
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A diarsagermylene and a diarsastannylene stabilised by arene⋯Ge/Sn interactions†
Keith
Izod
 *,
Peter
Evans
and
Paul G.
Waddell
*,
Peter
Evans
and
Paul G.
Waddell
Main Group Chemistry laboratories, School of Chemistry, Bedson Building, Newcastle University, Newcastle upon Tyne, NE1 7RU, UK. E-mail: keith.izod@ncl.ac.uk
First published on 20th February 2018
Abstract
The synthesis and structures of two new diarsatetrylenes {(Dipp)2As}2E are presented [E = Ge, Sn; Dipp = 2,6-diisopropylphenyl]. The high barrier to planarisation of As prevents stabilisation by As–E π-interactions; however, arene⋯Ge/Sn interactions stabilise these compounds by up to 181.4 kJ mol−1. This represents a new stabilisation mode for this class of compounds.
Currently, there is intense interest in low oxidation state main group compounds, particularly with regard to their ability to activate small molecules such as H2, CO2 and NH3.1,2 This reactivity has been compared to that of transition metal complexes and, consequently, low oxidation state main group compounds have been posited as inexpensive and earth abundant alternatives to precious metal catalysts.3
One of the simplest, and most versatile, classes of low oxidation state main group compounds is the tetrylenes R2E (E = Si, Ge, Sn, Pb). These electron-deficient compounds possess both a lone pair at the tetrel centre and a vacant p-orbital which lies perpendicular to the plane of the molecule and which act as the donor and acceptor orbitals, respectively, in small molecule activations. The energy difference between these donor and acceptor orbitals plays a key role in the reactivity of tetrylenes and, as a consequence, new methods to modulate this frontier orbital energy gap are of great interest. This energy gap is significantly increased by adjacent heteroatoms (e.g. N, O, S), which stabilise the tetrel lone pair through σ-withdrawing effects and destabilise the acceptor orbital through π-interactions between the vacant p-orbital at the tetrel centre and the heteroatom lone pairs, resulting in lower reactivity.
The chemistry of diaminotetrylenes, (R2N)2E (E = Si, Ge, Sn, Pb), the heavier group 14 element analogues of N-heterocyclic carbenes and their acyclic analogues, is well established.4 In contrast, far less is known about their phosphorus- and arsenic-substituted homologues, i.e. diphosphatetrylenes, (R2P)2E, and diarsatetrylenes (R2As)2E.5–8 Indeed, only three arsatetrylenes have been crystallographically authenticated (1–3), and, of these, compound 3 is the only example of a monomeric diarsatetrylene possessing a two-coordinate tetrel centre.6a
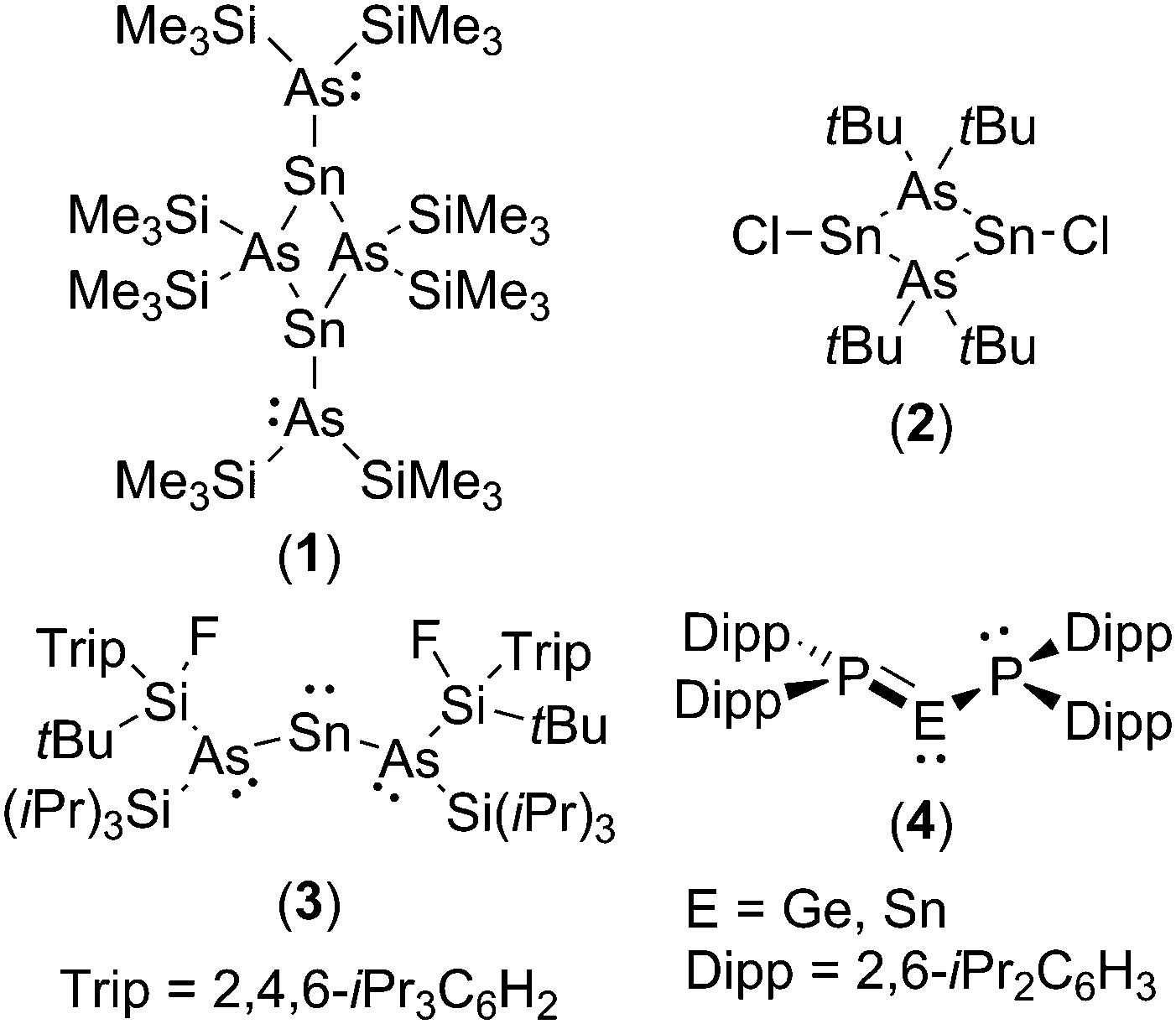
The low number of reported diphospha- and diarsatetrylenes may, at least in part, be attributed to the high barriers to planarisation of the heavier pnictogens, which prevents stabilisation of the electron-deficient tetrel centre through P–E/As–E π-interactions. Given the foregoing it is perhaps unsurprising that the arsenic centres in 3 adopt a pyramidal configuration with no evidence for As–Sn π-interactions.6a Nonetheless, it has been calculated that the heavier group 15 elements are as good π-donors as nitrogen if this barrier can be overcome.9
The barrier to planarisation of the heavier pnictogens may be lowered by several strategies, including (i) the use of sterically demanding substituents, (ii) the presence of electropositive substituents directly adjacent to the tetrel centre, and (iii) incorporation of the pnictogen centre into a ring.10,11 In this regard, we recently reported the synthesis, solid state structures and dynamic behaviour of the unusual diphosphatetrylenes 4, in which the phosphorus centres both possess sterically demanding substituents and lie directly adjacent to an electropositive germanium or tin centre12,13 Compounds 4 adopt a configuration with one planar and one pyramidal phosphorus centre, with a single, stabilising P–E π-interaction.
Based on our successful synthesis of P–E π-stabilised diphosphatetrylenes, we conjectured that the steric and electronic properties of our ligands might be sufficient to enable the synthesis of a similarly stabilised diarsatetrylene. However, we anticipated that the higher barrier to planarisation of the arsenic centres in the target compounds compared to the corresponding phosphorus centres in 4 might have a significant impact on any propensity for As–E π-interactions.14 We also anticipated that this, combined with the lower electronegativity of arsenic in comparison to phosphorus, would lead to a smaller frontier orbital gap in the resulting diarsatetrylenes and a consequently enhanced reactivity.
The reaction between amine-protected (iPr2N)AsCl2 and two equivalents of DippLi(OEt2), followed by two equivalents of ethereal HCl gives the secondary chloroarsane (Dipp)2AsCl (5) (Scheme 1) [Dipp = 2,6-iPr2C6H3]. Treatment of 5 with LiAlH4 gives the secondary arsane (Dipp)2AsH (6) in excellent yield. Compound 6 is readily metalated by nBuLi in THF to give the sterically hindered arsanide [(Dipp)2As]Li(THF)2.75(Et2O)0.25 (7), after recrystallisation from diethyl ether, as a yellow crystalline solid; the constitution of 7 was confirmed by 1H and 13C{1H} NMR spectroscopy and X-ray crystallography (see ESI†).
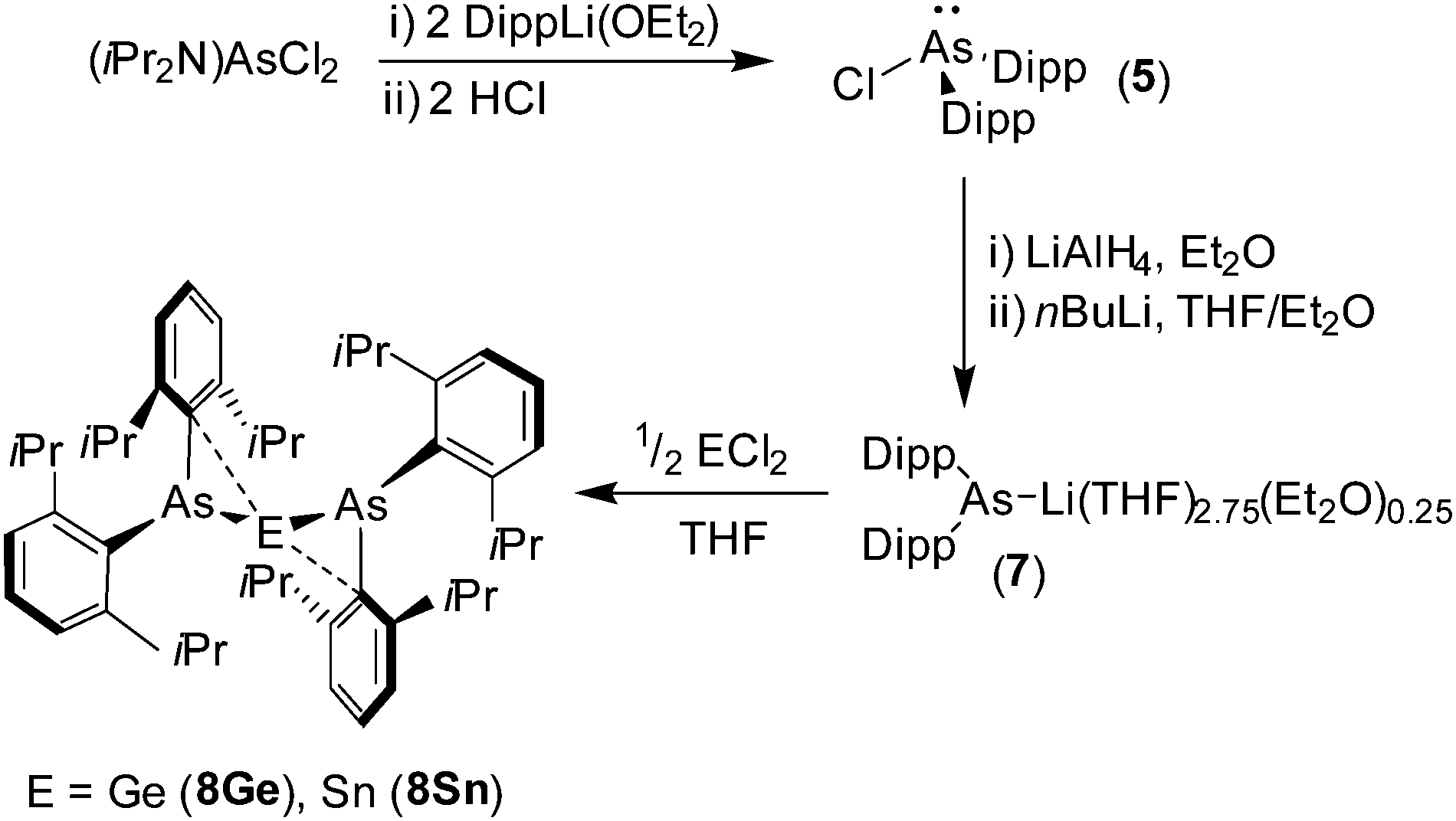 | ||
| Scheme 1 Synthesis of compounds 5–8. | ||
The reaction between two equivalents of in situ generated 7 and GeCl2(1,4-dioxane) in cold THF gives the diarsagermylene {(Dipp)2As}2Ge·PhMe (8Ge) as dark red prisms after crystallisation from toluene (Scheme 1). A similar reaction between 7 and SnCl2 gives the diarsastannylene {(Dipp)2As}2Sn·PhMe (8Sn) as red-purple blocks. Compounds 8Ge and 8Sn are sensitive to air and moisture and solutions of these compounds decompose slowly at room temperature, or more rapidly on exposure to ambient light; in the solid state both 8Ge and 8Sn are reasonably stable at room temperature and can be manipulated without significant decomposition when exposure to light is kept to a minimum.
Compounds 8Ge and 8Sn are isostructural and isomorphous; each compound crystallises as a discrete molecular species with a molecule of toluene in the asymmetric unit (Fig. 1). Compound 8Ge represents the first diarsagermylene to be crystallographically characterised, while 8Sn is only the second monomeric diarsastannylene to be structurally authenticated.
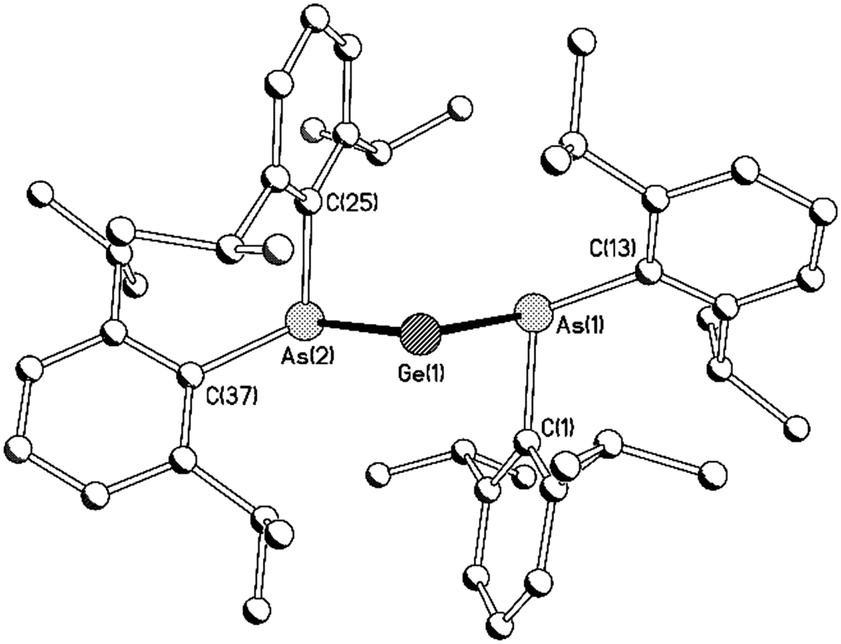 | ||
| Fig. 1 Molecular structure of 8Ge with H atoms and solvent of crystallisation omitted for clarity [compound 8Sn is isostructural]. Selected bond lengths (Å) and angles (°): 8Ge As(1)–Ge(1) 2.4851(4), As(2)–Ge(1) 2.4771(4), As(1)–C(1) 2.020(3), As(1)–C(13) 2.000(3), As(2)–C(37) 2.023(3), AS(2)–C(37) 2.001(3), Ge(1)⋯C(1) 2.666(3), Ge(1)⋯C(25) 2.710(2), As(1)–Ge–As(2) 90.808(13), Ge(1)–As(1)–C(1) 71.75(7), Ge(1)–As(1)–C(13) 108.70(8), Ge(1)–As(2)–C(25) 73.28(7), Ge(1)–As(2)–C(37) 109.94(8); 8Sn As(1)–Sn(1) 2.6579(4), As(2)–Sn(1) 2.6644(4), As(1)–C(1) 1.994(3), As(1)–C(13) 2.020(3), As(2)–C(37) 1.995(3), AS(2)–C(37) 2.014(3), Sn(1)⋯C(1) 2.858(2), Sn(1)⋯C(37) 2.762(3), As(1)–Sn(1)–As(2) 91.004(11), Sn(1)–As(1)–C(1) 110.57(8), Sn(1)–As(1)–C(13) 73.91(7), Sn(1)–As(2)–C(25) 108.49(8), Sn(1)–As(2)–C(37) 70.79(8). | ||
As expected, the As–Ge distances in 8Ge [2.4851(4) and 2.4771(4) Å] are significantly shorter than the corresponding distances in the tertiary arsane complexes {C6H4-1,2-(AsMe2)2}GeI2 [2.6210(11) and 2.6313(11) Å] and [{C6H4-1,2-(AsMe2)2}GeCl][GeCl3] [2.5847(5) Å],15 the only other reported examples of As–Ge(II) bond lengths. Examples of As–Sn(II) bonds are more numerous and the As–Sn distances in 8Sn [2.6579(4) and 2.6644(4) Å] are similar to the As–Sn distances in 3 [average 2.652(1) Å for the two crystallographically independent molecules in the asymmetric unit].6a The As–E–As angles [90.808(13) and 91.0004(11)° for 8Ge and 8Sn, respectively] suggest negligible hybridisation of the tetrel centres in these compounds and this is supported by DFT calculations (see below); the As–Sn–As angle in 8Sn is slightly smaller than the corresponding angle in 3 [94.64(4)°].
In marked contrast to the phosphorus centres in 4, both arsenic centres in 8Ge and 8Sn adopt a pyramidal geometry [sum of angles at As for 8Ge = 285.86 and 287.76°; 8Sn = 285.96 and 289.58°], consistent with the high barrier to planarisation of arsenic compared to phosphorus.14 The pyramidal geometries of the arsenic centres necessarily preclude any stabilisation of the tetrel centres by As–Ge/Sn π-interactions. However, in both 8Ge and 8Sn, the ipso-carbon of one phenyl ring from each arsanide ligand lies in close proximity to the tetrel centre [Ge⋯Cipso 2.710(2) and 2.666(3) Å; Sn⋯Cipso 2.762(3) and 2.858(2) Å]. These distances are well within the sums of the van der Waals radii of Ge and C (3.81 Å) and of Sn and C (3.87 Å),16 suggesting a significant interaction between the electron density at these ipso-carbon atoms and the vacant p-orbital at the tetrel centre. Consistent with this, the short Ge/Sn⋯Cipso distances are associated with rather acute Cipso–As–Ge/Sn angles [8Ge 71.75(7) and 73.28(7)°; 8Sn 70.79(8) and 73.91(7)°].
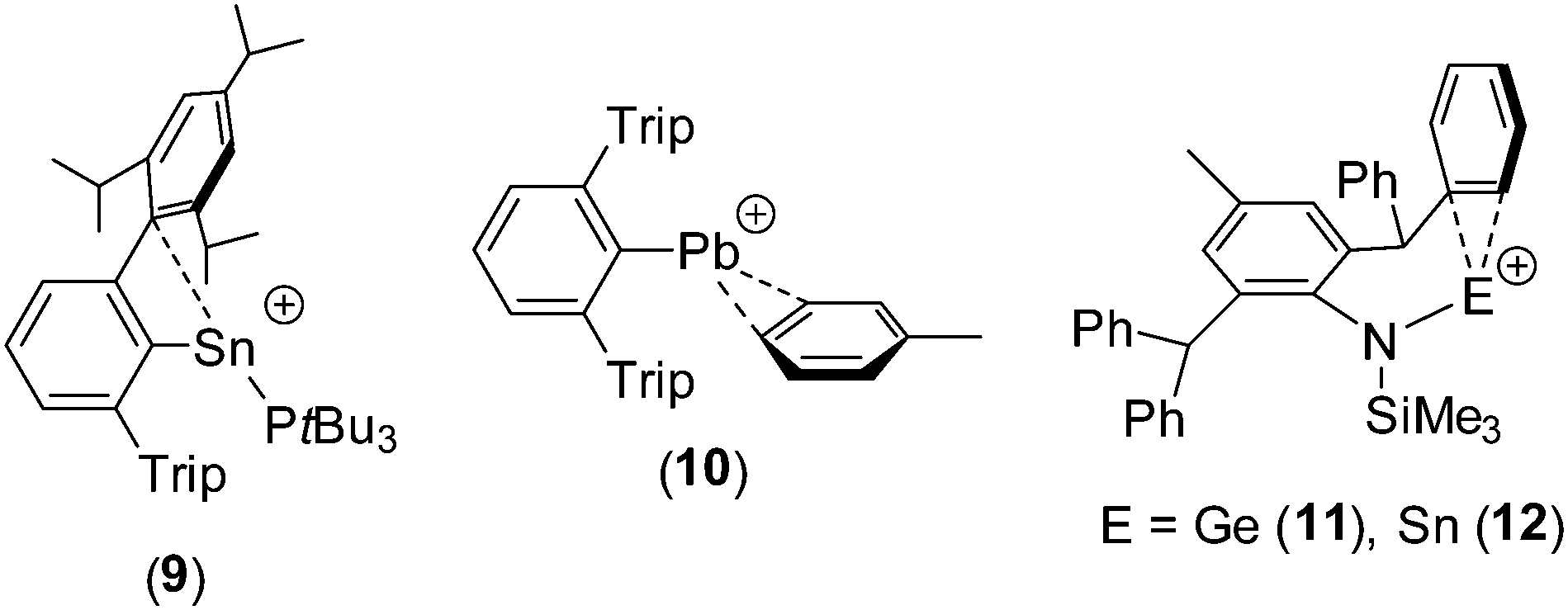
Group 14 element⋯arene interactions have been observed in a small number of Si(IV)-containing cations,17 and in a limited number of compounds in which the tetrel centre is in the +2 oxidation state (e.g.9–12).18–21 The Ge⋯C and Sn⋯C distances in 8Ge and 8Sn are similar to the corresponding distances in 11 [Ge⋯C 2.642(2) and 2.661(2) Å] and 12 [Sn⋯C 2.821(2) and 2.827(2) Å];21 for comparison, the closest Sn⋯C distances in 9 are 3.27 and 3.35 Å,19 suggesting a much weaker interaction in this case.
The room temperature 1H NMR spectra of 8Ge and 8Sn consist of a single set of extremely broad signals due to the iPr and aromatic protons of the ligands, indicating dynamic exchange between the aromatic rings engaged in the E⋯Cipso interactions and those that are not. At −60 °C the iPr peaks decoalesce into eight methyl signals (spanning the range 0.48–1.42 ppm for 8Ge and 0.51–1.52 ppm for 8Sn) and four methine signals (spanning the range 2.51–4.86 ppm for 8Ge and 2.80–4.93 ppm for 8Sn), consistent with the C2 symmetry of the solid-state structures. The large spread of methyl and methine signals in these spectra is due to the proximity (or not) of these protons to the aromatic rings or the arsenic lone pairs of the ligands.
We noted previously that, due to their inherently broad nature, the 119Sn signals for 4Sn and its more substituted analogue {(2,4,6-iPr3C6H2)2P}2Sn (13) could not be located at room temperature and only a very broad signal (FWHM ca 2000 Hz) could be observed for 13 even at −95 °C.12,13 This line-broadening is likely to be exacerbated by the presence of two adjacent quadrupolar 75As nuclei in 8Sn [75As natural abundance 100%, I = 3/2], however, while we could not observe a signal at room temperature, in part due to the thermal instability of this compound in solution, at −60 °C we were able to locate a broad 119Sn signal for 8Sn at 547 ppm (FWHM 500 Hz). Although no 119Sn NMR data are available for 2 and 3,6a,8b the 119Sn chemical shift of 8Sn is similar to that observed for 1 (475 and 671 ppm for the cis and trans isomers, respectively)8a and 13 (440 ppm).15
The presence of significant Ge/Sn⋯Cipso interactions in 8Ge and 8Sn is supported by DFT calculations. Geometry optimisations at the B97D/6-311G(2d,p) [LANL2DZ for Sn] level of theory give structures (8Gepyr and 8Snpyr) which correlate well with those obtained by X-ray crystallography (see Fig. 2 and ESI†). In particular, the calculated structures replicate the short E⋯Cipso distances rather nicely [Ge⋯Cipso 2.757/2.644 Å, Sn⋯Cipso 2.849/2.848 Å].
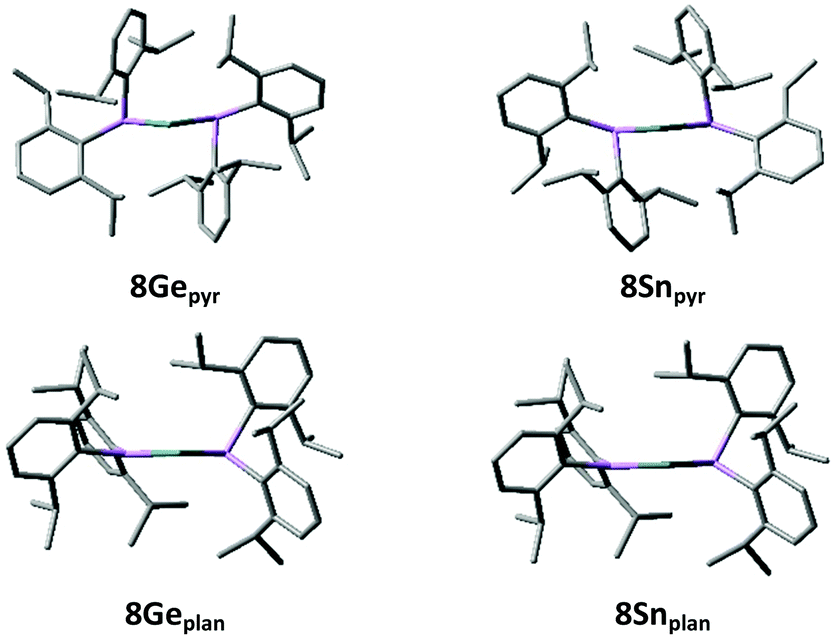 | ||
| Fig. 2 Optimised geometries for the pyramidal (8Gepyr and 8Snpyr) and planar (8Geplan and 8Snplan) forms of {(Dipp)2As}2E (E = Ge, Sn) [B97D/6-311G(2d,p) [LANL2DZ for Sn]]. | ||
The HOMO and LUMO for both 8Gepyr and 8Snpyr are largely based on the tetrel lone pair and the vacant p-orbital at the tetrel centre, respectively, although the LUMO has a significant component on the two nearby aromatic rings in each case (see ESI†). Natural Bond Orbital (NBO) analysis reveals that the tetrel lone pair in each case has >90% s-character, while the “vacant” orbital has essentially pure p-character. NBO analysis also reveals that this “vacant” orbital has an occupancy of 0.21 electrons in 8Gepyr and 0.18 electrons in 8Snpyr. This is associated with significant delocalisation of the As–Cipso σ-bonding electron density into this vacant orbital. Second order perturbation theory analysis shows that the approximate stabilisations afforded by these interactions (the E(2) energies) are 77.2 and 65.8 kJ mol−1 for 8Gepyr and 40.2 and 40.3 kJ mol−1 for 8Snpyr. Consistent with this, the Wiberg Bond Indices (WBIs) for the Ge⋯C interactions are 0.114 and 0.099, while the WBIs for the Sn⋯C interactions are both 0.094. Although small, these WBIs indicate a significant bonding interaction in each case (cf. WBIs of 0.763 and 0.771 for the As–Ge bonds in 8Gepyr and 0.726 for the two As–Sn bonds in 8Snpyr). The foregoing clearly indicates that these Cipso⋯E interactions stabilise the arsatetrylenes to a significant extent. In support of this, NBO deletions (in which the energy of the system is recalculated in the absence of the specified interactions) indicate that these interactions stabilise 8Gepyr and 8Snpyr by 181.4 and 106.2 kJ mol−1, respectively.
For comparison, we have located minimum energy geometries for both 8Ge and 8Sn in which one of the arsenic centres is planar (i.e. the direct analogues of 4, possessing Ge/Sn–As π-interactions), 8Geplan and 8Snplan. These lie 18.0 and 32.1 kJ mol−1, respectively, higher in free energy than the ground state forms 8Gepyr and 8Snpyr.
It has been suggested that the energy gap between the singlet and triplet states (ΔS–T) of tetrylenes correlates well with their reactivities, with smaller gaps indicating higher reactivities.22 For 8Gepyr and 8SnpyrΔS–T is calculated to be 79.8 and 90.0 kJ mol−1, respectively, suggesting that these compounds should exhibit interesting reactivities [for comparison, ΔS–T for the prototypical N-heterocyclic germylene {CHNH}2Ge has been calculated to be 195.0 kJ mol−1,22c consistent with the low reactivity of N-heterocyclic germylenes].
In summary, we have shown that the high barrier to planarisation of arsenic prevents the stabilisation of the diarsatetrylenes {(Dipp)2As}2E through As–E π-interactions. However, canting of the aromatic rings in these compounds leads to a significant interaction between the ipso carbon atoms of the rings and the tetrel centres which stabilises the tetrylenes by up to 181.4 kJ mol−1.
This work was partly funded by the U.K. Engineering and Physical Sciences Research Council (EPSRC; Grant EP/L5048281).
Conflicts of interest
There are no conflicts to declare.Notes and references
- For recent reviews see: (a) S. Yadev, S. Saha and S. S. Sen, ChemCatChem, 2016, 8, 486–501 CrossRef; (b) S. Yao, Y. Xiong and M. Driess, Organometallics, 2011, 30, 1748–1767 CrossRef CAS.
- For selected recent examples see: (a) Y. Peng, J.-D. Guo, B. D. Ellis, Z. Zhu, J. C. Fettinger, S. Nagase and P. P. Power, J. Am. Chem. Soc., 2009, 131, 16272–16282 CrossRef CAS PubMed; (b) J. W. Dube, Z. D. Brown, C. A. Caputo, P. P. Power and P. J. Ragogna, Chem. Commun., 2014, 50, 1944–1946 RSC; (c) R. Rodriguez, Y. Contie, R. Nougue, A. Bacereido, N. Saffon-Merceron, J.-M. Sotiropoulos and T. Kato, Angew. Chem., Int. Ed., 2016, 55, 14355–14358 CrossRef CAS PubMed.
- P. P. Power, Nature, 2010, 463, 171–177 CrossRef CAS PubMed.
- For recent reviews see: (a) Y. Mizuhata, T. Sasamori and N. Tokitoh, Chem. Rev., 2009, 109, 3479 CrossRef CAS PubMed; (b) M. F. Lappert, P. P. Power, A. Protchenko and A. Seeber, Metal Amide Chemistry, Wiley, Chichester, UK, 2009 Search PubMed; (c) A. V. Zabula and F. E. Hahn, Eur. J. Inorg. Chem., 2008, 5165–5179 CrossRef CAS.
- For a review see: K. Izod, Coord. Chem. Rev., 2012, 256, 2972–2997 CrossRef CAS.
- Homoleptic diphosphatetrylenes: (a) M. Driess, R. Janoschek, H. Pritzkow, S. Rell and U. Winkler, Angew. Chem., Int. Ed., 1995, 34, 1614–1616 CrossRef CAS; (b) A. H. Cowley, D. M. Giolando, R. A. Jones, C. M. Nunn and J. M. Power, Polyhedron, 1988, 7, 1909–1910 CrossRef CAS; (c) S. C. Goel, M. Y. Chiang, D. J. Rauscher and W. E. Buhro, J. Am. Chem. Soc., 1993, 115, 160 CrossRef CAS; (d) C. Druckenbrodt, W.-W. du Mont, F. Ruthe and P. G. Jones, Z. Anorg. Allg. Chem., 1998, 624, 590 CrossRef CAS; (e) E. Rivard, A. D. Sutton, J. C. Fettinger and P. P. Power, Inorg. Chim. Acta, 2007, 360, 1278 CrossRef CAS; (f) K. Izod, W. McFarlane, B. Allen, W. Clegg and R. W. Harrington, Organometallics, 2005, 24, 2157–2167 CrossRef CAS; (g) K. Izod, J. Stewart, E. R. Clark, W. McFarlane, B. Allen, W. Clegg and R. W. Harrington, Organometallics, 2009, 28, 3327–3337 CrossRef CAS; (h) K. Izod, J. Stewart, W. Clegg and R. W. Harrington, Organometallics, 2010, 29, 108–116 CrossRef CAS; (i) K. Izod, J. Stewart, E. R. Clark, W. Clegg and R. W. Harrington, Inorg. Chem., 2010, 49, 4698 CrossRef CAS PubMed; (j) K. Izod, E. R. Clark, W. Clegg and R. W. Harrington, Organometallics, 2012, 31, 246–255 CrossRef CAS.
- Heteroleptic phosphatetrylenes: (a) S. Yao, M. Brym, K. Merz and M. Driess, Organometallics, 2008, 27, 3601 CrossRef CAS; (b) S. Yao, S. Block, M. Brym and M. Driess, Chem. Commun., 2007, 3844 RSC; (c) E. C. Y. Tam, N. A. Maynard, D. C. Apperley, J. D. Smith, M. P. Coles and J. R. Fulton, Inorg. Chem., 2012, 51, 9403 CrossRef CAS PubMed; (d) B. P. Johnson, S. Almstätter, F. Dielmann, M. Bodensteiner and M. Scheer, Z. Anorg. Allg. Chem., 2010, 636, 1275 CrossRef CAS.
- (a) M. Arsatetrylenes, M. M. Westerhausen, W. Enzelberger and J. Schwarz, Organomet. Chem., 1995, 491, 83–90 CrossRef; (b) A. H. Cowley, D. M. Giolando, R. A. Jones, C. M. Nunn, J. M. Power and W.-W. duMont, Polyhedron, 1988, 7, 1317–1319 CrossRef CAS.
- J. Kapp, C. Schade, A. M. El-Nahasa, P. von and R. Schleyer, Angew. Chem., Int. Ed. Engl., 1996, 35, 2236 CrossRef.
- (a) G. Bouhadir and D. Borissou, Chem. Soc. Rev., 2004, 33, 210 RSC; (b) M. Driess, K. Merz and C. Monsé, Z. Anorg. Allg. Chem., 2000, 626, 2264 CrossRef CAS; (c) F. G. N. Cloke, P. B. Hitchcock, P. Hunnable, J. F. Nixon, L. Nyulászi, E. Niecke and V. Thelen, Angew. Chem., Int. Ed., 1998, 37, 1083 CrossRef CAS.
- (a) R. D. Beachler, J. P. Casey, R. J. Cook, G. H. Senkler Jr. and K. Mislow, J. Am. Chem. Soc., 1987, 109, 338 CrossRef; (b) R. D. Beachler and K. Mislow, J. Am. Chem. Soc., 1971, 93, 773 CrossRef; (c) R. D. Beachler, J. D. Andose, J. Stackhouse and K. Mislow, J. Am. Chem. Soc., 1972, 94, 8060 CrossRef; (d) K. Izod, E. R. Clark and J. Stewart, Inorg. Chem., 2011, 50, 3651 CrossRef CAS PubMed.
- K. Izod, D. G. Rayner, S. M. El-Hamruni, R. W. Harrington and U. Baisch, Angew. Chem., Int. Ed., 2014, 53, 3636–3640 CrossRef CAS PubMed.
- K. Izod, P. Evans, P. G. Waddell and M. R. Probert, Inorg. Chem., 2016, 55, 10510–10522 CrossRef CAS PubMed.
- (a) S. Pelzer, K. Wichmann, R. Wesendrup and P. Schwerdtfeger, J. Phys. Chem. A, 2002, 106, 6387–6394 CrossRef CAS; (b) D. A. Dixon and A. J. Arduengo III, J. Am. Chem. Soc., 1987, 109, 338–341 CrossRef CAS; (c) A. Clotet, J. Rubio and F. Illas, THEOCHEM, 1988, 164, 351–361 CrossRef; (d) D. A. Dixon and D. S. Marynick, J. Am. Chem. Soc., 1977, 99, 6101–6103 CrossRef CAS.
- F. Cheng, A. L. Hector, W. Levason, G. Reid, M. Webster and W. Zhang, Inorg. Chem., 2010, 49, 752–760 CrossRef CAS PubMed.
- M. Mantina, A. C. Chamberlain, R. Valero, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. A, 2009, 113, 5806–5812 CrossRef CAS PubMed.
- (a) S. Duttwyler, Q.-Q. Do, A. Linden, K. M. Baldride and J. S. Siegel, Angew. Chem., Int. Ed., 2008, 47, 1719–1722 ( Angew. Chem. , 2008 , 120 , 1743–1746 ) CrossRef CAS PubMed; (b) C. Gerdes, W. Saak, D. Haase and T. Muller, J. Am. Chem. Soc., 2013, 135, 10353–10361 CrossRef CAS PubMed.
- For selected examples see: (a) M. S. Weininger, P. F. Rodesiler and A. L. Amma, Inorg. Chem., 1979, 18, 751–755 CrossRef CAS; (b) P. F. Rodesiler, Th. Auel and E. L. Amma, J. Am. Chem. Soc., 1975, 97, 7405–7410 CrossRef CAS; (c) A. G. Gash, P. F. Rodesiler and E. L. Amma, Inorg. Chem., 1974, 13, 2429–2434 CrossRef CAS; (d) T. Probst, O. Steigelmann, J. Riede and H. Schmidbaur, Angew. Chem., Int. Ed., 1990, 29, 1397–1398 CrossRef; (e) A. Schäfer, F. Winter, W. Saak, D. Haase, R. Pöttgen and T. Müller, Chem. – Eur. J., 2011, 17, 10979–10984 CrossRef PubMed; (f) J. L. Lefferts, M. B. Hossain, K. C. Molloy, D. van der Helm and J. J. Zuckerman, Angew. Chem., Int. Ed., 1980, 19, 309–310 CrossRef.
- C. Sindlinger, F. S. W. Aicher, H. Schubert and L. Wesemann, Angew. Chem., Int. Ed., 2017, 56, 2198–2202 CrossRef CAS PubMed.
- S. Hino, M. Brynda, A. D. Philips and P. P. Power, Angew. Chem., Int. Ed., 2004, 43, 2655–2658 CrossRef CAS PubMed.
- J. Li, C. Schenk, F. Winter, H. Scherer, N. Trap, A. Higelin, S. Keller, R. Pottgen, I. Krossing and C. Jones, Angew. Chem., Int. Ed., 2012, 51, 9557–9561 CrossRef CAS PubMed.
- (a) Y. Wang and J. Ma, J. Organomet. Chem., 2009, 694, 2567–2575 CrossRef CAS; (b) Y. Peng, B. D. Ellis, X. Wang and P. P. Power, J. Am. Chem. Soc., 2008, 130, 12268–12269 CrossRef CAS PubMed; (c) A. V. Protchenko, J. I. Bates, L. M. A. Saleh, M. P. Blake, A. D. Schwarz, E. L. Kolychev, A. L. Thompson, C. Jones, P. Mountford and S. Aldridge, J. Am. Chem. Soc., 2016, 138, 4555–4564 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterisation of all compounds. Details of the crystal structure of 7 and crystallographic details for 7, 8Ge, and 8Sn. NMR spectra for all compounds. Details of DFT studies. CCDC 1810821–1810823. Data supporting this publication is openly available under an ‘Open Data Commons Open Database License’. Additional metadata are available at: http://dx.doi.org/10.17634/154300-77. Please contact Newcastle Research Data Service at rdm@ncl.ac.uk for access instructions. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7cc09564c |
| This journal is © The Royal Society of Chemistry 2018 |