
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A diversity of recently reported methodology for asymmetric imine reduction
Jonathan
Barrios-Rivera
 a,
Yingjian
Xu
*b,
Martin
Wills
*a and
Vijyesh K.
Vyas
a
a,
Yingjian
Xu
*b,
Martin
Wills
*a and
Vijyesh K.
Vyas
a
aDepartment of Chemistry, The University of Warwick, Coventry, CV4 7AL, UK
bGoldenKeys High-tech Materials Co., Ltd, Building B, Innovation & Entrepreneurship Park, Guian New Area, Guizhou Province, China. E-mail: m.wills@warwick.ac.uk
First published on 3rd September 2020
Abstract
This review contains an account of recent developments in catalytic, asymmetric processes reported for the reduction of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonds to amines, in which we have attempted to communicate the remarkable diversity of methods which have been reported in recent years, including organometallic and organocatalytic processes.
N bonds to amines, in which we have attempted to communicate the remarkable diversity of methods which have been reported in recent years, including organometallic and organocatalytic processes.
 Jonathan Barrios-Rivera | Jonathan Barrios-Rivera completed an MSci at Queen Mary University London in 2017 and is currently a PhD student at Warwick working under the supervision of Professor Wills. His project is concerned with the development of novel asymmetric catalysts based on Ru(II) complexes. |
 Yingjian Xu | Dr Yingjain Xu is a former PhD student of Professor Wills, having completed a PhD at Warwick University in 2005. He worked with Professor John Brown at Oxford University before joining Phosphonics. Recently he founded, and is currently the CEO of, GoldenKeys High Tech Materials Co. Ltd in Guiyang, China. |
 Martin Wills | Martin Wills is Professor of Organic Chemistry at the University of Warwick, UK He leads a group working on a combination of organic and organometallic asymmetric catalysis and the development of novel synthetic chemistry methodology. |
 Vijyesh K. Vyas | Dr Vijyesh Vyas received a Master's in Chemistry (2012) from the Institute of Chemical Technology (ICT). He obtained a PhD in organic chemistry from ICT in 2018, studying application of Ru-tethered catalyst to generate optically active value-added chiral alcohols with Prof. Bhalchandra M. Bhanage. Currently, he is doing his postdoctoral studies in the field of asymmetric catalysis with Prof. Martin Wills at University of Warwick. |
1. Introduction
Optically active amines are essential functional groups in many biological molecules. Hence, considerable amount of efforts have been made to develop strategies to obtain these. Imine reductions are some of the most fundamental transformations in organic synthesis and are considered key reactions in fine chemical and pharmaceutical industries for the production of valuable amines. It is evident that a considerable amount of research has been done on reducing CN bonds enantioselectively using various strategies. These include pressure hydrogenation using H2 gas, asymmetric transfer hydrogenation (ATH) using organic molecules as the reducing agent and organocatalytic reduction by using chiral reagents and these are probably the most common methods used to achieve reduction of imines to optically active chiral amines. All of these are important methods as sometimes one is preferred over the other depending on the situation. For instance, although ATH is a convenient and versatile method for imine reduction which avoids the use of a pressure reactor, most industries often prefer to use pressure hydrogenation over ATH since it utilises relatively inexpensive hydrogen gas as a hydrogen source and is potentially 100% atom efficient. Moreover, AH and ATH generally involve the use of a metal to deliver a hydrogen from a respective source whereas organocatalytic reduction avoids metals and utilises a chiral organic molecule to deliver the hydrogen to the imine.
This review aims to cover recent developments in the field of imine reduction by using AH, ATH and organocatalytic reductions. We will focus on the recent advancements in the last five years including 2016, since earlier results are widely reported by some recent reviews.1 This review primarily focuses on recent reports on catalysts based on iridium, ruthenium and rhodium, for AH and ATH for imines and subsequently organocatalytic reduction using chiral phosphoric acid and borane are covered. This review does not include non-asymmetric reports although interesting reports of the use less widely used approaches or reagents have been published in recent years.2
A striking aspect of the chemistry of imine reduction is the diversity of methods which have been employed, including metal-based and metal-free, and the use of a wide range of reducing agents. In this review we have tried to capture the remarkable range of exciting new synthetic chemistry methodology which has been developed during the course of these internationally-ranging studies.
2. Asymmetric hydrogenation (AH) of imines
2.1. Iridium-catalysed imine AH
A simple and efficient method for the synthesis of chiral tertiary amines was developed by Zhou and co-workers. A chiral iridium-diphosphine ligand catalyst was employed to produce chiral N-alkyl tetrahydroisoquinolines and N-alkyl tetrahydro-β-carbolines via asymmetric hydrogenation in up to 96% ee and 88% ee, respectively (Fig. 1).3 The protocol gives access to the synthesis of a class of drug molecule containing the N-alkyl tetrahydroisoquinoline moiety in a single step which does not need further alkylation to obtain the enantiomerically enriched product.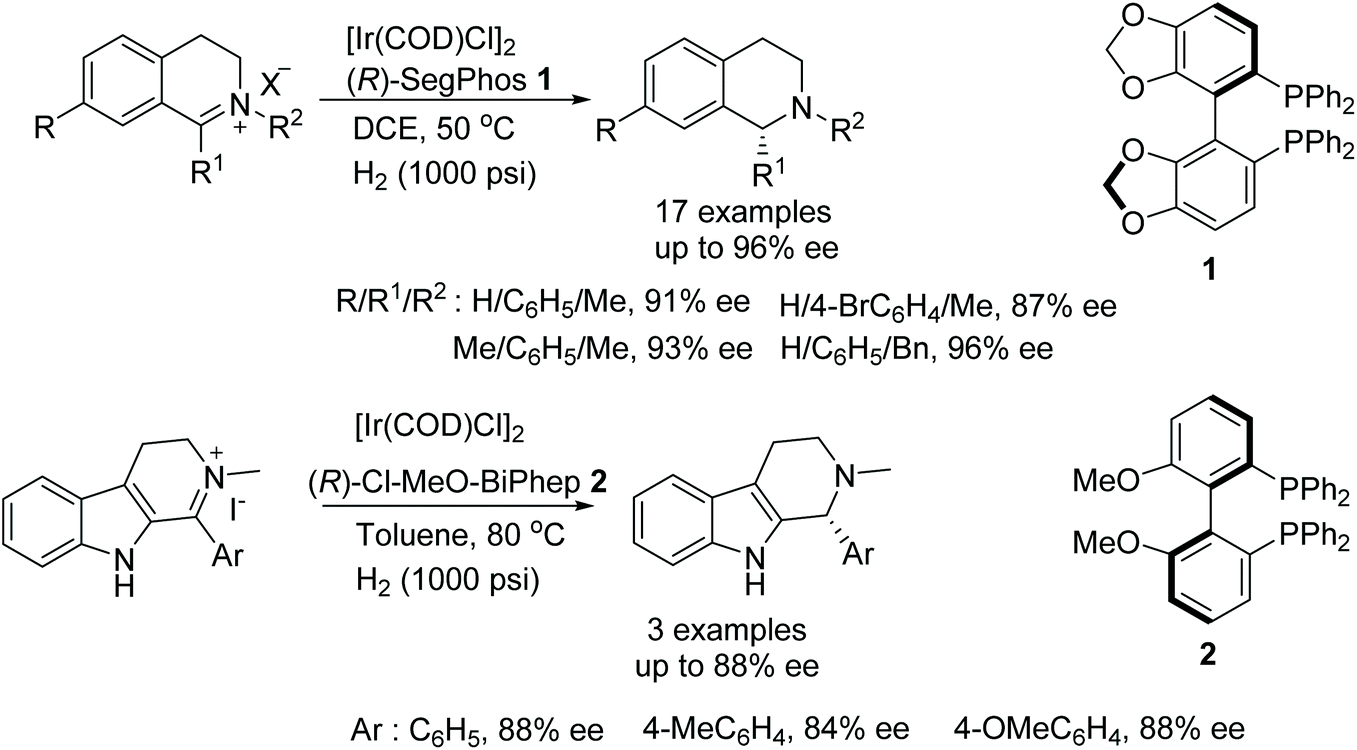 | ||
| Fig. 1 AH of N-alkyl tetrahydroisoquinolines and N-alkyl tetrahydro-β-carbolines. | ||
Studies on the use of an Ir-catalyst with a diphosphine ligand and I2 as additive in THF led to the AH of 4-phenylquinazolin-2(1H)-one in 77% ee (Fig. 2). Better enantioselectivities were obtained when the additives were changed to NIS (69% ee), NBS (95% ee) or BCMDH (96% ee). Upon finding the best additive, the diphosphine ligand was replaced with (R)-SegPhos 1, increasing the ee to 97%. The developed catalytic system was then applied to a library of substrates (Fig. 2). It was found that there were no decreases in yields or ee when either methyl or chloro groups were introduced to the fused-aryl ring.4
 | ||
| Fig. 2 Iridium-catalysed AH of 4-phenylquinazolin-2(1H)-one. | ||
A large amount of research has been conducted on the reductions of 1-aryl-3,4-dihydroisoquinolines using iridium-catalysed asymmetric hydrogenation. Chang and co-workers have reported the synthesis of bioactive chiral-1-substituted tetrahydroisoquinolines via a one pot N-deprotection and catalytic intramolecular asymmetric reductive amination.5 In this protocol, the iodine bridged dimeric [{Ir(H)[(R)-SegPhos]}2(μ-I)3]+I− complexes have been utilised in the presence of the Lewis acid titanium(IV) isopropoxide, molecular iodine, and p-toluenesulfonic acid as additives to activate the catalyst. This particular catalysis system proved to be effective in producing various enantiomerically pure tetrahydroisoquinoline alkaloids including the pharmaceutical Solifenacin, in high ee (Fig. 3).
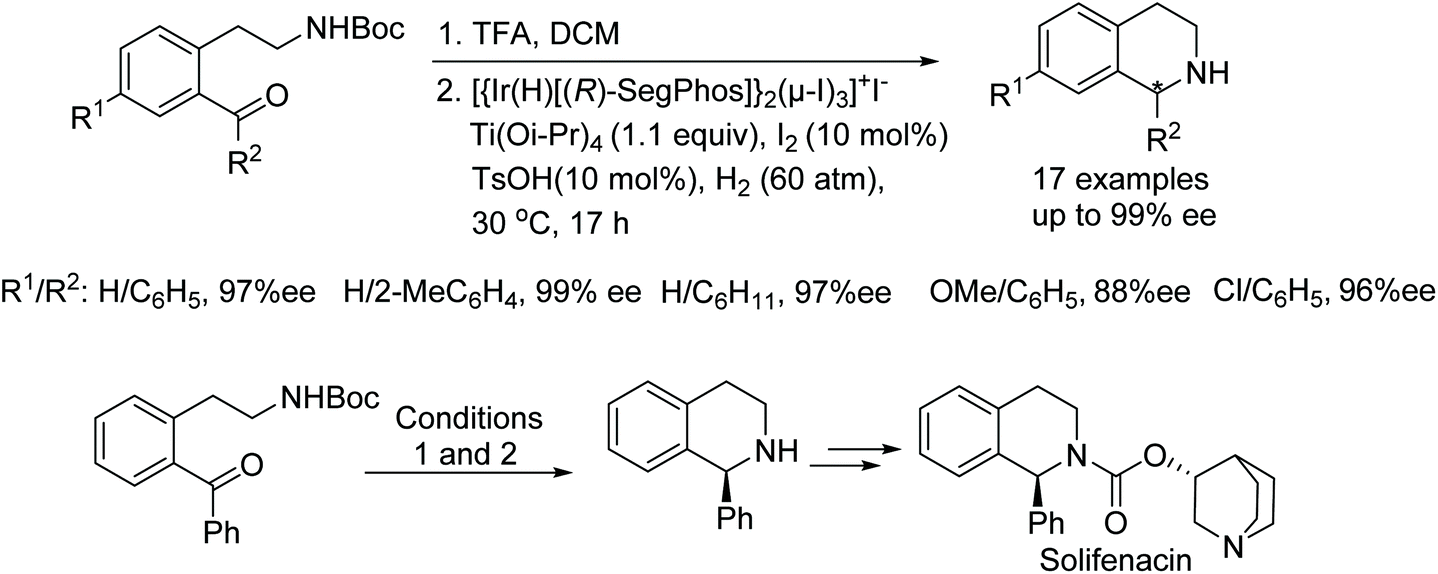 | ||
| Fig. 3 Reductions of 1-aryl-3,4-dihydroisoquinolines and enantioselective synthesis of Solifenacin. | ||
In another recent study on the AH of 1-aryl-3,4-dihydroisoquinolines, the Josiphos ligand 3 was shown, by Zhang et al. to exhibit turnover numbers of up to 4000 and with excellent enantioselectivities (up to 99% ee) in the AH of 1-phenyl-3,4-dihydroisoquinoline. AH of a library of isoquinolines substrates as depicted in Fig. 4, where R2 is a substituted aryl, gave products in 85–99% ee. The addition of HBr was essential for high reactivity and enantioselectivity in this reaction. The procedure was also applied at the gram-scale hydrogenation to yield the asymmetric precursor of Solifenacin.6
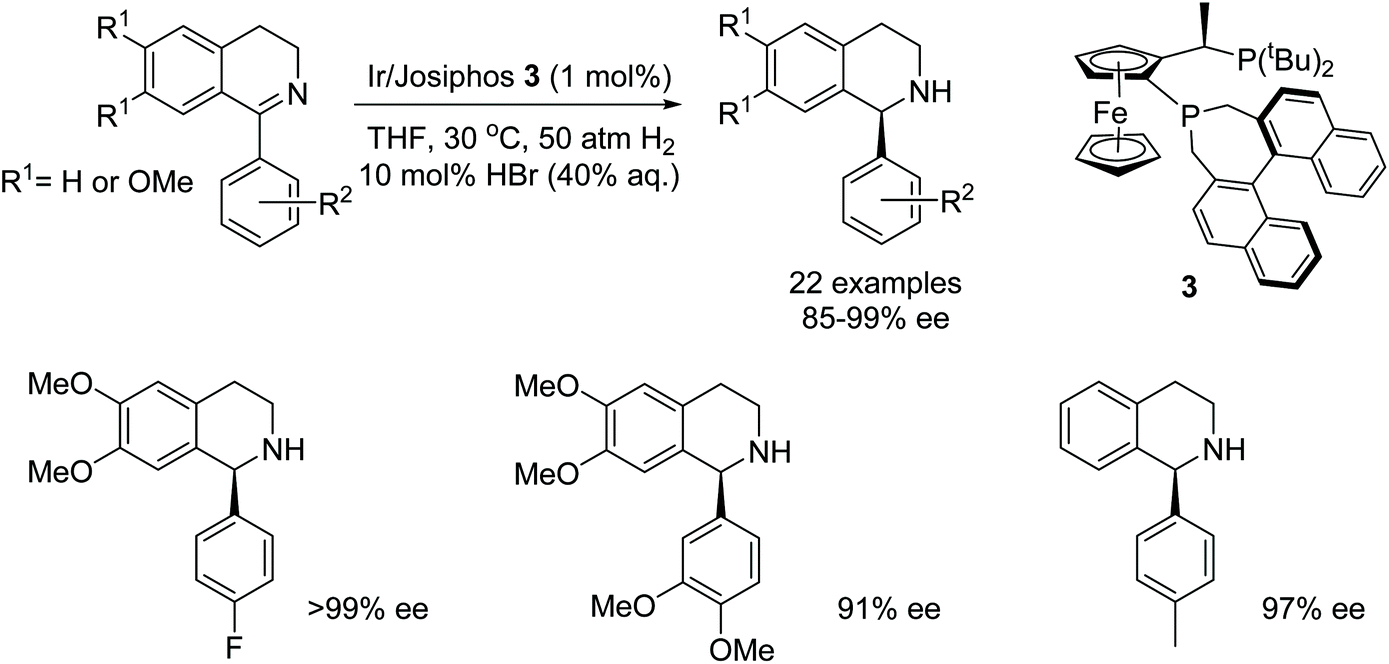 | ||
| Fig. 4 AH of 1-aryl-3,4-dihydroisoquinolines by Ir-Josiphos 3 complex. | ||
Schwenk and Togni reported the applications of P-chiral diphosphine ligands containing a trifluoromethyl substituent in the Ir(I)-catalysed asymmetric hydrogenation of dihydroisoquinoline hydrochlorides. The use of the hydrochlorides was important for high selectivity, with products formed in up to 96% ee.7
A one-pot N-Boc deprotection/intramolecular asymmetric reductive amination to synthesise chiral tetrahydroquinolines and tetrahydroisoquinolines has been demonstrated. This Brønsted acid protocol utilises Ir/ZhaoPhos catalytic system to furnish wide range of chiral tetrahydroquinolines and tetrahydroisoquinolines in excellent ees (Fig. 5).8
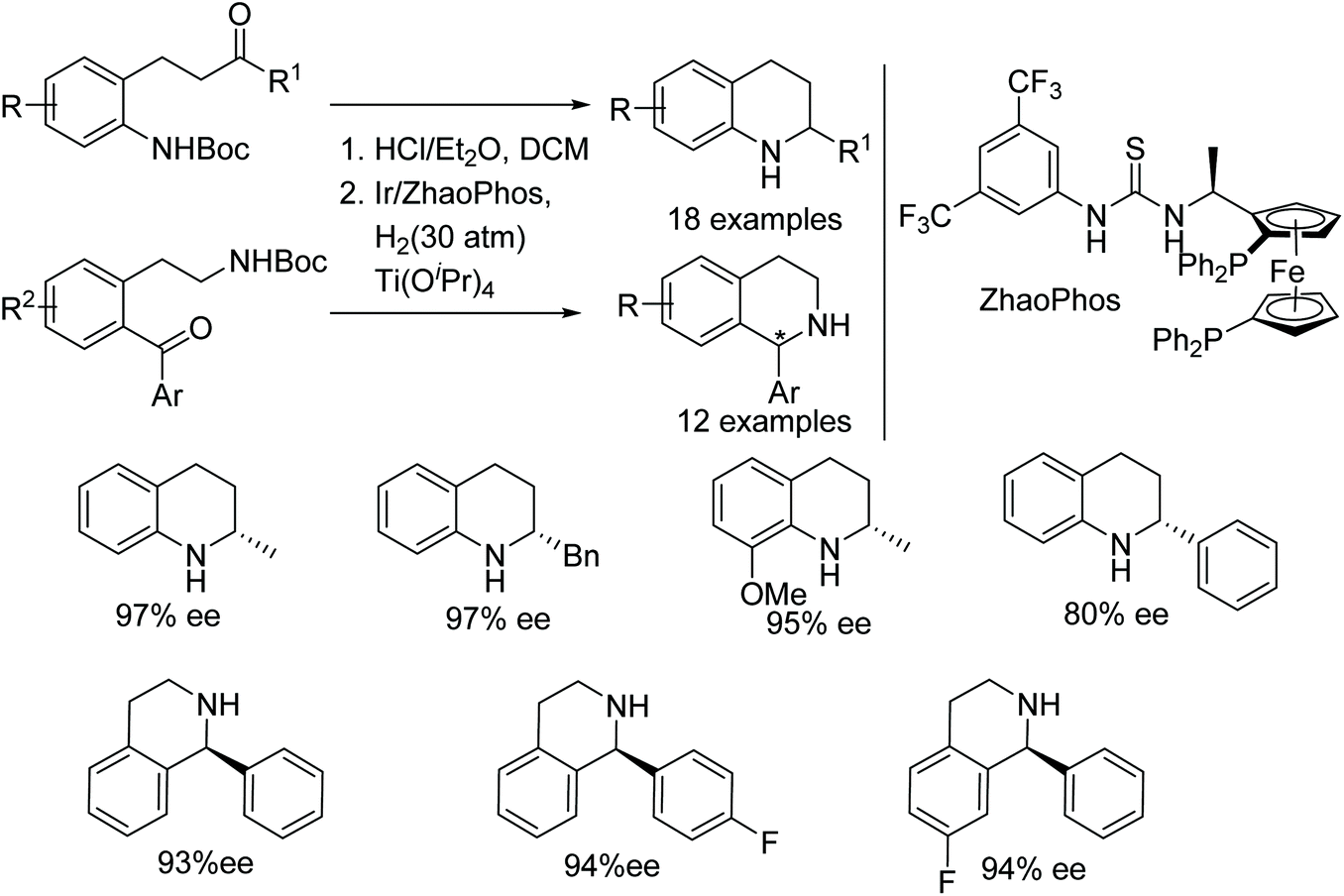 | ||
| Fig. 5 Intramolecular asymmetric reductive amination. | ||
After publishing a successful AH on cyclic imines with Josiphos-type binaphane ligands, Zhang et al. applied the same catalytic system to the AH of acyclic aromatic N-arylimines.9 A variety of acyclic imines were hydrogenated to the corresponding optically active amines in good to excellent ee. The utility of this protocol has been demonstrated by synthesizing a key chiral intermediates to calcium sensing receptor modulators on a gram scale (Fig. 6).
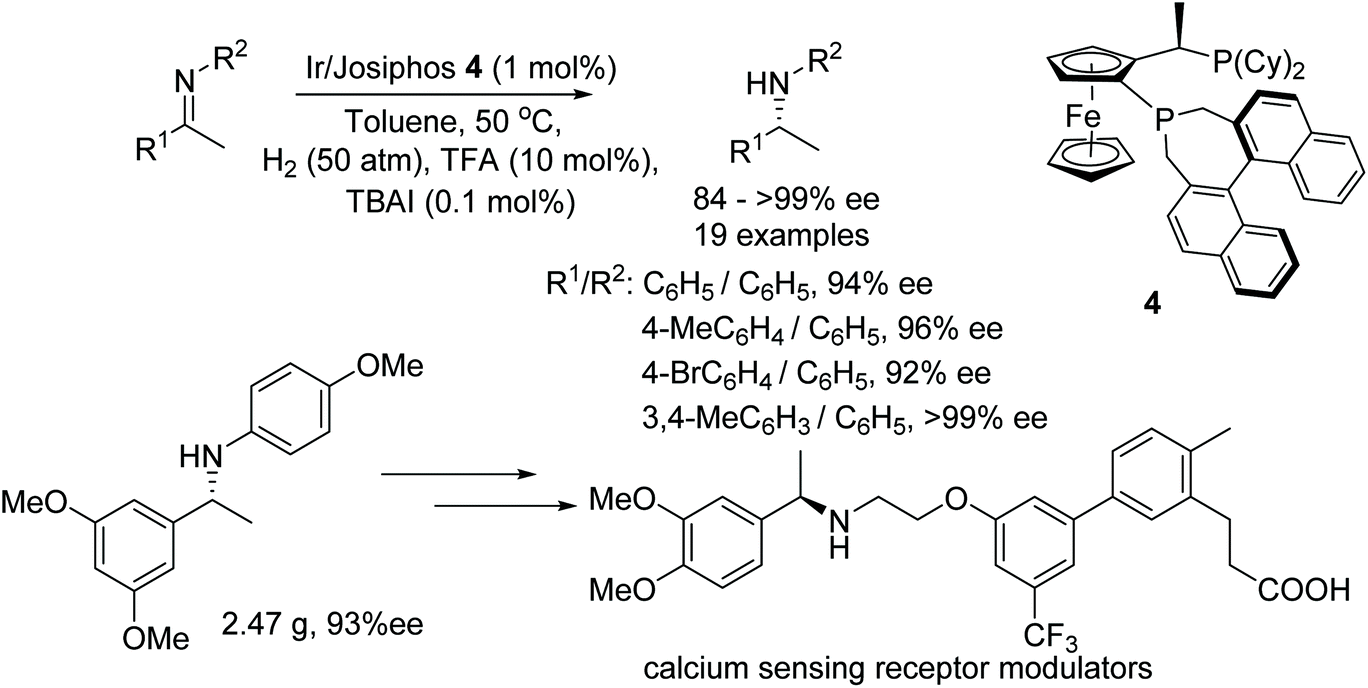 | ||
| Fig. 6 AH of acyclic aromatic N-arylimines and synthesis of optically active calcium sensing receptor modulators. | ||
Johansson et al. investigated the asymmetric reduction of α-arylfuryl-containing imines.10 Initial studies found the ATH of α-phenylfuryl-containing imines with Noyori-type catalysts gave the amine products in 60–74% ee. By contrast, pressure hydrogenation using an Ir-based catalyst with a diphosphine ligand, f-BINAPHANE 5, resulted in formation of the asymmetric amine product in 82% ee. Upon finding the optimum catalyst and conditions, a range of N-methyl imines with varying groups on the aryl functionality were reduced with 80–90% ee to the corresponding amine products (Fig. 7). A separate study revealed that the N-alkylated group significantly affects the ee of the product, where either distal branching or longer-chained alkyl groups slightly decreased the enantioselectivity when compared to its N-methylated derivative. However, the greatest effect on ee was with proximal branching where an i-Pr chain gave the amine product in 34% ee. The reduction products could potentially undergo oxidative cleavage of the furyl moiety to provide non-naturally occurring amino acids.
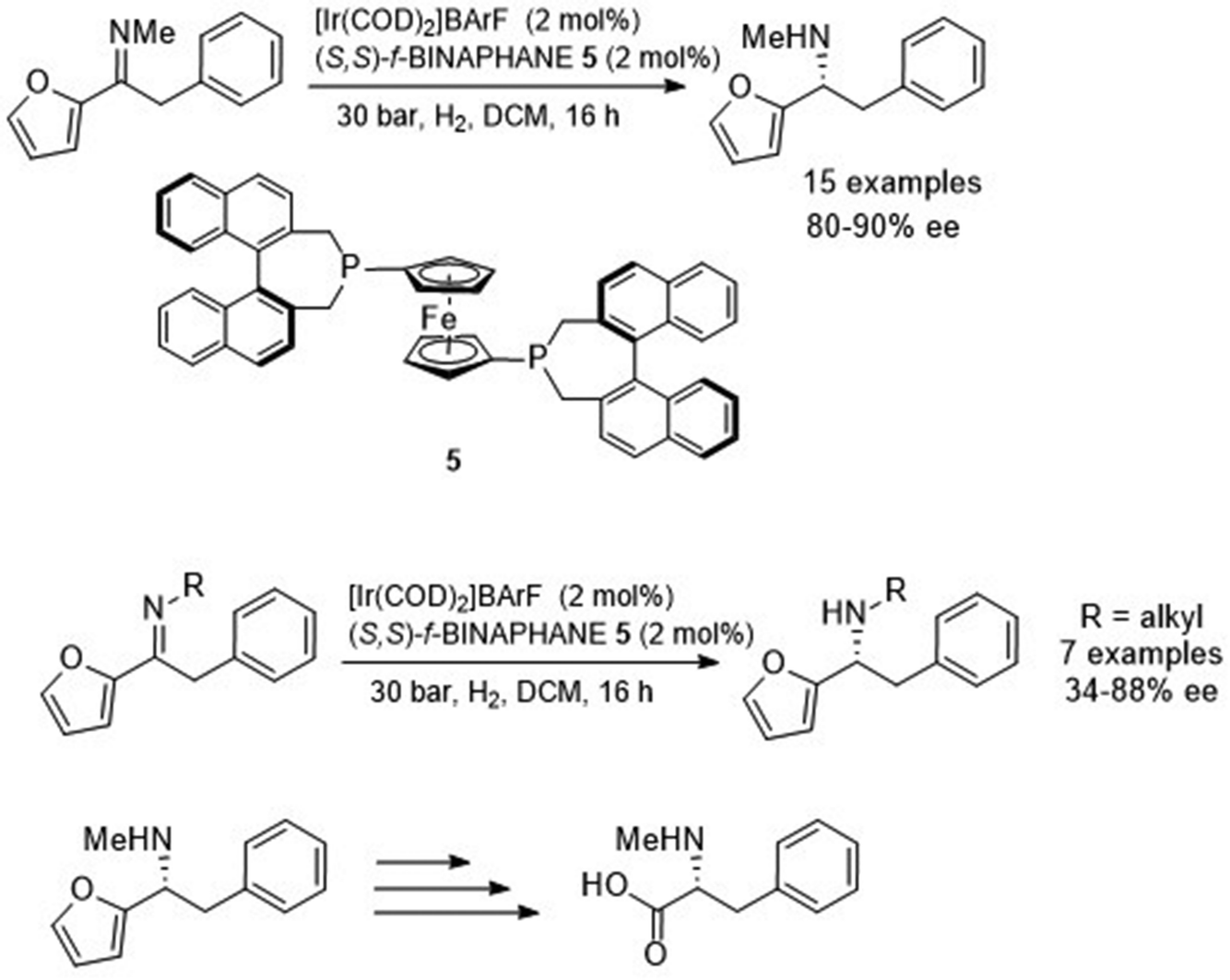 | ||
| Fig. 7 Asymmetric reduction of α-arylfuryl-containing imines and conversion into a chiral amino acid by oxidative cleavage. | ||
In another application of BINAPHANE 5, Zhou et al. have found a convenient method for the iridium-catalysed asymmetric hydrogenation of 4,6-disubstituted 2-hydroxypyrimidines in high ee (Fig. 8).11 Both electron-donating and electron-withdrawing groups could be tolerated on the substituted 2-hydroxypyrimidines, with the method giving products in up 96% ee. In the reaction, the presence of TCCA was suggested to favour the oxo form, from the lactam–lactim tautomerism, of the 2-hydroxypyrimidines which in turn facilitates the hydrogenation. Additionally the methodology was applied to the more challenging trisubstituted pyrimidines. Under the conditions, the substrates undergo partial hydrogenation to give 3,4-dihydropyrimidin-2-(1H)-ones in up to 83% ee.
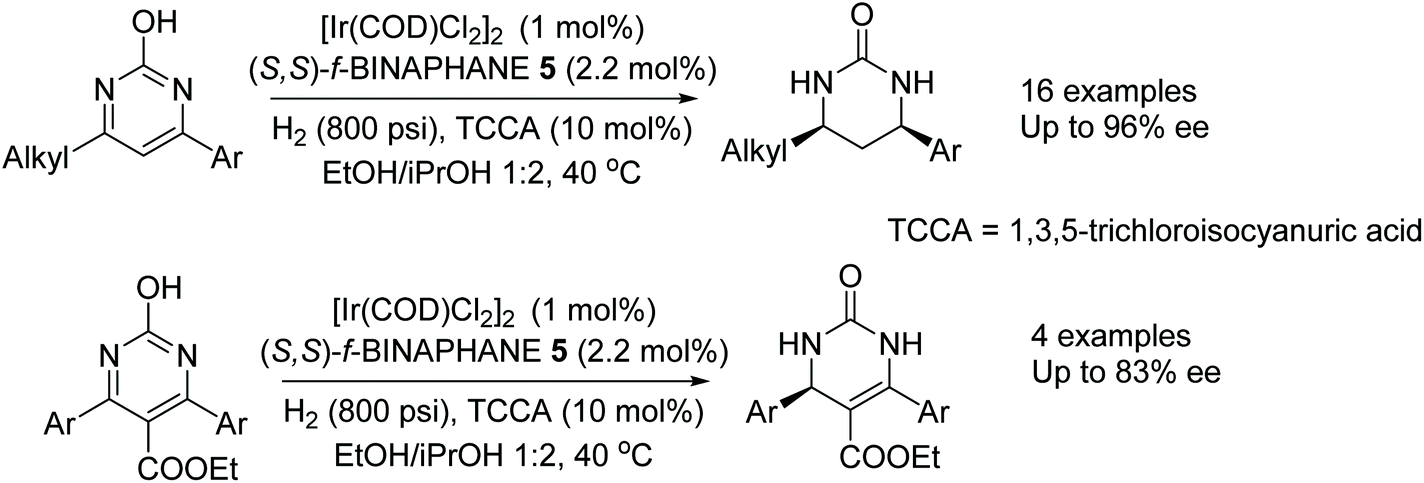 | ||
| Fig. 8 Iridium-catalysed asymmetric hydrogenation of 4,6-disubstituted 2-hydroxypyrimidines. | ||
Hou and co-workers have demonstrated an additive free protocol for the synthesis of optically active cyclic amines.12 This methodology involves the use of an iridium precursor with (R,R)-f-SpiroPhos ligand 6 as a catalyst-ligand combination to effect asymmetric hydrogenation of a variety of cyclic 2-aryl imines under mind conditions. Through this approach, optically active free amines can be produced in high yields and enantioselectivities (Fig. 9). This methodology was further applied to the synthesis of biological active molecular skeleton, (+)-(6S,10bR)-McN-4612-Z.
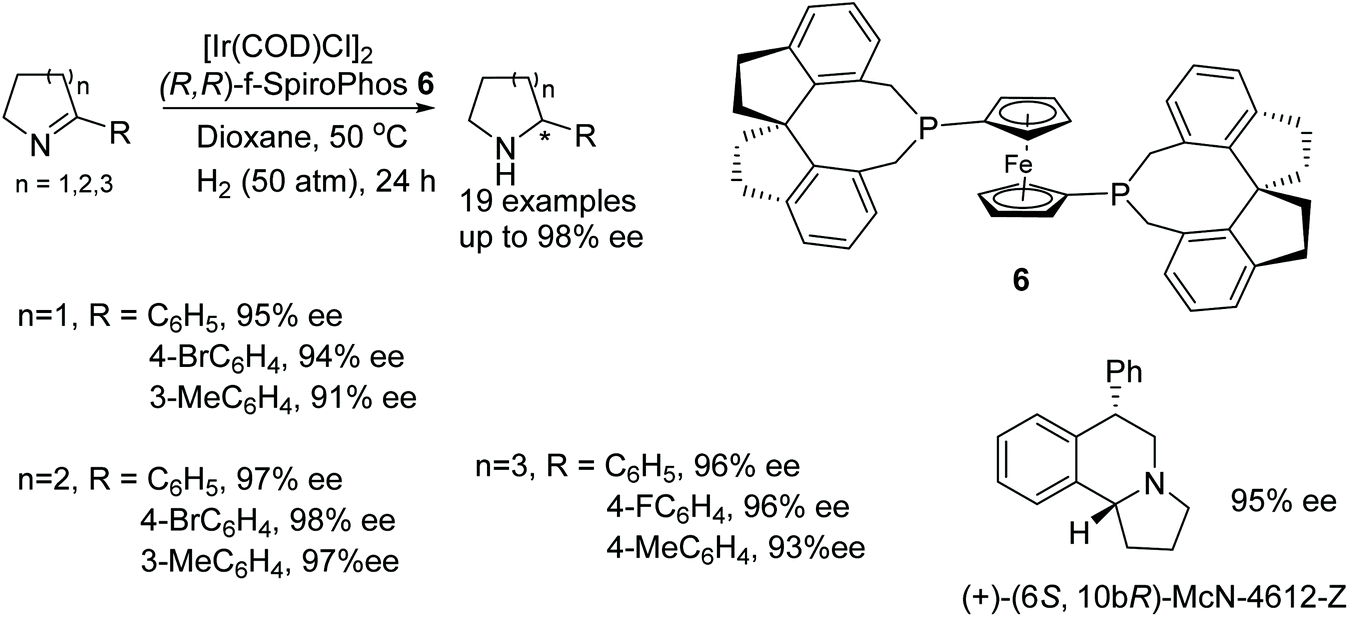 | ||
| Fig. 9 Additive free protocol for the synthesis of chiral cyclic amines. | ||
Hou and co-workers also developed an additive free Ir-f-SpiroPhos 6-catalysed reductive amination methodology to obtain chiral cyclic amines. A variety of chiral cyclic amines synthesised through intramolecular reductive amination of N-Boc-protected amino ketones were formed in excellent enantioselectivity (up to 97% ee).13 Further, the method was applied in the synthesis κ-opioid receptor selective antagonist (Fig. 10).
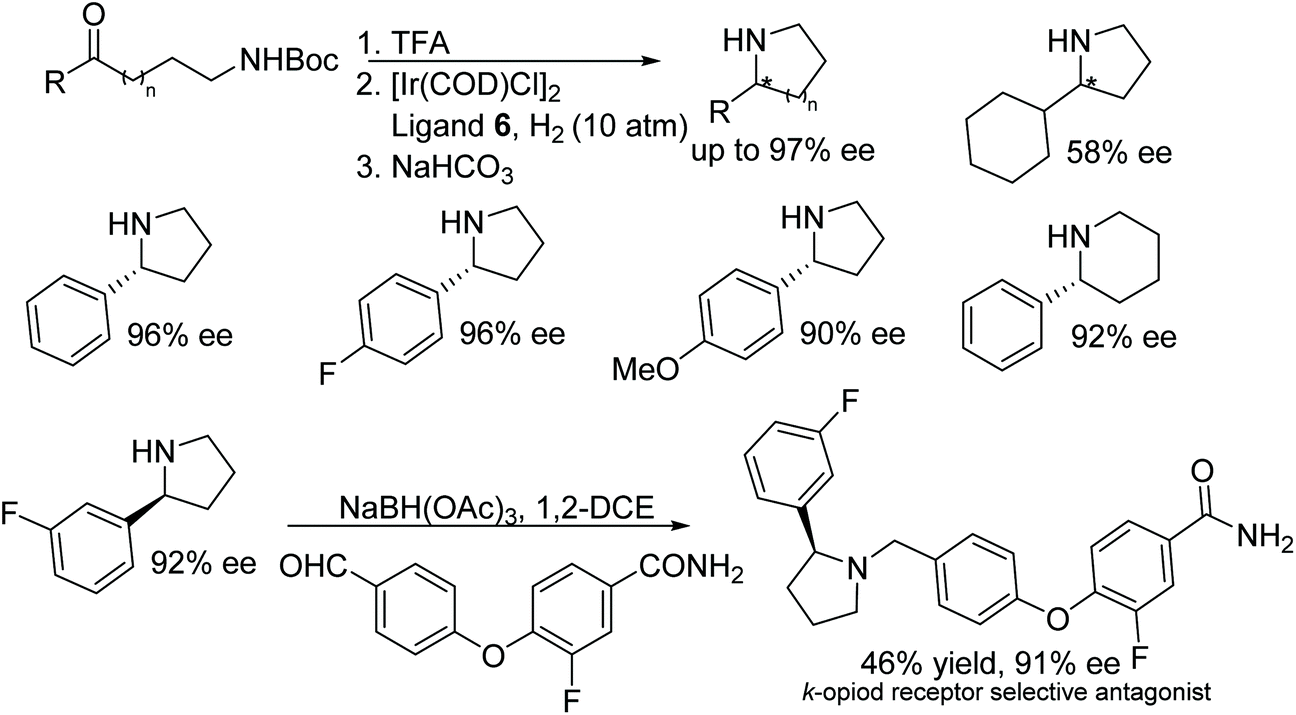 | ||
| Fig. 10 Intramolecular asymmetric reductive amination and application to a pharmaceutical target. | ||
A one pot N-Boc deprotection and intramolecular hydrogenative asymmetric reductive amination approach was developed to synthesise 2-substituted pyrrolidines. The current protocol utilises in situ generated Ir-chiral ferrocene ligand as catalyst. A wide range of chiral pyrrolidines were prepared in ee up to 92% (Fig. 11).14
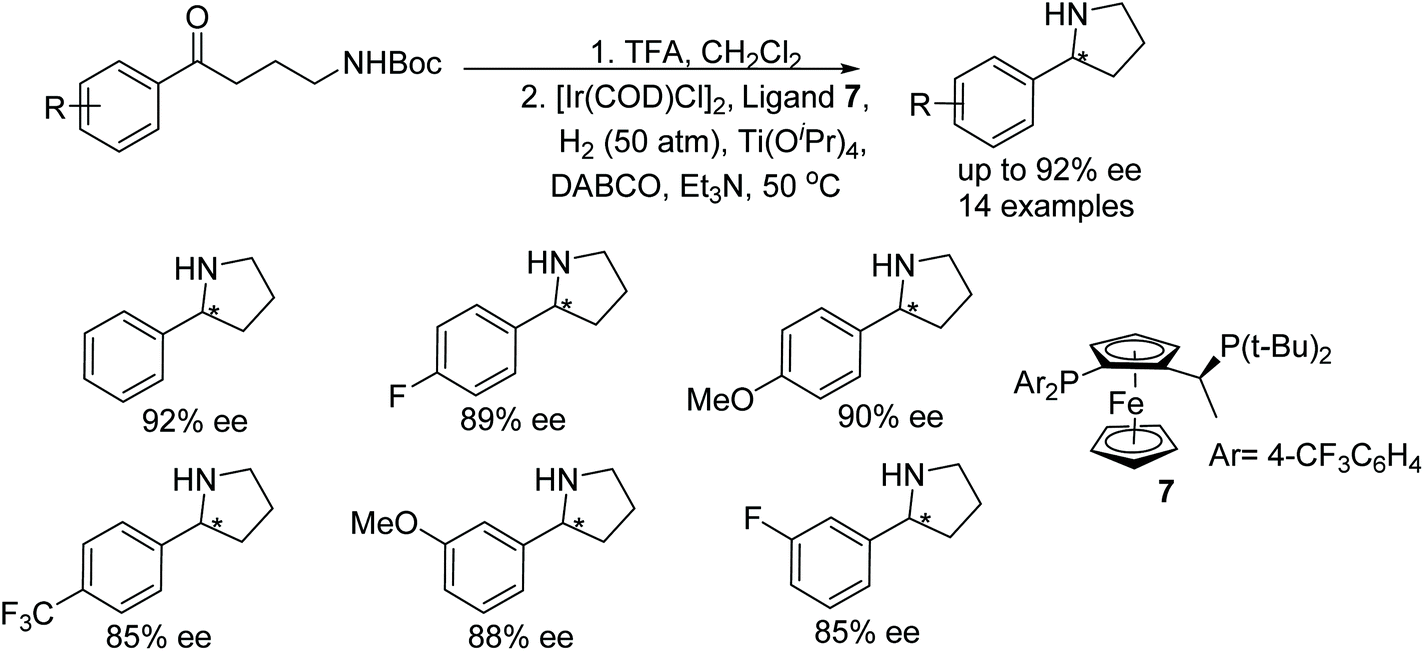 | ||
| Fig. 11 Asymmetric synthesis of 2-substituted pyrrolidines. | ||
He et al. found that diarylmethanimines could be asymmetrically hydrogenated using the f-spirophos ligand 6 in an iridium-catalysed reaction (Fig. 12).15 Excellent enantioselectivities were obtained when an ortho-substituted phenyl was present. Substrates containing 2-Cl and 2-Br substituents could also be dehalogenated after the asymmetric hydrogenation to provide phenyl containing substrates without loss in enantioselectivity. The method was applied at a gram scale and was found to hydrogenate at a lower catalyst loading when higher pressures were employed providing turnover numbers up to 4000.
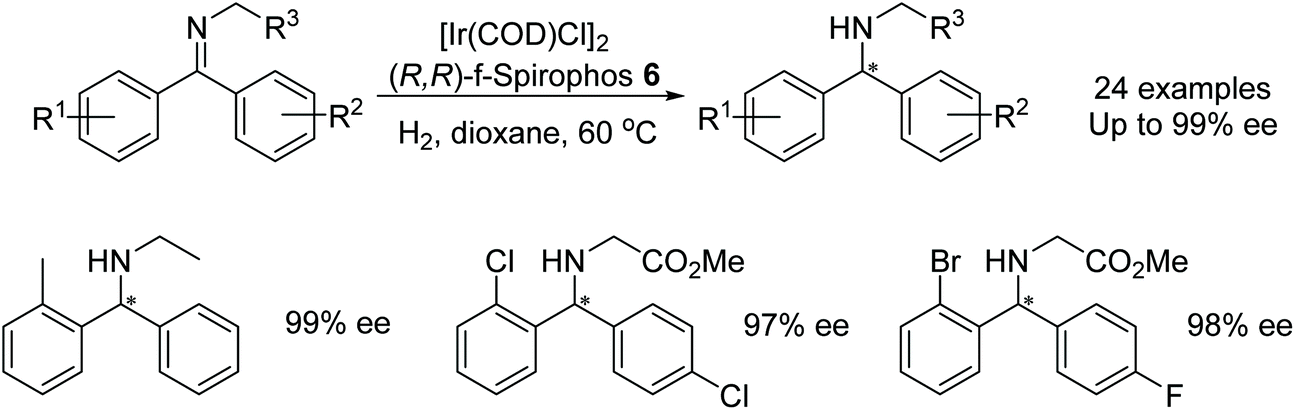 | ||
| Fig. 12 Asymmetric hydrogenation of diarylmethanimines. | ||
Quinoxaline-type compounds were reduced in iridium-catalysed AH with ligand 8 however the catalytic system proved to be reversible via a dehydrogenative rearomatization. Zhou et al. found that the reversible reaction was prevented by addition of Ac2O to afford the acetylated amine products (Fig. 13). Further application of the catalyst system to a variety of mono-substituted phenyl-quinoxaline substrates was undertaken and, when R was aromatic, reductions were achieved in 95–97% ee. The catalysts saw a loss in enantioselectivity when the R group was changed to an alkyl group, in which case, products of 37–87% ee were formed. Additionally, phenanthridine-type substrates were found to be compatible and gave the corresponding amine products in 62–98% ee.16
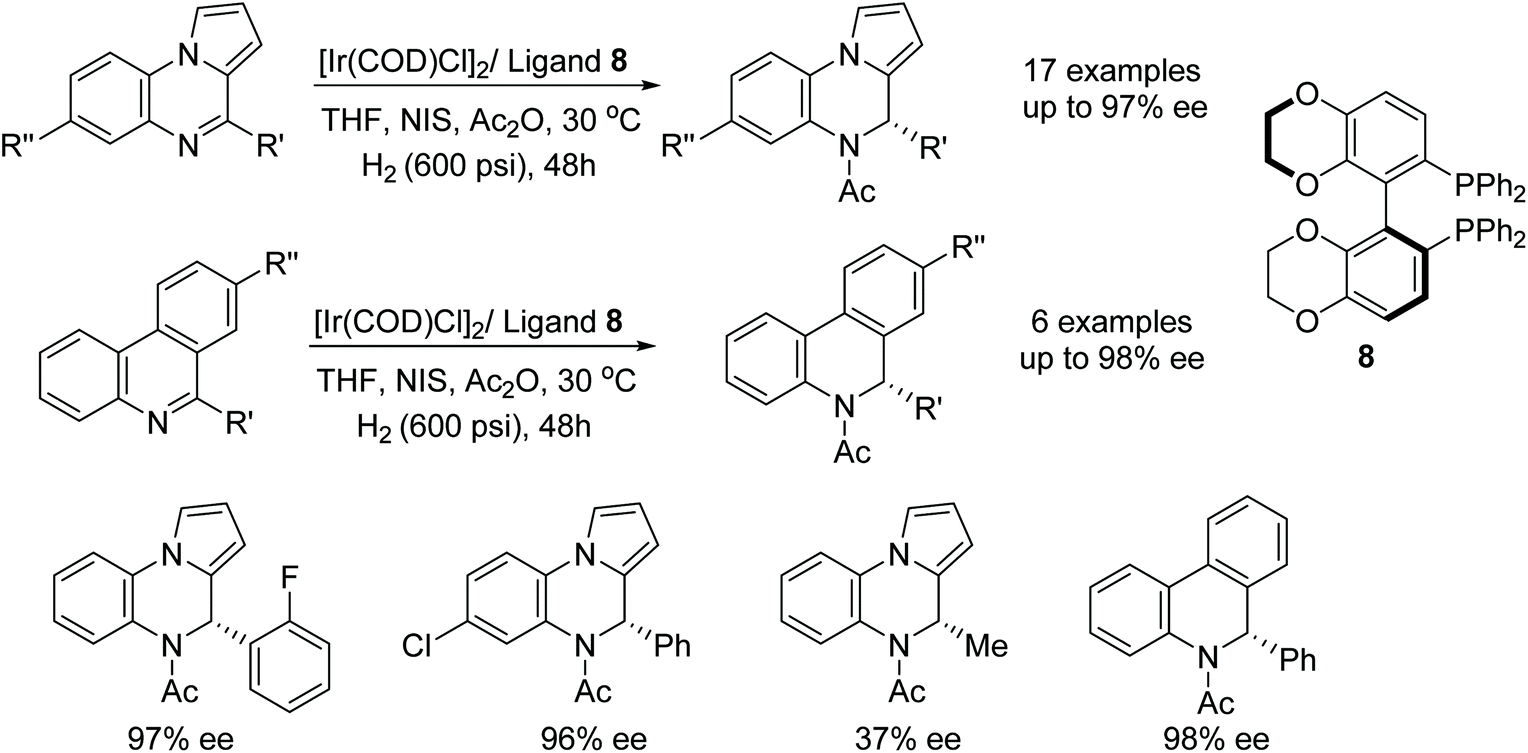 | ||
| Fig. 13 Iridium-catalysed AH of quinoxaline and phenanthridine. | ||
In a study by Verdaguer et al., four diastereomers of the ligand in complex 9a were synthesised alongside their respective complexes. AH of acetophenone-N-methyl imines with the complexes gave products in low enantioselectivities of 9–71% ee.17 Later studies revealed that the addition of the additives acetophenone-N-phenyl imine and THF yielded complex 9b which in turn provided a greater enantioselectivity of 91% ee for the reduction product of acetophenone-N-methyl imine. Catalyst 9b was found to work effectively at low pressures; 3 bar of H2 gas, and low temperatures of −10–0 °C. A range of imines were asymmetrically reduced as shown in the scheme in Fig. 14, and the different substituents on the phenyl group of the substrates were tolerated without major changes in ee. Further applications demonstrated that different N-alkyl groups were also compatible, without significant change in product ee.
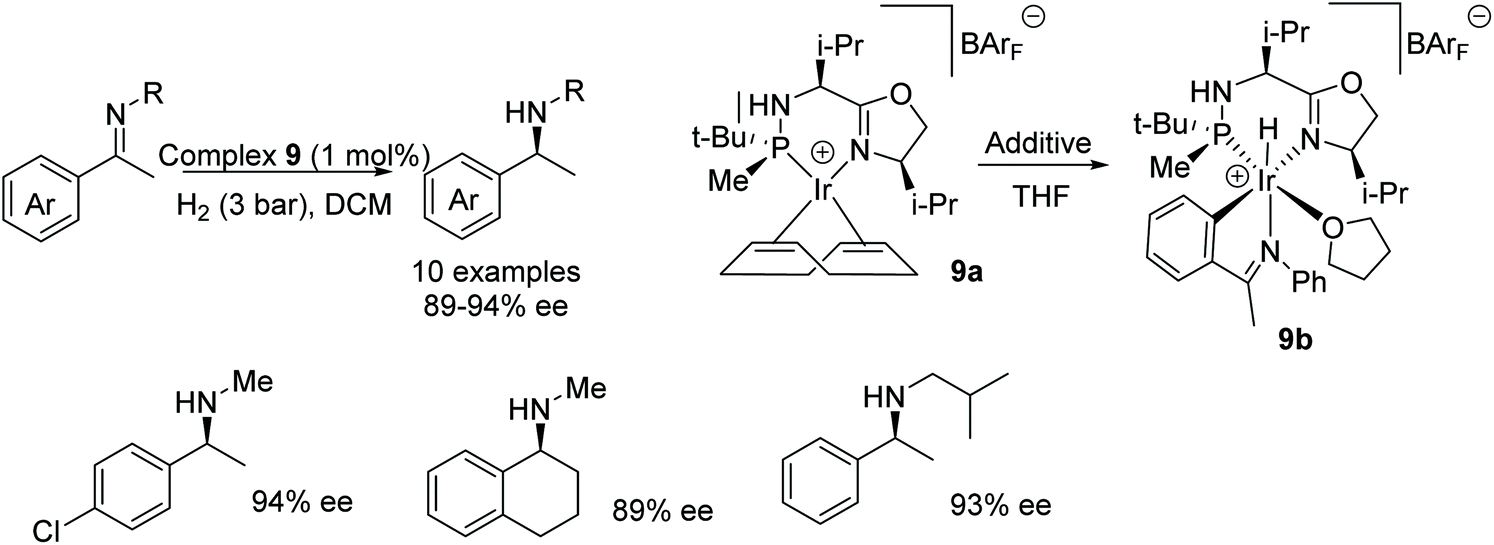 | ||
| Fig. 14 Iridium catalysed AH of acetophenone-N-alkyl imines. | ||
In a further application of 9a, the catalyst was found to be effective in the AH of acetophenone N-aryl imines. A range of substituents on the phenyl ring were found to be tolerable by the catalysts with amines formed in 74–96% ee however a substrate containing an ortho substituent was reduced in just 28% ee (Fig. 15).18
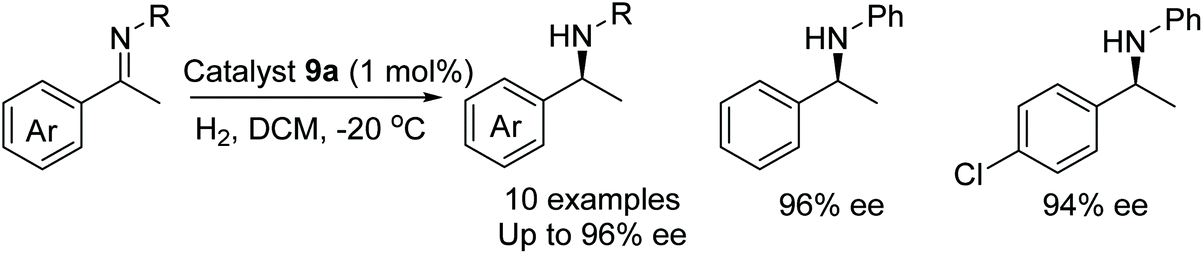 | ||
| Fig. 15 Ir-Catalysed AH using catalyst 9a. | ||
Recent advances by Zhang include the use of the N-methyated ligand 10 where the parent ligand has previously been reported as an effective ligand for Pd-based hydrogenations.19 Zhang's unmethylated derivative was unsuccessful in AH with [Ir(COD)Cl2] however the methylated derivative 10 formed effective catalysts in the AH of imines of the type shown in Fig. 16. A range of substituted phenyl (R = Ar) derivatives of these imines were reduced in 92–99% ee with naphthalene and thiophene derivatives also reduced in 97% and 96% ee respectively. The aromatic moieties in the substrates proved to be crucial when using the catalytic system; a benzyl derivative gave a lower enantioselectivity of 73%.
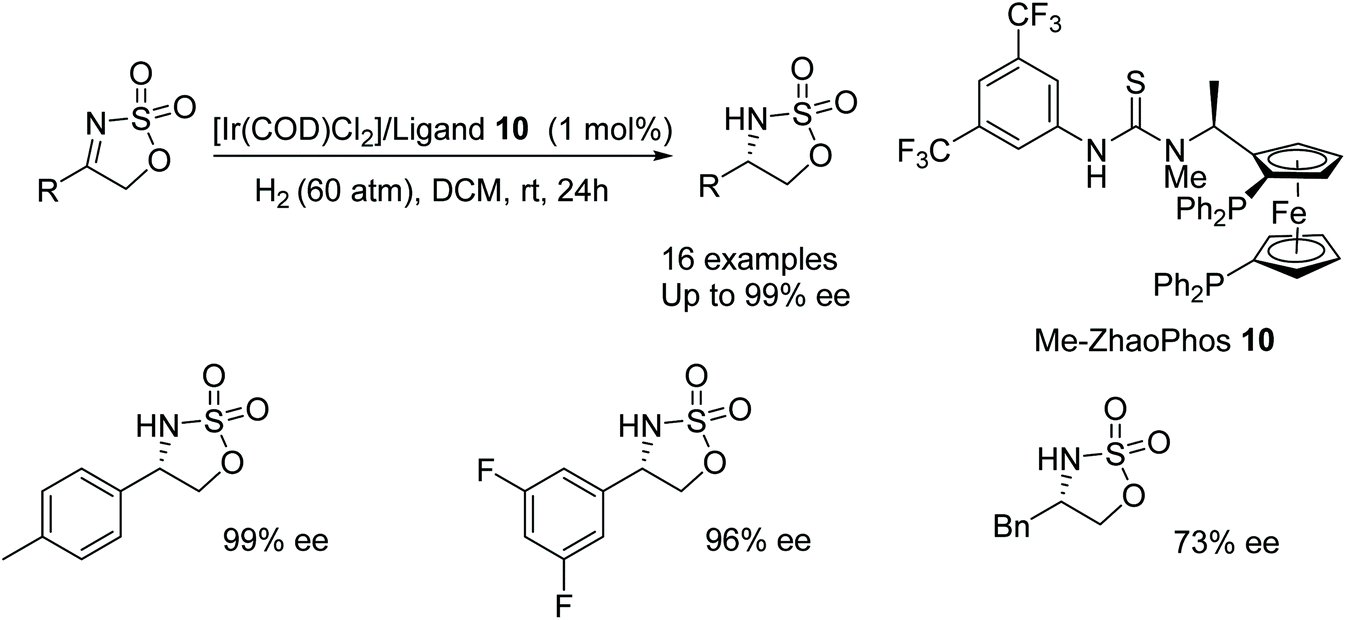 | ||
| Fig. 16 AH of cyclic sulfamidate imines. | ||
A wide range of chiral N,N′-diaryl vicinal diamines have been synthesised by Fan and co-workers.20 This ruthenium or iridium-catalysed protocol follows intermolecular reductive amination/asymmetric hydrogenation with a 2-quinoline aldehyde and aryl amines. The optically active chiral diamine could be obtained in excellent enantioselectivities and were subsequently easily converted into sterically hindered N-heterocyclic carbenes. The iridium complex 11 was found to be effective for the synthesis of non hindered chiral vicinal diamines with high enantioselectivities whereas the ruthenium-catalysed complex 12 was found suitable for the synthesis of hindered vicinal chiral diamines (Fig. 17). Furthermore, the utility of the process was demonstrated by applying the optically active chiral diamine in an asymmetric palladium-catalysed Suzuki–Miyaura cross-coupling reaction.
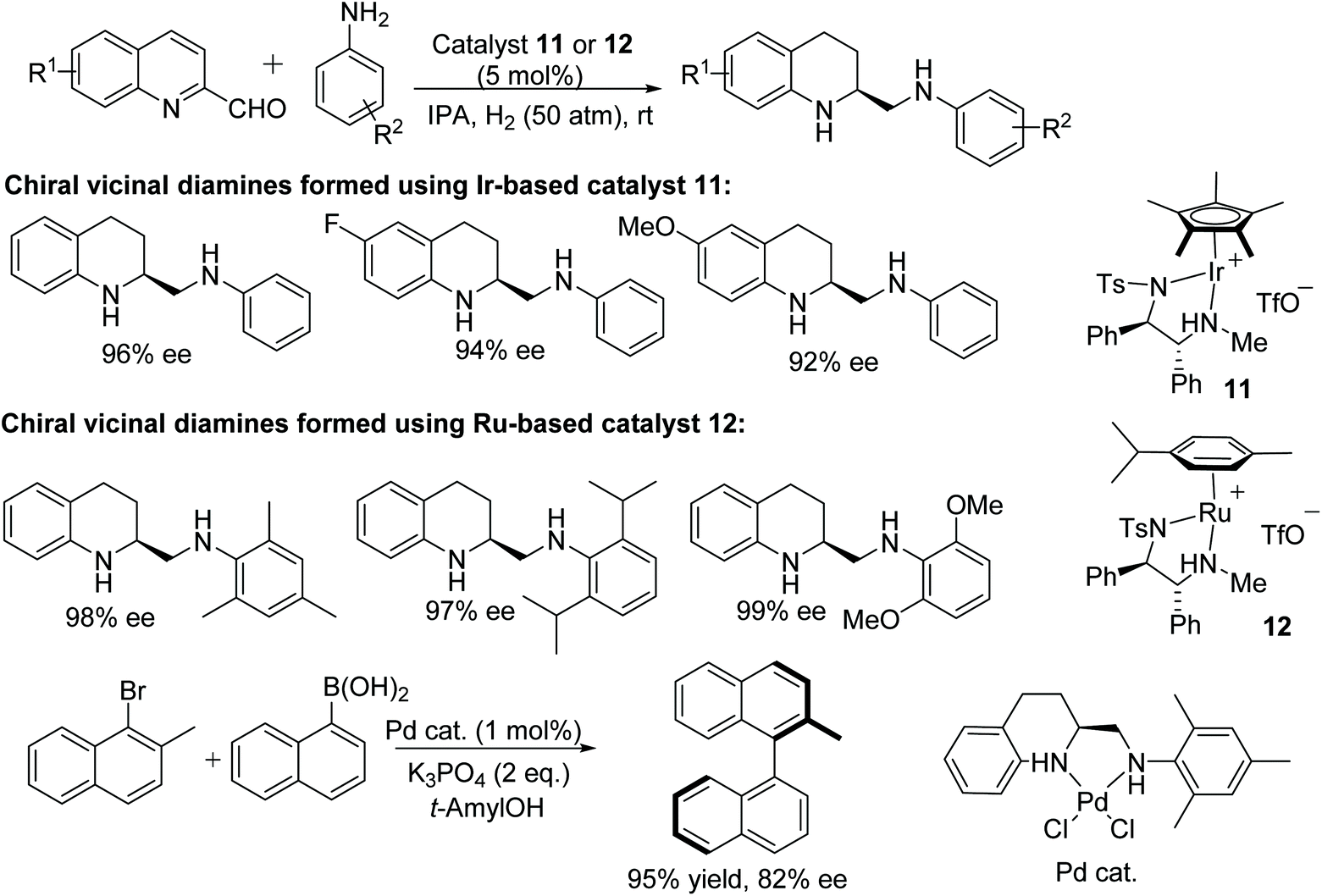 | ||
| Fig. 17 Synthesis of chiral N,N′-diaryl vicinal diamines by AH and application of the diamine product in Suzuki coupling. | ||
A facile and efficient approach to the hydrogenation of 2,4-diaryl-3H-benzo[b]azepines has been developed by Fan and co-workers.21 Dendritic phosphinooxazoline iridium complexes have been applied to synthesise optically active 2,4-diaryl-2,3-dihydro-1H-benzo[b]azepine derivatives (Fig. 18). This chemoselective and enantioselective approach converts the imine into the corresponding product in full conversions with good to excellent enantioselectivities without affecting the CC bond in the structure.
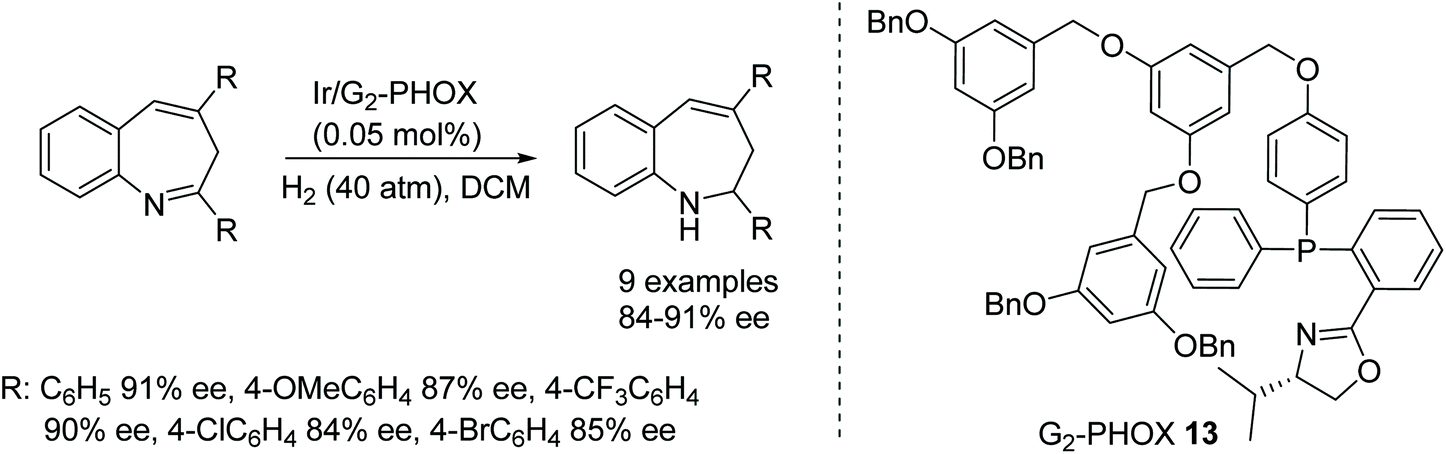 | ||
| Fig. 18 Chemoselective and enantioselective AH of 2,4-diaryl-3H-benzo[b]azepines. | ||
An additional study into CN reductions has been carried out using DFT calculations to investigate the mechanism of hydrogenation for the catalyst system shown in Fig. 19. The results suggest that the iridium catalysed reaction proceeds through an outer-sphere pathway.22
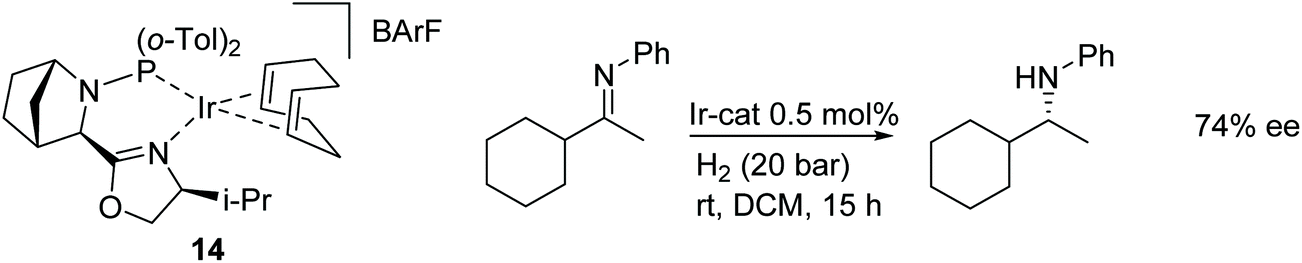 | ||
| Fig. 19 Iridium precatalyst with phosphine-oxazoline ligand for AH of N-alkylated imines. | ||
Yang et al. have applied a similar approach of N-Boc deprotection/intramolecular asymmetric reductive amination to synthesise bridged biaryl derivatives (Fig. 20).23 The Ir-catalysed protocol furnishes synthetically important dibenz[c,e]azepines containing both centre and axial chiralities in excellent enantioselectivities (up to 97% ee). A variety of chiral dibenz[c,e]azepines have been obtained through this methodology utilising (S)-DifuorPhos and (S)-SegPhos ligands. The application of the methodology has been shown by synthesizing an analogue of allocolchicine using the ZhaoPhos ligand (Fig. 20).
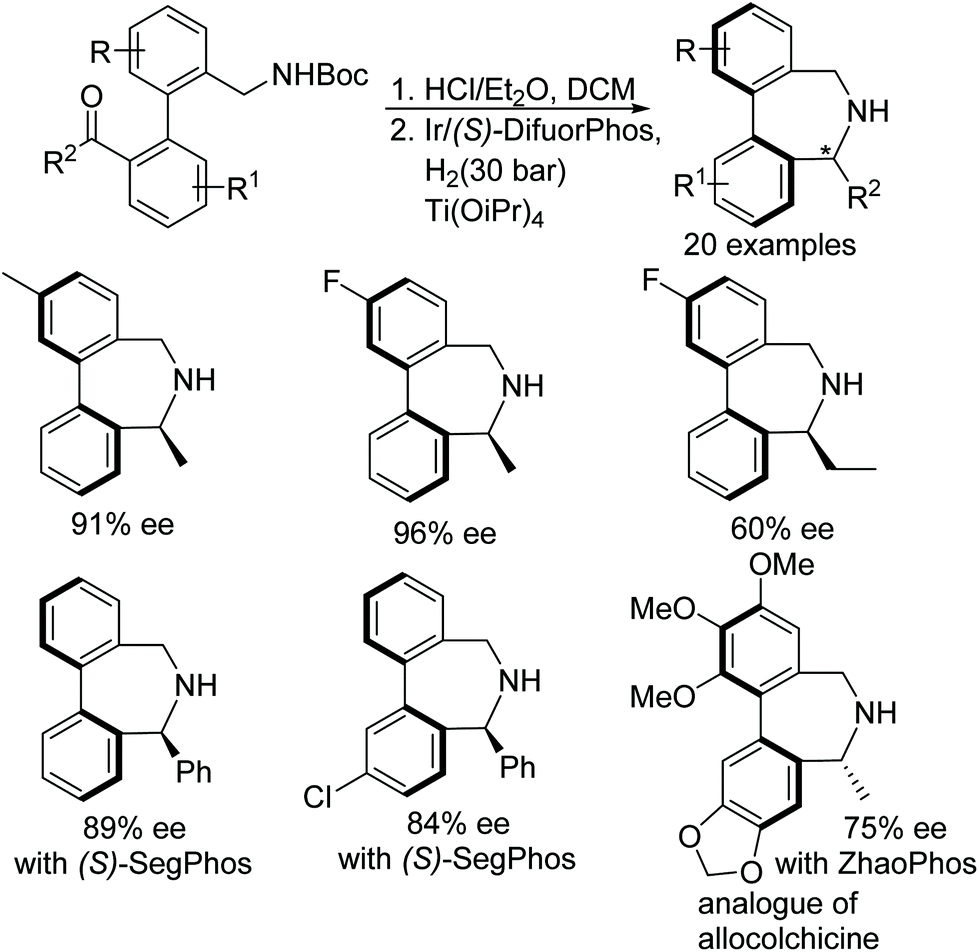 | ||
| Fig. 20 Synthesis of dibenz[c,e]azepines containing both centre and axial chirality. | ||
Chang and co-workers developed a highly efficient and novel approach to tertiary chiral amines via direct catalytic asymmetric reductive amination.24 In this protocol, secondary amines undergo direct reductive amination with ketones catalysed by Ir-phosphoramidite ligand complexes. Rivastigmine, which is used in the treatment of Alzheimer's and Parkinson's type diseases, was synthesised by this facile and scalable method (Fig. 21).
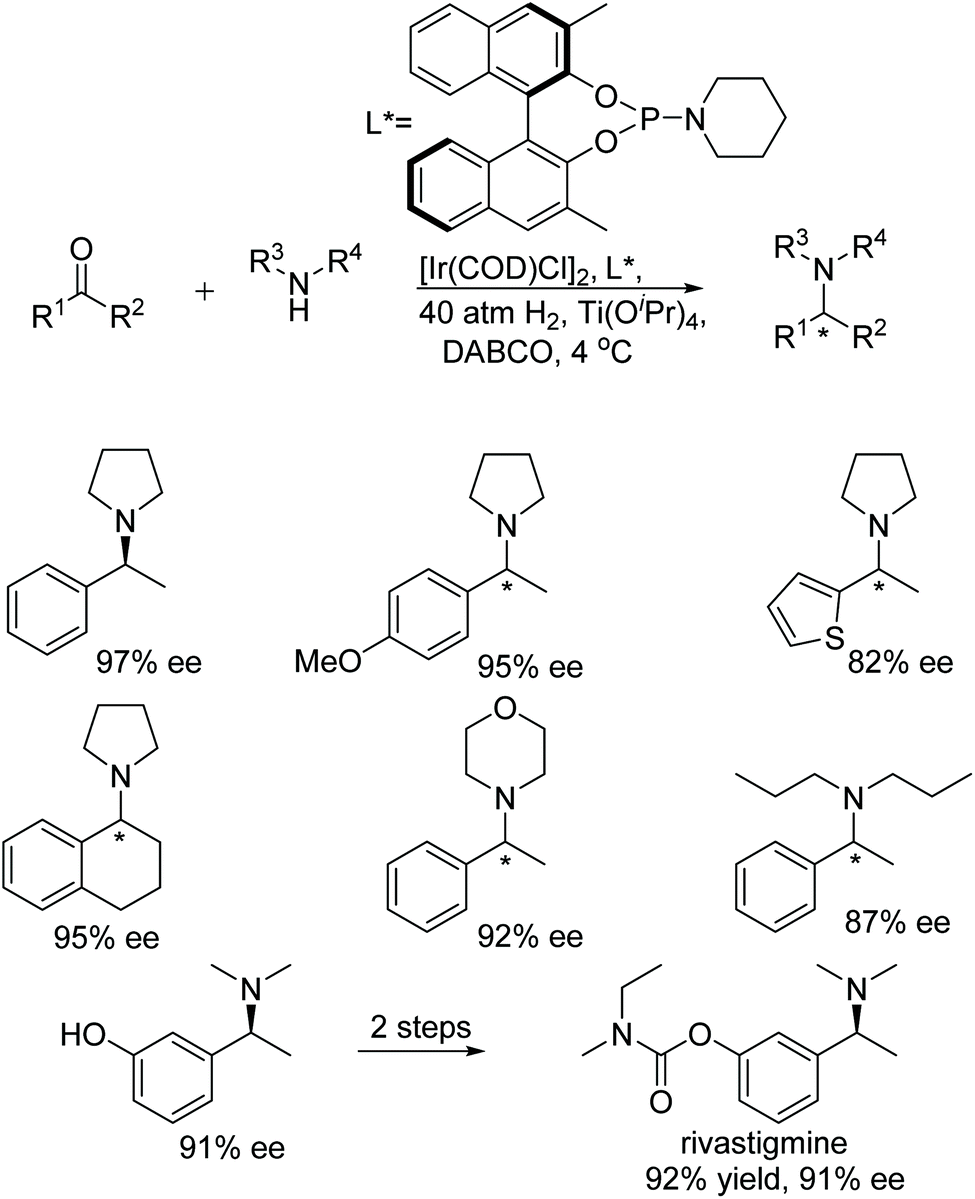 | ||
| Fig. 21 Tertiary amine synthesis via intermolecular reductive amination catalysed by Ir-phosphoramidite complexes. | ||
1-Alkyl dihydroisoquinolines have been successfully hydrogenated in high ee using Ir(I) complexes containing chiral spiro iridium phosphoramidite ligands. Two equivalents of the ligands were required, relative to Ir(I).25 Ir(I) complexes containing an (R)-synphos ligand were employed in a redox deracemisation of a range of amines, including tetrahydroisoquinolines, in up to 98% ee. In this process, NBS was used as an oxidant to create an imine intermediate in the reaction.26
2.2. Rhodium-catalysed imine AH
In a Rh-catalysed AH, a dibenzo[b,e]azepine substrate was reduced in up to 96% ee using ligand 15 (Fig. 22), whereas other ligands were less effective, e.g. SegPhos and JosiPhos which delivered products of 40% and 60% ee respectively. Zhang proposed that the thiourea moiety in ligand 15 activates the substrate via an anion-bonding interaction.27 The catalyst system was then applied to a range of substrates and it was found that when R3 was a methyl, all substrates were reduced with excellent enantioselectivities of 92–99% irrespective of the groups on R1 and R2. However, an ethyl group (R3 = Et) proved to be a poorer substrate with only a 71% ee achieved for the reduced amine product. Further application of the catalyst system using a gram-scale of substrate also proved effective. | ||
| Fig. 22 Rhodium catalysed AH of dibenzo[b,e]azepine and gram scale reduction. | ||
Rhodium-catalysed asymmetric hydrogenations of substituted 1,5-benzodiazepinones has been achieved by Rueping and co-workers (Fig. 23). The basis of the success in this protocol lies in the fruitful combination of the weakly coordinating tetrakis-[3,5-bis(trifluoromethyl)phenyl]borate (BArF) anion and ligand 16 in the rhodium-catalyzed hydrogenation thereby producing the optically active pharmacologically relevant class of dihydro-1,5-benzodiazepines in high yields and ee (up to 92% ee).28
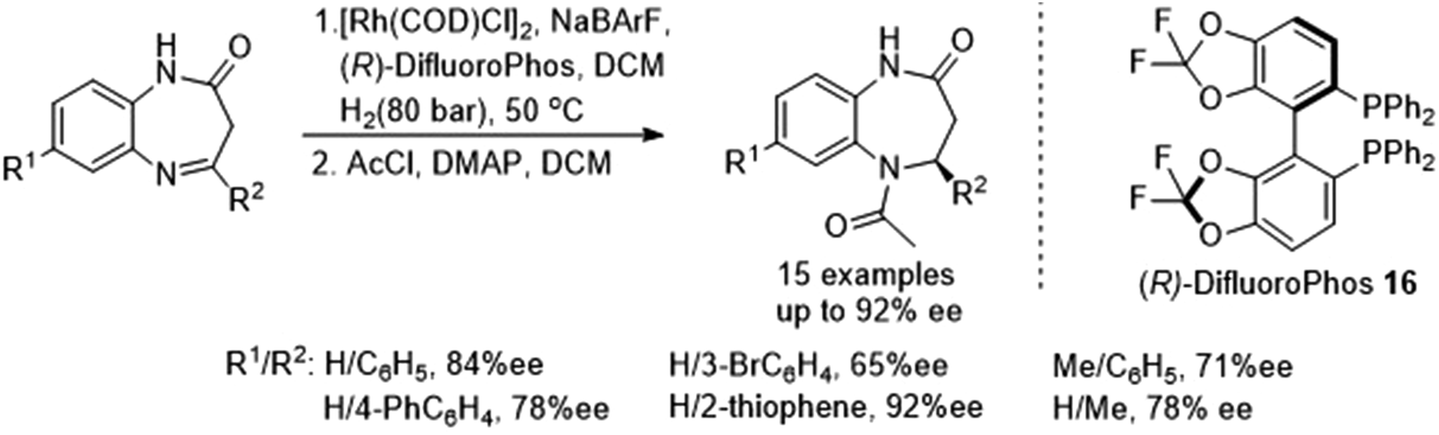 | ||
| Fig. 23 AH of substituted 1,5-benzodiazepinones. | ||
Zhang has developed a rhodium-based protocol for asymmetric hydrogenation of alkynyl-aryl hydrazones. A rhodium complex [Rh((R,Sp)-JosiPhos)(cod)]SbF6 allows access to a chemoselective and enantioselective approach to optically active propargyl hydrazines in good conversion and excellent ee (Fig. 24).29 A variety of substrates containing triple bonds were converted to chiral amines without affecting the alkyne. Furthermore, this protocol gives easy access to chemically important structure motifs, i.e. chiral propargylamines, from chiral propargyl hydrazines.
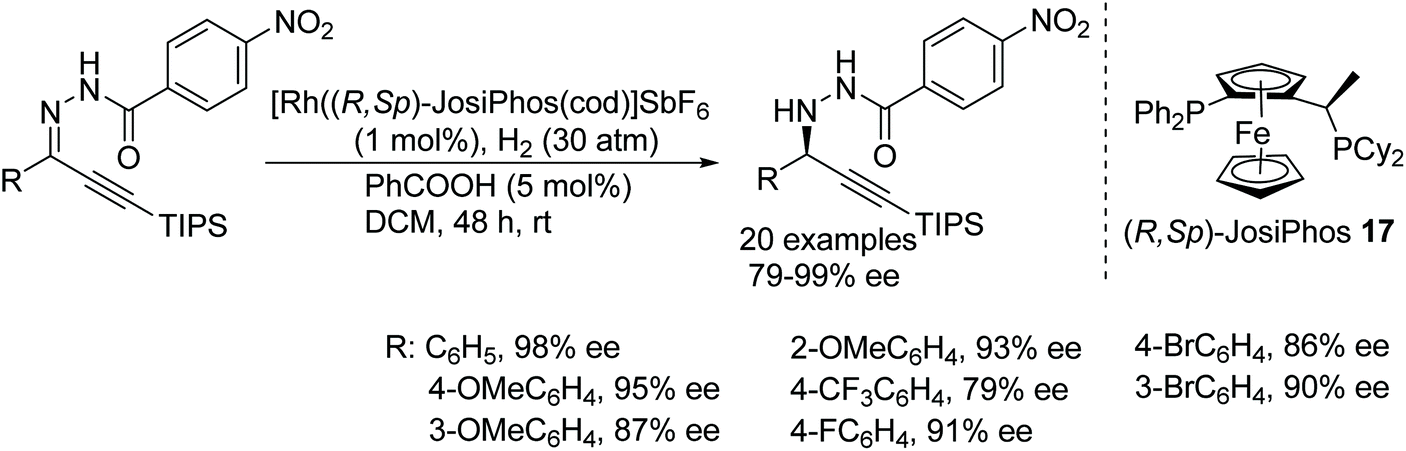 | ||
| Fig. 24 Chemoselective and enantioselective synthesis of chiral propargyl hydrazines. | ||
2.3. Ruthenium-catalysed imine AH
Kačer et al. have developed a method to asymmetrically hydrogenate dihydroisoquinolines and β-carboline-type compounds Fig. 25.30 The method was found to be compatible with a total of eight Ru and Rh half-sandwich complexes where trifluoroacetic acid is employed for substrate activation. In a separate study, catalysts 18a, b and c were found to reduce the dihydroisoquinolines and β-carboline-type compounds asymmetrically in a transfer hydrogenation reaction using formic acid–triethylamine. A kinetic study comparing the ATH reaction with catalysts 18a, b and c showed that the hydroxybutyl catalyst 18b, reduced imines most rapidly, followed by 18c then 18a, all of which gave similar enantioselectivities.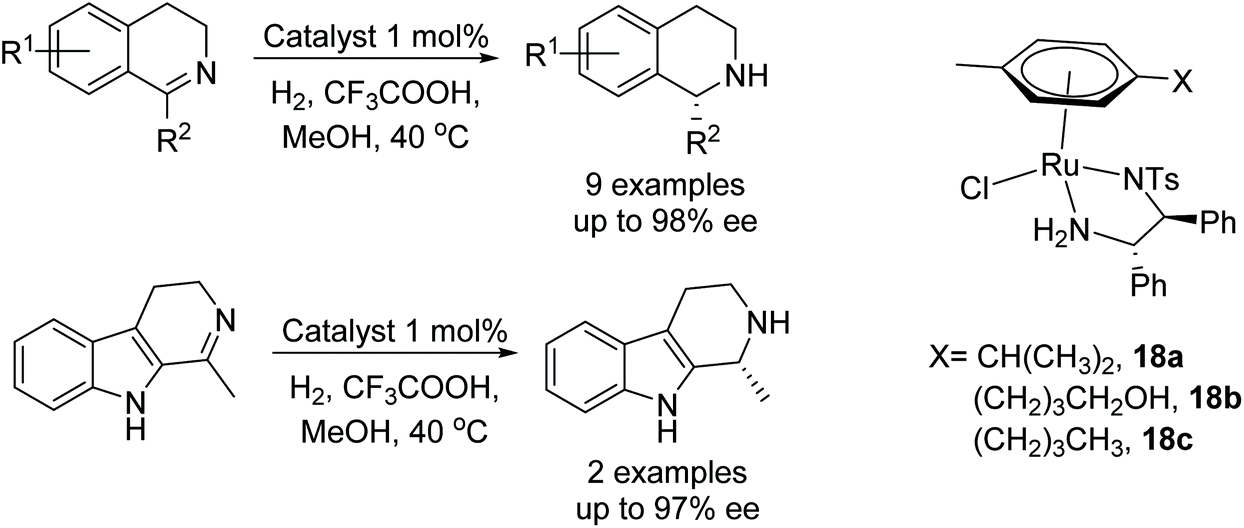 | ||
| Fig. 25 Ru-Catalysed hydrogenate dihydroisoquinolines and β-carboline hydrogenation. | ||
Biologically important pharmacophores such as benzoazepines, benzodiazepines, and benzodiazepinones have been synthesised by cationic ruthenium–diamine catalysts 18d/18e. This class of seven-membered N-containing heterocyclic imines have been hydrogenated in up to 99% ee (Fig. 26). The stereochemical outcome in this study was governed by the counter anion of the cationic complex, and complete reversal of enantioselectivity was observed when ruthenium catalyst (R,R)-18e bearing a phosphate anion was employed. Hence, all of these imines could be converted to both product enantiomers simply by changing the counteranion with the same configuration of ligand.31
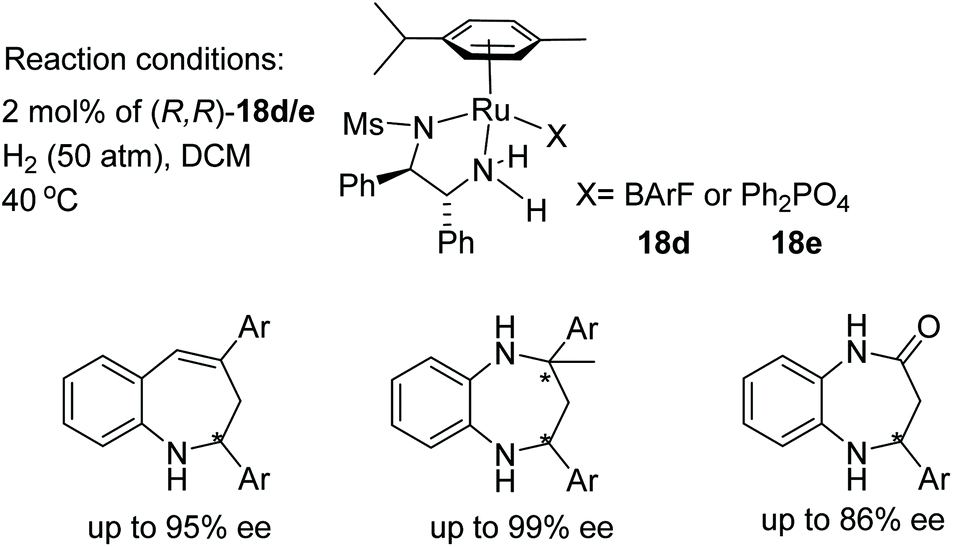 | ||
| Fig. 26 AH of seven-membered N-containing heterocyclic imines. | ||
The construction of julolidine derivatives was achieved by a ruthenium-catalysed cascade double reduction strategy. This ruthenium-catalysed protocol proceeds via enantioselective hydrogenation followed by reductive amination of 2-(quinolin-8-yl)ethyl ketones which are converted into optically active julolidines. The chiral product being obtained in excellent diastereo- and enantioselectivity. Furthermore, the methodology was applied to the preparation of the new chiral fluorescent molecular rotor (Fig. 27).32
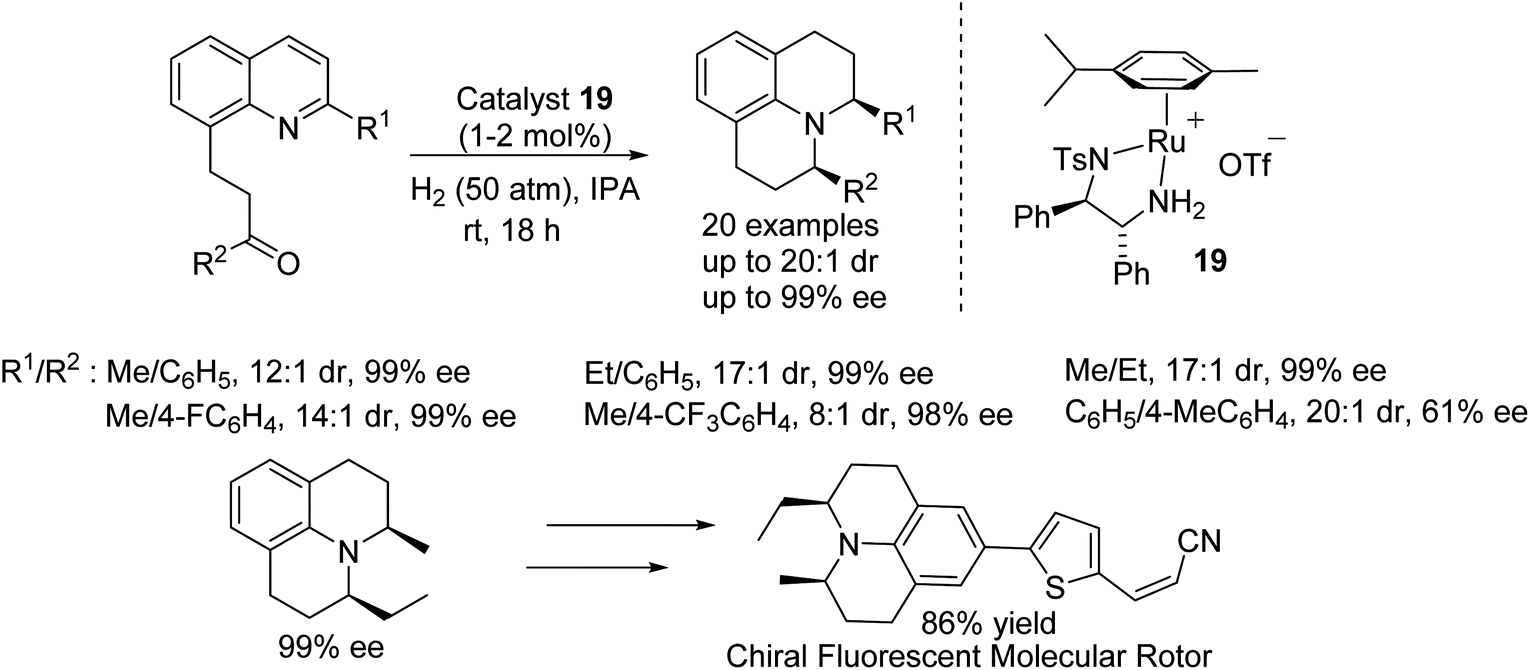 | ||
| Fig. 27 AH of 2-(quinolin-8-yl)ethyl ketones and conversion of product into a chiral fluorescent molecular rotor. | ||
Chiral cationic ruthenium complexes have been applied to the AH of bis(quinolin-2-yl)methanes to synthesise optically active 1,3-diamines.33 The corresponding chiral bis(tetrahydroquinolin-2-yl)methanes were obtained in excellent diastereo- and enantioselectivity (Fig. 28). The given protocol gives easy access to 6-membered chiral NHC ligands from optically active 1,3-diamines which otherwise difficult to prepare by any other route.
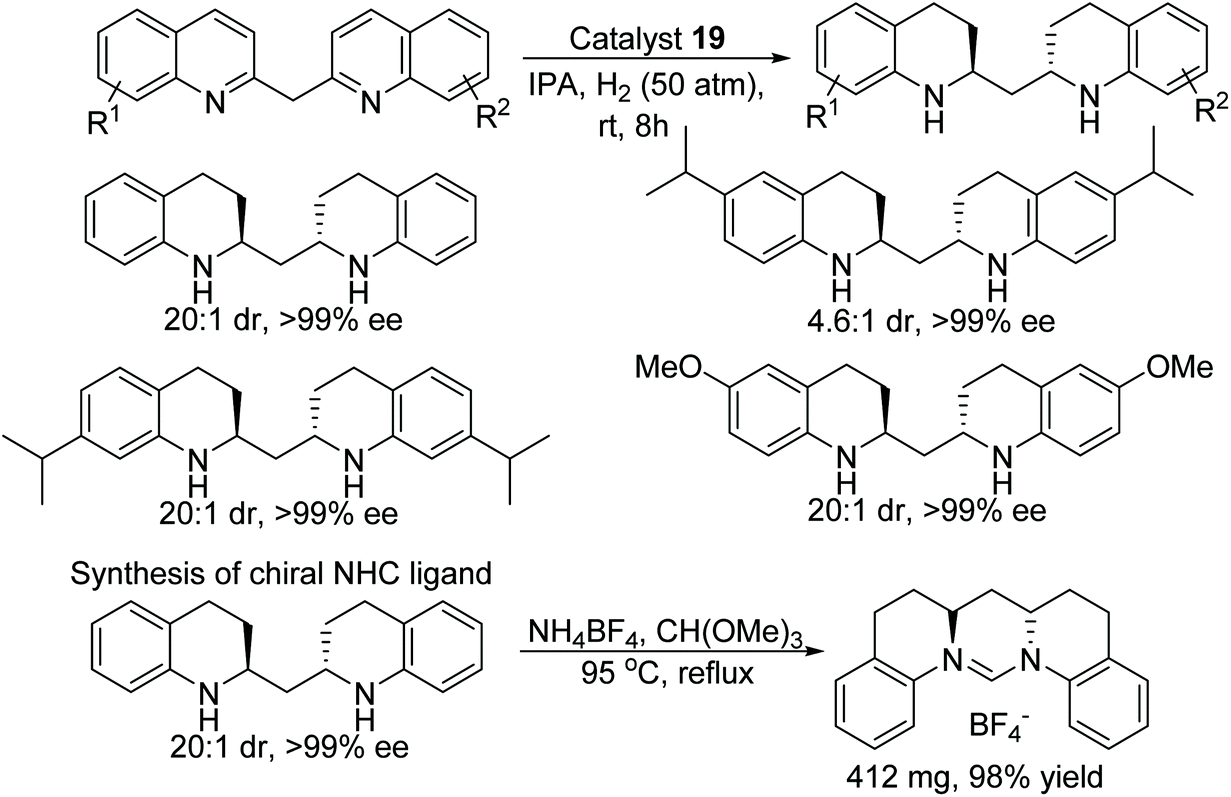 | ||
| Fig. 28 AH of bis(quinolin-2yl)methanes and subsequent conversion into chiral NHC ligand. | ||
A novel method to obtain optically pure chiral endocyclic vicinal diamines have been demonstrated by Fan and co-workers.34 In this protocol, the chiral cationic ruthenium diamine catalyst 19 has been applied to the AH of 2,2′-bisquinoline and 2,2′-bisquinoxaline derivatives to give the corresponding chiral amines in good diastereoselectivity (up to 93![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7) and excellent enantioselectivity (>99% ee) (Fig. 29). These chiral diamines provided an easy access to chiral N-heterocyclic carbenes which would otherwise be difficult to prepare by another route.
:7) and excellent enantioselectivity (>99% ee) (Fig. 29). These chiral diamines provided an easy access to chiral N-heterocyclic carbenes which would otherwise be difficult to prepare by another route.
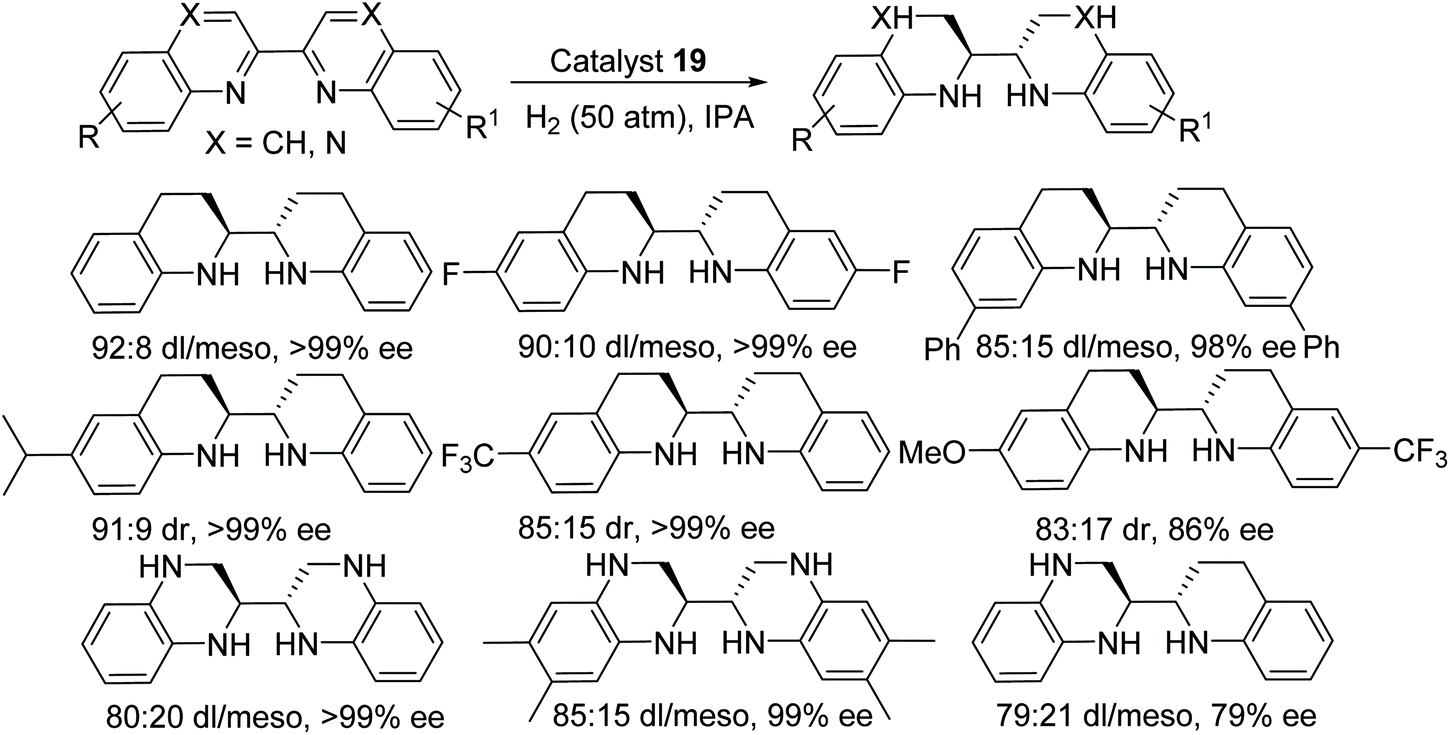 | ||
| Fig. 29 AH of 2,2′-bisquinoline and 2,2′-bisquinoxaline derivatives. | ||
The effect of solvents such as oligo(ethylene glycols) (OEGs) and poly(ethylene glycols) (PEGs) has been studied for asymmetric hydrogenation of quinoline derivatives using chiral cationic ruthenium complexes. The hydrogenation failed in PEG or long chain OEG, whereas 2-substituted quinoline derivatives were easily reduced in short chain OEG, providing the corresponding products in excellent enantioselectivities (Fig. 30).35
 | ||
| Fig. 30 AH of substituted quinoline in short chain OEG. | ||
A range of seven-membered cyclic imines was reduced by cationic ruthenium diamine catalysts. The counter anion of the ruthenium catalyst played a major role in the chiral induction and dibenzo[c,e]azepines were reduced in moderate to excellent ees (Fig. 31). The corresponding optically active amine product was obtained in up to 96% ee. Biologically important chiral 6,7-dihydro-5H-dibenz[c,e]azepines were synthesised in one step through a reductive amination/asymmetric hydrogenation strategy by applying this method.36 Fan et al. have also, in earlier research, reported highly enantioselective AH of both dihydro isoquinolines and quinolines to form chiral tetrahydroquinolines and tetrahydroisoquinolines respectively, using Ru(II) Noyori catalysts in ionic liquids.37
 | ||
| Fig. 31 AH of seven-membered cyclic imines derivatives and one step synthesis of chiral 6,7-dihydro-5H-dibenz[c,e]azepines. | ||
Fan et al. have also applied ruthenium catalysts to cascade reductive amination/asymmetric hydrogenation of range of quinolinyl, indolinyl and quinoxalinyl-containing ketones. This highly efficient protocol converts ketones into optically active products with excellent diastereo- and enantioselectivity. The counter anion of the catalyst plays a crucial role hence a diverse range of ruthenium complexes have been applied to this transformation and a particular type of ruthenium complex was found suitable for each class of substrates (Fig. 32). The applicability of this methodology has demonstrated by the formal synthesis of (+)-gephyrotoxin.38
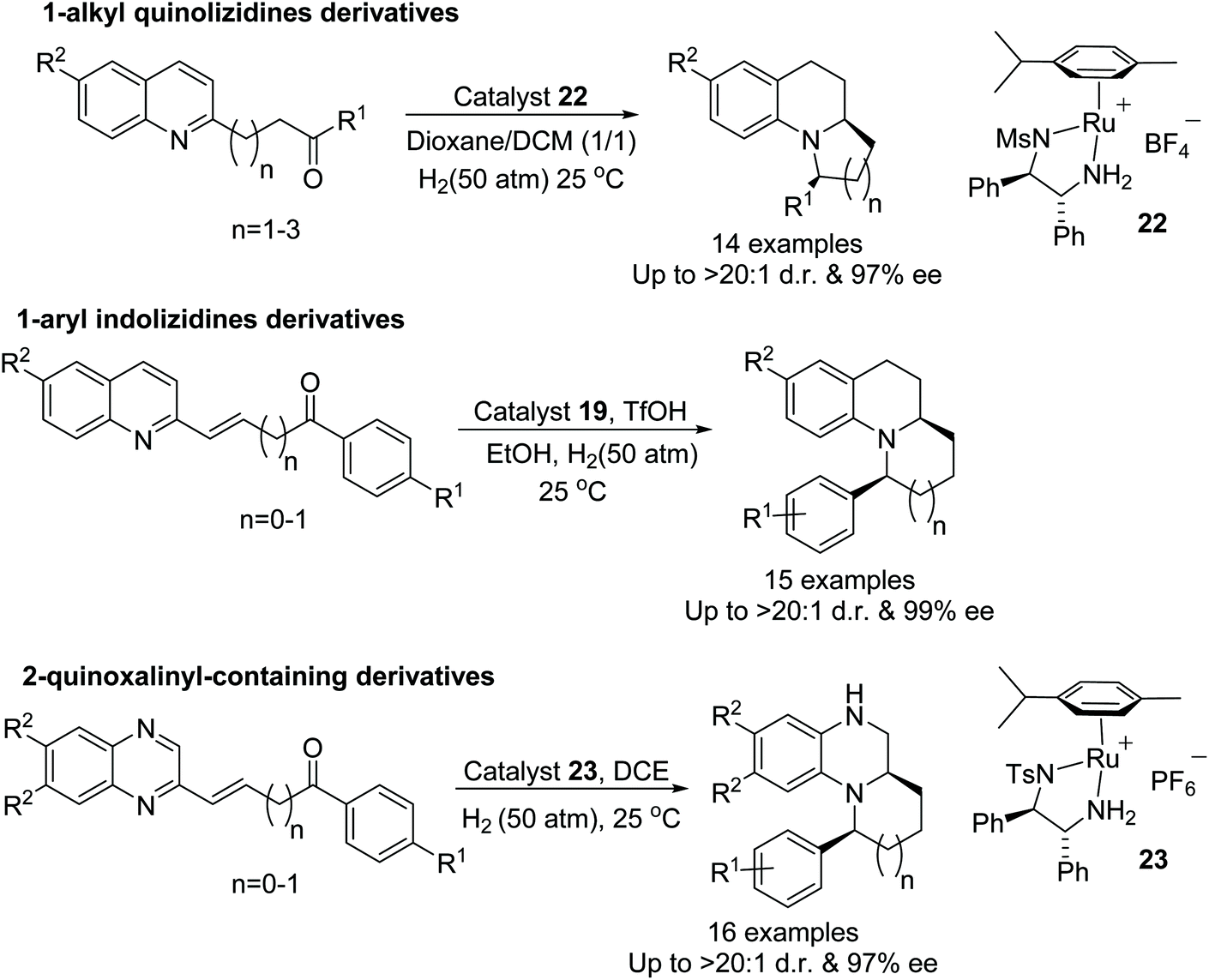 | ||
| Fig. 32 Ruthenium catalysed cascade reductive amination/asymmetric hydrogenation of range of quinolinyl, indolinyl and quinoxalinyl-containing ketones. | ||
Fan and co-workers have also developed a one-pot methodology to construct enantiomerically-pure benzo-fused N-heterocycles via sequential intramolecular hydroamination and asymmetric hydrogenation.39 This ruthenium catalysed protocol furnishes optically active 1,2,3,4-tetrahydroquinoline, indoline, and 2,3,4,5-tetrahydro-1H-benzo[b]-azepine derivatives in moderate to excellent ee. 2-Alkyl-1,2,3,4-tetrahydroquinolines derivatives were obtained in high ee with chiral ruthenium catalyst alone, whereas 2-phenyl 1,2,3,4-tetrahydroquinoline and indoline derivatives were obtained in high ee using a binary system consisting of an achiral gold complex and chiral ruthenium complex (Fig. 33). The utility of this methodology have shown by synthesizing the tetrahydroquinoline alkaloid (−)-Angustureine.
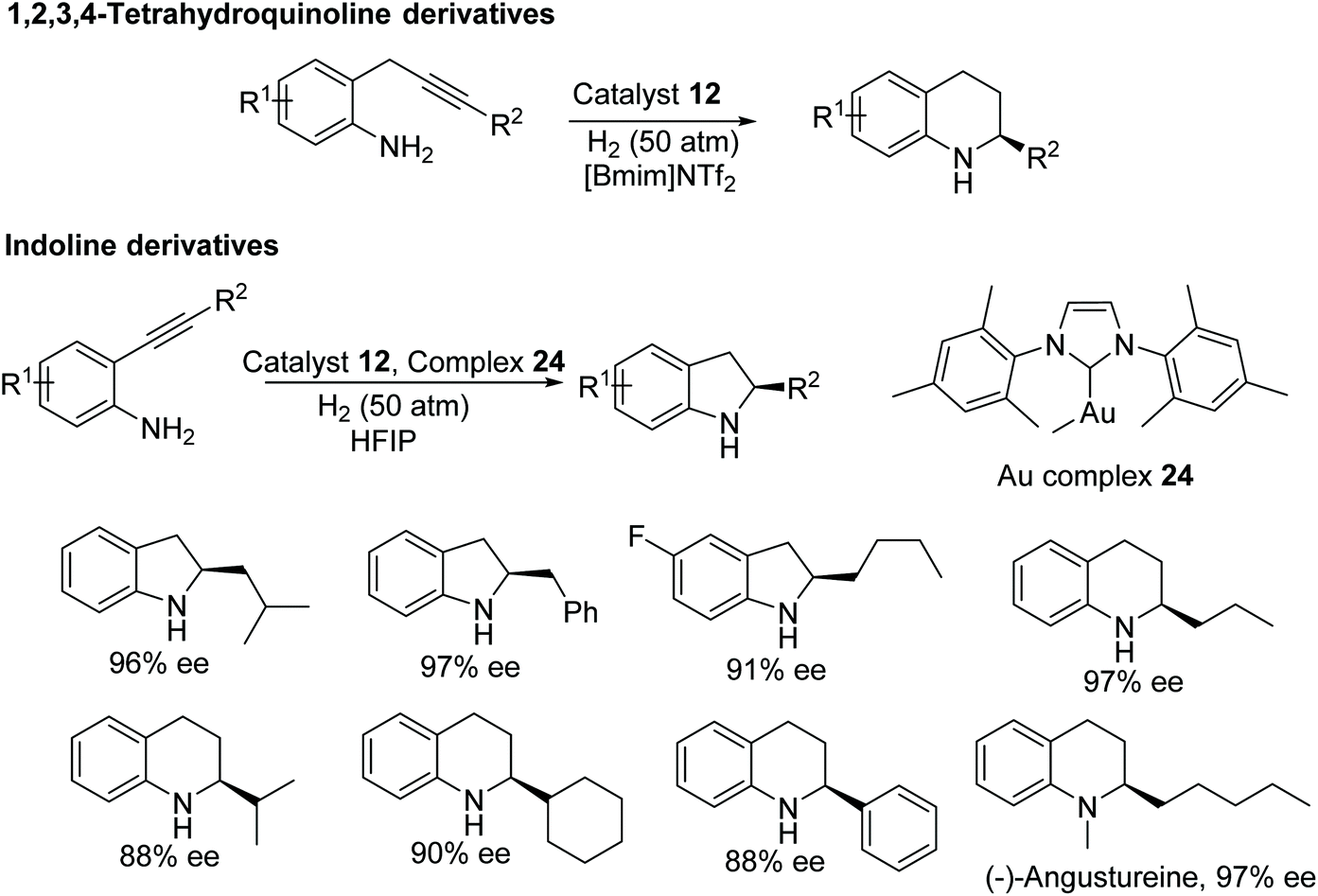 | ||
| Fig. 33 One-pot protocol to construct chiral benzo-fused N-heterocycles via sequential intramolecular hydroamination and asymmetric hydrogenation. | ||
Optically active 5,6-dihydrophenanthridines have been prepared by using the chiral cationic ruthenium complex 25. In this methodology, the selection of counter anion again proved crucial in order to achieve high enantioselectivities (Fig. 34). A wide product range was generated, providing corresponding amines in the range of 75–92% ee.40
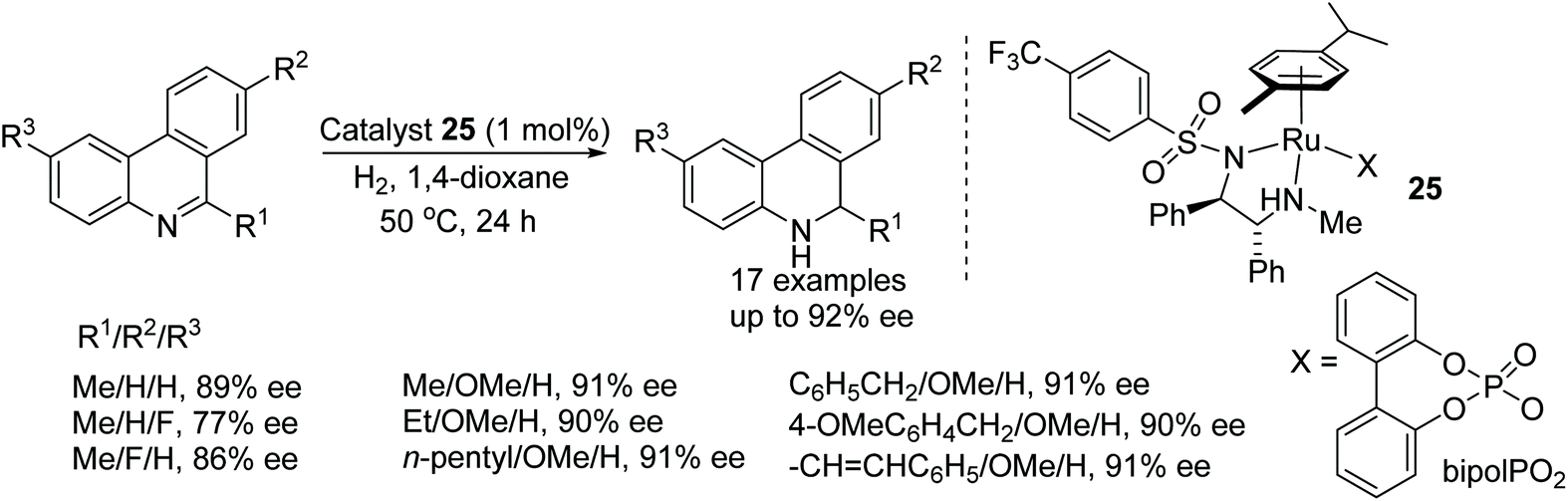 | ||
| Fig. 34 Ruthenium catalyzed asymmetric reduction of phenanthridine derivatives. | ||
A class of biologically and pharmaceutically important substructures have been synthesised by ruthenium-catalysed enantioselective hydrogenation. Optically enriched 1,2,3,4-tetrahydro-1,8-naphthyridines have been prepared by using chiral cationic ruthenium complexes (Fig. 35). A wide range of substrates were converted into the products in ees up to 99%. Furthermore, chiral products prepared by this method have a potential to develop into modular P–N ligands.41
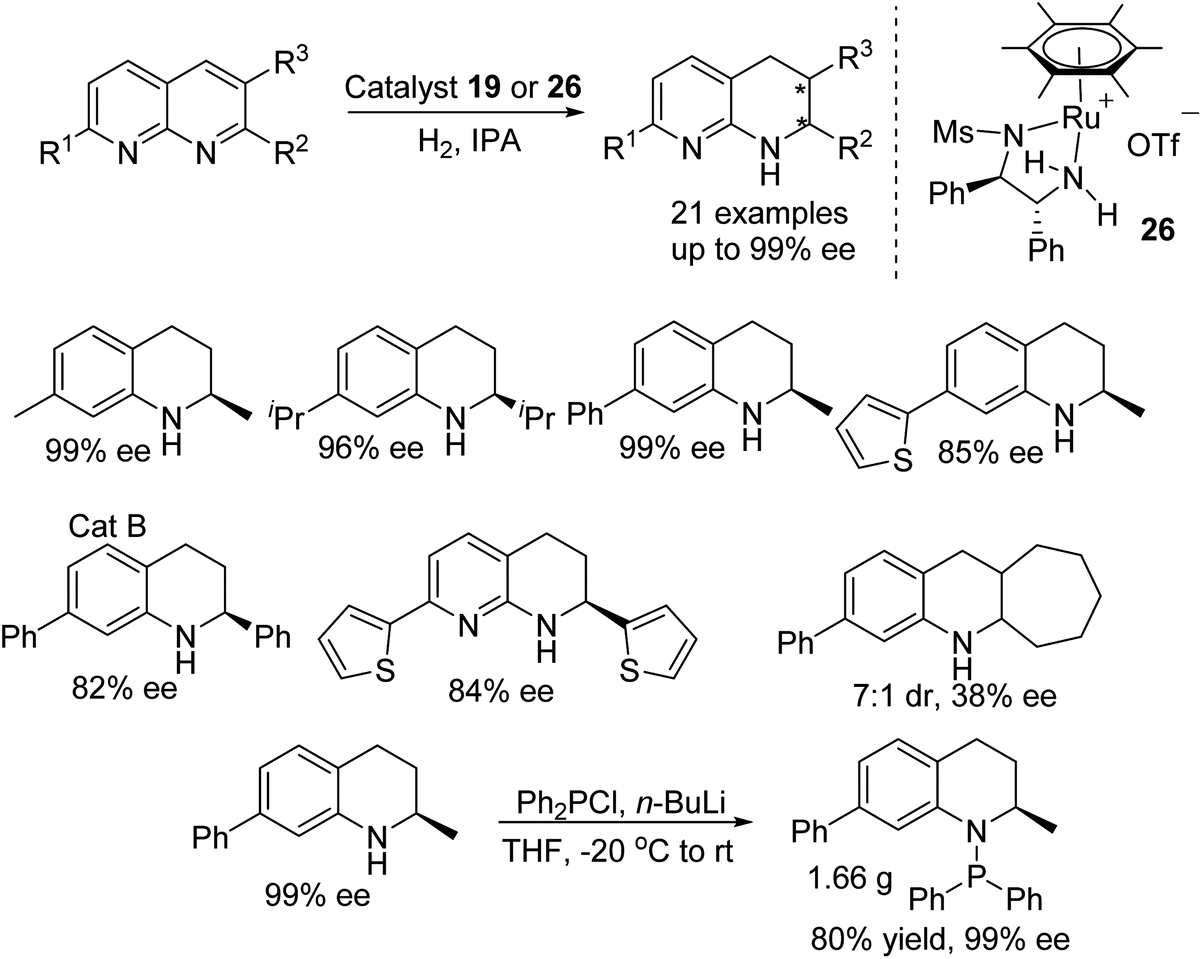 | ||
| Fig. 35 Ruthenium catalyzed asymmetric reduction of phenanthridine derivatives. | ||
Zhang and co-workers developed direct reductive amination approach to obtain chiral primary amines. A new system identified, where ruthenium C3-TunePhos catalytic system was utilised to affect the following transformation, which makes use of ammonium acetate as the amine source and H2 as the hydrogen source (Fig. 36).42 A wide range of substrates was screened, containing a diverse array of tolerable functional groups and chiral primary amines were obtained in up to 98% ee. Further, the utility of the current protocol was demonstrated by synthesizing a drug intermediate, tecalcet hydrochloride, on a gram scale.
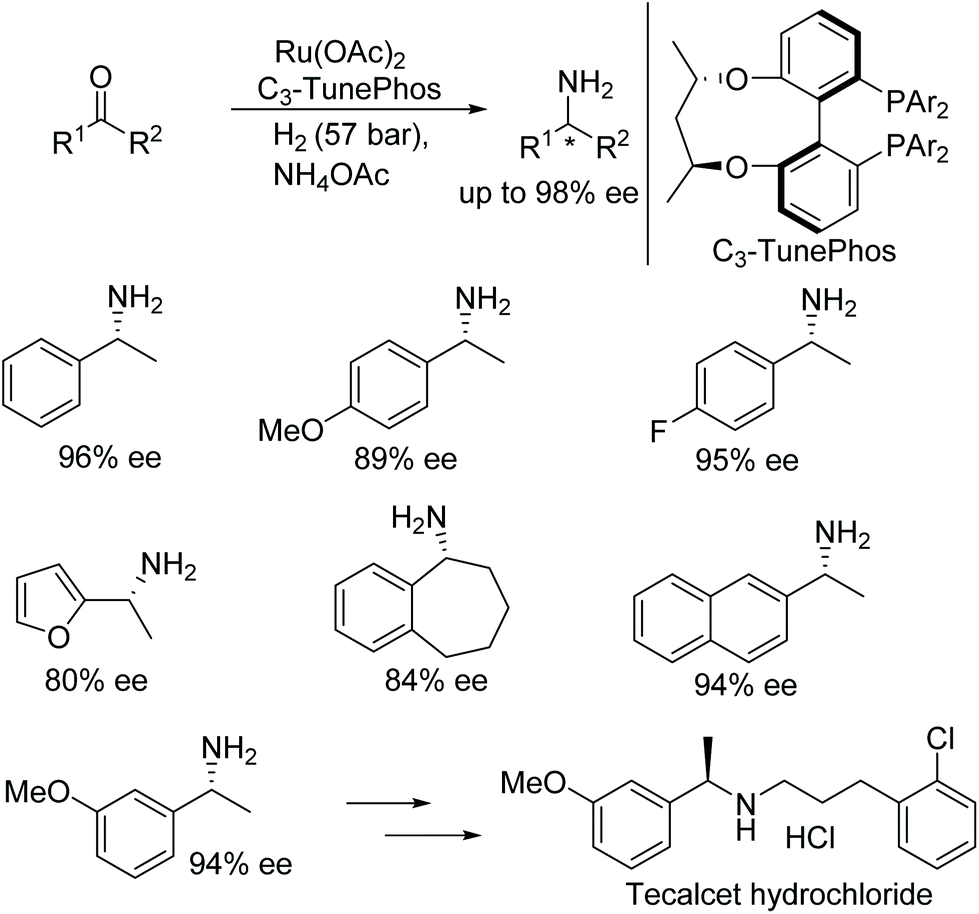 | ||
| Fig. 36 Asymmetric primary amine synthesis via reductive amination. | ||
2.4. Other metal-catalysed imine AH
Zhang et al. have screened a diverse range of phosphine-based ligands able to form active nickel catalysts in the AH of N-sulfonyl imines.43 Exhibiting one of the highest catalytic activities and an ee of 97% was the complex of the (R,R)-QuinoxP* ligand 27 (Fig. 37) in the reduction of 2-methyl-N-(1-phenylethylide-ne)propane-2-sulfonamides. Exploring the substrate scope; ortho-substitution of the aromatic ring with either electron-donating or electron-withdrawing groups gave products in 97–99% ee. On the other hand, meta-substituted substrates proved challenging with substrates containing electron-withdrawing groups affording lower yields and lower ees of 86–89%. Similar results were also exhibited by the para-substituted substrates. Changing the alkyl group in the substrate to an ethyl or i-propyl group gave products in lower enantioselectivities of 80% and 78% respectively and the use of fused cyclic rings provided the asymmetric products with 92–97% ee.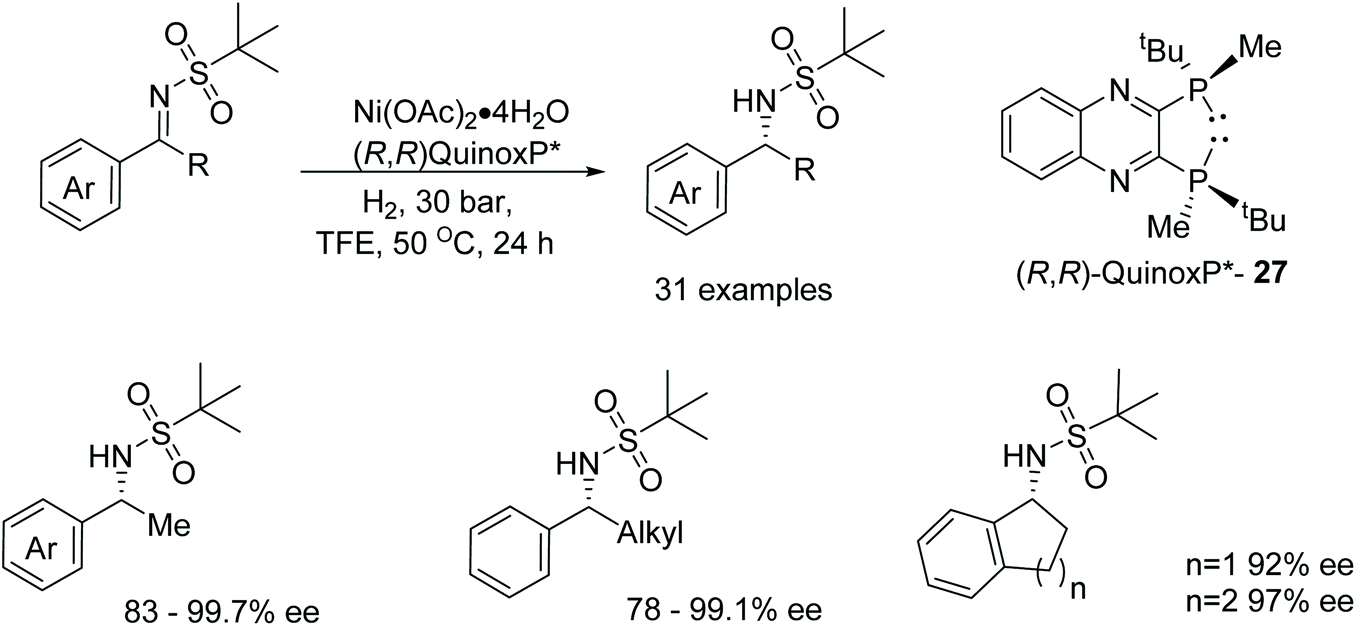 | ||
| Fig. 37 Nickel catalysed AH of N-sulfonylimines. | ||
Zhang et al. have also investigated nickel-catalysed asymmetric hydrogenations with the employment of a range of chiral diphosphane ligands. Cyclic sulfamidate imines could be reduced asymmetrically with a nickel/Ph-BPE complex (Fig. 38). Initial studies with a range of chiral diphosphane ligands revealed BINAP and SegPhos ligands to be ineffective in the nickel-catalysed hydrogenation however the use of Ph-PBE 28 proved effective in the AH of the phenyl-cyclic sulfamidate imine, giving products in up to 92% ee. Focusing on the substrate scope it was found that substituted phenyl groups affords improved results in some cases and enantioselectivities of 83–99%.44
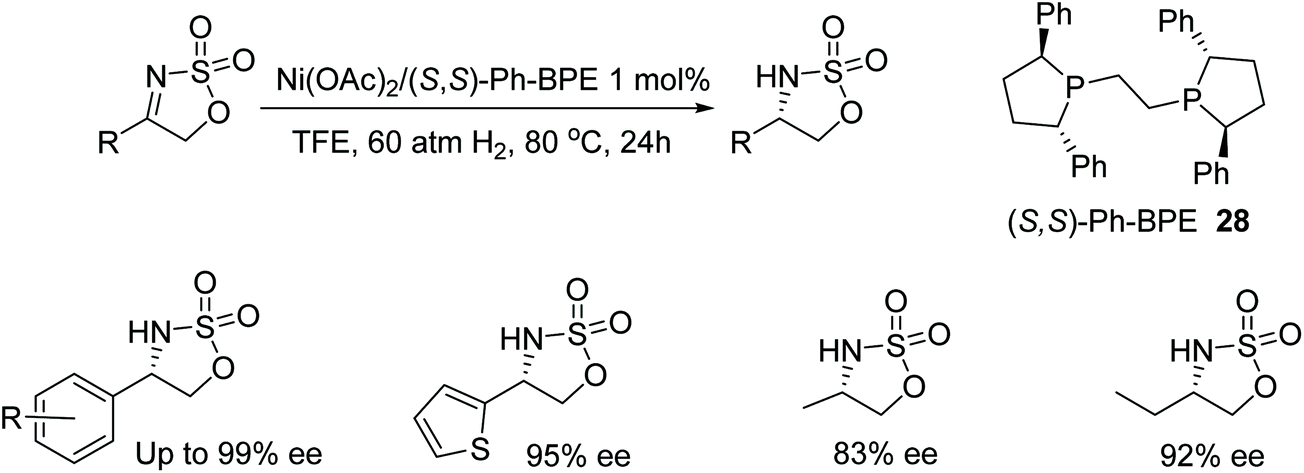 | ||
| Fig. 38 Nickel catalysed AH of cyclic sulfamidate imines. | ||
Diphosphine ligand 2 was also found to be effective in the Pd-catalysed AH of sulfonyl imines. The system proved effective under a low hydrogen pressure and in the presence of Lewis acids. AH of imines presented in Fig. 39 gave products in 95–99% ee however the catalyst system was less enantioselective for non-activated imines, which gave products of 0–75% ee. The procedure was further applied to cyclic sulfonyl imines that provided good enantioselectivity (59–90% ee) in the reductions.45
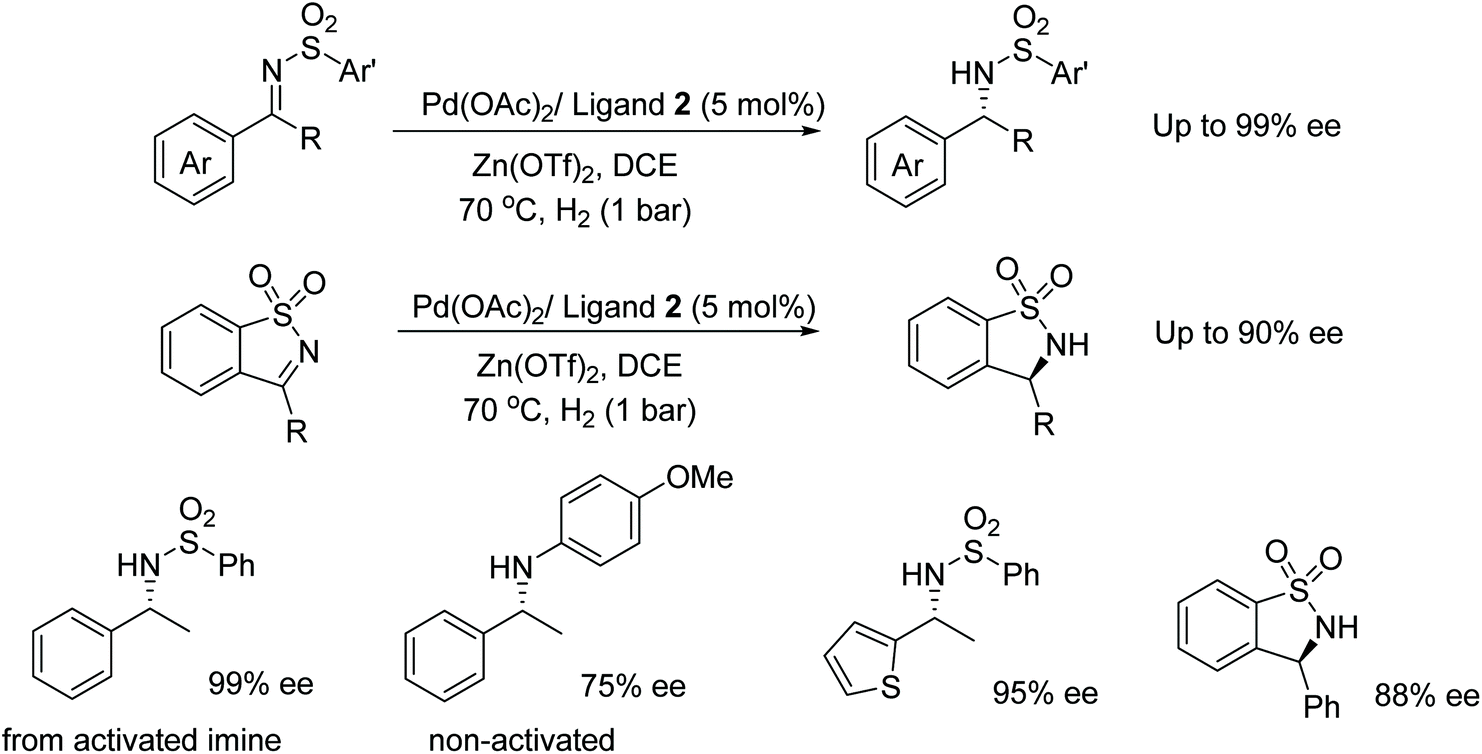 | ||
| Fig. 39 Palladium catalysed AH of sulfonylimines. | ||
The iron catalyst 29 was employed in the diastereoselective hydrogenation of N-alkylated chiral imines.46 Reduction of a phenyl derivative of the imine with the catalyst system yielded the amine product in 96% yield and 93:7 dr. Electron-donating substituents on the phenyl groups (electron-rich imines) made ideal substrates with the catalyst system giving products of up to 98:2 dr. On the other hand, electron-poor imines gave products in lower enantioselectivity, e.g. with an imine containing a para-trifluoromethyl group giving a dr of 67:33. DFT calculations on the reaction mechanism showed there to be a lower energy in the transition state for hydrogenation of the Re-face as shown in Fig. 40 compared to the Si-face. Proving that most likely a concerted hydride transfer occurs through the Re-face of the imines.
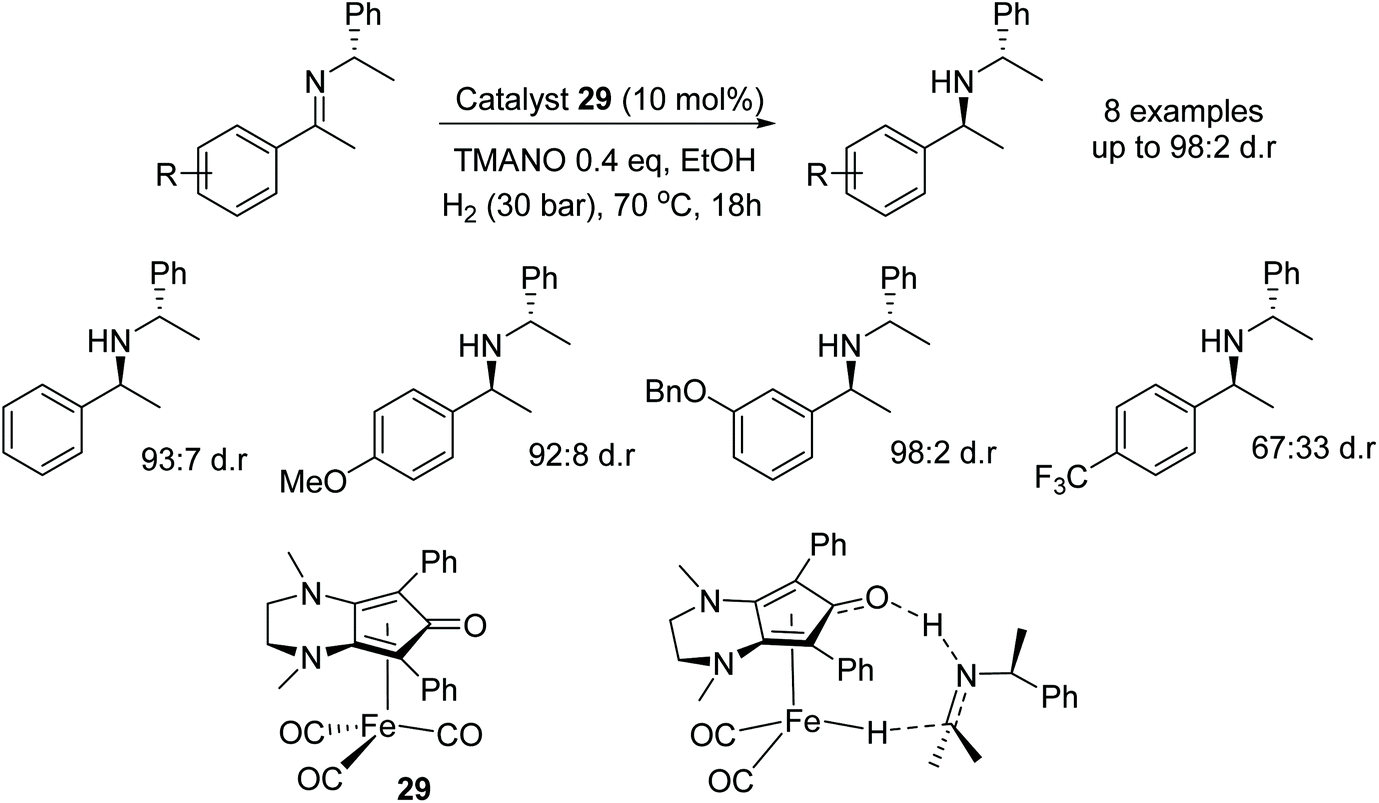 | ||
| Fig. 40 Iron catalysed diastereoselective hydrogenation of N-alkylated chiral imines. | ||
Morris and co-workers have investigated the ability of the unsymmetrical iron P-NH-P′ catalyst 30 in the asymmetric hydrogenation of activated imines. The base–metal precatalyst has been found to reduce a diverse range of N-phosphinoyl and N-tosyl aryl amines in good to excellent ee (Fig. 41). A DFT study showed that the role of the oxygen atom of the substituent on the activated imine nitrogen is significant and makes a hydrogen bond with the N–H of the iron catalyst.47
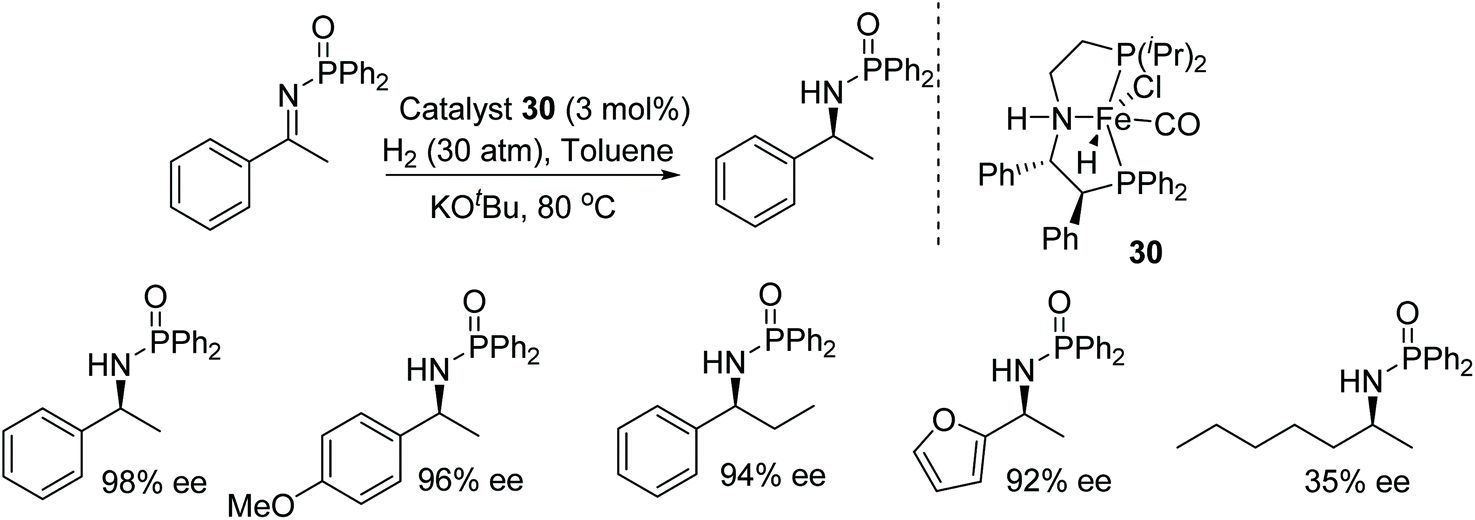 | ||
| Fig. 41 Iron catalysed AH of activated imines. | ||
An iron-based ferrocene phosphonium/hydridoborate catalyst 31, was developed using Piers’ borane [HB(C6F5)2] followed by H2 splitting. The obtained frustrated Lewis acid catalyst was applied to the asymmetric hydrogenation of various imines to generate chiral amines in moderate to good ee (Fig. 42).48
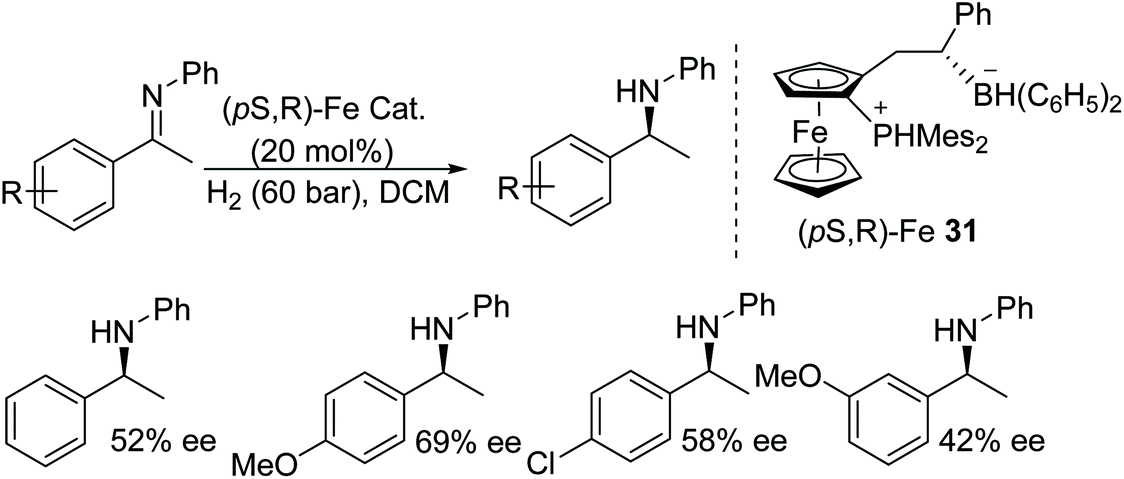 | ||
| Fig. 42 Iron catalysed AH of N-alkylated imines. | ||
Zhou et al. have demonstrated a rare application of palladium metal in the asymmetric hydrogenation of imines.49 Even though significant research has been conducted on the synthesis of chiral cyclic ureas using a multicomponent strategy, this asymmetric hydrogenation protocol benefits from direct access to chiral cyclic ureas. Following this protocol, asymmetric hydrogenation of 2-hydroxypyrimidine was achieved to furnish optically active chiral cyclic ureas in up to 99% ee (Fig. 43). A wide substrate scope has been demonstated, including mono- and disubstituted 2-hydroxypyrimidines. Furthermore, the chiral cyclic ureas can be converted to 1,3-diamines by hydrolysing the urea.
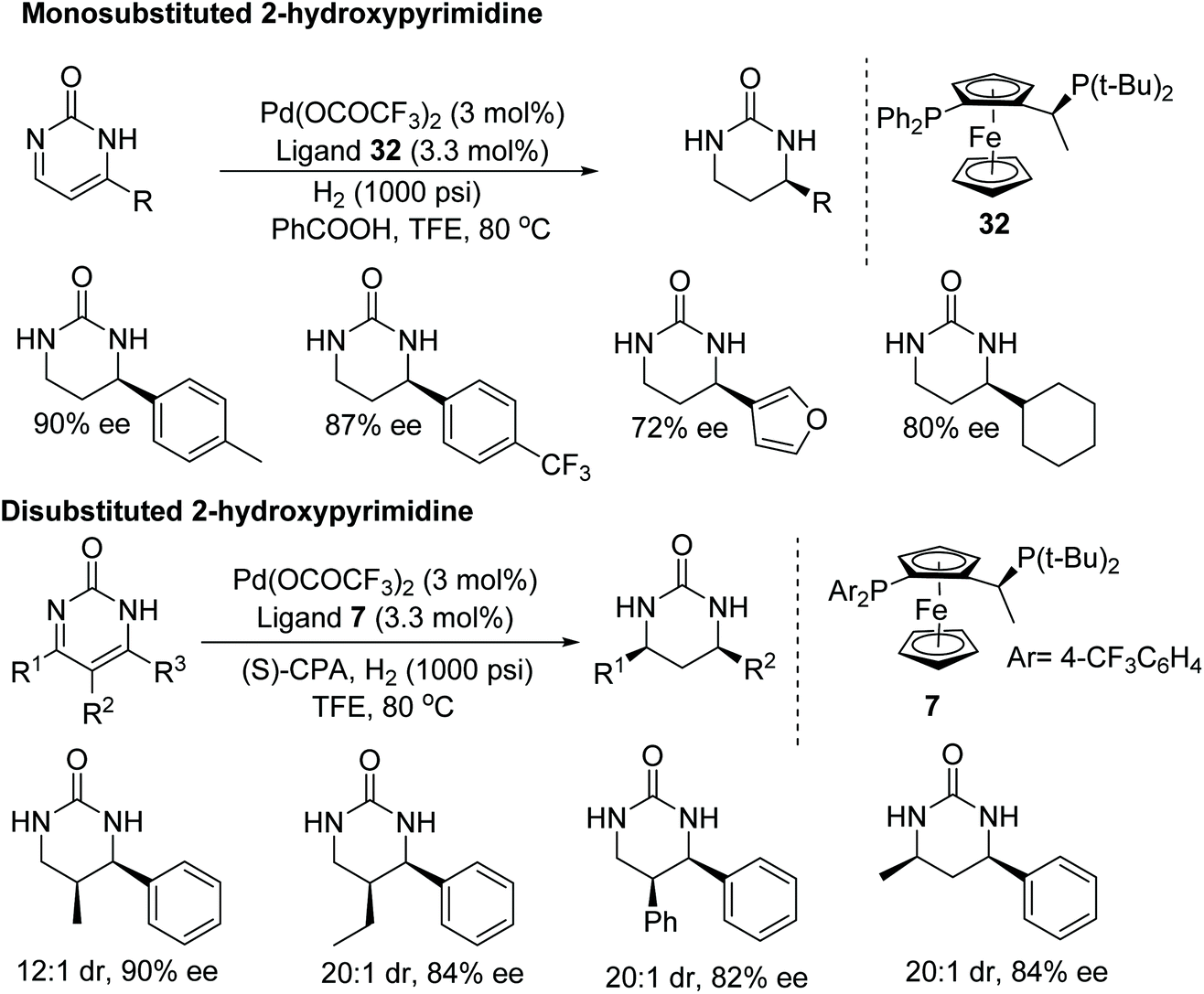 | ||
| Fig. 43 Palladium catalysed AH of mono- and disubstituted 2-hydroxypyrimidines. | ||
3. Asymmetric transfer hydrogenation (ATH) of imines
3.1. Iridium-catalysed imine ATH
A very recent and detailed study of the ATH of CN bonds in dihydroisoquinolines using chiral Cp*Ir(III) complexes revealed remarkably complex behaviour, including the observation of different kinetic orders of formation for each enantiomeric product.50 The investigators were also able to conclude, from a very careful series of studies, that the iminium cation was the reduction substrate rather than the free imine. An IrCp* based hybrid catalyst of 33 was prepared using the chiral structure of the antibiotic vancomycin. The novel vancomycin–iridium(III) interaction was proved by using techniques such as MALDI-TOF, UV, circular dichroism (CD), Raman IR, and NMR. The hybrid complex was further applied to the ATH of various cyclic amines in aqueous media under mild reaction conditions which furnished the corresponding cyclic amines in good to moderate ee in the respective buffer solutions (Fig. 44).51 Quinaldine was reduced in a MES (2-(4-morpholino)ethanesulfonic acid) 1.2 M buffer at pH 5 with a impressing 61% ee, whereas inversion of configuration was observed in the case of 3-methylbenzo[d]isothiazole 1,1-dioxide where a product of 42% ee was observed in phosphate buffer 0.1 M pH 8.
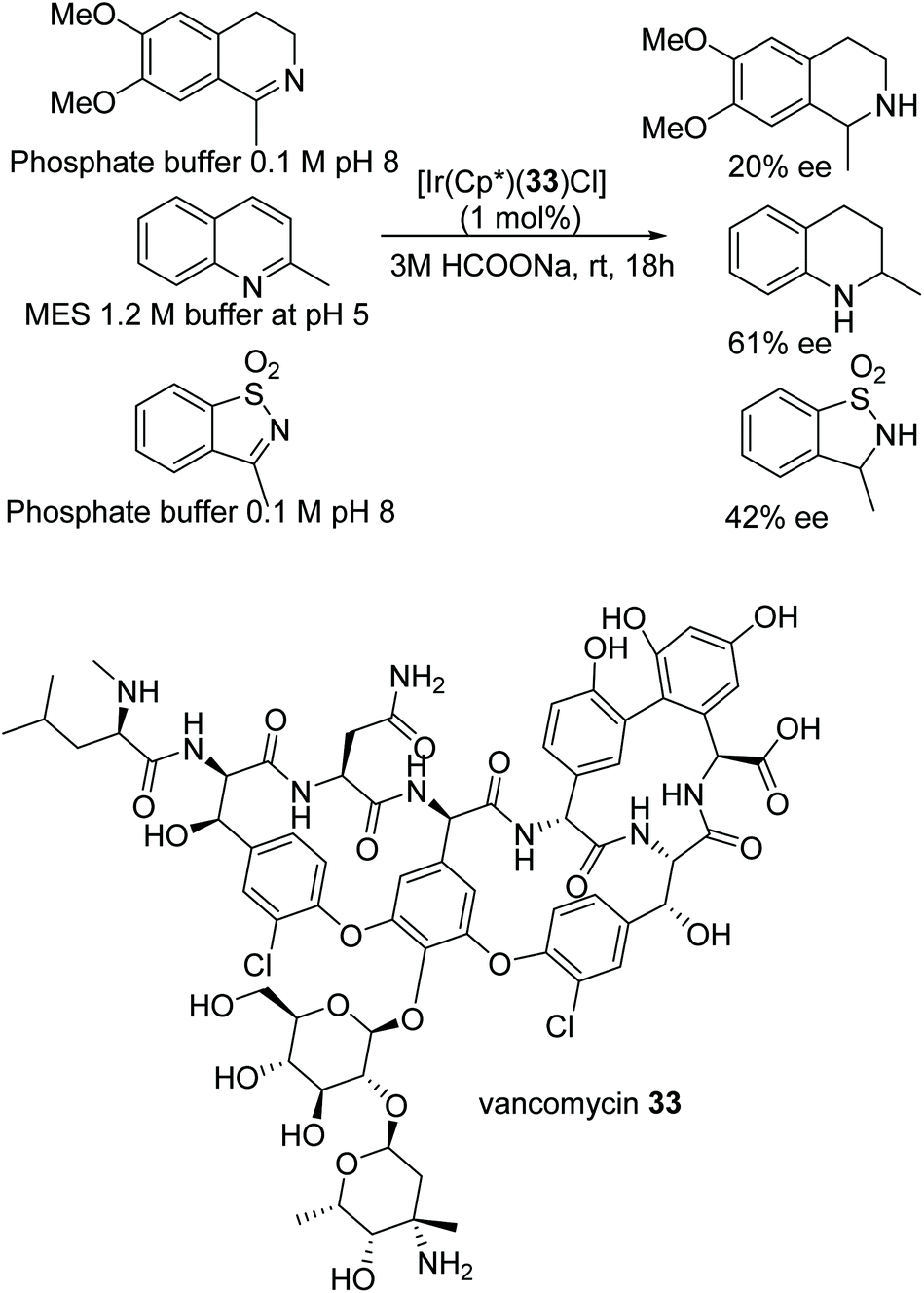 | ||
| Fig. 44 ATH of substituted amines using vancomycin as a chiral ligand. | ||
Gong et al. studied N-heterocyclic carbene-iridum catalysts to reveal that catalyst 34 exhibited metal-centred chirality that could be exploited in asymmetric reactions.52 Using catalyst 34 in an ATH reaction with ammonium formate as the hydrogen source facilitated reduction of a cyclic N-sulfonylimine in 99% ee. Under sodium formate or formic acid/triethylamine conditions, the reactions both proceeded slower and it was rationalised that the ammonium ion was crucial in the reaction. Applying the catalyst system to a range of cyclic N-sulfonylimines, as in Fig. 45, the ATH of these revealed that in cases where R1 was a phenyl group and R2 a substituted-phenyl group, the catalyst system gave products in 96 to 98% ee. Also when R2 was a methyl and R1 were aryl groups, the products were obtained in 94 to 98% ee. The same trends were also seen with a range of alkyl groups at the R2 position. With the results in hand, the method was applied to a half-gram scale of a bioactive compound with anti-HIV activity in 97% ee.
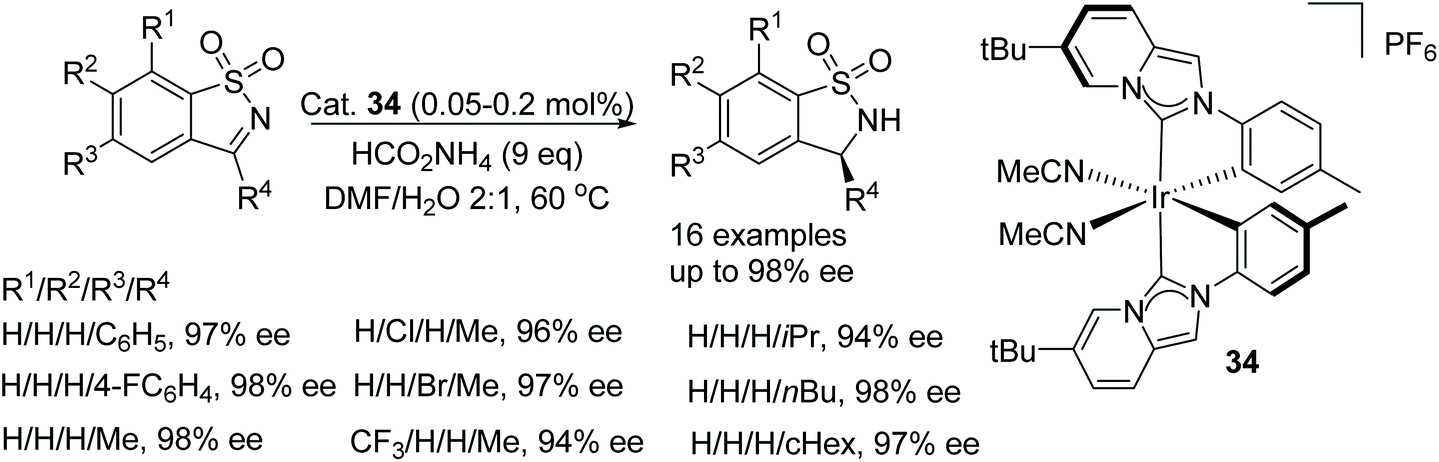 | ||
| Fig. 45 ATH of cyclic N-sulfonylimines. | ||
Rimoldi and co-workers have completed an interesting comparison of AH and ATH for the iridium-catalysed reduction of various imine derivatives (Fig. 46).53 This approach makes use of atropoisomeric diphosphines and cyclic diamines as ligands for AH and ATH respectively. Four challenging classes of cyclic imine substrates were selected and studied under optimised reaction conditions using both the protocols. ATH proved to be a superior method for the synthesis of optically active cyclic amines with 3,4 dihydroquinolines and sulfonyl amines being formed in up to 86% ee whereas AH was found to be a method of choice in the case of isoquinolines and quinolines which gave chiral amines in ee up to 64% ee.
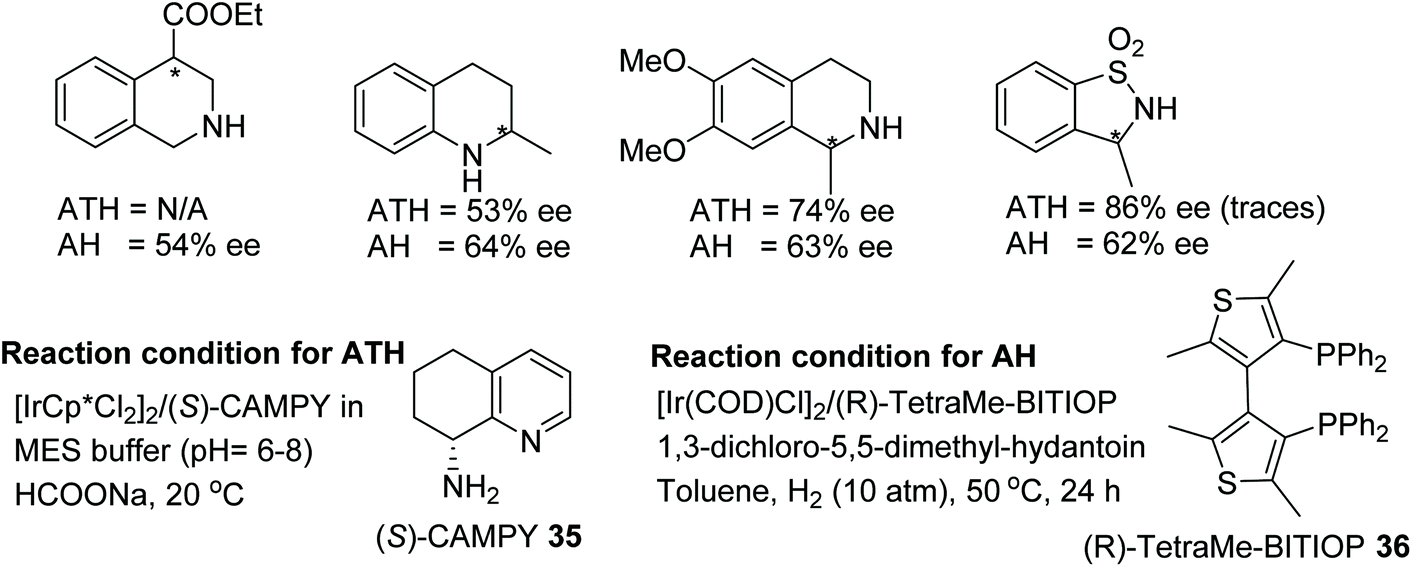 | ||
| Fig. 46 Comparison of AH and ATH for the iridium-catalysed enantioselective reductions of various imine derivatives. | ||
Catalyst 37 was found to reduce 1-phenyl-3,4-dihydroisoquinoline in 83% conversion and 63% ee whereas the rhodium analogue under the same conditions carried out the ATH in 66% conversion and just 3% ee. With the results in hand, Červený and co-workers explored the addition of Lewis and Brønsted acids to the Ir-catalysed system and found that the addition of anhydrous phosphoric acid (APA) increased both conversion and enantioselectivity to 99% and 86% respectively.54 The substrate scope of catalyst 37 was expanded to substrates of the type shown in Fig. 47 with varying substituted-phenyl groups giving rise to products of 64–86% ee. Applying the methodology however to a dihydro-β-carboline derivative was less effective; providing the reduced product in just 50% ee.
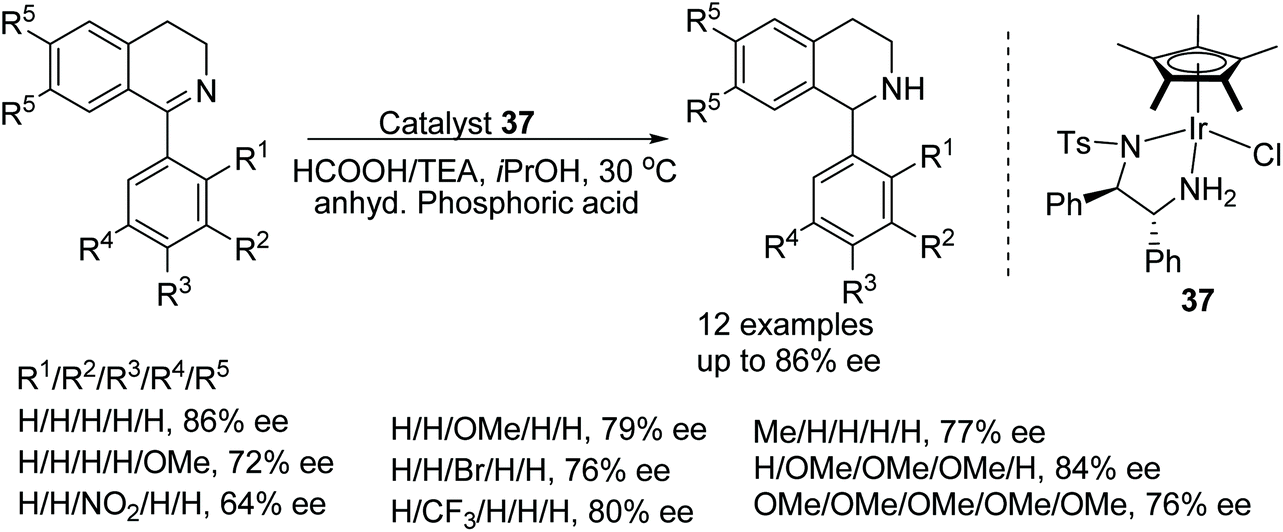 | ||
| Fig. 47 ATH of 1-phenyl-3,4-dihydroisoquinoline derivatives. | ||
Zhao et al. have developed a highly enantioselective method for the ATH of N-aryl and N-alkyl ketamines (Fig. 48).55 The iridium catalyst 38a aided by a chiral phosphoric acid ligand 38b catalyses the ATH of ketimines using an alcohol as the hydrogen source. The diol was found to provide the highest enantioselectivity from a series of alcohols, as hydrogen sources, with DFT studies providing evidence for the formation of an iridium alkoxide as the reducing agent. High enantioselectivities were obtained when R1 was alkyl or aryl and the N-group was either alkyl or aryl.
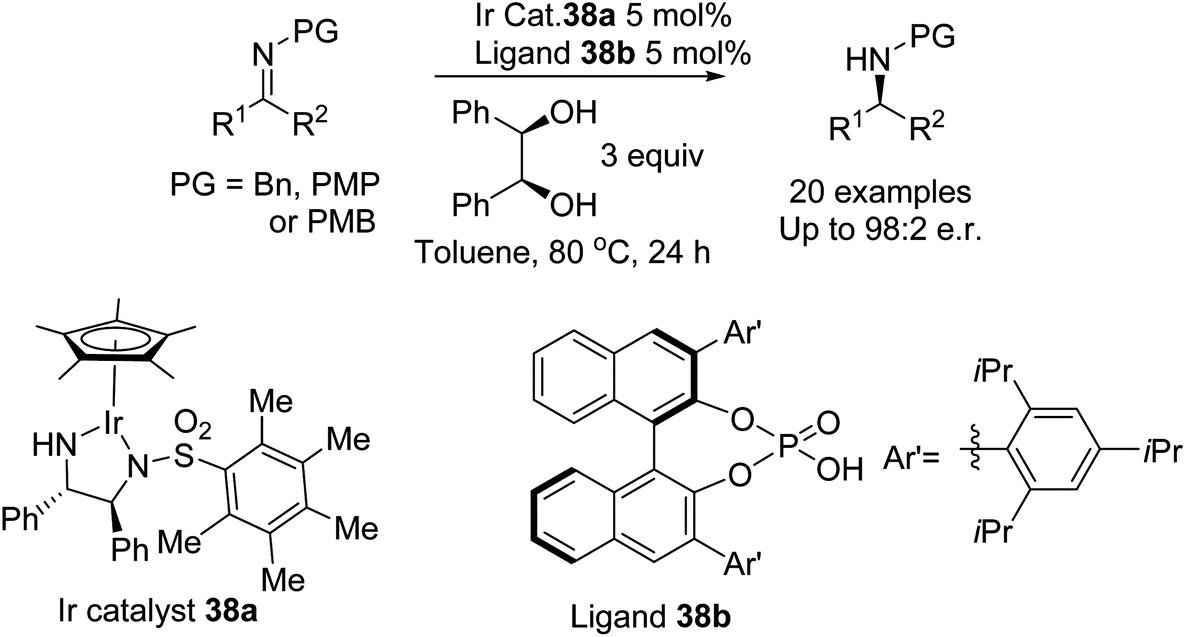 | ||
| Fig. 48 ATH of N-aryl and N-alkyl ketamines. | ||
Over an extended period of intense investigations, Ward et al. have completed the highly successful optimisation of an Ir(III)-based catalyst system in which the environment of the protein streptavidin serves to direct the asymmetric transfer hydrogenation in an aqueous environment. Selective modification of the streptavidin structure permits fine-tuning of the resulting new synthetic metalloenzymes towards their substrates, and notably tetrahydroisoquinolines in particular.56
3.2. Rhodium-catalysed imine ATH
DKR-ATH of 5-alkyl cyclic sulfamidate imines have been reported by Lee and co-workers. This DBU promoted protocol utilises a Noyori-type chiral rhodium catalyst and HCOOH/DBU as a hydrogen source.57 This particular process takes place through DBU promoted rapid racemisation of the cyclic imine substrates. Through this methodology a wide range of sulfamidates have been prepared in excellent diastereo- and enantioselectivities (Fig. 49).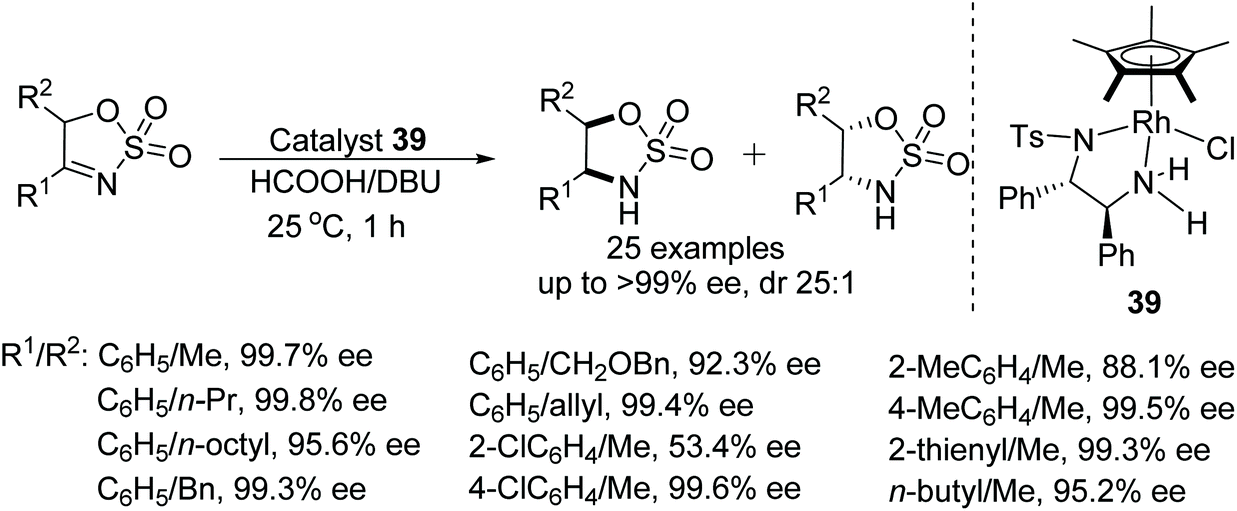 | ||
| Fig. 49 DKR-ATH of 5-alkyl cyclic sulfamidate imines. | ||
A detailed computational study of the mechanism of transfer hydrogenation by Cp*Rh(III) complexes has been reported, revealing the importance of different substituents on the chelating ligand for optimal efficiency.58
3.3. Ruthenium-catalysed imine ATH
An efficient methodology to synthesise optically active allylic amines has been demonstrated by Guijarro and co-workers. This protocol involves distereoselective transfer hydrogenation of enantiomerically-pure α,β-unsaturated N-(tert-butylsulfinyl) ketimines which were then converted to allyl amines by removal of the sulfinyl group. Through this methodology, the enantiomer of interest can be synthesised in 97 to 99% ee simply by selecting the sulfinyl group with the appropriate absolute configuration (Fig. 50).59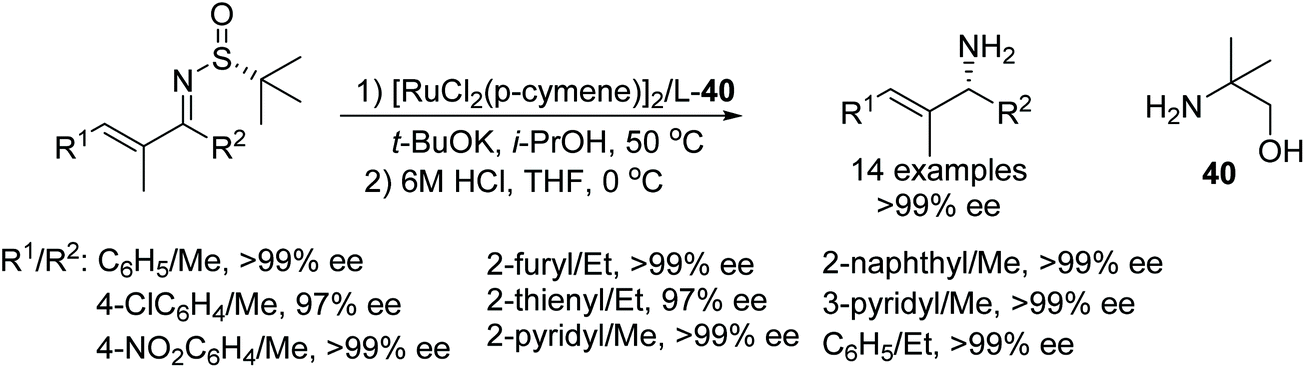 | ||
| Fig. 50 Distereoselective transfer hydrogenation of enantiomerically-pure α,β-unsaturated N-(tert-butylsulfinyl) ketimines. | ||
Noyori's complex 18a was applied to the ATH of derivative of imines in the total synthesis of a range of dysoxylum alkaloids. The all-important stereogenic centre in both enantiomeric forms were obtained at by reducing the CN bond (Fig. 54). Ruthenium catalysed asymmetric induction was affected by using sodium formate as a hydrogen source and the dysoxylum alkaloids were obtained in ees of up to 97% after methylation at the nitrogen. With the results at hand the mechanism was proposed to occur via the substrate approaching the catalyst through an N+–H⋯OS interaction between the protonated imine and the oxygen on the sulfonyl group of the catalyst which is then further aided by a CH⋯π interaction as shown in Fig. 51.60
 | ||
| Fig. 51 ATH of dysoxylum alkaloids and a possible transition state. | ||
The particular role of the η6-arene ligand in the ATH of dihydroisoquinolines has previously been the subject of detailed investigations.61 The effect of the level of electron-richness of the substituents on the fused aromatic ring of dihydroisoquinolines on rates of reduction by Noyori catalysts, studied by 1H NMR spectroscopy has also been studied in depth.62
Several papers have been published in the last decade describing the use of Ru(II) Noyori catalysts for the ATH of 1-aryl dihydroisoquinolines which, in contrast to 1-alkyl derivatives, are regarded as quite challenging substrates. Generally, either an ortho-substituent is required to be present on the 1-aryl ring of the DHIQ for best results, or the fused ring must be electron-rich.63 A recent solution to the challenge presented by DHIQs lacking both requirements was achieved b the addition of a methylfuran group to the basic N atom of the TsDPEN.64 Novel derivatives of the C2-symmetric diamine used in arene/Ru(II) ATH catalysts continue to be developed, including a series of recently reported, pyridinium-containing examples which facilitate ATH in aqueous solution.65
A further application of catalyst 18a in ATH was studied on dibenzo[b,f][1,4]oxazepines substrates by Bhanage and More.66 Preliminary studies revealed the catalyst system to be dependent on the pH of the reaction, in cases where the pH was greater than 5 the conversion decreased drastically and it was found that at pH 4 the conversion reached 99% and afforded the reduced product in 93% ee. The substrate scope was expanded with R1 as a methyl group that allowed the reduction of the mono substituted R4 group to yield products of 80–91% ee. Groups on R2 gave products of 71–93% ee and di-substituted aryl rings gave products of 77–91% ee. Substrates with R1 as an ethyl in turn produced products of 80–82% ee whilst a substrate with a phenyl group gave a product of 80% ee (Fig. 52).
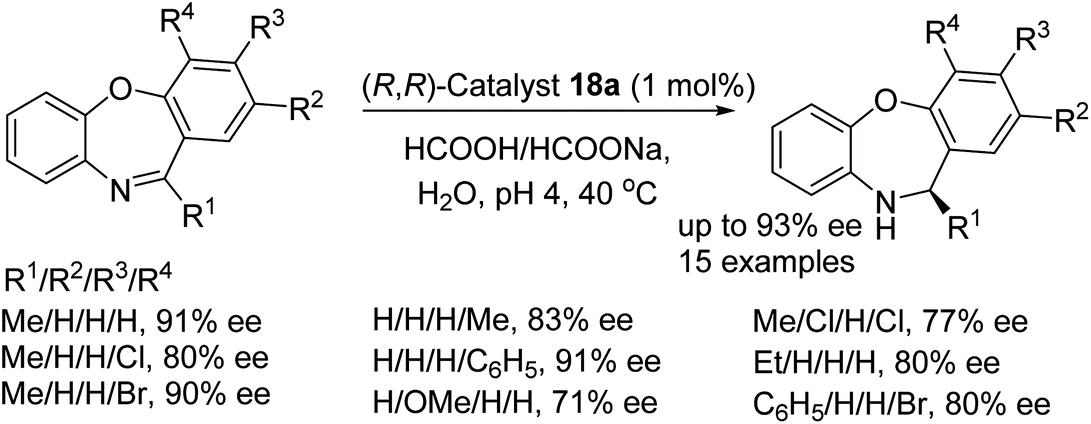 | ||
| Fig. 52 ATH of dibenzo[b,f][1,4]oxazepines derivatives. | ||
A ruthenium catalysed chemo- and stereoselective synthesis of Δ4-isoxazolines have been demonstrated by Wills and Chew.67 This challenging class of cyclic amines have been prepared using a mild reaction protocol and no side reaction owing to ring opening was observed. The protocol makes use of tethered ruthenium MsDPEN as a catalyst using HCOOH/TEA as a hydrogen source which gave the corresponding products in good to moderate ee (Fig. 53).
 | ||
| Fig. 53 ATH of isoxazolium Salts. | ||
A new method of obtaining N,N-diamine ligands derived from a natural source ((−)-menthol) was investigated by Czarnocki et al. A newly formed ligand proved to act as chiral ligand in a ruthenium catalysed asymmetric transfer hydrogenation of the a range of ketone and endocyclic imine substrates e.g. 1-methyl-3,4-dihydro-b-carboline, which were subsequently converted to amines with excellent conversion and 72% ee Fig. 54.68
 | ||
| Fig. 54 Application of a derivative of (−)-menthol in ruthenium catalysed ATH of 1-methyl-3,4-dihydro-β-carboline. | ||
3.4. Other metal-catalysed ATH
Fan et al. have used a variety of alcohols as a hydrogen source in the asymmetric transfer hydrogenation of N-sulfonylimines (Fig. 55).69 This palladium/zinc co-catalyst protocol required screening of wide range of primary and secondary alcohols as a hydrogen donor out of which MeOH was found to be the best. In this methodology, a Pd-hydride intermediate reduces N-sulfonylimines to the corresponding optically active amines in high ee.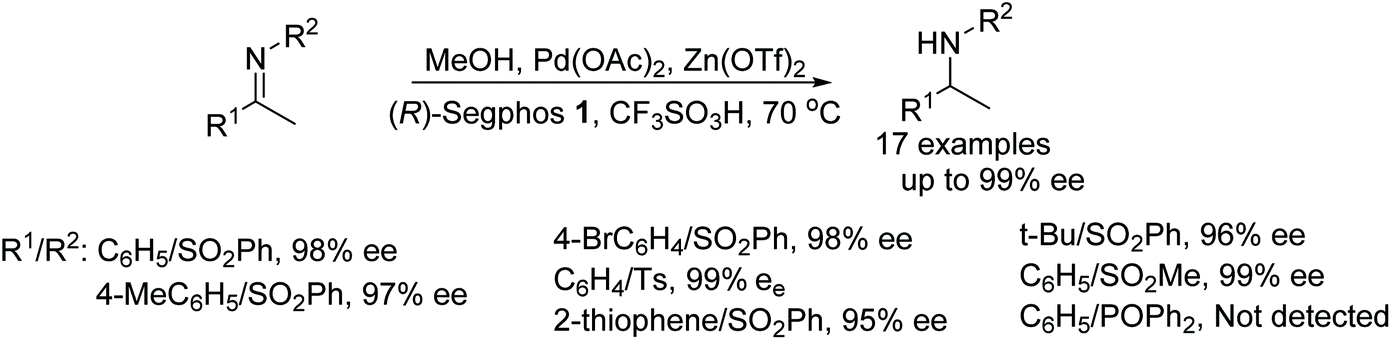 | ||
| Fig. 55 Palladium catalysed ATH of N-sulfonylimines derivatives. | ||
4. Organocatalysed asymmetric imine reductions
The chiral alkene 43 reacted with a boron species to form a chiral boron-based Lewis acid able to catalyse the asymmetric hydrogenation of imines as illustrated in Fig. 56. Employment of the chiral boron species at 10 mol% could asymmetrically reduce imine substrates in 45–89% ee.70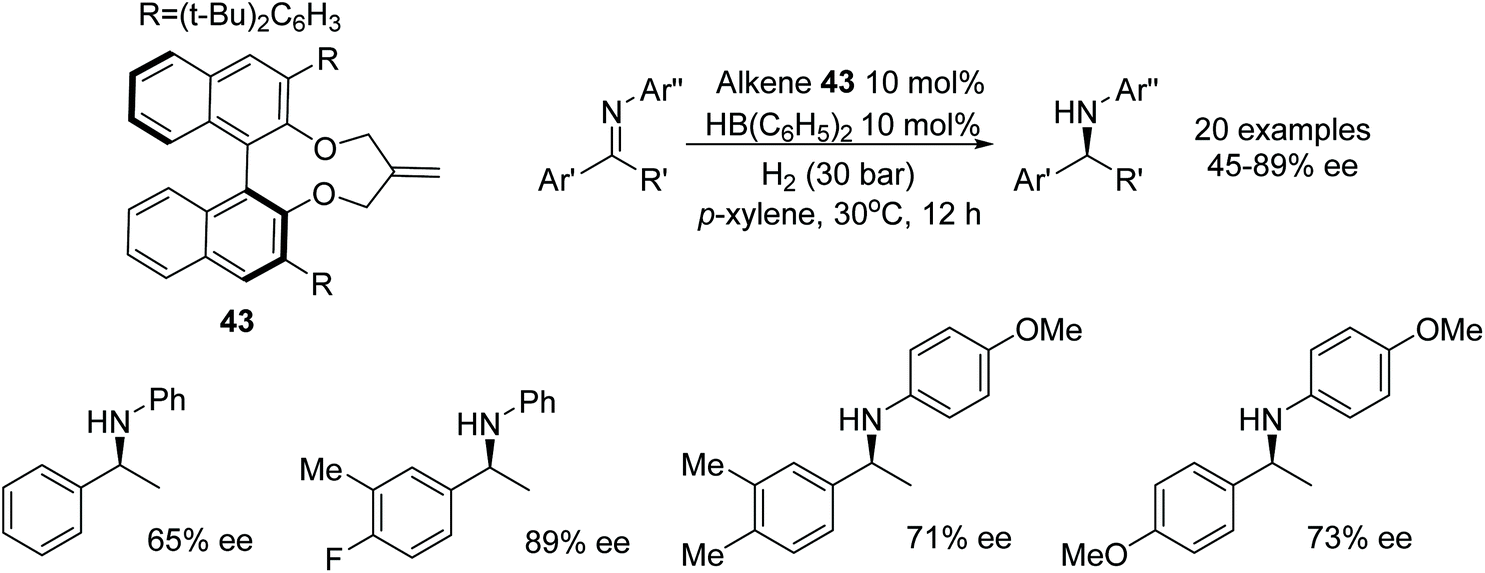 | ||
| Fig. 56 Chiral alkene borane catalysed AH of N-alkylated imines. | ||
A frustrated Lewis pair of Piers’ borane and (S)-tertbutylsulfinamide 44 has been applied to the ATH of the 2,3-disubstituted quinoxalines by Du and co-workers.71 Utilising ammonia borane as a hydrogen source this chiral Lewis acid pair reduced a variety of 2,3-disubstituted quinoxalines (Fig. 57). Interestingly, 2-alkyl-3-arylquinoxalines derivatives solely gave the cis products in up to 86% ee, whereas 2,3-dialkylquinoxalines often furnished trans products with ees of up to >99%.
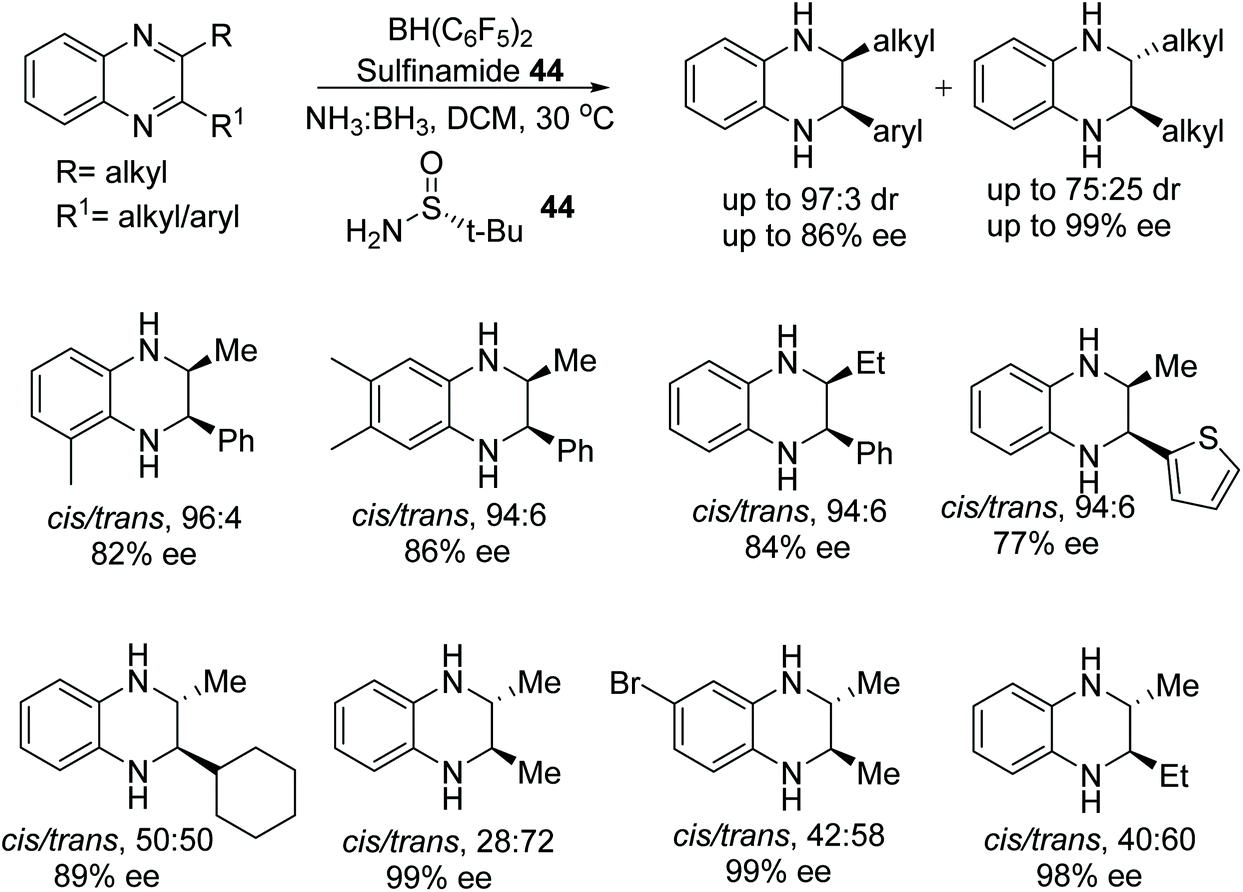 | ||
| Fig. 57 ATH of the 2,3-disubstituted quinoxalines using a frustrated Lewis pair of Piers’ borane and (S)-tertbutylsulfinamide. | ||
In a further application, the catalytic system using 44 was applied to the ATH of β-N-substituted enamino esters. Utilising ammonia borane as a hydrogen source, this chiral Lewis acid pair reduced a variety of β-N-substituted enamino esters in up to 91% ee. Further, the product was subjected to cyclisation to furnish the optically active β-lactam which produced >99% ee after recrystallization with hexane (Fig. 58).72
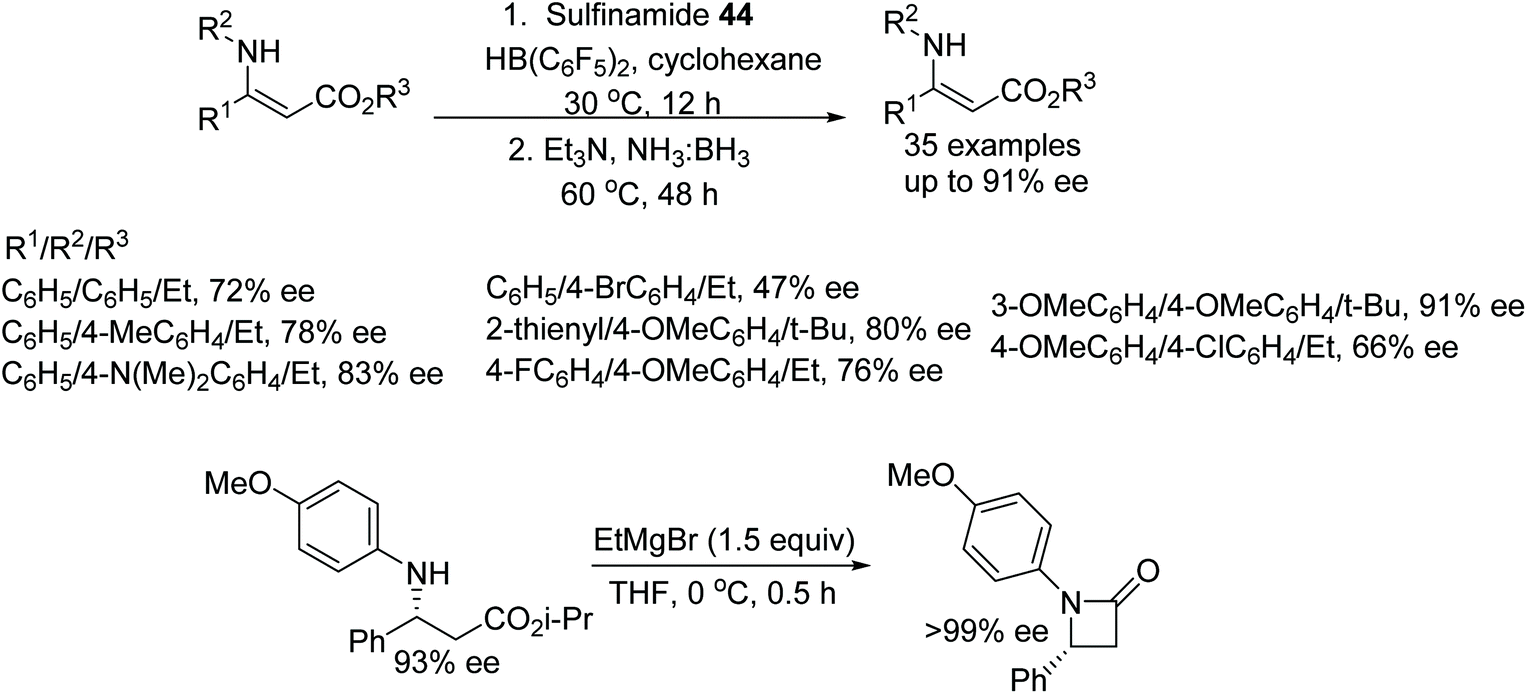 | ||
| Fig. 58 CPA catalysed ATH of β-N-substituted enamino esters and construction of chiral β-lactam. | ||
A newly designed chiral ammonia borane catalyst was synthesised by Du and co-workers and applied to the ATH of imines and β-enamino esters (Fig. 59).73 This protocol works through the dehydrogenation of the ammonia borane by the chiral phosphoric acid 45 which acts as a Brønsted acid. The chiral ammonia borane complex of 45 can be regenerated through the addition of ammonia borane and water which allows the use of the relatively more expensive CPA at very low loading i.e. 0.1%. The hydrogen transfer in this reaction from chiral borane to imine works through a 6-membered transition state which gave optically active imines in moderate to excellent ee.
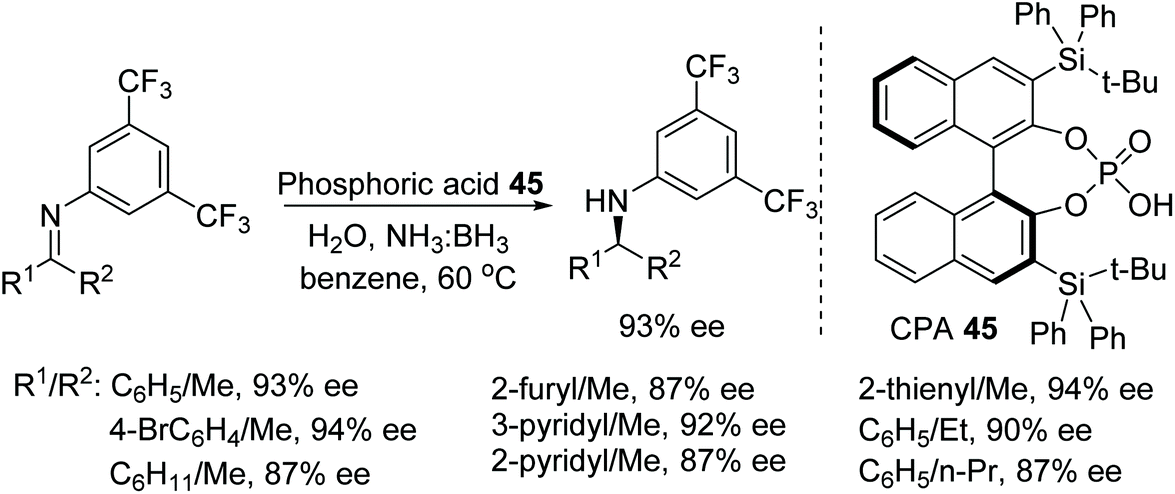 | ||
| Fig. 59 CPA catalysed ATH of N-alkylated imines. | ||
Another application of a chiral Brønsted acid for the ATH of indoles proceeding through an iminium ion intermediate has been reported (Fig. 60). A wide range of functional groups are tolerated by this in situ generated chiral phosphoric acid borane catalyst to obtain chiral indolines in the range of 64–96% ee. Theoretical studies support protonation at the C3 carbon followed by asymmetric transfer hydrogenation.74
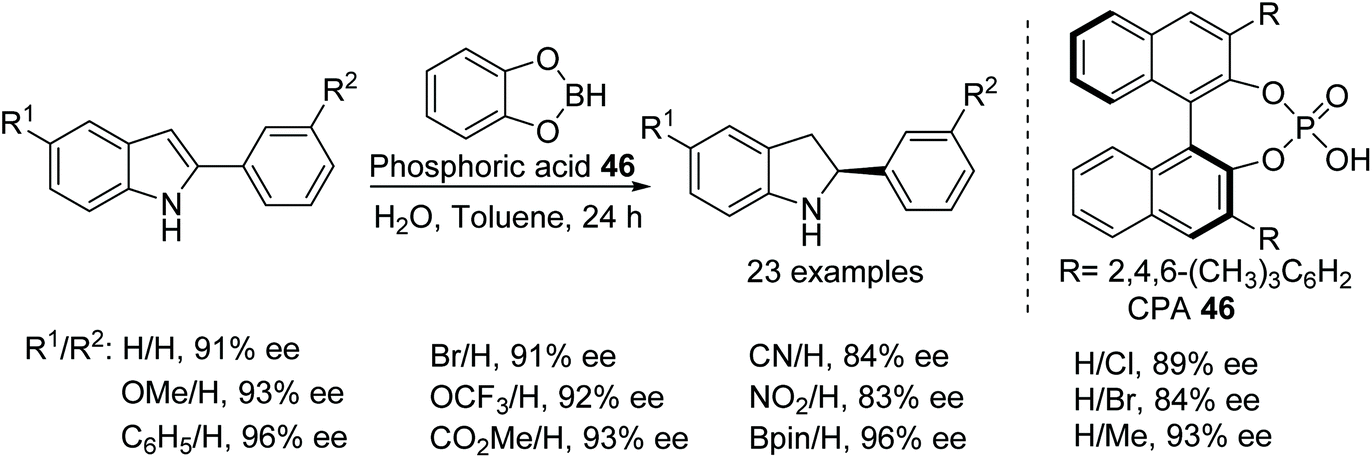 | ||
| Fig. 60 CPA catalysed ATH of indole derivatives. | ||
Peng and Akiyama have achieved a chiral phosphoric acid-catalysed asymmetric reduction of fluorinated alkynyl ketimines.75 The mild reaction conditions allow a chemoselective reaction, resulting in reduction of only the imine bond without affecting the triple bond. The chiral phosphoric acid catalysed approach involves a benzothiazole derivative as a hydrogen source and produces optically active fluorinated propargylamines in excellent ee (Fig. 61). Furthermore, the obtained optically active fluorinated propargylamines have been transformed into biologically active 2-(trifluoromethyl)-1,2-dihydroquinolines with selective COX-2 inhibitory activity.
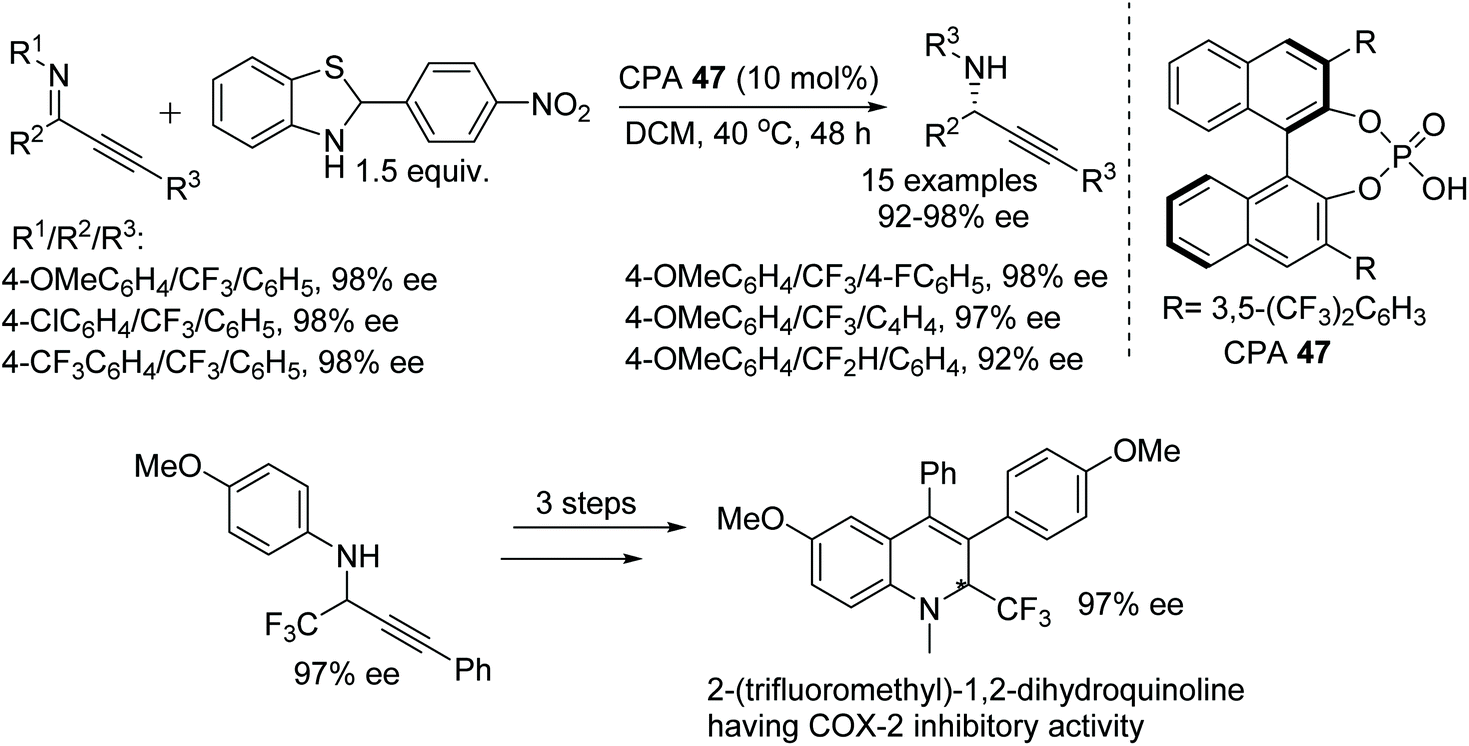 | ||
| Fig. 61 CPA catalysed asymmetric reductions of fluorinated alkynyl ketimines and conversion of product into 2-(trifluoromethyl)-1,2-dihydroquinoline. | ||
Speed et al. have synthesised a newly designed diazaphosphenium triflate catalyst 48 and applied it to the synthesis of chiral cyclic amines. A phosphenium cation-catalysed reaction proceeds through hydroboration or hydrosilylation of cyclic imines which gave the corresponding optically active heteroaryl pyrrolidines and piperidines in up to 94% ee at as low as 0.2% catalyst loading. This organocatalytic process tolerates imine substrates with thiophenes or pyridyl rings which in some cases are one of the limitations of metal catalysed ATH (Fig. 62).76
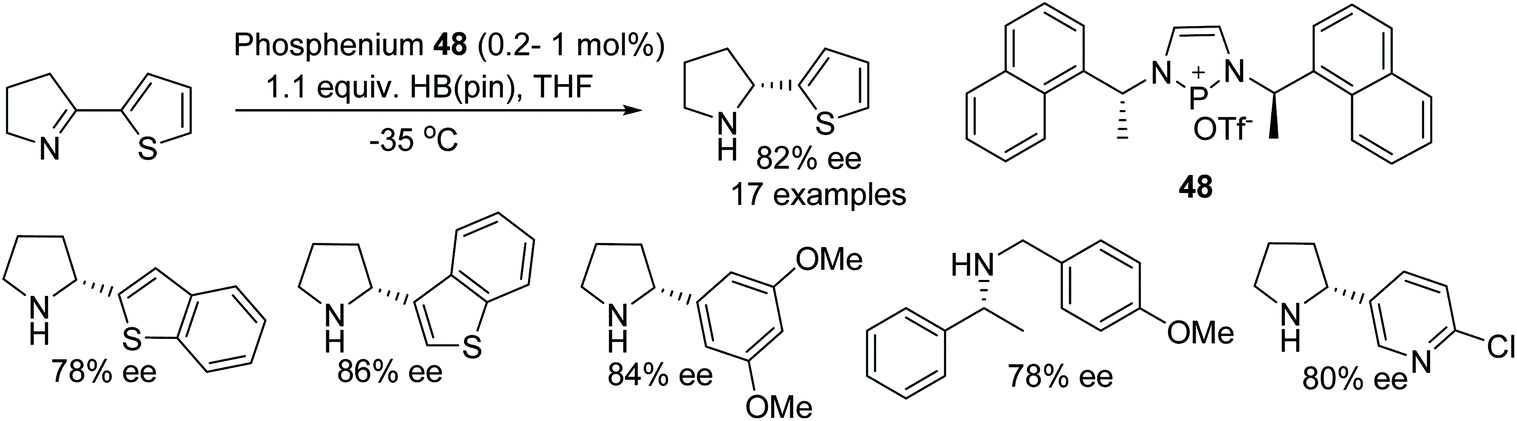 | ||
| Fig. 62 Phosphenium cation-catalysed ATH of cyclic imines. | ||
Wang et al. have enantioselectively hydrogenated 2-substituted quinoline-type compounds using newly developed chiral bicyclic bisborane catalysts.77 The borane 49 was employed in the hydrogenation of quinolines bearing an alkyl substituent (R2 = alkyl), Fig. 63, giving products with high enantioselectivity. The method was found to also tolerate both conjugated and unconjugated olefins, alkynes and consecutive double and triple bond substituents at R2. For 2-substituted quinolines with aryl groups, Borane 50 was found to give products in up to 93% ee. The reaction system in this case is assisted by the addition of [3,5-(CF3)2C6H3]3P and tolerates phenyls with both electron-donating and electron-withdrawing groups.
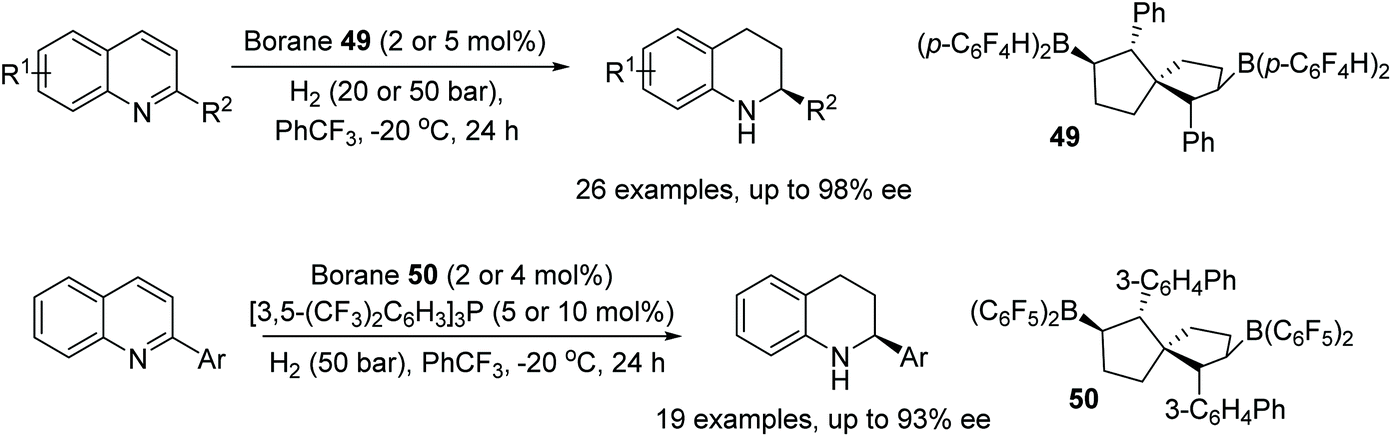 | ||
| Fig. 63 Chiral bicyclic bisborane catalysts for asymmetric quinoline hydrogenation. | ||
In a further application, Wang et al. applied chiral bisborane catalysts in the asymmetric reduction of 2-vinyl-substituted pyridines.78 In this procedure, hydroboration of the substituted pyridine occurs first to give a dihydropyridine intermediate which is subsequently converted to the enantiopure product through asymmetric transfer hydrogenation using borane 51. A range of 2-styryl-substituted pyridines were asymmetrically reduced in high ee, Fig. 64, as well as both conjugated and unconjugated double and triple bonds. In a comparative study with metal-catalysed hydrogenations it was found that these resulted in the reduction of both the pyridine and olefin. The method was also applied on a gram scale of two 2-vinyl-substituted pyridines.
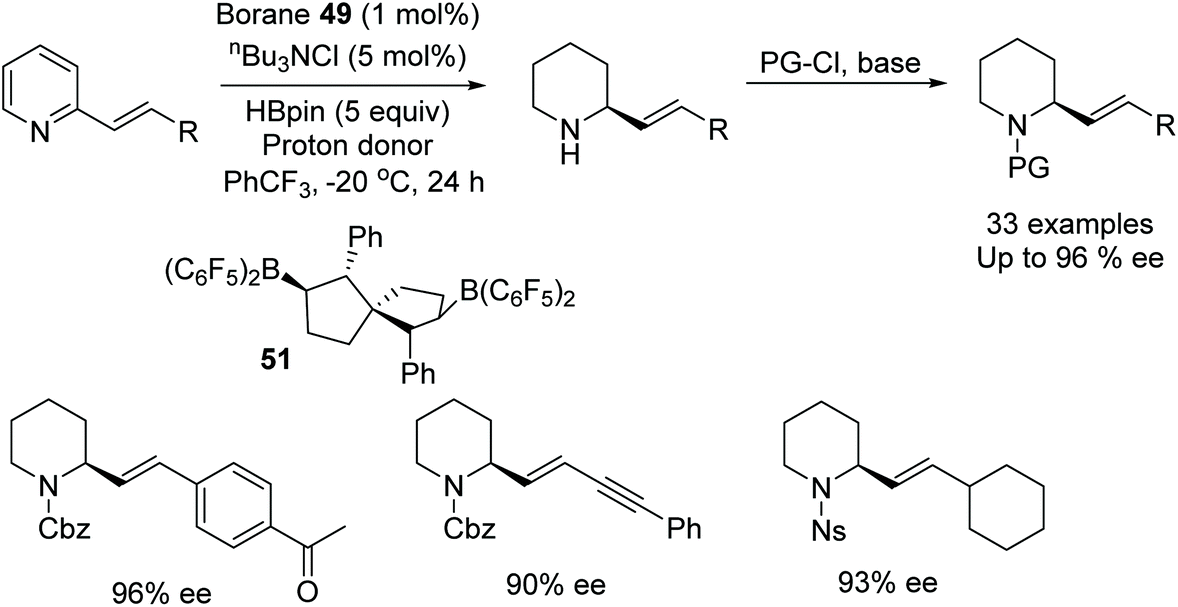 | ||
| Fig. 64 Chiral bisborane catalysts applied to the asymmetric reduction of 2-vinyl-substituted pyridines. | ||
5. Conclusions
In summary, a variety of methods have been applied to the reduction of CN bonds using AH, ATH and organocatalytic reagents. To date, significant breakthroughs have been achieved in the reduction of CN bonds using a wide range of catalysts, ligands, hydrogen sources and solvents. Asymmetric reductions benefits from the right design of catalyst, choice of hydrogen source, solvents etc. and the diversity of methods which has been applied to this transformation is very impressive, and highly innovative. Several approaches are now available to the synthetic chemist and many of them are very general. The application of asymmetric CN reduction has a major role in obtaining chiral fine chemicals, bioactive pharmaceuticals intermediated and many target molecules. Although significant research has been carried out on imine reduction using a variety of methods, the combination of this with other processes including cascade reactions could offer new discoveries in the near future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
GoldenKeys High-tech Materials Co., Ltd (Guizhou Science & Technology Department, Key Project No.: QianKeHeJiChu [2018]1406) and Warwick University are thanked for support of Jonathan Barrios-Rivera. We thank The Royal Society for funding VKV through an SERB Newton International Fellowship.References
- (a) S. E. Clapham, A. Hadzovic and R. H. Morris, Mechanisms of the H2-hydrogenation and transfer hydrogenation of polar bonds catalyzed by ruthenium hydride complexes, Coord. Chem. Rev., 2004, 248, 2201–2237 CrossRef CAS; (b) S. Gladiali and E. Alberico, Asymmetric transfer hydrogenation: chiral ligands and applications, Chem. Soc. Rev., 2006, 35, 226–236 RSC; (c) T. Ikariya, K. Murata and R. Noyori, Bifunctional transition metal-based molecular catalysts for asymmetric syntheses, Org. Biomol. Chem., 2006, 4, 393–406 RSC; (d) J. S. M. Samec, J.-E. Bäckvall, P. G. Andersson and P. Brandt, Mechanistic aspects of transition metal-catalyzed hydrogen transfer reactions, Chem. Soc. Rev., 2006, 35, 237–248 RSC; (e) M. Wills, in Modern reduction methods, ed. P. G. Andersson and I. J. Munslow, Wiley-VCH, Weinheim, 2008, ch. 11, p. 271 Search PubMed; (f) C. Wang, X. Wu and J. Xiao, Broader, Greener, and More Efficient: Recent Advances in Asymmetric Transfer Hydrogenation, Chem. – Asian J., 2008, 3, 1750–1770 CrossRef CAS; (g) T. C. Nugent and M. El-Shazly, Chiral Amine Synthesis- Recent Developments and Trends for Enamide Reduction, Reductive Amination, and Imine Reduction, Adv. Synth. Catal., 2010, 352, 753–819 CrossRef CAS; (h) C. Wang, B. Villa-Marcos and J. Xiao, Hydrogenation of imino bonds with half-sandwich metal catalysts, Chem. Commun., 2011, 47, 9773–9785 RSC; (i) J. Václavik, P. Kačer, M. Kuzma and L. Červený, Opportunities Offered by Chiral η6-Arene/N-Arylsulfonyl-diamine-RuII Catalysts in the Asymmetric Transfer Hydrogenation of Ketones and Imines, Molecules, 2011, 16, 5460–5495 CrossRef; (j) J. Václavik, P. Šot, B. Vilhanová, J. Pecháček, M. Kuzma and P. Kačer, Practical Aspects and Mechanism of Asymmetric Hydrogenation with Chiral Half-Sandwich Complexes, Molecules, 2013, 18, 6804–6828 CrossRef; (k) A. Bartoszewicz, N. Ahlsten and B. Martín-Matute, Enantioselective Synthesis of Alcohols and Amines by Iridium-Catalyzed Hydrogenation, Transfer Hydrogenation, and Related Processes, Chem. – Eur. J., 2013, 19, 7274–7302 CrossRef CAS; (l) J. Václavik, P. Šot, J. Pecháček, B. Vilhanová, O. Matuška, M. Kuzma and P. Kačer, Experimental and Theoretical Perspectives of the Noyori-Ikariya Asymmetric Transfer Hydrogenation of Imines, Molecules, 2014, 19, 6987–7007 CrossRef; (m) J. I. Ito and H. Nishiyama, Recent topics of transfer hydrogenation, Tetrahedron Lett., 2014, 55, 3153–3146 CAS; (n) D. Wang and D. Astruc, The Golden Age of Transfer Hydrogenation, Chem. Rev., 2015, 115, 6621–6686 CrossRef CAS; (o) K. Mashima, K. Higashida, A. Iimuro, H. Nagae and Y. Kita, Triply Halide-Bridged Dinuclear Iridium(III) Complexes with Chiral Diphosphine Ligands as New Easy-to-Handle Iridium Catalysts for Asymmetric Hydrogenation of Imines and N-Heteroaromatics, Chem. Rec., 2016, 16, 2585–2598 CrossRef CAS; (p) M. Wills, Imino Transfer Hydrogenation Reductions, Top. Curr. Chem., 2016, 374, 69–104 CrossRef; (q) H. G. Nedden, A. Zanotti-Gerosa and M. Wills, The Development of Phosphine-Free “Tethered” Ruthenium(II) Catalysts for the Asymmetric Reduction of Ketones and Imines, Chem. Rec., 2016, 16, 2619–2639 CrossRef CAS; (r) Y.-E. Luo, Y.-M. He and Q.-H. Fan, Asymmetric Hydrogenation of Quinoline Derivatives Catalyzed by Cationic Transition Metal Complexes of Chiral Diamine Ligands: Scope, Mechanism and Catalyst Recycling, Chem. Rec., 2016, 16, 2697–2711 CrossRef; (s) J. G. de Vries and N. Mršić, Organocatalytic asymmetric transfer hydrogenation of imines, Catal. Sci. Technol., 2011, 1, 727–735 RSC; (t) C. Zheng and S.-L. You, Transfer hydrogenation with Hantzsch esters and related organic hydride donors, Chem. Soc. Rev., 2012, 41, 2498–2518 RSC; (u) P. G. Echeverria, T. Ayad, P. Phansavath and V. Ratovelomanana-Vidal, Recent Developments in Asymmetric Hydrogenation and Transfer Hydrogenation of Ketones and Imines through Dynamic Kinetic Resolution, Synthesis, 2016, 48, 2523–2539 CrossRef CAS.
- (a) Z.-J. Yao, N. Lin, X.-C. Qiao, J.-W. Zhu and W. Deng, Cyclometalated half-sandwich iridium complex for catalytic hydrogenation of imines and quinolines, Organometallics, 2018, 37, 3883–3892 CrossRef CAS; (b) W. Wu, T. Seki, K. L. Walker and R. M. Waymouth, Transfer hydrogenation of aldehydes, allylic alcohols, ketones, and imines using molybdenum cyclopentadienone complexes, Organometallics, 2018, 37, 1428–1431 CrossRef CAS; (c) D. Wei, A. Bruneau-Voisine, M. Dubois, S. Bastin and J.-B. Sortais, Manganese-catalyzed transfer hydrogenation of aldimines, ChemCatChem, 2019, 11, 5256–5259 CrossRef CAS.
- Y. Ji, G.-S. Feng, M.-W. Chen, L. Shi, H. Du and Y.-G. Zhou, Iridium-catalyzed asymmetric hydrogenation of cyclic iminium salts, Org. Chem. Front., 2017, 4, 1125–1129 RSC.
- G. Feng, Z.-B. Zhao, L. Shi and Y.-G. Zhou, Iridium-catalyzed asymmetric hydrogenation of quinazolinones, Org. Chem. Front., 2019, 6, 2250–2253 RSC.
- H. Zhou, Y. Liu, S. Yang, L. Zhou and M. Chang, One-Pot N-Deprotection and Catalytic Intramolecular Asymmetric Reductive Amination for the Synthesis of Tetrahydroisoquinolines, Angew. Chem., Int. Ed., 2017, 56, 2725–2729 CrossRef CAS.
- H. Nie, Y. Zhu, X. Hu, Z. Wei, L. Yao, G. Zhou, P. Wang, R. Jiang and S. Zhang, Josiphos-Type Binaphane Ligands for Iridium-Catalyzed Enantioselective Hydrogenation of 1-Aryl-Substituted Dihydroisoquinolines, Org. Lett., 2019, 21, 8641–8645 CrossRef CAS.
- R. Schwenk and A. Togni, P-Trifluoromethyl ligands derived from Josiphos in the Ir-catalysed hydrogenation of 3,4-dihydroisoquinoline hydrochlorides, Dalton Trans., 2015, 44, 19566–19575 RSC.
- T. Yang, Q. Yin, G. Gu and X. Zhang, A one-pot process for the enantioselective synthesis of tetrahydroquinolines and tetrahydroisoquinolines via asymmetric reductive amination (ARA), Chem. Commun., 2018, 54, 7247–7250 RSC.
- D. Zhang, X. Zhou, R. Liu, M. Li, X. Li, R. Jiang, H. Nie and S. Zhang, Josiphos-type binaphane ligands for the asymmetric Ir-catalyzed hydrogenation of acyclic aromatic N-aryl imines, Catal. Commun., 2020, 136, 105906 CrossRef.
- J. Mazuela, T. Antonsson, L. Knerr, S. P. Marsden, R. H. Munday and M. J. Johansson, Iridium-Catalyzed Asymmetric Hydrogenation of N-Alkyl α-Aryl Furan-Containing Imines: an Efficient Route to Unnatural N-Alkyl Arylalanines and Related Derivatives, Adv. Synth. Catal., 2019, 361, 578–584 CrossRef CAS.
- G.-S. Feng, L. Shi, F.-J. Meng, M.-W. Chen and Y.-G. Zhou, Iridium-Catalyzed Asymmetric Hydrogenation of 4,6-Disubstituted 2-Hydroxypyrimidines, Org. Lett., 2018, 20, 6415–6419 CrossRef CAS.
- Y. Zhang, D. Kong, R. Wang and G. Hou, Synthesis of chiral cyclic amines via Ir-catalyzed enantioselective hydrogenation of cyclic imines, Org. Biomol. Chem., 2017, 15, 3006–3012 RSC.
- Y. Zhang, Q. Yan, G. Zi and G. Hou, Enantioselective Direct Synthesis of Free Cyclic Amines via Intramolecular Reductive Amination, Org. Lett., 2017, 19, 4215–4218 CrossRef CAS.
- H. Zhou, W. Zhao, T. Zhang, H. Guo, H. Huang and M. Chang, Synthesis, 2019, 51, 2713–2719 CrossRef CAS.
- D. Kong, M. Li, G. Zi, G. Hou and Y. He, Enantioselective Hydrogenation of Diarylmethanimines for Synthesis of Chiral Diarylmethylamines, J. Org. Chem., 2016, 81, 6640–6648 CrossRef CAS.
- S. B. Hu, X.-Y. Zhai, H.-Q. Shen and Y.-G. Zhou, Iridium-catalyzed Asymmetric Hydrogenation of Polycyclic Pyrrolo/Indolo[1,2-a]quinoxalines and Phenanthridines, Adv. Synth. Catal., 2018, 360, 1334–1339 CrossRef CAS.
- E. Salomó, A. Gallen, G. Sciortino, G. Ujaque, A. Grabulosa, A. Lledós, A. Riera and X. Verdaguer, Direct Asymmetric Hydrogenation of N-Methyl and N-Alkyl Imines with an Ir(III)H Catalyst, J. Am. Chem. Soc., 2018, 140, 16967–16970 CrossRef.
- E. Salomó, P. Rojo, P. Hernández-Lladó, A. Riera and X. Verdaguer, P-Stereogenic and Non-P-Stereogenic Ir-MaxPHOX in the Asymmetric Hydrogenation of N-Aryl Imines. Isolation and X-ray Analysis of Imine Iridacycles, J. Org. Chem., 2018, 83, 4618–4627 CrossRef.
- Y. Liu, Y. Huang, Z. Yi, G. Liu, X.-Q. Dong and X. Zhang, Enantioselective Access to Chiral Cyclic Sulfamidates Through Iridium-Catalyzed Asymmetric Hydrogenation, Adv. Synth. Catal., 2019, 361, 1582–1586 CrossRef CAS.
- Y. Chen, Y. Pan, Y.-M. He and Q.-H. Fan, Consecutive Intermolecular Reductive Amination/Asymmetric Hydrogenation: Facile Access to Sterically Tunable Chiral Vicinal Diamines and N-Heterocyclic Carbenes, Angew. Chem., 2019, 131, 16987–16990 CrossRef.
- T. Miao, B. Ma, Z. Ding, Y. Liu, Y.-M. He and Q.-H. Fan, Chemoselective and Enantioselective Hydrogenation of 2,4-Diaryl-3H-benzo[b]azepines Catalyzed by Dendritic Phosphinooxazoline Iridium Complexes, Asian J. Org. Chem., 2017, 6, 1219–1221 CrossRef CAS.
- B. Tutkowski, S. Kerdphon, E. Limé, P. Helquist, P. G. Andersson, O. Wiest and P.-O. Norrby, Revisiting the stereodetermining step in enantioselective iridium-catalyzed imine hydrogenation, ACS Catal., 2018, 8, 615–623 CrossRef CAS.
- T. Yang, X. Guo, Q. Yin and Z. Zhang, Intramolecular asymmetric reductive amination: synthesis of enantioenriched dibenz[c,e]azepines, Chem. Sci., 2019, 19, 2473–2477 RSC.
- Z. Wu, S. Du, G. Gao, W. Yang, X. Yang, H. Huang and M. Chang, Secondary amines as coupling partners in direct catalytic asymmetric reductive amination, Chem. Sci., 2019, 19, 4509–4514 RSC.
- J.-H. Xie, P.-C. Yan, Q.-Q. Zhang, K.-X. Yuan and Q.-L. Zhou, Asymmetric Hydrogenation of Cyclic Imines Catalyzed by Chiral Spiro Iridium Phosphoramidite Complexes for Enantioselective Synthesis of Tetrahydroisoquinolines, ACS Catal., 2012, 2, 561–564 CrossRef CAS.
- Y. Ji, L. Shi, M.-W. Chen, G.-S. Feng and Y.-G. Zhou, Concise Redox Deracemization of Secondary and Tertiary Amines with a Tetrahydroisoquinoline Core via a Nonenzymatic Process, J. Am. Chem. Soc., 2015, 137, 10496–10499 CrossRef CAS.
- P. Li, Y. Huang, X. Hu, X.-Q. Dong and X. Zhang, Access to chiral seven-member cyclic amines via Rh-catalyzed asymmetric hydrogenation, Org. Lett., 2017, 19, 3855–3858 CrossRef CAS.
- R. Borrmann, R. M. Koenigs, J. Zoller and M. Rueping, Asymmetric Hydrogenation of Cyclic Imines and Enamines: Access to 1, 5-Benzodiazepine Pharmacophores, Synthesis, 2017, 49, 310–318 CAS.
- D. Fan, Y. Hu, F. Jiang, Z. Zhang and W. Zhang, Rhodium-Catalyzed Chemo- and Enantioselective Hydrogenation of Alkynyl-Aryl Hydrazones, Adv. Synth. Catal., 2019, 360, 2228–2232 CrossRef.
- B. Vilhanová, J. Václavík, P. Šot, J. Pecháček, J. Zápal, R. Pažout, J. Maixner, M. Kuzma and P. Kačer, Enantioselective hydrogenation of cyclic imines catalysed by Noyori-Ikariya half-sandwich complexes and their analogues, Chem. Commun., 2016, 52, 362–365 RSC.
- Z. Yang, Z. Ding, F. Chen, Y.-M. He, N. Yang and Q.-H. Fan, Asymmetric Hydrogenation of Cyclic Imines of Benzoazepines and Benzodiazepines with Chiral, Cationic Ruthenium-Diamine Catalysts, Eur. J. Org. Chem., 2017, 1973–1977 CrossRef CAS.
- L.-R. Wang, D. Chang, Y. Feng, Y.-M. He, G.-J. Deng and Q.-H. Fan, Highly Enantioselective Ruthenium-Catalyzed Cascade Double Reduction Strategy: Construction of Structurally Diverse Julolidines and Their Analogues, Org. Lett., 2020, 22, 2251–2255 CrossRef CAS.
- B. Li, C. Xu, Y.-M. He, G.-J. Deng and Q.-H. Fan, Asymmetric Hydrogenation of Bis (quinolin-2-yl) methanes: A Direct Access to Chiral 1, 3-Diamines, Chin. J. Chem., 2018, 36, 1169–1173 CrossRef CAS.
- W. Ma, J. Zhang, C. Xu, F. Chen, Y.-M. He and Q.-H. Fan, Highly Enantioselective Direct Synthesis of Endocyclic Vicinal Diamines through Chiral Ru (diamine)-Catalyzed Hydrogenation of 2, 2′-Bisquinoline Derivatives, Angew. Chem., Int. Ed., 2016, 55, 12891–12894 CrossRef CAS.
- T. Wang, Y. Chen, G. Ouyang, Y.-M. He, Z. Li and Q.-H. Fan, Solvent-Regulated Asymmetric Hydrogenation of Quinoline Derivatives in Oligo (Ethylene Glycol) s through Host-Guest Interactions, Chem. – Asian J., 2016, 11, 2773–2777 CrossRef CAS.
- S. Zhang, F. Chen, Y.-M. He and Q.-H. Fan, Asymmetric Hydrogenation of Dibenzo[c,e]azepine Derivatives with Chiral Cationic Ruthenium Diamine Catalysts, Org. Lett., 2019, 21, 5538–5541 CrossRef CAS.
- Z.-Y. Ding, T. Wang, Y.-M. He, F. Chen, H.-F. Zhou, Q.-H. Fan, Q. Guo and A. S. C. Chan, Highly Enantioselective Synthesis of Chiral Tetrahydroquinolines and Tetrahydroisoquinolines by Ruthenium-Catalyzed Asymmetric Hydrogenation in Ionic Liquid, Adv. Synth. Catal., 2013, 355, 3727–3735 CrossRef CAS.
- Y. Chen, Y.-M. He, S. Zhang, T. Miao and Q.-H. Fan, Rapid Construction of Structurally Diverse Quinolizidines, Indolizidines, and Their Analogues via Ruthenium-Catalyzed Asymmetric Cascade Hydrogenation/Reductive Amination, Angew. Chem., Int. Ed., 2019, 58, 3809–3813 CrossRef CAS.
- C. Xu, Y. Feng, F. Li, J. Han, Y.-M. He and Q.-H. Fan, A synthetic route to chiral benzo-fused N-heterocycles via sequential intramolecular hydroamination and asymmetric hydrogenation of anilino-alkynes, Organometallics, 2019, 38, 3979–3990 CrossRef CAS.
- Z. Yang, F. Chen, S. Zhang, Y. He, N. Yang and Q.-H. Fan, Ruthenium-catalyzed enantioselective hydrogenation of phenanthridine derivatives, Org. Lett., 2017, 19, 1458–1461 CrossRef CAS.
- W. Ma, F. Chen, Y. Liu, Y.-M. He and Q.-H. Fan, Ruthenium-catalyzed enantioselective hydrogenation of 1,8-naphthyridine derivatives, Org. Lett., 2016, 18, 2730–2733 CrossRef CAS.
- X. Tan, S. Gao, W. Zeng, S. Xin, Q. Yin and X. Zhang, Asymmetric Synthesis of Chiral Primary Amines by Ruthenium-Catalyzed Direct Reductive Amination of Alkyl Aryl Ketones with Ammonium Salts and Molecular H2, J. Am. Chem. Soc., 2018, 140, 2024–2027 CrossRef CAS.
- B. Li, J. Chen, Z. Zhang, I. D. Gridnev and W. Zhang, Nickel-Catalyzed Asymmetric Hydrogenation of N-Sulfonyl Imines, Angew. Chem., 2019, 58, 7329–7334 CrossRef CAS.
- Y. Liu, Z. Yi, X. Tan, X.-Q. Dong and X. Zhang, Nickel-Catalyzed Asymmetric Hydrogenation of Cyclic Sulfamidate Imines: Efficient Synthesis of Chiral Cyclic Sulfamidates, iScience, 2019, 19, 63–73 CrossRef CAS.
- Y. Gao, F. Yang, D. Pu, R. D. Laishram, R. Fan, G. Shen, X. Zhang, J. Chen and B. Fan, Pd/Zn(OTf)2 C o-Catalyzed Asymmetric Hydrogenation of Imines under Normal Pressure of Hydrogen, Eur. J. Org. Chem., 2018, 6274–6279 CrossRef CAS.
- D. Brenna, S. Rossi, F. Cozzia and M. Benaglia, Iron catalyzed diastereoselective hydrogenation of chiral imines, Org. Biomol. Chem., 2017, 15, 5685–5688 RSC.
- C. S. G. Seo, T. Tannoux, S. A. M. Smith, A. J. Lough and R. H. Morris, Enantioselective Hydrogenation of Activated Aryl Imines Catalyzed by an Iron(II) P-NH-P′ Complex, J. Org. Chem., 2019, 84, 12040–12049 CrossRef CAS.
- K.-Y. Ye, X. Wang, C. G. Daniliuc, G. Kehr and G. Erker, A Ferrocene-Based Phosphane/Borane Frustrated Lewis Pair for Asymmetric Imine Reduction, Eur. J. Inorg. Chem., 2017, 2, 368–371 CrossRef.
- G.-S. Feng, M.-W. Chen, L. Shi and Y.-G. Zhou, Facile Synthesis of Chiral Cyclic Ureas through Hydrogenation of 2-Hydroxypyrimidine/Pyrimidin-2(1H)-one Tautomers, Angew. Chem., Int. Ed., 2018, 57, 5853–5857 CrossRef CAS.
- J. M. Mwansa, M. J. Stirling and M. I. Page, Changing the kinetic order of enantiomer formation and distinguishing between iminium ion and imine as the reactive species in the asymmetric transfer hydrogenation of substituted imines using a cyclopentadienyl iridium(III) complex, Pure Appl. Chem., 2020, 92, 107–121 CAS.
- G. Facchetti, S. Pellegrino, R. Bucci, D. Nava, R. Gandolfi, M. S. Christodoulou and I. Rimoldi, Vancomycin-Iridium(III) Interaction: An Unexplored Route for Enantioselective Imine Reduction, Molecules, 2019, 24, 2771 CrossRef.
- Y. Li, M. Lei, W. Yuan, E. Meggers and L. Gong, An N-heterocyclic carbene iridium catalyst with metal-centered chirality for enantioselective transfer hydrogenation of imines, Chem. Commun., 2017, 53, 8089–8092 RSC.
- G. Facchetti, R. Bucci, M. Fusè and I. Rimoldi, Asymmetric Hydrogenation vs Transfer Hydrogenation in the Reduction of Cyclic Imines, ChemistrySelect, 2018, 3, 8797–8800 CrossRef CAS.
- B. V. Vilhanová, A. Budinská, J. Václavík, V. Matouše, M. Kuzma and L. Červený, Asymmetric Transfer Hydrogenation of 1-Aryl-3,4-Dihydroisoquinolines Using a Cp*Ir(TsDPEN) Complex, Eur. J. Org. Chem., 2017, 5131–5134 CrossRef.
- H.-J. Pan, Y. Zhang, C. Shan, Z. Yu, Y. Lan and Y. Zhao, Asymmetric Transfer Hydrogenation of Imines using Alcohol: Efficiency and Selectivity are Influenced by the Hydrogen Donor, Angew. Chem., Int. Ed., 2016, 55, 9615–9619 CrossRef CAS.
- (a) T. Quinto, F. Schwizer, J. M. Zimbron, A. Morina, V. Koehler and T. R. Ward, Expanding the Chemical Diversity in Artificial Imine Reductases Based on the Biotin-Streptavidin Technology, ChemCatChem, 2014, 6, 1010–1014 CrossRef CAS; (b) F. Schwizer, V. Kohler, M. Durrenberger, L. Knorr and T. R. Ward, Genetic Optimization of the Catalytic Efficiency of Artificial Imine Reductases Based on Biotin-Streptavidin Technology, ACS Catal., 2013, 3, 1752–1755 CrossRef CAS.
- H. R. Kim, R. Achary and H.-K. Lee, DBU-Promoted Dynamic Kinetic Resolution in Rh-Catalyzed Asymmetric Transfer Hydrogenation of 5-Alkyl Cyclic Sulfamidate Imines: Stereoselective Synthesis of Functionalized 1,2-Amino Alcohols, J. Org. Chem., 2018, 83, 11987–11999 CrossRef CAS.
- A. Nova, D. J. Taylor, A. J. Blacker, S. B. Duckett, R. N. Perutzmand and O. Eisenstein, Computational Studies Explain the Importance of Two Different Substituents on the Chelating Bis(amido) Ligand for Transfer Hydrogenation by Bifunctional Cp*Rh(III) Catalysts, Organometallics, 2014, 33, 3433–3442 CrossRef CAS.
- E. Selva, Y. Sempere, D. Ruiz-Martínez, Ó. Pablo and D. Guijarro, Synthesis of Allylic Amines by Asymmetric Transfer Hydrogenation of α,β-Unsaturated N-(tert-Butylsulfinyl)imines, J. Org. Chem., 2017, 82, 13693–13699 CrossRef CAS.
- C. E. Puerto Galvis and V. V. Kouznetsov, Biomimetic Total Synthesis of Dysoxylum Alkaloids, J. Org. Chem., 2019, 84, 15294–15308 CrossRef CAS.
- P. Šot, B. Vilhanova, B. J. Pecháček, J. Václavík, J. Zápal, M. Kuzma and P. Kačer, The role of the aromatic ligand in the asymmetric transfer hydrogenation of the CN bond on Noyori's chiral Ru catalysts, Tetrahedron: Asymmetry, 2014, 25, 1346–1351 CrossRef.
- R. Hrdličková, J. Zápal, B. V. Vilhanová, M. Bugáňová, K. Truhlářová, M. Kuzma and L. Červený, Competitive asymmetric transfer hydrogenation of 3,4-dihydroisoquinolines employing Noyori-Ikariya catalytic complexes, React. Kinet., Mech. Catal., 2018, 124, 701–710 CrossRef.
- (a) J. Přech, J. Václavík, P. Šot, J. Pecháček, B. Vilhanová, J. Januščák, K. Syslová, R. Pažout, J. Maixner, J. Zápal, M. Kuzma and P. Kačer, Asymmetric transfer hydrogenation of 1-phenyl dihydroisoquinolines using Ru(II) diamine catalysts, Catal. Commun., 2013, 36, 67–70 CrossRef; (b) Z. Wu, M. Perez, M. Scalone, T. Ayad and V. Ratovelomanana-Vidal, Ruthenium-Catalyzed Asymmetric Transfer Hydrogenation of 1-Aryl-Substituted Dihydroisoquinolines: Access to Valuable Chiral 1-Aryl-Tetrahydroisoquinoline Scaffolds, Angew. Chem., Int. Ed., 2013, 52, 4925–4928 CrossRef CAS; (c) M. Perez, Z. Wu, M. Scalone, T. Ayad and V. Ratovelomanana-Vidal, Enantioselective Synthesis of 1-Aryl-Substituted Tetrahydroisoquinolines Through Ru-Catalyzed Asymmetric Transfer Hydrogenation, Eur. J. Org. Chem., 2015, 6503–6514 CrossRef CAS.
- J. Barrios-Rivera, Y. Xu and M. Wills, Asymmetric Transfer Hydrogenation of Unhindered and Non-Electron-Rich 1-Aryl Dihydroisoquinolines with High Enantioselectivity, Org. Lett., 2020, 22, 6283–6287 CrossRef CAS.
- A. M. Palvoelgyi, J. Bitai, V. Zeindlhofer, C. Schroeder and K. Bica, Ion-Tagged Chiral Ligands for Asymmetric Transfer Hydrogenations in Aqueous Medium, ACS Sustainable Chem. Eng., 2019, 7, 3414–3423 CrossRef CAS.
- G. V. More and B. M. Bhanage, Ru-Catalyzed asymmetric transfer hydrogenation of substituted dibenzo [b, f][1, 4] oxazepines in water, Org. Biomol. Chem., 2017, 15, 5263–5267 RSC.
- R. J. Chew and M. Wills, Exploitation of differential electronic densities for the stereoselective reduction of ketones bearing a masked amino surrogate, J. Catal., 2018, 361, 40–44 CrossRef CAS.
- P. Roszkowski, J. K. Maurin and Z. Czarnocki, (-)-Menthol as a source of new N, N-diamine ligands for asymmetric transfer hydrogenation, Tetrahedron Lett., 2018, 59, 2184–2188 CrossRef CAS.
- F. Yang, J. Chen, G. Shen, X. Zhanga and B. Fan, Asymmetric transfer hydrogenation reactions of N-sulfonylimines by using alcohols as hydrogen sources, Chem. Commun., 2018, 54, 4963–4966 RSC.
- X. Liu, T. Liu, W. Meng and H. Du, Asymmetric hydrogenation of imines with chiral alkene-derived boron Lewis acids, Org. Biomol. Chem., 2018, 16, 8686–8689 RSC.
- S. Li, W. Meng and H. Du, Asymmetric transfer hydrogenations of 2, 3-disubstituted quinoxalines with ammonia borane, Org. Lett., 2017, 19, 2604–2606 CrossRef CAS.
- W. Zhao, X. Feng, J. Yang and H. Du, Asymmetric transfer hydrogenations of β-N-substituted enamino esters with ammonia borane, Tetrahedron Lett., 2019, 60, 1193–1196 CrossRef CAS.
- Q. Zhou, W. Meng, J. Yang and H. Du, A Continuously Regenerable Chiral Ammonia Borane for Asymmetric Transfer Hydrogenations, Angew. Chem., Int. Ed., 2018, 57, 12111–12115 CrossRef CAS.
- K. Yang, Y. Lou, C. Wang, L.-W. Qi, T. Fang, F. Zhang, H. Xu, L. Zhou, W. Li, G. Zhang, P. Yu and Q. Song, Chiral Brønsted Acid from Chiral Phosphoric Acid Boron Complex and Water: Asymmetric Reduction of Indoles, Angew. Chem., 2020, 59, 3294–3299 CrossRef CAS.
- M. Miyagawa, K. Takashima and T. Akiyama, Asymmetric reduction of trifluoromethyl alkynyl ketimines by chiral phosphoric acid and benzothiazoline, Synlett, 2018, 29, 1607–1610 CrossRef CAS.
- T. Lundrigan, E. N. Welsh, T. Hynes, C.-H. Tien, M. R. Adams, K. R. Roy, K. N. Robertson and A. W. H. Speed, Enantioselective Imine Reduction Catalyzed by Phosphenium Ions, J. Am. Chem. Soc., 2019, 141, 14083–14088 CrossRef CAS.
- X. Li, J.-J. Tian, N. Liu, X.-S. Tu, N.-N. Zeng and X.-C. Wang, Spiro-Bicyclic Bisborane Catalysts for Metal-Free Chemoselective and Enantioselective Hydrogenation of Quinolines, Angew. Chem., Int. Ed., 2019, 58, 4664–4668 CrossRef CAS.
- J.-J. Tian, Z.-Y. Yang, X.-S. Liang, N. Liu, C.-Y. Hu, X.-S. Tu, X. Li and X.-C. Wang, Borane-Catalyzed Chemoselective and Enantioselective Reduction of 2-Vinyl-Substituted Pyridines, Angew. Chem., Int. Ed., 2020, 59 DOI:10.1002/anie.202007352.
| This journal is © the Partner Organisations 2020 |