
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Measuring multiple 17O–13C J-couplings in naphthalaldehydic acid: a combined solid state NMR and density functional theory approach†‡
Gregory J.
Rees
 a,
Stephen P.
Day
a,
Kristian E.
Barnsley
a,
Dinu
Iuga
a,
Jonathan R.
Yates
b,
John D.
Wallis
c and
John V.
Hanna
*a
a,
Stephen P.
Day
a,
Kristian E.
Barnsley
a,
Dinu
Iuga
a,
Jonathan R.
Yates
b,
John D.
Wallis
c and
John V.
Hanna
*a
aDepartment of Physics, University of Warwick, Gibbet Hill Road, Coventry, CV4 7AL, UK. E-mail: j.v.hanna@warwick.ac.uk
bDepartment of Materials, Oxford University, 16 Parks Rd, Oxford, OX1 3PH, UK
cSchool of Science and Technology, Nottingham Trent University, Clifton Lane, Nottingham, NG11 8NS, UK
First published on 24th January 2020
Abstract
A combined multinuclear solid state NMR and gauge included projected augmented wave, density functional theory (GIPAW DFT) computational approach is evaluated to determine the four heteronuclear 1J(13C,17O) couplings in solid 17O enriched naphthalaldehydic acid. Direct multi-field 17O magic angle spinning (MAS), triple quantum MAS (3QMAS) and double rotation (DOR) experiments are initially utilised to evaluate the accuracy of the DFT approximations used in the calculation of the isotropic chemical shifts (δiso), quadrupole coupling constants (CQ) and asymmetry (ηQ) parameters. These combined approaches give δiso values of 313, 200 and 66 ppm for the carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), ether (–O–) and hydroxyl (–OH) environments, respectively, with the corresponding measured quadrupole products (PQ) being 8.2, 9.0 and 10.6 MHz. The geometry optimised DFT structure derived using the CASTEP code gives firm agreement with the shifts observed for the ether (δiso = 223, PQ = 9.4 MHz) and hydroxyl (δiso = 62, PQ = 10.5 MHz) environments but the unoptimised experimental XRD structure has better agreement for the carbonyl group (δiso = 320, PQ = 8.3 MHz). The determined δiso and ηQ values are shown to be consistent with bond lengths closer to 1.222 Å (experimental length) rather than the geometry optimised length of 1.238 Å. The geometry optimised DFT 1J(13C,17O) coupling to the hydroxyl is calculated as 20 Hz and the couplings to the ether were calculated to be 37 (O–CO) and 32 (O–C–OH) Hz. The scalar coupling parameters for the unoptimised experimental carbonyl group predict a 1J(13C,17O) value of 28 Hz, whilst optimisation gives a value of 27 Hz. These calculated 1J(13C,17O) couplings, together with estimations of the probability of each O environment being isotopically labelled (determined by electrospray ionisation mass spectrometry) and the measured refocussable transverse dephasing (T2′) behaviour, are combined to simulate the experimental decay behaviour. Good agreement between the measured and calculated decay behaviour is observed.
O), ether (–O–) and hydroxyl (–OH) environments, respectively, with the corresponding measured quadrupole products (PQ) being 8.2, 9.0 and 10.6 MHz. The geometry optimised DFT structure derived using the CASTEP code gives firm agreement with the shifts observed for the ether (δiso = 223, PQ = 9.4 MHz) and hydroxyl (δiso = 62, PQ = 10.5 MHz) environments but the unoptimised experimental XRD structure has better agreement for the carbonyl group (δiso = 320, PQ = 8.3 MHz). The determined δiso and ηQ values are shown to be consistent with bond lengths closer to 1.222 Å (experimental length) rather than the geometry optimised length of 1.238 Å. The geometry optimised DFT 1J(13C,17O) coupling to the hydroxyl is calculated as 20 Hz and the couplings to the ether were calculated to be 37 (O–CO) and 32 (O–C–OH) Hz. The scalar coupling parameters for the unoptimised experimental carbonyl group predict a 1J(13C,17O) value of 28 Hz, whilst optimisation gives a value of 27 Hz. These calculated 1J(13C,17O) couplings, together with estimations of the probability of each O environment being isotopically labelled (determined by electrospray ionisation mass spectrometry) and the measured refocussable transverse dephasing (T2′) behaviour, are combined to simulate the experimental decay behaviour. Good agreement between the measured and calculated decay behaviour is observed.
Introduction
In comparison to other prominent organic functionalities, the inception and formation of the C–O bond is a relatively poorly understood process despite its fundamental prominence in many branches of organic, organometallic and industrial chemistry, and chemical engineering.1 The production of ethers, acetals and ketals, and the initiation of hydrolysis reactions rely intrinsically on the relative strength of the C–O bond in its particular environment.2 Some well-established industrial processes are underpinned by the formation of C–O bonds to yield industrial scale commercial products. The Paternò–Büchi reaction is a photochemical process that forms C–O bonds in natural products syntheses, and the Williamson ether synthesis is a widely used industrial process for ether formation.3,4 Furthermore, hydrogen bonding to the oxygen atom in C–O bonds, (e.g. in esters) can affect their strengths, and may play a role in enzymatic processes pertaining to numerous biological and biomedical applications.5 More recently, initiatives on biomass utilization have employed homogeneous catalysis for the activation and functionalization of ether C–O sigma (σ) bonds for F capture and remediation.6The high chemical specificity and ability to probe short range interactions enables solid state NMR to investigate the relationship between a negative O-centred nucleophile and a sp2 hybridized electrophilic C centre. In this study solid naphthalaldehydic acid (1) and its 2H-naphthalaldehydic acid (2) counterpart, depicted in Fig. 1(a) and (b), respectively, will be examined using this technique to characterise C–O bonding arrangements with varying strength. This molecule represents a unique structural motif which possesses three O-based functionalities which are in close proximity within the six-membered lactone crown of the naphthalaldehydic structure, thus introducing numerous C–O bonding scenarios. A recent report describes a series of salts in which the lactone ring has opened to produce isolated aldehyde and carboxylate groups, and the n–π* C⋯O interactions and H-bonding scenarios between them.7 Some prominent examples of this overall phenomenon included the efficacy and structure–function properties of aspirin,8 and the stabilisation of chirality in protein and polymer structures.9–13
 | ||
| Fig. 1 (a) The structures of the naphthalaldehydic acid variants with the hydroxyl group (1) and the deuterated hydroxyl group (2) as evaluated in this study. The numbering scheme and labels defining the carbonyl (CO, C11) and hydroxyl (C–OH, C12) positions, and the colour scheme denoting the O environments is conserved throughout the manuscript. The experimental (orange) and CASTEP-DFT geometry optimized (purple) bond lengths are also illustrated and summarised in Table 3. (b) The crystal packing scheme of solid naphthalaldehydic acid showing the H-bonding between stacks of molecules related by a twofold screw axis. | ||
The measurement of scalar (or J-) couplings between directly bonded or closely proximate NMR active nuclei are routinely exploited to aid molecular structure determination.14 Scalar coupling values are quantifiable entities emanating from the hyperfine interactions between neighbouring nuclei and is transmitted through the bonding electrons, providing information about connectivity and dynamics between atomic positions.15 In typical solution state NMR experiments on organic and inorganic compounds, the determination of such couplings can be attained from basic 1D experiments; this is due to fast Brownian motion (on the timescale of the NMR experiment) removing the anisotropic components of all interactions present, thus resulting in high resolution isotropic data. However, it should be emphasised that J-couplings in solution involving the 17O nucleus (particularly 1J(13C,17O) couplings) are typically elusive due to the fast quadrupole relaxation of the 17O nucleus.16 In contrast, the residual inhomogeneous broadenings commonly experienced in solids usually preclude J couplings between common nuclei (1H, 13C, 15N, 31P, 29Si, 17O, etc.) from being measurable in direct observation experiments. Solid state NMR experiments rarely succeed in the achieving the spectral resolution attainable in the solution state because of the existence of higher order cross terms which cannot be completely averaged by MAS, residual dipolar interactions and distributions of isotropic chemical shifts due to structural disorder.17 However, developments in high speed magic angle spinning (MAS),18 effective heteronuclear decoupling and pulse sequence development,19 higher magnetic field strengths, and the GIPAW DFT computation of NMR parameters20–23 have all contributed to improved resolution and introduced new rationales for interpreting solid state NMR data. These advances have stimulated a renewed interest in undertaking J-coupling measurements in the solid state.
The aim of this study is to establish a combined methodology which uses first principles density functional theory (DFT) J-coupling calculations using the CASTEP code and heteronuclear (13C–17O) spin echo-based experiments to determine the 1J(13C,17O) couplings within the various C–O moieties of naphthalaldehydic acid which is a complex lactone arrangement with three O-containing functionalities in close proximity. This approach provides a natural complement to single crystal X-ray structure determinations and charge density studies that can collectively interrogate the relative strength of C–O bonds and could be extended to C⋯O interactions and bond formation scenarios. Solid state NMR J-coupling measurements are typically achieved using J-resolved or INADEQUATE experiments, or specific variants of these sequences.24–29 A key feature of these spin echo-based experiments is that they refocus the evolution of all terms that appear as offsets, including those that are directly induced by chemical shift distributions.30 This has critical implications for solid state NMR experimentation as the utilisation of these methods allows the indirect detection of J-couplings, even when inhomogeneous broadenings impede its direct observation and measurement from the more routinely acquired 1D data. As the 17O nucleus has an extremely low natural abundance (0.037%), there exists a dearth of measured 1J(13C,17O) couplings in both the solid and solution NMR literature. Hung et al. demonstrated 1J(13C,17O) measurements from a doubly labelled (13C,17O) solid glycine·HCl sample, however no further investigations have been reported from solid systems.16 This work represents the first study to implement a combined computational and experimental approach to measure multiple 1J(13C,17O) couplings in a more complex organic system to further investigate C–O bond characteristics.
Experimental section
Synthesis
Maximum 17O enrichment of naphthalaldehydic acid 1 with optimal homogeneity was achieved by dissolving the solid (200 mg, 1 mmol) in dioxane (4 ml) whilst stirring under nitrogen, with 90% 17O labelled water (Cortecnet, 0.45 ml) being added. Phosphorus pentachloride (∼2 mg) was added and the mixture warmed to 75 °C for 24 h. The reaction was cooled, the solvent evaporated in vacuo, and the residue left to dry at 45 °C for 1 h. Toluene (18 ml) was added and the mixture heated to reflux, filtered through a warm glass filter funnel, and the filtrate left to crystallise at room temperature to give white crystals (122 mg, 60%), m.p. 165–166 °C. To test the selectivity of the 17O enrichment process, a less vigorous process was used where the initial reflux stage was conducted in THF (bp 66 °C) instead of dioxane (which was heated to 75 °C), using a reaction time of only 6 h and a lower 17O concentration of 40% (Cortecnet, 0.45 ml).The naphthalaldehydic acid (100 mg) was deuterated by dissolving the product in dichloromethane (20 ml), and the filtered solution shaken with D2O (99%, 2 × 2 ml). The dichloromethane solution was dried with anhydrous sodium sulphate, filtered and evaporated to give the mono-deuterated material.
1H NMR (400 MHz, (CD3)2CO, 292.1 K): δ = 8.36 (1H, dd, 3J = 7.2, 1.0 Hz, Ar–H1), 8.31 (1H, dd, 3J = 8.4, 1.0 Hz, Ar–H1), 8.09 (1H, d, 3J = 8.2 Hz, Ar–H1), 7.71–7.81 (3H, m, Ar–H3), 7.20 (1H, d, 3J = 6.8 Hz, OH), 6.93 (1H, br d, 3J = 6.0 Hz, CHOH); 13C NMR (100 MHz, (CD3)2CO, 292.1 K): δ = 163.6 br (CO), 134.4, 132.9, 130.8, 129.8, 129.1, 128.0, 127.7, 127.2, 126.1, 121.2 (Ar–C10), 97.0 br (CHOH).
IR (ATR): 3270 br, 1663, 1623, 1597, 1588, 1516, 1407, 1373, 1356, 1316, 1263, 1248, 1189, 1179, 1109, 1070, 993, 984, 919, 904, 843, 771, 739, 728, 687, 652 cm−1; note that the CO stretch in unlabelled material is 1675 cm−1.
MS: (LTQ Orbitrap XL, nano-ESI-negative): found: 203.0555 (13), 202.0522 (100), 201.0484 (67), 200.0444 (15), corresponds to ca. 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4:1 ratio of triply, doubly and singly 17O labelled naphthalaldehydate anions. Calculated for C12H7[17O]3: 202.0527; for C12H7O1[17O]2: 201.0485; C12H7O2[17O]: 200.0443.
:4:1 ratio of triply, doubly and singly 17O labelled naphthalaldehydate anions. Calculated for C12H7[17O]3: 202.0527; for C12H7O1[17O]2: 201.0485; C12H7O2[17O]: 200.0443.
Solid state NMR
The solid state 1H MAS NMR measurement was undertaken at 14.1 T using a Bruker Avance-II+ 600 spectrometer operating at a 1H Larmor frequency of 599.4 MHz. The data was acquired using a Bruker 1.3 mm triple resonance probe spinning at 60.0 kHz. A single pulse experiment was implemented using a 1.25 μs pulse duration (flip angle of π/4) and a recycle delay of 180 s. This 1H MAS NMR data was referenced to the primary standard of tetramethylsilane (TMS, δiso = 0 ppm). A corresponding 2H MAS NMR measurement was undertaken at 11.7 T on a Bruker Avance-III 500 spectrometer operating at a 2H Larmor frequency of 76.8 MHz. This data was acquired using a 4 mm Bruker MAS double resonance probe operating at a 5 kHz spinning frequency. A single pulse experiment was implemented using a 1.75 μs pulse duration (flip angle of π/4) and a recycle delay of 3 s was determined. The 2H MAS spectrum was referenced to d-chloroform (CDCl3, δiso = 7.26 ppm) which was compared to the primary reference of TMS-d12 (δiso = 0 ppm).32The 13C MAS NMR data was acquired at 11.7 T using a Bruker Avance-III 500 spectrometer operating at a 13C Larmor frequency of 125.8 MHz. A 1H–13C cross-polarisation MAS (CPMAS) experiment was implemented using a Bruker 4 mm double resonance probe which enabled a rotation frequency of 10 kHz. A variable amplitude (70–100%) CPMAS experiment employed an initial 1H π/2 pulse of 2.5 μs, a contact period of 5 ms, a recycle delay of 600 s and a 1H decoupling field of ∼100 kHz during acquisition. The 13C CPMAS NMR data was referenced to the primary standard of tetramethylsilane (TMS, δiso = 0 ppm) via a secondary solid alanine reference (δiso(CH3) = 20.5 ppm, δiso(CH) = 50.5 ppm and δiso(COOH) = 177.8 ppm). The measurement of the 1J(13C,17O) coupling constants were performed using a modified spin-echo experiment as previously described by Hung et al.16 A 1H–13C CP preparation of magnetisation (see above) was followed by a refocussing π pulse (CP–τ–π–τ) on both the 13C and 17O channels thus forming an echo. The 13C and 17O π pulse duration on each channel was 6 μs. The τ delays were incremented from 0 to 15 ms, with 100 kHz of 1H decoupling applied throughout. The 2D 1H–13C Lee–Goldberg heteronuclear correlation experiment used the same cross polarisation preparation as described above and implemented homonuclear Lee–Goldberg 1H–1H decoupling during acquisition. No additional resolution enhancement in the 1H spectrum was observed with the implementation of homonuclear decoupling.
For the quadrupolar 17O (I = 5/2) nucleus MAS NMR studies were performed at 14.1, 16.5 and 20.0 T using Bruker Avance-II+ 600, Bruker Avance-III HD 700 and Bruker Avance-III 850 instruments operating at 17O Larmor frequencies of 81.3, 94.9 and 115.2 MHz, respectively. A Bruker 3.2 mm double resonance probe was used at each field which facilitated a MAS frequency of 20.0 kHz for each measurement. All 17O MAS NMR data were acquired using a rotor-synchronised Hahn echo experiment (θ–τ–2θ–τ–acquire). A ‘non-selective’ (solution) π/2 pulse time of 3 μs was calibrated using 10% H217O which represented a ‘selective’ π/2 (solid) pulse of 1 μs; a θ = π/2 = 1 μs/2θ = π = 2 μs combination was used for all measurements. A recycle delay of 5 s was used throughout; this was carefully checked against acquisitions using recycle delays of up to 60 s. All 17O apparent shifts (centre-of-gravity, δcg) and isotropic chemical shifts (δiso) are reported against a primary reference of H217O (δiso = 0 ppm).32 Additional 17O double rotation (DOR) experiments using odd order sideband suppression33 were undertaken at 9.4, 11.7 and 14.1 T using Bruker Avance-III HD 400, Bruker Avance-III 500 and Bruker Avance-II+ 600 spectrometers operating at the 17O Larmor frequencies of 54.1, 67.8 and 81.3 MHz, respectively. The measurements at each field were performed using Samoson designed DOR probes which functioned with external rotor spinning frequencies of 1.3–1.9 kHz and internal rotor spinning frequencies of 5.9–9.7 kHz.
All central transition spectra from the 17O MAS NMR data of 1 acquired at 14.1, 16.5 and 20.0 T were simulated using the DMFit34 simulation package to determine the experimental isotropic chemical shift (δiso) and quadrupole (CQ, ηQ) NMR parameters (see Table 1 and Fig. 3(a), (c) and (e), respectively). The CASTEP calculated NMR parameters for δiso, 1JOC, CQ and ηQ (see Tables 2 and 3) were used as input into the SIMPSON 4.2.135 simulation package to generate a direct comparison between experimental and calculated data. The SIMPSON input files are given in Table S3 (ESI‡).
| Site | 17O MAS NMR | 17O DOR NMR | 17O(2H) DOR NMR | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| δ iso /ppm | C Q /MHz | C Q /MHz | η Q | η Q | P Q /MHz | P Q /MHz | δ cg /ppm | δ cg/ppm | P Q /MHz | P Q/MHz | δ cg /ppm | δ cg/ppm | P Q /MHz | P Q/MHz | |
| Error | Error | Error | Error | Error | Error | Error | |||||||||
| a IUPAC isotropic shift convention averaged from the multiple field simulations: δiso = (δ11 + δ22 + δ33)/3. b Error derived from average of deconvolutions at three magnetic fields. c EFG tensor conventions: |V33| ≥ |V22| ≥ |V11|, CQ = e2qQ/h = eQV33/h, ηQ = (V11 − V22)/V33 (1 ≥ ηQ ≥ 0). d Quadrupole parameter: δcg (ppm) = δiso − (3/500) (PQ2/ν02) × 106 and PQ (MHz) = CQ (1 + ηQ2/3)1/2. | |||||||||||||||
| CO |
313 | 8.2 | 0.1 | 0.03 | 0.02 | 8.2 | 0.1 | 285 | 19 | 7.3 | 1.7 | 308 | 2 | 8.1 | 0.1 |
| –O– | 200 | 8.9 | 0.1 | 0.25 | 0.01 | 9.0 | 0.1 | 173 | 20 | 8.2 | 1.5 | 199 | 1 | 9.0 | 0.1 |
| –OH/–OD | 66 | 9.6 | 0.4 | 0.81 | 0.08 | 10.6 | 0.4 | 63 | 19 | 10.4 | 0.1 | 16 | 32 | 8.6 | 2.5 |
| O site | GIPAW DFT calculated 17O NMR parameters (experimental (XRD) structure) | GIPAW DFT calculated 17O NMR parameters (geometry optimised structure) | ||||||
|---|---|---|---|---|---|---|---|---|
| δ iso/ppm | C Q/MHz | η Q | P Q/MHZ | δ iso/ppm | C Q/MHz | η Q | P Q/MHz | |
| CO |
320 | 8.3 | 0.09 | 8.3 | 307 | 8.1 | 0.31 | 8.2 |
| –O– | 210 | 9.3 | 0.30 | 9.4 | 223 | 9.3 | 0.25 | 9.4 |
| –OH/–OD | 26 | 9.7 | 0.65 | 10.4 | 62 | 9.5 | 0.82 | 10.5 |
O, –O– and –OH oxo functionalities as determined by GIPAW DFT calculations using the CASTEP code from the geometry optimised and experimental versions of the naphthalaldehydic acid structure 1. These calculated 1J(13C,17O) coupling constants are compared to experiment via the dephasing behaviour shown in Fig. 6 which was measured using a heteronuclear spin echo experimental technique
| Coupling constant | 13C nuclei | 17O nuclei | Bond length/Å | Bond angle/°O–C–O | 1 J co/Hz | Optimised bond length/Å | Optimised bond angle/°O–C–O | Optimised 1Jco/Hz |
|---|---|---|---|---|---|---|---|---|
| 1 J CO1 | C11 | C17O |
1.222 | 117.5 | 28 | 1.238 | 117.4 | 32 |
| 1 J CO2 | –17O– | 1.342 | 117.5 | 37 | 1.338 | 117.4 | 37 | |
| 1 J CO3 | C12 | –17O– | 1.477 | 106.7 | 32 | 1.490 | 107.4 | 32 |
| 1 J CO4 | –17OH | 1.386 | 106.7 | 21 | 1.379 | 107.4 | 20 | |
First principle DFT geometry optimisation and magnetic resonance calculations
All density functional theory (DFT) calculations were performed using the CASTEP 16.121 code which employs Kohn–Sham DFT methodology using periodic plane-waves under the ultrasoft pseudopotential approximation. The generalized gradient approximation for the exchange correlation energy was employed using the Perdew–Burke–Ernzerhof solids functional (PBE-sol). Pseudopotentials were generated ‘on-the-fly’ using the standard Accelrys Software Materials Studio pseudo-atom definitions. Each calculation was converged with respect to basis-set size and Brillouin zone k point sampling to at least an accuracy of 0.4 mH (∼0.000002% of total energy) per atom for each of the systems under investigation. To confirm this level of energy convergence was sufficient to produce accurate ionic forces, energy minimisation with respect to ionic positions was repeated with increasing plane wave cut off energy and density of k points within the Monkhorst–Pack Brillouin zone grid. This level of convergence was achieved using a plane wave cut-off energy of 900 eV for system, and by invoking k point Monkhorst–Pack grids of 3 × 2 × 2.Geometry optimisation and H position optimisation calculations were performed on all systems using the over-converged values described previously to ensure accurate forces. For these structural optimisations, the lattice parameters were kept fixed whilst the atomic positions were optimised to a force tolerance of 0.05 eV Å−1, a maximum ionic displacement of 1 × 10−3 Å and a total energy change of 2 × 10−5 eV per atom. NMR parameter calculation of the chemical shift,22,36 electric field gradient tensors20 and J-couplings23,37 invoked the gauge included projector augmented wave (GIPAW) DFT method which extended the pseudopotential (valence electron approximation) approximation to recover all electron charge densities. This approach demonstrated that the NMR parameters depend more sensitively upon the electron density than the total energy, so each NMR parameter will be more sensitive to basis set truncation errors than to total energies. This is compensated for by the over-convergence of the basis set to a level which experience suggests will produce fully converged NMR parameters. Consequently, these calculations were completed using the plane wave cut-off and k-point Monkhorst–Pack grids described earlier.
In order to calculate isotropic chemical shifts for the 17O nucleus the relationship δiso = −[σ − σref] was employed, where σ is the calculated isotopic shielding calculated against a bare atom, and σref is the previously determined reference isotropic shielding of 262 ppm when compared against a bare atom.38 These calculations used the same basis set convergence (0.4 mH) as the original calculations to minimise the propagation of errors.
Results and discussion
The homogenous 17O enrichment of solid naphthalaldehydic acid 1 was achieved by heating to 75 °C in 90% 17O labelled water under acid catalytic conditions in hot dioxane for 24 h, with the resulting product recrystallized from toluene. The electrospray ionisation mass spectrometry (ESI-MS) of the resultant anion suggested that it was characterised by a ratio of triply labelled: doubly labelled: mono labelled molecules of ∼6:4:1. A recently reported single crystal X-ray structure determination of 1 shows that the head group is essentially a closed lactone ring arrangement supporting the three proximate O positions referred to as carbonyl (CO), ether (–O–) and hydroxyl (–OH) groups for the purpose of the remaining discussion. However, open ring configurations can be realised when molecule 1 is deprotonated to give salts with various cations which exhibit interactions between the negatively charged carboxylate (CO2−) anion and the aldehyde (CHO) functionality (see Scheme 1 below).7
 | ||
| Scheme 1 | ||
As observed in Fig. 1(b), the crystalline salt naphthalaldehydic acid 1 has a cyclic lactone structure with only one crystallographically unique molecule comprising the asymmetric unit of the crystal. There exists intermolecular H-bonding between the CO functionality and the –OH moiety on an adjacent molecule constrained by a twofold screw axis, with the two symmetry related H-bonds associated with each molecule assuming an O⋯O distance of 2.754(4) Å.7 There is an anomeric interaction between the two O atoms attached to the methine C leading to a short C–OH bond length of 1.387 Å and a long C–O(CO) bond length of 1.477 Å due to donation of lone pair electron density from the hydroxyl O atom into the adjacent methine CH–O(CO) bond.7 In comparison, a typical unperturbed C–O bond length is usually observed to fall between these values at ∼1.43 Å.39
The 1H MAS NMR data from naphthalaldehydic acid measured at 14.1 T under fast MAS conditions (νr = 60 kHz) is shown in Fig. 2(a). This spectrum shows a single broad resonance with no individual proton environments able to be resolved from this signal, despite the acquisition of this data under high field and fast MAS conditions. The aromatic, hydroxyl and methine 1H species are all expected to appear at similar isotropic chemical shifts (δ 6.8–8.4 ppm in solution), and the strong homogeneous 1H–1H dipolar interaction broadens all resonances thus resulting in a single unresolved feature. It is possible that both the 1H and 13C resonances suffer from anisotropic bulk magnetic susceptibility (ABMS) broadening, however this broadening typically only increases the linewidth by ∼1 ppm.40 These problems are commonly observed in highly complex aromatic systems and heavy (but not complete) deuteration of the sample is required to significantly increase spectral resolution.41
 | ||
| Fig. 2 The characterisation of solid naphthalaldehydic acid 1 using (a) 1H MAS NMR (ν0 = 599.4 MHz, νr = 60 kHz), (b) 2H MAS NMR (ν0 = 76.8 MHz, νr = 5 kHz) with experimental data (black) and simulation (red), (c) 13C CPMAS (5 ms contact time) NMR (ν0 = 125.8 MHz, νr = 10 kHz) with the carbonyl, aromatic and alcohol regions labelled, and (d) a 2D 1H–13C Lee–Goldberg heteronuclear correlation experiment (5 ms contact time) which utilised homonuclear 1H decoupling throughout the evolution and acquisition periods. The asterisks (*) in the 13C CPMAS NMR data in (c) denote the presence of spinning sidebands. | ||
The 17O labelled naphthalaldehydic acid sample 1 was selectively deuterated at the hydroxy position to form a doubly 17O/2H enriched variant 2 (see Fig. 1(a)) to investigate (and reduce) the potential of 1H motional modulation effects on the 17O MAS and DOR NMR data (vide infra). The 2H MAS NMR data from this quadrupolar (I = 1) nucleus shown in Fig. 2(b) depicts a broad satellite transition spectrum with a 2H quadrupole coupling constant (CQ) of ∼200 kHz being measured from the simulation of this data. Previous measurements of 2H CQ values from various enriched –OD arrangements demonstrate that CQ occurs within the range of ∼190–290 kHz;42,43 the CQ characterising the –OD moiety in this hydroxyl-lactone species represents a value at the low end of this range thus suggesting that partial motional averaging of this interaction is evident. This value is significantly smaller than the predicted CQ value of ∼308 kHz as computed using the CASTEP code (see Table S1 in the ESI‡). These DFT calculations in their basic form do not account for rotation, vibration or libration of the –OD bond, hence any subsequent motional averaging of the static quadrupole interaction is not modelled. Recently, it has been suggested that dueteration can also reduce the point symmetry of the moiety.44 It is important to note that the δiso(2H) for the –OD species is ∼8.0 ppm, and this is corroborated by the 1D 1H MAS and 2D 1H–13C HETCOR MAS NMR data depicted in Fig. 2(a) and (d). The 2D 1H–13C HETCOR data of Fig. 2(d) clearly shows that all protonated C species are correlated with the broad 1H resonance at ∼8.0 ppm, indicating that the δiso(–OH) value is not resolved from the more intense 1H resonances associated with the aromatic protons. Collectively, the observations of a more deshielded 1H chemical shift (δiso(1H) ∼8.9 ppm) and a partial motional averaging of the 2H quadrupole interaction (CQ ∼200 kHz) indicates that the bond covalence characterising the –OH group is of intermediate strength, while the intermolecular H-bonding interaction inherent to the crystal structure of 1 (influencing the –OH proton) is weak.31,38
Solid state 17O MAS NMR data from 1 measured at 20.0 T (ν0 = 115.2 MHz), 16.5 (ν0 = 94.9 MHz) and 14.1 T (ν0 = 81.3 MHz) presented in Fig. 3(a)–(c), respectively, shows the three O species present in the six membered hydroxyl-lactone ring form of the naphthalaldehydic acid lactone crown are all influenced by substantial second order quadrupole interactions. The highly characteristic second order broadened quadrupole resonances are fully resolved at 20.0 T, however due to the 1/B0 dependence of the quadrupole interaction the observed linewidths increase with decreasing B0, consequently reducing the resolution of the lower field data. Corresponding 2D 17O 3QMAS data from 1 acquired at 16.5 T shown in Fig. 4 clearly demonstrates the chemical purity of this system, and that very high structural order is reflected by the absence of any distributions in the chemical shift and/or distributions of quadrupole NMR parameters. Previous studies have established that H-bond motion can modulate the multiple quantum excitation and evolution processes in the MQMAS experiment thus inducing detrimental effects on the resolution observed in the F1 dimension, however this is also not observed here.38
 | ||
| Fig. 3 The 17O MAS NMR data and spectral deconvolution from 17O enriched naphthalaldehydic acid 1 acquired at (a) 20.0 T (ν0 = 115.2 MHz, νr = 20 kHz), (c) 16.45 T (ν0 = 94.9 MHz, νr = 20 kHz), (e) 14.1 T (ν0 = 81.3 MHz, νr = 20 kHz), compared against their corresponding SIMPSON45 simulations (b), (d), (f) generated using GIPAW DFT calculations of the NMR parameters from the single crystal naphthalaldehydic acid 1 structure using the CASTEP code. In each case, the green resonance represents the carbonyl (CO) species, the red resonance represents the ether (–O–) species, the blue resonance represents the hydroxyl group (–OH), and the spectral sum shown in purple. | ||
 | ||
| Fig. 4 The (a) 2D 17O 3QMAS NMR data of naphthalaldehydic acid 1 (16.5 T, νr = 20 kHz), the individual 1D projections from 400 to −150 ppm are given along the F1 axis and the spinning sidebands are represented with asterisks (*), shown with 1D 17O MAS NMR data (14.1 T, νr = 20 kHz) for (a) a homogeneously and optimally enriched sample of 1 using 90% labelled H2O showing a ∼1:1:1 intensity distribution (refluxed in dioxane, 75 °C, 24 h), (b) an inhomogeneously and less enriched sample of 1 using 40% labelled H2O showing a ∼1:1:2 intensity distribution (refluxed in THF, 66 °C, 6 h), and a doubly labelled 2H/17O enriched sample 2 where the 2H enrichment is performed after the initial 17O labelling using the 90% H2O precursor (refluxed in dioxane, 75 °C, 24 h). Note the marked loss of 17O signal intensity in (d) compared to (b) upon further reaction required to achieve the 2D enrichment post the 17O enrichment, hence no 3QMAS data was achieved from the 2H-naphthalaldehydic acid 2 sample. | ||
Simulation of the 17O 1D data for the CO species (i.e. the most deshielded 17O environment) yields an isotropic chemical shift of δiso = 313 ppm and quadrupole parameters of CQ = 8.2 MHz/ηQ = 0.03. A 17O δiso value of this magnitude is low compared to a ketone group, but it is within the reported limits of typical carboxylic acid and lactone functionalities.46,47 Conversely, the observed 17O resonance from the hydroxyl (–OH) species is the most shielded (δiso = 66 ppm) and represents a larger quadrupole interaction with CQ = 9.6 MHz/ηQ = 0.81. Both sets of 17O quadrupole parameters measured for both the CO and the –OH groups are in firm agreement with previously reported data in the literature observed for amino acid·HCl salts.38,46,48 The high ηQ value for the –OH species, and corresponding low ηQ value for the CO species are typical for 17O nuclei bonded to larger conjugated aromatic moieties.38,49 In previous studies on amino acids, it has been reported that the 17O asymmetry parameter (ηQ) is close to zero for C⋯O distances in a CO functionality of ∼1.22 Å, while it appears to increase rapidly for longer bond lengths. Simulation of the 17O resonance from the ether (–O–) linkage (located between the CO and –OH resonances) in this lactone structure yields an isotropic chemical shift of δiso = 200 ppm and quadrupole parameters of CQ = 8.9 MHz/ηQ = 0.25. All isotropic chemical shift and quadrupole parameters characterising the three O species comprising this lactone structure are in agreement with previously reported values for the lactone of 2-[1,2,3-17O3]acetylbenzoic acid which is the only other reported labelled lactone structure studied by solid state NMR.47 Despite the use of high magnetic fields in this study, no chemical shift anisotropy (CSA) contributions were required to facilitate the accurate simulation the data in Fig. 3(a)–(c), suggesting that CSA influences are small in comparison to the dominant quadrupole interaction and subsequently eliminated by MAS.
For moderate-weak H-bonded systems, a previously established empirical relationship between the 17O quadrupole coupling constant of the O species involved in X–H⋯O (X = N, O) H bonds and the X⋯O distance across the whole X–H⋯OH bond takes the form:46,50,51
| CQ ≈ 1.5199(R(X⋯O)) + 3.9987 | (1) |
C–) bridging the H bond is 2.787 Å. Upon substitution of this O⋯O distance into eqn (1) a predicted CQ value of 8.23 MHz is obtained which compares very favourably with the measured CQ of 8.2 MHz as reported in Table 1, thus supporting moderate-weak H bond character in the structure of 1.
In addition, there is a strong correlation between the CO bond length and the carbonyl 17O isotropic chemical shift (∼1200 ppm Å−1), with a shift of ∼313 ppm representing a bond length of 1.222 Å.48 A comparison of the CO bond lengths to C17O δiso values compiled by Pike et al.,22 together with the GIPAW DFT data reported by Gervias et al.,52 produces an overall empirical correlation relating the 17O δiso value to the carbonyl bond length (R(CO)):
| δiso(CO) ≈ 1751–1177(R(CO)) | (2) |
O bond length of 1 measured in the single crystal X-ray structure determination at 100 K is 1.222 Å, and the DFT geometry optimisation yields a longer CO bond length of 1.238 Å. From Table 3, the measured 17O δiso value of the CO value is 313 ppm (from MAS NMR); when this value is introduced to eqn (2) a bond length of 1.223 Å is predicted, suggesting the room temperature NMR measurements are in firm agreement with the single crystal X-ray structure determination (performed at 100 K) despite the large temperature difference between the two measurements. Correspondingly, as the MAS NMR measured 17O ηQ value of the carbonyl is close to zero, then it can be presumed that the bond length is closer to the unoptimized 1.222 Å, as any CO bond length significantly above 1.2 Å gives a ηQ > 0 value.53 Support for the crystallographic derived structure by highly accurate 17O solid state NMR measurements is necessary to gauge the temperature variations between the NMR (298 K) and crystallographic (100 K) measurements. These observations infer that accurate calculated 1J(13C,17O) couplings can be obtained from DFT-CASTEP calculations (notionally undertaken at 0 K) on high quality crystal structure data.
From inspection of the spectral and computationally generated data exhibited in Fig. 3 and Tables 1 and 2, there exists a very strong correlation between the directly measured and simulated 17O MAS NMR data (see Fig. 3(a), (c) and (e), and Table 1) and the GIPAW DFT calculated 17O NMR interaction parameters from a geometry optimised structure of 1 using the CASTEP code (see Fig. 3(b), (d) and (f) and Table 2). In particular, the experimentally determined –OH data (δiso = 66 ppm, CQ = 9.6 MHz, ηQ = 0.81), –O– data (δiso = 200, CQ = 8.9, ηQ = 0.25) and CO data (δiso = 313 ppm, CQ = 8.2 MHz and ηQ = 0.03) show an excellent agreement with the corresponding DFT results (–OH, δiso = 62 ppm, CQ = 9.5 MHz, ηQ = 0.85; –O–, δiso = 223ppm, CQ = 9.3 MHz, ηQ = 0.25; CO, δiso = 307 ppm, CQ = 8.1 MHz and ηQ = 0.31) when geometry optimisation is implemented. These correlations are marginally stronger than those derived from calculations using non-geometry optimised structures; this is particularly evident when comparing the MAS NMR measured and DFT calculated –OH δiso values when geometry optimisation is omitted. In this case, a discrepancy of ∼40 ppm exists. Nevertheless, the strength of the above correlations suggests that the GIPAW DFT method represents a very sound approximation for describing the electronic configuration of O species in organic solids, and this approach thus presents a solid foundation on which to underpin 1J(13C,17O) coupling calculations in the naphthalaldehydic acid system 1. The DFT calculations also highlight that both geometry optimised and direct calculations on the X-ray derived structure need to be considered when calculating the 1J(13C,17O) couplings in this system.
Unlike the conventional MAS NMR technique, the DOuble Rotation (DOR) experiment involves spinning the sample about two angles to enable averaging of both the P2 and P4 terms of the 4th order Legendre polynomial, which describes the angular dependences of the second order quadrupole interaction. The first order quadrupole broadening is reduced by sample rotation around the familiar magic angle of 54.7° with respect to B0, while the second order quadrupole broadening is eliminated by simultaneous rotation at 54.7° and a second angle of 30.6° with respect to the magic angle. The two major advantages that the DOR NMR technique offers are, (a) the observed (featureless) centre-of-gravity shift (δcg) yields greater resolution than conventional MQMAS experiments which are limited by the number of attainable slices defining the F1 dimension, and (b) the DOR experiment uses a single pulse excitation experiment in comparison to two-pulse or three-pulse MQMAS experiments which possess limited pulse efficiencies. The greater resolution and superior signal-to-noise ratio afforded by the DOR technique is met with technical challenges; sample rotation at two angles simultaneously is difficult to stabilise, and the limited rotational rates achievable by the inner and outer rotors introduces large residual spinning sideband manifolds which necessitate acquisitions at multiple spinning rates to isolate the central transition.33,54
For a quadrupolar nucleus such as 17O, the apparent centre-of-gravity shift (δcg) is comprised of both field independent isotropic chemical shift (δiso) and field dependent second-order quadrupole shift (δ(2)Q,iso) components:
| δcg = δiso + δ(2)Q,iso(I,m) | (3) |
| δ(2)Q,iso(I,m) = (3CQ2/(40νo2I2(2I − 1)2))[I(I + 1) − 9m(m − 1) − 3](1 + ηQ2/3) | (4) |
| δcg(ppm) = δiso(ppm) – (3/500)(PQ2/ν02) × 106 | (5) |
| PQ = CQ√(1 + ηQ2/3) | (6) |
High resolution data afforded by the 17O DOR technique allows some subtle aspects of motion affecting the O positions to be detected. The characterisation of these properties is an important prerequisite to the measurement and calculation of 1JCO couplings. Table 1 summarises the δiso and PQ data elucidated from the variable field (14.1, 11.7 and 9.4 T) 17O DOR measurements and the graphical approach outlined above for 1 (see Fig. 5(a)–(d)) and 2 (see Fig. 5(e)–(h)). Each spectrum shows the centre-of-gravity shifts (δcg) identified; these were isolated from the spinning sideband manifolds by varying the spinning frequency of the outer rotor over a range of 1.3–1.9 kHz. From the undeuterated system 1 the determined 17O δiso values for the carbonyl (CO), ether (–O–) and hydroxyl (–OH) species are 285, 173 and 63 ppm, respectively, while the analogous δiso values of 308, 199 and 16 ppm are obtained from the deuterated version 2. Upon deuteration, a marked deshielding of ∼23–26 ppm in the 17O δiso values are observed for the CO and –O– moieties, which is in contrast with the increased shielding of ∼47 ppm observed for the –OH species.
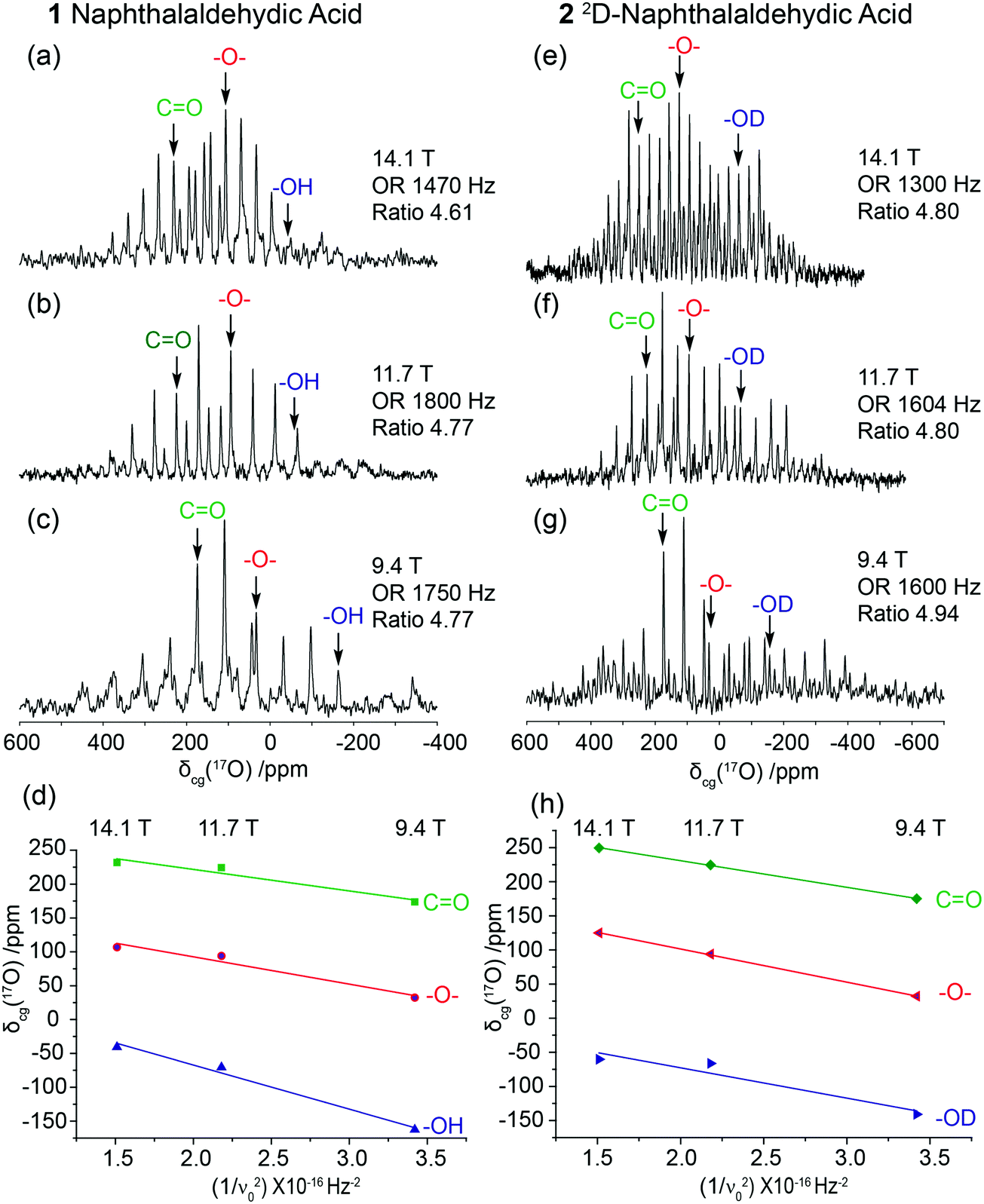 | ||
| Fig. 5 The solid state 17O DOR spectra of naphthalaldehydic acid at (a and e) 14.1 T (ν0 = 81.3 MHz), (b and f) 11.7 T (ν0 = 67.8 MHz) and (c and g) 9.4 T (ν0 = 54.1 MHz). The resonances for CO, –O– and –OH are labelled in each case and are determined by spinning the samples at a minimum of three different frequencies. Results from undeuterated naphthalaldehydic acid 1 are given in a, b and c, and results from deuterated naphthalaldehydic acid 2 are given in e, f and g. The outer rotor frequencies are denoted as ‘OR’, and the inner:outer rotor frequency ratios are labelled ‘ratio’. From plots of centre-of-gravity (δcg) for each DOR resonance against 1/ν02 (d, h), δcg is observed to be field (or ν0) dependent through second order quadrupole effects from the spin I = 5/217O nucleus such that δcg (ppm) = δiso (ppm) − (3/500 × PQ2/ν02) × 106, thus permitting graphical estimates for δiso (y intercept) and PQ (slope) for molecules 1 and 2. | ||
Previous studies have demonstrated that the deuteration of protonated oxo functionalities in solid organic systems improves the resolution obtained in the 17O DOR experiment. This is achieved by damping the heteronuclear dipolar proton coupling which homogenously broadens the 17O resonance.60–63 This is corroborated by the data in Fig. 5 where the 17O resonance for the –OD (see Fig. 5(e)–(h) for 2) moiety is significantly narrower and of greater intensity than its –OH counterpart (see Fig. 5(a)–(d) for 1), especially at the higher fields of 11.7 and 14.1 T. As observed from Table 1 and Fig. 5(d) and (h), the data for the CO and –O– moieties from the undeuterated sample 1 show an inferior overall correlation on the linear regression and greater errors on each individual measurement in comparison to their counterparts from the deuterated sample 2. This is a consequence of the broader 17O resonances due to proton motion in the H-bond which induces additional cooperative modes of motion throughout the other O functionalities. Furthermore, for the CO and –O– moieties the measured δiso and PQ values from the DOR experiment closely corroborate those reported from the MAS, MQMAS and geometry optimised DFT (GO-DFT) data. In comparison, for both samples 1 and 2 the overall correlations described in the linear regression for the –OH species are inferior to those described above for the CO and –O– moieties. As suggested above, a reduced correlation for the –OH data (sample 1) is expected to emanate from proton heteronuclear dipolar coupling in the H-bonded arrangement; however, the –OD data (sample 2) exhibits a more reduced correlation and much larger errors. This contradicts previously reported evidence that proposes sample deuteration reduces these errors, promoting more resolved 17O DOR data, and enables more accurate measurements of the δiso and PQ parameters.33 This anomaly suggests that the reduced correlations/larger errors from 2 originate from lower point energy of the OD bond and results in an isotope effect on the NMR parameters.62,63 The deuteration procedure of 17O enriched sample 1 (using D2O) to produce sample 2 does not yield a homogeneous doubly labelled 2D/17O system. Some proportion of the original 17O enrichment has exchanged with 16O producing a complex mixture of 2D/17O, 2D/16O, 1H/17O and 1H/16O species, thus producing a complex distribution of isotope effects. This phenomenon is demonstrated in Fig. 4(d) where 17O MAS NMR data from the deuterated sample 2 (which originated as sample 1) exhibits greatly inferior signal/noise in comparison to sample 1 shown in Fig. 4(b). These complex isotope affects clearly directly influence upon the 17O δiso and PQ parameters characterising each O environment, and the deuteration of molecule 1 can produce measured NMR parameters which are not directly comparable to those calculated from the single crystal derived structure. The process of deuteration of hydroxyl environments to reduce heteronuclear dipolar coupling, and potentially reduce the point symmetry of the moiety, is occasionally employed in the literature. Here, we show this is not best employed when trying to accurately compare diffraction derived DFT NMR parameters and the experimental MAS data. Overall, the GIPAW DFT-derived CQ and ηQ parameters show a strong correlation with the MAS and MQMAS achieved parameters, therefore for the following 1J determinations the experimentally determined parameters will be used.
The spin echo dephasing data from the (undeuterated) naphthalaldehydic acid system 1 is presented in Fig. 6, and the measured heteronuclear 1J(13C,17O) coupling constants are summarised in Table 3. This study represents only the second measurement of 1J(13C,17O) coupling constants in the solid state. While the first was demonstrated on a doubly 13C/17O labelled solid glycine·HCl sample,16 this work focusses on a uniformly labelled 17O system which possesses multiple O positions which are in close proximity.
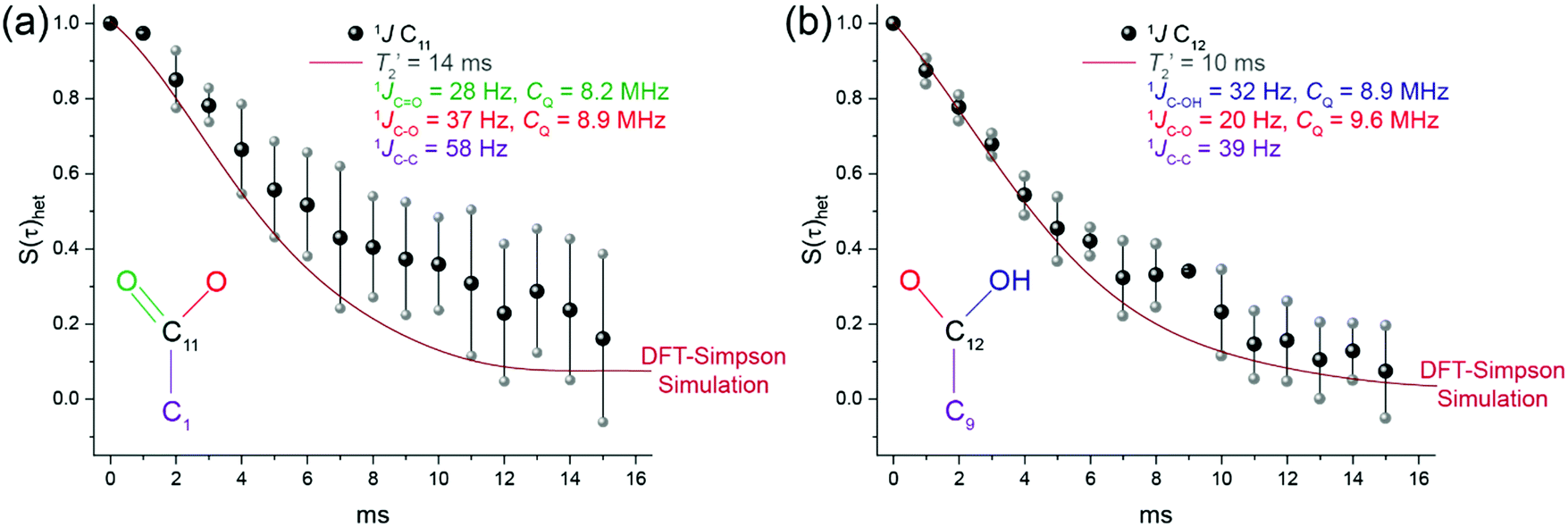 | ||
| Fig. 6 The experimental (black) and DFT predicted (red, SIMPSON simulated) 13C/17O heteronuclear spin echo dephasing behaviour for (a) C11 and (b) C12 acquired at 11.7 T (νr = 11 kHz) as a function of evolution time τ (0 to 16 ms). The predicted 13C/17O heteronuclear spin echo dephasing behaviour (red line) is derived using GIPAW DFT (CASTEP) calculations of the one-bond dominant 1J(13C,17O) and 1J(13C,13C) coupling constants (1JC11O, 1JC11–O, 1JC11–C1, 1JC12–O2, 1JC12–OH and 1JC12–C9) and simulated using the SIMPSON 4.2.1 package. The contributions from nJ-couplings (n > 2) are predicted by CASTEP to be minimal and are therefore ignored. The experimental quadrupole coupling constants, which are in good agreement with the DFT results, are used throughout. As SIMPSON does not consider T2′, then these were measured using a simple homonuclear spin-echo. The experimental and predicted behaviour S(τ)het = 0 ms is normalised to an intensity of 1. The individual compositions, derived from experimental quadrupole parameters, CASTEP 1J couplings and numerical SIMPSON simulations are deconvoluted in Fig. S1 and S2 (ESI‡), up to 32 ms, the results were multiplied with the T2′ results and their enrichment probabilities: pO1O2 = pO3O4 = 80%; probabilities that C11 or C12 is directly bonded to two enriched 17O species, pO1 = pO2 = pO3 = pO4 = 7.5%; probabilities that C11 or C12 is directly bonded to one enriched 17O species, pnone = 5%; probability that C11 or C12 are not directly bonded to any enriched 17O species. Therefore, C11 = 0.85 × SIMPSON output of 1JC11, 1JC11–O, 0.075 × 1JC11O, 0.075 × 1JC11–O, 1. 1% (natural abundance 13C) × 1JC11–C1, all multiplied by a T2′ of 14 ms. An example of the SIMPSON input file is given in the ESI,‡ Table S3. As the natural abundance of the 13C isotope is 1.1%, these species will induce minor contributions to the dephasing curve and therefore these were considered in the DFT derived SIMPSON simulations. | ||
A CT-selective 17O π-pulse in a heteroncuelar spin echo experiment only refocuses the central-transition (CT) spin states, consequently confining the dephasing behaviour of the 13C signals observed in Fig. 6 to only those13C spins bonded to 17O spins in the +1/2 or −1/2 CT spin states. Presuming 100% 17O enrichment of an isolated 13C–17O two spin system, only 1/3 of 13C spins attached to 17O CT (+1/2 or −1/2) spin states will show a cosine J-modulation, while the other 2/3 of the 13C spins will show no cosine modulation as they couple to the satellite transition (ST) +5/2, +3/2, −3/2 or −5/2 spin states. Hence, a J-resolved experiment utilising a CT-selective 17O pulse for a 13C–17O spin pair will exhibit a cosine modulation (presuming no T2′ contribution) that goes from an initial signal intensity of 1, down to a minimum signal of S(τ)het = 0.33, with a 0.66 signal intensity for a J-evolution time of 1/2J. However, the relatively broad nature (∼41 kHz) of the multiple 17O environments in naphthalaldehydic acid means uniformly exciting the three oxygen environments required non-selective pulses, therefore accurate SIMPSON simulations are required to evaluate the data with respect to the pulse lengths used, and fitting the decay using a function is challenging.
All data presented in Fig. 6 were acquired using a modified spin echo sequence as detailed by Hung et al.,16 with the simulation of each multi-component spin echo dephasing curve being achieved using SIMPSON.45 An analytical expressions for deriving isolated 1J-couplings have been developed by Duma et al., with the quadrupolar contributions to the scalar coupling explored by Perras and Bryce.64,65 SIMPSON 4.2.1 cannot simulate multiple 1J-couplings with multiple quadrupole coupling constants. Therefore, the simulation was broken up into their individual one-bond components and multiplied together. The π pulse length (6 μs, 41 kHz), was also considered in the SIMPSON calculation to account for any errors or issues caused by finite pulse lengths, this pulse length was chosen as it is the same as the linewidth of the three oxygen environments.
The output of the SIMPSON simulations were then scaled to represent the oxygen enrichment, which was determined by high-resolution ESI-MS. This gave the following probabilities, p:
| pO1O2 = pO3O4 = 80%; probabilities that C11 or C12 is directly bonded to two enriched 17O species, |
| pO1 = pO2 = pO3 = pO4 = 7.5%; probabilities that C11 or C12 is directly bonded to one enriched 17O species, |
| pnone = 5%; probability that C11 or C12 are not directly bonded to any enriched 17O species. |
17O), O2 (C11–17O–), O3 (–17O–C12) or O4 (C12–17OH) species within 1, it is assumed that their contributions are equal. This assumption is supported by the ∼1:1:1 ratio of the three O environments in the multiple field 17O MAS and MQMAS NMR data represented in Fig. 3, 4(a) and (b). It should be noted that the overnight refluxing method (dioxane, 75 °C, 24 h) for 17O enrichment (see Fig. 3 and 4(b)) provides a uniformly labelled product for 1 which is thus preferred for this study, in comparison to the labelled product derived from shorter and milder (THF, 66 °C, 6 h) reaction conditions (see Fig. 4(c)) which highlights a preferential 17O enrichment of the –OH position. The DFT derived SIMPSON outputs do not consider the transverse relaxation contribution to the decays, therefore T2C11′ and T2C12′ were measured using a homonuclear spin echo sequence and upon these the 1J(13C,17O) coupling modulation is superimposed. These times are estimated by conventional homonuclear 13C spin echo (π/2–τ–π–τ–acquire) measurements to be 14 ± 2 ms and 10 ± 4 ms for the C11 and C12 positions, respectively.
The GIPAW DFT calculated 1J(13C,17O) coupling constants for the O positions are derived from the single crystal structure determination of 1,7 with these calculations being performed on both the geometry optimised and the experimental structures, coupled with associated dispersion corrections. These results are summarised in Table 3. Key to the effectiveness of this computational approach for these 1J(13C,17O) coupling constants is the highly accurate characterisation of 1 involving the more routinely GIPAW DFT calculated NMR parameters such as δiso, CQ and ηQ as presented in Fig. 3, and Tables 1 and 2, which demonstrates that the ultrasoft ‘on-the-fly’ O pseudopotential is particularly optimised for 17O NMR measurements in organic solids. This provides the simulation of the dephasing characteristics for C11 and C12 shown in Fig. 6(a) and (b) using the calculated 1J(13C,17O) coupling constants (1JCO1, 1JCO2, 1JCO3 and 1JCO4) with greater statistical importance.
As the C11 and C12 positions are not 13C labelled, the spin echo intensities at larger evolution times are difficult to determine, as they cannot be accurately discriminated from the noise. The error in each data point grows cumulatively for increasingly longer evolution times; as observed from Fig. 6, these dephasing measurements were truncated at a maximum evolution time of 15 ms. The contributions from neighbouring proton species are removed by high power 1H (100 kHz) decoupling during the evolution and acquisition periods; however, the XJ couplings to neighbouring 13C spins are not suppressed. The 1J-coupling from C11 to C1 and C12 to C9 are 58 and 39 Hz, respectively. As the natural abundance of the 13C isotope is 1.1%, these species will induce minor contributions to the dephasing curve and therefore these were considered in the DFT derived SIMPSON simulations. Extended homonuclear 1J(13C,13C) contributions are predicted by CASTEP to have an insignificant contribution to the decay (Table S2, ESI‡), and as there is no 13C labelling in the structure, the maximum contribution of this 1J(13C,13C) coupling is 1.1% of cos(πJCCτ) to the overall result, these may account for a minor source of error in the observed decay. Using high power decoupling limits the acquisition times you can safely record the data, therefore the cosine modulation feature which is predicted at a τ between 20 to 24 ms was not possible to observe. This would also be challenging to reproduce accurately due to reduced signal-to-noise at these larger τ delays and the need for high power decoupling at these extreme lengths is likely to cause damage to the probe.
Despite these sources of error, the results presented in Fig. 6 suggest that overall methodology invoked above appears to be a robust approach for calculating multiple 1J(13C,17O) couplings in a complex arrangement containing multiple oxo functionalities in the solid state. In particular, it is evident that GIPAW DFT calculations of 1J(13C,17O) coupling constants are accurate as observed by the strong correlation between the experimental dephasing behaviour and the simulated S(τ)het behaviour (adopting the isolated spin system approximations above) using calculated values of 1JCO1, 1JCO2, 1JCO3 and 1JCO4 from CASTEP. The correlation between the calculated data using the proposed model and the experimental data is excellent for the three nuclear spin system involving C12 which is directly bonded to the methine (–O–) and hydroxyl (–OH) moieties (i.e.1JCO3 and 1JCO4). It should be noted that the reduced correlation describing C11 involves 1JCO1 coupling to the CO moiety which is influenced by intramolecular H-bonding to the –OH species in an adjacent molecule. Furthermore, the errors in the C11 heteronuclear spin echo experiment were larger, due to insufficient signal to noise, possibly due to less efficient cross polarisation.
It is interesting to note that the magnitudes of the two 1J(13C,17O) couplings to the central ether –O– linkage reflect the corresponding bond lengths involved. The 1J(13C,17O) coupling constant for the shorter bond length CO species is 37 Hz (carbonyl C–O bond length: 1.338 Å), which is larger than the coupling constant of 32 Hz to the more distant CH–OH (methine C–O bond length: 1.490 Å). In contrast, the 1J(13C,17O) coupling constant between the methine C and the –OH species is smaller than that to the ether –O– atom (20 Hz cf. 32 Hz) despite having a significantly shorter bond length (1.379 Å cf. 1.490 Å). As mentioned above, –OH motion within the weak H-bonding arrangement can modulate and diminish the magnitude of this 1J(13C,17O) coupling, however the bond angle disposition as proposed by the Karplus relationship may also contribute to this marked reversal in this trend.15,66 Although calculations67 and measurements68 of 1J(13C,17O) coupling constants characterising the CO and CO2 gas phase systems have been reported (measured/calculated; 16.4/17.2 Hz and 16.1/17.1 Hz, respectively), the only existing measurement of 1J(13C,17O) couplings ascertained from a real solid state system pertains to glycine·HCl where 1J(CO) = 25 Hz and 1J(C–OH) = 28 Hz.16 The determined 1J (13C,17O) coupling constants that characterise solid naphthalaldehydic acid 1 summarised in Table 3 are comparable to these previous values, and this study reports the first 1J(13C,17O) measurements on ether –O– linkages. This work has demonstrated that measurements of 1J(13C,17O) coupling constants is not limited to selectively labelled highly isolated spin systems in small molecules, and that this information can be elucidated from larger and more complex spin systems which are isotopically enriched by more routine synthetic methods.
Conclusions
A combined 17O MAS, 3QMAS and DOR NMR approach has been utilised to accurately characterise the O positions in naphthalaldehydic acid 1 and to assess the accuracy of the first principles GIPAW DFT calculations for deriving the associated 17O NMR parameters. The initial phase of this study demonstrated that 17O NMR parameters such as the isotropic chemical shift (δiso) and quadrupole parameters (CQ, ηQ) were well correlated with those emanating from the GIPAW DFT calculations using the CASTEP code. Heteronuclear 1J(13C,17O) measurements were undertaken on this system which possesses multiple O functionalities in the hydroxy lactone head group of the molecule. These measurements represent the first assessment of multiple 1J(13C,17O) couplings from a single molecule in the solid state, and they were also evaluated against the scalar coupling calculations resulting from first principles GIPAW DFT calculations and SIMPSON simulations. For the modified spin-echo dephasing experiment focussing on the C11 and C12 positions in 1, the dephasing of the magnetisation in each case relates to two three-spin (17O–13C–17O) systems in which the O positions are enriched. This experiment cannot isolate the individual 1J(13C,17O) contributions and a weighted average dephasing curve is produced which can only be deconvoluted by simulation using 1J(13C,17O) couplings from the GIPAW DFT calculations as the SIMPSON input, hence the reliance on accurate DFT prediction in this study. The resultant simulated spin-echo dephasing curves produced are in good agreement (within error) with those experimentally derived.The geometry optimised DFT structure derived using the CASTEP code gives a firm agreement with the experimentally measured shifts observed for the ether (δiso = 223, PQ = 9.4 MHz) and hydroxyl (δiso = 62, PQ = 10.5 MHz) groups, however the unoptimised experimental structure has better agreement for the carbonyl group (δiso = 320, PQ = 8.3 MHz). The determined δiso and ηQ values for the latter are shown to be consistent with bond lengths closer to 1.222 Å emanating from the unoptimised experimental XRD structure, rather than the geometry optimised length of 1.238 Å. The geometry optimised DFT 1J(13C,17O) coupling to the hydroxyl is calculated as 20 Hz and the 1JCO couplings to the ether oxygen were calculated to be 37 (O–CO) and 32 (O–C–OH) Hz. These measurements are all within error of the experimental echo decays.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
JVH acknowledges support for the solid state NMR instrumentation at Warwick used in this research which was funded by EPSRC (grants EP/M028186/1 and EP/K024418/1) and the University of Warwick, with additional partial funding being provided through the Birmingham Science City Advanced Materials Projects 1 and 2, which were supported by Advantage West Midlands (AWM) and the European Regional Development Fund (ERDF). The UK 850 MHz National Solid State NMR Facility used in this research is also acknowledged; this was funded by the EPSRC, BBSRC and the University of Warwick which included partial funding via the Birmingham Science City Advanced Materials Projects 1 and 2, supported by Advantage West Midlands (AWM) and the European Regional Development Fund (ERDF). JVH also acknowledges the University of Warwick Scientific Computing Research Technology Platform (RTP), and EPSRC grant EP/K000128/1, for access to the TINUS and MINERVA high performance computing clusters, respectively, that facilitated the computational effort undertaken in this work. JDW thanks the EPSRC for grant EP/E018203/1 from the Physical Organic Chemistry Initiative, and Nottingham Trent University for financial support.References
- J. Clayden, N. Greeves and S. Warren, Organic Chemistry, OUP Oxford, 2012 Search PubMed.
- S. Mandal, S. Mandal, S. K. Ghosh, P. Sar, A. Ghosh, R. Saha and B. Saha, RSC Adv., 2016, 6, 69605–69614 RSC.
- A. Williamson, London, Edinburgh Dublin Philos. Mag. J. Sci., 1850, 37, 350–356 CrossRef.
- Z. Wang, Comprehensive Organic Name Reactions and Reagents, John Wiley & Sons, Inc., 2010 DOI:10.1002/9780470638859.conrr480.
- M. T. Scerba, A. F. DeBlase, S. Bloom, T. Dudding, M. A. Johnson and T. Lectka, J. Phys. Chem. A, 2012, 116, 3556–3560 CrossRef CAS PubMed.
- S. Lau, B. Ward, X. Zhou, A. J. P. White, I. J. Casely, S. A. Macgregor and M. R. Crimmin, Organometallics, 2017, 36, 3654–3663 CrossRef CAS PubMed.
- G. Saritemur, L. Nomen Miralles, D. Husson, M. B. Pitak, S. J. Coles and J. D. Wallis, CrystEngComm, 2016, 18, 948–961 RSC.
- A. Choudhary, K. J. Kamer and R. T. Raines, J. Org. Chem., 2011, 76, 7933–7937 CrossRef CAS PubMed.
- K. J. Kamer, A. Choudhary and R. T. Raines, J. Org. Chem., 2013, 78, 2099–2103 CrossRef CAS PubMed.
- A. Choudhary, C. G. Fry, K. J. Kamer and R. T. Raines, Chem. Commun., 2013, 49, 8166–8168 RSC.
- A. Choudhary, R. W. Newberry and R. T. Raines, Org. Lett., 2014, 16, 3421–3423 CrossRef CAS PubMed.
- R. W. Newberry, G. J. Bartlett, B. VanVeller, D. N. Woolfson and R. T. Raines, Protein Sci., 2014, 23, 284–288 CrossRef CAS PubMed.
- G. J. Bartlett, R. W. Newberry, B. VanVeller, R. T. Raines and D. N. Woolfson, J. Am. Chem. Soc., 2013, 135, 18682–18688 CrossRef CAS PubMed.
- H. Kessler, C. Griesinger, J. Lautz, A. Mueller, W. F. Van Gunsteren and H. J. C. Berendsen, J. Am. Chem. Soc., 1988, 110, 3393–3396 CrossRef CAS.
- M. Karplus, J. Am. Chem. Soc., 1963, 85, 2870–2871 CrossRef CAS.
- I. Hung, A.-C. Uldry, J. Becker-Baldus, A. L. Webber, A. Wong, M. E. Smith, S. A. Joyce, J. R. Yates, C. J. Pickard, R. Dupree and S. P. Brown, J. Am. Chem. Soc., 2009, 131, 1820–1834 CrossRef CAS PubMed.
- D. H. Barich, J. M. Davis, L. J. Schieber, M. T. Zell and E. J. Munson, J. Pharm. Sci., 2006, 95, 1586–1594 CrossRef CAS PubMed.
- S. Penzel, A. A. Smith, V. Agarwal, A. Hunkeler, M.-L. Org, A. Samoson, A. Böckmann, M. Ernst and B. H. Meier, J. Biomol. NMR, 2015, 63, 165–186 CrossRef CAS PubMed.
- K. Sharma, P. K. Madhu and V. Agarwal, J. Magn. Reson., 2016, 270, 136–141 CrossRef CAS PubMed.
- M. Profeta, F. Mauri and C. J. Pickard, J. Am. Chem. Soc., 2003, 125, 541–548 CrossRef CAS PubMed.
- J. Clark Stewart, D. Segall Matthew, J. Pickard Chris, J. Hasnip Phil, I. J. Probert Matt, K. Refson and C. Payne Mike, Z. Kristallogr., 2005, 220, 567 Search PubMed.
- J. R. Yates, C. J. Pickard and F. Mauri, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 024401 CrossRef.
- T. F. G. Green and J. R. Yates, J. Chem. Phys., 2014, 140, 234106 CrossRef PubMed.
- A. P. M. Kentgens, W. S. Veeman and J. Van Bree, Macromolecules, 1987, 20, 1234–1237 CrossRef CAS.
- A. G. Stepanov, V. N. Zudin and K. I. Zamaraev, Solid State Nucl. Magn. Reson., 1993, 2, 89–93 CrossRef CAS PubMed.
- D. Massiot, F. Fayon, M. Deschamps, S. Cadars, P. Florian, V. Montouillout, N. Pellerin, J. Hiet, A. Rakhmatullin and C. Bessada, C. R. Chim., 2010, 13, 117–129 CrossRef CAS.
- A. Lesage, M. Bardet and L. Emsley, J. Am. Chem. Soc., 1999, 121, 10987–10993 CrossRef CAS.
- A. Lesage, S. Steuernagel and L. Emsley, J. Am. Chem. Soc., 1998, 120, 7095–7100 CrossRef CAS.
- A. Lesage, C. Auger, S. Caldarelli and L. Emsley, J. Am. Chem. Soc., 1997, 119, 7867–7868 CrossRef CAS.
- S. P. Brown and L. Emsley, J. Magn. Reson., 2004, 171, 43–47 CrossRef CAS PubMed.
- S. P. Brown, Solid State Nucl. Magn. Reson., 2012, 41, 1–27 CrossRef CAS PubMed.
- R. K. Harris, E. D. Becker, S. M. Cabral de Menezes, R. Goodfellow and P. Granger, Magn. Reson. Chem., 2002, 40, 489–505 CrossRef CAS.
- R. Dupree, eMagRes, John Wiley & Sons, Ltd, 2007 DOI:10.1002/9780470034590.emrstm1203.
- D. Massiot, F. Fayon, M. Capron, I. King, S. Le Calve, B. Alonso, J. O. Durand, B. Bujoli, Z. H. Gan and G. Hoatson, Magn. Reson. Chem., 2002, 40, 70–76 CrossRef CAS.
- M. Bak, J. T. Rasmussen and N. C. Nielsen, J. Magn. Reson., 2000, 147, 296–330 CrossRef CAS PubMed.
- C. J. Pickard and F. Mauri, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 245101 CrossRef.
- S. A. Joyce, J. R. Yates, C. J. Pickard and F. Mauri, J. Chem. Phys., 2007, 127, 204107 CrossRef PubMed.
- G. J. Rees, S. P. Day, A. Lari, A. P. Howes, D. Iuga, M. B. Pitak, S. J. Coles, T. L. Threlfall, M. E. Light, M. E. Smith, D. Quigley, J. D. Wallis and J. V. Hanna, CrystEngComm, 2013, 15, 8823–8839 RSC.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, S1–S19, 10.1039/P298700000S1.
- M. P. Hanrahan, A. Venkatesh, S. L. Carnahan, J. L. Calahan, J. W. Lubach, E. J. Munson and A. J. Rossini, Phys. Chem. Chem. Phys., 2017, 19, 28153–28162 RSC.
- J. K. Williams, K. Schmidt-Rohr and M. Hong, Solid State Nucl. Magn. Reson., 2015, 72, 118–126 CrossRef CAS PubMed.
- T. D. Ferris and T. C. Farrar, Mol. Phys., 2002, 100, 303–309 CrossRef CAS.
- L. S. Batchelder, J. Clymer and J. L. Ragle, J. Chem. Phys., 1981, 74, 4791–4799 CrossRef CAS.
- G. Wu, I. Hung, Z. Gan, V. V. Terskikh and X. Kong, J. Phys. Chem. A, 2019, 123, 8243–8253 CrossRef CAS PubMed.
- M. Bak, J. T. Rasmussen and N. C. Nielsen, J. Magn. Reson., 2011, 213, 366–400 CrossRef CAS PubMed.
- V. Lemaître, M. E. Smith and A. Watts, Solid State Nucl. Magn. Reson., 2004, 26, 215–235 CrossRef PubMed.
- X. Kong, Y. Dai and G. Wu, Solid State Nucl. Magn. Reson., 2017, 84, 59–64 CrossRef CAS PubMed.
- K. J. Pike, V. Lemaitre, A. Kukol, T. Anupõld, A. Samoson, A. P. Howes, A. Watts, M. E. Smith and R. Dupree, J. Phys. Chem. B, 2004, 108, 9256–9263 CrossRef CAS.
- X. Kong, V. V. Terskikh, R. L. Khade, L. Yang, A. Rorick, Y. Zhang, P. He, Y. Huang and G. Wu, Angew. Chem., Int. Ed., 2015, 54, 4753–4757 CrossRef CAS PubMed.
- K. Yamauchi, S. Kuroki and I. Ando, J. Mol. Struct., 2002, 602–603, 171–175 CrossRef CAS.
- K. Yamauchi, S. Kuroki, I. Ando, T. Ozaki and A. Shoji, Chem. Phys. Lett., 1999, 302, 331–336 CrossRef CAS.
- C. Gervais, R. Dupree, K. J. Pike, C. Bonhomme, M. Profeta, C. J. Pickard and F. Mauri, J. Phys. Chem. A, 2005, 109, 6960–6969 CrossRef CAS PubMed.
- K. J. D. MacKenzie and M. E. Smith, Multinuclear Solid-State Nuclear Magnetic Resonance of Inorganic Materials, Pergamon Press, Oxford, 2002 Search PubMed.
- A. Samoson and E. Lippmaa, J. Magn. Reson., 1989, 84, 410–416 CAS.
- A. Samoson, Chem. Phys. Lett., 1985, 119, 29–32 CrossRef CAS.
- B. P. de Laune, G. J. Rees, J. F. Marco, H.-Y. Hah, C. E. Johnson, J. A. Johnson, F. J. Berry, J. V. Hanna and C. Greaves, Inorg. Chem., 2017, 56, 10078–10089 CrossRef CAS PubMed.
- J. V. Hanna, K. J. Pike, T. Charpentier, T. F. Kemp, M. E. Smith, B. E. G. Lucier, R. W. Schurko and L. S. Cahill, Chem. – Eur. J., 2010, 16, 3222–3239 CrossRef CAS PubMed.
- C. S. Griffith, V. Luca, J. V. Hanna, K. J. Pike, M. E. Smith and G. S. Thorogood, Inorg. Chem., 2009, 48, 5648–5662 CrossRef CAS PubMed.
- C. Jager, NMR Basic Principles and Progress, Springer Verlag, Berlin, 1994, p. 135 Search PubMed.
- A. Wong, I. Hung, A. P. Howes, T. Anupõld, J. Past, A. Samoson, S. P. Brown, M. E. Smith and R. Dupree, Magn. Reson. Chem., 2007, 45, S68–S72 CrossRef CAS PubMed.
- A. Wong, A. P. Howes, K. J. Pike, V. Lemaître, A. Watts, T. Anupõld, J. Past, A. Samoson, R. Dupree and M. E. Smith, J. Am. Chem. Soc., 2006, 128, 7744–7745 CrossRef CAS PubMed.
- X. Kong, A. Brinkmann, V. Terskikh, R. E. Wasylishen, G. M. Bernard, Z. Duan, Q. Wu and G. Wu, J. Phys. Chem. B, 2016, 120, 11692–11704 CrossRef CAS PubMed.
- J. Lu, I. Hung, A. Brinkmann, Z. Gan, X. Kong and G. Wu, Angew. Chem., Int. Ed., 2017, 56, 6166–6170 CrossRef CAS PubMed.
- L. Duma, W. C. Lai, M. Carravetta, L. Emsley, S. P. Brown and M. H. Levitt, ChemPhysChem, 2004, 5, 815–833 CrossRef CAS PubMed.
- F. A. Perras and D. L. Bryce, J. Magn. Reson., 2014, 242, 23–32 CrossRef CAS PubMed.
- M. Karplus, J. Chem. Phys., 1959, 30, 11–15 CrossRef CAS.
- B. Wrackmeyer, Z. Naturforsch., B: J. Chem. Sci., 2004, 59, 286–290 CAS.
- R. E. Wasylishen, J. O. Friedrich, S. Mooibroek and J. B. Macdonald, J. Chem. Phys., 1985, 83, 548–551 CrossRef CAS.
Footnotes |
| † The experimental data for this study are provided as a supporting dataset from WRAP, the Warwick Research Archive Portal at http://wrap.warwick.ac.uk/130520. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cp03977e |
| This journal is © the Owner Societies 2020 |