Sub-stoichiometric inhibition of IAPP aggregation: a peptidomimetic approach to anti-amyloid agents†
Received
4th June 2020
, Accepted 3rd August 2020
First published on 19th August 2020
Abstract
Membrane-catalysed misfolding of islet amyloid polypeptide is associated with the death of β-cells in type II diabetes (T2D). Most active compounds so far reported require high doses for inhibition of membrane bound IAPP fibrillation. Here, we describe a naphthalimide-appended oligopyridylamide-based α-helical mimetic, DM 1, for targeting membrane bound IAPP. DM 1 completely inhibits the aggregation of IAPP at doses of 0.2 equivalents. DM 1 is also effective at similarly low doses for inhibition of seed-catalyzed secondary nucleation. An NMR based study demonstrates that DM 1 modulates IAPP self-assembly by stabilizing and/or perturbing the N-terminus helix conformation. DM 1 at substoichiometric doses rescues rat insulinoma cells from IAPP-mediated cytotoxicity. Most importantly, 0.2 equivalents of DM 1 disaggregate preformed oligomers and fibrils and can reverse cytotoxicity by modulating toxic preformed oligomers and fibrils of IAPP into non-toxic conformations.
Introduction
The amyloidogenesis of various proteins leads to toxic intermediates which are associated with multiple pathological disorders including Alzheimer's and Parkinson's disease, and non-neuropathic conditions such as type II diabetes (T2D).1,2 These disorders are characterized by insoluble β-sheet rich protein or peptide assemblies, which form via a series of conformational switches from soluble, functional proteins.3 Islet amyloid polypeptide is one such peptide whose amyloidogenesis is implicated in T2D.4 IAPP is a 37 residue natively unstructured hormonal peptide cosecreted with insulin by the pancreatic β-cells (Fig. 1a).5 The peptide has a disulfide bond between Cys-2 and Cys-7 and an amidated C terminus.6,7 Its physiological role is still unknown but it has been suggested to play a role in the control of adiposity, gastric emptying, glucose homeostasis, and other metabolic activities.8 There is increasing evidence emphasizing a role for β-cell loss in T2D, making islet amyloid deposition clinically relevant as it is an important contributor to the decline in β-cell mass.9,10 IAPP adopts a helical state after interaction with the cell membrane through a presentation of the positive charges on one surface.11 The membrane further templates the oligomerization of IAPP which eventually converts to an amyloid structure.12–14 The origin of the toxicity of IAPP is still under debate; however, a growing body of evidence suggests that the membrane bound helical oligomeric intermediates are key cytotoxic species.15,16 Therefore, inhibition of the membrane catalysed oligomerization and amyloidogenesis of IAPP using small molecules offers a valuable strategy to new therapeutics. Some examples include peptides,17N-methyl peptides,18,19 antimicrobial cathelicidin,20 polyphenols,21–25 engineered nanoparticles,26 acid fuchsin,27 tweezers,28,29 and helical mimetic foldamers.30–32 We have pioneered the development of a synthetic oligopyridylamide-based scaffold that mimics the side chain residues of an α helix at positions i, i + 3/i + 4, and i + 7 and modulates protein–protein interactions (Fig. 1c).33,34 Oligopyridylamides containing carboxylate functionalities were successfully used at high doses as antagonists of membrane catalysed self-assembly of IAPP via charge complementarity with basic side chain residues presented on the IAPP helical surface.30,31 Most of the earlier reported oligopyridylamides were effective inhibitors of IAPP self-assembly in the presence of lipid membranes but were agonists in their absence.30,31 IAPP fibrillation is facilitated in part by hydrophobic interactions35–38 and aromatic dyes based on a rhodanine scaffold, phenol red, or phenolsulfonphthalein have been shown to inhibit the aggregation process.39–41 A plausible generic mechanism for such inhibition involves π-stacking of the aromatic dye with the hydrophobic amino-acid rich core of the developing IAPP amyloid, thus inhibiting further propagation. A similar approach has been demonstrated for the disruption of other amyloid protein aggregates.42–45 Naphthalimide based hydrophobic dye is also known for aromatic interaction with different biomolecules.46–49 In this report, we have installed a naphthalimide on an oligopyridylamide scaffold along with carboxylate functionalities for targeting hydrophobic and cationic residues on IAPP. This generated a potent antagonist, DM 1, of de novo and membrane catalysed IAPP assembly (Fig. 2 and 3). The synthesis of DM 1 and its analogs was carried out using a straightforward strategy.30,31DM 1 contains three pyridyl units, one tagged with a 4-morpholine-1,8-naphthalic anhydride dye via imide formation; the other two pyridyl units contain carboxylate groups. In comparison to our earlier reported oligopyridylamides, DM 1 shows a dramatic improvement not only as a sub-stoichiometric antagonist of the aggregation, but also in its ability to disintegrate preformed fibers of IAPP.
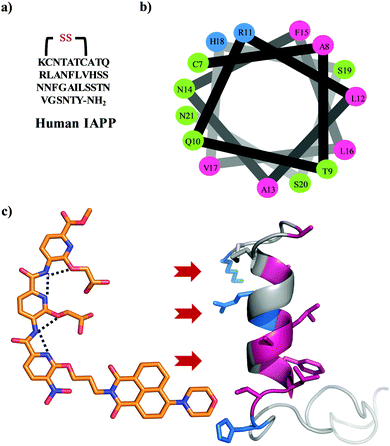 |
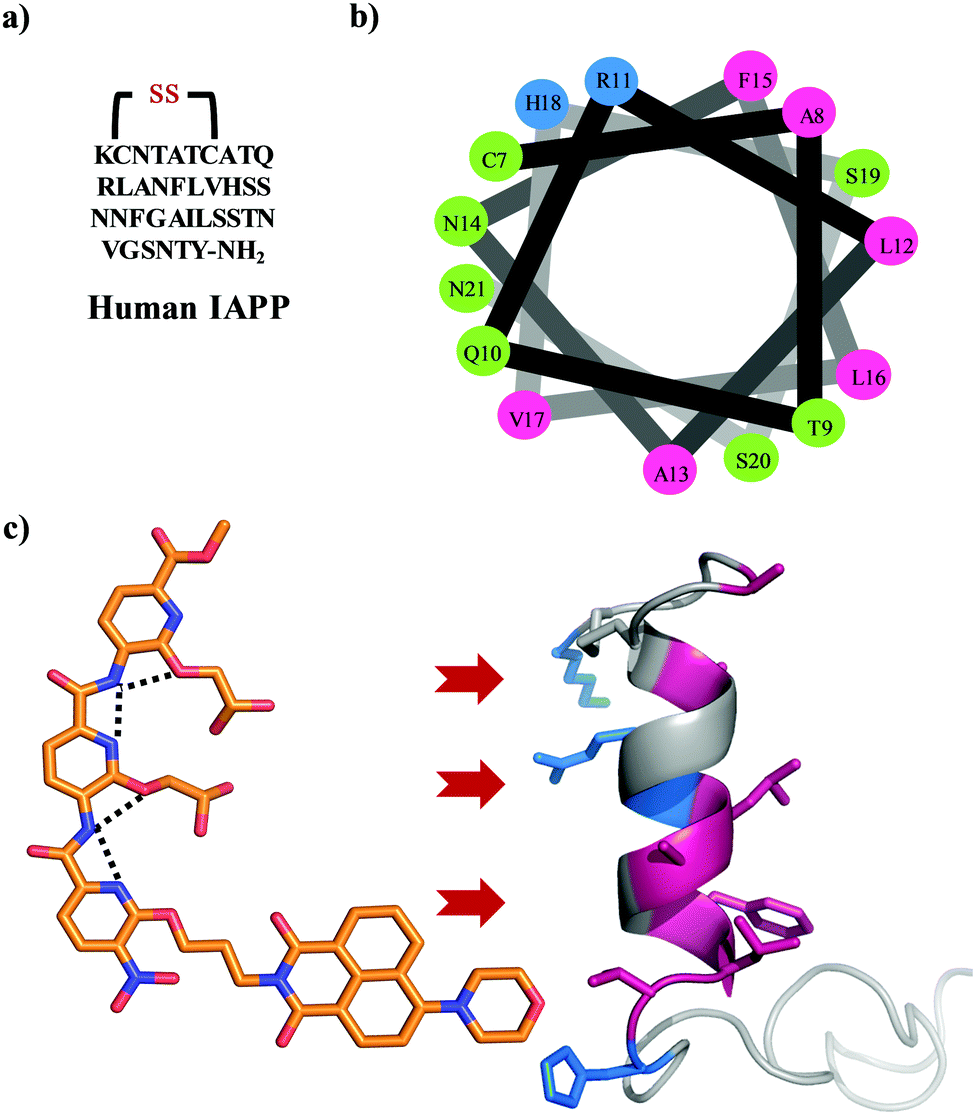
| | Fig. 1 (a) The primary sequence of IAPP with amidation at the C-terminal and a conserved disulfide bond between cysteines at positions 2 and 7. (b) A helical wheel representation of the helical subdomain of IAPP. The blue, green, and pink colours represent the positively charged, polar, and hydrophobic residues, respectively. (c) α-Helical mimetic oligopyridylamide structure depicting a single conformation stabilized by intramolecular hydrogen bonding network (black dotted lines). It is representing the potential electrostatic and hydrophobic interactions between oligopyridylamide and IAPP. The IAPP peptide domain was extracted from PDB: 5MGQ.6,7 | |
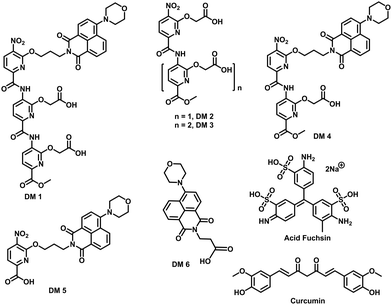 |
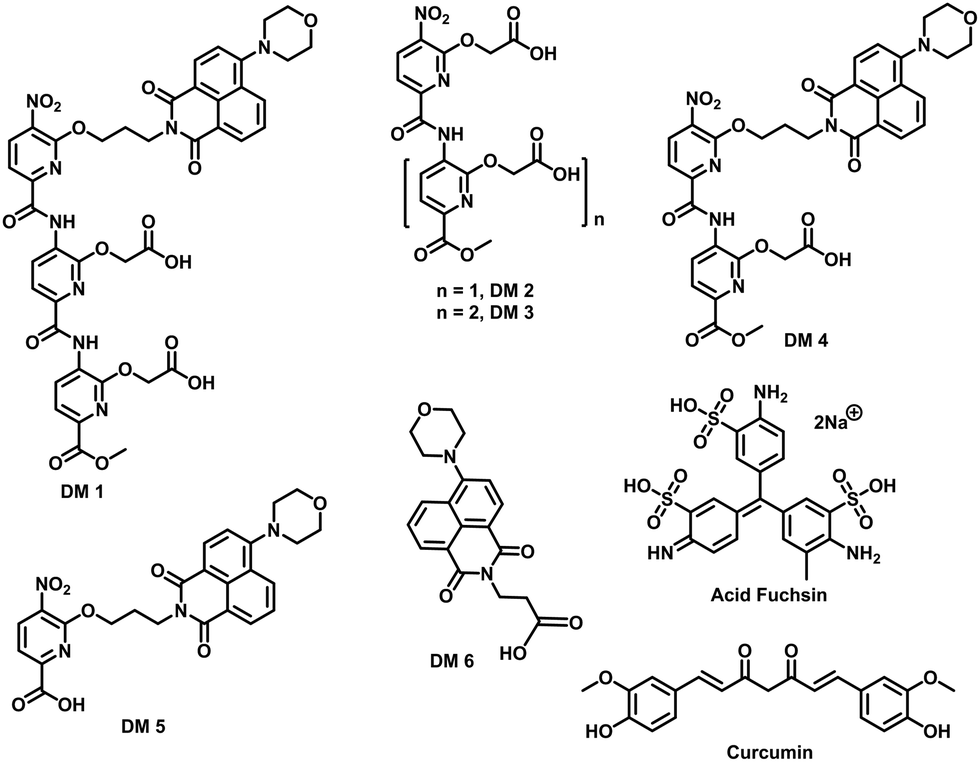
| | Fig. 2 Chemical structure of compounds used in this study. | |
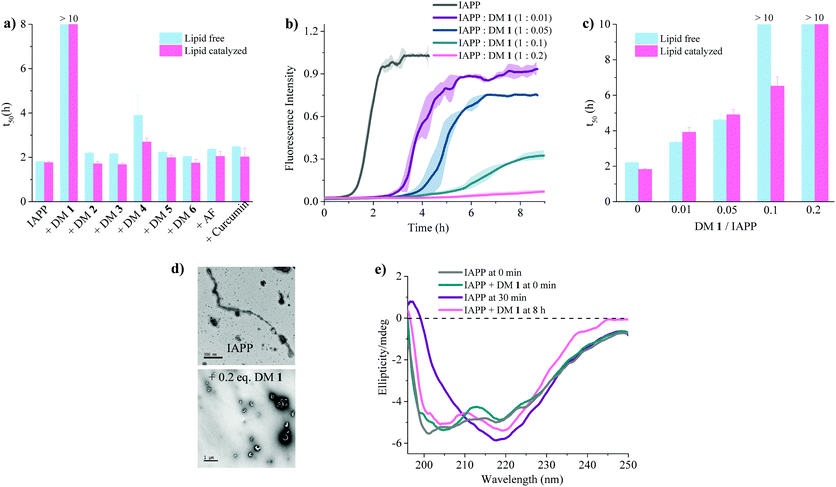 |
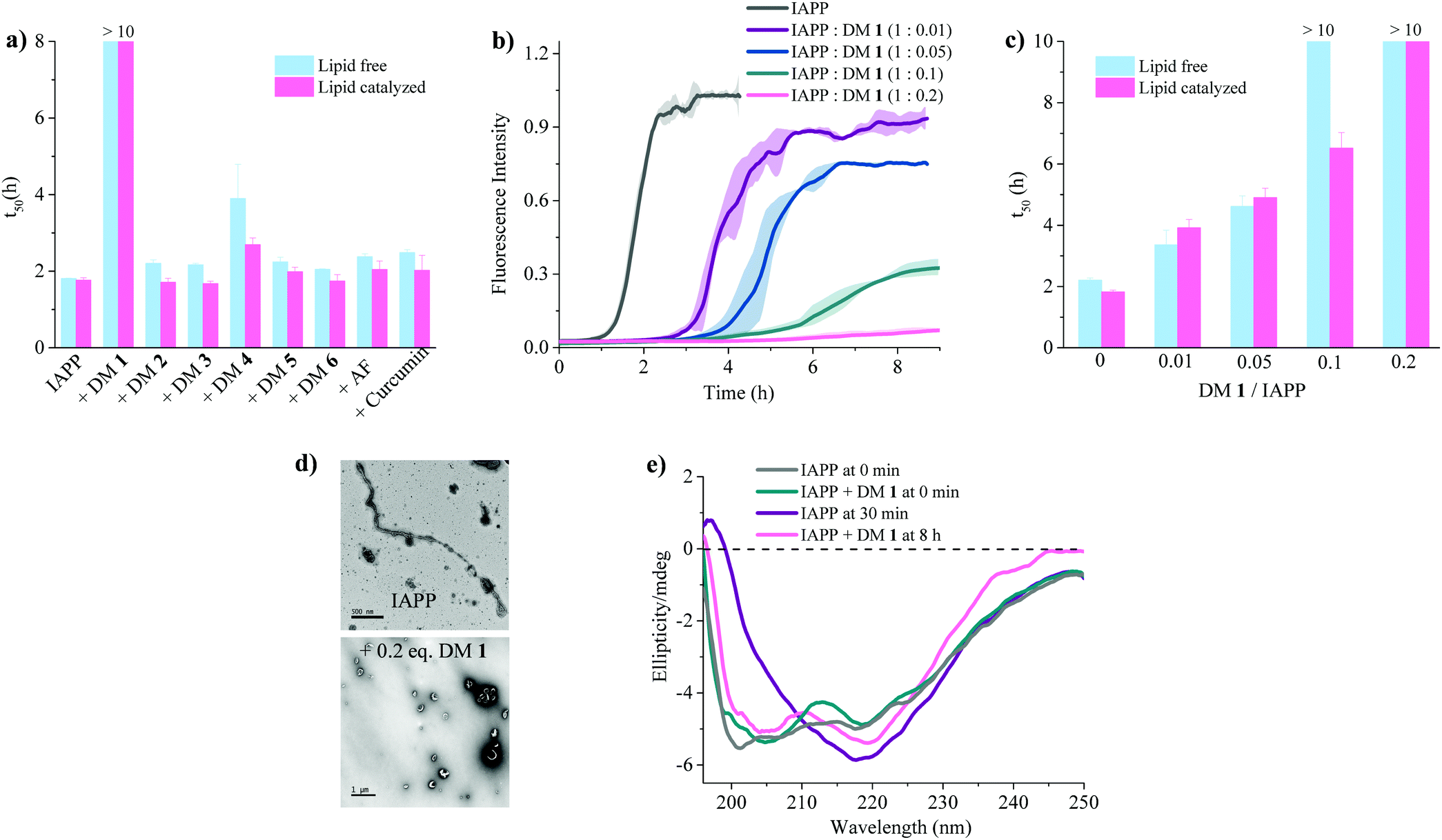
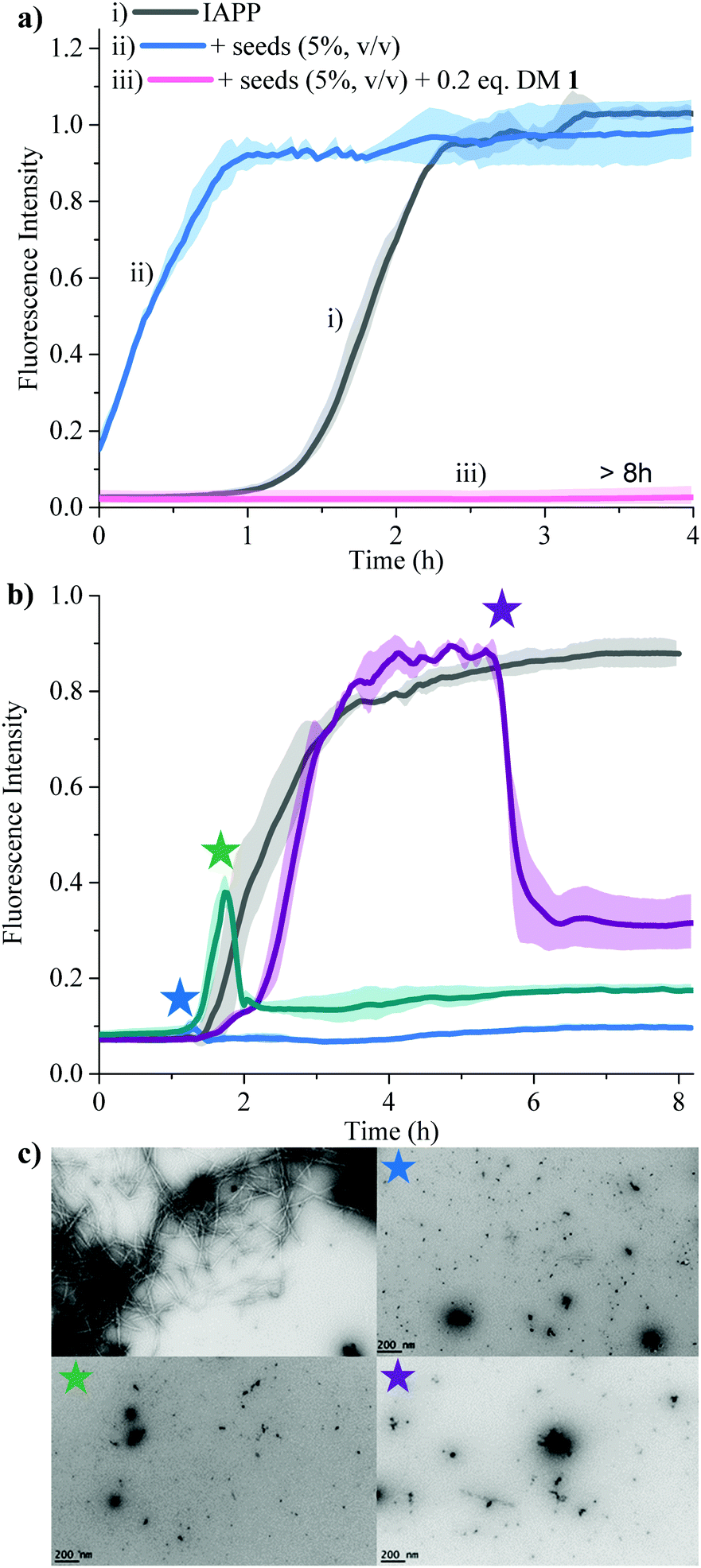
| | Fig. 3 Anti-amyloidogenic activity of DM 1 against IAPP self-assembly. (a) Comparison of t50's of IAPP aggregation in presence of the indicated ligands at a stoichiometric ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.2 (IAPP:ligand). Lipid free condition: IAPP = 25 μM in phosphate buffer (50 mM NaPi, 150 mM KCl, pH 7.4) [ThT] = 12.5 μM; lipid catalysed condition: IAPP = 15 μM in phosphate buffer (50 mM NaPi, 150 mM KCl, pH 7.4) including LUVs (DOPG:DOPC, 3:7, 750 μM, d = 100 nm). [ThT] = 7.5 μM. (b) ThT-based kinetic profile of lipid catalysed IAPP aggregation in the absence and presence of DM 1 at the indicated sub-stoichiometric ratios. Solid curves represent the average of three independent trials while the shaded regions represent the standard deviations of those measurements. (c) Statistical analysis of t50's of IAPP aggregation in the presence of different concentrations of DM 1. (d) TEM images of 15 μM IAPP after 2 h (top) and in presence of DM 1 (3 μM) after 8 h (bottom) under lipid catalysed condition. (e) Circular dichroism spectra of 15 μM IAPP in the absence (gray and purple curves) and presence (green and pink curves) of DM 1 in lipid catalysed conditions at different time intervals depicted in the figure. :0.2 (IAPP:ligand). Lipid free condition: IAPP = 25 μM in phosphate buffer (50 mM NaPi, 150 mM KCl, pH 7.4) [ThT] = 12.5 μM; lipid catalysed condition: IAPP = 15 μM in phosphate buffer (50 mM NaPi, 150 mM KCl, pH 7.4) including LUVs (DOPG:DOPC, 3:7, 750 μM, d = 100 nm). [ThT] = 7.5 μM. (b) ThT-based kinetic profile of lipid catalysed IAPP aggregation in the absence and presence of DM 1 at the indicated sub-stoichiometric ratios. Solid curves represent the average of three independent trials while the shaded regions represent the standard deviations of those measurements. (c) Statistical analysis of t50's of IAPP aggregation in the presence of different concentrations of DM 1. (d) TEM images of 15 μM IAPP after 2 h (top) and in presence of DM 1 (3 μM) after 8 h (bottom) under lipid catalysed condition. (e) Circular dichroism spectra of 15 μM IAPP in the absence (gray and purple curves) and presence (green and pink curves) of DM 1 in lipid catalysed conditions at different time intervals depicted in the figure. | |
Results and discussion
The fibrillation of IAPP was monitored using a thioflavin T (ThT)-based amyloid kinetic assay in the presence of a membrane model system relevant to the cell plasma membranes [DOPG (dioleoylphosphatidylglycerol):DOPC (dioleoylphosphatidylcholine), 3:7 molar ratio]. These kinetics were quantified using the reaction midpoint, t50, which was 1.8 ± 0.1 h for 15 μM IAPP in 750 μM LUVs (DOPG:DOPC, 3:7, d = 100 nm) (Fig. 3a). A library of compounds was screened against IAPP aggregation at substoichiometric concentration (0.2 equivalents) (Fig. 2 and 3a). Bispyridylamide DM 2 and trispyridylamide DM 3, having two and three acidic groups respectively, did not have any effect on IAPP aggregation. However, bispyridylamide DM 4, containing a naphthalimide and a carboxylate group, delays IAPP aggregation to a lesser extent suggesting naphthalimide mediated hydrophobic interaction seems to be crucial for inhibition of the IAPP self-assembly. But simple monopyridyl DM 5 and carboxylated naphthalimide DM 6 has no effect on IAPP aggregation, suggesting that the chemical composition and the spatial location of the side chains are crucial factors in achieving potent antagonism of IAPP aggregation. Known inhibitors of IAPP aggregation such as acid fuchsin27 and curcumin50–52 showed little inhibition under identical experimental conditions. Addition of as little as 0.01 equivalents of DM 1 delayed the aggregation with a significantly longer t50 = 3.8 ± 0.2 h, while 0.2 equivalents of DM 1 completely abolished IAPP fiber formation for the entire time course of the experiment (>8 h) (Fig. 3b and c). Similarly, the potent antagonist behaviour of DM 1 was observed for IAPP aggregation under de novo condition (Fig. 3c and Fig. S1, ESI†) with complete suppression at 0.2 equivalents. We also assessed the effect of DM 1 on IAPP amyloid formation under relevant physiological conditions where LUVs were prepared using cholesterol (LUVs, DOPG:DOPC, 1:1, 30% cholesterol, d = 100 nm). This condition is closely related to mammalian plasma cell membranes (30–50 mol% cholesterol).53–55DM 1 was found to be equally effective in inhibiting IAPP fibrillation under these conditions (Fig. S2, ESI†). Clearly, DM 1 is a potent inhibitor of IAPP fibrillation under a range of conditions including de novo, lipid membranes, and lipid membranes with cholesterol. As a negative control, no significant change in the ThT fluorescence intensity was observed with DM 1, which suggests that no interaction occurs between DM 1 and ThT (Fig. S3, ESI†). To exclude the possibility of DM 1 self-aggregation causing non-specific inhibition of amyloid formation we tested the self-aggregation of DM 1. The absorbance of DM 1 was proportional to its concentration (Fig. S4, ESI†) up to 20 μM, which is a much higher concentration than used for IAPP aggregation assays. To validate the ThT based assays, transmission electron microscopy56 was employed. The negatively stained TEM image of 15 μM IAPP showed lipid membrane tethered mature amyloid fiber formation within 2 h incubation under lipid membrane conditions; however, no mature fibers were observed in the presence of 0.2 equivalents DM 1 (Fig. 3d). Similarly, under lipid free conditions, the TEM image of IAPP is rich in fibers and no fibers are observed in the presence of sub-stoichiometric DM 1 (Fig. S5, ESI†). Circular dichroism was employed to monitor the effect of DM 1 on the conformation of IAPP under lipid membrane conditions (Fig. 3e). In the presence of LUVs, IAPP (15 μM) adopts an α-helical structure characterized by two minima at ∼208 and ∼222 nm. The conformation of IAPP switches from an α-helix to a β-sheet, characterized by a single minimum around 218 nm, within 30 min. However, IAPP stays in an α-helical state after 8 h in the presence of DM 1. Under lipid free conditions IAPP also showed transition from a random coil to a β-sheet conformation within 8 h (Fig. S6, ESI†). In marked contrast, in the presence of DM 1, IAPP transitioned from random coil to an α-helical state immediately and stayed in a helical state even after 8 h. It appears that DM 1 constrains IAPP into an α-helical state both in de novo and lipid membrane conditions and does not allow its conversion to the β-sheet conformation, which is necessary for amyloid formation.
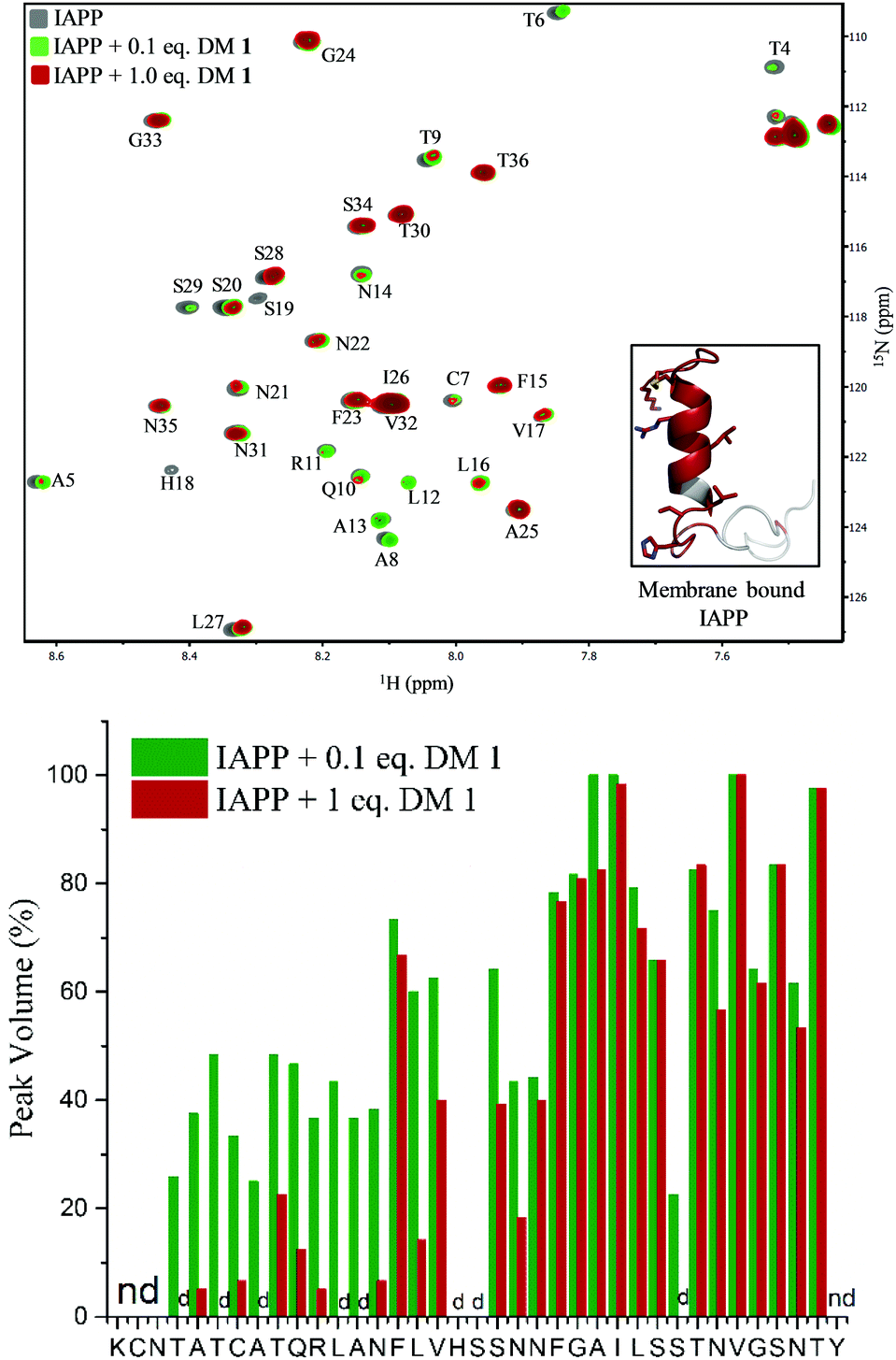
To gain insights into the binding site of DM 1 on IAPP, we employed two-dimensional HSQC (heteronuclear single quantum coherence) NMR. Recombinant IAPP with a free C-terminus, was titrated with DM 1 at two doses (Fig. 4). Using previously published assignments for the residues of recombinant IAPP, we observed a decrease in the intensities of residues in the region of the N-terminal domain in the presence of 0.1 equivalents DM 1. The largest changes were noticed for residues Thr4, Thr6, Ala8, Ala13, His18, and Ser19 in terms of the peak disappearance at an equimolar ratio (IAPP:DM 1, 1:1). There was a significant decrease in the peak volumes for other residues present at the N-terminus at an equimolar ratio compared to a 0.1 equivalent concentration, while the signals of residues towards the C-terminus (except Ser29) were mostly unchanged (Fig. 4). These results suggest that the N-terminus of recombinant IAPP is the potential binding site for DM 1. N-terminal residues of IAPP have been proposed to undergo a transition from an unstructured state to a helical conformation in the presence of lipid membranes.11 The formation of parallel helical oligomers facilitates amyloid nucleation in the non-helical C-terminal domain and subsequently leads to mature amyloid fibers.12–14 Likely, two carboxylate groups of DM 1 interact with positively charged residues (Lys1, Arg11) on recombinant IAPP while the naphthalimide forms strong hydrophobic interactions with the hydrophobic domain (Leu12–Val17) at the N-terminus of the protein. Therefore, amphiphilic DM 1, by interacting with cationic and hydrophobic residues, probably modulates IAPP aggregation by stabilizing and/or perturbing the helix conformation at the N-terminus.
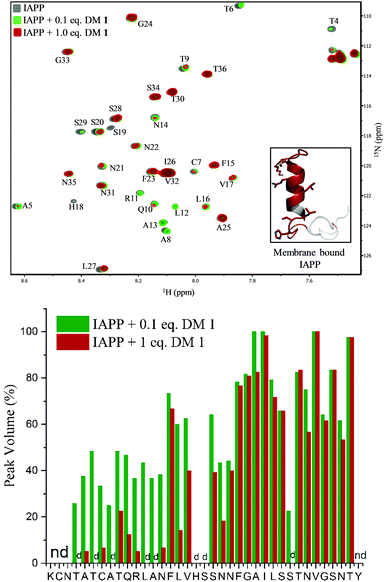 |
| | Fig. 4 Top: HSQC NMR based binding characterization between DM 1 and IAPP. Overlay of 15N-IAPP (25 μM) in the absence (gray) and presence of 0.1 eq. and 1 eq. of DM 1 represented by green, dark red colour, respectively. Residues with highest changes, in terms of peak volume, are highlighted in dark red in membrane bound IAPP structure (inset). Bottom: Peak volume of different residues of recombinant 15N-IAPP (25 μM) at different concentration of DM 1. nd = peak not determined; d = peak disappeared upon addition of DM 1 to 15N-IAPP. | |
Secondary nucleation can be inhibited by perturbing interactions between soluble monomers and amyloid fibrils. We added freshly prepared IAPP fibrils to IAPP monomer solution and incubated in the absence and presence of 0.2 equivalents of DM 1 (Fig. 5a). Fibrils of IAPP can accelerate the fibrillation by serving as seeds for elongation and secondary nucleation.
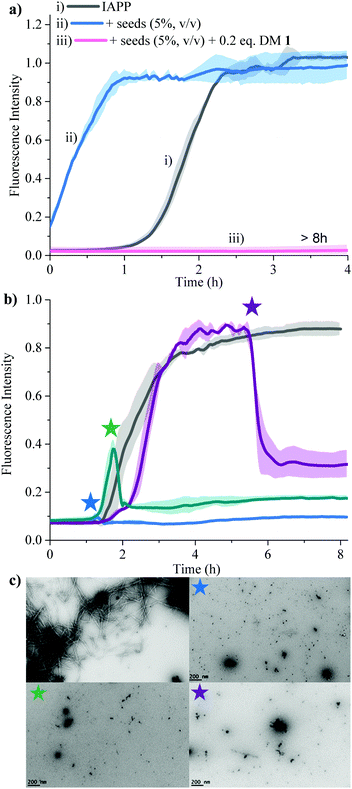 |
| | Fig. 5 Effect of DM 1 on the seed-catalysed processes, oligomerization, fibrillation of IAPP. (a) Inhibition of the secondary nucleation of IAPP aggregation by DM 1 under lipid catalysed conditions. ThT fluorescence based kinetic profiles of (i) 15 μM IAPP; (ii) 15 μM IAPP in presence of IAPP seed (5%, v/v) and (iii) 15 μM IAPP in presence of IAPP seed (5%, v/v) and 0.2 equivalents DM 1. (b) The effect of DM 1 on the preformed IAPP amyloid fibrils. ThT fluorescence based amyloid profile of 25 μM IAPP in the absence (gray) and presence of 0.2 equivalents of DM 1 added at different time points indicated by stars. Solid curves represent the average of three independent trials while the shaded regions represent the standard deviations of those measurements. (c) The TEM images were taken of all the samples after the completion of the reaction (at 8 h). | |
Addition of preformed seeds of IAPP (5%, v/v), to IAPP (15 μM) accelerated the fibrillation and eliminated the lag phase (t50 of 0.23 ± 0.1 h). However, 0.2 equivalents of DM 1 completely arrest seed-catalysed IAPP fibril propagation by interrupting interaction between seeds and IAPP monomer. Disaggregation of preformed IAPP oligomers and fibrils is an important goal because both species accelerate the formation of the toxic oligomers of IAPP via elongation or secondary nucleation. In order to assess its ability to disaggregate IAPP oligomers and fibrils, DM 1 was added at different stages of IAPP fibrillization both under de novo and lipid catalysed conditions (Fig. 5b and Fig. S8, ESI†). In both cases, addition of 0.2 equivalents of DM 1 at different points along the curve completely perturbed IAPP amyloidogenesis. Especially noteworthy is the ability of DM 1 to disaggregate mature IAPP fibrils. TEM images of the de novo -DM 1 treated samples lacked any mature fibrils (Fig. 5c). EGCG21 and N-methylated IAPP18 derivatives have been reported to show similar behaviour but at equimolar concentration under only de novo conditions. Recently, a di-phenyl pyrazole based compound showed similar behaviour at a lower concentration.57 Control compound DM 6 failed to show such amyloid disaggregation behaviour, nullifying the possibility of ThT displacement from IAPP fibrils by DM 1.
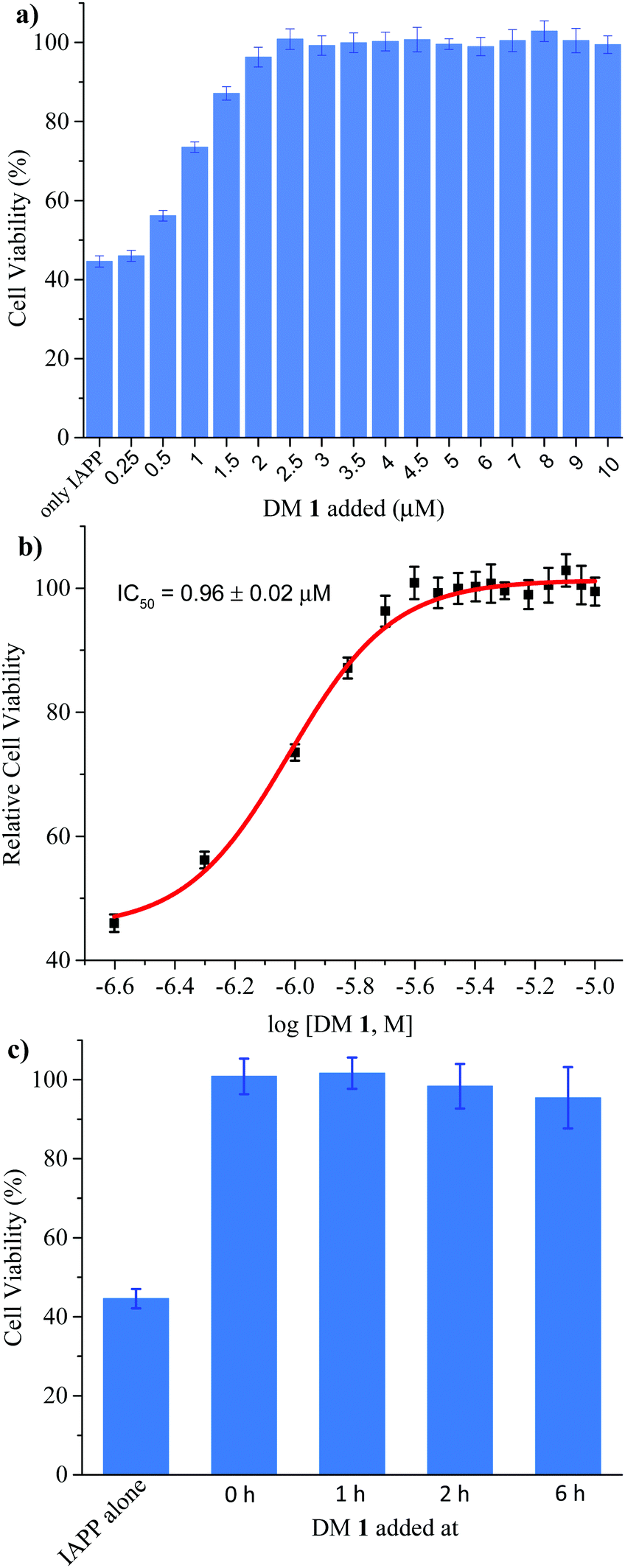
Finally, we investigated the effect of DM 1 on IAPP self-assembly mediated cytotoxicity (Fig. 6a). The cell-based experiments were conducted using rat insulinoma cells (RIN-m) as a good model for pancreatic β-cells,58 and cell viability was quantified using the CellTiter Blue fluorescence-based assay. Treatment of RIN-m cells with 10 μM IAPP for 72 h decreased viability to 44 ± 2%. Co-mixing IAPP with increasing concentrations of DM 1 revealed that the oligopyridylamide rescues toxicity at sub-stoichiometric doses (IC50 = 0.96 ± 0.02 μM) (Fig. 6b). A molar ratio of 0.25:1 (DM 1:IAPP) was sufficient to completely rescue IAPP induced toxicity (viability was 100 ± 4%). DM 1 did not demonstrate any inherent cytotoxicity to RIN-m cells under the conditions used for the cell viability assays (Fig. S9, ESI†). The cell-based assay demonstrates that DM 1 was able to modulate IAPP aggregation to a less toxic state. As DM 1 is capable of disaggregating IAPP oligomers and fibrils, we probed cell toxicity of the preformed IAPP oligomers and fibrils in the presence of DM 1 (Fig. 6c). IAPP (10 μM) was allowed to aggregate in buffer for 1–6 h, before DM 1 (2.5 μM) was added and the mixtures were incubated for an additional 2–7 h (total duration of IAPP aggregation was 8 h similar to Fig. 5b). The IAPP/DM 1 mixtures were then added to the RIN-m cells and incubated for an additional 48 h. Remarkably, complete reversal of IAPP cytotoxicity was observed as ∼100% cell viability was measured for all IAPP/DM 1 mixtures. Therefore, DM 1 effectively disaggregates IAPP oligomers and fibrils into non-toxic conformations.
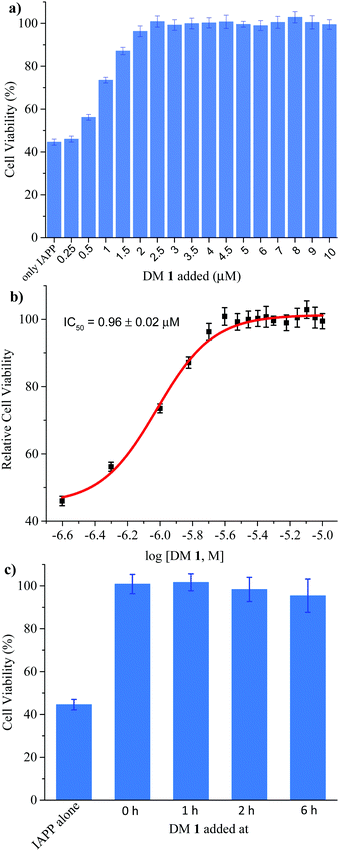 |
| | Fig. 6 Effect of DM 1 on the cytotoxicity mediated by IAPP. (a) Rescue of IAPP-mediated cytotoxicity by DM 1. Cytotoxicity of 10 μM IAPP applied to RIN-m cells in the absence and presence of different concentration of DM 1, measured using an MTS assay. Each experiment is the average of four on-plate repeats from each of three independently performed replicates. (b) Dose-dependent effect of DM 1 on 10 μM IAPP-induced toxicity in RIN-m cells. (c) The cell toxicity of IAPP (10 μM) fibrillation, mitigated by addition of DM 1 (2.5 μM) at different stages respectively. | |
Conclusions
In summary, we report a small molecule peptidomimetic DM 1 capable of the inhibition of membrane assisted IAPP toxic pathways at sub-stoichiometric doses. Earlier evidence suggests that the membrane stabilizes the N-terminal helical intermediates of IAPP and, consequently, promotes IAPP amyloidogenesis. The N-terminus of IAPP, which has a tendency to sample helical conformations, contains cationic and hydrophobic residues. Inhibitor designs that complement the chemical characteristics of this helical structure could manipulate the toxic behaviour of IAPP aggregation. Oligopyridylamide-based α-helical peptidomimetics have emerged as powerful tools for modulating the kinetic pathways of amyloidogenic proteins. We have identified a very potent antagonist of the toxic states of IAPP self-assembly, which is able to function at sub-stoichiometric concentrations. The ligand was also able to disintegrate preformed fibers as well as disrupt the elongation and secondary nucleation processes of IAPP aggregation. Research is underway in our laboratory to investigate the mechanism of action of this scaffold on IAPP amyloidogenesis at a molecular level. We believe that a better understanding of the mechanism of the activity of DM 1 will aid in developing potential therapeutic agents for the treatment of T2D.
Materials and methods
Materials
Thioflavin T (ThT) was purchased from Acros Organics (Fair Lawn, NJ). Lipids [dioleoylphosphatidylglycerol (DOPG) and dioleoylphosphatidylcholine (DOPC)] were purchased from Avanti Polar Lipids, Inc. (Alabaster, AL). The 96-well plates (black, w/flat bottom) were bought from Greiner Bio-One (Monroe, NC). All the chemicals were purchased from Sigma Aldrich (St. Louis, MI) and used without further purification. Silica plates (w/UV254, aluminum backed, 200 micron) and silica gel (standard grade, particle size = 40–63 micron, 230 × 400 mesh) for flash column chromatography were purchased from Sorbent Technologies (Atlanta, GA). Dry solvents were purchased from Sigma Aldrich (St. Louis, MI). Human islet amyloid polypeptide was purchased from Anaspec (Fremont, CA) with >98% purity. IAPP was re-purified using in-house purification method.
Preparation of IAPP
IAPP (∼2 mg) was solubilized in 8 M guanidinium hydrochloride. The solution was filtered (0.2 micron) and transferred to a C-18 spin column, washed twice with water (400 μL each) followed by 10% acetonitrile in water (0.1% formic acid (v/v)) and then eluted into 200 μL of 50% acetonitrile in water (0.1% formic acid (v/v)). The concentration of IAPP (oxidized form) was calculated using absorbance measurements at 280 nm (ε = 1400 M−1 cm−1). The IAPP solution was divided into several aliquots (20–50 μL, 1–2 mM), lyophilized, and stored as a white solid at −80 °C. A fresh stock solution of IAPP was prepared in water for each experiment.
Preparation of large unilamellar vesicles (LUVs)
LUVs were prepared using DOPG and DOPC at a stoichiometric ratio of 3:7 (DOPG:DOPC). The solution of DOPG and DOPC (6 mg and 14 mg) in chloroform (10 mg L−1) was mixed, dried over a stream of argon (g) for 2 h, followed by drying in high vacuum and then lyophilized for 12 h (0.1 × 10−3 bar). The solid was rehydrated in 1 mL phosphate buffer for 30 min. The turbid solution (6 mg:14 mg, 3:7, DOPG:DOPC) was then extruded (21 times) through 100 nm diameter filters (Whatman, GE Healthcare, Marlborough, MA). The concentration of the S4 phospholipid content in the extruded material was confirmed by calculating total phosphorus using a total phosphate assay.59
ThT-based kinetic assay
Kinetic assays were conducted on a FlexStation 3 Multi-Mode Microplate reader from Molecular Devices (Sunnyvale, CA). Experiments were conducted in triplicate in a 96-well plate with a final volume of 200 μL per well. The aggregation of IAPP was initiated by its addition from a stock solution (1 mM in filtered water) to phosphate buffer with or without lipid catalysed conditions. The stoichiometry ratio for ThT to IAPP was 0.5:1. Peptide aggregation was monitored by ThT fluorescence (λex = 445 nm and λem = 485 nm). The blank sample contained everything except peptide. The sample data were processed by subtracting the blank and renormalizing the fluorescence intensity by setting the maximum value to one. Kinetic assays in the presence of small molecules were conducted under the same conditions except that the small molecules were added from a stock solution (1 mM or 10 mM in DMSO) to keep the final concentration of DMSO less than 1.0% (v/v). Small molecules were added to the wells with ThT and buffer and mixed gently with a pipette before adding IAPP. To keep the conditions identical, an equal amount of DMSO was added to the wells with IAPP only reactions. Kinetic profiles were processed using Origin (version 9.1). Kinetic curves were fit using the sigmoidal fit. Each run was fit independently to extract the t50 (time required to reach 50% of the maximum fluorescence intensity). Error bars represent standard deviations from the mean of three independent experiments.
Seed-catalysed kinetic assay
Seeds of IAPP were prepared by incubating 100 μM of IAPP in phosphate buffer at room temperature and aged for 24 h. The formation of fibers was confirmed by TEM and ThT experiment. For the seed catalysed aggregation kinetics of IAPP, 5% (based on the monomeric IAPP, v/v) seeds were added with ThT in phosphate buffer to the 96-well plate. The aggregation was initiated by the addition of fresh IAPP followed by gentle mixing. Kinetic assay in the presence of DM 1 was conducted under the same condition except that the small molecule was added from a stock solution (10 mM in DMSO) to keep the final concentration of DMSO less than 1.0% (v/v).
Transmission electron microscopy56 analysis
IAPP was incubated in the absence and presence of DM 1 both in de novo and lipid catalysed conditions. Aliquots of these samples were then applied to glow-discharged carbon-coated 300-mesh copper grids for 2 min and dried. Grids were negatively stained with uranyl acetate (2%, w/v) and dried. Micrographs of grids were examined on a Phillips CM12 Cryoelectron Microscope equipped with Gatan 4k × 2.7k CCD camera at 120 kV accelerating voltage.
Circular dichroism (CD) spectroscopy
A freshly prepared stock solution of IAPP (500 μM in water) was diluted to 15 μM and 20 μM in phosphate buffer containing DOPG:DOPC (3:7, 750 μM, d = 100 nm) and in phosphate buffer, respectively, for CD measurements. The spectra of IAPP were recorded at 0.5 nm intervals from 190 to 260 nm with an averaging time of 10 s and an average of three repeats on a Jasco CD Spectropolarimeter. Spectra were recorded in presence of DM 1 using the identical method as described above.
HSQC NMR experiment
Recombinant human IAPP was produced using a cleavable fusion construct as described.60 Two-dimensional HSQC NMR experiments were performed on a 600 MHz Bruker instrument at 12 °C. The NMR sample (350 μL) contained 15N-IAPP at a concentration of 25 μM in 20 mM sodium phosphate, pH 6.2, with 10% D2O in Shigemi NMR tube (Shigemi Inc., Allison Park). A stock solution of 20 mM DM 1 was prepared in DMSO-d6 (pure, HPLC grade). For each NMR experiment, a freshly prepared aliquot of 15N-IAPP was used to avoid potential complication from amyloid formation. NMR spectra were processed using the software Mnova.
Cell culture
Rat insulinoma RIN-m cells (ATCC, Manassas, VA) were cultured in RPMI medium supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin, 100 mM sodium pyruvate, and 50 mM β-mercaptoethanol (Life Technologies, Carlsbad, CA) at 37 °C and 5% CO2. Upon reaching ∼95% confluence, cells were washed with phosphate buffered saline (VWR, Radnor, PA), split using 0.25% trypsin–EDTA (Life Technologies), and plated in clear 96-well plates (Corning, Glenview, IL) for cell viability assays.
Cell viability
Cell viability was measured using the CellTiter Blue (CTB, Promega, Madison, WI) fluorescence-based assay. Cells were plated at a density of 5 × 103 cells per well in 100 μL complete RPMI growth medium in 96-well plates. After culturing for 24 h in 5% CO2 at 37 °C, the medium was removed, and the cells were washed with PBS (100 μL per well). IAPP and DM 1 were pre-mixed in buffer (50 mM NaPi, 150 mM KCl, pH 7.4) and the mixture was added to the cells in complete medium. The cells were then incubated for an additional 48 h. For the IAPP fiber disaggregation experiments, IAPP was allowed to aggregate in buffer for 1–6 h before DM 1 was added and the mixture incubated for an additional 2 h. The mixture was then added to the cells in complete medium and incubated for an additional 48 h. Finally, 20 μL CellTiter Blue reagent was added to each well and incubated for 4 h. Fluorescence of the resorufin product (λex/em = 560/620 nm) of CTB reduction was measured on a Synergy H1MF Multi-Mode microplate reader (BioTek, Winooski, Vermont). Positive control wells contained 10% DMSO, whereas negative control wells contained water and 0.2% DMSO to account for the peptide and DM 1 vehicles, respectively. Percent cell viability was calculated as per the following equation:
| % Viability = 100 × [(〈S〉 − 〈P〉)/(〈N〉 − 〈P〉)] |
where 〈S〉, 〈P〉, and 〈N〉 are the average fluorescence intensities of the sample, positive control, and negative control, respectively. Error bars represent the S.E.M. of 4 independent triplet-well trials.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by funding from New York University to A. D. H. and from New York University Abu Dhabi to M. M.
Notes and references
- D. J. Selkoe, Nature, 2003, 426, 900–904 CrossRef CAS PubMed.
- F. Chiti and C. M. Dobson, Annu. Rev. Biochem., 2006, 75, 333–366 CrossRef CAS PubMed.
- F. Chiti and C. M. Dobson, Nat. Chem. Biol., 2009, 5, 15–22 CrossRef CAS PubMed.
- R. L. Hull, G. T. Westermark, P. Westermark and S. E. Kahn, J. Clin. Endocrinol. Metab., 2004, 89, 3629–3643 CrossRef CAS PubMed.
- P. Westermark, A. Andersson and G. T. Westermark, Physiol. Rev., 2011, 91, 795–826 CrossRef CAS PubMed.
- D. C. Rodriguez Camargo, K. Tripsianes, K. Buday, A. Franko, C. Göbl, C. Hartlmüller, R. Sarkar, M. Aichler, G. Mettenleiter, M. Schulz, A. Böddrich, C. Erck, H. Martens, A. K. Walch, T. Madl, E. E. Wanker, M. Conrad, M. H. de Angelis and B. Reif, Sci. Rep., 2017, 7, 44041 CrossRef CAS PubMed.
- G. J. Cooper, A. C. Willis, A. Clark, R. C. Turner, R. B. Sim and K. B. Reid, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 8628–8632 CrossRef CAS PubMed.
- R. A. DeFronzo, J. A. Davidson and S. Del Prato, Diabetes, Obes. Metab., 2012, 14, 5–14 CrossRef CAS PubMed.
- F. M. Ashcroft and P. Rorsman, Cell, 2012, 148, 1160–1171 CrossRef CAS PubMed.
- J. W. M. Höppener, B. Ahrén and C. J. M. Lips, N. Engl. J. Med., 2000, 343, 411–419 CrossRef PubMed.
- J. D. Knight and A. D. Miranker, J. Mol. Biol., 2004, 341, 1175–1187 CrossRef CAS PubMed.
- J. A. Williamson, J. P. Loria and A. D. Miranker, J. Mol. Biol., 2009, 393, 383–396 CrossRef CAS PubMed.
- J. A. Williamson and A. D. Miranker, Protein Sci., 2007, 16, 110–117 CrossRef CAS PubMed.
- J. A. Hebda and A. D. Miranker, Annu. Rev. Biophys., 2009, 38, 125–152 CrossRef CAS PubMed.
- J. Janson, R. H. Ashley, D. Harrison, S. McIntyre and P. C. Butler, Diabetes, 1999, 48, 491–498 CrossRef CAS PubMed.
- J. R. Brender, K. Hartman, K. R. Reid, R. T. Kennedy and A. Ramamoorthy, Biochemistry, 2008, 47, 12680–12688 CrossRef CAS PubMed.
- A. Abedini, F. Meng and D. P. Raleigh, J. Am. Chem. Soc., 2007, 129, 11300–11301 CrossRef CAS PubMed.
- L.-M. Yan, M. Tatarek-Nossol, A. Velkova, A. Kazantzis and A. Kapurniotu, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 2046–2051 CrossRef CAS PubMed.
- A. Spanopoulou, L. Heidrich, H.-R. Chen, C. Frost, D. Hrle, E. Malideli, K. Hille, A. Grammatikopoulos, J. Bernhagen, M. Zacharias, G. Rammes and A. Kapurniotu, Angew. Chem., Int. Ed., 2018, 57, 14503–14508 CrossRef CAS PubMed.
- V. Armiento, K. Hille, D. Naltsas, J. S. Lin, A. E. Barron and A. Kapurniotu, Angew. Chem., Int. Ed., 2020, 59(31), 12837–12841 CrossRef CAS PubMed.
- F. Meng, A. Abedini, A. Plesner, C. B. Verchere and D. P. Raleigh, Biochemistry, 2010, 49, 8127–8133 CrossRef CAS PubMed.
- A. Pithadia, J. R. Brender, C. A. Fierke and A. Ramamoorthy, J. Diabetes Res., 2016, 2016, 12 Search PubMed.
- D. E. Ehrnhoefer, J. Bieschke, A. Boeddrich, M. Herbst, L. Masino, R. Lurz, S. Engemann, A. Pastore and E. E. Wanker, Nat. Struct. Mol. Biol., 2008, 15, 558–566 CrossRef CAS PubMed.
- A. R. A. Ladiwala, J. C. Lin, S. S. Bale, A. M. Marcelino-Cruz, M. Bhattacharya, J. S. Dordick and P. M. Tessier, J. Biol. Chem., 2010, 285, 24228–24237 CrossRef CAS PubMed.
- P. Nedumpully-Govindan, A. Kakinen, E. H. Pilkington, T. P. Davis, P. Chun Ke and F. Ding, Sci. Rep., 2016, 6, 19463 CrossRef CAS PubMed.
- P. C. Ke, E. H. Pilkington, Y. Sun, I. Javed, A. Kakinen, G. Peng, F. Ding and T. P. Davis, Adv. Mater., 2020, 32(18), 1901690 CrossRef CAS PubMed.
- F. Meng, A. Abedini, A. Plesner, C. T. Middleton, K. J. Potter, M. T. Zanni, C. B. Verchere and D. P. Raleigh, J. Mol. Biol., 2010, 400, 555–566 CrossRef CAS PubMed.
- S. Sinha, D. H. J. Lopes, Z. Du, E. S. Pang, A. Shanmugam, A. Lomakin, P. Talbiersky, A. Tennstaedt, K. McDaniel, R. Bakshi, P.-Y. Kuo, M. Ehrmann, G. B. Benedek, J. A. Loo, F.-G. Klärner, T. Schrader, C. Wang and G. Bitan, J. Am. Chem. Soc., 2011, 133, 16958–16969 CrossRef CAS PubMed.
- D. H. J. Lopes, A. Attar, G. Nair, E. Y. Hayden, Z. Du, K. McDaniel, S. Dutt, H. Bandmann, K. Bravo-Rodriguez, S. Mittal, F.-G. Klärner, C. Wang, E. Sanchez-Garcia, T. Schrader and G. Bitan, ACS Chem. Biol., 2015, 10, 1555–1569 CrossRef CAS PubMed.
- I. Saraogi, J. A. Hebda, J. Becerril, L. A. Estroff, A. D. Miranker and A. D. Hamilton, Angew. Chem., Int. Ed., 2010, 49, 736–739 CrossRef CAS PubMed.
- S. Kumar, D. E. Schlamadinger, M. A. Brown, J. M. Dunn, B. Mercado, J. A. Hebda, I. Saraogi, E. Rhoades, A. D. Hamilton and A. D. Miranker, Chem. Biol., 2015, 22, 369–378 CrossRef CAS PubMed.
- S. Kumar, M. Birol, D. E. Schlamadinger, S. P. Wojcik, E. Rhoades and A. D. Miranker, Nat. Commun., 2016, 7, 11412 CrossRef CAS PubMed.
- M. K. P. Jayatunga, S. Thompson and A. D. Hamilton, Bioorg. Med. Chem. Lett., 2014, 24, 717–724 CrossRef CAS PubMed.
- D. Maity, S. Kumar, F. Curreli, A. K. Debnath and A. D. Hamilton, Chem. – Eur. J., 2019, 25, 7265–7269 CrossRef CAS PubMed.
- J. J. W. Wiltzius, S. A. Sievers, M. R. Sawaya, D. Cascio, D. Popov, C. Riekel and D. Eisenberg, Protein Sci., 2008, 17, 1467–1474 CrossRef CAS PubMed.
- K. Tenidis, M. Waldner, J. Bernhagen, W. Fischle, M. Bergmann, M. Weber, M.-L. Merkle, W. Voelter, H. Brunner and A. Kapurniotu, J. Mol. Biol., 2000, 295, 1055–1071 CrossRef CAS PubMed.
- P. Westermark, U. Engström, K. H. Johnson, G. T. Westermark and C. Betsholtz, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 5036–5040 CrossRef CAS PubMed.
- E. T. A. S. Jaikaran, C. E. Higham, L. C. Serpell, J. Zurdo, M. Gross, A. Clark and P. E. Fraser, J. Mol. Biol., 2001, 308, 515–525 CrossRef CAS PubMed.
- R. Mishra, B. Bulic, D. Sellin, S. Jha, H. Waldmann and R. Winter, Angew. Chem., Int. Ed., 2008, 47, 4679–4682 CrossRef CAS PubMed.
- Y. Porat, Y. Mazor, S. Efrat and E. Gazit, Biochemistry, 2004, 43, 14454–14462 CrossRef CAS PubMed.
- M. Levy, Y. Porat, E. Bacharach, D. E. Shalev and E. Gazit, Biochemistry, 2008, 47, 5896–5904 CrossRef CAS PubMed.
- J. E. Gestwicki, G. R. Crabtree and I. A. Graef, Science, 2004, 306, 865–869 CrossRef CAS PubMed.
- J. F. Aitken, K. M. Loomes, B. Konarkowska and G. J. Cooper, Biochem. J., 2003, 374, 779–784 CrossRef CAS PubMed.
- Y. Porat, A. Abramowitz and E. Gazit, Chem. Biol. Drug Des., 2006, 67, 27–37 CrossRef CAS PubMed.
- E. Gazit, FASEB J., 2002, 16, 77–83 CrossRef CAS PubMed.
- D. Maity, M. Matković, S. Li, M. Ehlers, J. Wu, I. Piantanida and C. Schmuck, Chem. – Eur. J., 2017, 23, 17356–17362 CrossRef CAS PubMed.
- D. Maity, M. Li, M. Ehlers and C. Schmuck, Chem. Commun., 2017, 53, 208–211 RSC.
- D. Maity, A. Gigante, P. A. Sánchez-Murcia, E. Sijbesma, M. Li, D. Bier, S. Mosel, S. Knauer, C. Ottmann and C. Schmuck, Org. Biomol. Chem., 2019, 17, 4359–4363 RSC.
- S. Banerjee, E. B. Veale, C. M. Phelan, S. A. Murphy, G. M. Tocci, L. J. Gillespie, D. O. Frimannsson, J. M. Kelly and T. Gunnlaugsson, Chem. Soc. Rev., 2013, 42, 1601–1618 RSC.
- M. Daval, S. Bedrood, T. Gurlo, C.-J. Huang, S. Costes, P. C. Butler and R. Langen, Amyloid, 2010, 17, 118–128 CrossRef CAS PubMed.
- S. Sparks, G. Liu, K. J. Robbins and N. D. Lazo, Biochem. Biophys. Res. Commun., 2012, 422, 551–555 CrossRef CAS PubMed.
- A. S. Pithadia, A. Bhunia, R. Sribalan, V. Padmini, C. A. Fierke and A. Ramamoorthy, Chem. Commun., 2016, 52, 942–945 RSC.
- M. Hao, S. X. Lin, O. J. Karylowski, D. Wüstner, T. E. McGraw and F. R. Maxfield, J. Biol. Chem., 2002, 277, 609–617 CrossRef CAS PubMed.
- D. E. Warnock, C. Roberts, M. S. Lutz, W. A. Blackburn, W. W. Young and J. U. Baenziger, J. Biol. Chem., 1993, 268, 10145–10153 CAS.
- M. F. M. Sciacca, F. Lolicato, G. Di Mauro, D. Milardi, L. D’Urso, C. Satriano, A. Ramamoorthy and C. La Rosa, Biophys. J., 2016, 111, 140–151 CrossRef CAS PubMed.
- K. G. Mawuenyega, W. Sigurdson, V. Ovod, L. Munsell, T. Kasten, J. C. Morris, K. E. Yarasheski and R. J. Bateman, Science, 2010, 330, 1774 CrossRef CAS PubMed.
- M. S. Saravanan, S. Ryazanov, A. Leonov, J. Nicolai, P. Praest, A. Giese, R. Winter, L. Khemtemourian, C. Griesinger and J. A. Killian, Sci. Rep., 2019, 9, 19023 CrossRef CAS.
- C.-j. Huang, C.-y. Lin, L. Haataja, T. Gurlo, A. E. Butler, R. A. Rizza and P. C. Butler, Diabetes, 2007, 56, 2016–2027 CrossRef CAS PubMed.
- P. S. Chen, T. Y. Toribara and H. Warner, Anal. Chem., 1956, 28, 1756–1758 CrossRef CAS.
- E. A. Mirecka, L. Gremer, S. Schiefer, F. Oesterhelt, M. Stoldt, D. Willbold and W. Hoyer, J. Biotechnol., 2014, 191, 221–227 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cb00086h |
| ‡ Present address: Department of Chemistry and Biochemistry, and KIHA, University of Denver, Denver, CO 80210, USA. |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Sunil
Kumar‡
*a,
Sunil
Kumar‡