
Rhodium(II)-catalyzed divergent intramolecular tandem cyclization of N- or O-tethered cyclohexa-2,5-dienones with 1-sulfonyl-1,2,3-triazole: synthesis of cyclopropa[cd]indole and benzofuran derivatives†
Cheng-Zhi
Zhu
a,
Yin
Wei
 *a and
Min
Shi
*abc
*a and
Min
Shi
*abc
aState Key Laboratory of Organometallic Chemistry, Center for Excellence in Molecular Synthesis, University of Chinese Academy of Sciences, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China. E-mail: weiyin@sioc.ac.cn; mshi@mail.sioc.ac.cn
bKey Laboratory for Advanced Materials and Institute of Fine Chemicals, School of Chemistry & Molecular Engineering, East China University of Science and Technology, Meilong Road No. 130, Shanghai 200237, China
cShenzhen Grubbs Institute, Southern University of Science and Technology, Shenzhen, Guangdong 518000, China
First published on 25th June 2019
Abstract
A rhodium(II)-catalyzed divergent intramolecular tandem cyclization of N- or O-tethered cyclohexa-2,5-dienones with 1-sulfonyl-1,2,3-triazole has been disclosed in this paper. When the connecting atom between the cyclohexa-2,5-dienone unit and the 1-sulfonyl-1,2,3-triazole moiety was a N-sulfonated group, a cyclopropanation of the olefinic unit in the cyclohexa-2,5-dienone moiety could take place, affording cyclopropa[cd]indole derivatives in moderate to good yields. This is the first example of the cyclopropanation of α-imino rhodium carbenes with electron-deficient intra-annular olefins. If the connecting linker was an oxygen atom, an oxy–Cope rearrangement could be triggered under similar reaction conditions, giving benzofuran derivatives. The reaction mechanisms have also been proposed along with a synthetic transformation.
Introduction
1-Sulfonyl-1,2,3-triazoles, easily prepared through copper(I)-catalyzed azide–alkyne cycloadditions (CuAAC),1 have recently received much attention from synthetic organic chemists. Owing to the elegant work of Fokin, Gevorgyan, Davies, Murakami, Sarpong, our group2 and others,3 it has been well known that α-imino rhodium carbenes generated from readily available 1-sulfonyl-1,2,3-triazoles could be easily utilized for a variety of novel carbene-induced transformations. For example, transannulations of 1-sulfonyl-1,2,3-triazoles with a variety of organic compounds containing unsaturated functional groups such as nitriles, alkynes, allenes, isocyanates, furans, aldehydes, and imines could provide a series of useful heterocyclic compounds upon Rh(II) catalysis, which are privileged structural motifs in many biologically active and medicinally valuable molecules.4 Besides, α-imino rhodium carbenes could also undergo C–H, O–H or N–H insertion,5 and arylation with boronic acids6 or could be trapped by a heteroatom, such as sulfur, nitrogen, bromine or oxygen to realize further transformations.7 The cyclopropanation of metal carbene complexes with olefins has been extensively researched in recent years.8 More importantly, in 2009, Fokin and co-workers reported that α-imino rhodium carbenes could undergo cyclopropanation with linear alkenes to afford cyclopropanecarbaldehydes in good yields (Scheme 1).9 Furthermore, in recent years, Lee, Anbarasan, Li and co-workers successively disclosed that the reactions of α-imino rhodium carbenes with electron-rich olefins could generate α-imino cyclopropyl intermediates, which subsequently furnished dihydropyrroles through a Cloke–Wilson rearrangement (Scheme 1).10 Inspired by these brilliant studies, we attempted to investigate the intramolecular tandem cyclization of intra-annular olefins with α-imino rhodium carbenes to rapidly generate polycyclic or spirocyclic frameworks. Recently, we reported a Rh(II)-catalyzed intramolecular transannulation of 4-methoxycyclohexa-2,5-dienone tethered 1-sulfonyl-1,2,3-triazoles for the facile synthesis of azaspiro[5.5]undecane derivatives.11 This interesting tandem cyclization reaction proceeded through an oxonium ylide generated from trapping a rhodium(II)-carbene by a methoxy group, a methoxy group migration, and C–N bond formation, providing a novel synthetic strategy for the construction of spirocyclic frameworks. We envisioned that N-tethered cyclohexa-2,5-dienones with 1-sulfonyl-1,2,3-triazole might yield an α-imino rhodium carbene in the presence of rhodium(II) catalysts, which could undergo an intramolecular cyclopropanation with an electron-deficient intra-annular olefin in the cyclohexa-2,5-dienone moiety to deliver cyclopropa[cd]indole derivatives (Scheme 1). Herein, we wish to report our findings on this novel protocol to construct these N-heterocyclic compounds that may have potential applications in many biologically active and medicinally valuable molecules. Moreover, Rh(II)-catalyzed O-tethered cyclohexa-2,5-dienones with 1-sulfonyl-1,2,3-triazole to give benzofuran derivatives will also be disclosed (Scheme 1).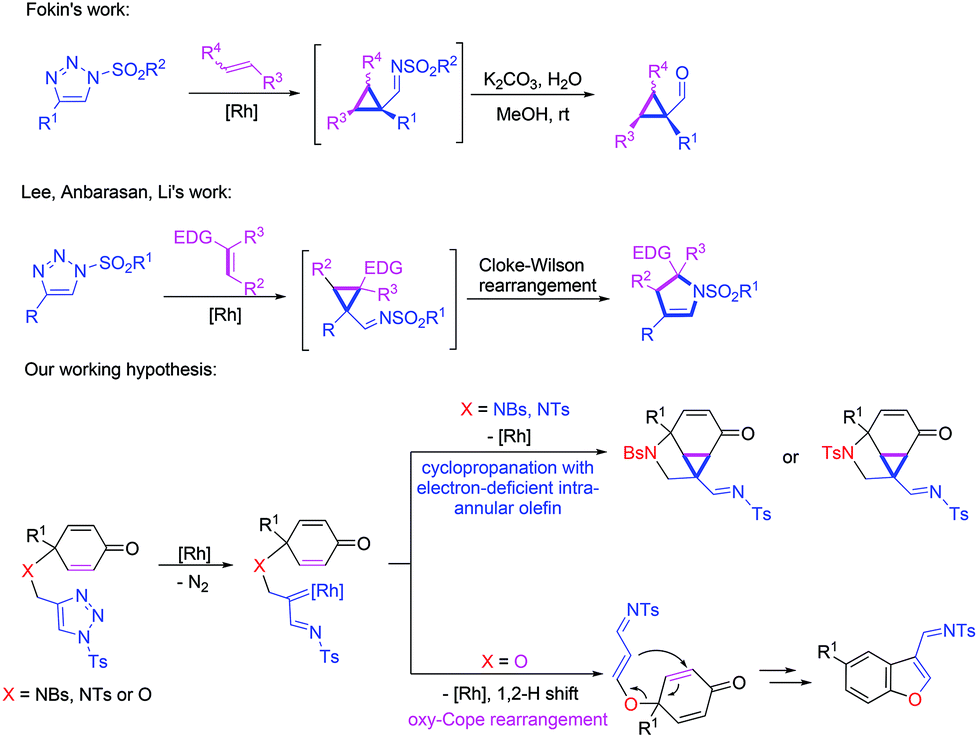 | ||
| Scheme 1 Previous work and our working hypothesis. | ||
Results and discussion
Experimental investigations
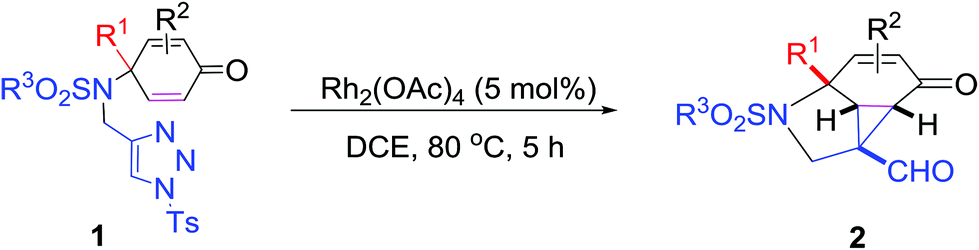
Our studies were initiated by examining the reactivity of NBs (4-bromobenzene sulfonamide)-tethered cyclohexa-2,5-dienone with 1-sulfonyl-1,2,3-triazole 1a in the presence of a variety of rhodium(II) catalysts, and the results are shown in Table 1. It was found that the desired cyclopropanation product 2a could be obtained in 95% yield when 1a was treated with 5 mol% Rh2(OAc)4 in dry 1,2-DCE (1,2-dichloroethane) at 80 °C (Table 1, entry 1). The structure of 2a has been unequivocally assigned by X-ray diffraction. The ORTEP drawing is shown in Table 1 and the CIF data are presented in the ESI.†![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 12 By changing the Rh(II) catalyst to Rh2(Oct)4 and Rh2(Piv)4, 2a could be obtained in 94% yield, respectively (Table 1, entries 2 and 4). Using Rh2(esp)2 as the catalyst gave 2a in 56% yield, but the use of Rh2(tfa)4 as the catalyst only afforded 2a in lower yield (9%) (Table 1, entries 3 and 5). When the reaction was carried out in the absence of a rhodium catalyst, no reaction occurred and the starting material was recovered, indicating that a rhodium catalyst was essential in this reaction (Table 1, entry 6). The examination of solvent effects revealed that when the reaction was conducted in DCM (dichloromethane) or toluene, 2a could be given in 94% and 87% yields, respectively (Table 1, entries 7 and 8). Using THF (tetrahydrofuran) and CH3CN (acetonitrile) as the solvents provided 2a in 59% and 20% yields under otherwise identical conditions (Table 1, entries 9 and 10). Thus, Rh2(OAc)4 was the best catalyst and DCE was the solvent of choice for this transformation to give 2a in higher yield.
a,b,c,d
12 By changing the Rh(II) catalyst to Rh2(Oct)4 and Rh2(Piv)4, 2a could be obtained in 94% yield, respectively (Table 1, entries 2 and 4). Using Rh2(esp)2 as the catalyst gave 2a in 56% yield, but the use of Rh2(tfa)4 as the catalyst only afforded 2a in lower yield (9%) (Table 1, entries 3 and 5). When the reaction was carried out in the absence of a rhodium catalyst, no reaction occurred and the starting material was recovered, indicating that a rhodium catalyst was essential in this reaction (Table 1, entry 6). The examination of solvent effects revealed that when the reaction was conducted in DCM (dichloromethane) or toluene, 2a could be given in 94% and 87% yields, respectively (Table 1, entries 7 and 8). Using THF (tetrahydrofuran) and CH3CN (acetonitrile) as the solvents provided 2a in 59% and 20% yields under otherwise identical conditions (Table 1, entries 9 and 10). Thus, Rh2(OAc)4 was the best catalyst and DCE was the solvent of choice for this transformation to give 2a in higher yield.
a,b,c,d
|
|
||||||
|---|---|---|---|---|---|---|
| Entrya | Catalyst | Solvent | Additive | Temp. (°C) | Time (h) | Yieldb (%) |
| a Reaction conditions: 1a (0.5 mmol), cat. (0.025 mmol), solvent (3 mL). b Yields of the isolated product. c NMR yield using 1,3,5-trimethoxybenzene as an internal standard. d No reaction and the starting material was recovered. Oct = octanoate, tfa = trifluoroacetic acid, Piv = pivalate, esp = a,a,a′,a′-tetramethyl-1,3-benzenedipropionic acid. | ||||||
| 1 | Rh2(OAc)4 | DCE | — | 80 | 5 | 95 |
| 2 | Rh2(Oct)4 | DCE | — | 80 | 5 | 94 |
| 3 | Rh2(tfa)4 | DCE | — | 80 | 5 | 9c |
| 4 | Rh2(Piv)4 | DCE | — | 80 | 5 | 94 |
| 5 | Rh2(esp)2 | DCE | — | 80 | 5 | 56c |
| 6 | — | DCE | — | 80 | 5 | —d |
| 7 | Rh2(OAc)4 | DCM | — | 80 | 5 | 94 |
| 8 | Rh2(OAc)4 | Toluene | — | 80 | 5 | 87 |
| 9 | Rh2(OAc)4 | THF | — | 80 | 5 | 59 |
| 10 | Rh2(OAc)4 | CH3CN | — | 80 | 5 | 20c |

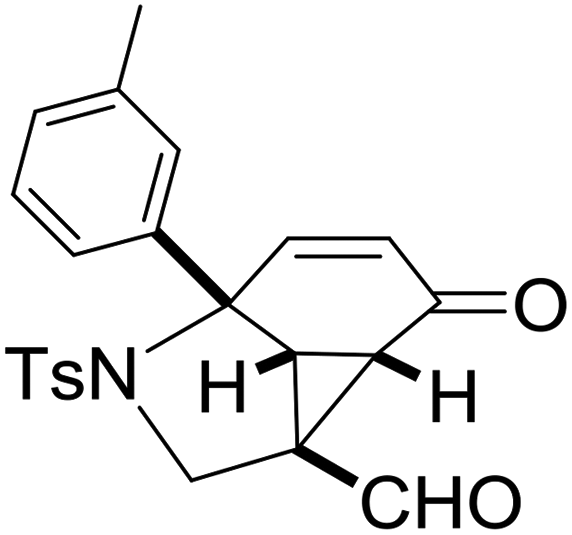
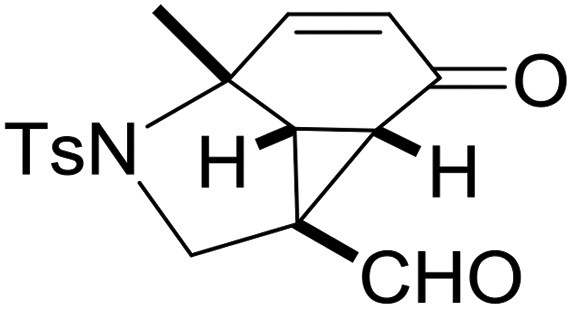
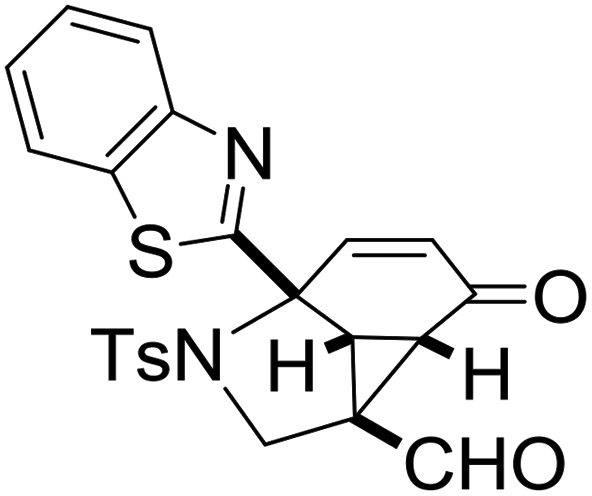
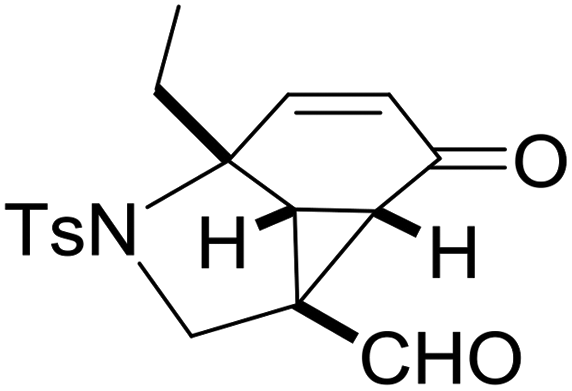
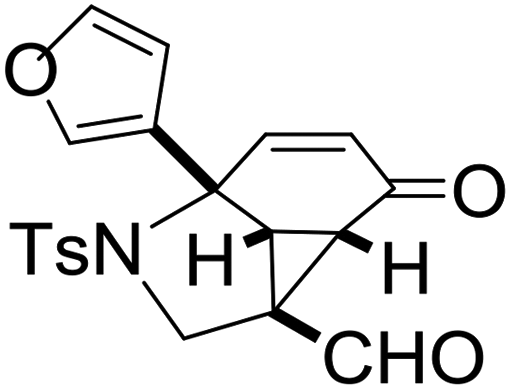
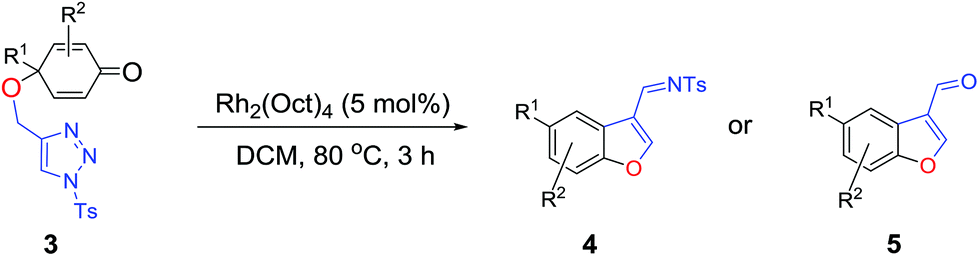
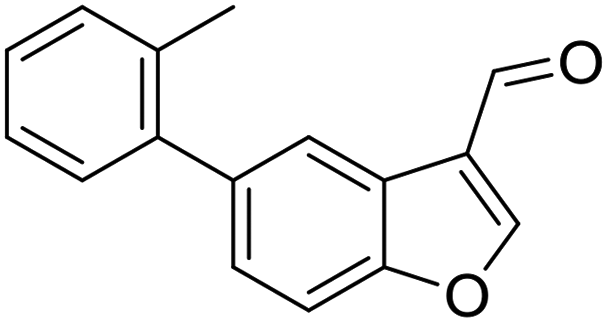
With the optimized reaction conditions in hand, we next examined the substrate scope of this reaction, and the results are summarized in Table 2. With NBs as the linker and the R1 group being a methyl group, the desired product 2b was obtained in 99% yield. Then, we changed the linker to NTs-(4-toluene sulfonamide), a variety of R1 groups have been investigated and the results are outlined in Table 2. In the cases of substrates 1c–1f, in which R1 was a methyl, ethyl, iPr or nbutyl group, the reactions proceeded efficiently to give the desired products 2c–2f in 85–98% yields. Substrates 1g and 1h bearing a cyclopentyl group and a cyclohexyl group also gave the corresponding products 2g and 2h in 87% and 86% yields, respectively. These results suggested that a variety of aliphatic groups could be tolerated in this transformation without steric or electronic influence. Substrates 1i–1j, in which R1 was an aromatic group, were also prepared and used for this transformation. When a methoxyl group, a fluorine atom, or a methyl group was introduced at the 4- or 3-position of the benzene ring, the reactions still proceeded smoothly to afford the desired products 2i–2k in 92–98% yields. Unfortunately, when a MeO group was introduced at the 2-position of the benzene ring, the desired product 2l was not obtained and a complex mixture was formed, presumably due to which the increased steric bulkiness impaired the cyclopropanation process or the rhodium carbene could be captured by the adjacent MeO group to change the reaction pathway.13 In addition, the heteroaromatic group was also compatible in this reaction. When R1 was a benzothiazolyl or a furyl group, the desired products 2m and 2n could be afforded in 56% and 75% yields, respectively. Substrate 1o containing a functionalized alkenyl group such as a 3-butenyl group was also suitable in this reaction, furnishing 2o in 85% yield. To further evaluate the generality of this method, some substituents were introduced in the cyclohexa-2,5-dienone skeleton. When the α- or β-position of the carbonyl group was occupied by a bromine atom, the corresponding products 2p and 2q were produced in 55% and 63% yields, in which cyclopropanation exclusively took place at the non-substituted olefinic unit of cyclohexa-2,5-dienone. We assumed that the steric bulkiness of the bromine atom was a possible reason for this observation. Another reasonable explanation was that the bromine atom could coordinate with the rhodium metal center, leading to the proposition that the non-substituted double bond could be closer to the carbon atom of the rhodium carbene center. As for substrates 1r and 1t bearing a fluorine atom or a methoxyl group at the β-position of the carbonyl group, cyclopropanation exclusively occurred at the substituted olefinic unit, giving the corresponding products 2r and 2t in 67% and 54% yields, respectively. In these cases, the steric influence did not have an impact on the proceeding reaction, but the electronic properties of the double bond may play a leading role. When an electron-neutral group such as a methyl group was introduced at the β-position of the carbonyl group, this cyclopropanation could occur on each side of the olefinic unit, affording the corresponding products 2sa and 2sb in 36% and 34% yields, respectively.
|
|
|||
|---|---|---|---|
| Product | No./yield (%) | Product | No./yield (%) |
| a Reaction conditions: 1 (1.0 mmol), [Rh2(OAc)4](0.05 mmol), DCE (5.0 mL). b Yields are those of the isolated products. | |||
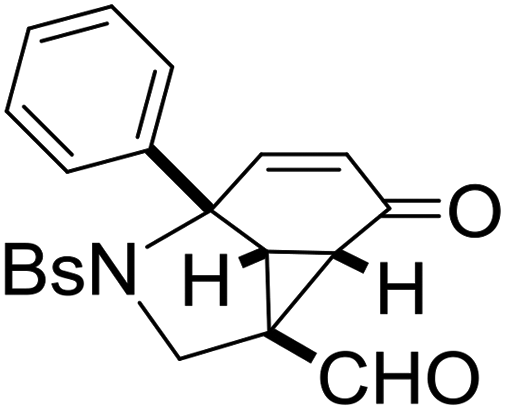
|
2a, 95 |
|
2k, 94 |

|
2b, 99 |
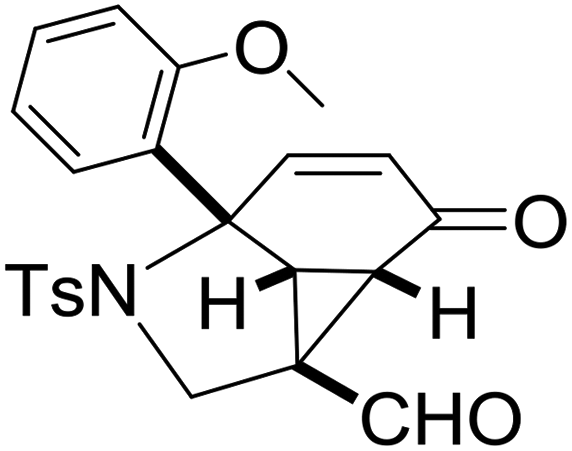
|
2l, — |
|
2c, 98 |
|
2m, 56 |
|
2d, 86 |
|
2n, 75 |

|
2e, 93 |
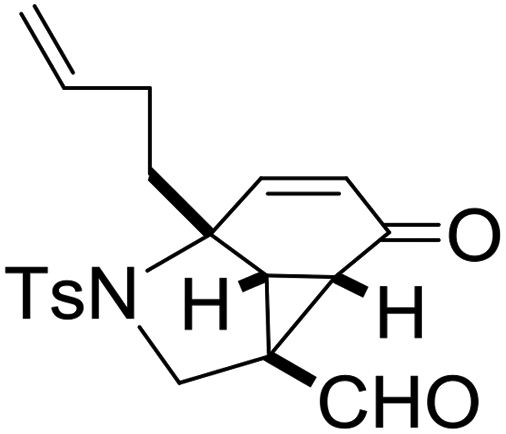
|
2o, 85 |
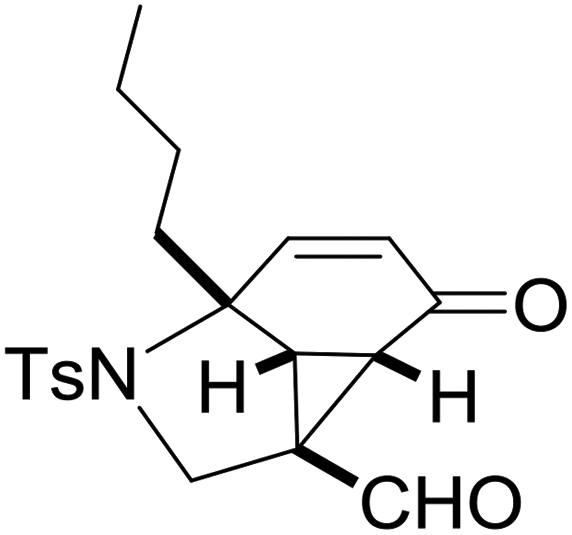
|
2f, 85 |
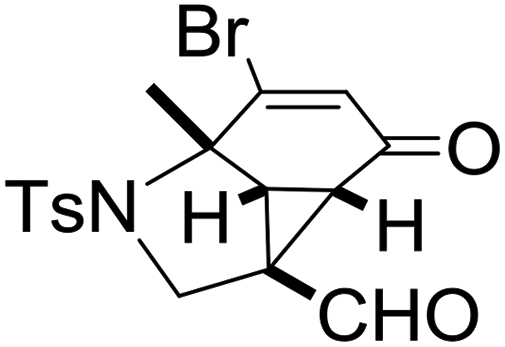
|
2p, 55 |
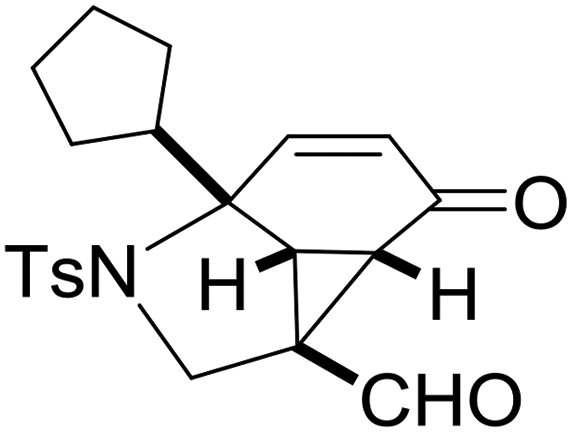
|
2g, 87 |

|
2q, 63 |

|
2h, 86 |

|
2r, 67 |

|
2i, 92 |
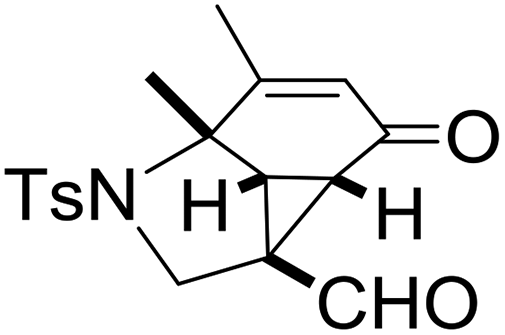
|
2sa, 36 |
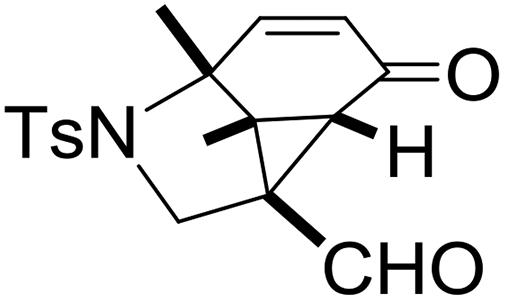
|
2sb, 34 | ||
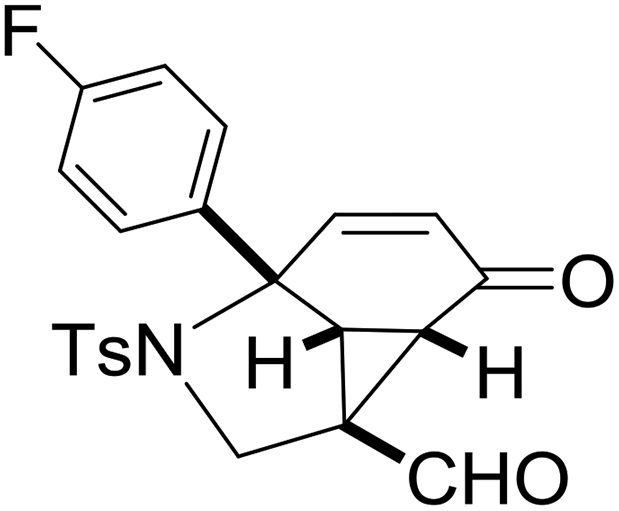
|
2j, 98 |
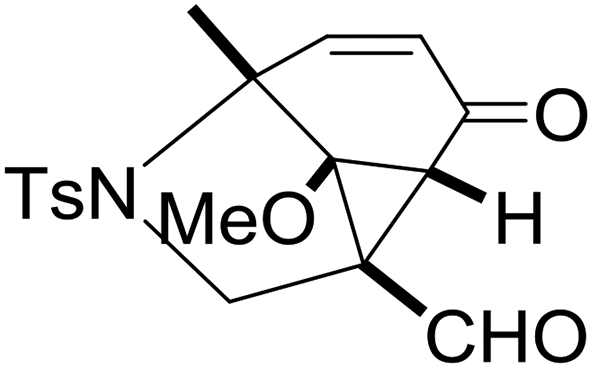
|
2t, 54 |
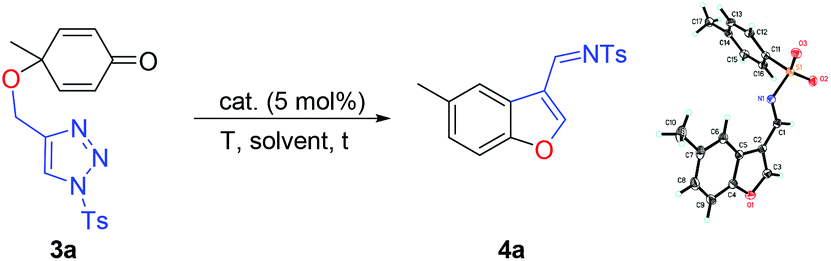
To further expand the substrate scope of this reaction, O-tethered cyclohexa-2,5-dienones with 1-sulfonyl-1,2,3-triazole 3a were synthesized and utilized to process the reaction under similar conditions. An unexpected benzofuran derivative 4a was isolated as a sole product in 48% yield upon treating 3a with Rh2(OAc)4 in DCE at 70 °C for 5 h (Table 3, entry 1). The structure of 4a has been unambiguously assigned by X-ray diffraction. The ORTEP drawing is shown in Table 3 and the CIF data are presented in the ESI.†14 Since benzofurans are important heterocyclic compounds occurring in a variety of natural products and synthetic pharmaceuticals, developing new methods for the preparation of these scaffolds is meaningful.15 Next we optimized the cascade cyclization reaction conditions using a variety of rhodium(II) catalysts in various solvents as well as examining the reaction temperatures (Table 3). Carrying out the reaction in toluene, THF, CH3CN and DCM revealed that DCM was the best solvent in this transformation, affording 4a in 75% yield and the desired product 4a was not formed in THF (Table 3, entries 2–5). Next, we investigated rhodium(II) catalysts in DCM at 70 °C and identified that Rh2(Oct)4 gave a better result, affording 4a in 86% yield (Table 3, entries 6–9). A quick solvent screening utilizing Rh2(Oct)4 as the catalyst demonstrated that DCM was still the solvent of choice (Table 3, entries 10–13). Moreover, when the reaction was conducted at 80 °C or at 90 °C in DCM in a sealed tube, 4a was obtained in 89% or 83% yield, respectively (Table 3, entries 14 and 15). In the absence of the rhodium(II) catalyst, none of the desired products was obtained and the substrate 3a was fully recovered (Table 3, entry 16).
a,b,c
|
|
||||||
|---|---|---|---|---|---|---|
| Entrya | Catalyst | Solvent | Additive | Temp (°C) | Time (h) | Yieldb (%) |
| a Reaction conditions: 3a (0.2 mmol) and Rh cat. (5 mol%) were stirred in 2 mL of solvent. b NMR yield using 1,3,5-trimethoxybenzene as an internal standard. c The starting material was recovered. | ||||||
| 1 | Rh2(OAc)4 | DCE | — | 70 | 5 | 48 |
| 2 | Rh2(OAc)4 | Toluene | — | 70 | 5 | 36 |
| 3 | Rh2(OAc)4 | THF | — | 70 | 5 | N.D |
| 4 | Rh2(OAc)4 | CH3CN | — | 70 | 5 | 21 |
| 5 | Rh2(OAc)4 | DCM | — | 70 | 5 | 75 |
| 6 | Rh2(Piv)4 | DCM | — | 70 | 5 | 67 |
| 7 | Rh2(tfa)4 | DCM | — | 70 | 5 | 13 |
| 8 | Rh2(Oct)4 | DCM | — | 70 | 5 | 86 |
| 9 | Rh2(esp)2 | DCM | — | 70 | 5 | 34 |
| 10 | Rh2(Oct)4 | DCE | — | 70 | 5 | 58 |
| 11 | Rh2(Oct)4 | Toluene | — | 70 | 5 | 46 |
| 12 | Rh2(Oct)4 | THF | — | 70 | 5 | N.D |
| 13 | Rh2(Oct)4 | CH3CN | — | 70 | 5 | 24 |
| 14 | Rh2(Oct)4 | DCM | — | 80 | 3 | 89 |
| 15 | Rh2(Oct)4 | DCM | — | 90 | 3 | 83 |
| 16 | — | DCM | — | 90 | 3 | —c |
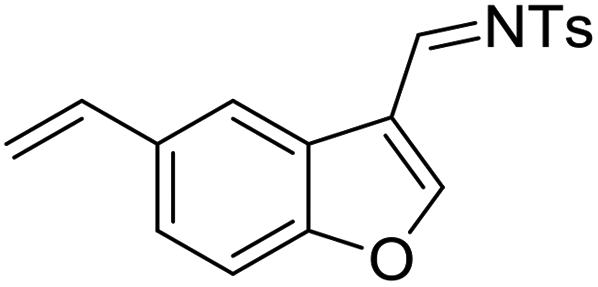
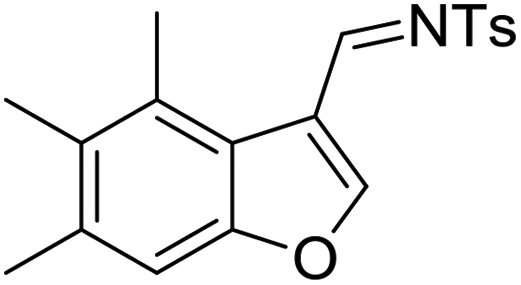
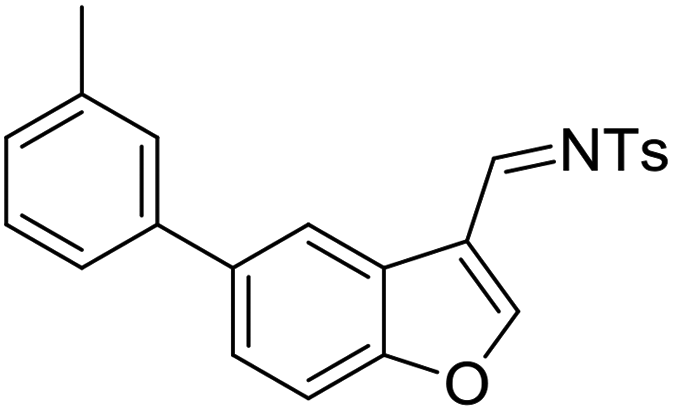
Having the optimized reaction conditions in hand, we next investigated the generality of this reaction and the results are shown in Table 4. When R1 was an ethyl or isopropyl group (R2 = H), the corresponding products 4b and 4c could be obtained in 76% and 82% yields, respectively. We also investigated aromatic group substituted substrates in this reaction and found that when R1 was a phenyl group or a 3-MeC6H4 group, the corresponding products 4d and 4e could also be afforded in 81% or 76% yield, respectively. It is noteworthy that when R1 was a 2-MeC6H4 group (substrate 3f), product 5f could be obtained in only 11% yield as an imine-hydrolyzed product in the purification process. In this case, another cyclopropanation product 6f was also isolated in 72% yield as the major product (Scheme 2). It was probably due to the fact that the steric bulkiness of substrate 3f affected the 1,2-H shift and intramolecular cyclopropanation became a prior process in this case (also see Scheme 3). When R1 was a vinyl group, the corresponding product 4g was obtained in 37% yield, probably due to the fact that the vinyl group influenced the oxy–Cope rearrangement. Then, we introduced two methyl groups at the two β-positions of the carbonyl group in the cyclohexa-2,5-dienone skeleton, and identified that the desired product 4h could be obtained in 86% yield. When only one methyl group was introduced at the β-position, the products 4i and 5i were isolated in 35% and 27% yields, respectively. It should be noted that 5i was also the hydrolyzed product of the imine during the purification process.
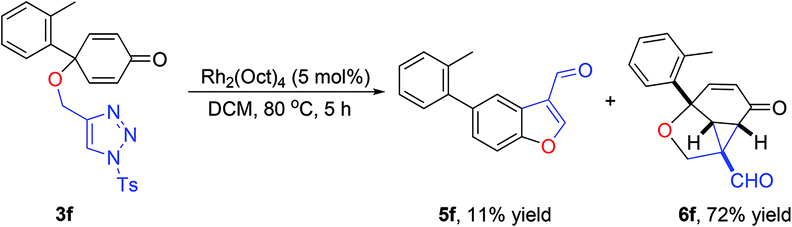 | ||
| Scheme 2 Rhodium(II)-catalyzed intramolecular tandem cyclization reaction of 3f. | ||
 | ||
| Scheme 3 Proposed reaction mechanisms. | ||



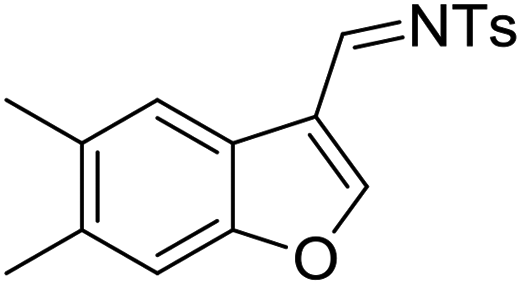

Mechanistic proposal
Further synthetic transformation of the polycyclic cyclopropa[cd]indole product 2k was also conducted. Subjecting 2k into DIBAL-H in THF at 0 °C afforded the corresponding reduction product 7k in 64% yield. While treating 2k with CeCl3·7H2O and NaBH4 in MeOH, both the aldehyde unit and the α,β-unsaturated ketone moiety could be reduced to afford the corresponding products 8ka and 8kb in 42% and 46% yields, respectively (Scheme 4).
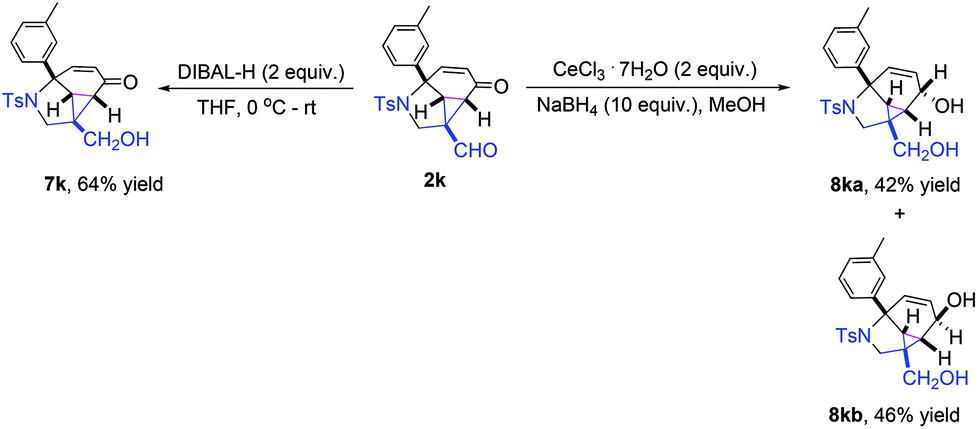 | ||
| Scheme 4 Synthetic transformation. | ||
Conclusions
In conclusion, we have developed an unprecedented and highly efficient rhodium(II)-catalyzed divergent intramolecular tandem cyclization of N- or O-tethered cyclohexa-2,5-dienones with 1-sulfonyl-1,2,3-triazole bearing a variety of functional groups. This synthetic strategy provided a powerful and rapid access to polycyclic cyclopropa[cd]indole and benzofuran skeletons via α-imino rhodium carbene and subsequently enriched the chemistry of 1-sulfonyl-1,2,3-triazoles. The substrate scope of this rhodium(II)-catalyzed intramolecular tandem cyclization is broad and diversified. The connecting atom (a linker such as the N or O atom) between the cyclohexa-2,5-dienone unit and the 1-sulfonyl-1,2,3-triazole moiety can significantly change the reaction pathway. These reaction mechanisms have also been proposed on the basis of the reaction outcomes and the previously reported literature. It should be emphasized here that we first disclosed in this paper that the cyclopropanation of α-imino rhodium carbene derived from 1-sulfonyl-1,2,3-triazole with an electron-deficient intra-annular olefin could take place smoothly. Some useful synthetic transformations were also conducted to set up a stage for further synthetic applications. The utilization of this strategy for the synthesis of heterocycles existing in natural products or pharmaceutical molecules is currently under investigation.Experimental section
General procedure for the synthesis of product 2
Under an argon atmosphere, Rh2(OAc)4 (0.05 mmol, 0.05 equiv.), dry 1,2-dichloroethane (5.0 mL), and substrate 1 (1.0 mmol, 1.0 equiv.) were added to a Schlenk tube. The reaction mixture was stirred for about 5 h at 80 °C until 1 was completely consumed by TLC monitoring. Then the solvent was removed under reduced pressure and the residue was purified by silica gel flash column chromatography (eluent: petroleum ether/EtOAc, 4/1–2/1) to give the product 2.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We are grateful for the financial support from the National Basic Research Program of China [(973)-2015CB856603], the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDB20000000 and sioczz201808), the National Natural Science Foundation of China (No. 20472096, 21372241, 21572052, 20672127, 21421091, 21372250, 21121062, 21302203, 20732008, 21772037, 21772226 and 21861132014), the Fundamental Research Funds for the Central Universities 222201717003, and the Shenzhen Nobel Prize Scientists Laboratory Project.References
-
(a) V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS
; (b) E. J. Yoo, M. Ahlquist, S. H. Kim, I. Bae, V. V. Fokin, K. B. Sharpless and S. Chang, Angew. Chem., Int. Ed., 2007, 46, 1730–1733 CrossRef CAS PubMed
-
(a) T. Horneff, S. Chuprakov, N. Chernyak, V. Gevorgyan and V. V. Fokin, J. Am. Chem. Soc., 2008, 130, 14972–14974 CrossRef CAS
-
(a) H. Shang, Y. H. Wang, Y. Tian, J. Feng and Y. F. Tang, Angew. Chem., Int. Ed., 2014, 53, 5662–5666 CrossRef CAS PubMed
- For transannulations, see:
(a) S. Chuprakov, F. W. Hwang and V. Gevorgyan, Angew. Chem., Int. Ed., 2007, 46, 4757–4759 CrossRef CAS
-
(a) T. Miura, T. Biyajima, T. Fujii and M. Murakami, J. Am. Chem. Soc., 2012, 134, 194–196 CrossRef CAS
- N. Selander, B. T. Worrell, S. Chuprakov, S. Velaparthi and V. V. Fokin, J. Am. Chem. Soc., 2012, 134, 14670–14673 CrossRef CAS
- For the ylide reaction starting from triazole, see:
(a) T. Miura, T. Tanaka, A. Yada and M. Murakami, Chem. Lett., 2013, 42, 1308–1310 CrossRef CAS
-
(a) M. P. Doyle, Angew. Chem., Int. Ed., 2009, 48, 850–852 CrossRef CAS
- S. Chuprakov, S. W. Kwok, L. Zhang, L. Lercher and V. V. Fokin, J. Am. Chem. Soc., 2009, 131, 18034–18035 CrossRef CAS PubMed
-
(a) C.-E. Kim, S. Park, D. Eom, B. Seo and P. H. Lee, Org. Lett., 2014, 16, 1900–1903 CrossRef CAS
- C.-Z. Zhu, Y. Wei and M. Shi, Adv. Synth. Catal., 2019 DOI:10.1002/adsc.201900290
- The crystal data of 2a have been deposited in CCDC with number 1839382.†.
-
(a) H. J. Shen, J. K. Fu, J. X. Gong and Z. Yang, Org. Lett., 2014, 16, 5588–5591 CrossRef CAS PubMed
- The crystal data of 4a have been deposited in CCDC with number 1812836.†.
-
(a) L. Arias, Y. Vara and F. P. Cosso, J. Org. Chem., 2012, 77, 266–275 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data of new compounds.CCDC 1839382 and 1812836. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c9qo00714h |
| This journal is © the Partner Organisations 2019 |