
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMicrofluidic systems for rapid antibiotic susceptibility tests (ASTs) at the single-cell level
Kaixiang
Zhang†
ab,
Shangshang
Qin†
a,
Sixuan
Wu
a,
Yan
Liang
a and
Jinghong
Li
 *b
*b
aSchool of Pharmaceutical Sciences, Key Laboratory of Targeting Therapy and Diagnosis for Critical Diseases, Zhengzhou University, Zhengzhou 450001, China
bDepartment of Chemistry, Key Laboratory of Bioorganic Phosphorus Chemistry & Chemical Biology, Tsinghua University, Beijing 100084, China. E-mail: jhli@mail.tsinghua.edu.cn
First published on 1st April 2020
Abstract
Infectious diseases caused by multidrug resistant (MDR) bacterial pathogens are impending threats to global health. Since delays in identifying drug resistance would significantly increase mortality, fast and accurate antibiotic susceptibility tests (ASTs) are critical for addressing the antibiotic resistance issue. However, the conventional methods for ASTs are always labor-intensive, imprecise, complex and slow (taking 2–3 days). To address these issues, some advanced microfluidic systems have been designed for rapid phenotypic and genotypic analysis of antibiotic resistance. This review highlights the recent development of microfluidics-based ASTs at the single-cell or single-molecule level for guiding antibiotic treatment decisions and predicting therapeutic outcomes.
Introduction
Overuse of antibiotics has led to worldwide development of antibiotic resistance, which is a huge threat to human health.1,2 Multidrug resistant (MDR) bacteria are widely found in many healthcare settings, leading to a wide range of acute infections associated with high mortality rates.3 If left unchecked, they will cause 10 million deaths and a more than $1 trillion economic impact by 2050.4 Considering the life-threatening conditions, the rapid detection and quantification of antibiotic resistance, combined with appropriate antimicrobial stewardship, are key clinical tasks for correcting the treatment of infectious disease and reducing the use of broad-spectrum drugs.5Antibiotic susceptibility tests (ASTs) are widely used clinically to determine the antibiotic resistance profiles of bacterial isolates.6,7 Current gold-standard AST assays are based on measurement of bacterial growth in the presence of an antibiotic, which may take several days.7 Specifically, after collection of patient samples, the bacteria are isolated by streaking the samples on selective culture media and incubating overnight. Then, the isolated colonies are used to perform ASTs via disk diffusion or agar and broth dilution.8 To determine if the bacteria are susceptible or resistant to an antibiotic, a key factor, the minimum inhibitory concentration (MIC), is measured to estimate the lowest concentration of the antibiotic required to prevent bacterial growth. If the determined MIC is less than or equal to a breakpoint, the bacterial isolate is considered susceptible to the antibiotic. Clinical breakpoints for different bacteria and antibiotics are updated annually by national organizations, such as the Clinical and Laboratory Standards Institute (CLSI) in the USA.9
The MIC value serves as an important parameter to analyze phenotypic resistance in bacteria, determine the effectiveness of new antibiotics and monitor the global drug resistance status. Even though it is slow, the method can directly answer the key question of whether the antibiotic would inhibit pathogen growth. However, this delay prolongs the time to make decisions for appropriate and effective antibiotic therapy, which leads to increased patient mortality and poor clinical outcomes. To address this issue, technologies that can rapidly identify antibiotic susceptibility at the earliest possible treatment stage are urgently needed (Fig. 1).
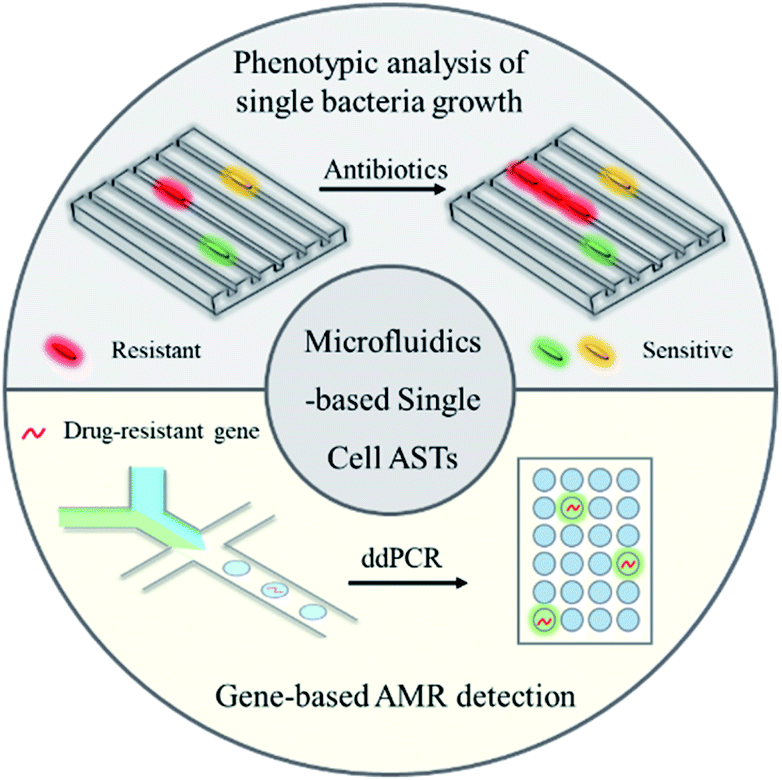 | ||
| Fig. 1 Microfluidic technologies for rapid antibiotic susceptibility tests (ASTs) at the single-cell level, including both phenotypic analysis (microfluidic-based single bacterial culture) and gene-based antimicrobial resistance (AMR) detection (droplet digital analysis). | ||
Except for phenotypic ASTs, some genotypic ASTs have also been developed for rapid detection of drug resistance genes in bacterial pathogens by molecular methods.10 There are some FDA-cleared panels that incorporate genotypic resistance detection alongside pathogen identification, such as the mecA gene for detection of methicillin-resistant Staphylococcus aureus (MRSA).11 Rapid detection of a drug resistance gene may allow improved antimicrobial therapy, especially in the case of infections by Gram-positive bacteria.12 Notably, the absence of a drug resistance gene does not necessarily predict susceptibility to a particular drug, since there are some other drug resistance mechanisms to be considered, such as porin loss and efflux pumps.13 Therefore, rather than replacing phenotypic susceptibility testing, genotypic testing acts as a supplementary tool which could help to exclude the use of some specific types of antibiotics by detection of corresponding drug resistance genes.
Recently developed microfluidic systems hold some advanced features of miniaturization and automation, which provide promising solutions for rapid ASTs at the single-cell level.14 As shown in Fig. 1, in this review, we summarize the newly developed microfluidic technologies for rapid bacterial ASTs at the single-cell level, including both phenotypic (microfluidic-based single bacterial culture) and genotypic methods (droplet digital systems for genotypic AMR detection). The point-of-care (POC) systems that integrate sample processing, fluid handling and ASTs are also discussed. At the end, remaining challenges in microfluidics-based rapid identification of antibiotic resistant bacteria are discussed for future development.
Microfluidic-based single bacterial culture for phenotypic ASTs
By manipulating small volumes of fluids in an integrated microchannel, a microfluidic lab-on-a-chip device is able to combine various steps of bacterial analysis together, including single bacterial culture, cell lysis, nucleic acid purification, sequence amplification and target detection.15–17 Since confining bacteria in a small and discrete volume at the single-cell level can minimize cross-contamination, potentially accelerate biochemical reactions and make the marker concentration under the isolated conditions increase quickly, this integration system could achieve high sensitivity with a low sample volume and offer automation and high-throughput analysis.18–20Culture-based methods remain the gold standard for determining antibiotic susceptibility.21 Notably, the development of microfluidic systems has made it possible to push the time requirements for culture-based ASTs to 1–3 h by minimizing the bacterial incubation chamber to the single-cell level and increasing the signal to background ratio in detecting the variation of proteomes, metabolomes, genomes and/or transcriptomes.22,23 Since the conventional AST systems only sense the change of bacterial population by measuring the optical density (OD) of the antibiotic-dosed pathogen culture or the zone of inhibition, the major limitation for the delay of conventional ASTs is the low sensitivity.24 For example, the limit of detection (LOD) for OD in a conventional AST is 107 colony-forming units (CFU) per mL,25 and it would take 16 to 20 h for the bacterial population to reach the minimum detectable growth level. However, the only information needed to determine antibiotic susceptibility is whether the pathogen is dividing after the antibiotic is added.
Therefore, some microfluidic-based methods have been developed to observe bacterial division at early stages.26 The most commonly used microfluidic chips for bacterial culture are the microchannels. The physical confinement of the pathogen allows rapid ASTs on a time scale comparable to the doubling time of the bacteria.27 For example, Lu et al. demonstrated that by confining individual bacteria in gas permeable microchannels with dimensions comparable to those of a single bacterium, the antibiotic resistance of the bacteria can be monitored in real-time at the single cell level (Fig. 2).27
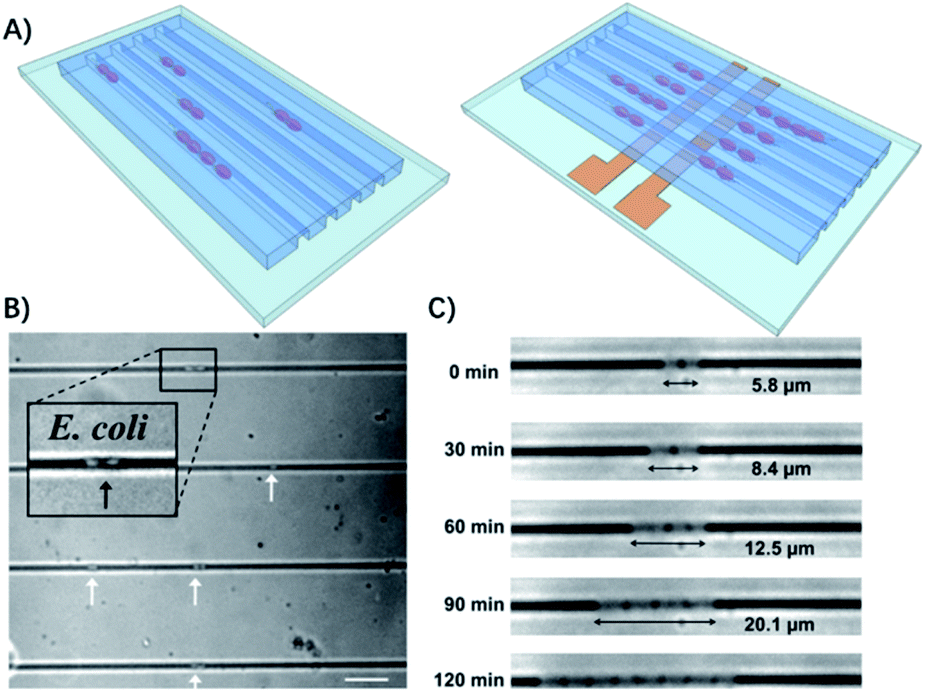 | ||
| Fig. 2 Single cell AST using confined microchannels. (A) Bacteria trapped in confined microchannels for the single cell AST with or without electrokinetic loading. (B) E. coli loaded at different locations in confined microchannels. The white arrows indicate the position of the bacteria trapped in the channels. The scale bar is 10 μm. (c) Time lapse images of E. coli growing in a microchannel for 2 h. Reproduced from ref. 27 with permission from the American Chemical Society, Copyright 2013. | ||
In another case, by tracking single bacterial growth and calculating the bacteria-occupying area in a microfluidic channel, Choi et al. were able to determine antimicrobial susceptibility by the simple observation of whether the bacteria are growing.28 Furthermore, they demonstrated the clinical application of imaging-based single-cell morphological analysis (SCMA) for rapid ASTs by automatically analyzing and categorizing morphological changes in single bacterial cells under various antimicrobial conditions (Fig. 3).29 Four standard bacterial strains and 189 clinical samples from hospitals were tested using SCMA. The results were compared with the gold standard broth microdilution test. In less than 4 h, SCMA can provide rapid and accurate antimicrobial susceptibility data that satisfy the recommended performance by the FDA.29
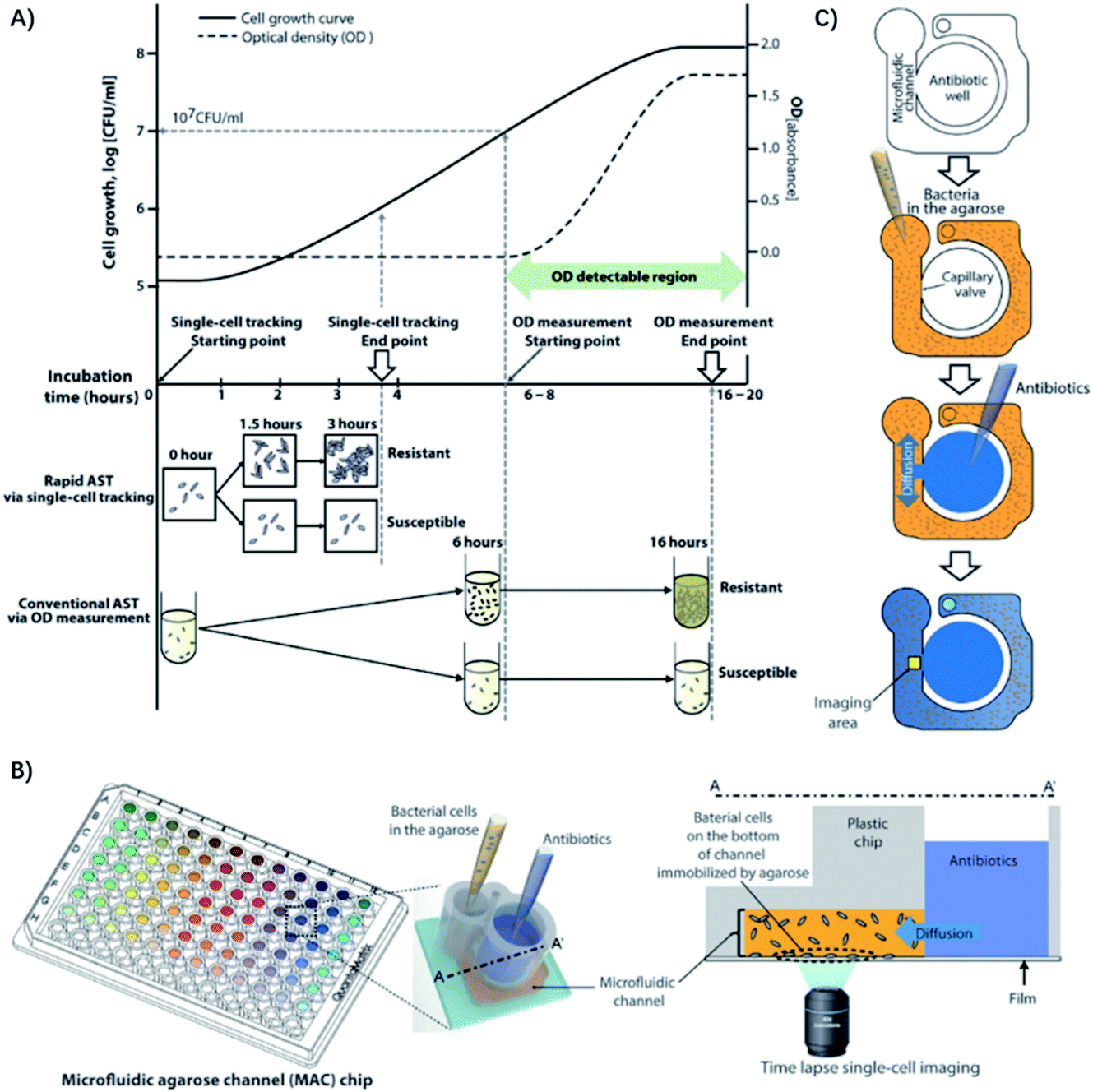 | ||
| Fig. 3 Single-cell morphological analysis (SCMA) in microfluidic agarose channels. (A) Comparison of an AST based on SCMA with the conventional method using OD measurements. In conventional ASTs by OD measurement, the OD value does not change until the bacterial concentration reaches 107/mL. However, in single-cell tracking using a microscope, changes in bacterial cells can be detected as soon as cells divide, so antibiotic susceptibility can be determined in 3 to 4 h. (B) Schematic of the microfluidic agarose channel (MAC) chip integrated with a 96-well platform for high-throughput analysis. The MAC chip is composed of microfluidic channels containing bacteria in agarose and a well to supply antibiotics and nutrients. The imaging region is the interface between the liquid medium and the microfluidic channel. The immobilized bacterial cells on the bottoms of channels are monitored for SCMA by time-lapse bright-field microscopy. (C) Experimental procedure for the MAC chip. Bacterial cells were mixed with agarose and loaded into a microfluidic channel, where the cells were immobilized by gel solidification. Liquid nutrients, some spiked with antimicrobials, were then loaded into the wells. These liquid samples diffused into the agarose through openings between the channels and the wells. Time-lapse imaging was performed in the imaging region (yellow box). Reproduced from ref. 29 with permission from AAAS, Copyright 2014. | ||
Recently, Baltekin and colleagues developed another single-cell imaging method to further shorten the analysis time. The total time for the test can be reduced to less than 30 min (including the time for loading a dilute sample), which is ideal for point-of-care (POC) applications.30 They used the method for urinary tract infections (UTIs), which is a disease that 100 million women suffer from annually and usually exhibits widespread antibiotic resistance.31 For UTIs, a fast AST could greatly improve medical practice by making it possible to prescribe an efficient antibiotic to the infecting bacteria before the patient leaves the primary care unit.32 Using a microfluidic chip composed of silicon elastomer and glass, they designed parallel “cell traps” with the size optimized to trap Escherichia coli, which can directly capture bacteria from samples with low bacterial counts (104 CFU mL−1). After loading, fluids flowed through these cell traps, but the bacteria were trapped in a line by a constriction at the “out” end of the trap. Alternating cell traps received one of two fluid streams, which contained growth media with or without antibiotics. Two-dimensional phase contrast images were obtained at intervals to determine if the cell trap was being filled with E. coli during the experiment. By averaging the growth rate of individual bacteria in response to 9 antibiotics that are used to treat urinary tract infections, they correctly classified susceptible and resistant uropathogenic Escherichia coli (UPEC) isolates in 49 clinical samples in less than 10 min. The total time for the AST, from loading of the sample to diagnostic readout, was less than 30 min.30
The diameter of the microfluidic chamber is critical for proper alignment of the bacteria, while the above-mentioned microfluidic device can only be optimized for a single type of pathogen, and lacks the ability for distinguishing polymicrobial samples and is only optimized for a small panel of pathogens, limiting its general applicability for diagnosis of different infectious diseases. To address these issues, Li et al. reported an adaptable microfluidic system for rapid bacterial classification and ASTs at the single-cell level (Fig. 4).33 By incorporating tunable microfluidic valves along with real-time optical detection, the bacteria can be trapped and classified according to their sizes and physical shapes for pathogen classification. By monitoring the bacterial growth in the presence of antibiotics at the single-cell level, the antimicrobial susceptibility can be determined in 30 min. Besides, the system can be widely applied for detecting bacterial pathogens and analyzing polymicrobial samples in urine, blood cultures, and whole blood. In a study of 25 clinical urine samples, the system was able to achieve a sensitivity of 100%, specificity of 83.33% for pathogen classification and 100% concordance for the AST.
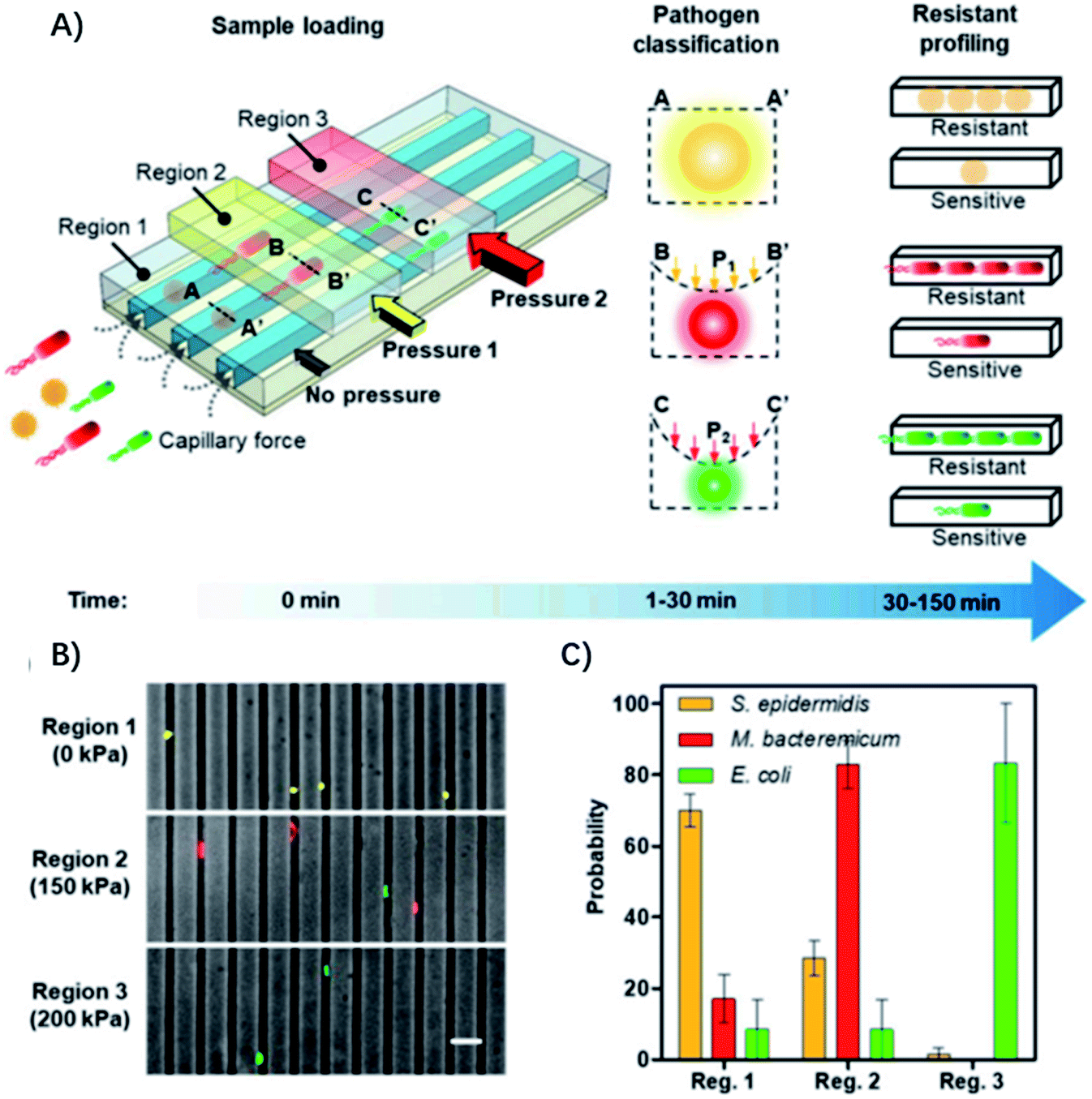 | ||
| Fig. 4 Single-cell pathogen classification and ASTs. (A) Schematic of the adaptable microfluidic device for pathogen classification and the AST at the single-cell level. Bacterial pathogens are loaded into the channels automatically by capillary force. Bacteria are trapped in different regions of the channels and classified according to the applied pressure, which dynamically adjusts the height of the channel. Antimicrobial susceptibility is determined by monitoring phenotypic growth of the bacteria in the presence of antibiotics. (B) Microfluidic separation of three bacterial species using the tunable microfluidic device. S. epidermidis, M. bacteremicum, and E. coli were fluorescently stained, mixed, and loaded into the microfluidic system to demonstrate pathogen separation. Images are representative of three independent experiments (scale bar, 10 μm). (C) Distributions of the bacteria in regions with 0, 150, and 200 kPa applied pressure in the microchannels. Data represent mean ± SEM (n = 3). Reproduced from ref. 33 with permission from the National Academy of Sciences, Copyright 2019. | ||
Another important category of microfluidic chips for bacterial analysis is droplet-based microfluidics.34,35 Droplet microfluidic systems enable the formation and analysis of large amounts of discrete microdroplets in an immiscible continuous phase.36 A microdroplet reactor is a tiny volume of liquid that could envelop samples and reagents for an analytical assay. Droplet reactors are fundamentally different from continuous flow reactors. Each droplet is separated from each other by the liquid–liquid interface, which is usually stabilized by amphiphilic surfactants.37 The main advantages of droplet technologies for bacterial analysis include (1) generation of a large number of microreactors for single bacterial analysis, (2) confinement of the reaction in tiny volumes for high sensitivity, and (3) high throughput analysis. For example, an integrated microfluidic droplet system has been used for confinement of single bacteria in picoliter droplets to accelerate bacterial growth and determine E. coli susceptibility to gentamicin in 1 h of incubation.38
Droplet microfluidic systems can also be used for analysis of metabolites from pathogenic bacteria. For example, Kang, et al. developed a technique termed ‘Integrated Comprehensive Droplet Digital Detection’ (IC 3D) that can selectively detect bacteria directly from blood at the single-cell level in a one-step and culture- and amplification-free process within 4 h (Fig. 5).39 Specifically, they used a DNAzyme sensor for reaction with the metabolites of target bacteria, which is able to rapidly generate a fluorescence signal.40 By mixing the blood sample with the DNAzyme sensor solution and bacterial lysis buffer within a microfluidic channel, they were able to generate trillions of individual picoliter droplets. The microdroplet reactor significantly increased the concentration of metabolites and minimized the interference from nonspecific background in whole blood. Importantly, in the IC 3D system, the generated droplets were collected in a vial and analyzed using a high-throughput 3D particle counting system, which can robustly and accurately detect single-fluorescent droplets from milliliter volumes within minutes.41 This detection system solved an important challenge of 1D on-chip counting systems which have limited detection efficiency and cannot be used for analysis of a large volume sample. Since blood stream infection always occurs at low concentration, it's critical to analyze a large amount of the sample to achieve enough sensitivity.
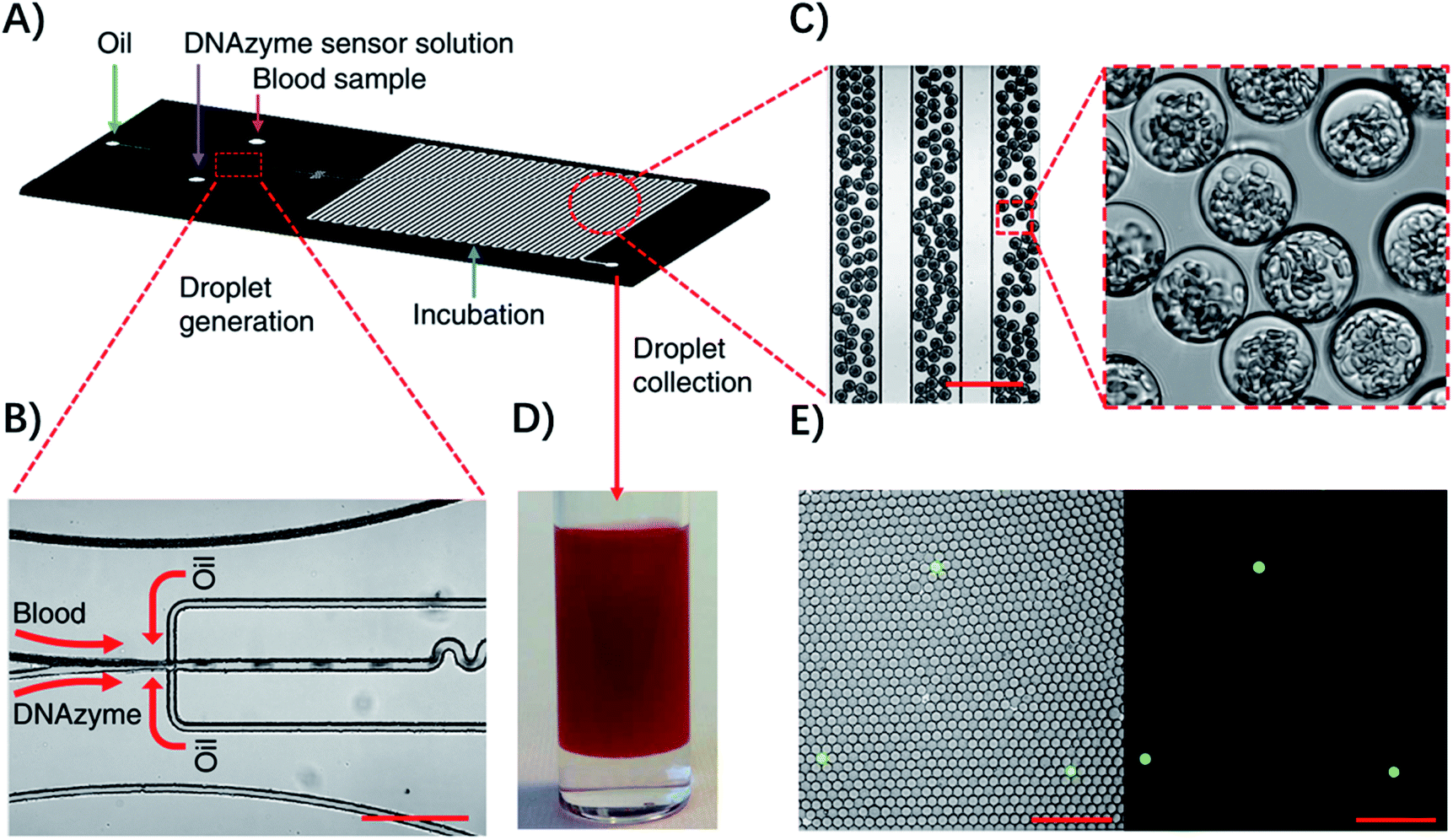 | ||
| Fig. 5 Detection of single bacteria in unprocessed blood using droplet microfluidics. (A) Design of the droplet-based microfluidic chip. (B) Representative microscopic images of the flow focusing droplet generation part on the microfluidic chip. (C) Images of the uniform 30 μm size droplets containing 10% blood. (D) Droplets collected in a cuvette. (E) Representative fluorescence images of DNAzyme sensors (250 nM) lighting up the droplets that contain single E. coli K12 cells in 10% blood after 3 h of reaction. Left panel: overlay of fluorescence and brightfield images. Right panel: fluorescence. Scale bar, 200 μm. Reproduced from ref. 39 with permission from the Nature Publishing Group, Copyright 2014. | ||
Digital PCR and digital LAMP for rapid ASTs
Genotypic ASTs are based on detection of a specific genetic marker (genes, plasmids, or mutations) associated with resistance phenotypes using genetic analysis methods, such as sequence specific amplification by PCR, whole genome sequencing and padlock probe-mediated rolling circle amplification.42–47 Detecting genes responsible for known mechanisms of drug resistance is more rapid than culture-based approaches, where the detection time is only limited by what is needed to amplify selected DNA sequences to a detectable level.48,49 For example, Yeh et al. developed a self-powered integrated microfluidic chip for quantitative analysis of the methicillin-resistance gene directly from human blood samples within 30 min.48 Abram et al. reported a one-step ddPCR assay for rapid detection of multiple antibiotic resistance genes directly from whole blood specimens, which can achieve a sensitivity of 10 CFU mL−1 with a rapid sample-to-answer assay time (1 hour).50 However, for genotypic ASTs, detailed knowledge of which resistance genes to test is required, which makes the methods not feasible for detecting unknown antibiotic resistant species. Since new forms of resistance evolve quickly, predicting resistance by analyzing a few known resistance genes is not a general solution.51 Moreover, the presence of certain resistance genes/mutations does not necessarily translate into phenotypic resistance,52 which further makes genotypic analysis a supplementary approach.Recently, some hybrid strategies have been proposed to develop more rapid and specific ASTs. The quantification of nucleic acids is conducted after a short antibiotic exposure to determine the susceptibility or resistance phenotype. Since mRNA encodes genotypic information in its sequence and phenotypic information in its abundance, it's an ideal target for analysis. For example, quantification of mRNA has allowed determination of susceptibility of pathogenic bacteria to ciprofloxacin and rifampin, after exposure times as short as 15 min.53 Yet, these methods generally require longer incubation times when using antibiotics with different mechanisms of action, but an ideal exposure time would be shorter than one cell division. To address this issue, Schoepp et al. applied digital PCR (dPCR) for measuring DNA replication of target bacteria through single-molecule counting, which greatly shortened the required time of antibiotic exposure (Fig. 6A).54
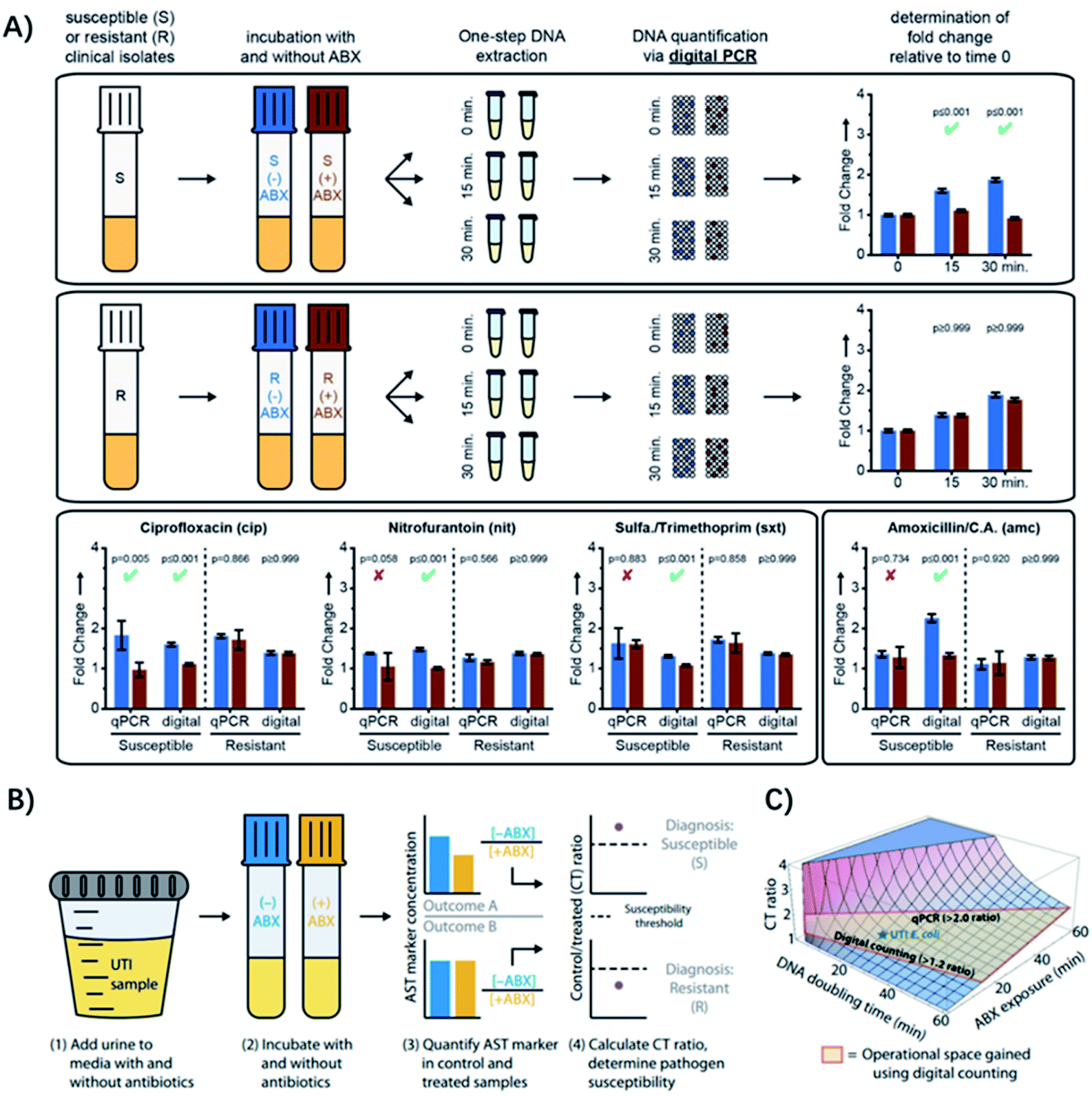 | ||
| Fig. 6 Digital PCR (dPCR) and digital LAMP for rapid pathogen-specific ASTs in clinical samples. (A) AST results analyzed by dPCR. Exposure of susceptible and resistant E. coli isolates to 4 antibiotics. Partitioning bacterial chromosomal DNA into many small volumes enables performing the AST for urinary tract infections in 15 min. (B) Digital LAMP for detecting antibiotic susceptibility by measuring the quantity of a specific AST marker sequence. Urine samples were incubated with or w/o antibiotics (ABX) (steps 1 and 2), AST markers were quantified in the control (−ABX) and treated (+ABX) samples (step 3), and the CT ratios were analyzed (step 4). (C) The theoretical model that predicts the CT ratio as a function of pathogen DNA doubling time and antibiotic exposure time. The operational space gained by using digital counting compared with qPCR is outlined in red. Reproduced from ref. 54 with permission from John Wiley & Sons, Inc., Copyright 2016; and ref. 59 with permission from AAAS, Copyright 2017. | ||
Digital PCR is an absolute quantification method, where DNA samples are partitioned into picoliter droplets, resulting in one or no molecules in each droplet reaction.55 Compared with qPCR, dPCR has greater precision and improved reproducibility because it uses digital counting (1 or 0) without the need for normalization.56 Digital PCR enables more precise and robust measurements of bacterial DNA, achieving higher statistical power with fewer replicates relative to qPCR.57,58 Besides, it has been found that the partitioning could provide unique capabilities for ASTs when analyzing the delay in chromosome segregation.54 The high resolution of digital quantification enables measurement of small (less than two-fold) changes in chromosome replication and segregation after an antibiotic exposure shorter than the average time of cell division (Fig. 6A). Therefore, dPCR adds chromosome segregation to the list of phenotypic markers suitable for rapid antibiotic susceptibility testing, which has been used in clinical isolates from UTIs after 15 min of exposure to 4 antibiotic classes relevant to UTIs (Fig. 6A).54
To make the test even faster, ideally within a single patient visit (30 min), Schoepp et al. further applied an ultrafast (∼7 min) digital real-time loop-mediated isothermal amplification (dLAMP) assay for the AST, which was much faster than dPCR (require ∼2 h).59 The rapid dLAMP assay can be used with SlipChip microfluidic devices to determine the phenotypic antibiotic susceptibility of E. coli directly from clinical urine samples, and the entire workflow can be finished in less than 30 min (Fig. 6B). 51 clinical UTI urine samples were analyzed using this method and the AUC for the generated ROC curve was 0.96,59 demonstrating that the quantification of transcriptional responses within minutes of antibiotic exposure can distinguish susceptible bacteria from resistant ones.
Recently, Bhattacharyya et al. described a rapid assay for combined genotypic and phenotypic ASTs through RNA detection (termed GoPhAST-R) that classifies strains with 94–99% accuracy by coupling machine learning analysis of early antibiotic-induced transcriptional changes with simultaneous detection of key genetic resistance determinants to increase the accuracy of resistance detection (Fig. 7).60 The method facilitates molecular epidemiology and enables early detection of emerging resistance mechanisms by integration of all genotype and phenotype information. Using a next-generation nucleic acid detection platform (NanoString Hyb & Seq), GoPhAST-R detects specific messenger RNA expression signatures in bacteria after brief antibiotic exposure, while the susceptible cells that are stressed would be transcriptionally distinct from resistant cells. Notably, this process can be generalizable to any pathogen-antibiotic pair of interest by a simple biological phenomenon that an antibiotic would elicit a differential transcriptional response in susceptible versus resistant isolates.60 Besides, GoPhAST-R can be directly applied in positive blood culture bottles, which shows great promise for clinical applications.
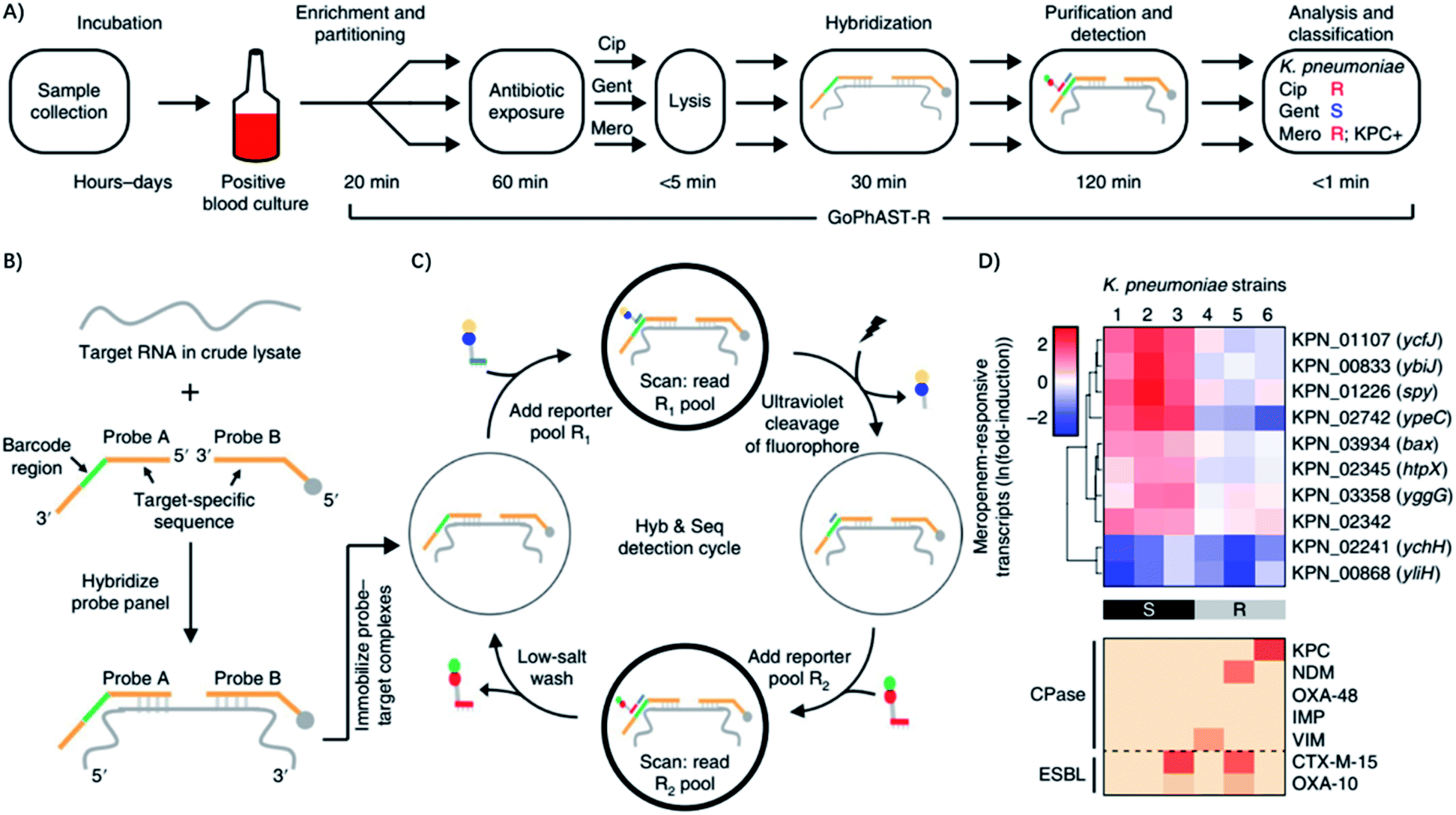 | ||
| Fig. 7 GoPhAST-R workflow with the NanoString Hyb & Seq platform distinguishes phenotypically susceptible from resistant strains and detects genetic resistance determinants in less than 4 h. (A) The GoPhAST-R workflow begins once growth is detected in a blood culture bottle. Pathogen identification could be performed either before this process or in parallel by multiplexing mRNA targets from multiple organisms. (B) Hyb & Seq hybridization scheme: probe pairs targeting each RNA transcript are hybridized in the crude lysate. Each probe A contains a unique barcode sequence (green) for detection and a shared 3′ capture sequence; each probe B contains a biotin group (gray circle) for surface immobilization and a shared 5′ capture sequence. (C) Hyb & Seq detection strategy: immobilized probe–target complexes undergo sequential cycles of multistep imaging for spatially resolved single-molecule detection. Each cycle consists of reporter probe binding and detection, ultraviolet cleavage, a second round of reporter probe binding and detection, and a low-salt wash to regenerate the unbound probe–target complex. 5 Hyb & Seq cycles were used to generate the data shown. (D) Pilot studies for accelerated meropenem susceptibility testing of 6 clinical K. pneumoniae isolates. Reproduced from ref. 60 with permission from the Nature Publishing Group, Copyright 2019. | ||
Conclusion and perspectives
The application of microfluidic systems has made it possible to perform antibiotic susceptibility tests at the single-cell level, which shows great promise for fast and accurate determination of antimicrobial susceptibility profiles for guiding antibiotic treatment decisions. In particular, physical confinement of the pathogenic bacteria in microchannels allows rapid ASTs on a time scale comparable to the doubling time of the bacteria. Besides, by incorporating dPCR or dLAMP-based nucleic acid analysis, the antibiotic exposure time can be further reduced and the whole process would take less than 30 min, which is within the time span of a patient visit. The collected AST information can be used to guide infection treatment and facilitate antimicrobial stewardship.However, most of the advanced technologies for single-cell ASTs are still validated with clinical isolates of pathogens, which are grown in culture to a high density before the assay (taking 5–6 h). For direct clinical sample analysis, a standardized and fast sample processing protocol would be necessary, since the clinical sample matrices, such as blood and urine, present a huge challenge for rapid microscopy-based ASTs.61 Besides, most of the currently developed microfluidic systems are not fully automatic, requiring open pipetting and handling steps, which may cause batch-to-batch variation, cross-contamination and even potential biohazard risks. Ideally, the AST should be performed in an integrated, automatic device. For POC application, the device also needs to be inexpensive and simple-to-use.
To make the process of ASTs more rapid, the future development of microfluidics-based ASTs may not directly measure the phenotypic cell viability, but could focus on some more subtle biological information alterations, which are related to the molecular mechanism of antibiotic resistance.62 For this purpose, some ultrasensitive nanotechnology-based analytical methods would play a crucial role. For example, the microfluidic cantilever system has already been well applied for rapid ASTs.63,64 In addition, nanoparticle-based fluorescence or surface plasmon resonance technology may also be applied to improve the sensitivity of ASTs.
Other than nanotechnology, the isothermal nucleic acid amplification strategy is another category of toolbox. For example, some newly developed CRISPR systems (such as Cas12a and Cas13a) have now be used for genotypic and phenotypic analysis of bacterial pathogens.47,65–70 Specifically, a CRISPR-Cas13a/C2c2-based SHERLOCK platform has been applied for ultrafast and sensitive RNA/DNA quantitation, which is suitable for rapid identification of bacterial pathogens by detection of specific bacterial genes.66 Recently, an allosteric probe-initiated catalysis and CRISPR-Cas13a amplification reaction (termed ‘APC-Cas’) was developed for detecting very low numbers of a bacterial pathogens without isolation71 and can selectively quantify bacterial cells with a concentration from 1 to 105 CFU mL−1 in various types of samples, and the cost of reagents can be reduced to below $1 per test.66 Therefore, we think that incorporating the powerful CRISPR-based signal amplification with the microfluidic-based single-cell AST may provide a more rapid and cost-effective point-of-care diagnostic and therapeutic tool for clinical applications.
Except for clinical test applications, microfluidics-based single-cell ASTs can also be used as a powerful tool for new antibiotic discovery.72 Recently, some deep learning approaches have been developed for predicting antibiotic activity in molecules that are structurally different from known antibiotics.73 To rapidly test the performance of these newly identified drug candidates, the high throughput microfluidic systems show great potential.74,75
The single-cell AST also holds huge potential for high throughput single bacterial heterogeneity analysis.76 Since the antimicrobial susceptibility may differ among individual cells, the adequate detection of single bacterial heterogeneity is of great clinical importance.77 Besides, microfluidic-based AST systems naturally fit the requirement of on-site quantitative and multiplexed testing with high simplicity and portability, which may not only be applied for medical uses, but also for environmental and agricultural analysis applications.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The work was supported by grants from the National Natural Science Foundation of China (No. 81601597 and 21904119) and Key Scientific Research Projects (Science and Technology Department of Henan Province, No. 192102310147).References
- S. B. Levy and B. Marshall, Nat. Med., 2004, 10, 122–129 CrossRef PubMed.
- A. van Belkum, C. D. Burnham, J. W. A. Rossen, F. Mallard, O. Rochas and W. M. Dunne, Jr, Nat. Rev. Microbiol., 2020, 18 DOI:10.1038/s41579-020-0327-x.
- R. Daniels, J. Antimicrob. Chemother., 2011, 66, 11–23 CrossRef PubMed.
- R. Laxminarayan, A. Duse, C. Wattal, A. K. Zaidi, H. F. Wertheim, N. Sumpradit, E. Vlieghe, G. L. Hara, I. M. Gould, H. Goossens, C. Greko, A. D. So, M. Bigdeli, G. Tomson, W. Woodhouse, E. Ombaka, A. Q. Peralta, F. N. Qamar, F. Mir, S. Kariuki, Z. A. Bhutta, A. Coates, R. Bergstrom, G. D. Wright, E. D. Brown and O. Cars, Lancet Infect. Dis., 2013, 13, 1057–1098 CrossRef PubMed.
- J. J. Kerremans, P. Verboom, T. Stijnen, L. Hakkaart-van Roijen, W. Goessens, H. A. Verbrugh and M. C. Vos, J. Antimicrob. Chemother., 2008, 61, 428–435 CrossRef CAS PubMed.
- J. Barenfanger, C. Drake and G. Kacich, J. Clin. Microbiol., 1999, 37, 1415–1418 CrossRef CAS PubMed.
- J. H. Jorgensen and M. J. Ferraro, Clin. Infect. Dis., 2009, 49, 1749–1755 CrossRef CAS PubMed.
- I. Wiegand, K. Hilpert and R. E. Hancock, Nat. Protoc., 2008, 3, 163–175 CrossRef CAS PubMed.
- K. Syal, M. Mo, H. Yu, R. Iriya, W. Jing, S. Guodong, S. Wang, T. E. Grys, S. E. Haydel and N. Tao, Theranostics, 2017, 7, 1795–1805 CrossRef CAS PubMed.
- O. Altun, M. Almuhayawi, M. Ullberg and V. Ozenci, J. Clin. Microbiol., 2013, 51, 4130–4136 CrossRef PubMed.
- A. S. Rossney, C. M. Herra, G. I. Brennan, P. M. Morgan and B. O'Connell, J. Clin. Microbiol., 2008, 46, 3285–3290 CrossRef CAS PubMed.
- J. D. Bard and F. Lee, Clin. Microbiol. Newsl., 2018, 40, 87–95 CrossRef.
- A. M. Nicasio, J. L. Kuti and D. P. Nicolau, Pharmacotherapy, 2008, 28, 235–249 CrossRef CAS PubMed.
- Y. Li, X. Yang and W. Zhao, SLAS Technol., 2017, 22, 585–608 Search PubMed.
- T. S. Kaminski and P. Garstecki, Chem. Soc. Rev., 2017, 46, 6210–6226 RSC.
- L. F. Harris and A. J. Killard, Anal. Methods, 2018, 10, 3714–3719 RSC.
- J. D. Besant, E. H. Sargent and S. O. Kelley, Lab Chip, 2015, 15, 2799–2807 RSC.
- S. Park, Y. Zhang, S. Lin, T. H. Wang and S. Yang, Biotechnol. Adv., 2011, 29, 830–839 CrossRef CAS PubMed.
- W. B. Lee, C. Y. Fu, W. H. Chang, H. L. You, C. H. Wang, M. S. Lee and G. B. Lee, Biosens. Bioelectron., 2017, 87, 669–678 CrossRef CAS PubMed.
- K. Zhang, H. Gao, R. Deng and J. Li, Angew. Chem., Int. Ed., 2019, 58, 4790–4799 CrossRef CAS PubMed.
- L. Varadi, J. L. Luo, D. E. Hibbs, J. D. Perry, R. J. Anderson, S. Orenga and P. W. Groundwater, Chem. Soc. Rev., 2017, 46, 4818–4832 RSC.
- A. Reece, B. Xia, Z. Jiang, B. Noren, R. McBride and J. Oakey, Curr. Opin. Biotechnol., 2016, 40, 90–96 CrossRef CAS PubMed.
- A. Charnot-Katsikas, V. Tesic, N. Love, B. Hill, C. Bethel, S. Boonlayangoor and K. G. Beavis, J. Clin. Microbiol., 2018, 56, 21–27 Search PubMed.
- M. R. Pulido, M. Garcia-Quintanilla, R. Martin-Pena, J. M. Cisneros and M. J. McConnell, J. Antimicrob. Chemother., 2013, 68, 2710–2717 CrossRef CAS PubMed.
- P. Dalgaard, T. Ross, L. Kamperman, K. Neumeyer and T. A. McMeekin, Int. J. Food Microbiol., 1994, 23, 391–404 CrossRef CAS PubMed.
- N. Cermak, S. Olcum, F. F. Delgado, S. C. Wasserman, K. R. Payer, A. M. Murakami, S. M. Knudsen, R. J. Kimmerling, M. M. Stevens, Y. Kikuchi, A. Sandikci, M. Ogawa, V. Agache, F. Baleras, D. M. Weinstock and S. R. Manalis, Nat. Biotechnol., 2016, 34, 1052–1059 CrossRef CAS PubMed.
- Y. Lu, J. Gao, D. D. Zhang, V. Gau, J. C. Liao and P. K. Wong, Anal. Chem., 2013, 85, 3971–3976 CrossRef CAS PubMed.
- J. Choi, Y. G. Jung, J. Kim, S. Kim, Y. Jung, H. Na and S. Kwon, Lab Chip, 2013, 13, 280–287 RSC.
- J. Choi, J. Yoo, M. Lee, E. G. Kim, J. S. Lee, S. Lee, S. Joo, S. H. Song, E. C. Kim, J. C. Lee, H. C. Kim, Y. G. Jung and S. Kwon, Sci. Transl. Med., 2014, 6, 267ra174 CrossRef PubMed.
- O. Baltekin, A. Boucharin, E. Tano, D. I. Andersson and J. Elf, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 9170–9175 CrossRef CAS PubMed.
- T. M. Hooton, N. Engl. J. Med., 2012, 366, 1028–1037 CrossRef CAS PubMed.
- W. E. Stamm and S. R. Norrby, J. Infect. Dis., 2001, 183(suppl 1), 1–4 CrossRef PubMed.
- H. Li, P. Torab, K. E. Mach, C. Surrette, M. R. England, D. W. Craft, N. J. Thomas, J. C. Liao, C. Puleo and P. K. Wong, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 10270–10279 CrossRef CAS PubMed.
- A. B. Theberge, F. Courtois, Y. Schaerli, M. Fischlechner, C. Abell, F. Hollfelder and W. T. Huck, Angew. Chem., Int. Ed. Engl., 2010, 49, 5846–5868 CrossRef CAS PubMed.
- L. Mazutis, J. Gilbert, W. L. Ung, D. A. Weitz, A. D. Griffiths and J. A. Heyman, Nat. Protoc., 2013, 8, 870–891 CrossRef CAS PubMed.
- D. K. Kang, M. M. Ali, K. X. Zhang, E. J. Pone and W. A. Zhao, TrAC, Trends Anal. Chem., 2014, 58, 145–153 CrossRef CAS.
- M. T. Guo, A. Rotem, J. A. Heyman and D. A. Weitz, Lab Chip, 2012, 12, 2146–2155 RSC.
- A. M. Kaushik, K. Hsieh, L. Chen, D. J. Shin, J. C. Liao and T. H. Wang, Biosens. Bioelectron., 2017, 97, 260–266 CrossRef CAS PubMed.
- D. K. Kang, M. M. Ali, K. Zhang, S. S. Huang, E. Peterson, M. A. Digman, E. Gratton and W. Zhao, Nat. Commun., 2014, 5, 5427–5433 CrossRef CAS PubMed.
- M. M. Ali, S. D. Aguirre, H. Lazim and Y. F. Li, Angew. Chem., Int. Ed., 2011, 50, 3751–3754 CrossRef CAS PubMed.
- I. Altamore, L. Lanzano and E. Gratton, Meas. Sci. Technol., 2013, 24, 65702–65707 CrossRef CAS PubMed.
- H. Frickmann, W. O. Masanta and A. E. Zautner, BioMed Res. Int., 2014, 2014, 375681–375686 Search PubMed.
- A. van Belkum, G. Durand, M. Peyret, S. Chatellier, G. Zambardi, J. Schrenzel, D. Shortridge, A. Engelhardt and W. M. Dunne, Ann. Lab. Med., 2013, 33, 14–27 CrossRef CAS PubMed.
- H. Gao, K. Zhang, X. Teng and J. Li, TrAC, Trends Anal. Chem., 2019, 121, 115700 CrossRef CAS.
- K. Zhang, J. Liu, Q. Song, X. Yang, D. Wang, W. Liu, J. Shi and Z. Zhang, ACS Appl. Mater. Interfaces, 2019, 11, 46604–46613 CrossRef CAS PubMed.
- K. Zhang, Y. Yue, S. Wu, W. Liu, J. Shi and Z. Zhang, ACS Sens., 2019, 4, 1245–1251 CrossRef CAS PubMed.
- K. Zhang, R. Deng, H. Gao, X. Teng and J. Li, Chem. Soc. Rev., 2020, 49, 1932–1954 RSC.
- E. C. Yeh, C. C. Fu, L. Hu, R. Thakur, J. Feng and L. P. Lee, Sci. Adv., 2017, 3, e1501645 CrossRef PubMed.
- J. T. Connelly, J. P. Rolland and G. M. Whitesides, Anal. Chem., 2015, 87, 7595–7601 CrossRef CAS PubMed.
- T. J. Abram, H. Cherukury, C. Y. Ou, T. Vu, M. Toledano, Y. Li, J. T. Grunwald, M. N. Toosky, D. F. Tifrea, A. Slepenkin, J. Chong, L. Kong, D. V. Del Pozo, K. T. La, L. Labanieh, J. Zimak, B. Shen, S. S. Huang, E. Gratton, E. M. Peterson and W. Zhao, Lab Chip, 2020, 20, 477–489 RSC.
- M. A. Webber and L. J. Piddock, J. Antimicrob. Chemother., 2003, 51, 9–11 CrossRef CAS PubMed.
- R. T. Cirz, J. K. Chin, D. R. Andes, V. de Crecy-Lagard, W. A. Craig and F. E. Romesberg, PLoS Biol., 2005, 3, e176 CrossRef PubMed.
- C. Halford, R. Gonzalez, S. Campuzano, B. Hu, J. T. Babbitt, J. Liu, J. Wang, B. M. Churchill and D. A. Haake, Antimicrob. Agents Chemother., 2013, 57, 936–943 CrossRef CAS PubMed.
- N. G. Schoepp, E. M. Khorosheva, T. S. Schlappi, M. S. Curtis, R. M. Humphries, J. A. Hindler and R. F. Ismagilov, Angew. Chem., Int. Ed. Engl., 2016, 55, 9557–9561 CrossRef CAS PubMed.
- C. M. Hindson, J. R. Chevillet, H. A. Briggs, E. N. Gallichotte, I. K. Ruf, B. J. Hindson, R. L. Vessella and M. Tewari, Nat. Methods, 2013, 10, 1003–1005 CrossRef CAS PubMed.
- K. Zhang, D. K. Kang, M. M. Ali, L. Liu, L. Labanieh, M. Lu, H. Riazifar, T. N. Nguyen, J. A. Zell, M. A. Digman, E. Gratton, J. Li and W. Zhao, Lab Chip, 2015, 15, 4217–4226 RSC.
- A. S. Whale, J. F. Huggett, S. Cowen, V. Speirs, J. Shaw, S. Ellison, C. A. Foy and D. J. Scott, Nucleic Acids Res., 2012, 40, e82 CrossRef CAS PubMed.
- S. Weaver, S. Dube, A. Mir, J. Qin, G. Sun, R. Ramakrishnan, R. C. Jones and K. J. Livak, Methods, 2010, 50, 271–276 CrossRef CAS PubMed.
- N. G. Schoepp, T. S. Schlappi, M. S. Curtis, S. S. Butkovich, S. Miller, R. M. Humphries and R. F. Ismagilov, Sci. Transl. Med., 2017, 9, eaal3693 CrossRef PubMed.
- R. P. Bhattacharyya, N. Bandyopadhyay, P. Ma, S. S. Son, J. Liu, L. L. He, L. Wu, R. Khafizov, R. Boykin, G. C. Cerqueira, A. Pironti, R. F. Rudy, M. M. Patel, R. Yang, J. Skerry, E. Nazarian, K. A. Musser, J. Taylor, V. M. Pierce, A. M. Earl, L. A. Cosimi, N. Shoresh, J. Beechem, J. Livny and D. T. Hung, Nat. Med., 2019, 25, 1858–1864 CrossRef CAS PubMed.
- M. Mo, Y. Yang, F. Zhang, W. Jing, R. Iriya, J. Popovich, S. Wang, T. Grys, S. E. Haydel and N. Tao, Anal. Chem., 2019, 91, 10164–10171 CrossRef CAS PubMed.
- J. M. Blair, M. A. Webber, A. J. Baylay, D. O. Ogbolu and L. J. Piddock, Nat. Rev. Microbiol., 2015, 13, 42–51 CrossRef CAS PubMed.
- G. Longo, L. Alonso-Sarduy, L. M. Rio, A. Bizzini, A. Trampuz, J. Notz, G. Dietler and S. Kasas, Nat. Nanotechnol., 2013, 8, 522–526 CrossRef CAS PubMed.
- H. Etayash, M. F. Khan, K. Kaur and T. Thundat, Nat. Commun., 2016, 7, 12947 CrossRef CAS PubMed.
- D. S. Chertow, Science, 2018, 360, 381–382 CrossRef PubMed.
- J. S. Gootenberg, O. O. Abudayyeh, J. W. Lee, P. Essletzbichler, A. J. Dy, J. Joung, V. Verdine, N. Donghia, N. M. Daringer, C. A. Freije, C. Myhrvold, R. P. Bhattacharyya, J. Livny, A. Regev, E. V. Koonin, D. T. Hung, P. C. Sabeti, J. J. Collins and F. Zhang, Science, 2017, 356, 438–442 CrossRef CAS PubMed.
- J. S. Chen, E. B. Ma, L. B. Harrington, M. Da Costa, X. R. Tian, J. M. Palefsky and J. A. Doudna, Science, 2018, 360, 436–439 CrossRef CAS PubMed.
- K. Zhang, R. Deng, X. Teng, Y. Li, Y. Sun, X. Ren and J. Li, J. Am. Chem. Soc., 2018, 140, 11293–11301 CrossRef CAS PubMed.
- K. X. Zhang, R. J. Deng, Y. Li, L. Zhang and J. H. Li, Chem. Sci., 2016, 7, 4951–4957 RSC.
- Y. Li, X. Teng, K. Zhang, R. Deng and J. Li, Anal. Chem., 2019, 91, 3989–3996 CrossRef CAS PubMed.
- J. Shen, X. Zhou, Y. Shan, H. Yue, R. Huang, J. Hu and D. Xing, Nat. Commun., 2020, 11, 267–272 CrossRef CAS PubMed.
- J. Dai, M. Hamon and S. Jambovane, Bioengineering, 2016, 3, 25–38 CrossRef PubMed.
- J. M. Stokes, K. Yang, K. Swanson, W. Jin, A. Cubillos-Ruiz, N. M. Donghia, C. R. MacNair, S. French, L. A. Carfrae, Z. Bloom-Ackerman, V. M. Tran, A. Chiappino-Pepe, A. H. Badran, I. W. Andrews, E. J. Chory, G. M. Church, E. D. Brown, T. S. Jaakkola, R. Barzilay and J. J. Collins, Cell, 2020, 180, 688–702 CrossRef PubMed.
- J. Hage-Hulsmann, A. Grunberger, S. Thies, B. Santiago-Schubel, A. S. Klein, J. Pietruszka, D. Binder, F. Hilgers, A. Domrose, T. Drepper, D. Kohlheyer, K. E. Jaeger and A. Loeschcke, PLoS One, 2018, 13, e0200940 CrossRef PubMed.
- J. Dai, S. J. Suh, M. Hamon and J. W. Hong, Biotechnol. J., 2015, 10, 1783–1791 CrossRef CAS PubMed.
- O. Scheler, K. Makuch, P. R. Debski, M. Horka, A. Ruszczak, N. Pacocha, K. Sozanski, O. P. Smolander, W. Postek and P. Garstecki, Sci. Rep., 2020, 10, 3282 CrossRef CAS PubMed.
- H. Nicoloff, K. Hjort, B. R. Levin and D. I. Andersson, Nat. Microbiol., 2019, 4, 504–514 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |