
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Matching precursor kinetics to afford a more robust CVD chemistry: a case study of the C chemistry for silicon carbide using SiF4 as Si precursor†
Pontus
Stenberg
,
Örjan
Danielsson
 ,
Edvin
Erdtman
,
Pitsiri
Sukkaew
,
Lars
Ojamäe
,
Erik
Janzén
and
Henrik
Pedersen
*
,
Edvin
Erdtman
,
Pitsiri
Sukkaew
,
Lars
Ojamäe
,
Erik
Janzén
and
Henrik
Pedersen
*
Department of Physics, Chemistry and Biology, Linköping University, SE-581 83 Linköping, Sweden. E-mail: henrik.pedersen@liu.se
First published on 10th May 2017
Abstract
Chemical vapor deposition (CVD) is one of the technology platforms forming the backbone of the semiconductor industry and is vital in the production of electronic devices. To upscale a CVD process from the lab to the fab, large-area uniformity and high run-to-run reproducibility are needed. We show by a combination of experiments and gas phase kinetics modeling that the combinations of Si and C precursors with the most well-matched gas phase chemistry kinetics gives the largest area of homoepitaxial growth of SiC. Comparing CH4, C2H4 and C3H8 as carbon precursors to the SiF4 silicon precursor, CH4 with the slowest kinetics renders the most robust CVD chemistry with large-area epitaxial growth and low-temperature sensitivity. We further show by quantum chemical modeling how the surface chemistry is impeded by the presence of F in the system, which limits the amount of available surface sites for the C to adsorb onto.
Introduction
Chemical vapor deposition (CVD) is one of the most important techniques in semiconductor manufacturing, typically employed to make the active region of the device structures, where a very high control over material quality and precise control over the incorporation of impurities is required. Of paramount importance for upscaling any CVD process in a device production are large-scale uniformity and run-to-run reproducibility, both of which depend on the spatial and temporal uniformity of the CVD chemistry. The CVD chemistry for most semiconductor materials relies on gas phase chemical reactions to form reactive species from the precursor molecules and subsequent surface chemical reactions to form the material.1 The uniformity in a CVD process is therefore dependent on both the gas phase and surface chemistry of the employed precursor system. The uniformity in an industrial CVD reactor is typically controlled by carefully designing the gas flow patterns in the CVD reactor. However, a well-chosen overall robust CVD chemistry, which is not affected by minor temperature fluctuations and gas flow disturbances, is also a prerequisite for highly uniform CVD. The CVD chemistry should, ideally, render high concentrations of the reactive species with very similar spatial and temporal distributions.Here we use silicon carbide (SiC) as a model system to explore the effect of precursor kinetics on the overall CVD chemistry. SiC is a semiconductor with a wide band gap, high saturation electron drift velocity and high energy of the optical phonons which renders a high electric field breakdown strength, which makes SiC very well suited for high-power devices.2 A CVD process with large-area uniformity is required for the commercial realization of SiC devices. The growth of homoepitaxial SiC layers using CVD – based on SiH4 and C2H4 or C3H8 – has already been commercialized,3 but lately much interest has been directed towards using a halogenated CVD chemistry, mainly by adding chlorine, to allow a higher growth rate on account of the eliminated gas phase nucleation.4 Since the halogens F, Cl and Br bind more strongly to Si than Si itself – the binding strengths for F, Cl, Br and Si to Si are 576, 417, 358 and 310 kJ mol−1, respectively5 – higher partial pressures of Si species can be used in SiC CVD with a halogenated CVD chemistry without the formation of Si droplets in the gas phase.
Both chlorinated4 and brominated6 chemistries have been shown to behave very similarly to the standard, non-halogenated chemistry, albeit with a 5–20 times higher deposition rate. SiC CVD using a fluorinated chemistry was first demonstrated in 2012 by Rana et al.7 Lately, it has been shown that a fluorinated chemistry, using SiF4 and C2H4 as precursors, renders a CVD chemistry that is very sensitive to the C/Si ratio of the growth species impinging the surface and with a deposition rate of only around 30 μm h−1 compared to 100 μm h−1 or more for a chlorinated or brominated chemistry.8 We hypothesize that this behavior is due to the low reactivity of the SiF4 molecule given its very strong Si–F bonds and the slower kinetics of the Si precursor, which is not matched to the faster kinetics of the C2H4 precursor, rendering a less robust CVD chemistry, as manifested by the sensitivity to the impinging C/Si ratio. We test this hypothesis by kinetic modeling combined with experimental SiC deposition from SiF4 and different hydrocarbons and show that the best-matched gas phase chemical kinetics renders the most robust SiC CVD.
Methods
Experimental details
A horizontal hot wall CVD reactor was used for the growth of epitaxial SiC on 4H-SiC substrates with 4° off-cut from the (0001) axis in the (11−20) direction. As a carrier gas, palladium membrane purified hydrogen (H2) was used. The precursors used were SiF4 or a mixture of SiF4 and SiH4 as Si precursor and either CH4, C2H4 or C3H8 as C precursors, which were diluted in the carrier gas to the inlet concentrations Si/H2 = 0.25% and C/H2 = 0.125%, respectively. The use of SiF4 renders F/Si = 4 but when a mixture of SiF4 and SiH4 was used the F/Si ratio varied from 0 to 4. 25 standard liters per minute (SLM) of carrier gas was used at the process pressure p = 100 mbar, except when growth rate was of interest when 37.5 SLM was used, as well as Si/H2 = 0.125%. The temperatures were either T = 1550 or 1600 °C. The SiC coated graphite susceptor used in the experiments was 190 mm long with a gas cross section of 78 mm in width and 24 mm in height, in which 16 × 16 mm pieces of SiC substrates were placed at 3, 6 and 9 cm from the inlet. The upstream direction was connected to a broadened SiC coated graphite inlet connected to a quartz liner for the process gases. Heating of the susceptor was made via an RF heated graphite cylinder surrounding the susceptor. The graphite cylinder and susceptor were insulated with rigid graphite insulation, which was purged with argon – instead of H2 – to improve its thermal insulation properties. The whole setup was encapsulated in a quartz tube. The grown SiC films were characterized using a Nomarski differential interference contrast optical microscope from which the surface morphology could be studied. It should be noted that the optical microscopy images cannot at the same time show both the surface of the whole 16 × 16 mm samples and the surface features in detail. As the surface features are indicative of epitaxial film growth, we show only detailed micrographs to point out surface features.Modeling
Simulations of chemical kinetics were done using a one-dimensional transient model to evaluate the gas phase composition with respect to time at constant temperature and constant pressure. The initial gas mixture at time t = 0 was set to the same concentrations as in the experiments, and calculations were done for the three different carbon precursors. The kinetic model consisted of 89 elementary reaction steps, whereof 57 steps were for hydrocarbon reactions. No reactions between silicon and carbon species are accounted for in the model. This is motivated by the low amounts of Si–C species found at thermodynamic equilibrium in the Si–C–H–F system.8 Apart from H2, H and HF, 11 hydrocarbon species and 10 fluorosilanes were taken into account, plus SiH4, SiH2 and Si. The full reaction scheme is listed in ESI,† Table S1. Backward rates were calculated from the equilibrium constant, if not explicitly written out. Hydrocarbon reactions are taken from the literature, while reactions of fluorosilanes were obtained using quantum chemical calculations. The ground state and transition state structures were optimized using the hybrid density functional theory (DFT) B3LYP9,10 and Dunning's cc-pVTZ basis set11 together with the dispersion energy corrected by D3 correction from Grimme et al.12 Harmonic frequencies were calculated at the same level of theory. Transition states were verified by either visualizing the displacement of the imaginary frequencies or by tracing the reaction path from the transition state to the product and reactant states using the intrinsic reaction coordinate (IRC) computation.13 The electronic energies were calculated using CCSD(T) single point calculations14,15 and cc-pVnZ basis sets from Dunning11,16 for n = D, T and Q before extrapolation to the values at the complete basis set limit by,17| E(n) = E(∞) + Be−(n−1) + Ce−(n−1)2. | (1) |
Surface vacancies on SiC were probed by quantum-chemical calculations of SiC clusters consisting of 13 carbon and 13 silicon atoms, terminated by hydrogen atoms. The B3LYP functional9 was used together with the LANL2DZ basis set18–20 with Grimme dispersion correction12,21 for the optimization. Single point calculations were done with the M062X Minnesota functional,22 using the GTLargeXP basis set.23 The thermodynamic data from the DFT calculations have been utilized to compute the total Gibbs free energy, which was minimized with respect to the distribution of the species to get the thermodynamically most stable (i.e. the equilibrium) composition of the system24 using a MATLAB code.25
Results
It has previously been shown that epitaxial growth of SiC using a fluorinated chemistry is possible if the effective C/Si impingement ratio, i.e. the C/Si impingement ratio from the growth species, which changes along the susceptor, is low enough.8 The position at which the C/Si impingement ratio is low enough and the epitaxial growth begins is referred to as the onset of epitaxial growth. When comparing three different carbon precursors (C2H4, CH4 and C3H8) at the growth temperature T = 1600 °C, and otherwise the same process parameters, it can be seen (Fig. 1) that the onset of epitaxial growth differs between the precursors. C2H4 shows no onset of epitaxial growth within 10 cm from the inlet, while both CH4 and C3H8 had their onset of epitaxial growth upstream of the most upstream sample, placed 3 cm from the inlet. | ||
| Fig. 1 Optical microscopy images of the SiC surface grown with T = 1600 °C and different C precursors. (a) 10 cm from the inlet using C2H4 showing a rough surface indicative of polycrystalline growth with no onset of epitaxial growth. (b) 3 cm from the inlet using CH4 showing a smooth surface with a triangular surface defect (circled) indicative of epitaxial growth. (c) 3 cm from the inlet using C3H8 showing a smooth surface with a triangular surface defect (circled) indicative of epitaxial growth. | ||
The results shown in Fig. 1 display no difference between CH4 and C3H8. Lowering the temperature to T = 1550 °C (Fig. 2) reveals that C3H8 has no onset of epitaxial growth within 10 cm from the inlet, while CH4 shows epitaxial growth at the most upstream sample placed 3 cm from the inlet.
 | ||
| Fig. 2 Optical microscopy images of the SiC surface grown with T = 1550 °C and different C precursors. (a) 10 cm from the inlet using C3H8 showing a rough surface indicative of no growth and no onset of epitaxial growth, (b) 3 cm from the inlet using CH4 showing a smooth surface with a triangular surface defect (circled) indicative of epitaxial growth. | ||
Simulations of the gas phase composition for the three different carbon precursors in combination with SiF4 were done using kinetic modelling. Concentrations of the main gas phase species with respect to time are shown in Fig. 3 and 4 at two different temperatures. The time frame of interest is in the range 0.01–0.1 s, as it is in this time range that the precursors pass through the susceptor.
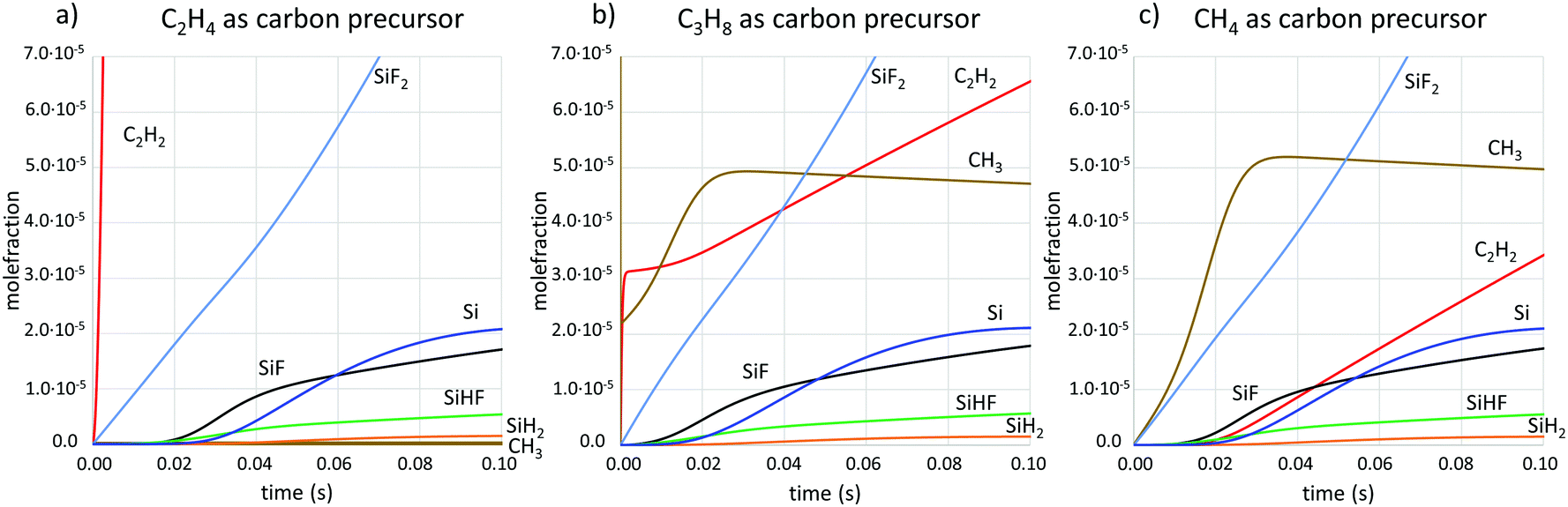 | ||
| Fig. 3 Concentrations of main gas phase species at T = 1600 °C from kinetic simulations when the carbon precursor is (a) C2H4, (b) C3H8, (c) CH4. | ||
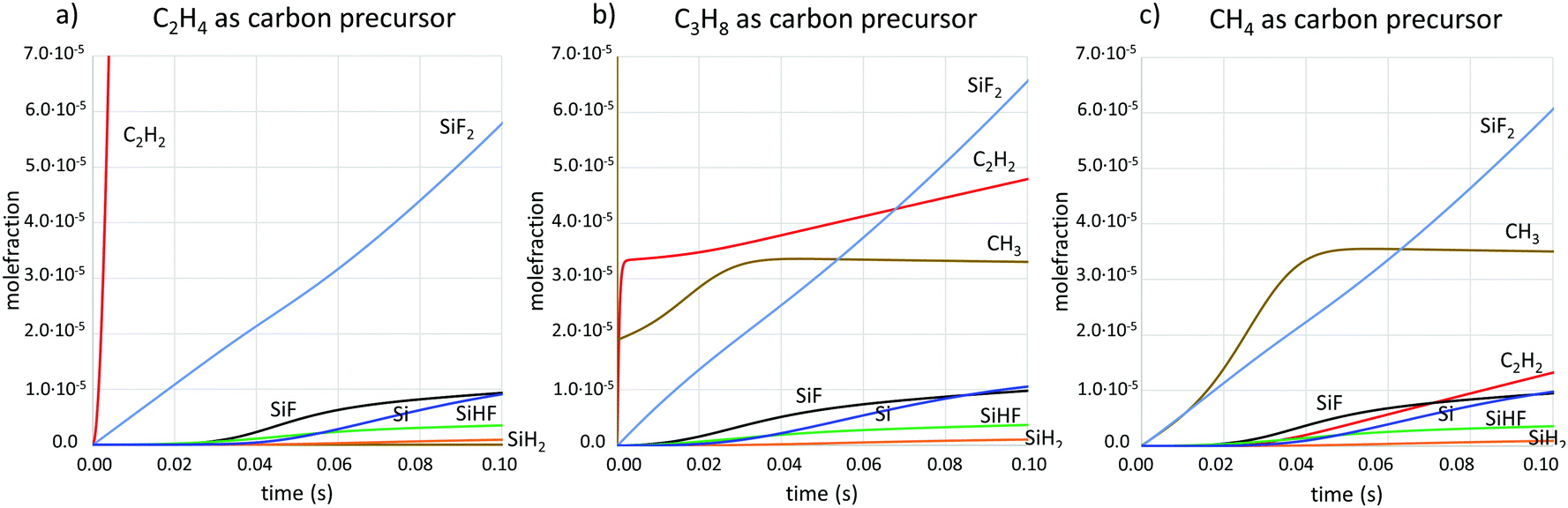 | ||
| Fig. 4 Concentrations of main gas phase species at T = 1550 °C from kinetic simulations when the carbon precursor is (a) C2H4, (b) C3H8, (c) CH4. | ||
CH3 and C2H2 have been identified as the carbon-containing growth species in SiC CVD26 and SiF has previously been suggested to be the main Si growth species in fluorinated SiC CVD.8 Here we can also see that the amount of atomic silicon is available at similar concentrations as SiF. Atomic silicon should be very reactive and is most likely also contributing to the growth. Comparing the concentrations of the two hydrocarbon growth species for the three different carbon precursors, we note that the concentration of C2H2 is highest when using C2H4, and lowest when using CH4. The CH3 concentration is very similar for C3H8 and CH4 as precursors, while it is at least three orders of magnitude lower when C2H4 is used.
Discussion
The decomposition chemistry of SiF4, leading to more usable Si-containing species for SiC growth, is slow compared to the decomposition of C3H8 and C2H4 and their usable C-containing decomposition products (Fig. 3a and b). This leads to unmatched concentrations of carbon and silicon in the susceptor, with potentially large C/Si ratios upstream that do not promote epitaxial growth conditions, and with a constantly changing C/Si ratio along the flow direction as the SiF4 precursor decomposes on its way through the susceptor. Changes in the C/Si ratio will influence the growth conditions for e.g. dopant incorporation, which is dependent on the amount of available C on the surface,27 or shift the growth mode from C limited to Si limited, thereby changing the spatial growth rate profile and thickness of the final layer. We have recently shown that a too-high C/Si leads to a surface poisoning of carbon, which prevents epitaxial growth.8 To achieve a robust CVD chemistry that gives epitaxial growth over large areas, SiF4 needs to be matched to a carbon precursor with an equally slow chemistry, so that the C/Si impingement ratio can be kept at low enough levels. CH4, which is not normally used in the CVD of SiC just because of its slow decomposition chemistry, is an excellent partner in this respect.The experimental results (Fig. 1 and 2) show that the kinetics for forming carbon-bearing growth species for SiC differs between different carbon precursors. When using SiF4 together with C2H4, the combination of resulting growth species does not lead to epitaxial growth along the studied area in the susceptor. With C3H8 or CH4, on the other hand, the growth is already epitaxial at the most upstream part of the susceptor at T = 1600 °C, suggesting that the C/Si impingement ratio is low enough for epitaxial growth. The difference in the kinetics between precursors is further demonstrated by the results at T = 1550 °C, when the case where C3H8 is used does not lead to epitaxial growth even at the most downstream position studied, while CH4 renders epitaxial growth at all positions at this temperature.
The experimental results are supported by the chemical kinetic calculations, where it can be seen that the production of C2H2 is very fast when C2H4 is used as precursor, while it is relatively slow for CH4 as precursor. Indeed, the decomposition paths for the hydrocarbons differ significantly. While C2H4 breaks two C–H bonds and forms a third C–C bond to form C2H2, the decomposition product of CH4 is mainly CH3, which may then react to form C2H6, which in turn will undergo further reactions that eventually lead to C2H2. The latter reaction path contains more reaction steps, each with some energy barrier to climb, which makes the production of C2H2 slow in comparison to the first reaction path. The production rate of C2H2 in the CH4 case matches well the production rate of SiF and Si, which would help keep the C/Si ratio at the SiC surface at levels suitable for epitaxial growth.
Unterminated surface sites are necessary for an efficient chemisorption of carbon growth species.26 Without halogen addition to the SiC CVD process, the SiC surface is only partially terminated by hydrogen at the growth conditions studied, and there are unterminated surface sites available for the growth to proceed. When SiF4 is used as precursor, HF is formed in the gas phase as a by-product from the SiF4 decomposition. The presence of HF could lead to Si–H bonds on the surface being replaced by Si–F bonds, and unterminated surface sites may be readily saturated by fluorine, considering the very strong bond energy between Si and F. A SiC surface saturated by Si–F bonds, rather than Si–H bonds, would lead to an inhibited growth due to the stronger Si–F bond. The surface saturation has been studied by quantum-chemical computations using a SiC cluster model (Fig. 5). The amount of hydrogen termination, H (s), unterminated surface sites, X (s), and fluorine termination, F (s), with increasing F gas phase concentration are shown in Fig. 6. The fluorine termination increases with fluorine concentration at the expense of hydrogen termination and unterminated surface sites. Due to the slow kinetics of the SiF4 molecule, the gas phase concentration of HF increases further into the susceptor, which then would also increase the amount of fluorine-terminated surface sites downstream.
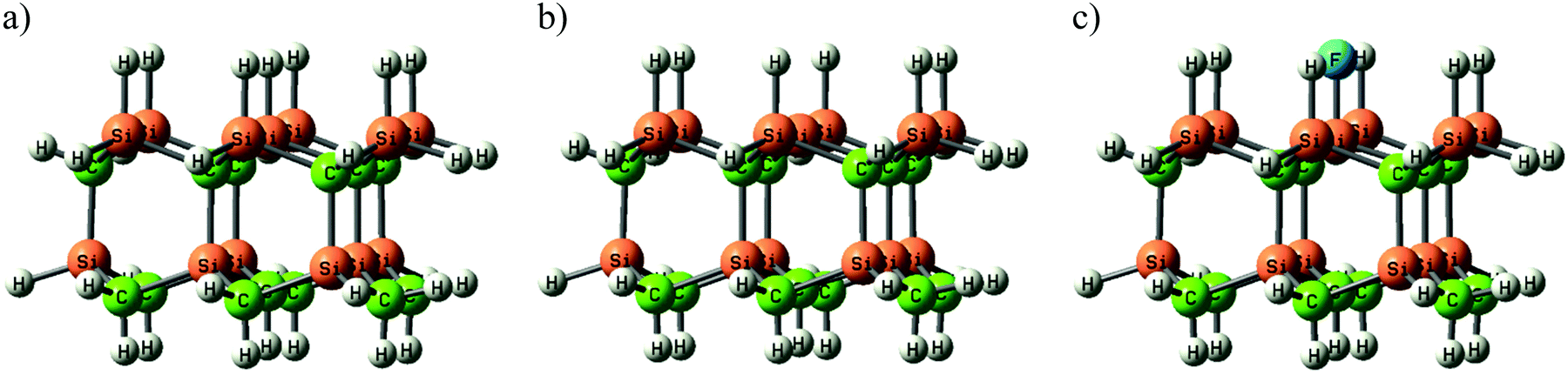 | ||
| Fig. 5 The SiC cluster models used in this study of (a) the completely hydrogen-terminated surface – H (s), (b) the cluster with a dangling bond – X (s) and (c) the single fluorine-terminated structure – F (s). | ||
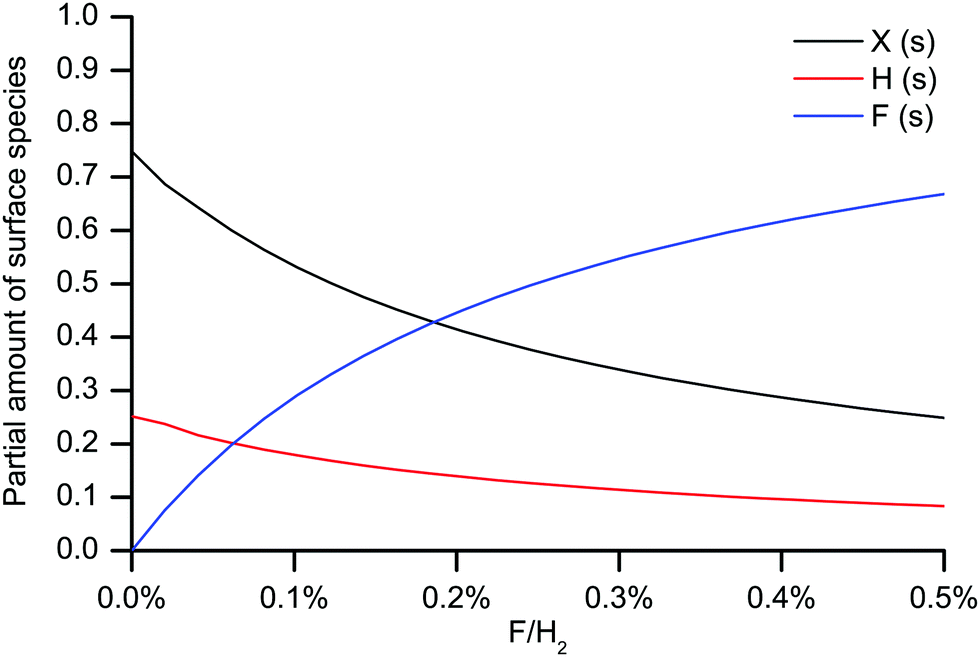 | ||
| Fig. 6 Equilibrium composition of the surface species at different F/H2 ratios from quantum-chemical modeling. H (s) denotes hydrogen termination, X (s) denotes surface vacancy, F (s) denotes fluorine termination. | ||
To verify the dependence of the amount of surface vacancies on the SiC growth, experiments were conducted where the F/Si ratio was controlled by mixing SiH4 and SiF4 as Si precursors. The F/Si ratio could then be varied between 0 and 4. As for the other CVD experiments in this study, H2 was in great excess in the experiments, with an inlet ratio of the SiH4 + SiF4 and H2 of 0.125%. Hence, the F/H2 for the experiments were between 0 and 0.5%. The temperature was T = 1600 °C and the carrier gas flow was 37.5 SLM. In Fig. 7 the maximum growth rate is plotted against the F/H2 ratio and compared with the amount of surface vacancies, X (s). The growth rate declines with the F/H2 ratio, following the decline in surface vacancies. We therefore speculate that the growth rate is limited by the growth of the C layers due to the F-terminated surface from the use of SiF4 as Si precursor. But also the amount of possible Si-bearing growth species (SiF, SiHF and Si) decreases with increasing F/H2. Increasing the Si/H2 ratio could therefore give a high growth rate upstream before more F is released, terminating the surface downstream, and thereby reducing the growth rate even more than if a lower Si/H2 ratio had been used. That the growth rate decreases faster along the flow direction with a higher Si/H2 using SiF4 has been shown previously.8
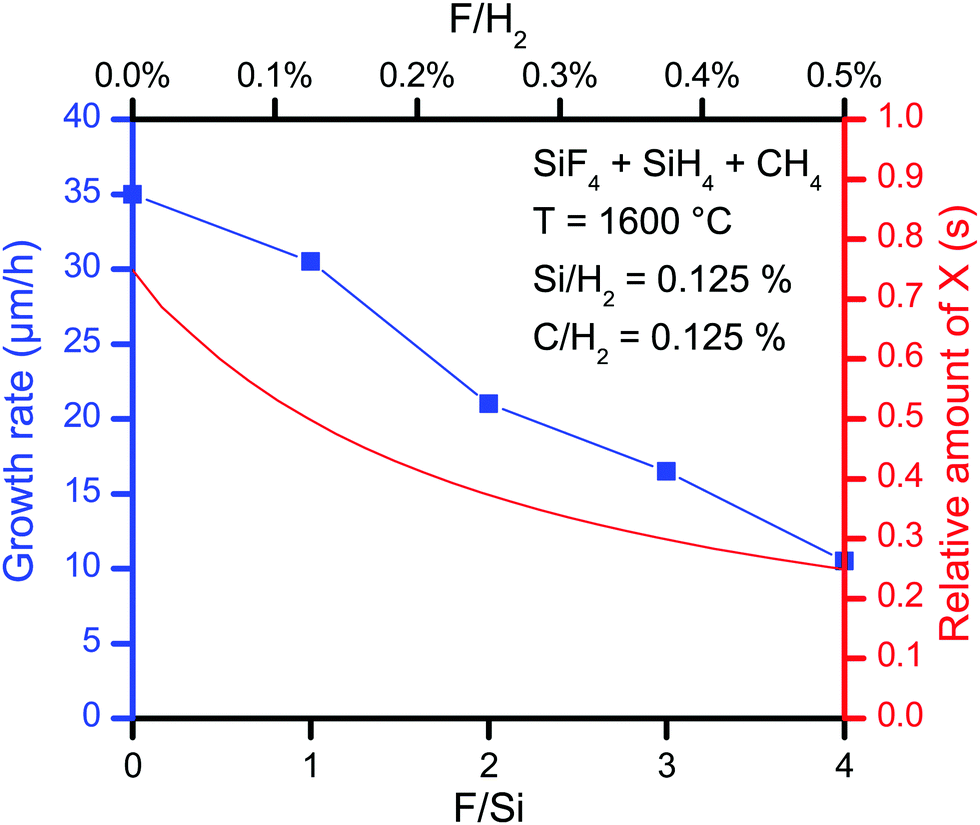 | ||
| Fig. 7 The relative amount of surface vacancies available for chemisorption of C-bearing growth species and the growth rate obtained for various F/Si or F/H2 ratios. | ||
So while the fluorinated CVD chemistry for SiC is problematic, we show that there is a better match between SiF4 and CH4 than between SiF4 and C2H4. This is opposite to when CH4 is used in chlorinated SiC CVD, where good growth conditions are confined to a narrower C/Si ratio window than when using C2H4 as the C precursor.28 The different rates at which growth species are formed from the precursors also suggests that combining different amounts of different precursors could be a way of controlling the spatial profile of the growth species impingement rate, and thereby the spatial profile of the C/Si impingement ratio.
Conclusions
We show that matching the kinetics of the silicon and carbon precursors in a SiC CVD process is necessary for achieving large-area homoepitaxial growth of SiC. We show this by comparing CH4, C2H4 and C3H8 as carbon precursors to the SiF4 silicon precursor where CH4 with the slowest kinetics renders the most robust CVD chemistry with large-area epitaxial growth and low-temperature sensitivity. It is also shown how the amount of F in the system affects the surface for further growth and that the growth rate could be limited by the amount of available surface sites for the C to adsorb onto. We show that the slow kinetics of the CH4 molecule matches the slow kinetics of the SiF4 molecule while the C2H4 and C3H8 molecules have faster kinetics, leading to a reduced process temperature window. We suggest that this precursor kinetics matching is a generic approach to choosing suitable combinations of CVD chemistries.Acknowledgements
Shortly after the original submission of this paper one of the authors, Erik Janzén, unexpectedly passed away. The other authors wish to express their deepest appreciation to Erik Janzén for giving them the chance to work with him in the field of SiC CVD and for all scientific discussions they had with him. His passion and devotion to science will forever serve as a guiding light. This work was supported by the Knut & Alice Wallenberg Foundation (KAW) project “Isotopic Control for Ultimate Material Properties” and the Swedish Foundation for Strategic Research project “SiC – the Material for Energy-Saving Power Electronics” (EM11-0034). L. O. acknowledges financial support from the Swedish Government Strategic Research Area in Materials Science on Functional Materials at Linköping University (Faculty Grant SFO Mat LiU No. 2009 00971). Supercomputing resources were provided by the Swedish National Infrastructure for Computing (SNIC) and the Swedish National Supercomputer Centre (NSC).References
- H. Pedersen and S. D. Elliott, Theor. Chem. Acc., 2014, 133, 1476 CrossRef.
- H. Matsunami and T. Kimoto, Mater. Sci. Eng., R, 1997, 20, 125 CrossRef.
- A. A. Burk, Chem. Vap. Deposition, 2006, 12, 465 CrossRef CAS.
- H. Pedersen, S. Leone, A. Henry, O. Kordina, S. Nishizawa, Y. Koshka and E. Janzén, Chem. Rev., 2012, 112, 2434 CrossRef CAS PubMed.
- CRC Handbook of Chemistry and Physics, ed. W. M. Haynes, Internet Version 2015, CRC Press/Taylor and Francis, Boca Raton, FL, 95th edn, 2015 Search PubMed.
- M. Yazdanfar, Ö. Danielsson, E. Kalered, P. Sukkaew, O. Kordina, D. Nilsson, I. G. Ivanov, L. Ojamäe, E. Janzén and H. Pedersen, Chem. Mater., 2015, 27, 793 CrossRef CAS.
- T. Rana, M. V. S. Chandrashekhar and T. S. Sudarshan, Phys. Status Solidi A, 2012, 209, 2455 CrossRef CAS.
- P. Stenberg, P. Sukkaew, I. Farkas, O. Kordina, E. Janzén, L. Ojamäe, Ö. Danielsson and H. Pedersen, J. Phys. Chem. C, 2017, 121, 2711 CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. A. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. A. Peterssonet al., Gaussian 09, Gaussian, Inc., Wallingford, CT, USA, 2009 Search PubMed.
- R. J. Bartlett, J. Phys. Chem., 1989, 93, 1697 CrossRef CAS.
- K. Raghavachari, G. W. Trucks, J. A. Pople and M. A. Head-Gordon, Chem. Phys. Lett., 1989, 157, 479 CrossRef CAS.
- D. E. Woon and T. H. Dunning, J. Chem. Phys., 1993, 98, 1358 CrossRef CAS.
- K. A. Peterson, D. E. Woon and T. H. Dunning, J. Chem. Phys., 1994, 100, 7410 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 85, 270 CrossRef.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 85, 299 CrossRef.
- W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 85, 284 CrossRef.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 CrossRef CAS.
- L. A. Curtiss, P. C. Redfern and K. Raghavachari, J. Chem. Phys., 2007, 126, 084108 CrossRef PubMed.
- E. Erdtman, M. Andersson, A. Lloyd Spetz and L. Ojamäe, Surf. Sci., 2017, 656, 77 CrossRef CAS.
- MATLAB R2015a, The MathWorks Inc., Natick, MA, USA, 2015 Search PubMed.
- P. Sukkaew, Ö. Danielsson, O. Kordina, E. Janzén and L. Ojamäe, J. Phys. Chem. C, 2017, 121, 1249 CAS.
- D. J. Larkin, P. G. Neudeck, J. A. Powell and L. G. Matus, Appl. Phys. Lett., 1994, 65, 1659 CrossRef CAS.
- M. Yazdanfar, H. Pedersen, P. Sukkaew, I. G. Ivanov, Ö. Danielsson, O. Kordina and E. Janzén, J. Cryst. Growth, 2014, 390, 24 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7tc00138j |
| This journal is © The Royal Society of Chemistry 2017 |