DOI:
10.1039/D0NR02525A
(Paper)
Nanoscale, 2020,
12, 11908-11915
Hybrid phase 1T/2H-MoS2 with controllable 1T concentration and its promoted hydrogen evolution reaction†
Received
30th March 2020
, Accepted 6th May 2020
First published on 6th May 2020
Abstract
MoS2 has been investigated as a low-cost alternative to Pt in the electrochemical hydrogen evolution reaction. One of the promising methods to further activate MoS2 is phase engineering. MoS2 generally exhibits two kinds of crystalline phases: hexagonal 2H phase and octahedral 1T phase. 1T-MoS2 exhibits much better chemical/physical properties than natural semiconductor 2H-MoS2. However, 1T-MoS2 is metastable and its synthesis is still a challenge. Hybrid 1T/2H-MoS2 has been synthesized under relatively mild conditions, but controlling the 1T/2H ratio is still an issue which has not been discussed in detail. In this study, the synthesis methods of hybrid phase 1T/2H-MoS2 with controllable 1T concentration are investigated. The electrochemical hydrogen evolution reaction is then evaluated for 1T/2H-MoS2 with different 1T concentrations by performing both experiments and theoretical calculations.
Introduction
Molybdenum sulfide (MoS2) is a transition metal dichalcogenide that exhibits a graphite-like 2-dimensional structure, planar electric transportation and tunable bandgap between 1.3 V and 1.9 V.1–5 Recently, research studies have focused on the application of MoS2 in various fields such as energy storage, electro/photocatalysis and biotherapy.6–9 Natural MoS2 is composed of an S–Mo–S sandwich structure, known as hexagonal 2H phase (semiconductor phase). Besides natural 2H-MoS2, there is metastable octahedral 1T (metallic) phase MoS2 with higher catalytic activity than 2H-MoS2.1,3,10 However, pure 1T-MoS2 exhibits fatal defects: metastable state and requirement of extremely harsh synthesis conditions, which strictly limit its application.11–13 As a solution, research studies recently have found that by inserting 1T-MoS2 into 2H-MoS2, the number of catalytically active sites in MoS2 could be increased efficiently while maintaining the stability.
A promising application of 1T/2H-MoS2 is the electrochemical hydrogen evolution reaction (HER). To meet the growing demand for renewable energy, H2 has been regarded as an essential fuel candidate in the near future.14,15 Though the most active catalyst in the electrochemical HER in acidic media is Pt or Pt alloys,16,17 a cheaper substitute candidate is needed when considering the cost and scarcity of Pt. MoS2 has been researched as a promising candidate for HER catalyst, especially 1T-MoS2 which is active on both the basal and edge planes (natural 2H-MoS2 is only active on the edge plane).10,18–20 But the high synthesis cost and poor stability of 1T-MoS2 make it burdensome to use as a low-cost alternative to Pt. For this reason, 1T/2H-MoS2 has more potential for practical application due to its simple synthesis conditions and much better HER performance than 2H-MoS2.3,21,22
Latest studies have found that by inserting guest ions into MoS2 1T/2H-MoS2 could be synthesized. Wang et al.23 and Huang et al.24 have researched the crystalline structure distortion mechanism by inserting Li+ or Na+ ions. The charge transfers between the injected electron and Mo4+ ions result in S plane sliding and finally lead to a phase transition. Based on the above mechanism, researchers have further investigated that under milder synthesis conditions, NH4+ could permeate into MoS2 instead of Li+ or Na+.3,25–27 Using an ammonium-containing precursor through a hydrothermal process, NH4+ could be easily inserted into MoS2, thereby creating 1T/2H-MoS2.
Up to now, several methods have been developed to obtain 1T/2H-MoS2 usually at a fixed 1T concentration.26–29 Research on controlling and optimizing the 1T/2H ratio in the HER reaction is still unreported. In this work, we discuss the effects of different precursors used in the synthesis of hybrid phase MoS2 containing different concentrations of 1T phase. Their HER performances are then tested. In order to further understand the relationship between HER performances and 1T concentration, we have carried out a series of DFT calculations to clarify the different activities between 1T-MoS2 and 2H-MoS2 and for the first time, we have investigated the nature of 1T/2H interfacial reactions.
Experimental
Chemicals
(NH4)6Mo7O24·6H2O (ammonium molybdate tetrahydrate), NaMoO4·2H2O (sodium molybdate dihydrate), CH3CSNH2 (thioacetamide, TAA), N,N-dimethylformamide (DMF), NH4HCO3 (ammonium bicarbonate) and H2SO4 (sulphuric acid, 98%) were purchased from Nacalai Tesque Inc. Nafion 117 solution and 10% Pt on carbon were purchased from Sigma-Aldrich Co. Commercial MoS2 was purchased from Alfa Aesar Co. Ltd. All chemical reagents were used without any further purification.
Catalyst synthesis
Hybrid phase MoS2 was synthesized by a solvothermal method using precursors with different NH4+ contents. In this work, we discuss the effects of solvent (H2O or DMF) and guest ion agent (NH4HCO3).
Synthesis of MoS2.
In a typical synthesis process, 0.0028 mmol sodium molybdate was mixed with 0.025 mmol TAA and dissolved in 40 mL of distilled water, and then the solution was transferred to a 50 mL Teflon-lined stainless steel autoclave and placed in an oven. The hydrothermal reaction was carried out for 12 h at 200 °C. The black products were then collected by centrifugation and washed with water and ethanol. The sample was named MoS2.
Synthesis of 1T/2H-MoS2 (DMF).
In a typical synthesis process, 0.02 mmol ammonium molybdate and 0.025 mol TAA were mixed and then dissolved in an aqueous solution containing different DMF contents (25%, 50%, 75%, and 100%). The solution was then stirred for 15 min and transferred to a 50 mL Teflon-lined stainless steel autoclave and placed in an oven. The solvothermal reaction was carried out for 12 h at 200 °C. The black products were then collected by centrifugation and washed with water and ethanol. The samples were named 1T/2H-MoS2 (D25), 1T/2H-MoS2 (D50), 1T/2H-MoS2 (D75) and 1T/2H-MoS2 (D100), respectively.
Synthesis of 1T/2H-MoS2 (NH4+).
In a typical synthesis process, 0.02 mmol ammonium molybdate and different amounts (50 mg, 100 mg, 200 mg, 400 mg, 800 mg, and 1600 mg) of guest ion agent (ammonium bicarbonate) were mixed and dissolved in 20 mL of distilled water. The solution was then stirred for 15 min. 0.025 mol TAA was dissolved in 20 mL of distilled water and stirred for 15 min separately. After that, these two solutions were mixed and stirred for another 5 min and then transferred to a 50 mL Teflon-lined stainless steel autoclave and placed in an oven. The hydrothermal reaction was carried out for 12 h at 200 °C. The black products were then collected by centrifugation and washed with water and ethanol. The samples were named 1T/2H-MoS2 (Nx), where x = the mass of ammonium bicarbonate (mg).
Characterization
Thermogravimetric (TG) analysis was performed using a Bruker TG-DTA2010SA system from room temperature to 800 °C at a heating rate of 10 °C·min−1. X-ray photoelectron spectroscopy (XPS) spectra were recorded on a Shimadzu ESCA 3200 photoelectron spectrometer using Mg Kα radiation, and C 1s (284.6 eV) was used as the reference to calibrate the peak position of the element. X-ray diffraction (XRD) spectra were obtained using a Rigaku Ultima IV diffractometer using Cu Kα radiation (λ = 0.15406 nm). Mo K-edge X-ray absorption fine structure (XAFS) measurements were performed in a transmission mode at the beamline 01B1 station with an attached Si(111) monochromator at SPring-8, JASRI, Harima, Japan (prop. no. 2018A1144, 2018A1089). Field emission scanning electron microscopy (FE-SEM) was performed using a JSM-6500F (JEOL Ltd). High-resolution TEM micrographs were obtained with a TEM instrument (Hf-2000, Hitachi) equipped with an energy-dispersive X-ray detector (Kevex) operated at 200 kV. The specific surface area (SBET) and total pore volume (Vtotal) were determined using the N2 adsorption isotherm data obtained at −196 °C using a BELSORP-max system (MicrotracBEL Corp.)
Electrochemical measurements
Electrochemical measurements were performed using an HZ-7000 electrochemical workstation. The hydrogen evolution reactions were evaluated using a three-electrode system in 0.5 M H2SO4 with a graphite rod and a saturated Ag/AgCl electrode as the counter electrode and the reference electrode, respectively. The linear sweep potential tests run from 0.00 to −1.20 V (vs. Ag/AgCl) at a scanning rate of 10 mV s−1 in the three electrode system. The working electrode was prepared on a glassy carbon electrode. The catalyst ink was made by dissolving 5 mg of catalyst in 1 mL of the water–ethanol (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 volume ratio) solvent with 40 μL of 5% Nafion solution. The Pt/C ink was made by dissolving 5 mg of 10% Pt/C in 1 mL of the water–DMF (1:1 volume ratio) solvent with 40 μL of 5% Nafion solution.
:1 volume ratio) solvent with 40 μL of 5% Nafion solution. The Pt/C ink was made by dissolving 5 mg of 10% Pt/C in 1 mL of the water–DMF (1:1 volume ratio) solvent with 40 μL of 5% Nafion solution.
DFT calculations
All DFT calculations were performed using the Dmol3 program in the Materials Studio 17.2 software package using the B3LYP functional combined with the DND basis set. The (100) surface of 2H-MoS2, 1T-MoS2 and 1T/2H-MoS2 were modeled using a 4 × 1 supercell cluster model. The bottom S layer and Mo layer were fixed in their bulk positions, whereas the top layer of S atoms and adsorbates were allowed to relax during geometry optimization.
Results and discussion
In this work, we have tried to synthesize MoS2 with different 1T/2H ratios to achieve a high HER performance in a controllable way. The conception of 1T/2H-MoS2 synthesis can be summarized as the insertion of guest ions.26–28 Herein, two methods for the synthesis of 1T/2H-MoS2 with high 1T content are discussed.
Solvothermal process using N,N-dimethylformamide (DMF)
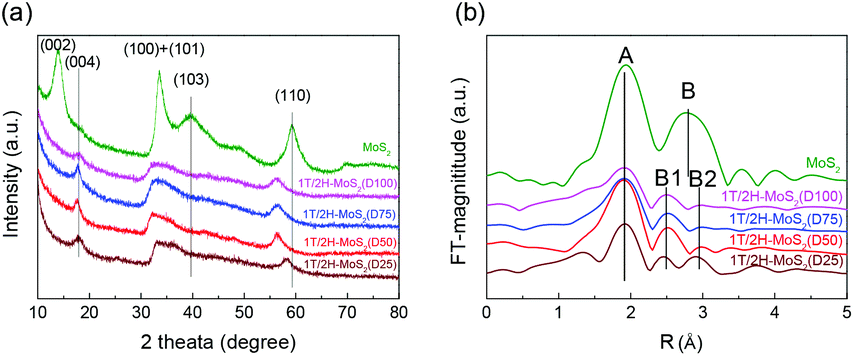
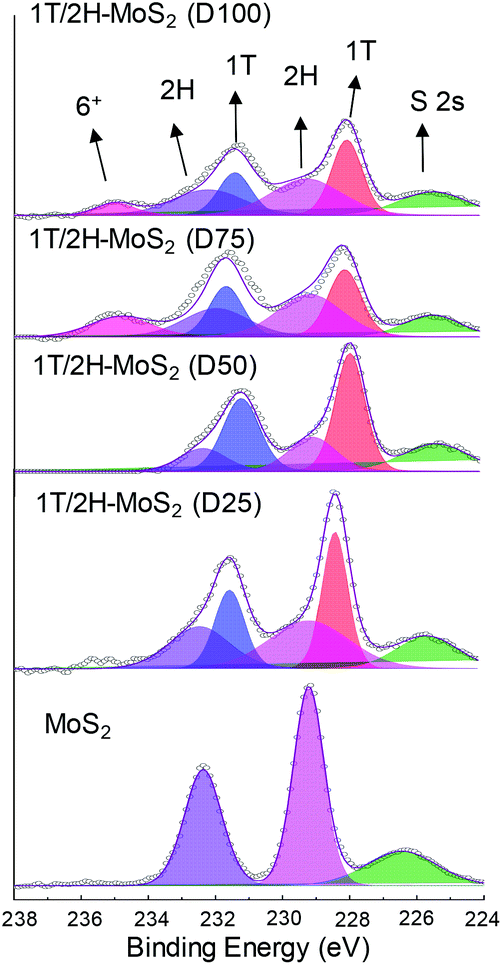
N,N-Dimethylformamide with an acyl group could provide NH4+ during the solvothermal process, and also its better solubility (than water) could be helpful for the reaction. Firstly, the 1T concentration is controlled by using different DMF/H2O solvents in a solvothermal process. Fig. 1 shows the XRD patterns and Mo K-edge FT-EXAFS spectra of the as-prepared samples. The characteristic peaks of 2H-MoS2 can be observed in the as-prepared MoS2 sample, including the (002) (2θ = 14.4°), (100) (2θ = 32.7°), (101) (2θ = 33.5°), (103) (2θ = 39.6°), and (110) (2θ = 58.3°) planes (MoS2 card from JCPDS 65-1951). For samples synthesized using DMF, the absence of the (002) and (103) planes, together with a newly-emerged (004) plane at 17.8° refers to the 1T-MoS2 pattern.11,29 Besides, a broadening of the (100) and (101) planes, and a shift to the lower angle of the (110) plane could be observed, which correspond to the distortion of the inserted 1T-MoS2 crystalline phase.3,27 The distortion level of the crystalline structure can be roughly judged by the amount of shift of the (110) plane. From MoS2 to 1T/2H-MoS2 (D50), the lattice distortion is gradually increased by increasing the amount of DMF. However, 1T/2H-MoS2 (D75) and 1T/2H-MoS2 (D100) almost exhibit the same shift level as 1T/2H-MoS2 (D50). X-ray absorption fine structure (XAFS) spectroscopy also provides evidence for the synthesized 1T/2H-MoS2. Mo K-edge X-ray absorption near edge structure (XANES) results have been provided in Fig. S1.† Samples synthesized using DMF show a similar absorption edge to MoS2, which indicates that the chemical valence of Mo is +4. A shoulder peak at 20100 eV can be observed in the MoS2 sample but disappears in samples synthesized using DMF, demonstrating that the lattice structure is changed owing to the inserted 1T phase.3,27,30–32 To investigate the detail of distortion, Fourier-transforms of extended X-ray absorption fine structure (FT-EXAFS) spectra are obtained (Fig. 1b). The FT magnitude of the nearest coordination sphere of the chemically bonded Mo–Mo (peak B) sphere split into two contributions in samples synthesized using DMF, which is due to the formation of a hybrid 1T/2H phase structure.30,33 It is worth noting that the intensity of peak B2 is gradually decreased by increasing the amount of DMF and finally the peak disappears in 1T/2H-MoS2 (D75) and 1T/2H-MoS2 (D100). Also, the intensity of peak A (corresponding to Mo-S) decreases, and the shape of peak A becomes wider by increasing the amount of DMF. These facts indicate that the crystalline structure would be destroyed by the excessive (more than 75%) DMF precursor. The TG/DTA is measured under air flow and the measured data are shown in Fig. S2.† The MoS2 sample showed only one endothermic peak with a mass loss at around 420 °C, which corresponds to the oxidation of MoS2.25 Different from MoS2, 1T/2H-MoS2 (D25) and 1T/2H-MoS2 (D50) samples showed a mass loss at around 200 °C with two DTA peaks at around 230 °C and 250 °C, which represent the deintercalation of NH4+ and a phase change from 1T to 2H, respectively.3,26,27 It should be noted that 1T/2H-MoS2 (D75) and 1T/2H-MoS2 (D100) exhibited only one endothermic peak around 230–250 °C, possibly due to the destroyed crystal lattice structure as shown in the FT-EXAFS spectra. The hybridized 1T/2H structure could also be identified in Mo 3d and S 2p XPS spectra as shown in Fig. 2 and Fig. S3.† In the Mo 3d spectrum of MoS2, resonance at 229.3 eV and 232.2 eV correspond to typical 3d5/2 and 3d3/2 peaks of 2H-MoS2, respectively. Nevertheless, two new peaks emerged at lower binding energy in samples synthesized using DMF. The new peaks at around 228.2 eV and 231.3 eV correspond to 1T-MoS2 in the hybrid 1T/2H structure.33–37 In addition, the Mo6+ 3d5/2 peak appeared in 1T/2H-MoS2 (D75) and 1T/2H-MoS2 (D100) spectra, indicating that MoS2 is partly oxidized under these conditions. Furthermore, the Mo6+ peak ratio is lower in 1T/2H-MoS2 (D100) than in 1T/2H-MoS2 (D75), which is because of the reducibility of DMF. S 2p spectra also show similar results to Mo 3d spectra. The existence of the 1T/2H hybrid structure could be directly observed by high-resolution TEM (HR-TEM). The HR-TEM image of 1T/2H-MoS2 is shown in Fig. 3(a) and its fast Fourier transform pattern is shown in Fig. 3(b). To identify the positions of the existing different lattice structures, the inverse Fourier transform images that emphasized the periodicity of the (110) plane were obtained at different frequencies, as shown in Fig. S4(c) and (d).† From these distribution images, area 1 and area 2 were chosen for further analysis. The hexagonal lattice region could be observed clearly in area 1 with a crystalline space of 0.27 nm, which is ascribed to the (100) plane of 2H-MoS2 (Fig. 3(c) and (e)). Otherwise, the trigonal lattice region in area 2 that showed a crystalline space of 0.24 nm is indexed to the (100) plane of 1T-MoS2 (Fig. 3(d) and (f)). In addition, to show the combination structure of the 1T/2H hybrid phase, dark-field images focused on the lattice space of 0.24 nm and 0.27 nm were obtained, as shown in Fig. S5.† The dark-field images were colored and stacked together so that the mixed state could be intuitively observed. From these pieces of evidence, the hybrid 1T/2H phase in MoS2 prepared using DMF was identified.
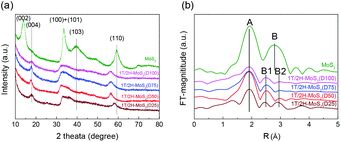 |
| | Fig. 1 (a) XRD patterns, (b) FT-EXAFS spectra of 1T/2H-MoS2 (D25), 1T/2H-MoS2 (D50), 1T/2H-MoS2 (D75), 1T/2H-MoS2 (D100) and MoS2. | |
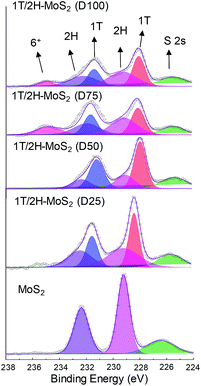 |
| | Fig. 2 Mo 3d XPS spectra of 1T/2H-MoS2 (D25), 1T/2H-MoS2 (D50), 1T/2H-MoS2 (D75), 1T/2H-MoS2 (D100) and MoS2. | |
 |
| | Fig. 3 (a), (b) High-resolution TEM bright field images of 1T/2H-MoS2, (c), (d) zoom in of area 1 and 2 respectively, and (e), (f) selected area electron diffraction (SAED) of area 1 and 2, respectively. | |
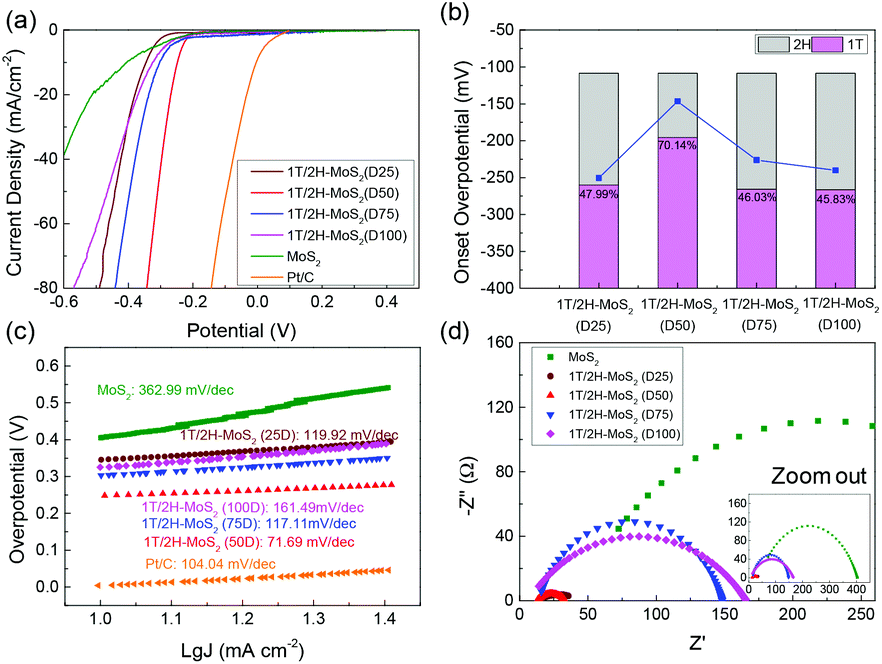
In order to quantify the concentrations of 1T-MoS2 in a hybrid sample, the multi-peak curve fitting of XPS spectra was carried out. The 1T and 2H concentration of samples are presented in Table 1. Among these samples, 1T/2H-MoS2 (D50) exhibited the highest 1T concentration, which was about 70.13% of 1T-MoS2. With an increase in the DMF amount, the 1T percentage initially increases and then decreases. The increase in the 1T concentration is consistent with the peak shift of the (110) plane in XRD and higher intensity ratio of peak B1:B2 in FT-EXAFS. Otherwise, the decrease in the 1T:2H ratio could be attributed to the destruction of the crystalline structure and oxidation, as discussed above. In order to further clarify the reason, a series of SEM images and N2 absorption–desorption isotherms have been obtained. From SEM images shown in Fig. 4, an open flower-like structure could be observed in the MoS2, 1T/2H-MoS2 (D25) and 1T/2H-MoS2 (D50) samples. These structures usually have large surface areas, as shown in Fig. S6 and Table S1† (SBET = 55.2 m2 g−1, 51.8 m2 g−1 and 31.6 m2 g−1, respectively), which facilitate electron transfer between the catalyst and the reaction compound.3,5 However, nonideal flake-like structures are observed in the 1T/2H-MoS2 (D75) sample, which result in a lower surface area (SBET = 10.6 m2 g−1). Besides, only a few flake-like structures appear in 1T/2H-MoS2 (D100), and hence this sample exhibits higher surface area (SBET = 28.3 m2 g−1) than 1T/2H-MoS2 (D75). Combining with the XPS data, the flake-like structures may originate from the oxidized molybdenum. In addition, compared with the open structures in Fig. 4(b) and (c), the surface structure of 1T/2H-MoS2 (D100) (Fig. 4(e)) seems closed due to the different solvent conditions.4 The destruction of the structure results in lower surface area (SBET = 28.3 m2 g−1) and total pore volume (Vtotal = 0.099 cm3 g−1) than 1T/2H-MoS2 (D50) (SBET = 31.6 m2 g−1 and Vtotal = 0.146 cm3 g−1).
 |
| | Fig. 4 FE-SEM images of (a) MoS2, (b) 1T/2H-MoS2 (D25), (c) 1T/2H-MoS2 (D50), (d) 1T/2H-MoS2 (D75) and (e) 1T/2H-MoS2 (D100). | |
Table 1 The 1T/2H concentration of samples synthesized using different amounts of DMF
| Sample |
1T (%) |
2H (%) |
| MoS2 |
— |
100 |
| 1T/2H-MoS2(D25) |
47.99 |
52.01 |
| 1T/2H-MoS2(D50) |
70.14 |
29.86 |
| 1T/2H-MoS2(D75) |
46.03 |
53.97 |
| 1T/2H-MoS2(D100) |
45.83 |
54.17 |
The electrocatalytic HER performance of the MoS2 sample is tested under acidic conditions (0.5 M H2SO4) using a three-electrode system by LSV method at a scan rate of 10 mV s−1. The LSV curves are shown in Fig. 5, and the onset overpotential and Tafel slope values are listed in Table S2.† Former research studies on the HER have reported that 2H-MoS2 is only active on the edge plane, while 1T-MoS2 is active on both the basal and edge planes.2,18 For this reason, the intrinsic activity of 1T/2H-MoS2 is predicted to depend on the 1T concentration. From Fig. 5(a), it can be observed that the 1T/2H-MoS2 (D50) sample exhibited the best HER activity with the lowest onset overpotential of 146.6 mV which is closer to that of the Pt/C reference electrode (onset overpotential: 45.5 mV). Among these samples, MoS2 exhibited a high onset overpotential of 270.5 mV. 1T/2H-MoS2 (D25) exhibited an onset overpotential of 243.6 mV, and 1T/2H-MoS2 (D75) and 1T/2H-MoS2 (D100) exhibited onset overpotentials of 226.2 mV and 240.2 mV, respectively. The 1T percentages and onset overpotential values are shown in Fig. 5(b). From this figure, a good positive correlation between HER activities and 1T concentrations could be observed. The specific activity normalized by the BET surface area could also be used to investigate the intrinsic activity. The specific activity is defined as j/Sr (mA cm−2), where Sr is accessible/electrochemical active surface area of the catalyst (obtained from the BET surface area) and j is the HER current. Fig. S7† shows the specific activity at −0.35 V. The 1T/2H-MoS2 (D50) sample shows the highest specific activity at this potential. Although 1T/2H-MoS2(D25) and 1T/2H-MoS2 (D75) exhibit similar 1T concentrations, the difference between their surface morphologies leads to different specific activities, which indicates that the morphology also plays an important role in intrinsic activity. Fig. 5(c) displays the Tafel plots of these samples. The Tafel slope is an important indicator of reaction kinetics. The Tafel plots are fit to the Tafel equation (η = blogj + a, where j is the current density and b is the Tafel slope) to get the Tafel slopes. Among these samples, 1T/2H-MoS2 (D50) exhibited the lowest Tafel slope owing to its faster electron transfer kinetics resulting from a higher content of 1T-MoS2. Although 1T/2H-MoS2 (D75) shows a similar 1T concentration to 1T/2H-MoS2 (D100), the HER performance of 1T/2H-MoS2 (D75) is slightly higher than that of 1T/2H-MoS2 (D100). This may be because the oxidized molybdenum in 1T/2H-MoS2 (D75) is devoted to the reaction, or the destroyed crystalline structure in 1T/2H-MoS2 (D100) hinders the reaction. Furthermore, the electrochemical impedance spectroscopy results are displayed in Fig. 5(d). The Rs and Rct values are listed in Table S2.† Among these samples, 1T/2H-MoS2 (D50) showed the smallest semicircle, suggesting its lower charge transfer resistance (Rct = 20.9 Ω). The lower Rct affords faster HER kinetics brought about by the higher content of 1T-MoS2, which we will discuss later in the section DFT calculations. The lower Rs of 1T/2H-MoS2 samples than MoS2 indicates that the metallic 1T phase contributed to the conductivity of catalysts. All the electrochemical results demonstrated that the HER performance and 1T concentration have a positive correlation. The electrochemical stability of 1T/2H-MoS2 (D50) was obtained using the chronoamperometric curve (I–t) at a static overpotential of 0.3 V, as shown in Fig. S8.† The current density decreased slightly during 4 hours of running, which indicated its good stability.
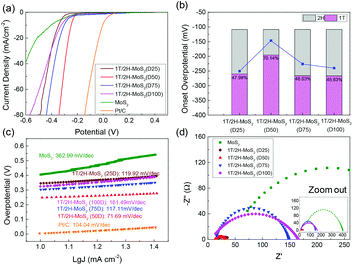 |
| | Fig. 5 (a) Linear sweep voltammetry (LSV) curves of 1T/2H-MoS2 (D25), 1T/2H-MoS2 (D50), 1T/2H-MoS2 (D75), 1T/2H-MoS2 (D100), MoS2, and Pt/C. (b) The relevant relation between onset overpotential and 1T concentration. (c) Tafel slopes of 1T/2H-MoS2 (D25), 1T/2H-MoS2 (D50), 1T/2H-MoS2 (D75), 1T/2H-MoS2 (D100), MoS2, and Pt/C. (d) Nyquist plots of 1T/2H-MoS2 (D25), 1T/2H-MoS2 (D50), 1T/2H-MoS2 (D75), 1T/2H-MoS2 (D100) and MoS2. | |
Hydrothermal process with NH4HCO3 as the guest ion agent
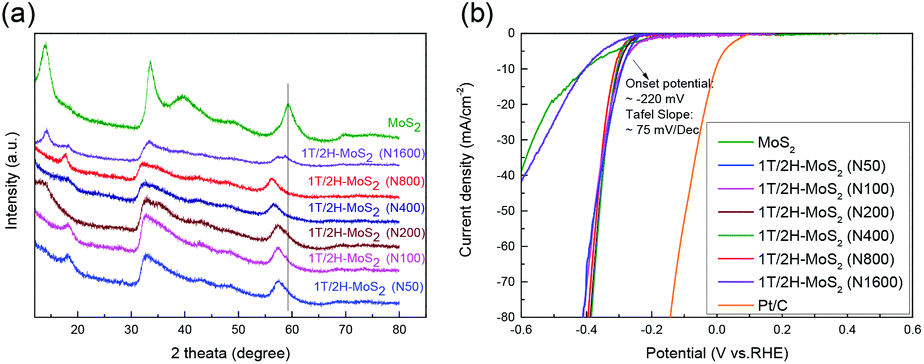
Another method to synthesize 1T/2H-MoS2 is to introduce a guest ion agent during a hydrothermal process. Different from creating a 1T phase using DMF, the hydrothermal process provides relatively mild conditions that are favorable for crystallinity. In order to control the 1T concentration, a series of catalysts are synthesized by varying the amount of guest ion agent. The mass of the guest ion agent is varied from 50 mg (about 5 mass% of other precursors) to 1600 mg (about 200 mass% of other precursors). The XRD patterns and HER performances are illustrated in Fig. 6. Firstly, the 1T concentration could be roughly judged by the shift of the (110) plane peak, as we discussed above. With the increasing amount of guest ion agent, the as-prepared samples showed almost the same XRD pattern, with the amount of guest ion agent ranging from 50 mg to 800 mg in the precursor. When the amount of precursor reached 1600 mg, the peak of the (110) plane emerged at around 58°, which was close to that of 2H-MoS2. The HER performances were tested and are shown in Fig. 6(b), and the Tafel slopes are shown in Fig. S9(a).† It is obvious that similar HER activity was exhibited by samples synthesized using different amounts of guest ion agent ranging from 50 mg to 800 mg in the precursor (with an onset overpotential of ∼220 mV and a Tafel slope of ∼75 mV dec−1, as summarized in Table S3†). Corresponding to the XRD patterns, when the precursor amount reached 1600 mg, it showed much worse HER performance than the other samples (with an onset overpotential of 264.1 mV and a Tafel slope of 241.9 mV dec−1). It should be noted that the charge transfer resistance shown by Nyquist plots (Fig. S9(b),† the values are listed in Table S3†) also follow the same tendency as those observed from the HER and XRD results. The above characterization and catalytic test suggest that by using a guest ion agent in a hydrothermal process, the hybrid 1T/2H phase with fixed concentration could be generated easily but difficult to be controlled. On the other hand, 1T/2H-MoS2 could be stably synthesized with a huge adjustable range of precursor amount. Since the samples showed similar properties, the 1T concentration was quantified by 1T/2H-MoS2 (N400), 1T/2H-MoS2 (N800), and 1T/2H-MoS2 (N1600). The XPS spectra of Mo 3d and S 2p are shown in Fig. S10† and the 1T concentrations are presented in Table 2. The samples synthesized with less than 800 mg of guest ion agent exhibited around 60% 1T concentration. This 1T concentration was lower than that of 1T/2H-MoS2 (D50) but was higher than those of the other samples synthesized using DMF. The 1T/2H-MoS2 (N1600) sample exhibited a small amount of 1T content (26.06%), maybe due to the existence of excessive ions or improper pH of the precursor solution. Although this method could not reach extremely high 1T concentration, it is a feasible synthesis process to reach a fixed content of 1T phase in 1T/2H-MoS2.
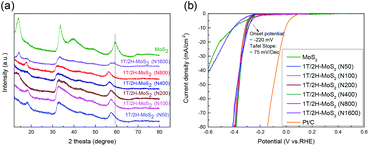 |
| | Fig. 6 (a) XRD patterns of 1T/2H-MoS2 (N50), 1T/2H-MoS2 (N100), 1T/2H-MoS2 (N200), 1T/2H-MoS2 (N400), 1T/2H-MoS2 (N800), 1T/2H-MoS2 (N1600) and MoS2. (b) Linear sweep voltammetry (LSV) curves of 1T/2H-MoS2 (N50), 1T/2H-MoS2 (N100), 1T/2H-MoS2 (N200), 1T/2H-MoS2 (N400), 1T/2H-MoS2 (N800), 1T/2H-MoS2 (N1600) MoS2, and Pt/C. | |
Table 2 The 1T/2H concentration of samples synthesized using different amounts of NH4HCO3
| Sample |
1T (%) |
2H (%) |
| MoS2 |
— |
100 |
| 1T/2H-MoS2(N400) |
60.45 |
39.55 |
| 1T/2H-MoS2(N800) |
58.49 |
41.51 |
| 1T/2H-MoS2(N1600) |
26.06 |
73.94 |
DFT calculations
From the above experimental results, it can be noted that the 1T concentration of hybrid 1T/2H-MoS2 is positively correlated with the HER performance. To better understand the reaction kinetics of 1T/2H-MoS2 in the HER process, the nature of the reaction on the 1T-MoS2, 2H-MoS2 and 1T/2H interface was investigated by DFT calculations. The reaction site was chosen as S atoms on the (100) surface because it has been reported to be an active site on a typically exposed surface of MoS2.38 The HER mechanism under acidic conditions is usually considered as follows:| | | H3O+ + e− + * → H* + H2O (Volmer reaction) | (R1) |
| | | H* + H3O+ + e− → H2 + H2O (Heyrovsky reaction) | (R2) |
| | | H* + H* → H2 (Tafel reaction) | (R3) |
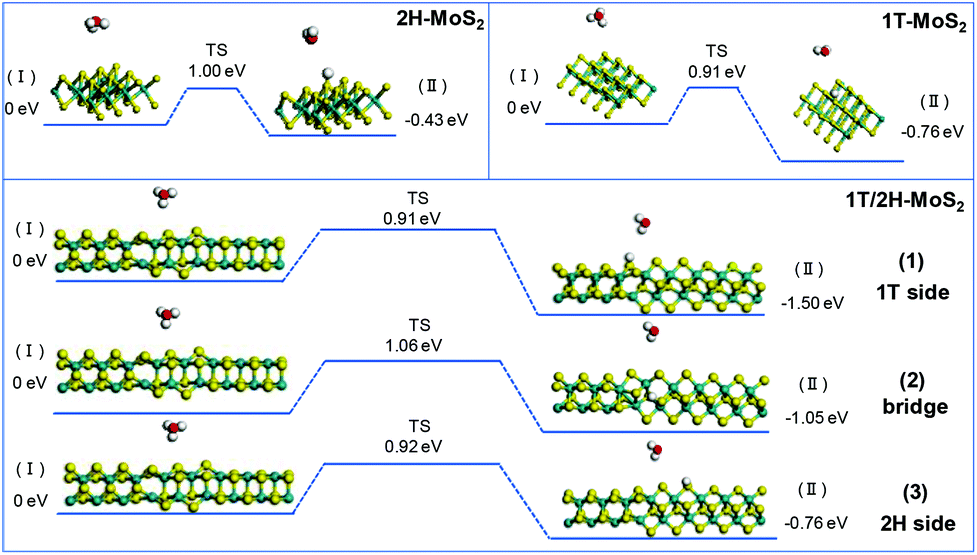
The initial adsorption of protons from an acidic solution to form adsorbed H (R1) is the first step of the HER process. The following two paths could also be considered. One is the reaction between adsorbed H* and an extra solvated proton to form H2 (Heyrovsky reaction). Another is the reaction of two adsorbed H* to form one molecule of H2 (Tafel reaction). In a DFT process, the energy barriers of the Volmer reaction and Heyrovsky reaction are approximated from the energy barriers of analogous chemical reactions by assuming that the transition state (TS) of analogous chemical reactions represents the transition state (TS) of the electrochemical reaction. This assumption allows us to evaluate the intrinsic activity of catalysts towards (R1) and (R2) without applying a potential. (R3) is originally a chemical (non-electrochemical) reaction that is independent of potential.38Fig. 7 shows the simulation results of the Volmer reaction on 1T-MoS2, 2H-MoS2, and at different sites on the 1T/2H interface. The barrier energy of all catalysts is about 0.90 eV, which originates from the dissociation of H3O+. The similar barrier energy of all catalysts suggests that catalysts contribute little to the dissociation, and this process will be completed by the applied potential. The total energy of the final product could represent the stability after catalyst binding with H*, which could be used to compare the binding abilities. 1T-MoS2 has a reaction energy of −0.76 eV, which is almost twice that of 2H-MoS2 (−0.43 eV). The high reaction energy of 1T-MoS2 indicates that it has a better binding ability than 2H-MoS2. Interestingly, the reaction energy on the 1T/2H interface is higher than or similar to that on 1T-MoS2, which indicates that the distorted structure could activate the S atom near the interface. Owing to the activation, the S atom close to the 1T side exhibits the highest reaction energy (−1.50 eV). Besides, the S atom close to the 2H side exhibits similar reaction energy to 1T-MoS2 (−0.76 eV vs. −0.76 eV).
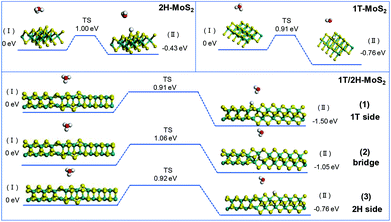 |
| | Fig. 7 Geometric structures of the initial state (I) and final state (II) of the Volmer reaction and its energetics on (100)-S atoms of 1T-MoS2 and 1T/2H interface. | |
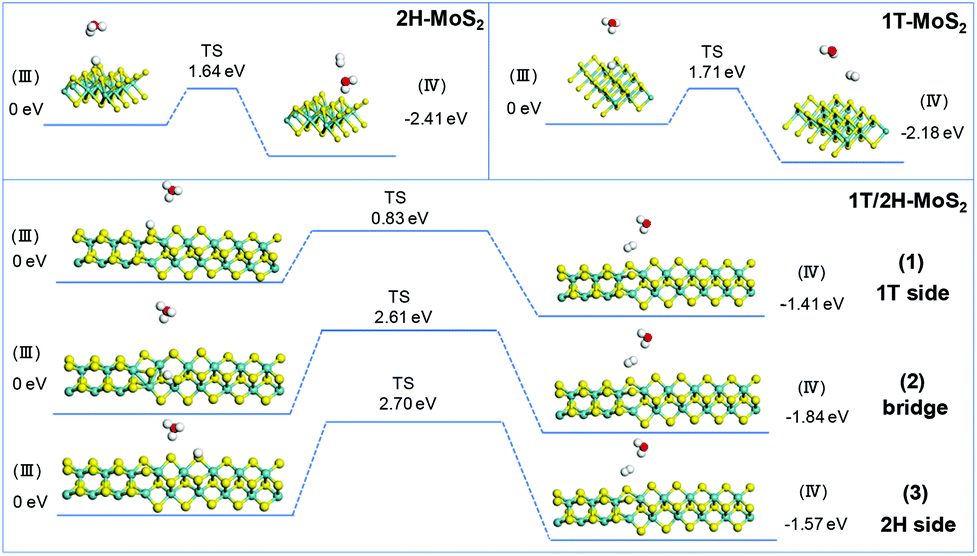
The simulation of Heyrovsky reactions (R2) is shown in Fig. 8. The barrier energy in this step represents the energy that surface H* desorption process under the effect of catalysts, and the dissociation of another H3O+. In this step, 2H-MoS2 and 1T-MoS2 show similar activities (1.64 eV vs. 1.71 eV) on the (100) S sites. On the interface of 1T/2H-MoS2, the barrier height is quite different. The S atom close to the 1T side shows a much lower barrier (0.83 eV) than 1T or 2H-MoS2, which indicates its much higher activity in the Heyrovsky reaction. However, the energy barrier on the bridge S atom and 2H side S atom is much higher than pure 1T or 2H-MoS2, which means they are relatively unfavorable for the Heyrovsky reaction. Based on the above analysis, the activity of Volmer–Heyrovsky process on the (100) surface could be concluded as follows: (1) the 2H-MoS2 sample exhibits the worst performance in the overall Volmer–Heyrovsky process; (2) the 1T/2H interfacial S atom near the 1T side exhibits the best simulated-activity in the overall Volmer–Heyrovsky process which is even better than a pure 1T-MoS2 sample; (3) other 1T/2H interfacial sites exhibit a better performance in the Volmer step, but worse performance in the Heyrovsky process than 1T-MoS2 which could hardly contribute to the overall Volmer–Heyrovsky process. Another process, the Volmer–Tafel process, was also simulated under four patterns of neighboring interfacial S sites together with 1T-MoS2. The results are shown in Fig. S11.† It should be noted that the model of 2H-MoS2 with two adsorbed H* could not be stabilized by geometry optimization, suggesting that the Volmer–Tafel process does not occur on the (100) neighboring S sites of 2H-MoS2. The simulation results of the Tafel reaction all show high barrier energy (above 2.5 eV), which indicates that the reaction is unfavorable on the catalysts. Among different adsorbed sites on the interface, 2H-bridge sites exhibit a reaction energy of 0.02 eV, which indicates that the reaction is even endothermic. Considering that the Tafel reaction is impossible on 2H-MoS2, the reaction is still exothermic on the other patterns of interfacial sites, which suggests the least possibility of the Tafel reaction. However, compared with the Volmer–Heryvosky process, the Volmer–Tafel process on MoS2 exhibits higher barrier energy and smaller total Gibbs free energy, which indicates that the main path of the HER catalyzed by MoS2 is the Volmer–Heyrovsky mechanism. In conclusion, 1T-MoS2 exhibits a better activity than 2H-MoS2. On the interface of 1T/2H-MoS2, the interfacial S atoms show even better H* adsorption ability than active 1T-MoS2, which demonstrates the activation of the interfacial S atoms by the distortion of the structure. The interfacial S atoms near the 1T side exhibit the best overall Volmer–Heyrovsky reaction activity among 1T-MoS2, 2H-MoS2, and other sites on the interface. As the conclusion, the HER performance is positively correlated with the 1T concentration, and the HER performance will also be affected by the amount of 1T/2H interface.
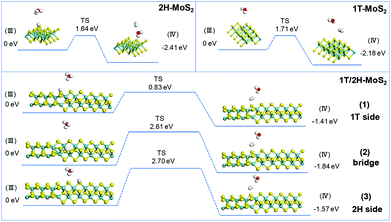 |
| | Fig. 8 Geometric structures of the initial state (III) and final state (IV) of the Heryvosky reaction and its energetics on (100)-S atoms of 1T-MoS2 and 1T/2H interface. | |
Conclusions
In conclusion, hybrid 1T/2H-MoS2 was synthesized and the 1T concentration was controlled by using different precursors. In a solvothermal method using DMF, the 1T content could be controlled by the different proportions of water and DMF. The highest 1T concentration reached was 70.13%. Another hydrothermal synthesis with a guest ion agent (NH4HCO3) could also create 1T/2H-MoS2 with a 1T concentration of around 60%. In this method, the fixed 1T content hybrid phase MoS2 could be synthesized using a wide range of precursor amount, and 60% 1T content 1T/2H-MoS2 could be stably synthesized. The hydrogen evolution reaction performance was positively correlated with the 1T concentration. By introducing a higher content of the 1T phase, MoS2 could act as highly-active HER catalysts, which was also confirmed by DFT calculations on the (100) surface of 1T-MoS2, 2H-MoS2 and 1T/2H interface. This work provides a scientific basis for applying hybrid phase MoS2 in the electrochemical hydrogen evolution reaction.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the International Joint Research Promotion Program of Osaka University. The present work was also supported by the Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan (no. 19H008380). Part of this work was supported by Element Strategy Initiative of MEXT, Japan (no. JPMXP0112101003). The synchrotron radiation experiments for XAFS measurements were performed at the BL01B1 beamline in SPring-8 with the approval of JASRI (no. 2018B1082 and 2018B1185).
References
- M. A. Lukowski, A. S. Daniel, F. Meng, A. Forticaux, L. Li and S. Jin, J. Am. Chem. Soc., 2013, 135, 10274–10277 CrossRef CAS PubMed.
- D. Voiry, M. Salehi, R. Silva, T. Fujita, M. Chen, T. Asefa, V. B. Shenoy, G. Eda and M. Chhowalla, Nano Lett., 2013, 13, 6222–6227 CrossRef CAS PubMed.
- Y. Zhang, Y. Kuwahara, K. Mori and H. Yamashita, Chem. – Asian J., 2019, 14, 278–285 CAS.
- Y. Zhang, P. Ju, C. Zhao and X. Qian, Electrochim. Acta, 2016, 219, 693–700 CrossRef CAS.
- C. Zhao, Y. Zhang and X. Qian, Electrochim. Acta, 2016, 198, 135–143 CrossRef CAS.
- U. Maitra, U. Gupta, M. De, R. Datta, A. Govindaraj and C. N. R. Rao, Angew. Chem., Int. Ed., 2013, 52, 13057–13061 CrossRef CAS PubMed.
- A. B. Laursen, S. Kegnæs, S. Dahl and I. Chorkendorff, Energy Environ. Sci., 2012, 5, 5577 RSC.
- Y. Li, H. Wang, L. Xie, Y. Liang, G. Hong and H. Dai, J. Am. Chem. Soc., 2011, 133, 7296–7299 CrossRef CAS PubMed.
- X. Lin, D. Xue, L. Zhao, F. Zong, X. Duan, X. Pan, J. Zhang and Q. Li, Chem. Eng. J., 2019, 356, 483–491 CrossRef CAS.
- K. Chang, X. Hai, H. Zhang, S. Li, G. Liu, H. Liu, G. Zhao, M. Li and J. Ye, Adv. Mater., 2016, 28, 10033–10041 CrossRef CAS PubMed.
- T. Xiang, Q. Fang, H. Xie, C. Wu, C. Wang, Y. Zhou, D. Liu, S. Chen, A. Khalil, S. Tao, Q. Liu and L. Song, Nanoscale, 2017, 9, 6975–6983 Search PubMed.
- X. Geng, Y. Jiao, Y. Han, A. Mukhopadhyay, L. Yang and H. Zhu, Adv. Funct. Mater., 2017, 27, 1702998 CrossRef.
- F. Wypych and R. Schöllhorn, J. Chem. Soc., Chem. Commun., 1992, 1386–1388 RSC.
- R. Miao, B. Dutta, S. Sahoo, J. He, W. Zhong, S. A. Cetegen, T. Jiang, S. P. Alpay and S. L. Suib, J. Am. Chem. Soc., 2017, 139, 13604–13607 Search PubMed.
- W. Lubitz and W. Tumas, Chem. Rev., 2007, 107, 3900–3903 CrossRef CAS PubMed.
- Z. Cao, Q. Chen, J. Zhang, H. Li, Y. Jiang, S. Shen, G. Fu, B. Lu, Z. Xie and L. Zheng, Nat. Commun., 2017, 8, 15131 CrossRef PubMed.
- Q. Tang and D. Jiang, ACS Catal., 2016, 6, 4953–4961 CrossRef CAS.
- M. A. Lukowski, A. S. Daniel, F. Meng, A. Forticaux, L. Li and S. Jin, J. Am. Chem. Soc., 2013, 135, 10274–10277 CrossRef CAS PubMed.
- Y. Jiao, A. M. Hafez, D. Cao, A. Mukhopadhyay, Y. Ma and H. Zhu, Small, 2018, 14, 1800640 CrossRef PubMed.
- S. Jayabal, G. Saranya, J. Wu, Y. Liu, D. Geng and X. Meng, J. Mater. Chem. A, 2017, 5, 24540–24563 RSC.
- S. Wang, D. Zhang, B. Li, C. Zhang, Z. Du, H. Yin, X. Bi and S. Yang, Adv. Energy Mater., 2018, 8, 1801345 Search PubMed.
- J. Yang, K. Wang, J. Zhu, C. Zhang and T. Liu, ACS Appl. Mater. Interfaces, 2016, 8, 31702–31708 CrossRef CAS PubMed.
- L. Wang, Z. Xu, W. Wang and X. Bai, J. Am. Chem. Soc., 2014, 136, 6693–6697 Search PubMed.
- Q. Huang, X. Li, M. Sun, L. Zhang, C. Song, L. Zhu, P. Chen, Z. Xu, W. Wang and X. Bai, Adv. Mater. Interfaces, 2017, 4, 1700171 CrossRef.
- Z. Wu, C. Tang, P. Zhou, Z. Liu, Y. Xu, D. Wang and B. Fang, J. Mater. Chem. A, 2015, 3, 13050–13056 RSC.
- D. Wang, Y. Xiao, X. Luo, Z. Wu, Y. Wang and B. Fang, ACS Sustainable Chem. Eng., 2017, 5, 2509–2515 CrossRef CAS.
- Y. Zhang, Y. Kuwahara, K. Mori and H. Yamashita, Langmuir, 2020, 36, 1174–1182 CrossRef CAS PubMed.
- Y. Liu, Y. Li, F. Peng, Y. Lin, S. Yang, S. Zhang, H. Wang, Y. Cao and H. Yu, Appl. Catal., B, 2019, 241, 236–245 Search PubMed.
- H. Fan, R. Wu, H. Liu, X. Yang, Y. Sun and C. Chen, J. Mater. Sci., 2018, 53, 10302–10312 CrossRef CAS.
- L. Cai, J. He, Q. Liu, T. Yao, L. Chen, W. Yan, F. Hu, Y. Jiang, Y. Zhao, T. Hu, Z. Sun and S. Wei, J. Am. Chem. Soc., 2015, 137, 2622–2627 CrossRef CAS PubMed.
- T. Yoshii, K. Nakatsuka, Y. Kuwahara, K. Mori and H. Yamashita, Chem. Lett., 2017, 46, 789–791 CrossRef CAS.
- K. Nakatsuka, T. Yoshii, Y. Kuwahara, K. Mori and H. Yamashita, Phys. Chem. Chem. Phys., 2017, 19, 4967–4974 RSC.
- D. Wang, X. Zhang, S. Bao, Z. Zhang, H. Fei and Z. Wu, J. Mater. Chem. A, 2017, 5, 2681–2688 RSC.
- M. Acerce, D. Voiry and M. Chhowalla, Nat. Nanotechnol., 2015, 10, 313 CrossRef CAS PubMed.
- G. Eda, H. Yamaguchi, D. Voiry, T. Fujita, M. Chen and M. Chhowalla, Nano Lett., 2011, 11, 5111–5116 CrossRef CAS PubMed.
- D. Ghim, Q. Jiang, S. Cao, S. Singamaneni and Y.-S. Jun, Nano Energy, 2018, 53, 949–957 CrossRef CAS.
- Y. Zhang, Z. Mu, C. Yang, Z. Xu, S. Zhang, X. Zhang, Y. Li, J. Lai, Z. Sun, Y. Yang, Y. Chao, C. Li, X. Ge, W. Yang and S. Guo, Adv. Funct. Mater., 2018, 28, 1707578 Search PubMed.
- W. Li, G. Liu, J. Li, Y. Wang, L. Ricardez-Sandoval, Y. Zhang and Z. Zhang, Appl. Surf. Sci., 2019, 498, 143869 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Textural properties, XAFS data, TG data, XPS data, HR-TEM data and electrochemical measurement data of MoS2 samples. See DOI: 10.1039/d0nr02525a |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Yasutaka
Kuwahara
a,
Yasutaka
Kuwahara