
Copper-catalysed ortho-selective C–H bond functionalization of phenols and naphthols with α-aryl-α-diazoesters†
Ben
Ma
 ab,
Zhiqiong
Tang
a,
Junliang
Zhang
*abc and
Lu
Liu
*ab
ab,
Zhiqiong
Tang
a,
Junliang
Zhang
*abc and
Lu
Liu
*ab
aSchool of Chemistry and Molecular Engineering, East China Normal University, 500 Dongchuan Road, Shanghai, 200241, P. R. China. E-mail: lliu@chem.ecnu.edu.cn
bShanghai Engineering Research Center of Molecular Therapeutics and New Drug Development, East China Normal University, Shanghai 200062, P. R. China
cDepartment of Chemistry, Fudan University, 2005 Songhu Road, Shanghai 200438, P. R. China. E-mail: junliangzhang@fudan.edu.cn
First published on 9th July 2020
Abstract
Herein, we reported the first copper-catalyzed highly efficient C(sp2)–H functionalization of unprotected naphthols and phenols with α-aryl-α-diazoesters. In this transformation, CuCl2 efficiently promoted the highly chemo- and site-selective C–H bond functionalization under mild conditions, furnishing diverse phenol derivatives in moderate to excellent yields from readily available starting materials.
Phenol and naphthol motifs are prevalent in natural products, dyes, medicines, bioactive compounds, functional materials, and privileged chiral ligands, and they also represent the most readily available chemical feedstocks and versatile building blocks for diverse transformations in chemical science.1 Thus, the development of straightforward strategies to synthesize phenol derivatives via the direct site-selective C–H bond functionalization of free phenols is highly attractive to the synthetic community. However, chemo- and site-selective C–H functionalization of unprotected phenols poses considerable challenges, because there are four possible reaction sites, including the chemoselectivity of oxygen or carbon and the site-selectivity of the ortho-, meta- or para-position.2
In the past decade, with the advance of transition-metal-catalysed directed C–H bond functionalization,3 many strategies have been developed to achieve ortho-selective functionalization of phenols. In this regard, a directing-group-assisted approach, which requires directing groups to be installed on the oxygen atom to ensure ortho selectivity, as well as to protect the hydroxyl group, has emerged as one of the most efficient and popular solutions to address the ortho-selectivity problem (Scheme 1a).4 However, this extra operation of installing and removing such directing groups limits the utilization of this approach in organic synthesis. Recently, Bedford and Ye et al. realized an alternative way for direct ortho-selective C–H bond arylation of phenols and naphthols by using P-containing ligands R2PX as transient directing groups (Scheme 1b).5 Unfortunately, this strategy is still limited to a few examples.
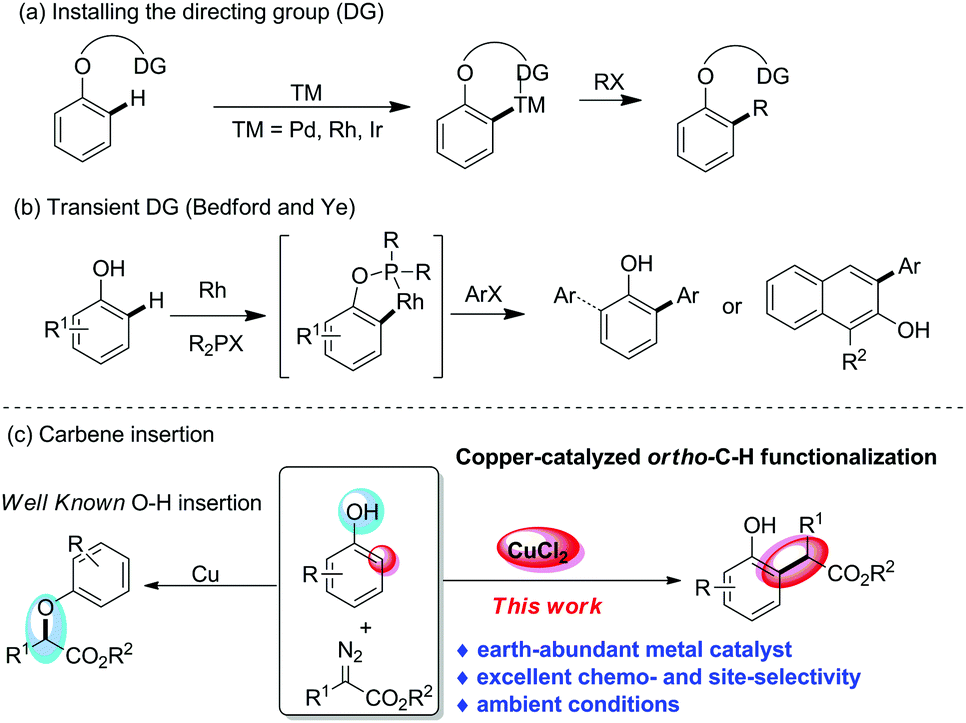 | ||
| Scheme 1 Ortho-selective C–H functionalization of phenols. | ||
Recently, the catalytic carbene transfer reaction, typically using diazo compounds,6 has emerged as one of the most powerful strategies for aromatic C(sp2)–H bond functionalization.7 Nonetheless, most of the commonly used catalysts are based on noble metals, including rhodium, gold, palladium and iridium, which are becoming more and more expensive and scarce because they are derived from dwindling resources.8 The development of effective and abundant catalysts to replace the rare and toxic transition metals is a long-term need. Copper, which is an earth abundant, readily available, inexpensive, environmentally benign, and less toxic metal, represents an ideal catalyst in organic synthesis.9 Although Cu-catalysed carbene transfer reactions have been in use for over half a century, compared with the well-established O–H bond insertion of phenols with copper–carbene,10 the analogous C(sp2)–H bond functionalization is still unknown.11 Very recently, Nemoto and we have reported the ortho-C–H functionalization of naphthols and phenols with diazoesters.12 However, these methods require the use of expensive catalysts, gold complexes or B(C6F5)3. In addition, gold catalysts12b,c were suitable for naphthols, but were not compatible with phenols, and B(C6F5)312a showed the opposite trend. A general approach to achieve ortho-alkylation of both phenols and naphthols has not been developed so far. Herein, we reported the unprecedented CuCl2-catalysed highly chemo- and ortho-selective C–H bond functionalization of phenols and naphthols with diazoesters (Scheme 1c).
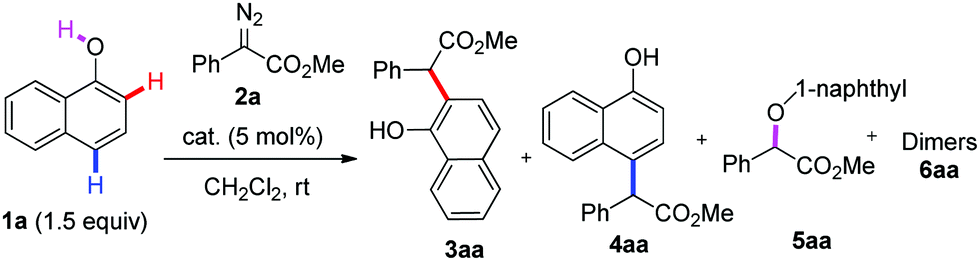
Initially, we performed the reaction of 1-naphthol 1a with α-phenyl-α-diazoacetate 2a in the presence of 5 mol% Cu(ClO4)2·6H2O in DCM at room temperature. To our delight, the desired ortho-C–H functionalization products, including 3aa and the corresponding lactone 11 from 3aa, were obtained in 60% yield with 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 C–H/O–H selectivity and regio-specific ortho-selectivity (Table 1, entry 1). Encouraged by this result, various copper salts were then screened, and finally CuCl2 was observed to be the best catalyst, affording the ortho-selective C–H bond alkylation product in 85% yield with excellent chemo- and site-selectivity after 6 h at room temperature (Table 1, entries 2–7). Solvent screening showed that DCE, toluene and THF could not improve the yield (Table 1, entries 10–12).
:1 C–H/O–H selectivity and regio-specific ortho-selectivity (Table 1, entry 1). Encouraged by this result, various copper salts were then screened, and finally CuCl2 was observed to be the best catalyst, affording the ortho-selective C–H bond alkylation product in 85% yield with excellent chemo- and site-selectivity after 6 h at room temperature (Table 1, entries 2–7). Solvent screening showed that DCE, toluene and THF could not improve the yield (Table 1, entries 10–12).
|
|
||
|---|---|---|
| Entry | Cat. (5 mol%) | Yielda (%) |
| 3/4/5/6 | ||
| Reaction conditions: a solution of 2a (0.4 mmol) in 1 mL of solvent was introduced into 1a (0.6 mmol) and the catalyst (5 mol%) in solvent (1 mL) at room temperature via a syringe over a period of 15 min.a NMR yield.b The corresponding lactone 11 from 3aa was detected.c DCE instead of DCM.d Toluene instead of DCM.e THF instead of DCM. | ||
| 1 | Cu(ClO4)2·6H2O | 60 (12)b/0/2/38 |
| 2 | Cu(CH3CN)4PF6 | 60 (18)b/0/2/38 |
| 3 | CuOAc | 68 (10)b/0/1/31 |
| 4 | CuOTf | 54 (24)b/0/0/46 |
| 5 | CuCl | 72(4)b/0/0/22 |
| 6 | CuCl2 | 85 (2) /0/2/3 |
| 7 | CuF2 | 70(12)b/0/2/24 |
| 8c | CuCl2 | 76 (2)b/0/5/5 |
| 9d | CuCl2 | 53/0/3/35 |
| 10e | CuCl2 | 25/0/5/31 |
Next, the scope of the copper-catalysed ortho-selective C–H functionalization of 1-naphthol with various α-aryl-α-diazoacetates was explored using CuCl2 (5 mol%) in DCM at room temperature. As shown in Scheme 2, this reaction proved to be applicable to a wide range of α-aryl-α-diazoacetates and the desired 2-alkylated 1-naphthols were obtained in good to excellent yields with excellent chemo- and site-selectivity. The steric hindrance of the ester group had no effect on this reaction (3ab–3ad). Various commonly encountered functional groups such as chloro, bromo, methyl, methoxyl, and CF3 at various positions of the phenyl rings of α-aryl-α-diazoacetates were well tolerated (3ae–3at). Strong electron-donating groups (OMe) could facilitate this reaction (3ah and 3ao), while strong electron-withdrawing groups (CF3) would slow down this reaction (3am). Furthermore, several substituted 1-naphthols 1b–1e were tested in this transformation, delivering the corresponding ortho-selective C–H functionalization products 3ba–3ea in moderate to excellent yields (68% to 97%). It must be noted that all the reactions exhibited 100% ortho-selectivity.
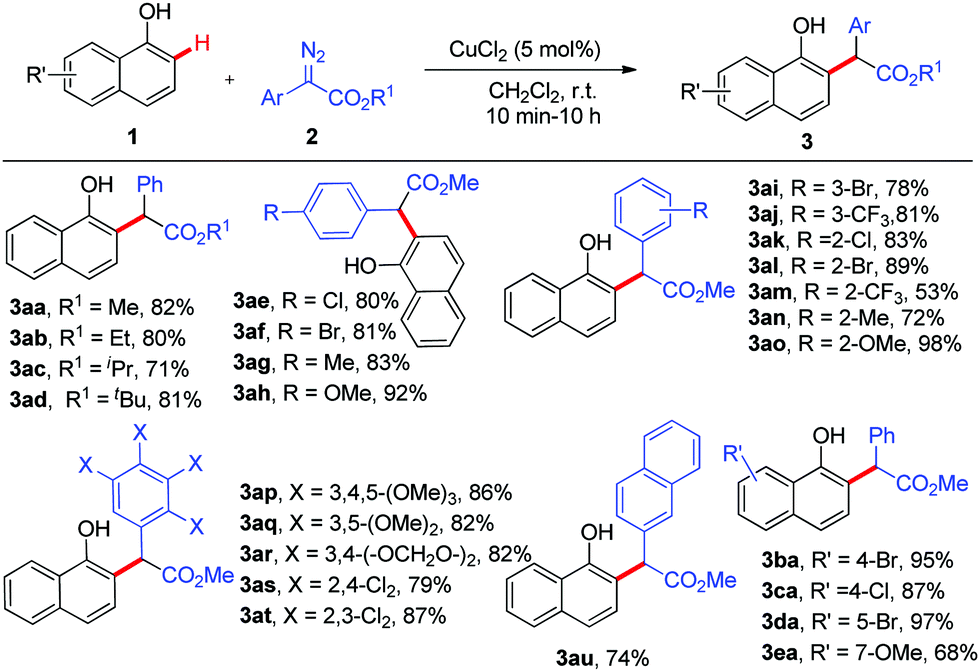 | ||
| Scheme 2 Ortho C–H bond functionalization of 1-naphthols. | ||
Subsequently, we investigated the substrate scope of the CuCl2-catalyzed ortho-selective C–H bond functionalization of 2-naphthols. All the reactions of 2-naphthols 7 with various diazoacetates 2 proceeded smoothly under standard conditions, affording the corresponding ortho-selective products in 58% to 98% yields with excellent chemo- and site-selectivity (Scheme 3).
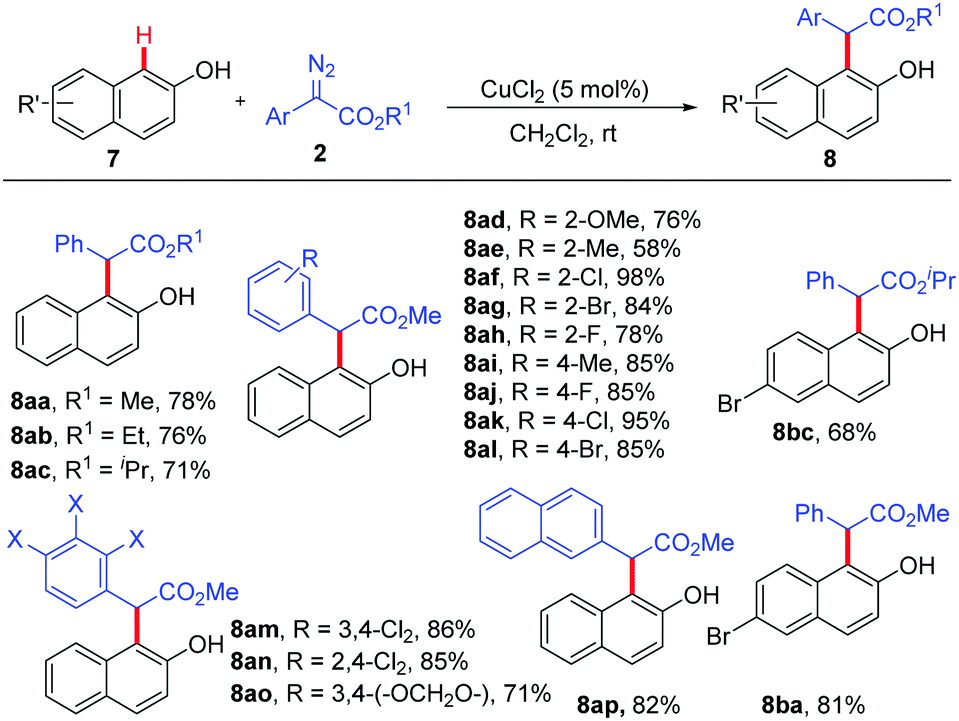 | ||
| Scheme 3 Ortho C–H bond functionalization of 2-naphthols. | ||
Furthermore, we wondered whether phenols were applicable to this copper-catalysed ortho-C–H functionalization, which was more challenging because the nucleophilicity of phenols was much lower than that of naphthols. Indeed, all attempts to realize the ortho-C–H alkylation of unsubstituted phenol failed. To our delight, CuCl2 could enable the ortho-C–H bond functionalization of alkoxyl-substituted phenols 9 with α-aryl-α-diazoacetates 2 to give the corresponding products 10a–10d in good to excellent yields with excellent site-selectivity (Scheme 4).
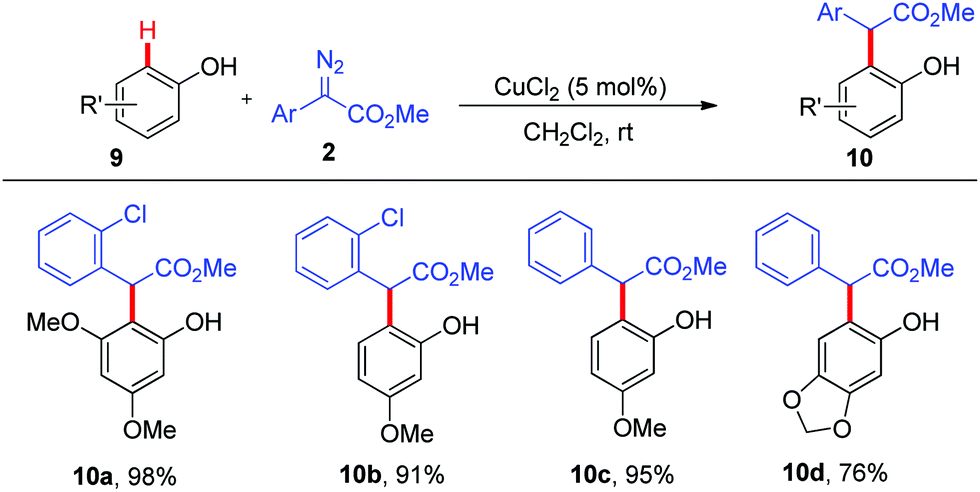 | ||
| Scheme 4 Ortho C–H bond functionalization of phenols. | ||
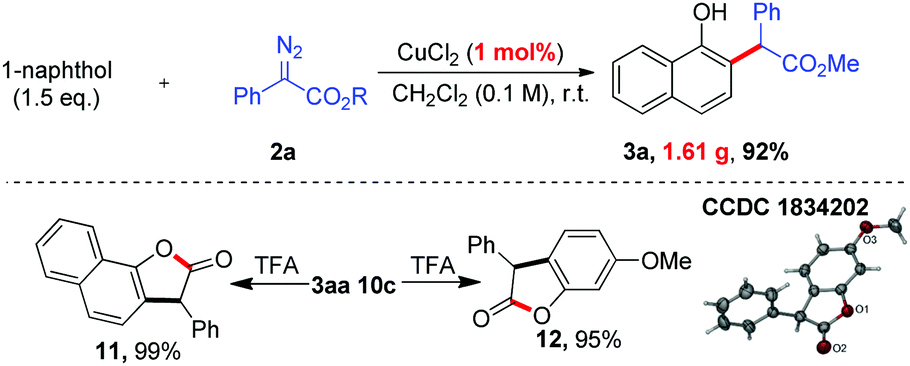
To demonstrate the practicality of this transformation, a gram-scale reaction was carried out (Scheme 5). To our delight, the ortho-selective alkylation was easy to scale up to a gram-scale with only 1 mol% catalyst loading, affording the desired product 3aa (1.61 g, 92%). To demonstrate the utility of this method further, transformations of the products were also performed. As shown in Scheme 5, TFA-catalyzed lactonization of 3aa and 10c could afford the corresponding cyclized products 11 and 12 in 99% and 95% yields, respectively. The structure of 12 was further confirmed by single-crystal X-ray analysis.13
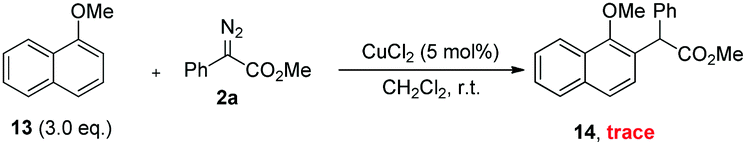 | (1) |
 | ||
| Scheme 5 Gram-scale reactions and transformations of products. | ||
To further gain mechanistic insight into this copper-catalysed ortho-C–H alkylation of phenols, 1-methoxynaphthalene 13 was reacted with diazoester 2a in the presence of CuCl2, which only delivered a trace amount of ortho-selective C–H bond functionalization products (eqn (1)). This result indicated that the hydroxyl group was vital not only for site-selectivity but also for reactivity, and the interaction between the hydroxyl group and the copper catalyst could give rise to the ortho-selectivity.
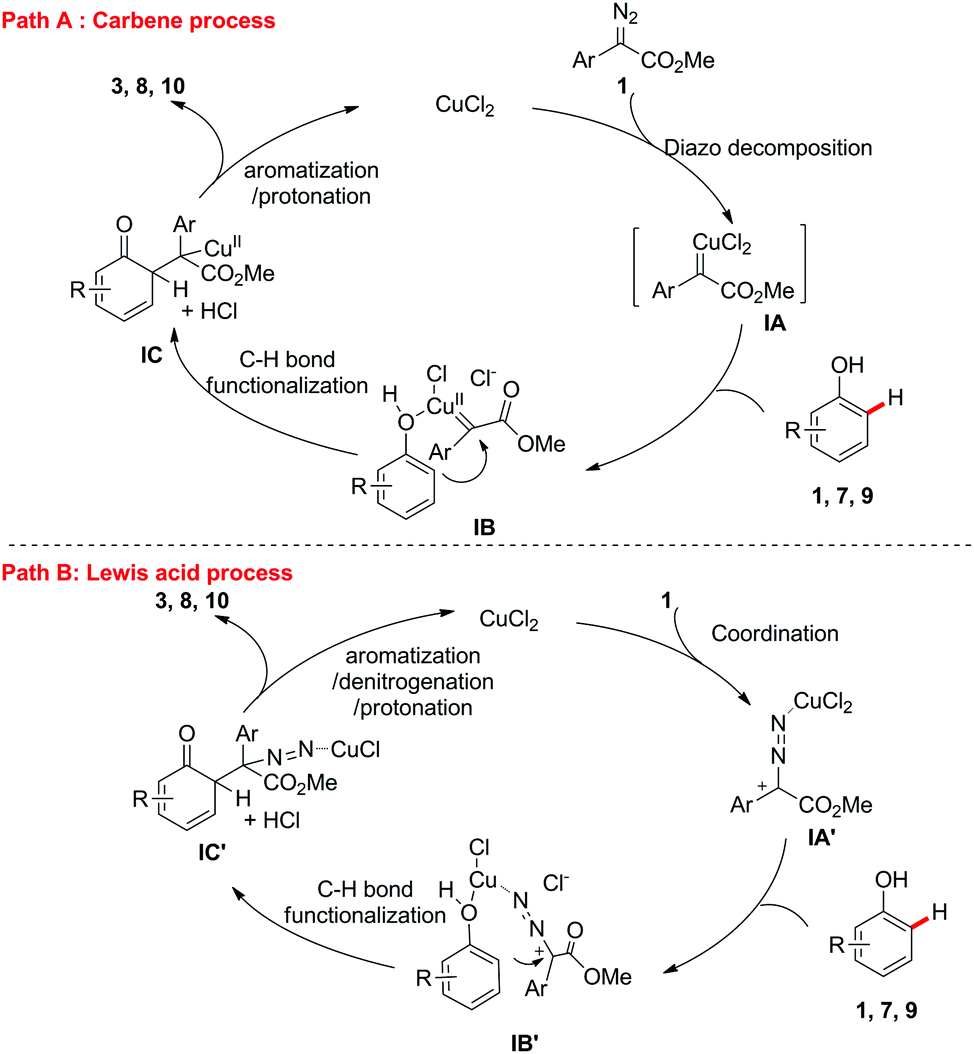
Based on the aforementioned results, two possible catalytic cycles accounting for this transformation are depicted in Scheme 6. Path A is the Cu-carbene process. The Cu(II)-carbene intermediate IA was formed from α-aryl-α-diazoesters 2 with CuCl2,14 which results in the formation of intermediate IBvia coordination. The electrophilic addition of copper-carbene at the ortho-position of the phenols generated IC, which then underwent aromatization and protonation to produce the target ortho-C–H bond functionalization products 3, 8 or 10 and regenerate the copper catalyst. In path B, CuCl2 serves as a Lewis acid to coordinate the nitrogen of α-aryl-α-diazoesters 2 to form the carbocation intermediate IA′, which would further coordinate the oxygen of the phenols to generate IB′. The following electrophilic addition of the copper-carbocation at the ortho-position of the phenols generated IC′, which then underwent aromatization, denitrogenation and protonation to produce the target ortho-C–H bond functionalization products and regenerate the copper catalyst. Further mechanistic studies indicated that these two processes occurred in this reaction.15
 | ||
| Scheme 6 Proposed catalytic cycle. | ||
To conclude, we have described the first example of a CuCl2-catalyzed direct C–H functionalization of unprotected phenols and naphthols with α-aryl-α-diazoacetates under mild conditions, leading to diverse phenol derivatives with convertible functional groups. This work broadens the application scope of copper catalysts in carbene transfer reactions. The salient features of this reaction include an inexpensive catalyst, readily available starting materials, unprecedented C–H functionalization rather than O–H insertion, a good substrate scope, mild conditions, and diverse convenient transformations of the products. Further studies on the mechanism and the application of this protocol in organic synthesis are currently underway in our laboratory.
We are grateful to the National Natural Science Foundation of China (No. 21971066, 21772042) and the STCSM (18JC1412300) for financial support.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) Synthetic and Natural Phenols, ed. J. H. P. Tyman, Elsevier, 1996 Search PubMed; (b) F. H. Fiege, H.-W. Voges, T. Hamamoto, S. Umemura, T. Iwata, H. Miki, Y. Fujita, H.-J. Buysch, D. Garbe and W. Paulus, Phenol derivatives, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley VCH, Weinheim, 2000 Search PubMed; (c) J. M. Brunel, Chem. Rev., 2005, 105, 857 CrossRef CAS PubMed.
- For recent reviews on phenol functionalization, see: (a) D.-G. Yu, F. de Azambuja and F. Glorius, Angew. Chem., Int. Ed., 2014, 53, 7710 CrossRef CAS PubMed; (b) G. Rousseau and B. Breit, Angew. Chem., Int. Ed., 2011, 50, 2450 CrossRef CAS PubMed; (c) Z. Huang and J.-P. Lumb, ACS Catal., 2019, 9, 521 CrossRef CAS ; for recent examples, see: ; (d) K. Kobayashi, M. Arisawa and M. Yamaguchi, J. Am. Chem. Soc., 2002, 124, 8528 CrossRef CAS PubMed; (e) D.-H. Lee, K.-H. Kwon and C. S. Yi, J. Am. Chem. Soc., 2012, 134, 7325 CrossRef CAS PubMed; (f) C. Yu and F. W. Patureau, Angew. Chem., Int. Ed., 2018, 57, 11807 CrossRef CAS PubMed; (g) G. Wang, L. Gao, H. Chen, X. Liu, J. Cao, S. Chen, X. Cheng and S. Li, Angew. Chem., Int. Ed., 2019, 58, 1694 CrossRef CAS PubMed; (h) J.-L. Dai, N.-Q. Shao, J. Zhang, R.-P. Jia and D.-H. Wang, J. Am. Chem. Soc., 2017, 139, 12390 CrossRef CAS PubMed; (i) S. Agasti, U. Sharma, T. Naveen and D. Maiti, Chem. Commun., 2015, 51, 5375 RSC; (j) P. Feng, G. Ma, X. Chen, X. Wu, L. Lin, P. Liu and T. Chen, Angew. Chem., Int. Ed., 2019, 58, 8400 CrossRef CAS PubMed; (k) M. Murai, M. Yamamoto and K. Takai, Org. Lett., 2019, 21, 3441 CrossRef CAS PubMed.
- (a) C. Cheng and J. F. Hartwig, Chem. Rev., 2015, 115, 8946 CrossRef CAS PubMed; (b) O. Daugulis, J. Roane and L. D. Tran, Acc. Chem. Res., 2015, 48, 1053 CrossRef CAS PubMed; (c) Y. Segawa, T. Maekawa and K. Itami, Angew. Chem., Int. Ed., 2015, 54, 66 CrossRef CAS PubMed; (d) S. A. Girard, T. Knauber and C.-J. Li, Angew. Chem., Int. Ed., 2014, 53, 74 CrossRef CAS PubMed; (e) P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879 CrossRef CAS PubMed; (f) B.-J. Li and Z.-J. Shi, Chem. Soc. Rev., 2012, 41, 5588 RSC.
- For selected recent examples, see: (a) T. A. Boebel and J. F. Hartwig, J. Am. Chem. Soc., 2008, 130, 7534 CrossRef CAS PubMed; (b) C. Huang and V. Gevorgyan, J. Am. Chem. Soc., 2009, 131, 10844 CrossRef CAS PubMed; (c) C. Huang, B. Chattopadhyay and V. Gevorgyan, J. Am. Chem. Soc., 2011, 133, 12406 CrossRef CAS PubMed; (d) B. Xiao, Y. Fu, J. Xu, T.-J. Gong, J.-J. Dai, J. Yi and L. Liu, J. Am. Chem. Soc., 2010, 132, 468 CrossRef CAS PubMed; (e) X. Zhao, C. S. Yeung and V. M. Dong, J. Am. Chem. Soc., 2010, 132, 5837 CrossRef CAS PubMed; (f) T. Patra, S. Bag, R. Kancherla, A. Mondal, A. Dey, S. Pimparkar, S. Agasti, A. Modak and D. Maiti, Angew. Chem., Int. Ed., 2016, 55, 7751–7755 CrossRef CAS PubMed; (g) Y. Wu, Z. Chen, Y. Yang, W. Zhu and B. Zhou, J. Am. Chem. Soc., 2018, 140, 42 CrossRef CAS PubMed.
- (a) R. B. Bedford, S. J. Coles, M. B. Hursthouse and M. E. Limmert, Angew. Chem., Int. Ed., 2003, 42, 112 CrossRef CAS PubMed; (b) R. B. Bedford and M. E. Limmert, J. Org. Chem., 2003, 68, 8669 CrossRef CAS PubMed; (c) J.-F. Yang, R.-H. Wang, Y.-X. Wang, W.-W. Yao, Q.-S. Liu and M. Ye, Angew. Chem., Int. Ed., 2016, 55, 14116 CrossRef CAS PubMed; (d) Q.-S. Liu, D.-Y. Wang, J.-F. Yang, Z.-Y. Ma and M. Ye, Tetrahedron, 2017, 73, 3591 CrossRef CAS.
- For selected reviews, see: (a) H. M. L. Davie and J. R. Manning, Nature, 2008, 451, 417 CrossRef PubMed; (b) M. M. Díaz-Requejo and P. J. Pérez, Chem. Rev., 2008, 108, 3379 CrossRef PubMed; (c) M. P. Doyle, R. Duffy, M. Ratnikov and L. Zhou, Chem. Rev., 2010, 110, 704 CrossRef CAS PubMed; (d) A. Ford, H. Miel, A. Ring, C. N. Slattery, A. R. Maguire and M. A. Mckervey, Chem. Rev., 2015, 115, 9981 CrossRef CAS PubMed; (e) F. Wei, C. Song, Y. Ma, L. Zhou, C.-H. Tung and Z. Xu, Sci. Bull., 2015, 60, 1479 CrossRef CAS; (f) M. R. Fructos, M. M. Díaz-Requejo and P. J. Pérez, Chem. Commun., 2016, 52, 7326 RSC; (g) Y. Xia, D. Qiu and J. Wang, Chem. Rev., 2017, 117, 13810 CrossRef CAS PubMed; (h) L. Liu and J. Zhang, Chem. Soc. Rev., 2016, 45, 506 RSC.
- For selected reviews on aromatic C–H bond functionalization with diazo compounds, see: (a) B. Ma, L. Liu and J. Zhang, Asian J. Org. Chem., 2018, 7, 2015 CrossRef CAS; (b) L. Liu and J. Zhang, Chin. J. Org. Chem., 2017, 37, 1117 CrossRef CAS ; for rhodium catalysts: ; (c) W. W. Chan, S. F. Lo, Z. Zhou and W.-Y. Yu, J. Am. Chem. Soc., 2012, 134, 13565 CrossRef CAS PubMed; (d) T. K. Hyster, K. E. Ruhl and T. Rovis, J. Am. Chem. Soc., 2013, 135, 5364 CrossRef CAS PubMed; (e) H. Qiu, M. Li, L.-Q. Jiang, F.-P. Lv, L. Zan, C.-W. Zhai, M. P. Doyle and W.-H. Hu, Nat. Chem., 2012, 4, 733 CrossRef CAS PubMed; (f) B. Xu, M.-L. Li, X.-D. Zuo, S.-F. Zhu and Q.-L. Zhou, J. Am. Chem. Soc., 2015, 137, 8700 CrossRef CAS PubMed; (g) A. DeAngelis, V. W. Shurtleff, O. Dmitrenko and J. M. Fox, J. Am. Chem. Soc., 2011, 133, 1650 CrossRef CAS PubMed; (h) C. M. Qin and H. M. L. Davies, J. Am. Chem. Soc., 2014, 136, 9792 CrossRef CAS PubMed; (i) T. Miura, Q. Zhao and M. Murakami, Angew. Chem., Int. Ed., 2017, 56, 16645 CrossRef CAS PubMed; (j) D. Best, D. J. Burns and H. W. Lam, Angew. Chem., Int. Ed., 2015, 54, 7410 CrossRef CAS PubMed ; for palladium catalysts: ; (k) D. Zhang, H. Qiu, L. Jiang, F. Lv, C. Ma and W. Hu, Angew. Chem., Int. Ed., 2013, 52, 13356 CrossRef CAS PubMed; (l) X. Gao, B. Wu, W.-X. Huang, M.-W. Chen and Y.-G. Zhou, Angew. Chem., Int. Ed., 2015, 54, 11956 CrossRef CAS PubMed ; for gold catalysts: ; (m) G. Xu, K. Liu and J. Sun, Org. Lett., 2018, 20, 72 CrossRef CAS PubMed; (n) Y. Xi, Y. Su, Z. Yu, B. Dong, E. J. McClain, Y. Lan and X. Shi, Angew. Chem., Int. Ed., 2014, 53, 9817 CrossRef CAS PubMed; (o) B. Ma, Z. Chu, B. Huang, Z. Liu, L. Liu and J. Zhang, Angew. Chem., Int. Ed., 2017, 56, 2749 CrossRef CAS PubMed; (p) B. Ma, J. Wu, L. Liu and J. Zhang, Chem. Commun., 2017, 53, 10164 RSC; (q) Y. Liu, Z. Yu, J. Z. Zhang, L. Liu, F. Xia and J. Zhang, Chem. Sci., 2016, 7, 1988 RSC; (r) Z. Yu, H. Qiu, L. Liu and J. Zhang, Chem. Commun., 2016, 52, 2257 RSC; (s) B. Ma, Z. Wu, B. Huang, L. Liu and J. Zhang, Chem. Commun., 2016, 52, 9351 RSC; (t) Y. Li, Z. Tang, J. Zhang and L. Liu, Chem. Commun., 2020, 41 10.1039/D0CC01118E; (u) Z. Yu, B. Ma, M. Chen, H.-H. Wu, L. Liu and J. Zhang, J. Am. Chem. Soc., 2014, 136, 6904 CrossRef CAS PubMed ; for iridium catalysts: ; (v) Y. N. Aher, D. M. Lade and A. B. Pawar, Chem. Commun., 2018, 54, 6288 RSC ; for others: ; (w) A. Conde, G. Sabenya, M. Rodríguez, V. Postils, J. S. Luis, M. M. Díaz-Requejo, M. Costas and P. J. Pérez, Angew. Chem., Int. Ed., 2016, 55, 6530 CrossRef CAS PubMed; (x) B. Wang, I. G. Howard, J. W. Pope, E. D. Conte and Y. Deng, Chem. Sci., 2019, 10, 7958 RSC.
- E. Nakamura and K. Sato, Nat. Mater., 2011, 10, 158 CrossRef CAS PubMed.
- For selected reviews, see: (a) X.-X. Guo, D.-W. Gu, Z. Wu and W. Zhang, Chem. Rev., 2015, 115, 1622 CrossRef CAS PubMed; (b) C. Zhang, C. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3464 RSC ; for selected examples, see: ; (c) T. Naveen, A. Deb and D. Maiti, Angew. Chem., Int. Ed., 2017, 56, 1111 CrossRef CAS PubMed; (d) S. Jana, A. Ashokan, S. Kumar, A. Verma and S. Kumar, Org. Biomol. Chem., 2015, 13, 8411 RSC.
- For selected books and reviews on O–H insertion reactions, see: (a) Modern Catalytic Methods for Organic Synthesis with Diazo Compounds, ed. M. P. Doyle and M. A. McKervey, Wiley, New York, 1998 Search PubMed; (b) S.-F. Zhu and Q.-L. Zhou, Acc. Chem. Res., 2012, 45, 1365 CrossRef CAS PubMed; (c) X. Guo and W. Hu, Acc. Chem. Res., 2013, 46, 2427 CrossRef CAS PubMed ; For recent Cu-catalyse O–H insertion of phenols and naphthols, see: ; (d) T. C. Maier and G. C. Fu, J. Am. Chem. Soc., 2006, 128, 4594 CrossRef CAS PubMed; (e) X.-G. Song, S.-F. Zhu, X.-L. Xie and Q.-L. Zhou, Angew. Chem., Int. Ed., 2013, 52, 2555 CrossRef CAS PubMed; (f) C. Chen, S.-F. Zhu, B. Liu, L.-X. Wang and Q.-L. Zhou, J. Am. Chem. Soc., 2007, 129, 12616 CrossRef CAS PubMed; (g) T. Osako, D. Panichakul and Y. Uozumi, Org. Lett., 2012, 14, 194 CrossRef CAS PubMed.
- P. Yates, J. Am. Chem. Soc., 1952, 74, 5376 CrossRef CAS . In Yates' report, the reaction of phenols with diazo compound catalyzed by copper gave the ortho C–H bond functionalization product as a minor one.
- For B(C6F5)3-catalysed ortho-selectivity of phenols, see: (a) Z. Yu, Y. Li, J. Shi, B. Ma, L. Liu and J. Zhang, Angew. Chem., Int. Ed., 2016, 55, 14807 CrossRef CAS PubMed ; for Au(I)-catalysed ortho-selectivity of naphthols, see: ; (b) Z. Yu, Y. Li, P. Zhang, L. Liu and J. Zhang, Chem. Sci., 2019, 10, 6553–6559 RSC; (c) S. Harada, C. Sakai, K. Tanikawa and T. Nemoto, Tetrahedron, 2019, 75, 3650 CrossRef CAS.
- CCDC 1834202 (12)†.
- F. Li, J. Z. Zhang and F. Xia, J. Phys. Chem. A, 2020, 124, 2029 CrossRef CAS PubMed.
- According to the reviewer's suggestion, we did several control experiments to understand the mechanism. Because of the page limitation, all the details were in the ESI.† We thank the reviewer for his/her helpful suggestion.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1834202. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cc04495d |
| This journal is © The Royal Society of Chemistry 2020 |