 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Characterising polar compounds using supercritical fluid chromatography–nuclear magnetic resonance spectroscopy (SFC–NMR)†
F. H. M.
van Zelst
ab,
S. G. J.
van Meerten
b and
A. P. M.
Kentgens
 *b
*b
aTI-COAST, Science Park 904, 1098 XH Amsterdam, The Netherlands
bInstitute for Molecules and Materials, Radboud University, Heyendaalseweg 135, 6525 AJ Nijmegen, The Netherlands. E-mail: A.Kentgens@nmr.ru.nl
First published on 3rd January 2019
Abstract
To detect and characterise compounds in complex matrices, it is often necessary to separate the compound of interest from the matrix before analysis. In our previous work, we have developed the coupling of supercritical fluid chromatography (SFC) with nuclear magnetic resonance (NMR) spectroscopy for the analysis of nonpolar samples [Van Zelst et al., Anal. Chem., 2018, 90, 10457]. In this work, the SFC–NMR setup was successfully adapted to analyse polar samples in complex matrices. In-line SFC–NMR analysis of two N-acetylhexosamine stereoisomers was demonstrated, namely N-acetyl-mannosamine (ManNAc) and N-acetyl-glucosamine (GlcNAc). ManNAc is a metabolite that is present at elevated concentrations in patients suffering from NANS-mediated disease. With our SFC–NMR setup it was possible to distinguish between the polar stereoisomers. Until now, this was not possible with the standard mass-based analysis techniques. The concentrations that are needed in the SFC–NMR setup are currently too high to be able to detect ManNAc in patient samples (1.7 mM vs. 0.7 mM). However, several adaptations to the current setup will make this possible in the future.
1 Introduction
Polar compounds make up a great part of all molecules and are of special interest in biochemistry. They are present in biological systems, for example in biofluids such as urine. Metabolomics is an emerging field in analytical science, which focusses on characterising small molecules in biological systems. These metabolites form the (intermediate) products of biochemical reactions in our metabolism. Metabolomics can be used to identify biomarkers for many diseases, such as inborn errors of metabolism. The standard workflow in untargeted metabolomics is analysis by liquid chromatography–mass spectrometry (LC-MS). In this LC-MS workflow, metabolites are first separated and their mass-to-charge-ratio (m/z) is determined. These m/z-values are then compared to databases, which often result in multiple hits. In this case, the chromatographic retention times and MS/MS-fragmentation patterns will be compared to a reference compound, often leading to the metabolite of interest.1 However, if the structure cannot be identified in the database, more extensive analyses have to be performed.Metabolites are very diverse in structure and functional groups, ranging from very hydrophilic to lipophilic. The metabolite composition also varies for different types of biofluids. In urine, very hydrophilic compounds are present, although trace amounts of lipids and fatty acids have also been detected. Serum, on the contrary, contains more lipids. Compared to cerebrospinal fluid and saliva, urine contains more, and more diverse metabolites. Blood contains all the metabolites present in urine, and more, but the concentrations differ substantially. The kidneys filter out some of the metabolites or toxins from the blood, which is why metabolite concentrations in urine are often higher than those in blood.2 The chemical diversity of the compounds, along with the broad range of concentrations, call for a variety of analytical techniques. As mentioned previously, LC-MS is generally used as the standard technique for metabolomics. Nuclear magnetic resonance (NMR) spectroscopy, which is unbiased and can be used for a broad range of metabolites, can be an attractive alternative. An important advantage of using NMR spectroscopy over MS is that the technique is quantitative. However, NMR spectroscopy is less sensitive than MS and can therefore not be used to identify metabolites at low concentrations (typically the lower limit is around 1 μM).
Due to the complex nature of urine samples, NMR spectra are usually very crowded and therefore especially metabolites present in lower concentrations are difficult to identify from the spectrum. As is the case in the LC-MS workflow, it would be beneficial to separate the sample into less complex fractions by chromatography before NMR analysis. It is however good to note that the sample gets diluted into the mobile phase during chromatography, which can make NMR detection difficult or even impossible. In our group, we have recently developed a new setup, which combines supercritical fluid chromatography (SFC) and NMR spectroscopy.3 In this work we demonstrated the separation by SFC of a mixture of tocopherol (vitamin E) isomers, followed by in-line concentration and NMR detection. Although tocopherol is a nonpolar molecule and therefore very different from the mostly polar molecules present in urine, we are adapting the setup to analyse polar metabolites in urine. SFC is a type of chromatography that uses supercritical fluids, typically supercritical CO2, as the mobile phase. Although CO2 is highly nonpolar, by adding a co-solvent to the supercritical fluid, such as methanol, the mobile phase becomes more polar. In this way, SFC is able to separate nonpolar as well as polar compounds, making it suitable for separating polar metabolites from urine. To be able to detect lower concentrations by NMR spectroscopy, a method for concentrating the sample in-line after SFC separation has been implemented as well. This was achieved by in-line expansion of the supercritical CO2 (scCO2), thus separating the mobile phase from the sample during transport from the SFC outlet to the NMR probehead.
Another approach to detect smaller amounts in NMR spectroscopy is by miniaturising the detection coil. In this way, smaller sample volumes can be detected at higher sensitivity. The challenge in miniaturisation is to maintain a high resolution. Different designs have been researched, such as tightly wound solenoid coils and planar helical coils.4–7 Our group has developed a design in which the conventional detection coil is replaced by a high resolution flat stripline radio-frequency inductor. The flat geometry of the stripline allows in-flow detection, by passing a capillary over the stripline.8
To see if our SFC–NMR setup can be adapted to analyse polar samples, we have chosen to study N-acetylhexosamines in urine as a concept application. These monosaccharides appear in different diastereomeric forms (shown in Fig. 1), which have equal m/z-values, and are therefore not distinguishable in the standard HPLC-MS workflow described previously. N-Acetylmannosamine (ManNAc) is a biomarker for NANS-mediated disease, an inborn error in the sialic acid metabolism.9 It is, however, difficult to distinguish N-acetylmannosamine from its stereoisomers, N-acetylglucosamine (GlcNAc) and N-acetylgalactosamine (GalNAc), using MS since they are equal in mass. For identification of ManNAc in body fluids, NMR spectroscopy has been employed.9 This, however, requires relatively large amounts of sample at a higher concentration compared to MS analyses. Martens et al. developed a new method to study ManNAc in biofluids using infrared ion spectroscopy after MS.10 This is a sensitive method, requiring only small amounts of sample. However, this technique either requires a reference for comparison, or computer simulations of the IR spectra, which do not always match the experimental spectra. In our approach we will separate the compound of interest (ManNAc) with SFC and shuttle it in-line to the NMR probehead, which is able to detect much smaller volumes (∼150 nL in this case) than the standard NMR approach (∼500 μL) and does not require a reference for structural characterisation. However, sensitivity remains an issue in NMR analysis.
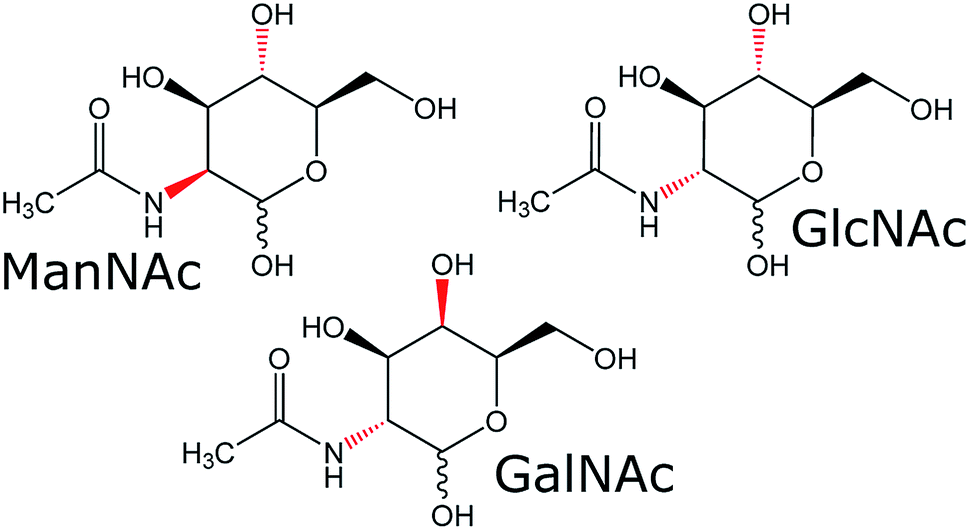 | ||
| Fig. 1 Molecular structures of the three N-acetylhexosamines. The difference in stereochemistry between the diastereomers is indicated in red. Each diastereomer has an α- and β-stereoisomer, indicated by the wavy bond of the alcohol group at the anomeric carbon atom. | ||
Due to the variations in urine samples, between different patients but also within patients, metabolite concentrations in urine are usually expressed in μmol per mmol creatinine. Van Karnebeek et al. found that the concentration of ManNAc was between 41 and 98 μmol mmol−1 creatinine in adult patients suffering from NANS-mediated disease (<10 μmol mmol−1 in the reference sample).9 The amount of creatinine within and between patients varies enormously. However according to the guidelines of the World Health Organization (WHO), a valid urine sample ranges between 30 and 300 mg creatinine per dL urine.11 Taking a value of 150 mg dL−1 urine, the concentration of ManNAc in the urine of patients suffering from NANS-mediated disease is estimated to be in the range of 0.3–0.7 mM. This concentration is sufficient in a standard NMR setup, however in our previous SFC–NMR setup we needed samples in the mM range to be able to detect them within 50 minutes.3 A higher signal can be gained by acquiring over a longer period of time, therefore lower concentrations may also be detectable.
2 Experimental
2.1 Setup
The SFC–NMR setup that was used has been introduced in our previous work3 and is shown in Fig. 2. An overview of the setup is given here; for further details the reader is referred to our previous paper.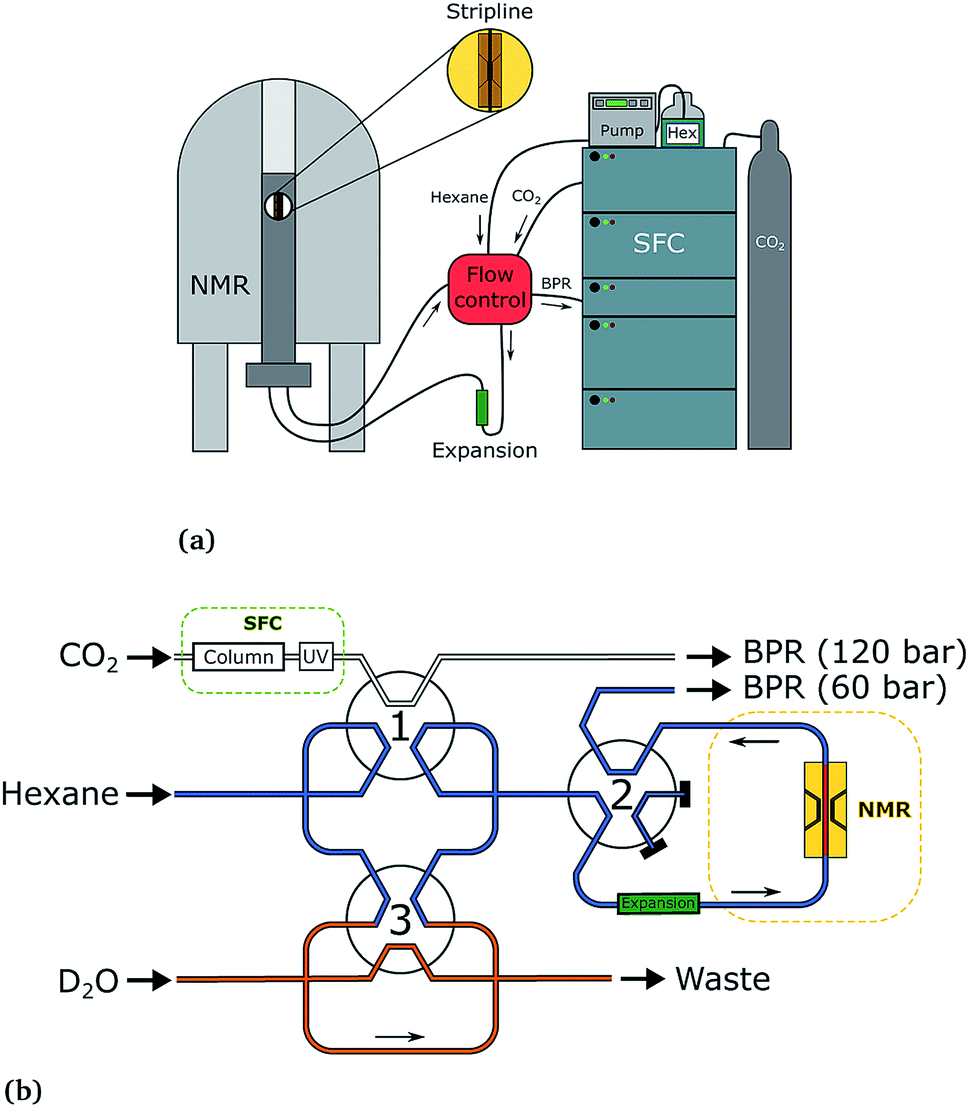 | ||
| Fig. 2 The SFC–NMR setup (a) and detailed flow control system (b). Adapted from ref. 3. Once a sample of interest is observed in the UV chromatogram, the first valve is switched, letting the sample into the sample loop. When the loop is filled, the first valve switches back. In this way, a sample plug is created in the hexane flow, which is kept at a lower pressure. Simultaneously, the third valve switches to inject a small amount of water in the sample plug. When the sample reaches the middle of the NMR probehead, the second valve switches to stop the flow. In this way, multiple scans can be acquired on the same sample, in order to increase the signal-to-noise ratio. | ||
The SFC is connected to the NMR probe with a flow control system. Once a peak of interest passes the UV detector, the first valve is switched, letting the sample including the mobile phase into a 100 μL sample loop. When the first valve switches back, the sample is injected into a n-hexane (AnalaR NORMAPUR, VWR Chemicals) flow-line, which is kept at a lower pressure than the SFC flow (60 bar compared to 120 bar). This pressure is regulated by an additional backpressure with an operating range between 20 and 103 bar (Vici Jour). Simultaneously, the third valve switches to inject 10 μL of D2O from the additional sample loop (orange in Fig. 2b), which ends up in the middle of the sample plug. The sample plug is transported in the hexane flow at a rate of 0.1 mL min−1 from the SFC to the NMR probe, through PEEK tubing. When the sample reaches the middle of the NMR stripline detector, the flow can be stopped by switching valve 2. In this way, multiple scans can be acquired, to obtain a better signal-to-noise ratio.
The three switching valves are 6-port binary-position valves (Vici Valco). A broad ‘expansion tube’ made of stainless steel, with an inner diameter of 2.159 mm and length of ∼20 cm, is placed in the hexane flow line to allow for the different phases in the sample plug to pass each other during flow. The CO2 from the trailing edge of the plug can then go past the methanol to the beginning of the plug. All PEEK tubing used in the system has an inner diameter of 0.508 mm, except for the tubing from the ‘expansion tube’ to the NMR probe, which has an inner diameter of 0.254 mm. In the stripline probe, a fused silica capillary with an inner diameter of 250 μm and outer diameter of 360 μm (Polymicro Technologies) transports the sample over the active detection volume of the chip. After the experiment, the sample flows back from the NMR spectrometer, over the additional backpressure regulator, to the waste.
2.2 Instruments
In the SFC–NMR setup, a Waters Acquity UPC2 instrument was coupled to a Varian VNMRS spectrometer at 600 MHz Larmor frequency (14.1 T). A home-built stripline probe was used,8 with a 600 μm wide and 5 mm long chip with split contacts, with an active detection volume of ∼150 nL. In-flow experiments were performed with a relaxation delay of 0.2 s and an acquisition time of 0.5 s (in total 0.7 s per spectrum), while stop-flow experiments and the three reference spectra were acquired with a relaxation delay of 10 s and an acquisition time of 1 s. All spectra were recorded with a receiver bandwidth of 10 kHz. The spectra are referenced to the water peak, which was set to 4.79 ppm. All spectra were processed using the ssNake software package.122.3 SFC chromatography
For the SFC optimisation, in Fig. 3, a sample of 0.78 M N-acetyl-D-mannosamine (>98%, Sigma Aldrich) in ultra pure water was prepared. For the separation in Fig. 4, a sample of 0.035 M N-acetyl-D-mannosamine was dissolved in synthetic urine (Surine Negative Urine Control, Cerilliant, Sigma Aldrich). In both cases, 0.5 μL of the sample was injected onto a 100 mm × 1.7 μm packed 2-PIC SFC column (Waters) at a backpressure of 120 bar, a flow rate of 1.5 mL min−1 and a temperature of 60 °C. For the optimal separation, an isocratic mobile phase of 30% methanol (BioSolve, SFC grade) in CO2 (Linde Gas Benelux, food grade) was used. All UV chromatograms were recorded at a wavelength of 210 nm.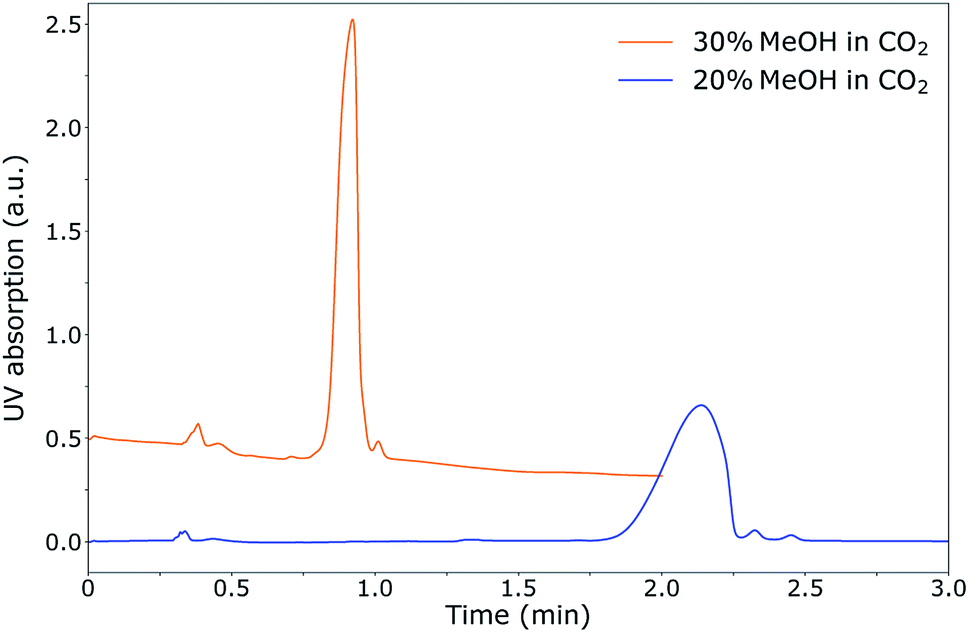 | ||
| Fig. 3 UV chromatogram of the SFC separation of ManNac in water (0.78 M, 0.5 μL injection volume) on a 2-PIC column (Waters) at 60 °C, 120 bar and a flow rate of 1.5 ml min−1. The top graph was separated with a mobile phase of 30% MeOH in CO2, and the bottom graph with 20% MeOH in CO2. The sharpest peak is obtained with 30% MeOH in CO2. | ||
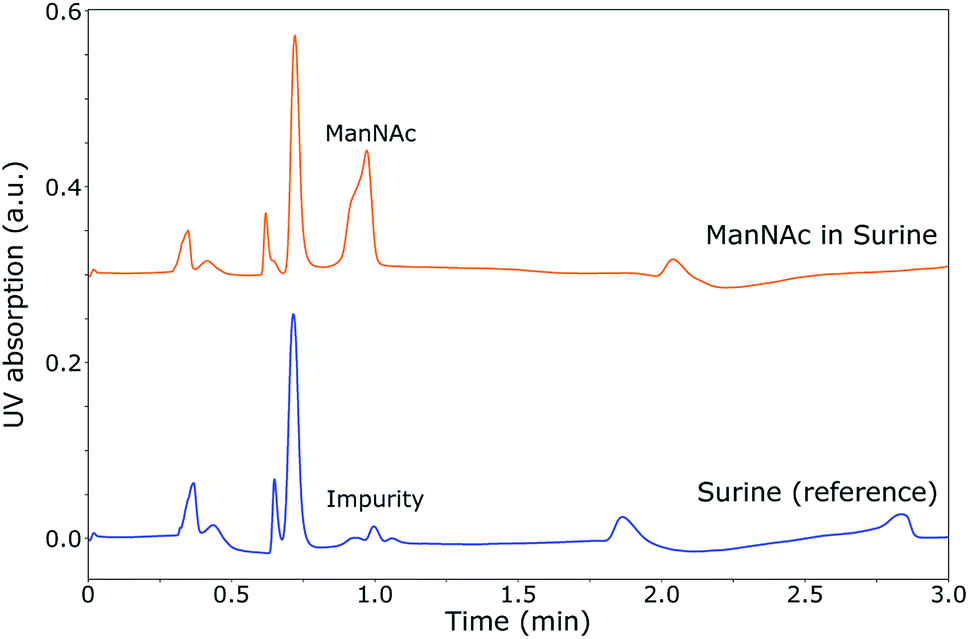 | ||
| Fig. 4 UV chromatogram of the SFC separation of ManNac in Surine (top) (0.035 M, 0.5 μL injected volume) and pure Surine (bottom) on a 2-PIC column at 60 °C, 120 bar and a flow rate of 1.5 ml min−1 for a mobile phase of 30% MeOH in CO2. A small impurity of a previous separation is present in the chromatogram of pure Surine. Under these conditions, ManNAc can be separated from synthetic urine. | ||
2.4 (SFC–)NMR
The 1H NMR reference spectra were obtained by dissolving N-acetylglucosamine, N-acetyl-D-mannosamine (>98%) and N-acetyl-D-galactosamine (∼98%) (all from Sigma Aldrich) in deuterated water (99.9%; Sigma Aldrich) at concentrations of 0.38 M, 0.38 M and 0.75 M, respectively. The samples were then inserted separately into a ∼15 cm long, 250 μm I.D./350 μm O.D. fused silica capillary by capillary suction, which was then sealed at both ends with optical glue. The three reference spectra were acquired using a stripline probe employing a relaxation delay of 10 s and an acquisition time of 1 s, and summing over 6400 scans (19 h 33 min), 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 scans (41 h 33 min) and 1500 scans (4 h 35 min), respectively.
600 scans (41 h 33 min) and 1500 scans (4 h 35 min), respectively.
For the SFC–NMR spectra, a ManNAc sample of 0.78 M and a GlcNAc sample of 0.73 M in ultra pure water were used, of which 4 μL was injected onto the SFC column (further details in the previous section). The 1H NMR spectra were acquired under the same conditions as the reference spectra, but this time averaging over 4860 scans (14 h 51 min) and 3520 scans (10 h 45 min) for ManNAc and GlcNAc, respectively.
3 Results and discussion
In this research, ManNAc in urine was chosen as a concept application to develop the SFC–NMR setup for polar samples. First, the SFC separation will be optimised for ManNAc in urine. Then, a method for in-line concentration of the sample will be discussed, which is needed to be able to detect the analyte by NMR spectroscopy. Finally, the SFC–NMR analysis of ManNAc (and its stereoisomers) is demonstrated.3.1 SFC of N-acetylhexosamines in synthetic urine
During the optimisation of the SFC separation of ManNAc from urine, we have worked with synthetic urine to eliminate sample variation between batches. Later on, this research could be extended to real urine samples. Several stationary phases were investigated for the SFC separation of ManNAc (BEH, BEH 2-EP and 2-PIC columns from Waters), of which the 2-PIC column showed the best results. The next parameter under investigation was the mobile phase composition. Methanol can be mixed with CO2 order to make the mobile phase more polar. In Fig. 3, the elutions of ManNAc in water from a 2-PIC SFC column with mobile phases containing 20% and 30% methanol in CO2 are shown. It is clear that the retention of ManNAc decreases with the increase in the polarity of the mobile phase, which was expected. For 30% methanol, the peak is much sharper. Using a higher concentration of methanol decreases the retention time further and an even sharper peak is obtained. However, 30% methanol in CO2 was chosen as the optimum, since a higher methanol volume means that the sample will be more diluted in the NMR analysis.With these optimised parameters, the separation of ManNAc from synthetic urine was performed. In Fig. 4, the UV chromatogram of the SFC separation of ManNAc in synthetic urine, on a 2-PIC column with a mobile phase of 30% methanol in CO2, is shown. The optimised parameters give a clear separation of ManNAc from synthetic urine. Due to this, the peak from ManNAc can be selected separately from the sample matrix for the SFC–NMR analysis.
3.2 In-line concentration by expansion of scCO2
Since the sample gets diluted in the mobile phase during SFC, it is difficult to detect this low concentration by NMR spectroscopy. This is why concentrating the sample, preferably in-line, after chromatography is required.In our previous work we have demonstrated that by expanding the sample plug in-line in a controlled fashion, the sample and co-solvent are separated from the mobile phase, CO2.3 In this way, the sample is concentrated in a smaller volume, which leads to a higher concentration, which is needed for NMR detection. This method will be employed here as well. However, in our previous work, our transportation medium was water, which did not mix with the nonpolar samples that were investigated. If water is used as the transportation medium in the case of polar samples, the compound will dissolve into the water, thus diluting the sample even more. This is why an alternative, nonpolar transportation solvent was chosen in this work, namely hexane. The sample plug, including CO2 and co-solvent methanol, is injected into the hexane flow through a valve switching system, and is transported to the NMR probehead. By keeping the hexane flow at a lower pressure, the plug is expected to expand and the sample and co-solvent will separate from the CO2.
Under the same conditions as employed in our previous work (hexane flow-line at 50 bar, injection of the sample plug at 120 bar), however, no phase separation occurred. No defined sample plug was observed. This was due to the mixing of hexane with CO2, which both have a similar polarity. Due to this mixing, the expansion did not take place at a pressure of 50 bar. Several pressures were investigated, after which it was found that the sample and methanol co-solvent separated best from the CO2 at a pressure of 60 bar for the hexane flow-line. Additionally, it was found that methanol has a strong affinity for the tubing walls, which are made out of PEEK (polyether ether ketone). Most of the methanol in the sample plug, and with it the sample, will stick to the tubing and will therefore not reach the NMR detector. In order to transport the methanol in the sample plug to the NMR probe, it is necessary to inject a small volume of water into the sample plug, which collects the methanol and sample and allows them to reach the NMR probe.
To follow the phase separation in the plug in-flow, NMR spectra were recorded each 0.7 s of an expanding mixture of 30% MeOH in CO2. 100 μL of this mixture at 120 bar, to which 10 μL D2O is added later on, is injected in the hexane flow-line at 60 bar. The sample plug, as observed in the NMR probe, is shown in Fig. 5. In this figure, the methyl peak of hexane (the transportation medium) and the methyl peak of methanol (part of the plug) were integrated separately in each of the NMR spectra and plotted over time.
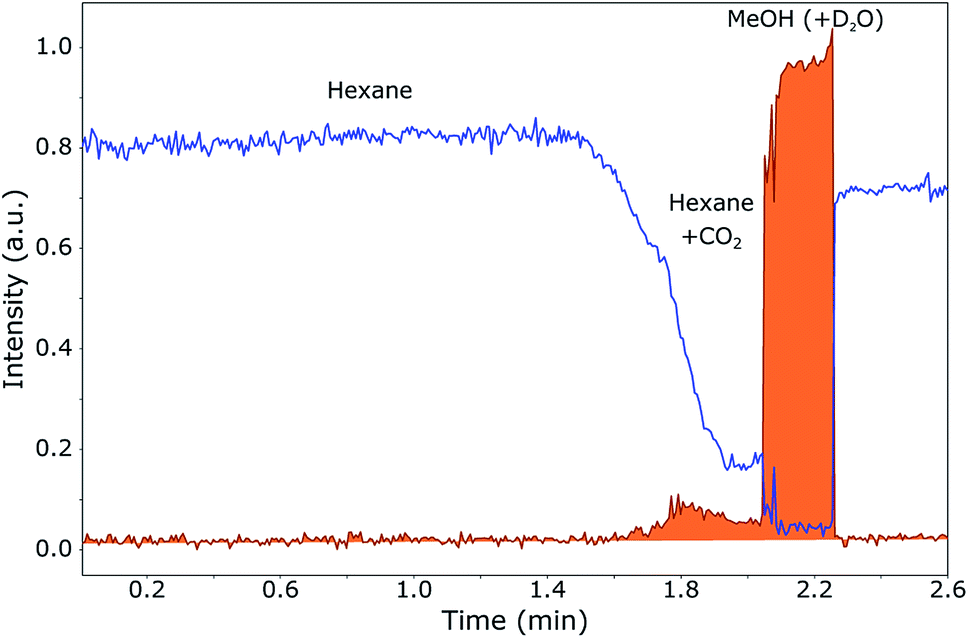 | ||
| Fig. 5 Sample plug of 30% MeOH in CO2 injected in a hexane flow, measured in-flow after in-line expansion to 60 bar. 1H NMR spectra were acquired every 0.7 s during the flow of the sample from the SFC towards the NMR probehead. The methyl peaks of hexane (blue) and methanol (orange) were integrated separately in each spectrum over time and normalised to the maximum methanol integral to give the graphs. A polar sample will dissolve in methanol/water and will therefore mainly be concentrated in the trailing edge of the plug. | ||
The most important result is that methanol and water are mainly collected at the trailing edge of the plug. When a polar sample is present, this will dissolve in methanol and water, not in hexane or CO2. When the pressure is kept at 120 bar instead of 60 bar, the methanol spreads out over the entire plug (1.55 min until 2.3 min), since it stays dissolved in the CO2. Without the expansion to 60 bar, the volume in which the sample is dissolved is much larger than when the CO2 is separated from the methanol, namely approximately 110 μL (CO2, D2O and MeOH) of 40 μL (D2O and MeOH). From Fig. 5, it can be seen that most of the CO2 is separated from the methanol and therefore the concentration method by in-line expansion is successful. It can be seen, however, that a small amount of methanol is not collected at the trailing edge of the plug, but remains dissolved in the CO2 at the start of the plug (1.6–2.0 min). This means that a small part of the sample will not be collected at the end, but will remain spread out over this part of the plug. This loss is, however, quite small, since most of the sample will be concentrated at the end.
Another interesting phenomenon is that hexane from the flow starts mixing in with CO2 at the beginning of the plug. This is due to the fact that hexane and CO2 have a similar polarity. At the trailing edge of the plug, this mixing does not occur since (almost) no CO2 is present in the highly concentrated methanol. However, a very small amount of hexane is present also in the methanol, probably due to the presence of a small remainder of CO2 in which the hexane can dissolve. This will be visible in the SFC–NMR spectra.
The in-line concentration method, which is performed by expanding the sample plug in-line to 60 bar and adding a small amount of water to collect the methanol, can be used successfully for polar samples. However, a small sample loss might occur due to the fact that a small amount of methanol remains dissolved in the CO2 and some methanol sticks to the tubing walls during transportation of the sample to the NMR probe. A small amount of hexane may also be observed in the SFC–NMR spectra that are recorded at the trailing edge of the plug, due to mixing of hexane with a small remainder of CO2 in the concentrated methanol.
3.3 Stripline NMR analysis of N-acetylhexosamines
To show that NMR spectroscopy is able to distinguish between the different isomers ManNAc, GlcNAc and GalNAc, reference 1H NMR spectra were acquired for these three compounds in deuterated water. The three spectra were acquired in the same stripline probe as was used for the in-flow SFC–NMR experiments. The samples were measured in sealed off capillaries. The three spectra are plotted in Fig. 6.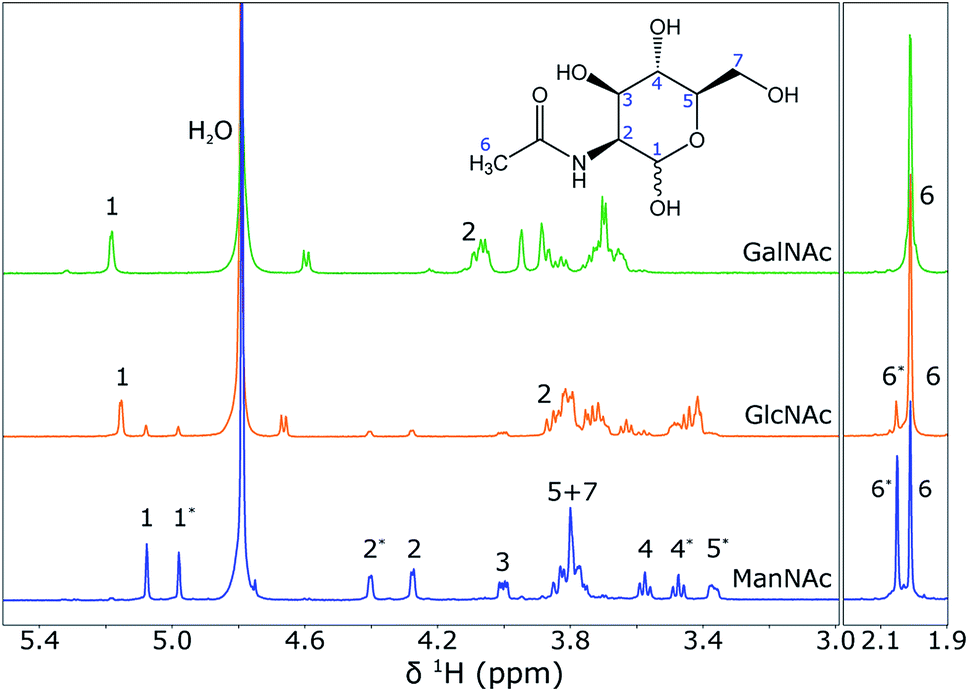 | ||
| Fig. 6 1H NMR spectra of GalNac (0.75 M), GlcNAc (0.38 M) and ManNAc (0.38 M) in D2O (from top to bottom), acquired using the stripline NMR probe. The spectra were scaled to the integrals of the peak(s) around 2 ppm. The structure of ManNAc is shown in the figure for assignment of the peaks. An asterisk (*) next to the peak number indicates the difference between the α- and β-isomer of the same molecule. A full assignment of the spectra can also be found in the Human Metabolome Database (HMDB).13 | ||
It can be seen that the different diastereomers have different 1H NMR spectra. This is due to the stereochemistry of the molecules. The chemical shift of a proton depends on its chemical environment. When an alcohol group or the amine group flips from the top to the bottom of the ring, the chemical environment of the proton attached to that group, but also of the surrounding protons, will change and with it their chemical shifts change. Between the three isomers, the stereochemistry of proton 2 and proton 4 is different, which is observed as a shift in the spectra. As explained before, however, the shifts of the other protons also change. An interesting observation, especially for the spectra of ManNAc and GlcNAc, is that many of the peaks come in pairs. This is due to the fact that within the molecule the alcohol group next to the heteroatom in the ring can change in axial/equatorial position, between α-ManNAc and β-ManNAc. Although the reference compounds that were purchased were 98% pure, in water the position of this alcohol group can change. Also, it can be seen that a small ManNAc contamination is present in the GlcNAc reference spectrum, since all of the peaks match, but are present at much lower intensities.
These reference spectra were acquired at a high concentration, averaging over a substantial number of scans to obtain a high signal-to-noise ratio. For the SFC–NMR analysis, the concentrations will be lower. The most intense peak will be the peak that can be detected in the least amount of time at lower concentrations. In this case, this will be the peak at 2.0 ppm, corresponding to the CH3 group. However, this peak has the same chemical shift for all three diastereomers and cannot be used to distinguish them. The peak around 5.1 ppm (proton 1) will therefore be the best to make a distinction between the three isomers.
3.4 SFC–NMR analysis of ManNAc and GlcNAc
In the previous sections, the SFC separation was optimised for separating ManNAc from water or synthetic urine and it was shown that a distinction between the isomers can be made by looking at the 1H NMR spectra. In this section, these two techniques will be combined in-line to obtain an SFC–NMR analysis of these polar stereoisomers. For this, 4 μL of ManNAc (0.78 M) and GlcNAc (0.73 M) in water were injected for SFC separation. The chromatograms can be found in the ESI (S1).† 100 μL of the peak of interest was selected through a valve switching system in a sample loop. 10 μL of D2O was injected in the middle of this sample loop. The whole plug was then placed into a hexane flow-line at a pressure of 60 bar and transported to the middle of the NMR stripline probe. There, the flow was stopped and multiple scans (taking 10 h 45 min for GlcNAc and 14 h 51 min for ManNAc) were acquired to obtain the SFC–NMR spectra shown in Fig. 7. The reference 1H NMR spectrum, already shown in Fig. 6, of each corresponding isomer is shown under each SFC–NMR spectrum for comparison.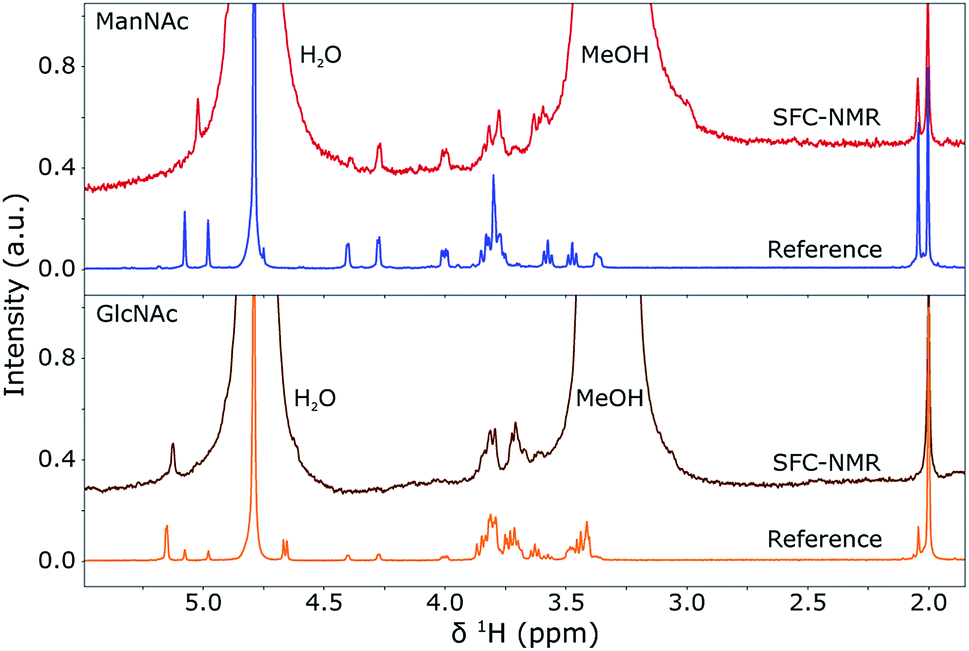 | ||
| Fig. 7 SFC-1H NMR spectra of GlcNAc (bottom) and ManNAc (top) in D2O and MeOH, compared to their respective 1H NMR reference spectra (shown before in Fig. 6). The spectra were scaled to the integrals of the peak(s) around 2 ppm. A Lorentzian line broadening of 1.5 Hz was applied to the SFC–NMR spectra. The distinction between the two isomers is clearly visible in the SFC–NMR spectra. | ||
From the SFC–NMR analysis it can be seen that the different stereoisomers can be distinguished, especially by the peak around 5.1 ppm, corresponding to proton 1 on the anomeric carbon atom next to the heteroatom in the ring. ManNAc can also be identified by the peak around 4.3 ppm, which is not present at this shift for the other two stereoisomers and corresponds to the proton at the carbon atom to which the amine group is attached. Some chemical shifts are slightly different in the SFC–NMR spectra compared to the reference spectra. This is due to the solvent, since the reference spectra were measured in D2O while the SFC–NMR spectra have a 1:3 ratio of D2O and methanol as the solvent. The latter comes from the SFC separation, since 30% methanol is used as the co-solvent. The water peak at 4.79 ppm and the methanol peak, here at 3.3 ppm, are therefore quite substantial in the spectra. D2O exchanges protons with the non-deuterated methanol, therefore the residual water peak is more intense than usual. A solution to this would be to use deuterated methanol, but this would be quite expensive since the whole SFC separation would have to be performed with MeOD. However, in the SFC–NMR spectra shown, the solvent peaks do not overlap too much with the peaks of interest. Two extra peaks are present at 0.8 and 1.2 ppm (not shown), which originate from the small amount of hexane that is still present in the sample plug (discussed in the previous section). These are, however, low in intensity, namely about the same intensity as the peak at 2.0 ppm, and do not overlap with the peaks of interest. If this causes a problem in the analysis of other compounds, an alternative might be to use fluorinated hexane as the transportation medium, which will not be visible in the 1H NMR spectra.
An interesting feature is the presence of the α- and β-stereoisomers. As can be seen in the figure, the ratios between these two are different in the SFC–NMR spectra compared to the reference spectra. This may have different causes. First, the SFC–NMR spectra contain more methanol than water, which might influence the transition from one stereoisomer into the other. Second, due to the SFC separation, only one part of the peak is selected, which possibly contains only one isomer. However, it would still be expected that the selected isomer transfers into the other stereoisomer, but the timescale of the transition might be longer than the time in which the SFC–NMR analysis took place. This is, however, less likely, due to the long timescale of the experiment.
An important point of interest is to see if this method will be a viable analysis technique for metabolites in urine. For ManNAc, the SFC–NMR analysis started with a 4 μL injection of a sample with a high concentration (0.78 M). The chromatogram of this separation is shown in the ESI (S1).† Although the concentration is high, the amount injected on the column is then only 3.1 μmol. Of this 3.1 μmol, only a part is selected for transportation towards the NMR spectrometer. Since the UV detector was overloaded due to the high concentration that was injected, it is not possible to determine which fraction of the peak was injected. It is, however, possible to determine this from the NMR spectra, since these are quantitative. By comparing the integral of the reference spectrum with a known concentration (0.35 M) to the integral of the SFC–NMR spectrum and correcting for the number of scans, it can be concluded that the ManNAc concentration in the SFC–NMR spectrum is 15 mM. The signal-to-noise (S/N) ratio of the peak at 5.1 ppm is higher than necessary, therefore a lower concentration can be handled. If the minimum S/N-ratio in the spectrum is set to 3, the concentration could be lowered from 15 mM to 1.7 mM. As determined in the introduction, the concentration of ManNAc in the urine of patients suffering from NANS-mediated disease is estimated to be in the range of 0.3–0.7 mM. This is a factor 2.5–5.5 lower than we can currently detect. One of the solutions to this problem is using a larger volume stripline detector. The volume in which our sample is dissolved is 40 μL, however our current NMR probe has an active detection volume of 150 nL. A significant fraction of the sample is therefore not detected. By scaling up the stripline probe by a factor of 10 to 1.5 μL detection volume, a gain of a factor 5 in concentration can be achieved,14 or a decrease of a factor 25 in experimental time at equal concentration. This will be enough to be able to detect ManNAc in the urine of patients suffering from NANS-mediated disease. Such a large-volume stripline probe is under development.
The calculations above were made assuming that there is no sample loss in the system. The concentration in the SFC–NMR spectrum of ManNAc was determined to be 15 mM. This corresponds to 0.61 μmol in 40 μL of solvent (10 μL D2O and 30 μL MeOH). The entire peak in the SFC chromatogram consisted of 3.1 μmol, of which only a fraction was sent to the NMR spectrometer. If we assume that half of the peak is selected, meaning 1.5 μmol is sent to the NMR detector, this indicates that more than half of the sample gets lost during transport. Probably this sticks to the tubing walls, since methanol binds strongly to the PEEK tubing which couples the SFC machine to the spectrometer. Changing the tubing material to a less hydrophilic material may help in getting a higher concentration to the NMR probe and reducing sample losses during transportation.
4 Conclusions
Our SFC–NMR setup was successfully adapted to analyse polar samples in complex matrices. By controlled expansion of the scCO2, the sample coming from the chromatograph can be concentrated in-line. Concentrating the sample in-line after chromatography is needed to detect the sample with NMR spectroscopy.SFC separation of N-acetylhexosamine stereoisomers from synthetic urine was optimised, using a 2-PIC column (Waters) as the stationary phase and 30% MeOH/CO2 as the mobile phase. After this, in-line SFC–NMR analysis of two N-acetylhexosamine isomers, namely N-acetyl-mannosamine (ManNAc) and N-acetyl-glucosamine (GlcNAc), was demonstrated. ManNAc is a metabolite that is present at elevated concentrations in patients suffering from NANS-mediated disease. With the SFC–NMR setup it is possible to distinguish between these polar stereoisomers. This is not possible with the current standard analysis techniques, which are based on mass detection.
The concentrations that are needed in the SFC–NMR setup are currently too high to be able to detect ManNAc in patient samples (1.7 mM vs. 0.7 mM) in a reasonable time. However, several adaptations to the current setup will make this possible. First, the detection volume of the stripline NMR probe needs to be increased by a factor 10 to better match the sample volumes coming from the chromatograph. It will then be possible to measure concentrations around 0.3 mM. Second, the tubing connecting the SFC to the NMR probe needs to be replaced with a less hydrophilic material, to decrease sample losses during transportation of the sample towards the NMR spectrometer. With these adaptations it will be possible to detect ManNAc in urine samples from patients suffering from NANS-mediated disease with in-line SFC–NMR. Due to the broad separation range of SFC and its coupling to NMR spectroscopy, it is expected that other polar and non-polar metabolites present at these, or higher, concentrations in biofluids can also be analysed with the same setup. By tuning the polarity of the mobile phase, SFC–NMR can be used to analyse a broad range of hydrophilic and lipophilic metabolites.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research received funding from The Netherlands Organization for Scientific Research (NWO) in the framework of Technology Area COAST (053.21.115). The authors thank Jan van Bentum, Julija Romanuka (Shell), Noud van den Borg (Waters), Peter Schoenmakers (UvA) and Ulrich Braumann (Bruker) for discussions and support, and Hans Janssen (RU) for technical support. The authors would also like to acknowledge Jonathan Martens (RU) for providing the N-acetylhexosamine samples and Arno Hoefnagels for optimising the SFC separations.Notes and references
- G. J. Patti, O. Yanes and G. Siuzdak, Nat. Rev. Mol. Cell Biol., 2012, 13, 263 CrossRef CAS PubMed.
- S. Bouatra, F. Aziat, R. Mandal, A. C. Guo, M. R. Wilson, C. Knox, T. C. Bjorndahl, R. Krishnamurthy, F. Saleem and P. Liu, et al. , PLoS One, 2013, 8, e73076 CrossRef CAS PubMed.
- F. H. M. van Zelst, S. G. J. van Meerten, P. J. M. van Bentum and A. P. M. Kentgens, Anal. Chem., 2018, 90, 10457–10464 CrossRef CAS PubMed.
- D. L. Olson, T. L. Peck, A. G. Webb, R. L. Magin and J. V. Sweedler, Science, 1995, 270, 1967–1970 CrossRef CAS.
- J. Dechow, A. Forchel, T. Lanz and A. Haase, Microelectron. Eng., 2000, 53, 517–519 CrossRef CAS.
- C. Massin, F. Vincent, A. Homsy, K. Ehrmann, G. Boero, P.-A. Besse, A. Daridon, E. Verpoorte, N. F. De Rooij and R. S. Popovic, J. Magn. Reson., 2003, 164, 242–255 CrossRef CAS PubMed.
- A. P. M. Kentgens, J. Bart, P. J. M. Van Bentum, A. Brinkmann, E. R. H. Van Eck, J. G. E. Gardeniers, J. W. G. Janssen, P. Knijn, S. Vasa and M. H. W. Verkuijlen, J. Chem. Phys., 2008, 128, 052202 CrossRef CAS PubMed.
- P. J. M. Van Bentum, J. W. G. Janssen, A. P. M. Kentgens, J. Bart and J. G. E. Gardeniers, J. Magn. Reson., 2007, 189, 104–113 CrossRef CAS PubMed.
- C. D. Van Karnebeek, L. Bonafé, X.-Y. Wen, M. Tarailo-Graovac, S. Balzano, B. Royer-Bertrand, A. Ashikov, L. Garavelli, I. Mammi and L. Turolla, et al. , Nat. Genet., 2016, 48, 777 CrossRef CAS PubMed.
- J. Martens, G. Berden, R. E. Outersterp, L. A. Kluijtmans, U. F. Engelke, C. D. Karnebeek, R. A. Wevers and J. Oomens, Sci. Rep., 2017, 7, 3363 CrossRef PubMed.
- World Health Organization, Biological monitoring of chemical exposure in the workplace: guidelines, World Health Organization, Geneva, 1996 Search PubMed.
- S. G. J. van Meerten, W. M. J. Franssen and A. P. M. Kentgens, J. Magn. Reson., 2019, 301, 56–66, http://https://www.ru.nl/science/magneticresonance/software/ssnake/ Search PubMed.
- D. S. Wishart, Y. D. Feunang, A. Marcu, A. C. Guo, K. Liang, R. Vázquez-Fresno, T. Sajed, D. Johnson, C. Li and N. Karu, et al. , Nucleic Acids Res., 2018, 46, D608–D617 CrossRef CAS PubMed.
- J. Bart, J. W. G. Janssen, P. J. M. Van Bentum, A. P. M. Kentgens and J. G. E. Gardeniers, J. Magn. Reson., 2009, 201, 175–185 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: SFC UV chromatograms of ManNAc and GlcNAC. See DOI: 10.1039/c8fd00237a |
| This journal is © The Royal Society of Chemistry 2019 |