
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
pH dependent binding in de novo hetero bimetallic coiled coils†
Paul
Teare
a,
Caitlin F.
Smith
a,
Samuel J.
Adams
a,
Sellamuthu
Anbu
 a,
Barbara
Ciani
b,
Lars J. C.
Jeuken
c and
Anna F. A.
Peacock
*a
a,
Barbara
Ciani
b,
Lars J. C.
Jeuken
c and
Anna F. A.
Peacock
*a
aSchool of Chemistry, University of Birmingham, Edgbaston, B15 2TT, UK. E-mail: a.f.a.peacock@bham.ac.uk
bCentre for Membrane Interactions and Dynamics, and Krebs Institute, Department of Chemistry, University of Sheffield, Sheffield, S3 7HF, UK
cSchool of Biomedical Sciences and the Astbury Centre for Structural Molecular Biology, University of Leeds, Leeds, LS2 9JT, UK
First published on 5th July 2018
Abstract
Herein the first example of a bimetallic coiled coil featuring a lanthanide binding site is reported, opening opportunities to exploit the attractive NMR and photophysical properties of the lanthanides in multi metallo protein design. In our efforts to fully characterise the system we identified for the first time that lanthanide binding to such sites is pH dependent, with optimal binding at neutral pH, and that the double AsnAsp site is more versatile in this regard than the single Asp site. Our second site featured the structural HgCys3 site, the chemistry of which was essentially unaltered by the presence of the lanthanide site. In fact, both metal binding sites within the hetero bimetallic coiled coil displayed the same properties as their mononuclear single binding site controls, and operated independently of each other. Finally, pH can be used as an external trigger to control the binding of Hg(II) and Tb(III) to the two distinct sites within this coiled coil, and offers the opportunity to “activate” metal binding sites within complex multi metallo and multi-functional designs.
Introduction
The de novo design (from “scratch”) of metallocoiled coils has been a productive area of research and has led to a large number of mononuclear metal ion sites having been engineered within relatively simple, synthetically accessible, coiled coil architectures. These include sites inspired by those found in nature, often in an effort to gain insight into the native site, and ultimately to recreate its function. Notable examples include mononuclear sites mimicking the active site of carbonic anhydrase;1 dinuclear sites, including dioxygen-activating di-iron sites;2 multinuclear clusters, such as the cubane-like [4Fe–4S] cluster;3 and binding of haem cofactors.4 But similarly, various mononuclear sites have been introduced, which though not found in nature, have appealing chemical properties for exploitation in protein design. These include, but are not limited to, lanthanide binding sites, where the attractive NMR and photophysical properties, such as their narrow emission lines, large Stokes shifts and long-lived emission lifetimes, of the lanthanide ion can be exploited.5–9 Lanthanides are therefore widely used as NMR shift probes and in luminescence resonance energy transfer, representing tools with which one can gain important insight into biomolecular structure, interactions and function.Though multinuclear sites have also been successfully engineered within coiled coils,2,3,10 the inclusion of two distinct mononuclear sites has great appeal. As the distinct chemical properties of these sites can be harnessed in a single system, either where they operate independently in isolation from one another, or in contrast, are sensitive to, and dependent on one another. Not surprisingly, Nature contains numerous examples of proteins with multiple distinct mononuclear metal ion sites.
Two distinct thiol binding sites have been introduced within three stranded coiled coil scaffolds, and the binding of [4Fe–4S] clusters3 and Cd(II)11 to these, has been reported. For the latter, various homo bimetallic coiled coils in which both sites feature either a trigonal CdCys3 or a tetrahedral CdCys3(OH/OH2) site have been prepared.12 Subsequent pseudo-hetero bimetallic coiled coils have been realized, which feature two Cd(II) bound to a single coiled coil, but one as trigonal CdCys3, and the second as tetrahedral CdCys3(OH2).13,14
Truly hetero bimetallic coiled coils, with different metal ions binding to two distinct sites, should be easier to design, as they exploit real intrinsic differences in metal ion coordination chemistry, such as hard–soft-acid–base (HSAB) theory, to achieve selectivity and control. The first example of these features a Ru(bipyridyl)3 site at one end of a coiled coil, which helps assemble and preorganise the second CuHis3 binding site located at the opposite end.15 Similarly, it has been reported that binding of Cu(II) and Ni(II) to Hisx sites, involves the first metal inducing folding and promoting the formation of the second metal ion binding site.16 A more recent example features a structural HgCys3 site17–19 site and a catalytic ZnHis3(OH/OH2) site, inspired by the metalloenzyme carbonic anhydrase, within a three stranded coiled coil.1,20
The advantage of including two distinct mononuclear metal ion sites with different properties within a single assembly is clear, as it allows multi functionality and complexity, relating to interdependence, to be introduced. And so it remains to expand the number and type of such sites that can be combined in designs such as this, if complex multi-metallo, multi-functional, systems are to be realised. Herein we explore the inclusion of a lanthanide binding site, of interest in biotechnology and protein design due to their appealing NMR and luminescent properties, into a hetero bimetallic coiled coil, alongside the previously well characterized structural, but pH dependent, HgCys3 site,17 see Fig. 1. Not only do we report the first example of a heterobimetallic coiled coil featuring a lanthanide binding site, we also report on the pH dependence of these lanthanide sites, and illustrate how pH can be used as to control and select for Hg(II) and Tb(III) binding, respectively.
 | ||
| Fig. 1 Cartoon representation showing our designed hetero bimetallic coiled coil, featuring LnAsp3 and HgCys3 sites towards the N- and C-termini, respectively. Shown are the main chain atoms represented as helical ribbons (green), the Asp, Trp and Cys side chains in stick form (oxygen in red, nitrogen in blue and sulfur in orange), and the metal ions as spheres (lanthanide in grey and mercury in cyan). | ||
Results and discussion
Design of a hetero bimetallic coiled coil
Given that the introduction of a metal binding site is a destabilizing modification to the coiled coil structure, the inclusion of two has to be carefully considered. Previously we reported a small family of mononuclear lanthanide binding sites within three stranded coiled coils, and found the location of the binding site to have a significant impact on coiled coil folding and stability. Terminal binding sites were significantly better tolerated than core sites, and the N-terminal site was better tolerated than the more buried C-terminal site. Furthermore, whereas the majority of binding sites required the substitution of two adjacent hydrophobic layers to accommodate the double AsnAsp binding site, when the binding site was introduced towards the N-terminus, the Asn was found to be non-essential. As such the least destabilizing site, which retains lanthanide binding, features a single Asp layer towards the N-terminus, CS1-1 and see Table 1.9 Our hetero bimetallic coiled coil therefore features this lanthanide binding site, with an adjacent Trp residue which reports on Tb(III) binding, and a second distinct metal binding site.| Peptide | Sequence (abcdefg) |
|---|---|
| CS1-1 | Ac-G IAAIE![[W with combining low line]](https://www.rsc.org/images/entities/b_char_0057_0332.gif) K K ![[D with combining low line]](https://www.rsc.org/images/entities/b_char_0044_0332.gif) AAIEQK IAAIEQK IAAIEQK IAAIEQK G-NH2 AAIEQK IAAIEQK IAAIEQK IAAIEQK G-NH2 |
| CS2-1,4 | Ac-G IAAIEK AAIEQK IAAIEQK IAAIEQK CAAIEQK G-NH2 |
| MB1-2 | Ac-G IAAIEQK IAA![[N with combining low line]](https://www.rsc.org/images/entities/b_char_004e_0332.gif) EK AAIEQK IAAIEQK IAAIEQK G-NH2 EK AAIEQK IAAIEQK IAAIEQK G-NH2 |
In order to achieve a high degree of metal selectivity for the two sites, we elected to generate a soft Cys3 metal binding site, to contrast with the hard lanthanide binding site generated by the Asp3 layer. The replacement of a core Ile with a Cys should be less destabilizing than the analogous mutation to Asp, but in an effort to minimize its impact, the Cys has again been introduced in a terminal rather than a core heptad, CS2-1,4 (and see Table 1, Fig. S1 and S2†). Though the binding of various soft metal ions has been reported to such a site,21,22 we selected the well-studied and previously reported, structural HgCys site located in an “a” (the first position within a “abcdefg” heptad) rather than the “d” position (also located within the core of a three stranded coiled coil).
Substitution of Ile in position 30 with a Cys, was anticipated to be destabilizing, with previous reports18,19,23 showing this to be the case. Circular dichroism (CD) spectroscopy was therefore used to establish the degree of peptide folding. Though spectra recorded for 30 μM monomer solutions of both CS1-1 and CS2-1,4 in 5 mM HEPES buffer pH 7.0 displayed the characteristic double minima around 210 and 223 nm, consistent with an α-helix, their intensities differed. CS1-1 was notably more folded (Θ222 = −31![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 320 deg dmol−1 cm2 res−1, ca. 81% folded) than CS2-1,4 (Θ222 = −23933 deg dmol−1 cm2 res−1, ca. 62% folded), see Fig. 2.
320 deg dmol−1 cm2 res−1, ca. 81% folded) than CS2-1,4 (Θ222 = −23933 deg dmol−1 cm2 res−1, ca. 62% folded), see Fig. 2.
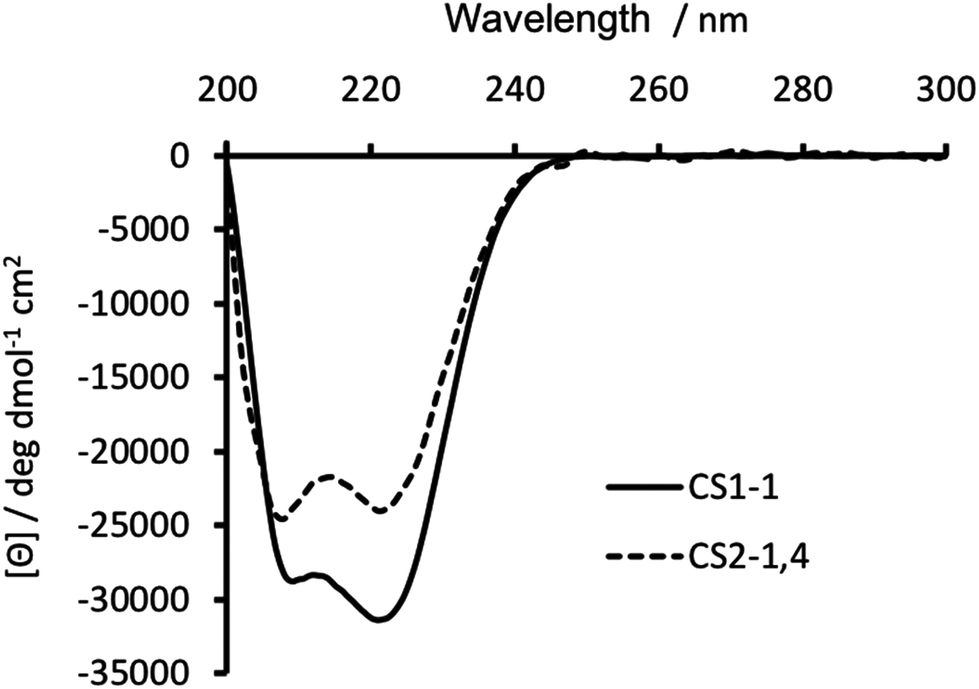 | ||
| Fig. 2 CD spectra of 30 μM CS1-1 (solid line) and CS2-1,4 (dashed line) peptide monomer, recorded at 293 K in 5 mM HEPES buffer pH 7.0. | ||
Retaining Tb(III) binding to Asp
Verification that Tb(III) binding is preserved in CS2-1,4, can be best obtained from luminescence spectroscopy, see Fig. 3A. Tb(III) emission is known to be sensitized on binding to the CS1-1 site, due to an adjacent Trp residue which is retained in the CS2-1,4 design. This sensitized Tb(III) emission is consistent with peptide binding, but more significantly, it is consistent with binding at the designed Asp3 lanthanide binding site. This is because Trp sensitized Tb(III) emission is highly distant dependent, and therefore rules out the possibility of a binding site further away, and certainly rules out the Cys location (Trp – LnAsp3 site ca. 12 Å vs. Trp – Cys3 site ca. 32 Å).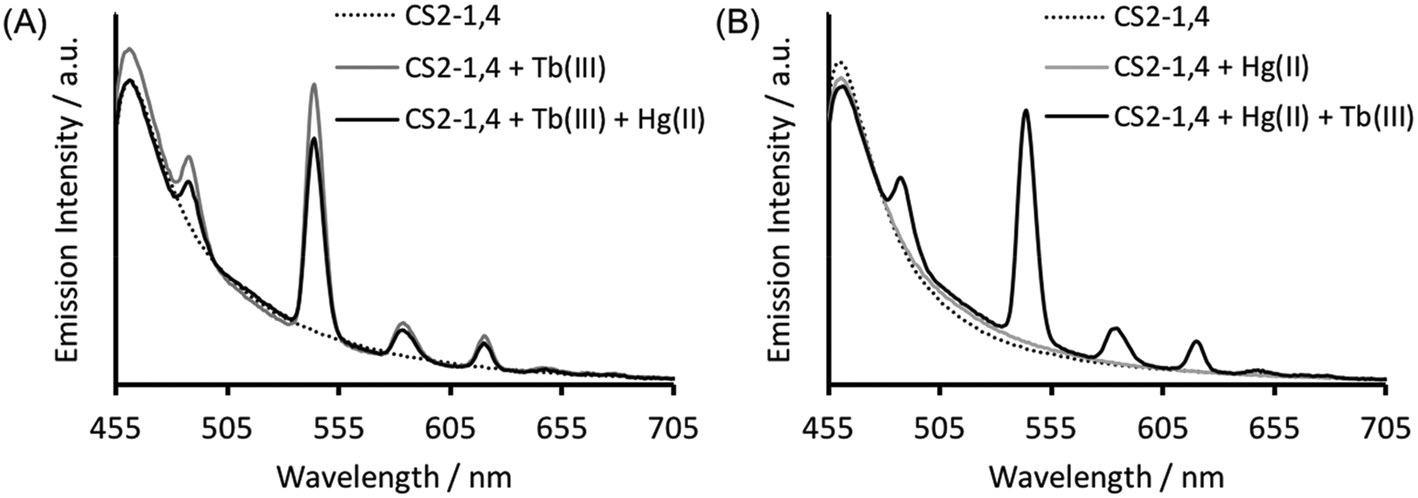 | ||
| Fig. 3 Steady state luminescence spectra of 30 μM CS2-1,4 monomer (dashed line) in the presence of 10 μM TbCl3 (grey line panel A), 10 μM HgCl2 (grey line panel B) or both 10 μM TbCl3 and 10 μM HgCl2 (black line) in 5 mM HEPES buffer pH 6.8 on order of addition of (A) TbCl3 or (B) HgCl2, first. λexc = 280 nm. | ||
Retaining Hg(II) binding to Cys
Similarly, in order to verify that Hg(II) binding to the Cys site was retained, ultraviolet-visible (UV-vis) absorption spectra were recorded of solutions containing 30 μM CS2-1,4 peptide monomer in 10 mM HEPES buffer. On addition of 10 μM HgCl2 at pH 7.0 we see a fairly featureless spectrum (see Fig. 4), but which is consistent with Hg(II) binding as HgCys2 to Cys located in “a” sites at pH 7.0,17 as would be the case here. However, as the pH is raised to 7.5 and ultimately 8.6, we observe the appearance of the characteristic signature for trigonal HgS3.17 This pH dependence is consistent with the previously reported apparent pKa of 7.6, associated with coordination of trigonal HgS3 to an “a” site.17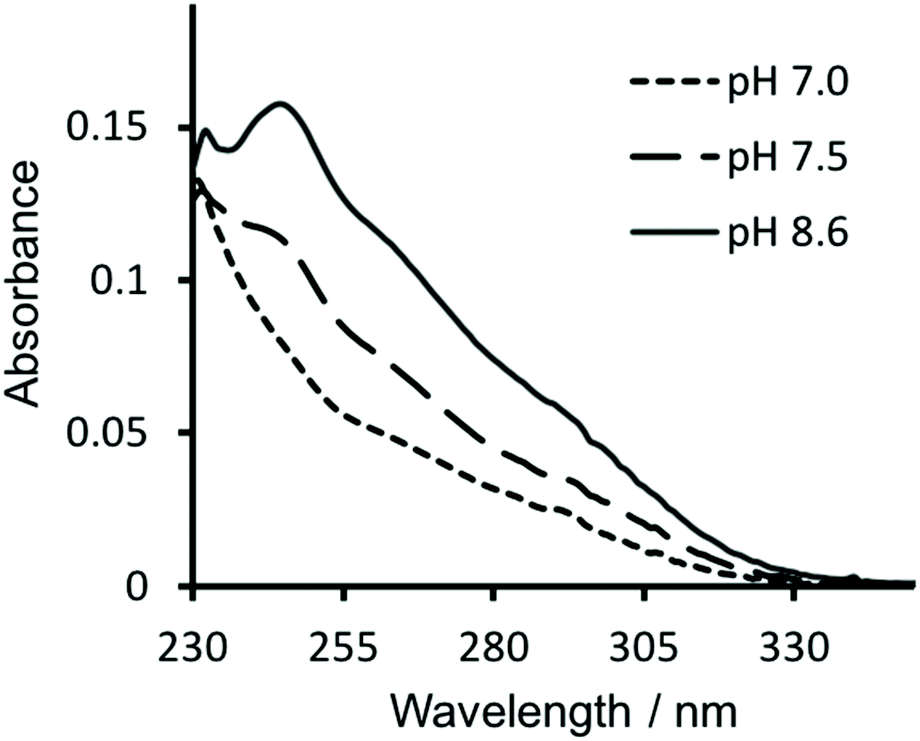 | ||
| Fig. 4 UV-visible difference absorption spectra of 30 μM CS2-1,4 monomer upon the addition of 10 μM HgCl2, in 10 mM HEPES buffer at pH 7.0 (––––), 7.5 (−−−−) and 8.6 (solid line). | ||
Hg(II) binding to Cys residues within the “a” site of a three stranded coiled coil has been extensively studied and found to be dependent on both the pH and the ratio of peptide monomer to Hg(II).17 Consistent with these reports, the CS2-1,4 peptide, at pH 8.6 where Hg(II) is able to bind as trigonal HgS3 to a Cys in an “a” site, does so up to the addition of a ca. 1:3 ratio of Hg(II):CS2-1,4 monomer. On addition of further Hg(II) we observe the disappearance of the characteristic signal for HgCys3, and instead observe the featureless spectrum previously assigned to HgCys2, see Fig. S3,† again consistent with the literature.17
pH dependence of Tb(III) binding in the presence of Hg(II)
The Tb(III) and Hg(II) binding data is consistent with selective binding to their respective Asp3 and Cys3 binding sites within our two binding site peptide, CS2-1,4, and with similar chemistries to when they bind to these sites in isolation. However, with Tb(III) binding only studied at pH 7.0, but with Hg(II) binding being pH dependent with HgCys3 forming above pH 8.6, it remained to be seen if Tb(III) could bind in the presence of Hg(II), and under the more basic conditions required for HgCys3 formation.Luminescence studies of 30 μM CS2-1,4 peptide monomer in the presence of both 10 μM TbCl3 and 10 μM HgCl2 at pH 6.8, still show enhanced emission, comparable to that recorded in the absence of Hg(II), consistent with similar Tb(III) binding when the Hg(II) is likely bound as HgCys2 (see Fig. 3).24 However, on raising the pH, the signal for the Tb(III) emission is significantly reduced, and lost entirely above pH 8, see Fig. 5. This could be due to reduced Tb(III) binding under these more basic conditions, due to competing formation of lanthanide hydroxide species. It could be a result of Hg(II) binding and distortion of the Asp3 site on formation of HgCys3. Alternatively, as the Tb(III) CS1-1 type site is highly hydrated,9 the lack of luminescence may be due to deprotonation of coordinated water and enhanced quenching by coordinated hydroxide.
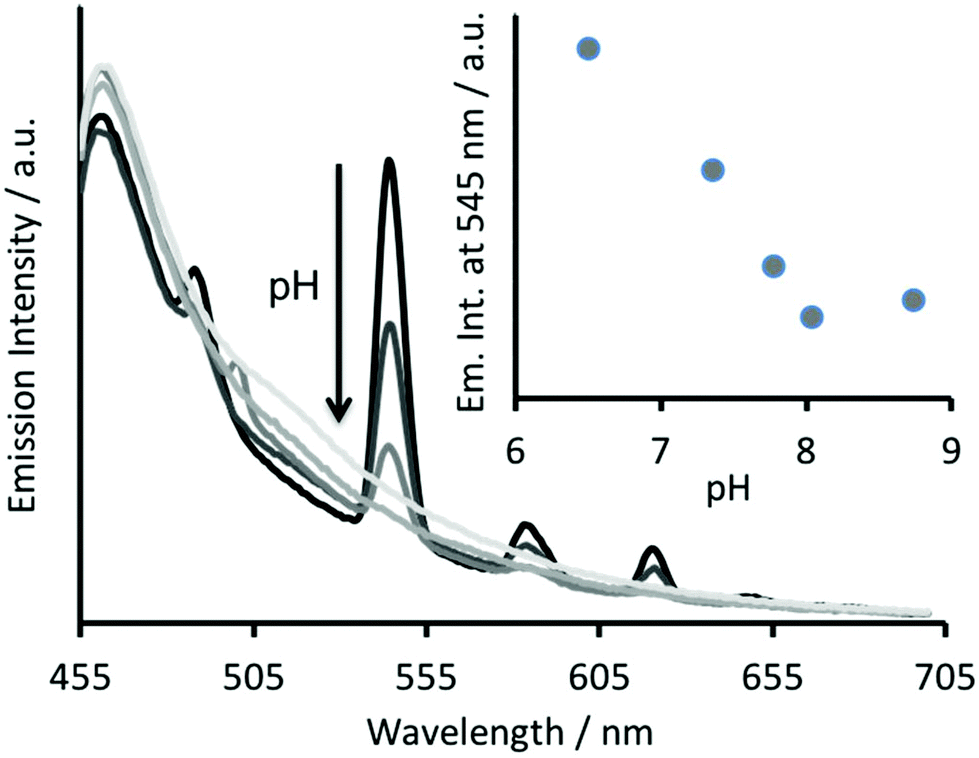 | ||
| Fig. 5 Steady state luminescence spectra of 30 μM CS2-1,4 monomer in the presence of 10 μM TbCl3 and 10 μM HgCl2, at increasing pH. λexc = 280 nm. Inset showing plot of emission intensity at 545 nm as a function of pH. The non-zero emission intensity above pH 8 is due to the contribution from the Trp residue. | ||
In order to determine if the lack of Tb(III) emission relates to Hg(II) binding at the Cys site, luminescence spectra were recorded of 30 μM CS2-1,4 monomer at pH 8.6 on addition of 10 μM TbCl3, but in the absence of Hg(II). However, this again showed a lack of emission at 545 nm, see Fig. S4.† Similar spectra were acquired in the presence of both 10 μM TbCl3 and 10 μM HgCl3, and the order of addition altered, so as to rule out the formation of a kinetic product, but again no notable Tb(III) emission was detected, see Fig. S4.† Therefore, we conclude that the lack of sensitized Tb(III) emission is not due to binding of Hg(II) at the Cys site, but rather is a feature of the TbAsp3 site at basic pH.
pH dependence of Tb(III) binding to single site peptides
In an effort to study the pH dependence of Tb(III) binding to the Asp3 site, we initially performed a pH titration on the control CS1-1 peptide, which lacks the Cys3 binding site, and monitored this by CD and luminescence spectroscopy. We also studied the pH dependence of Tb(III) binding to MB1-2, a single site coiled coil, which in contrast to the highly hydrated Tb(III) site generated on binding CS1-1, offers a coordinatively saturated Tb(III) site. This would also allow us to address whether a different Tb(III) binding site would be more attractive for use in designing hetero bimetallic coiled coils featuring a lanthanide binding site, as we have previously reported that the chemistry of Tb(III) sites within these coiled coils, can be very different depending on ligand set presented and the linear location along the coiled coil.9The CD titration of CS1-1 in the presence of Tb(III) revealed very little change in folding as a function of pH, see Fig. S5A.† However, as the change in folding associated with Tb(III) binding to CS1-1 is only modest,9 this is not sufficient to address Tb(III) de-complexation.
Performing the same pH CD titration for a buried Tb(III) binding site, as featured in MB1-2, should show a notable change in folding on de-complexation.8,9 Though the CD pH titration shows a steady decrease in MB1-2 folding as a function of pH, see Fig. S5B,† it is not consistent with full Tb(III) de-complexation and complete conversion to the apo form, which is only 21 ± 3% folded at pH 7.9 These findings suggest that though Tb(III) de-complexation may be occurring with increase in pH, it may not be complete in these single binding site control peptides. Therefore, Tb(III) may remain bound to some extent in CS2-1,4 at pH 8.6.
The analogous luminescence pH titration of Tb(CS1-1)3 revealed a loss in luminescence signal with increase in pH (see Fig. 6A), consistent with our previous observations with CS2-1,4. Again these findings are consistent with the Cys site, and its metallated HgCys3 derivative, not influencing the behaviour of the TbAsp3 site, and that the loss of luminescence signal relates to the chemistry of the TbAsp3 site in isolation. Intriguingly, the pH titration also revealed for the first time that Tb(III) luminescence associated with binding to the TbAsp3 site, is not observed under acidic conditions, and is in fact optimal across a very narrow pH range between pH 6 and 7, the very pH conditions under which all such lanthanide binding sites have been studied to date. Lack of binding under acidic conditions may result from protonation of the Asp residues, making them unavailable for Tb(III) complexation, and for which the pKa may be altered in the different peptide sequences due to the presence and absence of, for example, an adjacent Asn.
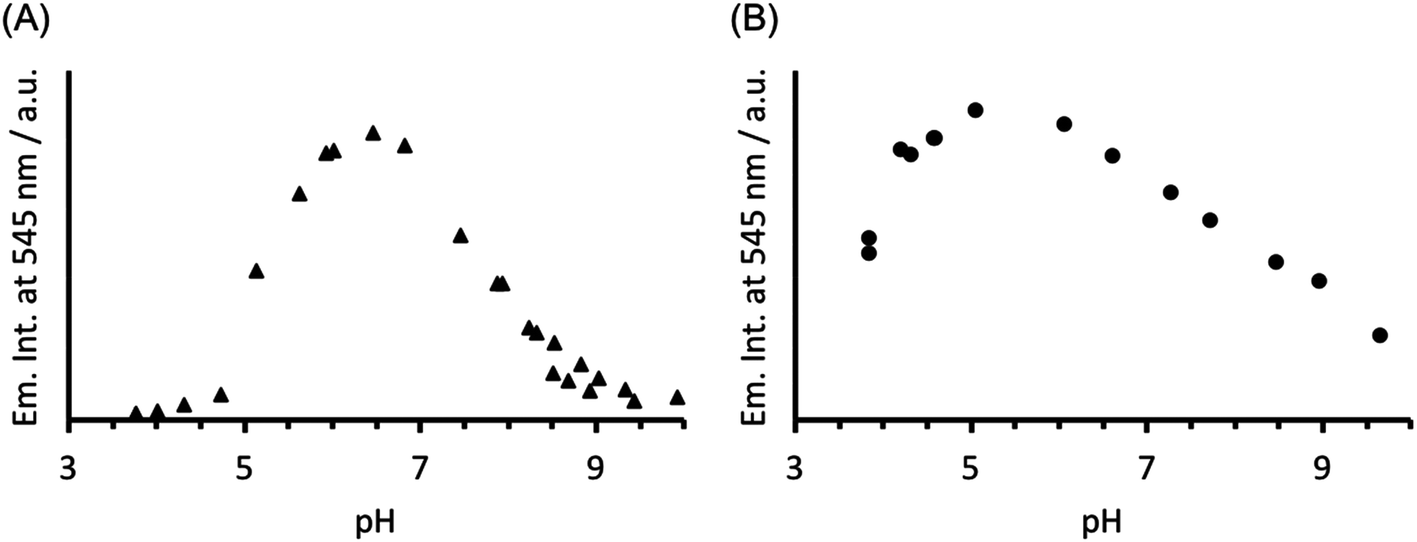 | ||
| Fig. 6 Plot of the change in emission intensity at 545 nm as a function of pH, for a solution containing 30 μM (A) CS1-1 and (B) MB1-2 monomer in the presence of 10 μM TbCl3. λexc = 280 nm. | ||
In order to establish if the lack of Tb(III) luminescence signal at basic pH, is in fact a result of enhanced quenching due to deprotonation of water bound to the TbAsp3 site, an analogous luminescence pH titration was performed with Tb(MB1-2)3, in which the Tb(III) is coordinatively saturated and is not bound directly to any water molecules. A similar profile is observed, though with a much larger pH range for optimal Tb(III) binding (ca. pH 4–7), with a steady decrease in luminescence on increasing the pH (see Fig. 6B). However, as was the case in the analogous CD titrations, the decrease is less extreme than for CS1-1 and is only consistent with partial Tb(III) de-complexation at pH 8.6.
pH as an external trigger to control metal binding to a hetero bimetallic coiled coil
Taken together, the CD and luminescence pH titrations of CS1-1, MB1-2 and CS2-1,4, indicate that an increase in pH leads to Tb(III) de-complexation, and that not much Tb(III) remains bound to CS2-1,4 at pH 8.6. Future hetero bimetallic designs which were to feature a lanthanide binding site, might therefore benefit from the use of the double AsnAsp binding site, as featured in MB1-2, as it is more versatile with respect to pH. However, at neutral pH where Tb(III) is well bound, the Hg(II) can still bind to the Cys site in CS2-1,4 as HgCys2, but does require more basic conditions to bind fully as HgCys3. Therefore, in systems such as CS2-1,4, it is possible to use pH as an external trigger to control, in the case of the Hg(II) site, in what form it binds, and which metal site provides a spectroscopically measurable output. This offers a way to activate these outputs in more complex multi functional assemblies.Conclusions
Herein, and to date, two distinct mononuclear sites have been successfully engineered into a single coiled coil, by exploiting clear differences in metal binding preferences, such as HSAB theory. The ability to do so offers protein designers the opportunity to develop systems with clear distinct functional sites, which may operate in isolation or in a dependent fashion. Herein we report the first inclusion of a lanthanide binding site alongside a second distinct metal binding site, and illustrate that pH can be used as an external trigger to induce different binding at the two distinct metal binding sites. Through our efforts, we have for the first time reported on the pH dependence of these lanthanide binding sites, and describe the different chemistries associated with hydrated and coordinatively saturated lanthanide binding sites.Though these findings further the metallo protein design field by contributing distinct and useful sites from which metallo protein designers can select from, a real challenge remains to selectively bind, and distinguish between, two very similar metal ions but with different desirable properties, for which differences in HSAB or coordination chemistry, cannot be so easily exploited. Efforts in our laboratory are on-going to achieve this ultimate goal.
Materials and methods
TbCl3·6H2O and HgCl2 were purchased from Sigma Aldrich. Urea (≥99% purity), xylenol orange indicator, glacial acetic acid and 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) were all purchased from Fisher Scientific Ltd. All Fmoc protected amino acids, dimethylformamide (DMF) and N,N,N′,N′-tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate (HBTU) were purchased from Pepceuticals Ltd, Leicester. The rink amide MBHA resin was obtained from AGTC Bioproducts Ltd, along with diisopropylethylamine (DIPEA) and trifluoroacetic acid (TFA). Gd(III) and Tb(III) standards were purchased from SCP Science, Quebec.Peptide synthesis and purification
Peptides were synthesized on a CEM Liberty Blue automated peptide synthesizer on rink amide MBHA resin (0.25 mmol scale, 0.65 mmol g−1), using standard Fmoc-amino acid solid-phase peptide synthesis protocols.25 Peptides were purified as previously reported26 and characterised by electrospray ionisation mass spectrometry recorded on a Waters Ltd. Synapt-G2-S, by direct injection with a mobile phase of 50% water: 50% acetonitrile and 0.05% formic acid, at a flow rate of 0.1 mL min−1. Data was processed with MassLynx. In the synthesis of CS2-1,4 the Fmoc-L-Cys(Trt)-OH was coupled twice and the temperature maintained below 50 °C.Sample preparation
Stock solutions of TbCl3 and HgCl2 (1 mM) were prepared in MilliQ water, and the Tb(III) concentrations determined spectroscopically using xylenol orange indicator and Tb(III) standard solutions as reported by Fedeli et al.27 Peptide concentrations were determined based on the tryptophan absorbance at 280 nm (ε280 = 5690 M−1 cm−1) in 7 M aqueous solutions of urea, and in the case of CS2-1,4, verified by determining the available thiol concentration via the published Ellman's assay,28 with experimental concentrations found to be within 10%.Circular dichroism (CD) spectroscopy
CD spectra for solutions containing 30 μM monomer in the absence or presence of 10 μM metal (TbCl3 or HgCl2), in 5 mM HEPES buffer pH 7 or 8.6, were recorded in a 1 mm pathlength quartz cuvettes on a Jasco J-810 spectropolarimeter. The observed ellipticity in millidegrees was converted into molar ellipticity, (Θ), and is reported in units of deg dmol−1 cm2. The percentage folding, %Folded, was calculated (eqn (1)) based on the theoretical maximum ellipticity, [Θ]max, determined from (−42500 × (1 − (3/n))), where n is the number of residues in the sequence, and [Θ]coil is the ellipticity of a random coil, based on reports by Scholtz et al.29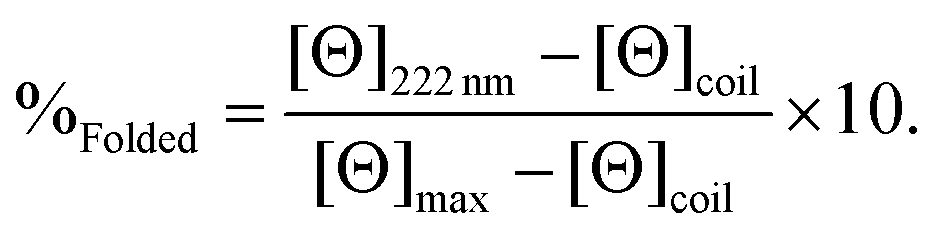 | (1) |
UV-visible spectroscopy
UV-visible spectra for solutions containing 30 μM CS2-1,4 monomer, 10 μM HgCl2 in 10 mM HEPES buffer pH 7.0, 7.5 or 8.6 were recorded in a 1 cm pathlength quartz cuvettes on a Cary50 spectrometer.Luminescence
Emission spectra were recorded in a 1 cm pathlength quartz cuvette using an Edinburgh Instruments Fluorescence FLS920 system with a 450 W Xenon arc lamp and a Hamamatsu R928 photomultiplier tube. The emission monochromator was fitted with two interchangeable gratings blazed at 500 nm and 1200 nm and the data was collected using F900 spectrometer analysis software. Aliquots of a 1 mM stock solution of TbCl3 or HgCl2 were titrated into 30 μM peptide monomer solutions in 5 mM HEPES buffer pH 7 or 8.6, and the emission profile recorded after 20 min equilibration. Solutions were excited at 280 nm and the emission was scanned from 475–750 nm using a 455 nm long pass filter. Spectra were corrected for instrument response (grating/PMT) in all cases. The integration of the Tb(III) emission at 545 nm was measured between 535–555 nm.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We thank the EPSRC Directed Assembly Network for S. J. A. (EP/K014382/1), Horizon 2020-Marie-Sklodowska Curie Fellowship for S. A. (H2020-MSCA-IF-2014-658843) and the University of Birmingham, School of Chemistry and the Defence and Science Technology Laboratory (DSTL) for a Ph.D. studentship for P. T., and for support of this research. We thank Matthew Berwick, Gemma Bullen, Louise Slope, Chi Tsang and Jonathon Snelling, for technical assistance. Some equipment used was obtained through Birmingham Science City: Innovative Uses for Advanced Materials in the Modern World (West Midlands Centre for Advanced Materials Project 2) and Birmingham Science City Translational Medicine: Experimental Medicine Network of Excellence project, with support from Advantage West Midlands (AWM) and part funded by European Regional Development fund (ERDF).References
- M. L. Zastrow, A. F. A. Peacock, J. A. Stuckey and V. L. Pecoraro, Nat. Chem., 2012, 4, 118 CrossRef PubMed.
- A. J. Reig, M. M. Pires, R. A. Snyder, Y. Wu, H. Jo, D. W. Kulp, S. E. Butch, J. R. Calhoun, T. Szyperski, T. G. Szyperski, E. I. Solomon and W. F. DeGrado, Nat. Chem., 2012, 4, 900 CrossRef PubMed.
- A. Roy, D. J. Sommer, R. A. Schmitz, C. L. Brown, D. Gust, A. Astashkin and G. Ghirlanda, J. Am. Chem. Soc., 2014, 136, 17343 CrossRef PubMed.
- R. L. Koder, J. L. R. Anderson, L. A. Solomon, K. S. Reddy, C. C. Moser and P. L. Dutton, Nature, 2009, 458, 305 CrossRef PubMed.
- W. D. Kohn, C. M. Kay and R. S. Hodges, J. Pept. Res., 1998, 51, 9 CrossRef PubMed.
- W. D. Kohn, C. M. Kay, B. D. Sykes and R. S. Hodges, J. Am. Chem. Soc., 1998, 120, 1124 CrossRef.
- A. Kashiwada, K. Ishida and K. Matsuda, Bull. Chem. Soc. Jpn., 2007, 80, 2203 CrossRef.
- M. R. Berwick, D. J. Lewis, Z. Pikramenou, A. W. Jones, H. J. Cooper, J. Wilkie, M. M. Britton and A. F. A. Peacock, J. Am. Chem. Soc., 2014, 136, 1166 CrossRef PubMed.
- M. R. Berwick, L. N. Slope, C. Smith, S. M. King, S. L. Newton, R. Gillis, G. Adams, A. Rowe, S. Harding, M. M. Britton and A. F. A. Peacock, Chem. Sci., 2016, 7, 2207 RSC.
- O. A. Kharenko and M. Y. Ogawa, J. Inorg. Biochem., 2004, 98, 1971 CrossRef PubMed.
- M. Matzapetakis and V. L. Pecoraro, J. Am. Chem. Soc., 2005, 127, 18229 CrossRef PubMed.
- O. Iranzo, S. Chakraborty, L. Hemmingsen and V. L. Pecoraro, J. Am. Chem. Soc., 2011, 133, 239 CrossRef PubMed.
- O. Iranzo, C. Cabello and V. L. Pecoraro, Angew. Chem., Int. Ed., 2007, 46, 6688 CrossRef PubMed.
- A. F. A. Peacock, L. Hemmingsen and V. L. Pecoraro, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 16566 CrossRef PubMed.
- M. R. Ghadiri and M. A. Case, Angew. Chem., Int. Ed., 1993, 32, 1594 CrossRef.
- T. Tanaka, T. Mizuno, S. Fukui, H. Hiroaki, J. Oku, K. Kanaori, K. Tajima and M. Shirakawa, J. Am. Chem. Soc., 2004, 126, 14023 CrossRef PubMed.
- V. L. Pecoraro, A. F. A. Peacock, O. Iranzo and M. Luczkowski, Understanding the Biological Chemistry of Hg(II) Using a De NovoDesign Strategy, in Bioinorgani Chemistry Cellular Systems and Synthetic Models, ACS Symposium Series 1012, ed. E. Long and M. J. Baldwin, American Chemical Society, Washington, 2009, ch. 12, pp. 183–197 Search PubMed.
- G. R. Dieckmann, D. K. McRorie, D. L. Tierney, L. M. Utschig, C. P. Singer, T. V. O'Halloran, J. E. Penner-Hahn, W. F. DeGrado and V. L. Pecoraro, J. Am. Chem. Soc., 1997, 119, 6195 CrossRef.
- G. R. Dieckmann, D. K. McRorie, J. D. Lear, K. A. Sharp, W. F. DeGrado and V. L. Pecoraro, J. Mol. Biol., 1998, 280, 897 CrossRef PubMed.
- M. L. Zastrow and V. L. Pecoraro, J. Am. Chem. Soc., 2013, 135, 5895 CrossRef PubMed.
- D. S. Touw, J. A. Stuckey, C. E. Nordman and V. L. Pecoraro, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 11969 CrossRef PubMed.
- M. Matzapetakis, D. Ghosh, T.-C. Weng, J. E. Penner-Hahn and V. L. Pecoraro, J. Biol. Inorg. Chem., 2006, 11, 876 CrossRef PubMed.
- N. E. Zhou, C. M. Kay and R. S. Hodges, Biochemistry, 1993, 32, 3178 CrossRef PubMed.
- The small reduction following Hg(II) addition to Tb(CS2-1,4)3 may be due to small structural changes to the TbAsp3(OH2)3 site, or the emission quenching effect of the Hg(II).
- W. C. Chan and P. D. White, Fmoc Solid Phase Peptide Synthesis: A Practical Approach, Oxford University, New York, 1st edn, 2000 Search PubMed.
- A. F. A. Peacock, G. A. Bullen, L. A. Gethings, J. P. Williams, F. H. Kriel and J. Coates, J. Inorg. Biochem., 2012, 117, 298 CrossRef PubMed.
- A. Barge, G. Cravotto, E. Gianolio and F. Fedeli, Contrast Media Mol. Imaging, 2006, 1, 184 CrossRef PubMed.
- M. Mantle, G. Stewart, G. Zayas and M. King, Biochem. J., 1990, 266, 597 Search PubMed.
- J. K. Myers, C. N. Pace and J. M. Scholtz, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 2833 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8dt01568f |
| This journal is © The Royal Society of Chemistry 2018 |