and a related acyclic diphosphine ligand L14; (b) SCXRD of nickelalactone complex 34. Colours: C grey, H white, Ni green (shown as a sphere), P magenta, O red. The nickelalactone ring is also shown using spheres; (c) the catalytic conversion of ethene and CO2 to acrylate by the Ni/L11 ligand system. | ||
Another exciting recent application has been with a 16-membered tetraphosphine macrocycle L15.82 This P4N4 ligand formed Cr(0) complexes that also bound two molecules of dinitrogen (35), which could be characterised by SCXRD (Fig. 21a). Complex 35 was able to achieve the reduction of dinitrogen in various ways (Fig. 21b). Treatment with a source of acid and electrons (from a reductant such as CoCp2) led to the stoichiometric production of ammonium (NH4+) and hydrazinium (N2H5+). This complex also enabled the first example of stoichiometric NH3 formation from the reaction of a terminally bound N2 ligand with the hydrogen atom (H˙) source TEMPOH. Alternatively, in the presence of Na and Me3SiCl, this complex mediated the catalytic silylation of dinitrogen to form N(SiMe3)3.83
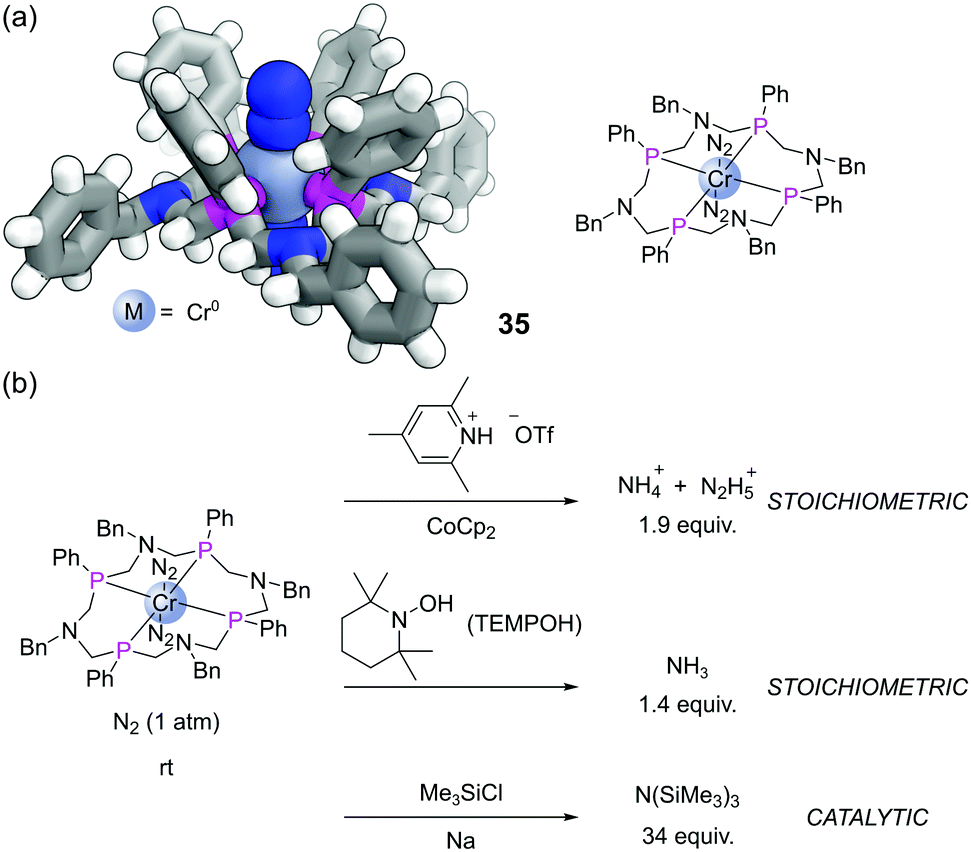 | ||
| Fig. 21 (a) Chemical structure and SCXRD of the bis-N2 chromium complex 35 of the sixteen membered P4N4 macrocycle. Colours: C grey, H white, Cr blue grey (shown as a sphere), N blue, P magenta. Bound dinitrogen also shown as spheres; (b) stoichiometric and catalytic activations of N2 promoted by complex 35. | ||
6.2 Phosphine-containing COFs as microenvironments.
Covalent Organic Frameworks (COFs) are permanently porous crystalline organic polymers with highly ordered structures.84 Like MOFs, the well-defined pores of COFs have been utilised for a variety of functions including adsorption/separation and in catalysis. Amongst this variety of applications, COFs have shown promise as supports for nanoparticles (NPs) for several reasons: (i) their highly ordered structures can be used to regularly distribute NP binding sites (e.g. bipyridyl, thioether, and phosphine) throughout their frameworks; (ii) site isolation of NP binding sites prevents aggregation of NPs resulting in enhanced stability of the heterogeneous catalyst; and (iii) well-defined pore sizes and dimensions allow for the confined growth of NPs with narrow size distributions.Zhang and co-workers reported a COF (Phos-COF-1) with triarylphosphine moieties distributed throughout its porous lattice (Fig. 22a). The COF was prepared by the condensation of 4,4′,4′′-phosphinetriyltribenzaldehyde with 1,4-phenylenediamine under solvothermal conditions.85Phos-COF-1 could be used as a scaffold to produce ultrafine (<2 nm) nanoparticles (PdNPs, PtNPs, AuNPs, and Pd/AuNPs) with narrow size distributions, since the triarylphosphine groups serve as nucleation sites, and the isolated, well-defined, pores template NP size and prevent agglomeration (Fig. 22b). These Phos-COF-1-supported nanoparticles demonstrated high catalytic activities for a range of reactions, function which is maintained after recycling. Phos-COF-1-supported PdNPs (PdNPs@Phos-COF-1) show excellent activity (90–99% yields) in the C–C bond forming Suzuki–Miyaura reaction with low catalyst loading (0.35 mol% Pd) under mild conditions (50 °C, 2 h), consistently outperforming commercially available Pd sources in terms of both yields and reaction rates. By comparison, significant agglomerations of PdNPs were observed by TEM following catalytic reactions when supported by a PPh3-based amorphous polymer.55 Significantly, COF-supported NPs benefit from enhanced stability offered by the framework and also demonstrate improved catalytic activity. For example, the cross-coupling of 4-iodotoluene with phenylboronic acid using PdNPs@Phos-COF-1 gave >99% yield with TOF of up to 1648 h−1, whilst PdNPs with no support performed considerably worse with yields of just 21% and TOF of 122 h−1 (Fig. 22c).
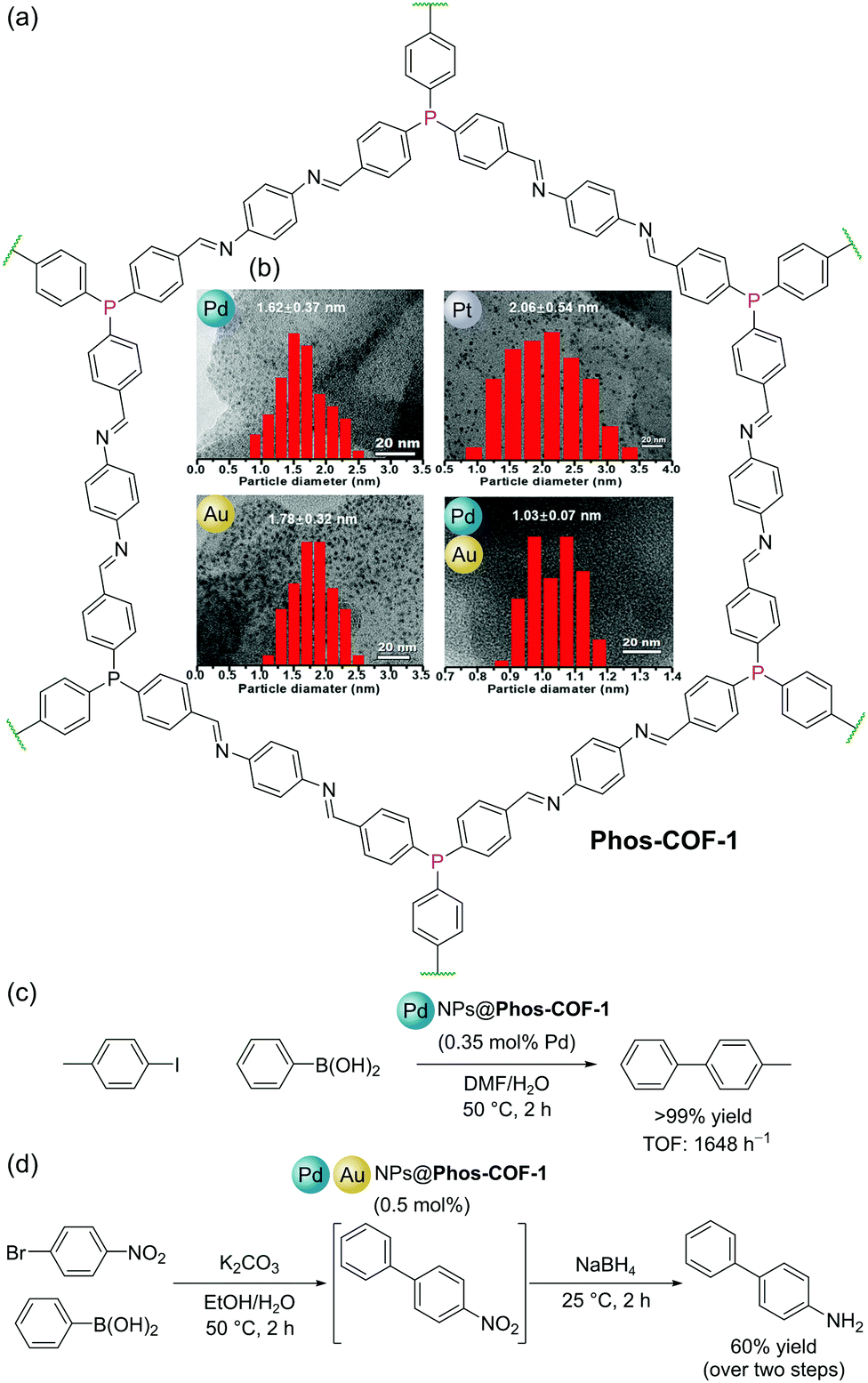 | ||
| Fig. 22 (a) Structure of Phos-COF-1; (b) (inset) TEM images and particle size distribution graphs for Pd, Pt, Au and Pd/Au nanoparticles (NPs), adapted from ref. 85; (c) Suzuki–Miyaura coupling catalysed by PdNPs@Phos-COF-1; (d) two step, one-pot Suzuki–Miyaura coupling and reduction catalysed by mixed Pd/AuNPs@Phos-COF-1. TEM images and distribution graphs in (b) reproduced from ref. 84 with permission from John Wiley & Sons, Inc., Copyright 2020. | ||
Since mixed metal NPs may be used to catalyse multiple reactions in one pot, Zhang and co-workers also extended their studies to the templated synthesis and catalytic activity of bimetallic Pd/AuNPs within Phos-COF-1, denoted PdAuNPs@Phos-COF-1. As with the examples described above, ultrafine Pd/AuNPs with an average size of 1.03 nm and a narrow size distribution (1.03 ± 0.07 nm) could be synthesised within the well-defined COF pores, while large clusters were formed in the absence of Phos-COF-1. The exemplary dispersion of uniform NPs throughout the material is supported by energy-dispersive X-ray spectroscopy (EDS) elemental mapping, which confirms an even distribution of C, N, O, Pd and Au over the material. Tandem Pd-catalysed cross-coupling and Au-catalysed nitro reduction could be performed using 1-bromo-4-nitrobenzene as the substrate and 0.5 mol% Pd/AuNPs@Phos-COF-1 as the catalyst to form 4-aminobiphenyl in a one-pot reaction with an isolated yield of 60% over the two steps (Fig. 22d).
Phosphine-containing COFs have also been developed to mimic behaviour widely investigated in the studies of Frustrated Lewis Pairs (FLPs).86 COFs 36 and 37, which feature both Lewis-basic phosphorus atoms and Lewis acidic boron centres, were produced via transesterification of a triboronated triphenylphosphine derivative with the esters 2,3,6,7,10,11-hexahydroxytriphenylene (HHTP) or 2,3,6,7-tetrahydroxy-9,10-dimethylanthracene (THDMA) respectively (Fig. 23).87 Although variable orientation of the phosphorus centres leads to significant disorder, and an absence of defined long-range order, across the material, the SPXRD or PXRD patterns of these materials do reveal some degree of shorter-range structural ordering. Comparison of PXRD patterns with calculated data suggests that the short-range order observed for 36 is consistent with the formation of a two-fold interpenetrated network. Gas uptake studies for both 36 and 37 reveal higher relative affinity for H2, CO2, and CH4 than exhibited by other reported boron-containing COFs. DFT calculations suggested that this improved gas adsorption performance could be attributed to the presence of adjacent boron and phosphorus centres in the framework structure. It was hypothesised that the proximity of boron and phosphorus generates local electric field gradients which facilitates the binding of polarisable molecules, reminiscent of the behaviour of FLPs. Here the effect is dominated by the interaction of the phosphorus donor and only mildly supported by the weakly acidic Lewis acidic boron atom, and thus is insufficient to promote bond cleavage commonly observed for FLPs.
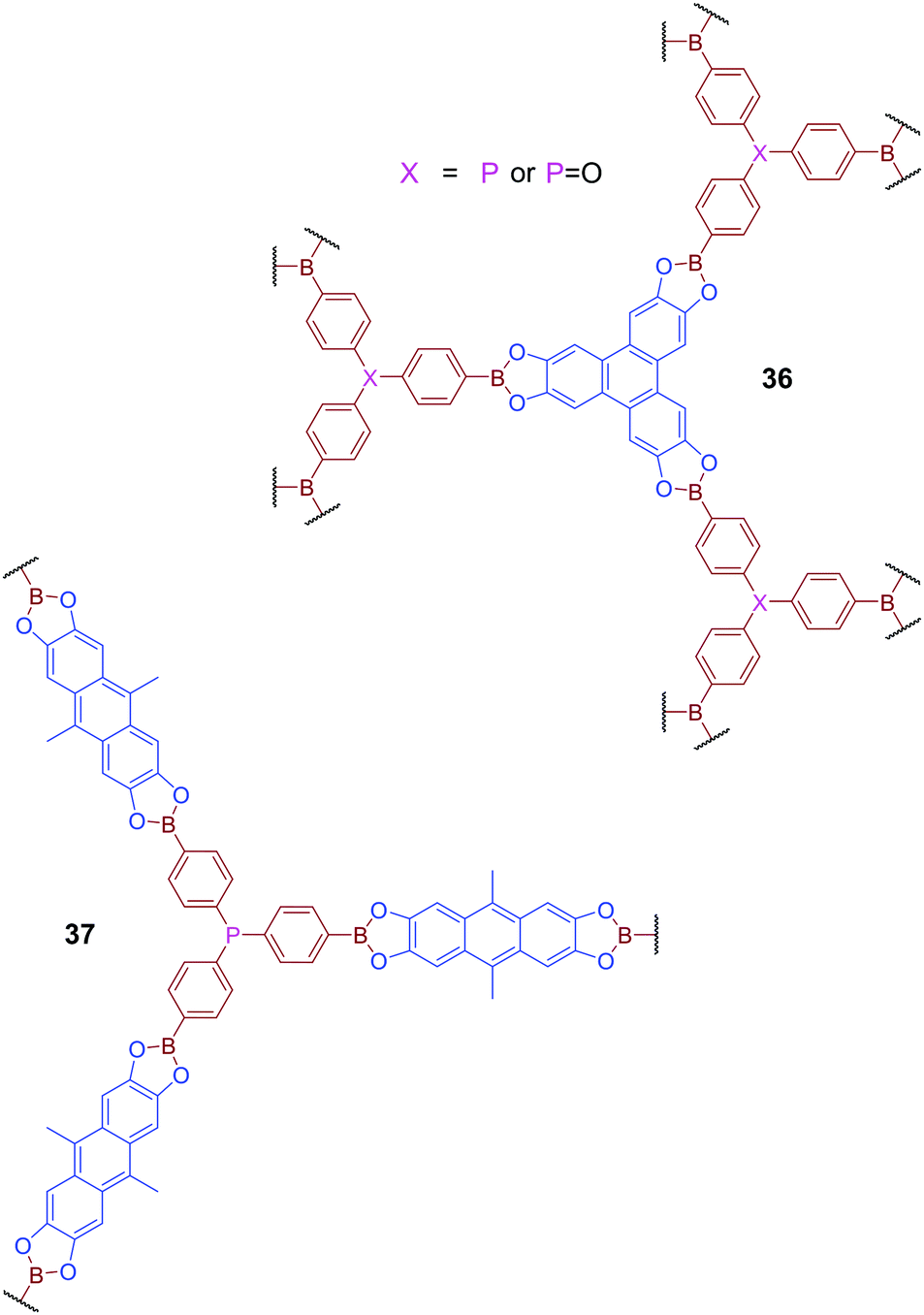 | ||
| Fig. 23 COFs 36 and 37 contain both Lewis basic phosphorus sites and Lewis acidic boron sites and have been designed to mimic the behaviour of Frustrated Lewis Pairs (FLPs). | ||
6.3 Mechanically interlocked molecules
Mechanically interlocked molecules such as rotaxanes and catenanes provide new opportunities for controlling the three-dimensional environment around specific atoms. There are relatively few examples where phosphines have been incorporated within catenanes.88 Hagiwara and co-workers synthesised a [2]catenane whereby each ring possessed a diphenylphosphine group.89 This catenane was shown to be active as a ligand for a Suzuki–Miyaura cross-coupling, although control experiments were not performed that enabled the comparison of this system to non-constrained analogues. Nevertheless, this is a promising area for further study given the potential of the mechanical bond of the catenane to control the bite angle of the bidentate ligand.Recent synthetic advances such as the ‘active template’ approach to rotaxane synthesis mean these systems can be considered for applications such as catalysis. Goldup and co-workers have developed a [2]rotaxane where the axle component is terminated by a diphenyl phosphine unit that both acts as the capping group and as a ligand for metals in catalytic reactions.90 Rotaxanes can exhibit interesting mechanical stereochemical properties in the absence of conventional stereochemical elements such as the point chirality of stereocentres.91 [2]Rotaxanes can exhibit mechanical chirality when subunits with appropriate symmetry properties are combined. In rotaxanes 39, 40 and 41 the macrocycle has a single plane of symmetry (Cs) perpendicular to the axle and the axle has a single plane of symmetry (Cs) along the plane of the axle (i.e. the two ends are different). In precursor rotaxane 38, which is constructed via a copper mediated azide–alkyne cycloaddition,92 the α-stereocentre on the azide results in two diastereomeric rotaxanes being formed (having both stereocentre and mechanical chirality) which could be separated (Fig. 24a). Alkylation of each separated diastereomer (to give 39) removes the stereocentre, converting the two separated diastereoisomers to two enantiomers differing just in their mechanical chirality. Subsequent reduction of the phosphine oxide gave 40, which could then be combined with a source of gold(I) to make the enantioenriched catalyst 41. The proximity of the macrocyclic ring to the phosphorus atom considerably restricts the space around the phosphine and resultant metal–phosphine complexes (Fig. 24b). Au(I)-Catalysed reactions are hard to render enantioselective due to the linear coordination of the gold. The considerable steric bulk of this mechanical stereogenic element was able to promote enantioselective cyclopropanations of propargylic esters (Fig. 24c) in comparable enantioselectivity to traditional covalent catalysts. This catalytic system also displays interesting stimuli-responsive regulation, in that a copper(I) cofactor is needed to switch the catalyst on. In its absence, the bipyridine unit in the macrocycle coordinates to the gold atom and turns off the catalysis. This raises interesting possibilities of incorporating these catalysts within more complex systems.
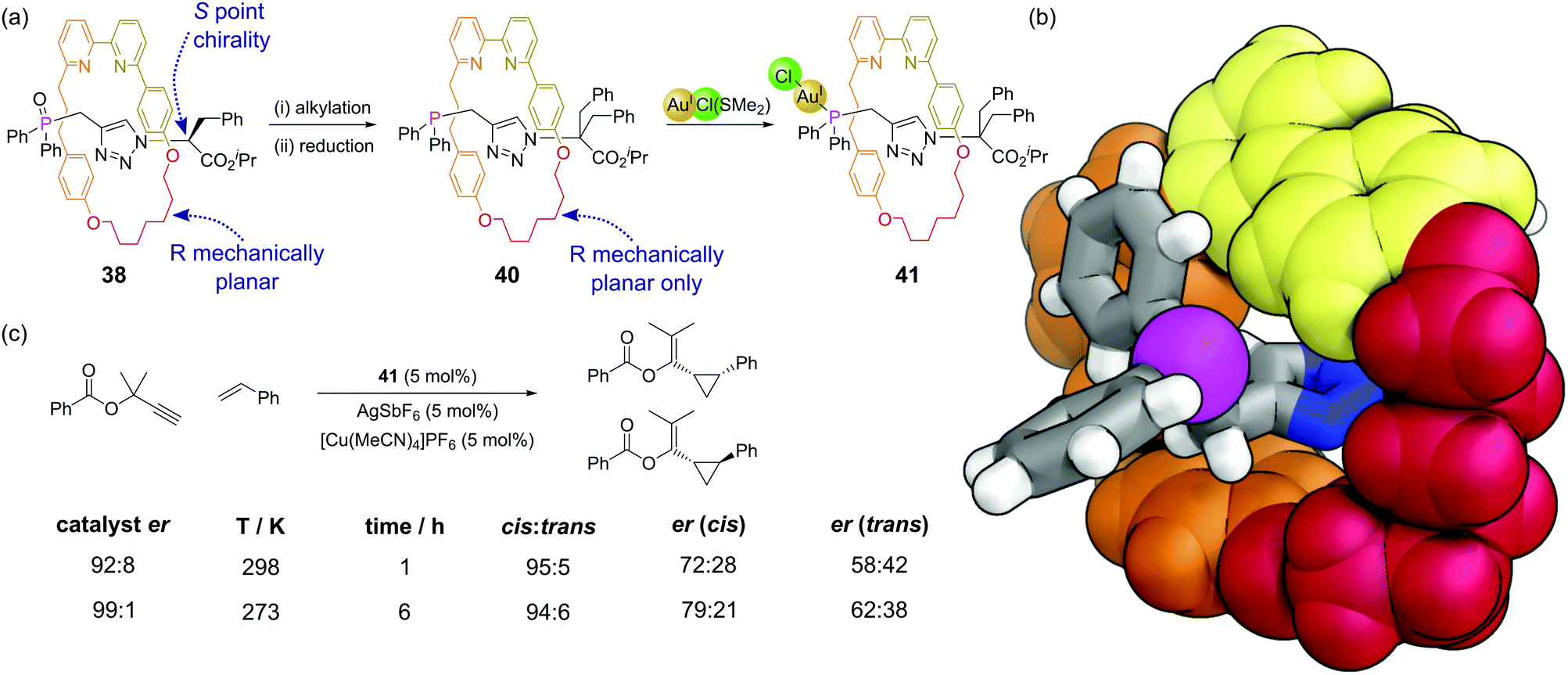 | ||
| Fig. 24 (a) Synthetic route for a mechanically planar chiral rotaxane catalyst 41, whereby the point chirality of a stereocentre in 38 enables subsequent separation based on the mechanical stereochemistry of the rotaxane before the stereocentre is removed; (b) visualisation of the environment around the phosphine in 41 based on a SCXRD of the phosphine oxide precursor with the oxygen atom removed. Colours: C grey, H white, N blue, P magenta (shown as a sphere). Macrocycle shown in yellow, orange, and red spheres to highlight mechanical chirality and constrained nature of the environment; (c) enantioselective gold-catalysed cyclopropanation of benzoate esters mediated by enantiomerically-enriched rotaxane 41 with the mechanical bond as the only stereogenic element. | ||
The creation of functional interlocked molecules is crucial to further the development of artificial molecular machines.93 Nature uses its own molecular machine, the ribosome, for the sequence selective synthesis of proteins from amino acids and this has inspired artificial machine mimics that move along a track, collecting monomeric building blocks, and then joining them in sequence.94 However, the functional groups that can be linked by such biological machines are limited. Synthesising carbon–carbon bonds is crucial to construct organic molecules and the Wittig reaction is one of the most well-known methods for this. Recently, a rotaxane-based molecular machine 42 containing four phosphonium salts was shown to encode the sequence selective synthesis of tetraene 44.95 The rotaxane thread contains four phosphonium salts separated by rigid spacer units, which are sufficiently large to stopper movement of the macrocyclic ring (Fig. 25a). Crucially, these stopper units also contain a pendant aldehyde themselves to enable propagation of the reaction. Upon treatment with base, four phosphonium ylides are formed, but the only Wittig reaction initially possible due to sterics is between the free aldehyde on the macrocycle and the ylide of the first stopper. This first Wittig reaction not only appends building block one, but also removes the first stopper, allowing the macrocycle to slide to the second stopper. At this point, the newly appended aldehyde (from the other end of the first stopper) is at the end of the growing chain and can reach the second phosphorus ylide and so the machine continues. This progresses until the fourth stopper is removed and the macrocycle tetraene product 44 can de-thread from the spent machine 43 (Fig. 25b). Phosphorus plays both a key functional and structural role here, as it is a core structural atom of the axle and the point at which the stoppers are attached, but also mediates the reaction which drives the machine. This work advances the state-of-the-art in molecular synthesisers, but as the authors point out, this machine only be used once, and a challenge for the field is that of constructing reusable machines.
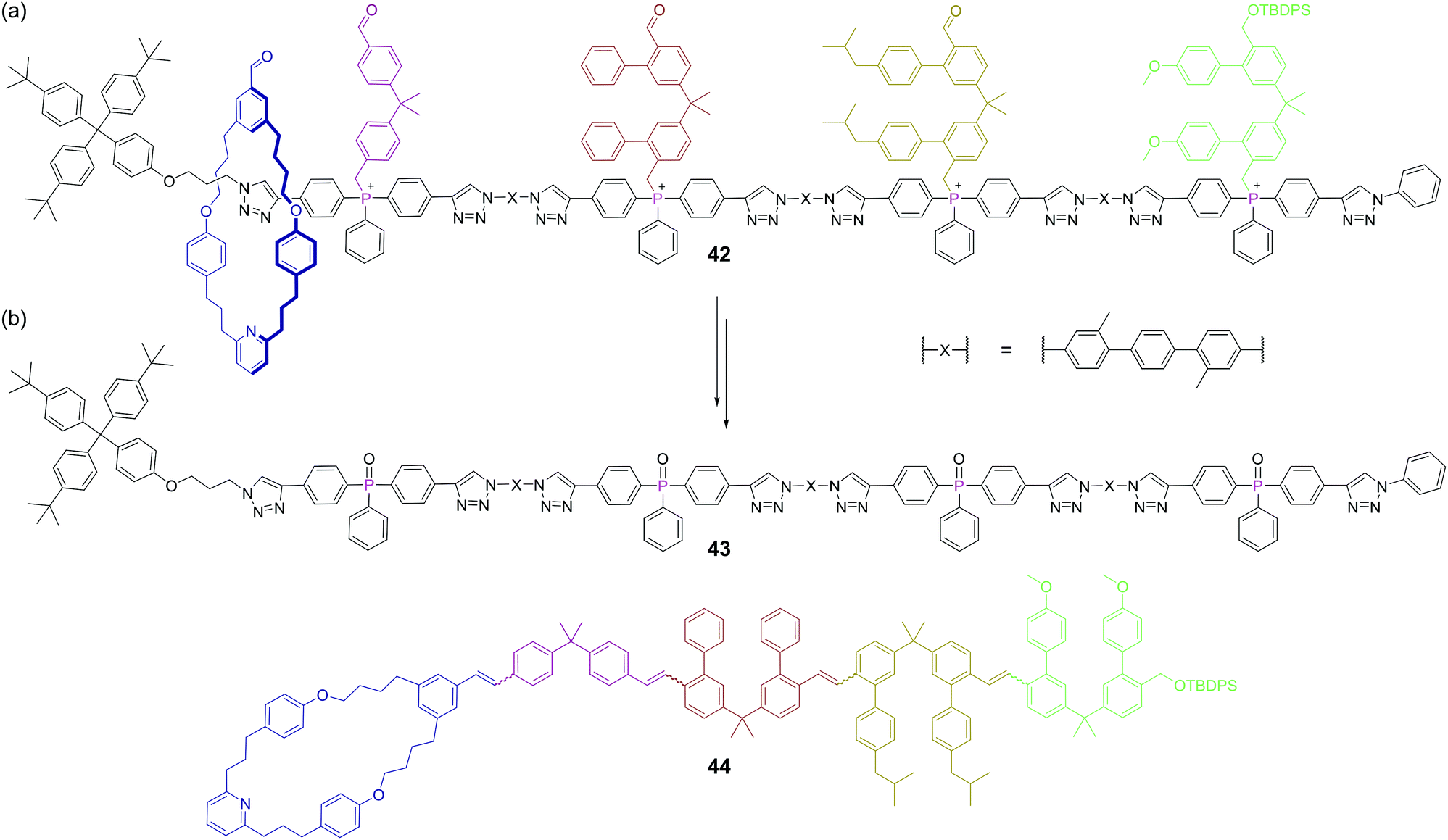 | ||
| Fig. 25 A molecular machine capable of performing four sequence selective Wittig reactions: (a) the precursor tetra phosphonium salt rotaxane 42, with residues separated by rigid spacer groups; (b) upon formation of the phosphonium ylides, each Wittig reaction attaches a new residue to the growing chain, whilst simultaneously removing a stopper, allowing the macrocycle to slide one further position along the rotaxane until it can de-thread after completion of the fourth Wittig reaction. | ||
7. A note on phosphines for further assembly
As noted above a major contributing factor to the widespread use of phosphines and their metal complexes is largely due to the versatility offered by their steric and electronic tunability. This review covers a diverse range of structures with functionality provided by a variety of phosphine ligands. Nevertheless, it may be noted that many systems featured here are based on triarylphosphines. This limited ligand diversity in constrained microenvironments is particularly notable in the synthesis of MOFs and MOCs, the synthesis of which introduces the additional hurdle of requiring multidentate phosphine ligands/linkers capable of further assembly. The attraction to structures incorporating triarylphosphines as initial targets is clear; they offer enhanced air stability and have more straightforward syntheses than phosphines with alkyl substituents. Their rigidity also aids crystallographic studies and ensures MOFs and MOCs have well-defined, uniform structures, with enhanced probability of permanent porosity.Where phosphines undergo further assembly, the ligands often require at least two types of functional group with significantly different reactivities to permit a greater degree of control over the resulting assembly process. For example, the reaction of ZrCl4 with L6 (see Section 4.2) produces a structure with coordinated carboxylate groups and non-coordinated phosphines due to incompatibility between the hard Zr(IV) centres and soft phosphine donors.44,45
We will briefly outline some of the key strategies and considerations when designing and synthesising phosphine ligands/linkers for further assembly. Avoiding oxidation of the phosphorus centre is perhaps the most obvious challenge in the synthesis of such ligands, and manipulations are almost exclusively carried out under inert conditions. Alternatively, the facile oxidation of the phosphorus centre may be exploited (Fig. 26a), whereby the phosphine is masked as a phosphine oxide during installation of other functional groups and only converted to the phosphine in a final step by reduction, for example using SiHCl3. This may be an attractive option due to reduction of air sensitivity, but two extra steps are added, and as SiHCl3 is pyrophoric, corrosive, and toxic, this route is not always a synthetically straightforward alternative.
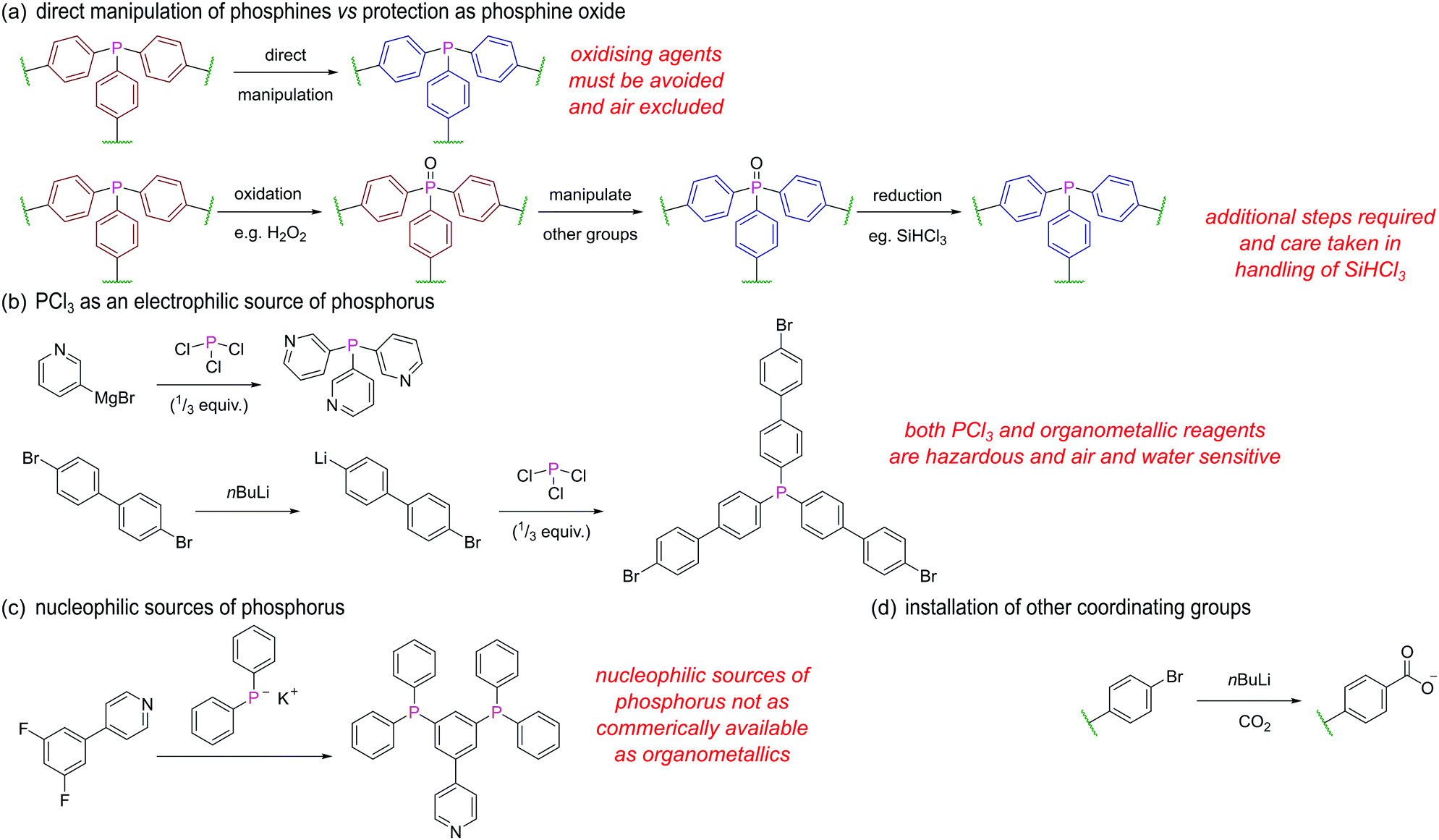 | ||
| Fig. 26 Synthetic strategies towards phosphine ligands suitable for further assembly (a) protection of the phosphine as a phosphine oxide; (b) use of PCl3 as an electrophilic source of phosphorus; (c) use of KPPh2 as a nucleophilic source of phosphorus; (d) a common installation route of other coordinating groups on phosphine ligands. | ||
PCl3 is a common electrophilic source of phosphorus as the chlorines can be displaced with suitable nucleophiles. For example, 3-pyridyl magnesium bromide reacts with PCl3 to yield the tetradentate ligand L12 (Fig. 26b).96 Similarly, treatment of 1,4-dibromobenzene or 4,4′-dibromo-1,1′-biphenyl with one equivalent of nBuLi, followed by a third of an equivalent of PCl3 gives tris(4-bromophenyl)phosphine and tris(4′-bromo[1,1′-biphenyl]-4-yl)phosphine43 respectively. The remaining bromines can then be converted into carboxylate groups for further assembly via reaction with nBuLi and CO2 (Fig. 26d).
Alternative pathways employ lithium or potassium diphenylphosphide which are both commercially available nucleophilic sources of phosphorus. Such reagents undergo substitution reactions with halogenated reactants possessing additional binding groups, such as in the conversion of 4-(3,5-difluorophenyl)-pyridine to 4-(3,5-bis(diphenylphosphino)-phenyl)pyridine (Fig. 26c).
The full potential of phosphines within confined spaces is a long way from being reached, with scope for incorporation of phosphines with a far greater degree of steric and electronic variation into the building blocks for supramolecular structures.
8. Conclusions
If one considers the widespread interest in phosphines and their metal complexes, then it is perhaps unsurprising that research continues to find new ways to improve their performance. Furthermore, considering the extensive history of phosphine chemistry it is apparent that there are many approaches to boost such performance, notably including steric control of phosphine centres. In this context the development of phosphine envelopment in constrained environments is a logical step to further exploit this highly useful class of molecules. Although enclosing phosphine in microenvironments may be an obvious development, the diversity of approaches is less predictable.It is noticeable that the methodology described in this review touches on many areas of modern supramolecular chemistry. Strategies that have a well-developed understanding of employing constrained environments such as macrocyclic systems (e.g. cyclodextrins and calixarenes) sit alongside larger porous species such as MOCs, MOFs and COFs. It is apparent why these materials have now been employed in attempts to constrain the environment of phosphine species. However, it is less obvious that these developments would have also been extended to other areas of supramolecular chemistry, such as mechanically interlocked structures and molecular machines.
It is apparent from the examples discussed above that the confinement of phosphines in microenvironments can lead to exciting new developments and enhanced performance of systems, such as catalytic processes. So where do the challenges in the field currently lie? One of the most useful features of phosphines, which has made them such a success story, is the ability to tune the electronic properties of the phosphorus donor. Typically, this is achieved by varying the aryl/alkyl substituents which enable tuning of the donor properties of the phosphine ligand. It is noticeable from the examples surveyed above that many of the phosphines studied, notably those in MOFs, COFs and MOCs, are based on triarylphosphines. Despite the synthetic considerations discussed in Section 7, it is understandable that triarylphosphines would be the preliminary targets at the start of this field due to the greater air-stability and easier synthesis of such compounds in comparison to trialkylphosphines. However, trialkylphosphines offer distinct donor properties, usually being more powerful σ-donors and weaker π-acceptors, and this distinction from triarylphosphines has driven the tuning of catalytic properties of metal–phosphine complexes, in combination with steric approaches. Although trialkylphosphines are commonly more synthetically challenging, and thus their incorporation into complex materials more daunting, clearly such chemistry would be an exciting addition to the field of confined phosphines. Indeed, once confined within a microenvironment it seems likely that trialkylphosphines may exhibit enhanced stability.
Steric properties of phosphine ligands are commonly discussed in terms of cone angles and bite angles. Whilst useful properties for comparing phosphines in purely covalent systems, they may not fully describe the subtle effects imparted by the constrained microenvironments mentioned here. This is true both of the ‘outer coordination sphere’ static effects that might be influenced by the larger structure of the MOF or MOC, or more dynamic effects such as the movement of a macrocyclic ring along the phosphine axle in a rotaxane. These dynamic effects could underpin new classes of phosphine ligands with switchable properties and stimuli-responsive behaviour.
Indeed, until the full breadth of phosphine chemistry, with all its functionality, is brought to the confined environments of MOCs, MOFs and related systems, the full potential of the field will not have been realised. Whilst many exciting developments have been made in the field, many more are yet to come.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
BSP thanks the Green Chemicals Beacon of Excellence, University of Nottingham, UK for financial support. GRFO and NRC thank the Engineering and Physical Sciences Research Council (EP/S002995/1) for financial support.References
- J. F. Young, J. A. Osborn, F. H. Jardine and G. Wilkinson, Chem. Commun., 1965, 131–132 RSC
.
- R. H. Crabtree, Acc. Chem. Res., 1979, 12, 331–337 CrossRef CAS
- T. M. Trnka and R. H. Grubbs, Acc. Chem. Res., 2001, 34, 18–29 CrossRef CAS
- L. Vaska and J. W. DiLuzio, J. Am. Chem. Soc., 1961, 83, 2784–2785 CrossRef CAS
- D. S. Surry and S. L. Buchwald, Chem. Sci., 2011, 2, 27–50 RSC
- J. F. Hartwig, Angew. Chem., Int. Ed., 1998, 37, 2046–2067 CrossRef CAS
- T. Barber, S. P. Argent and L. T. Ball, ACS Catal., 2020, 10, 5454–5461 CrossRef CAS
- A. Agarkov, S. Greenfield, D. Xie, R. Pawlick, G. Starkey and S. R. Gilbertson, Biopolymers, 2006, 84, 48–73 CrossRef CAS
- IUPAC. Compendium of Chemical Terminology, 2nd edn, the “Gold Book”, Compiled by A. D. McNaught and A. Wilkinson. Blackwell Scientific Publications, Oxford, 1997. Online version (2019-) created by S. J. Chalk. ISBN 0-9678550-9-8. https://doi.org/10.1351/goldbook.
- S. L. Griffin and N. R. Champness, Coord. Chem. Rev., 2020, 414, 213295 CrossRef CAS
- Q. Qian, P. A. Asinger, M. J. Lee, G. Han, K. M. Rodriguez, S. Lin, F. M. Benedetti, A. X. Wu, W. S. Chi and Z. P. Smith, Chem. Rev., 2020, 120, 8161–8266 CrossRef CAS
- R. J. Young, M. T. Huxley, E. Pardo, N. R. Champness, C. J. Sumby and C. J. Doonan, Chem. Sci., 2020, 11, 4031–4050 RSC
- K. E. Cordova and O. M. Yaghi, Mater. Chem. Front., 2017, 1, 1304–1309 RSC
- M. Bosch, M. Zhang and H.-C. Zhou, Adv. Chem., 2014, 182327 Search PubMed
- X. Xu, M. Nieuwenhuyzen and S. L. James, Angew. Chem., Int. Ed., 2002, 41, 764–767 CrossRef CAS
- X. Tan, L. Li, J. Zhang, X. Han, L. Jiang, F. Li and C.-Y. Su, Chem. Mater., 2012, 24, 480–485 CrossRef CAS
- J. Zhang, X. Xu and S. L. James, Chem. Commun., 2006, 4218–4220 RSC
- H. Ni, W.-L. Chan and Y. Lu, Chem. Rev., 2018, 118, 9344–9411 CrossRef CAS
- J. Feng, L. Yao, J. Zhang, Y. Mu, Z. Chi and C. Y. Su, Dalton Trans., 2016, 45, 1668–1673 RSC
- J. Zhang, M. Nieuwenhuyzen, J. P. H. Charmant and S. L. James, Chem. Commun., 2004, 2808–2809 RSC
- R. Lin, J. H. K. Yip, K. Zhang, L. L. Koh, K.-Y. Wong and K. P. Ho, J. Am. Chem. Soc., 2004, 126, 15852–15869 CrossRef CAS
- S. L. James, Chem. Soc. Rev., 2009, 38, 1744–1758 RSC
- S. H. Lim, Y. Su and S. M. Cohen, Angew. Chem., Int. Ed., 2012, 51, 5106–5109 RSC
- J. R. Shakirova, E. V. Grachova, A. J. Karttunen, V. V. Gurzhiy, S. P. Tunik and I. O. Koshevoy, Dalton Trans., 2014, 43, 6236–6243 CrossRef CAS
- X. Wang, J. Huang, S. Xiang, Y. Liu, J. Zhang, A. Eichhöfer, D. Fenske, S. Baic and C.-Y. Su, Chem. Commun., 2011, 47, 3849–3851 CrossRef CAS
- D. T. Walters, R. B. Aghakhanpour, X. B. Powers, K. B. Ghiassi, M. M. Olmstead and A. L. Balch, J. Am. Chem. Soc., 2018, 140, 7533–7542 CrossRef CAS
- B. S. Pilgrim and N. R. Champness, ChemPlusChem, 2020, 85, 1842–1856 CrossRef CAS
- S. Shanmugaraju, V. Vajpayee, S. Lee, K. W. Chi, P. J. Stang and P. S. Mukherjee, Inorg. Chem., 2012, 51, 4817–4823 CrossRef CAS
- S. Bivaud, S. Goeb, V. Croué, P. I. Dron, M. Allain and M. Sallé, J. Am. Chem. Soc., 2013, 135, 10018–10021 CrossRef CAS
- A. Grigoropoulos, A. I. McKay, A. P. Katsoulidis, R. P. Davies, A. Haynes, L. Brammer, J. Xiao, A. S. Weller and M. J. Rosseinsky, Angew. Chem., Int. Ed., 2018, 57, 4532–4537 CrossRef CAS
- K. Ikemoto, Y. Inokuma, K. Rissanen and M. Fujita, J. Am. Chem. Soc., 2014, 136, 6892–6895 RSC
- M. A. Nasalevich, R. Becker, E. V. Ramos-Fernandez, S. Castellanos, S. L. Veber, M. V. Fedin, F. Kapteijn, J. N. H. Reek, J. I. Van Der Vlugt and J. Gascon, Energy Environ. Sci., 2015, 8, 365–375 CrossRef CAS
- J. Ren, P. C. Lan, M. Chen, W. Zhang and S. Ma, Organometallics, 2019, 38, 3460–3465 CrossRef CAS
- S. Tarnowicz-Ligus, A. Augustyniak and A. M. Trzeciak, Eur. J. Inorg. Chem., 2019, 4282–4288 CrossRef CAS
- D. T. Genna, A. G. Wong-Foy, A. J. Matzger and M. S. Sanford, J. Am. Chem. Soc., 2013, 135, 10586–10589 RSC
- K. Harris, D. Fujita and M. Fujita, Chem. Commun., 2013, 49, 6703–6712 CrossRef CAS
- Q. Q. Wang, S. Gonell, S. H. A. M. Leenders, M. Dürr, I. Ivanovic-Burmazovic and J. N. H. Reek, Nat. Chem., 2016, 8, 225–230 CrossRef CAS
- T. A. Bender, R. G. Bergman, K. N. Raymond and F. D. Toste, J. Am. Chem. Soc., 2019, 141, 11806–11810 CrossRef CAS
- F. Hapiot, A. Ponchel, S. Tilloy and E. Monflier, C. R. Chim., 2011, 14, 149–166 CrossRef CAS
- F. Hapiot, H. Bricout, S. Tilloy and E. Monflier, Eur. J. Inorg. Chem., 2012, 1571–1578 CrossRef CAS
- V. Pascanu, G. González Miera, A. K. Inge and B. Martín-Matute, J. Am. Chem. Soc., 2019, 141, 7223–7234 CrossRef CAS
- A. A. Bezrukov, K. W. Törnroos and P. D. C. Dietzel, Cryst. Growth Des., 2017, 17, 3257–3266 CrossRef CAS
- A. A. Bezrukov and P. D. C. Dietzel, Inorg. Chem., 2017, 56, 12830–12838 CrossRef CAS
- T. Sawano, Z. Lin, D. Boures, B. An, C. Wang and W. Lin, J. Am. Chem. Soc., 2016, 138, 9783–9786 CrossRef CAS
- X. Xu, S. M. Rummelt, F. L. Morel, M. Ranocchiari and J. A. van Bokhoven, Chem. – Eur. J., 2014, 20, 15467–15472 CrossRef CAS
- S. G. Dunning, G. Nandra, A. D. Conn, W. Chai, R. E. Sikma, J. S. Lee, P. Kunal, J. E. Reynolds, J.-S. Chang, A. Steiner, G. Henkelman and S. M. Humphrey, Angew. Chem., Int. Ed., 2018, 57, 9295–9299 CrossRef CAS
- S. G. Dunning, J. E. Reynolds, K. M. Walsh, D. J. Kristek, V. M. Lynch, P. Kunal and S. M. Humphrey, Organometallics, 2019, 38, 3406–3411 CrossRef CAS
- S. A. Burgess, A. Kassie, S. A. Baranowski, K. J. Fritzsching, K. Schmidt-Rohr, C. M. Brown and C. R. Wade, J. Am. Chem. Soc., 2016, 138, 1780–1783 CrossRef CAS
- A. A. Kassie, P. Duan, E. T. McClure, K. Schmidt-Rohr, P. M. Woodward and C. R. Wade, Inorg. Chem., 2019, 58, 3227–3236 CrossRef CAS
- J. M. Falkowski, T. Sawano, T. Zhang, G. Tsun, Y. Chen, J. V. Lockard and W. Lin, J. Am. Chem. Soc., 2014, 136, 5213–5216 CrossRef
- C. De Wu, A. Hu, L. Zhang and W. Lin, J. Am. Chem. Soc., 2005, 127, 8940–8941 CrossRef CAS
- R. Noyori and H. Takaya, Acc. Chem. Res., 1990, 23, 345–350 CrossRef CAS
- R. R. R. Prasad, D. M. Dawson, P. A. Cox, S. E. Ashbrook, P. A. Wright and M. L. Clarke, Chem. – Eur. J., 2018, 24, 15309–15318 CrossRef CAS
- R. Gramage-Doria, J. Hessels, S. H. A. M. Leenders, O. Tröppner, M. Dürr, I. Ivanović-Burmazović and J. N. H. Reek, Angew. Chem., Int. Ed., 2014, 53, 13380–13384 CrossRef CAS
- S. H. A. M. Leenders, M. Dürr, I. Ivanovic-Burmazovic and J. N. H. Reek, Adv. Synth. Catal., 2016, 358, 1509–1518 RSC
- M. Jouffroy, D. Armspach and D. Matt, Dalton Trans., 2015, 44, 12942–12969 CrossRef CAS
- M. Jouffroy, R. Gramage-Doria, D. Armspach, D. Sémeril, W. Oberhauser, D. Matt and L. Toupet, Angew. Chem., Int. Ed., 2014, 53, 3937–3940 CrossRef CAS
- S. D. Ittel, L. K. Johnson and M. Brookhart, Chem. Rev., 2000, 100, 1169–1204 CrossRef CAS
- V. C. Gibson and S. K. Spitzmesser, Chem. Rev., 2003, 103, 283–316 CrossRef CAS
- M. Jouffroy, D. Armspach, D. Matt, K. Osakada and D. Takeuchi, Angew. Chem., Int. Ed., 2016, 55, 8367–8370 CrossRef CAS
- A. Nakamura, T. M. J. Anselment, J. Claverie, B. Goodall, R. F. Jordan, S. Mecking, B. Rieger, A. Sen, P. W. N. M. Van Leeuwen and K. Nozaki, Acc. Chem. Res., 2013, 46, 1438–1449 CrossRef CAS
- Y. Takashima, K. Uramatsu, D. Jomori, A. Harima, M. Otsubo, H. Yamaguchi and A. Harada, ACS Macro Lett., 2013, 2, 384–387 CrossRef CAS
- L. Poorters, D. Armspach, D. Matt, L. Toupet, S. Choua and P. Turek, Chem. – Eur. J., 2007, 13, 9448–9461 CrossRef CAS
- R. Gramage-Doria, D. Armspach and D. Matt, Coord. Chem. Rev., 2013, 257, 776–816 CrossRef CAS
- C. Wieser-Jeunesse, D. Matt and A. De Cian, Angew. Chem., Int. Ed., 1998, 37, 2861–2864 RSC
- C. Wieser, D. Matt, L. Toupet, H. C. Bourgeois and J.-P. Kintzinger, J. Chem. Soc., Dalton Trans., 1996, 4041–4043 CAS
- T. Besset, D. W. Norman and J. N. H. Reek, Adv. Synth. Catal., 2013, 355, 348–352 CrossRef CAS
- V. F. Slagt, J. N. H. Reek, P. C. J. Kamer and P. W. N. M. van Leeuwen, Angew. Chem., Int. Ed., 2001, 40, 4271–4274 CrossRef
- V. Bocokic, A. Kalkan, M. Lutz, A. L. Spek, D. T. Gryko and J. N. H. Reek, Nat. Commun., 2013, 4, 2670 CrossRef CAS
- M. Kuil, T. Soltner, P. W. N. M. van Leeuwen and J. N. H. Reek, J. Am. Chem. Soc., 2006, 128, 11344–11345 CrossRef CAS
- X. Wang, S. S. Nurttila, W. I. Dzik, R. Becker, J. Rodgers and J. N. H. Reek, Chem. – Eur. J., 2017, 23, 14769–14777 CrossRef CAS
- L. J. Jongkind and J. N. H. Reek, Chem. – Asian J., 2020, 15, 867–875 RSC
- D. A. Roberts, B. S. Pilgrim and J. R. Nitschke, Chem. Soc. Rev., 2018, 47, 626–644 CrossRef CAS
- B. S. Pilgrim, D. A. Roberts, T. G. Lohr, T. K. Ronson and J. R. Nitschke, Nat. Chem., 2017, 9, 1276–1281 CrossRef CAS
- C. T. McTernan, T. K. Ronson and J. R. Nitschke, J. Am. Chem. Soc., 2019, 141, 6837–6842 CrossRef
- A. I. D’Aquino, H. F. Cheng, J. Barroso-Flores, Z. S. Kean, J. Mendez-Arroyo, C. M. McGuirk and C. A. Mirkin, Inorg. Chem., 2018, 57, 3568–3578 CrossRef CAS
- J. Mendez-Arroyo, A. I. D’Aquino, A. B. Chinen, Y. D. Manraj and C. A. Mirkin, J. Am. Chem. Soc., 2017, 139, 1368–1371 CrossRef CAS
- N. C. Gianneschi, S. B. T. Nguyen and C. A. Mirkin, J. Am. Chem. Soc., 2005, 127, 1644–1645 CrossRef CAS
- C. D. Swor and D. R. Tyler, Coord. Chem. Rev., 2011, 255, 2860–2881 RSC
- I. Knopf, D. Tofan, D. Beetstra, A. Al-Nezari, K. Al-Bahily and C. C. Cummins, Chem. Sci., 2017, 8, 1463–1468 CrossRef CAS
- N. Huguet, I. Jevtovikj, A. Gordillo, M. L. Lejkowski, R. Lindner, M. Bru, A. Y. Khalimon, F. Rominger, S. A. Schunk, P. Hofmann and M. Limbach, Chem. – Eur. J., 2014, 20, 16858–16862 CrossRef CAS
- M. T. Mock, S. Chen, M. O’Hagan, R. Rousseau, W. G. Dougherty, W. S. Kassel and R. M. Bullock, J. Am. Chem. Soc., 2013, 135, 11493–11496 CrossRef CAS
- A. J. Kendall, S. I. Johnson, R. M. Bullock and M. T. Mock, J. Am. Chem. Soc., 2018, 140, 2528–2536 CrossRef CAS
- K. Geng, T. He, R. Liu, S. Dalapati, K. T. Tan, Z. Li, S. Tao, Y. Gong, Q. Jiang and D. Jiang, Chem. Rev., 2020, 120, 8814–8933 CrossRef CAS
- R. Tao, X. Shen, Y. Hu, K. Kang, Y. Zheng, S. Luo, S. Yang, W. Li, S. Lu, Y. Jin, L. Qiu and W. Zhang, Small, 2020, 16, 1906005 CrossRef CAS
- A. R. Jupp and D. W. Stephan, Trends Chem., 2019, 1, 35–48 CrossRef CAS
- P. Pacholak, K. Gontarczyk, R. Kamiński, K. Durka and S. Luliński, Chem. – Eur. J., 2020, 26, 12758–12768 CrossRef CAS
- C. P. McArdle, M. J. Irwin, M. C. Jennings and R. J. Puddephatt, Angew. Chem., Int. Ed., 1999, 38, 3376–3378 CrossRef CAS
- M. Yamazaki, T. Hagiwara, M. Sekiguchi, T. Sawaguchi and S. Yano, Synth. Commun., 2008, 38, 553–558 CrossRef CAS
- M. Galli, J. E. M. Lewis and S. M. Goldup, Angew. Chem., Int. Ed., 2015, 54, 13545–13549 CrossRef CAS
- R. J. Bordoli and S. M. Goldup, J. Am. Chem. Soc., 2014, 136, 4817–4820 CAS
- A. W. Heard and S. M. Goldup, Chem, 2020, 6, 994–1006 CrossRef CAS
- J. F. Stoddart, Angew. Chem., Int. Ed., 2017, 56, 11094–11125 CrossRef CAS
- B. Lewandowski, G. De Bo, J. W. Ward, M. Papmeyer, S. Kuschel, M. J. Aldegunde, P. M. E. Gramlich, D. Heckmann, S. M. Goldup, D. M. D’Souza, A. E. Fernandes and D. A. Leigh, Science, 2013, 339, 189–193 CAS
- C. T. McTernan, G. De Bo and D. A. Leigh, Chem, 2020, 6, 2964–2973 CrossRef CAS
- A. M. Kluwer, I. Ahmad and J. N. H. Reek, Tetrahedron Lett., 2007, 48, 2999–3001 RSC
| This journal is © The Royal Society of Chemistry 2021 |