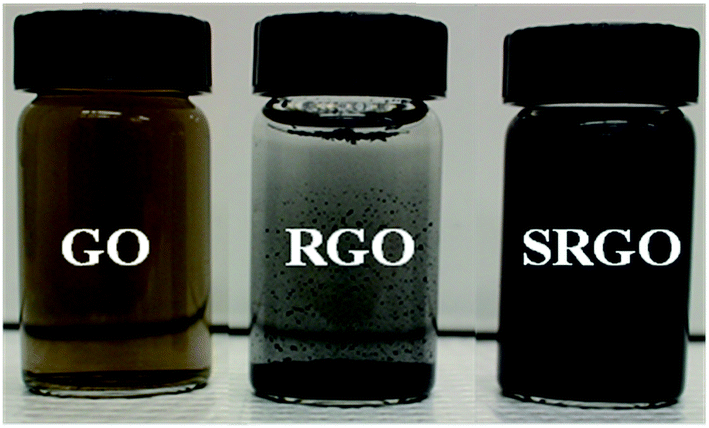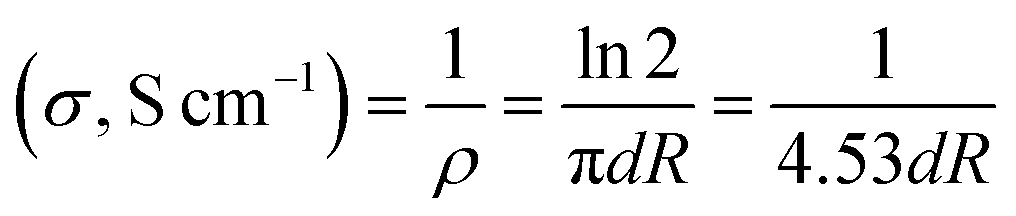DOI:
10.1039/C6RA28311J
(Paper)
RSC Adv., 2017,
7, 27224-27234
Synthesis and characterization of sulfophenyl-functionalized reduced graphene oxide sheets†
Received
16th December 2016
, Accepted 10th May 2017
First published on 23rd May 2017
Abstract
We report the modification of graphene oxide (GO) by thermal reduction to obtain reduced graphene oxide (RGO) and subsequent modification by sulfophenyl groups as well as the characterization of these materials by thermogravimetric analysis coupled with mass spectroscopy (TGA-MS). The chemical modification of RGO was carried out by the spontaneous reaction of RGO with in situ generated sulfophenyl diazonium ions. The three different types of materials were also characterized by elemental analysis, Fourier transform infrared spectroscopy (FTIR), Raman spectroscopy and X-ray photoelectron spectroscopy (XPS). The characteristic absorption band at 1034 and 1160 cm−1 in the FTIR spectrum of the sulfophenyl-modified RGO (SRGO), as well as Raman spectroscopy and TGA-MS data indicated that sulfophenyl groups were successfully grafted on RGO. The presence of organic molecules at the SRGO surface was also demonstrated by elemental analysis, transmission electron microscopy, energy dispersive X-ray spectroscopy and XPS. TGA data and elemental analysis results showed that the loading of sulfophenyl groups was about 12 wt% and UV-visible-near IR spectroscopy confirms the slight increase of the optical band gap of RGO after covalent grafting of sulfophenyl groups on its surface.
1 Introduction
Graphene is a two-dimensional carbon material consisting of a single-atom-thick graphitic layer that has been used in electronic devices,1,2 composite materials3,4 and energy storage systems.5–9 Graphene is commonly produced from natural graphite, which is widely available at low cost.10 However, graphene is not directly prepared from graphite. Instead it is obtained by the reduction of graphene oxide (GO), previously produced by the Hummers method.11 The reduction of GO to graphene restores the electronic properties of graphene and has been performed by using reducing agents such as hydrazine (N2H4),12,13 sodium borohydride (NaBH4),14,15 dimethyl hydrazine3 and hydriodic acid (HI).16,17 However, these reducing agents may be harmful to the environment or too expensive when used for mass production of graphene. Also, the quality of the reduced GO, RGO, strongly depends on the reducing agent and other experimental conditions.18 Alternatively, thermal reduction, which is considered a green method because no hazardous chemicals are required, can also afford RGO.
Graphene possesses a zero band gap that severely limits its applications due to its chemical inertness.4,19 Opening the band gap of graphene and its derivative by doping, intercalation or grafting by organic molecules would be useful for applications mentioned above.6,8 Importantly, functionalization with organic molecules led to a good dispersion of graphene in common organic solvents.20 For several large-scale applications, RGO is a more widely used and attractive material than graphene. Similarly, its functionalization is important to modify its properties and open up its applications to the areas.
Here we report, a detailed investigation of graphene oxide (GO), reduced graphene oxide (RGO) and sulfophenyl-modified RGO (SRGO) by thermogravimetric analysis coupled with mass spectrometry (TGA-MS) analysis. The TGA-MS data confirmed the covalent grafting of sulfophenyl groups on RGO. The three materials were also characterized by nitrogen gas adsorption, FTIR, four-point probe measurements as well as by Raman and X-ray photoelectron spectroscopy.
2 Experimental
Preparation of graphene oxide (GO)
Graphene oxide was synthesized from natural graphite (<44 μm, 99.99%, supplied by Sigma-Aldrich) through the Hummers method11 which has been improved to be more environmentally friendly and produce graphene in good yield (95%). The graphite is first pre-oxidized by mixing graphite powder (5 g) with concentrated sulfuric acid (H2SO4, 12.5 mL), potassium persulfate (K2S2O8, 2.5 g) and phosphorus pentoxide (P2O5, 2.5 g). The mixture was heated at 80 °C for 6 hours. After dilution with 500 mL of H2O, the mixture was stirred at room temperature overnight. After that, the product is recovered by centrifugation and washed thoroughly with Nanopure water until the filtrate has a pH close to 7 (neutral). The product obtained is then dried at room temperature for one day. Then, the pre-oxidized graphite is dispersed in H2SO4 (0 °C, 115 mL). The temperature of the mixture is carefully controlled to not exceed 10 °C. Subsequently, potassium permanganate (KMnO4, 15 g) is gradually added with constant stirring for 1 hour. The dispersion is then incubated at 35 °C for 2 h and this is followed by the addition of Nanopure water (225 mL) in small portions (15 mL) to control the temperature of the mixture, which must remain below 50 °C. In order to completely dissolve the KMnO4 remaining, hydrogen peroxide (H2O2 30%, 12.5 mL) was immediately added at the end of a second dilution (H2O, 700 mL), and the mixture is stirred for 48 hours. Finally, the suspension is filtered, washed first with HCl (10%) to remove residual metal ions, and repeatedly with Nanopure water until the pH of filtrate becomes neutral. The filtrate is quickly tested by adding a few drops of 1 M NaOH to verify the presence of metal ions in GO. The product obtained (graphite oxide) is then dried in air. The resultant graphite oxide was dispersed in Nanopure water kept in the ultrasonic bath for 24 hours to maximize exfoliation. A homogeneous and stable colloidal suspension for several months is obtained (Fig. 1, GO).
 |
| | Fig. 1 Optical images of GO, RGO and SRGO dispersed in water at the concentration of 0.5 mg mL−1. | |
Preparation of reduced graphene oxide (RGO)
The reduced graphene oxide (RGO) is obtained by thermal reduction of GO in Ar/5% H2 at various temperatures for 2 h. The resulting RGO can be dispersed in water and the dispersion stayed stable for few hours, as shown in Fig. 1.
Covalent attachment of 4-sulfophenyl groups by the diazonium chemistry on RGO surface
Typically, a mass of 100 mg of RGO is dispersed in 100 mL of an acetonitrile/H2O (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50, v/v) mixture and a homogeneous and stable colloidal suspension was obtained after sonication, for 30 min. Then, 15 mmol of amine (4-aminobenzenesulfonic acid) and an excess of sodium nitrite (22.5 mmol; 1.5 equiv. compared to the amine) was directly added to the dispersion. The mixture was dispersed by sonication during an additional 30 min to completely dissolve the reagents and this was followed by the addition of 10 mL of concentrated HCl. The reaction mixture remained under agitation for 24 hours at room temperature. The dispersion was finally vacuum filtered on a Nylon filtration membrane having a pore size diameter of 0.47 μm (Pall) and the resulting powder was successively washed with a mixture acetonitrile/Nanopure water, acetonitrile, DMF, methanol and acetone. Finally, the resulting modified-RGO (SRGO) was dried under vacuum at 80 °C overnight before being subjected to thermal annealing under Ar atmosphere at 250 °C for 1 h. The resulting powder is dispersible in water and remains stable for several days (Fig. 1, SRGO).
:50, v/v) mixture and a homogeneous and stable colloidal suspension was obtained after sonication, for 30 min. Then, 15 mmol of amine (4-aminobenzenesulfonic acid) and an excess of sodium nitrite (22.5 mmol; 1.5 equiv. compared to the amine) was directly added to the dispersion. The mixture was dispersed by sonication during an additional 30 min to completely dissolve the reagents and this was followed by the addition of 10 mL of concentrated HCl. The reaction mixture remained under agitation for 24 hours at room temperature. The dispersion was finally vacuum filtered on a Nylon filtration membrane having a pore size diameter of 0.47 μm (Pall) and the resulting powder was successively washed with a mixture acetonitrile/Nanopure water, acetonitrile, DMF, methanol and acetone. Finally, the resulting modified-RGO (SRGO) was dried under vacuum at 80 °C overnight before being subjected to thermal annealing under Ar atmosphere at 250 °C for 1 h. The resulting powder is dispersible in water and remains stable for several days (Fig. 1, SRGO).
Morphological, structural and optical characterization
The morphology of the graphene materials was investigated by transmission electron microscopy (TEM) using a 200 keV JOEL JEM-2100F model transmission electron microscope operated with a bright field image. The energy band gap of reduced graphene oxide (RGO) and sulfophenyl-modified reduced graphene oxide (SRGO) was determined at room temperature using UV/VIS/NIR Spectrophotometer Lambda 750. For these analyses, the graphene samples were dispersed in NMP (N-methyl-2-pyrrolidone) to form colloidal suspensions (0.03 mg mL−1). The optical band gap (Eg) of RGO and SRGO were estimated using the Tauc–David Mott equation (eqn (1)).21–25where hν is the photon energy (h is Planck's constant, ν is the light frequency), α the absorption coefficient, Eg is the optical gap, the nature of band transition characterized by n = 1/2, 2, 3/2 and 3 and the constant, which is different for each transition.25 Fourier transform infrared (FTIR) spectroscopy was employed to characterize the graphene materials using the Nicolet 6700 FTIR in the 3800–700 cm−1 region. XPS spectra were collected using the spectrophotometer PHI 5600-ci (Physical Electronics, Eden Prairie, MN, USA). The excitation source used for survey spectra was by standard Al Kα (1486.6 eV) X-rays at 400 W and for core level spectra, by Mg Kα (1253.6 eV) X-rays at 150 W. The analyses were performed without charge compensation at an angle of 45° with the surface. The detector aperture was set at 5 and the surface area analyzed was 0.016 cm2. The core level spectra were curve-fitted with the Casa XPS software by using mixed Gaussian–Lorentzian product function (70% Gaussian). Each spectrum was corrected with respect to C 1s at 284.5 eV (C sp2 graphite like carbon) and a Shirley type background subtraction was performed before curve-fitting. Thermogravimetric analysis coupled with mass spectroscopy (TGA-MS) was carried out with a thermal gravimetric analyzer (TA Instruments TGA (Q500)/Discovery MS). Samples of typically 2 mg were placed in Pt pans and heated from 30 to 900 °C with a temperature ramp of 5 °C min−1, under flowing helium (He) atmosphere. Raman spectroscopy measurements were performed using a micro-Raman system (UHTS300) with excitation from an argon ion laser beam (532 nm) at low power level (2 mW) in order to avoid damaging the organic functional groups. The surface area and pore volume were quantified using the Brunauer–Emmett–Teller (BET) method from the adsorption branch data set recorded for P/P0 values between 3 × 10−1 and 5 × 10−2. The volume of nitrogen adsorbed was recorded for relative pressures (P/P0) ranging from 1 × 10−6 to 1. The density functional theory (DFT) was used to provide a much more accurate approach to pore size analysis.26 Prior to measurements, the sample was degassed for 4 h under vacuum. Electronic conductivity of GO, RGO and SRGO films was obtained at room temperature by using a 4-point probe measurement by using a Keithley 6220 DC precision current source (US). The GO, RGO and SRGO films were obtained as follows. The samples were dispersed in water by ultrasonication for 30 min and the resulting suspensions were filtered through a polytetrafluoroethylene (PTFE) membrane filter by vacuum filtration and dried under vacuum at 70 °C overnight. The electrical conductivities were calculated by using the following equation:27| | 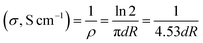 | (2) |
where ρ (Ω cm) is the resistivity, d (cm) is the sample thickness and R (Ω) is the resistance.
3 Results and discussion
Morphological characterization of RGO and SRGO
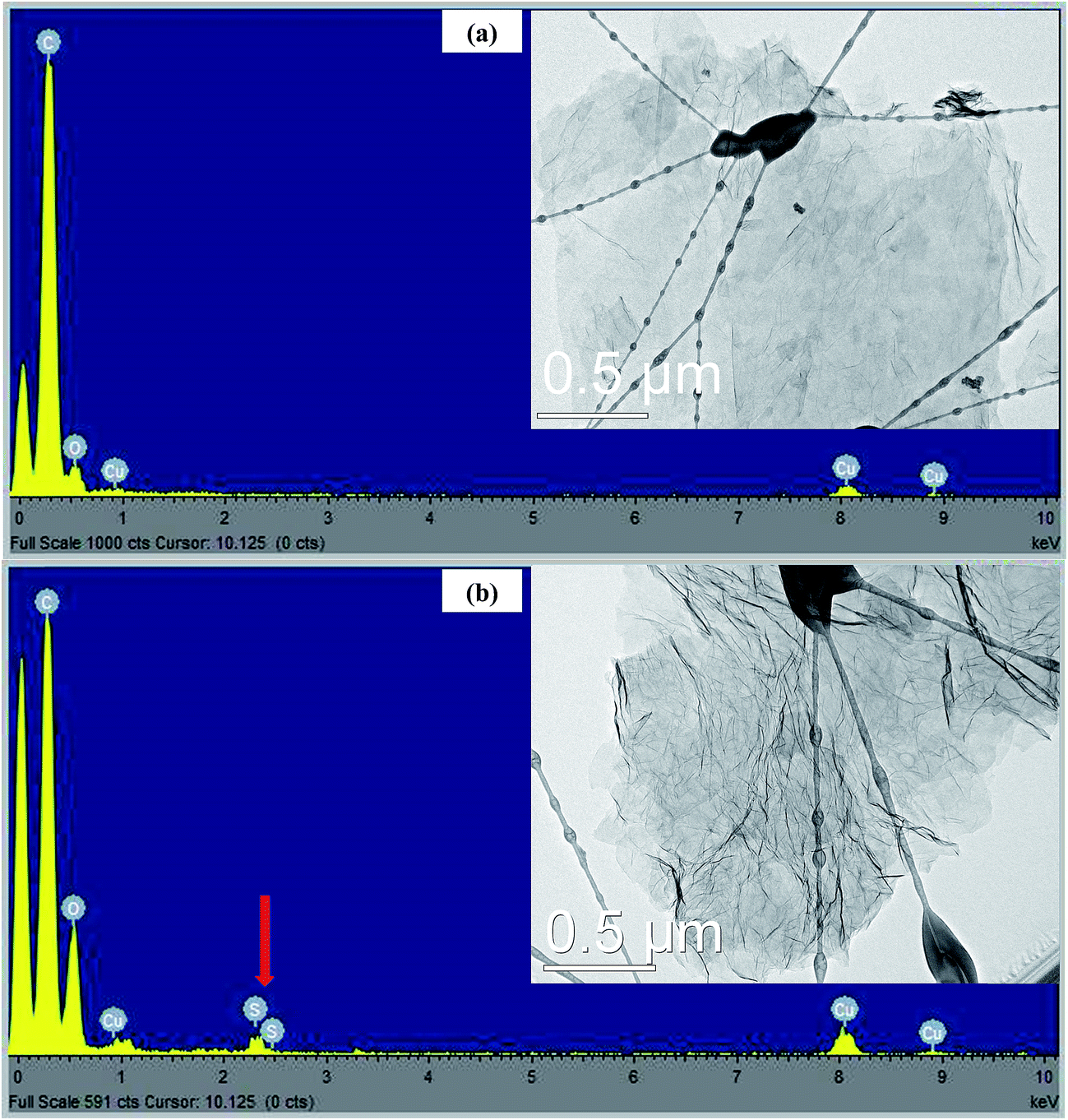
TEM images were employed to study the morphology of RGO and SRGO. Fig. 2a clearly shows that the RGO sheets are almost transparent, suggesting that they consist of a few layers. The EDX spectrum (Fig. 2a) of the RGO sample confirms the presence of only carbon and of a small amount of oxygen (O/C: 0.08). On the other hand, Fig. 2b exhibits aggregated and wrinkled sheets.
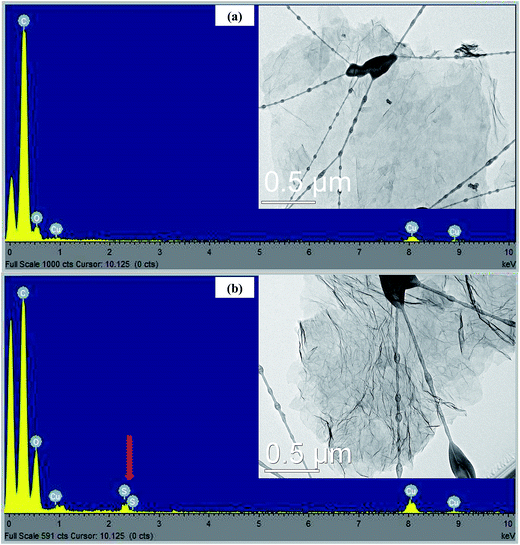 |
| | Fig. 2 EDX spectra and TEM images of (a) RGO and (b) SRGO. | |
This observation demonstrates that the attachment of organic groups on the surface of graphene sheets favour their overlapping and folding.20,28–30 It is an indirect way to confirm the functionalization of RGO sheets with organic molecules. Energy dispersive spectrum of SRGO (Fig. 2b) shows sulfur peaks that confirm the presence of sulfophenyl groups on reduced graphene oxide.
Optical band gap
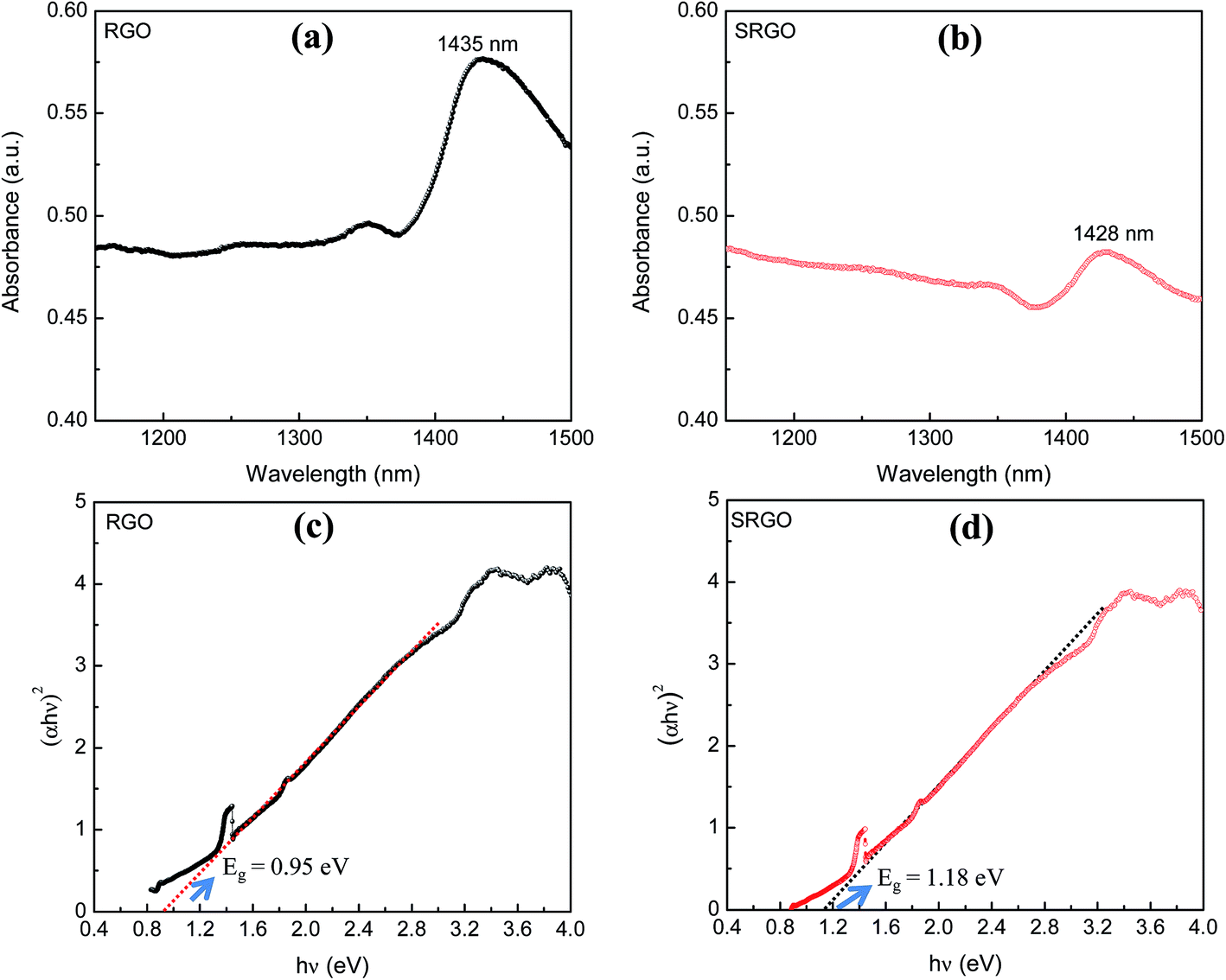
The visible/near-infrared absorption spectra of RGO and SRGO presented in Fig. 3a and b were measured to evaluate the optical band gap of graphene materials. The absorption spectra of RGO and SRGO show an absorption band around 1430 nm. The optical band gap (Eg) of the graphene materials was estimated from the absorption spectra (ESI, Fig. S1†) by plotting (αhν)2versus hν, as shown in Fig. 3c and d, and extrapolating the linear region of the curve to the x-axis.31 The band gap slightly increased following RGO functionalization, from 0.95 (ref. 32) to 1.18 eV SRGO, indicating that the optical properties of RGO have been changed after chemical modification with sulfophenyl groups.33
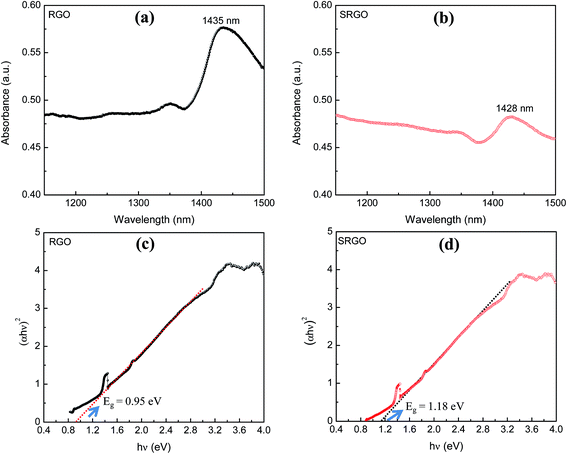 |
| | Fig. 3 Visible/near-infrared spectra of (a) RGO, (b) SRGO and the plot of (αhν)2 as a function of hν for (c) RGO and (d) SRGO (see ESI, Fig. S1† for complete absorption spectra). | |
Nitrogen gas adsorption
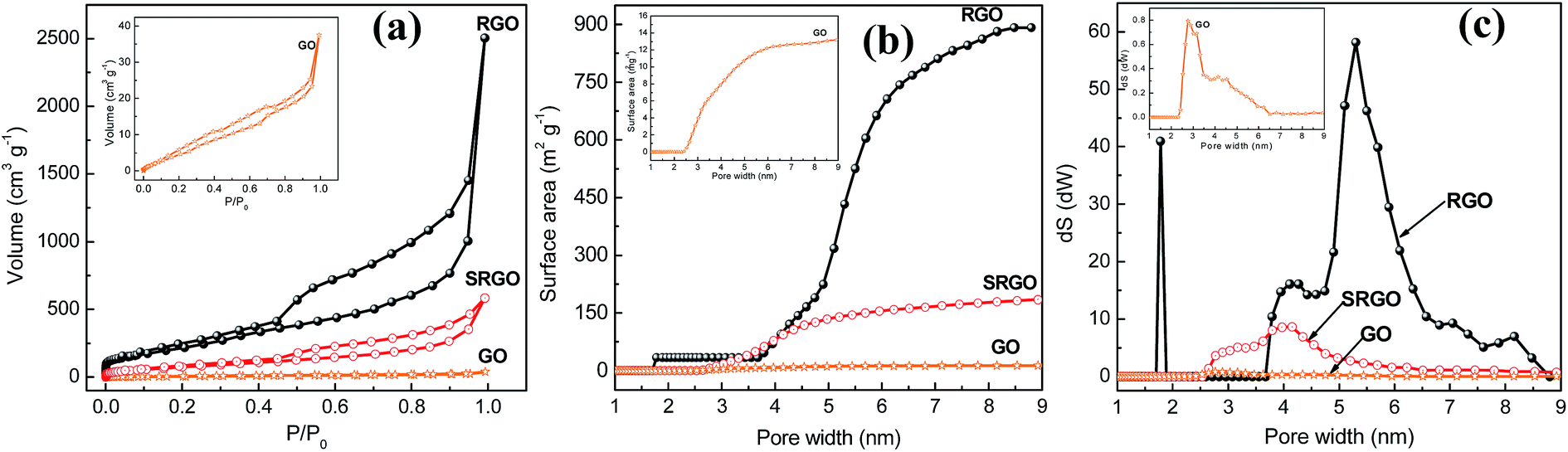
Fig. 4a shows the nitrogen adsorption/desorption isotherm at 77 K for GO, RGO and SRGO. The adsorption isotherm of GO is featureless and shows a small volume of adsorbed nitrogen gas at low relative pressure (Fig. 4a, GO and inset).34 This translates in a low specific surface areas of 25 and 15 m2 g−1 according to BET and DFT Monte Carlo approaches, respectively (Table 1), which is in agreement with that reported in the literature.35 Both unmodified and modified RGO present mixed type I and type II isotherms for low and high relative pressure (P/P0),9,36,37 respectively. At low P/P0 (0–0.5), the low adsorbed volume for RGO and SRGO is characteristic of mesoporous-like material, which is confirmed by the plateau and a H3 hysteresis loop.38,39 It can be seen that, after modification of RGO, a drop of the adsorbed volume is observed for SRGO a low relative pressure. The effect of organic molecules grafting on graphene sheets can be quantified by BET surface area (Table 1), the cumulated surface area (Fig. 4b) as well as the pore size distribution (Fig. 4c).
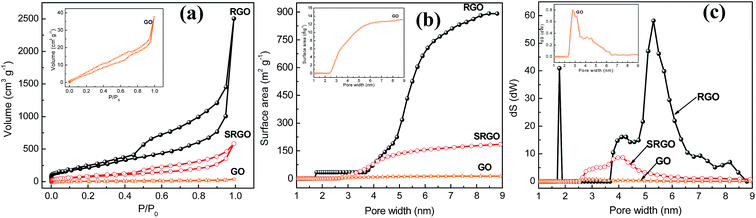 |
| | Fig. 4 (a) N2 adsorption isotherms of graphene oxide, reduced graphene oxide before (RGO) and after reaction with in situ-generated 4-sulfophenyl diazonium cations (SRGO), (b) cumulated surface area vs. pore width of GO, RGO and SRGO, (c) pore size distribution of GO, RGO and SRGO. The insets present the data for GO at a more sensitive scale. | |
Table 1 Specific surface areas and electronic conductivity of graphene oxide (GO), reduced graphene oxide (RGO) and sulfophenyl-modified reduced graphene oxide (SRGO)
| Sample |
BET surface area (m2 g−1) |
DFT surface area (m2 g−1) |
Electronic conductivity, σ (S cm−1) |
| GO |
25 |
15 |
0.5 |
| RGO |
900 |
895 |
7.7 |
| SRGO |
300 |
200 |
2.2 |
The BET surface area of RGO is about 900 m2 g−1 and the material consists of small mesopores (2–50 nm) (Fig. 4c). The high specific surface area demonstrates that the thermal reduction with loss of oxygen functional groups created porosity. Although, it is still far below to the theoretical value for completely exfoliated and isolated graphene sheets (2630 m2 g−1),4,40,41 it compares well with those published in the literature.12,41–43 The lower value could be due to the agglomeration/precipitation and partial overlapping of reduced sheets during the thermal reduction process, which could lead to inaccessible surface.42 Following grafting of sulfophenyl groups, the BET surface area decreased to 300 m2 g−1 for SRGO (Fig. 4b and Table 1). This decrease of the BET surface area provides indirect evidence for grafting. Obviously, the attachment of organic molecules at the RGO surface causes changes in the graphene structure and creates a similar situation to that of the oxides on the surface of the graphene sheets.44,45 The grafting block some pores of graphene sheets aggregates which make them inaccessible,9 which creates a significant decrease of the cumulated surface area (Fig. 4b) and a noticeable difference of the pore size distribution (Fig. 4c). Interestingly, grafting of sulfophenyl groups lead to smaller pores which might be formed by the decarboxylation of groups present on RGO.45,46
FTIR
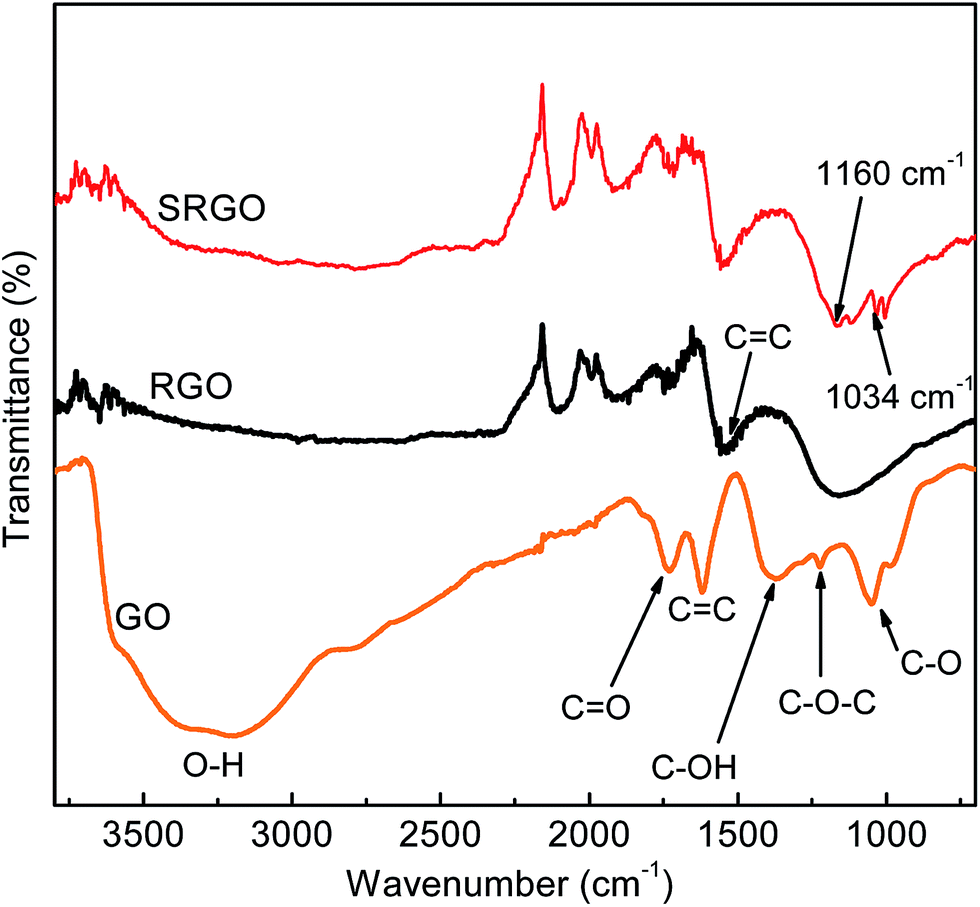
Fig. 5 shows the Fourier transform infrared spectroscopy (FTIR) spectra of GO, RGO and SRGO. The spectrum of GO (Fig. 5a) displays the presence of bands associated to C–O (υC–O at 1048 cm−1), C–O–C (υC–O–C at 1223 cm−1), C–OH (υC–O–H at 1376 cm−1), C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O in carboxylic acid and carbonyl moieties that are present mostly along sheet edges but also on the basal plane of graphene sheets (υCO at 1725 cm−1) and a broad peak between 3000 and 3500 cm−1 corresponding to O–H vibration.13,43,47 The RGO spectrum exhibits only two peaks at 1160 cm−1 (υC–O–H) and 1550 cm−1 (υCC), which suggests that the GO has been effectively reduced during the process. The slight shift of these two bands to higher energy indicates the restoration of the π-network.43 The SRGO spectrum shows new bands at 1034 and 1160 cm−1, which fall within the range of the symmetric and asymmetric stretching modes of –SO3H functional groups and demonstrate the presence of sulfophenyl groups on RGO.48–50
O in carboxylic acid and carbonyl moieties that are present mostly along sheet edges but also on the basal plane of graphene sheets (υCO at 1725 cm−1) and a broad peak between 3000 and 3500 cm−1 corresponding to O–H vibration.13,43,47 The RGO spectrum exhibits only two peaks at 1160 cm−1 (υC–O–H) and 1550 cm−1 (υCC), which suggests that the GO has been effectively reduced during the process. The slight shift of these two bands to higher energy indicates the restoration of the π-network.43 The SRGO spectrum shows new bands at 1034 and 1160 cm−1, which fall within the range of the symmetric and asymmetric stretching modes of –SO3H functional groups and demonstrate the presence of sulfophenyl groups on RGO.48–50
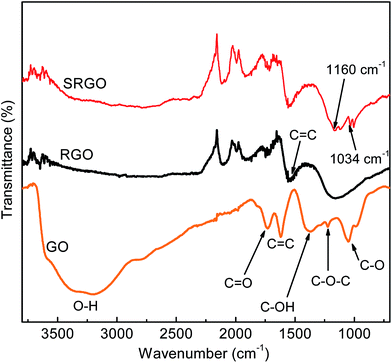 |
| | Fig. 5 FTIR curves of graphene oxide, reduced graphene oxide and sulfophenyl groups modified RGO. | |
Raman spectroscopy
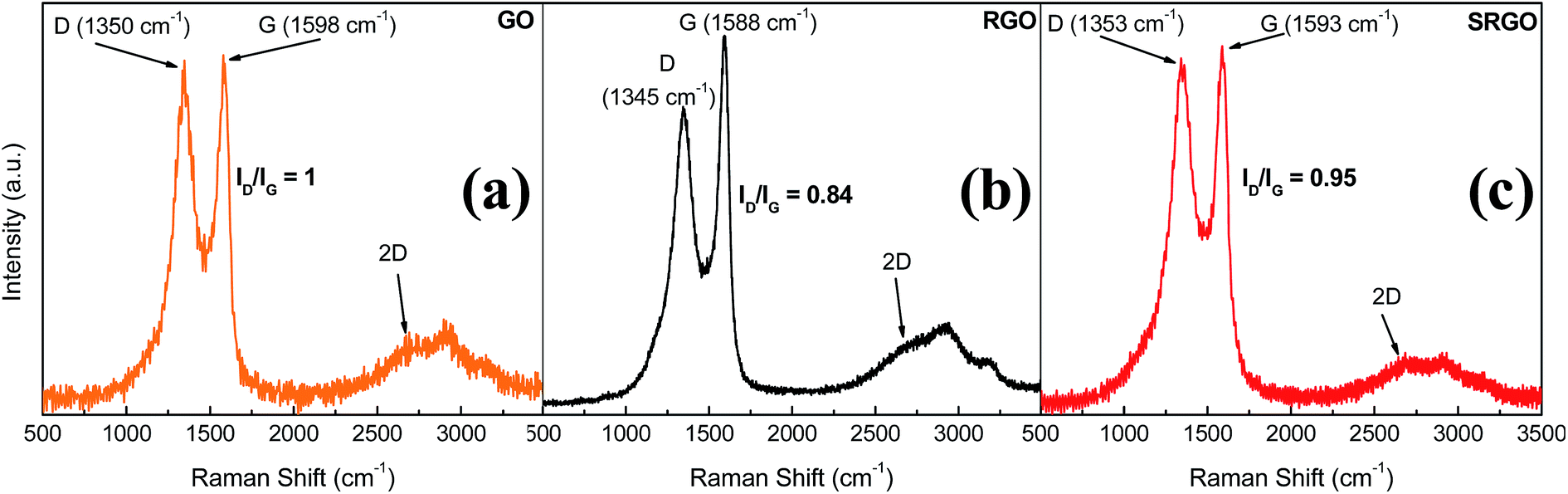
Raman spectroscopy provides valuable information for graphene and its derivates because it is very sensitive to the electronic structure of the carbon nanostructures, therefore the degree of hybridization, the crystal disorder and the extent of chemical modification.20,51Fig. 6 shows the Raman spectra obtained for GO, RGO and SRGO. Each spectrum exhibits a G band corresponding to the first-order scattering of the E2g mode42 around 1600 cm−1 and a D band arising from the doubly resonant disorder-induced mode at ∼1350 cm−1.52–56 The ratio of the intensity of these two bands (ID/IG) is included on the figure.
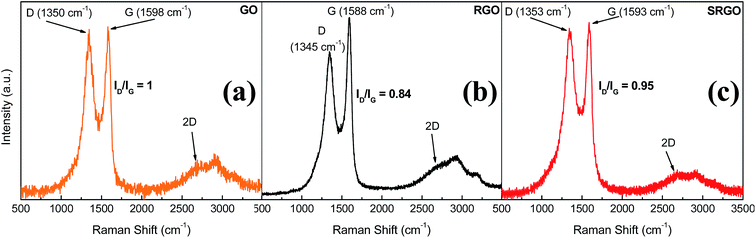 |
| | Fig. 6 Raman spectra of GO, RGO and SRGO recorded using 532 nm laser excitation. | |
The GO sample shows a prominent D peak with a ID/IG of 1 indicative of significant structural disorder created due to the presence of oxygen functional groups.57,58 Consequently, the sharp increase of the ID/IG ratio from ∼0.09 for pristine graphite (ESI, Fig. S2†) indicates a decrease in the in-plane crystal and a partial amorphization of graphite, by conversion of sp2 to sp3 carbon bonds. The G peak of GO is shifted to higher energy (∼19 cm−1) and broadened significantly compared to that of pristine graphite (ESI, Fig. S2†).52,56,57,59 Following thermal reduction of graphene oxide, the vibration frequency of the G band decreases to 1588 cm−1 (Fig. 6b), a value still slightly higher than that of pristine graphite. This phenomenon could be attributed to the influence of residual defects and isolated double bonds in RGO.56,60 Nevertheless, the ID/IG ratio in this case decreases to 0.84, indicating that there were some structural changes occurring during the thermal reduction process, which did not much altered the structure of RGO but partially restored the graphitic (sp2) network.58Fig. 6c shows the Raman spectrum of functionalized RGO (SRGO) by sulfophenyl groups. The increase of the ID/IG ratio from 0.84 to 0.95 reflects the enhancement of in disorder after grafting which is due to the transformation of sp2 carbon to sp3 during the covalent attachment of organic molecules on graphene sheets.20 The slight shift of G-band (∼5 cm−1) to higher energy confirms the covalent grafting of organic molecules, which often isolates sp2 C atoms.13,53,61
The shape and the position of 2D band of GO, RGO and SRGO spectra around 2680 cm−1 indicate that these graphene materials consist of few layers.62,63 Also, it provides information on the quality of graphene oxide initially synthesized. Typically, the low intensity of 2D peak and the profile recorded in this region are the signature of graphene oxide and its derivatives.58,64 This low intensity and broad 2D peak for GO compared with those of electrochemically exfoliated graphene (EG)20,54 or prepared by chemical vapor deposition (CVD)65–67 reflects the more important contribution of the steric effects of oxygen functionalities on the stacked layers as well as the partial amorphization and reduction in sp2 domains.43,52
Electrical conductivity measurements
The electronic conductivities of GO, RGO and SRGO films are collected in Table 1. The low conductivity of graphene oxide (0.5 S cm−1) is due to the lack of π-electronic conjugation caused by the extensive oxidation of graphite during the Hummers process.11 The thermal reduction increases the conductivity68,69 to 7.7 S cm−1. Theoretically, the reduction of GO should firstly remove the oxygen functionalities groups, secondly rehybridize the sp3 carbon atoms to sp2 C and finally leave the material defects free like pristine graphene. However, no reduction method could totally restore the sp2 structure of graphene. Then, the residual defects will affect the properties especially the electrical conductivity of RGO.17,69–71 The higher conductivity of RGO is in good agreement with its lower ID/IG ratio relative to that of GO. The significant decrease of electrical conductivity of SRGO (Table 1) is related to the covalent grafting of organic molecules that converts some sp2 C atoms to sp3 C atoms resulting in the increase of the ID/IG ratio (Fig. 6c) and disruption of the π-network.
X-ray photoelectron spectroscopy (XPS)
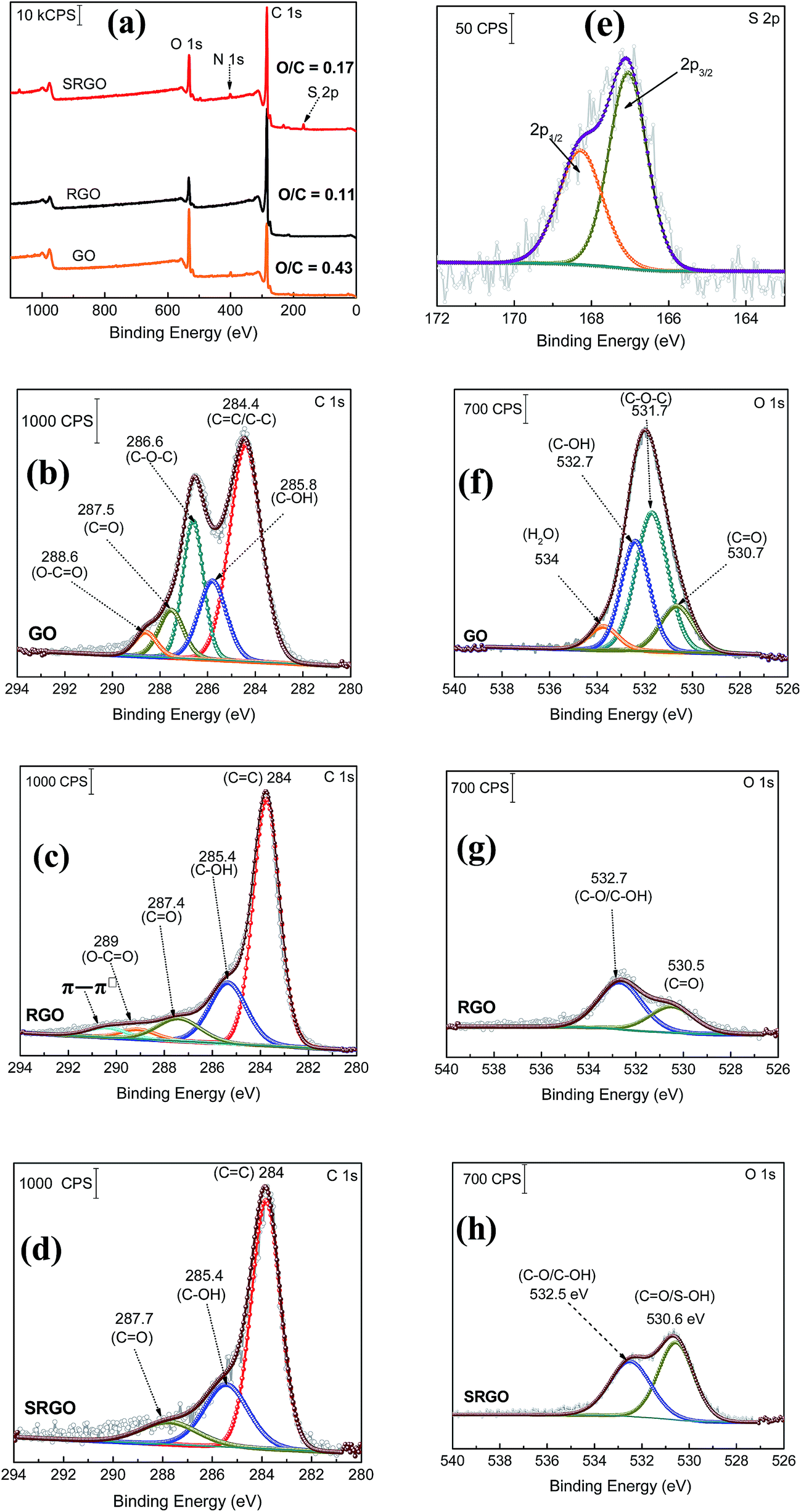
XPS is a valuable tool for the surface chemical analysis of carbon nanostructure materials and to confirm the immobilization of the different functional groups at the graphene surface during the synthesis or its functionalization.45,72,73Fig. 5 shows a set of XPS spectra for GO, RGO and SRGO. The survey spectra of GO and RGO exhibit the characteristic C 1s peak at 285 eV and O 1s at 533 eV and for GO an additional small N 1s peak at 400 eV which could be related the trapping of molecular nitrogen.62 The decrease of the O/C ratio from 0.43 to 0.11 after GO reduction to RGO demonstrates that thermal annealing eliminates most of the oxygen functionalities of graphene oxide.18,69,75 A decrease of the intensity of N 1s peak is also noticeable. Following RGO functionalization by sulfophenyl groups, the O/C ratio increases from 0.11 to 0.17 due to the sulfonate groups (–SO3−) immobilized on graphene sheets. In addition, the SRGO survey spectrum (Fig. 3a) displays additional peaks at 230 and 167.7 eV assigned to the sulfonate groups (S 2s and S 2p, respectively).76,77 The N 1s signal observed at 400 eV indicates the formation of azo bridges (C–NN–C) that are commonly present in grafted modified carbon materials by using diazonium cations.46,78–82 Their presence could be used as an indirect proof of grafting.83 Furthermore, core level spectra were recorded for the three samples (GO, RGO and SRGO) and curve-fitted spectra are shown in Fig. 7b–h.
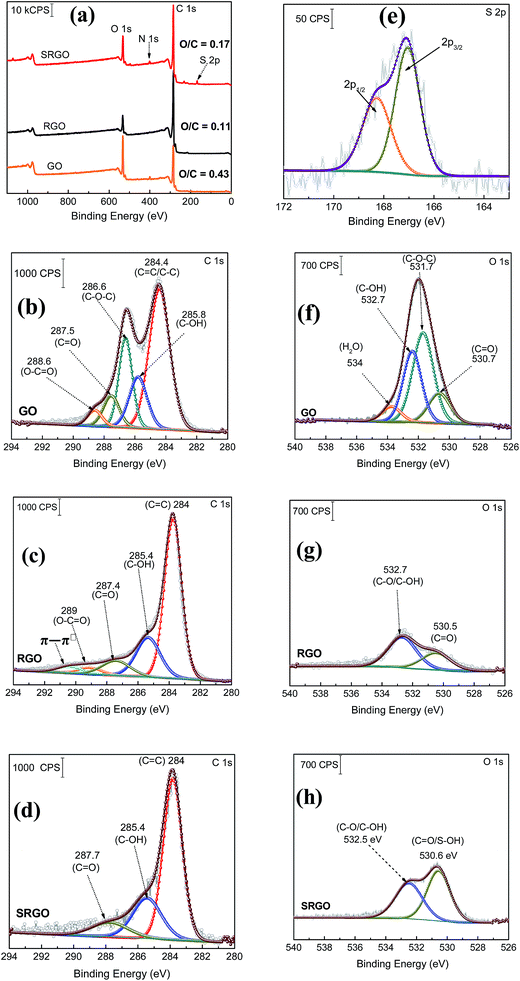 |
| | Fig. 7 (a) XPS survey spectra of graphene oxide (GO), reduced graphene oxide (RGO) and sulfophenyl-modified RGO (SRGO); XPS C 1s spectra of (b) GO, (c) RGO and (d) SRGO, (e) S 2p core level spectrum of SRGO and XPS O 1s spectra of (f) GO, (g) RGO and (h) SRGO. | |
C 1s region.
The C 1s core level spectrum of GO (Fig. 7b) shows a sp2 component (CC/C–C) in aromatic rings at 284.4 eV, followed by the surface oxides components (sp3) C–OH (285.8 eV), C–O–C (286.6 eV), CO (287.5 eV), and the carboxylate carbon (O–CO) at 288.6 eV. Their atomic concentration (at%) are given in ESI, Table S1.†17,42 The relative atomic concentration for different oxygenated carbon functional (C sp3) groups is also included in Table S1† together with their counter parts from the O 1s core level spectra. The C–O species represent 50% of the total carbon atoms (Table S1†). The high oxygen content of GO is essentially related to the use of KMnO4 as oxidizing agent during its preparation.84 For RGO, its C 1s core level spectrum was fitted with five components (Fig. 7c). The contribution at 284 eV, attributed to non-oxygenated ring C, shows a significant increase of its relative area and a decrease of the FWHM (ESI, Table S1†). This suggests that the thermal treatment partially restored the π-electron network by removing most of oxygen functional groups on graphene sheets.85 Indeed, the component observed at 286.6 eV (C–O–C) in GO spectrum (Fig. 7b), significantly decreased after thermal reduction as well as the one of C–OH at 285.8 eV (ESI, Table S1†).64,69,85,86 This result is in good agreement with the FTIR spectra of these two materials. The C 1s XPS spectrum of RGO (Fig. 3c) also exhibits the carbonyl (CO, 287.4 eV) and the carboxylate carbon (O–CO, 289 eV) with peak intensities noticeably reduced in comparison to GO. However, their relative atomic concentration in terms of oxygenated carbon species (ESI, Table S1†) slightly increased, probably due to CO2 blisters (especially from epoxy groups)85 which could be trapped between graphene sheets during the annealing.87,88 In addition, there is an additional component at 290.5 eV corresponding to shake-up satellite (π–π*) peak or to π-electrons delocalized in the aromatic network.89Fig. 5d displays the C 1s core level spectrum of modified RGO (SRGO) with sulfophenyl groups. The spectrum can be fitted with 3 major components. It can be seen that, after graphene functionalization the relative atomic concentration of C–OH (peak at 285.4 eV) increased from 66 to 78% (ESI, Table S1†), suggesting an increase of the contribution of graphene derived sp3 carbon (the C in C–N/C–O) bonds27,90,91 due to the reaction with sulfophenyl diazonium ions. It is also noted that the relative area of the peak around 287.7 eV decreased and the carboxylate component (O–CO) at 289 eV in RGO spectrum (Fig. 5c) disappeared after grafting of sulfophenyl groups. The departure of the carboxylic (COOH) groups can be attributed to the decarboxylation of the carboxylic functionalities present at the RGO surface during the reduction of the diazonium cations and subsequent grafting.45,46
S 2p region.
The core level spectrum of S 2p peak displayed in Fig. 3e can be curve-fitted with a doublet at 168.3 and 167 eV for S 2p1/2 and S 2p3/2, respectively (Fig. 7e). This confirms the presence of sulfonate groups (–SO3−) on the graphene sheets surface.92,93
O 1s region.
The O 1s core level spectrum of graphene oxide (GO) shown in Fig. 5f can be curve-fitted with two main contributions at 531.7 and 532.7 eV corresponding to C–O–C (46%)74,94,95 and C–O (37%), (ESI, Table S1†)72,74,85,95,96 bonds,85,97 respectively. A third component, located at 530.7 eV can be assigned to the ketone and quinone functionalities (CO) in lesser amount (16%, ESI, Table S1†), which arise at the edge or bonded to the basal plane of GO as carbonyl groups.85,98,99 The additional weaker contribution observed at 534 eV is related to water intercalation.43,85,89,95,96,99–102 After thermal reduction of GO, two components related to CO (530.5 eV) and C–OH (532.7 eV) became clearly visible with very low intensities (Fig. 5g)85,89 and a noticeable decrease of the relative areas (ESI, Table S1†) because of complete loss/conversion of C–O–C groups.103 Water molecules trapped between graphene layers, were removed during GO thermal reduction.85,104 Following RGO functionalization, the atomic concentration (ESI, Table S1†) and the peak intensity (Fig. 5g) of the component at 530.6 eV considerably increase. This observation is in good agreement with the introduction of the oxygenated groups attributed to S–O bonds.73
Thermogravimetric analysis coupled to mass spectrometry
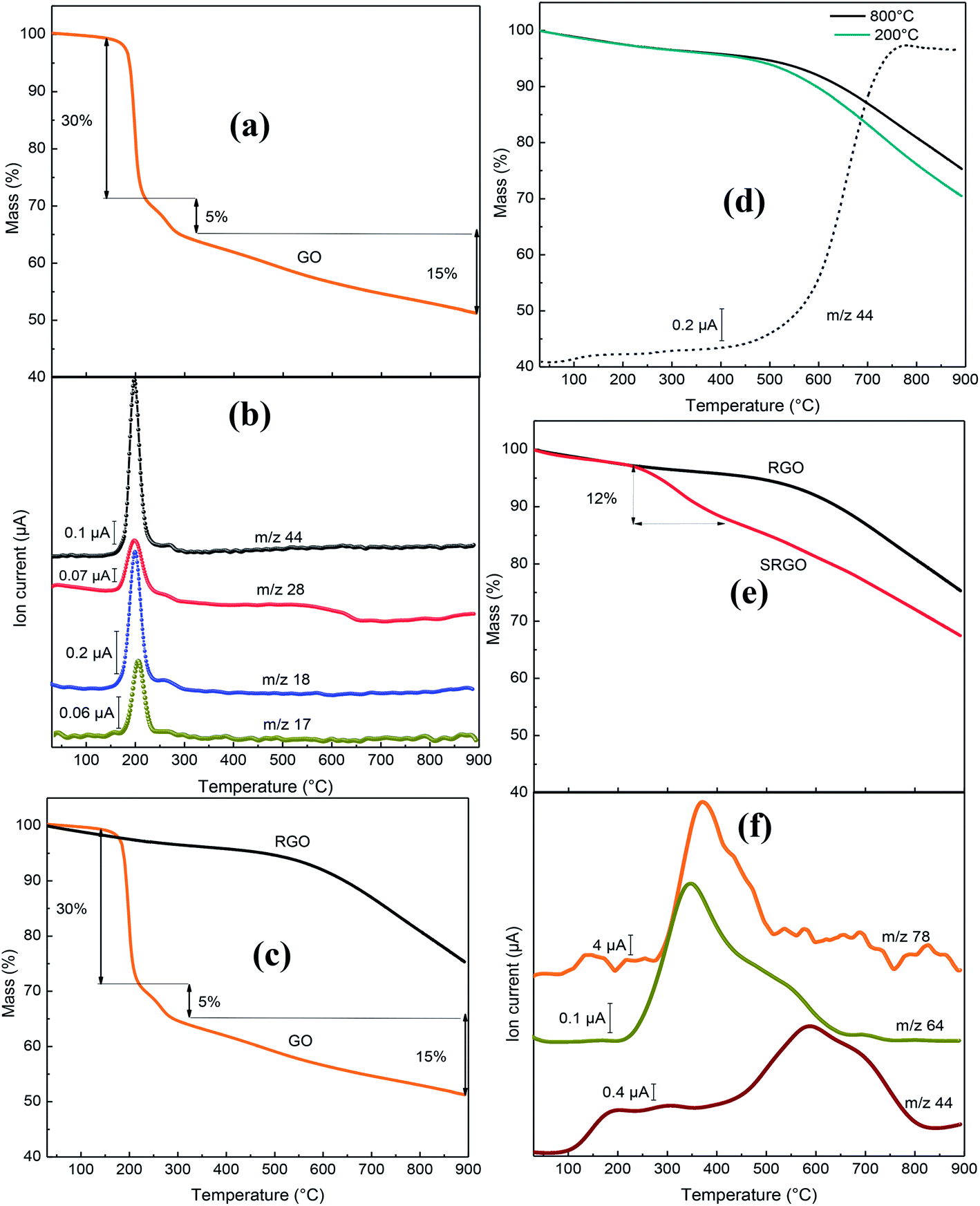
The novelty of our work relative the vast literature existing on functionalized graphene lies in the use of TGA-MS to characterize the sulfophenyl-modified RGO. Fig. 8 shows the TGA-MS profiles of GO, RGO and SRGO under He flow. The thermogram of graphene oxide (GO) (Fig. 8a) shows a major weight loss of 35% between 170 and 300 °C, with an inflection point at 200 °C. The thermogravimetric analysis simultaneously coupled with mass spectroscopy (TGA-MS) allowed to show that the mass loss is originating from OH (m/z 17), water (m/z 18), CO (m/z 28) and CO2 (m/z 44) release (Fig. 8b). This sudden mass loss is attributed to the removal of labile oxygen functional groups on GO sheets.42 A weaker mass loss occurs when the temperature is increased between 300 and 900 °C and is related to the gradual removal of more stable oxygen functionalities. The TGA-MS data of GO (Fig. 8a and b) indicate that the weight loss above 300 °C can essentially be assigned to CO (m/z 28) release. The significant mass loss (∼50 wt%) observed in the TGA analysis reflects the extent of the defects in the GO, which make the material thermally unstable. In contrast, RGO (obtained by thermal reduction of GO at 800 °C) show only a 5 wt% mass loss up to 500 °C, which suggests that a significant amount of labile oxygen groups were removed during heat treatment (Fig. 8c). The weight loss (20%) observed between 500 and 900 °C (Fig. 8c and d) is mostly associated to the departure of the carbonyl groups (CO2, m/z 44) (Fig. 8d), that have not been removed during the pre-heat treatment of the GO as shown on the mass spectrum in Fig. 8c. The elimination of oxygen functionalities from GO during the pre-thermal annealing enhances the van der Waals forces attraction between graphene layers, which makes RGO thermally more stable.17 The TGA curves of Fig. 8d show no significant difference for GO treated at 200 and 800 °C.
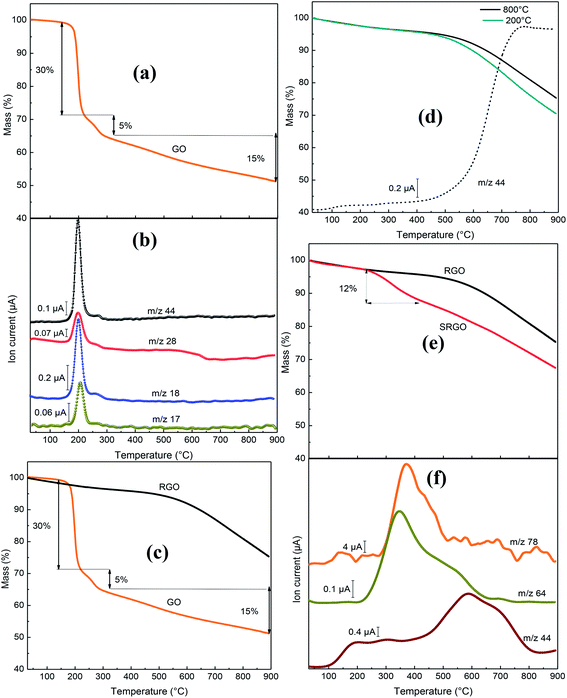 |
| | Fig. 8 (a) Mass variation for GO, (b) MS profile for GO, (c) mass variation for GO and RGO, (d) mass variation for GO following heat treatment at 200 and 800 °C, MS profile (–––) for GO following heat treatment at 800 °C, (e) mass variation for RGO and SRGO and (f) MS profiles for SRGO. | |
However, after sulfophenyl functionalization of RGO, SRGO displays a different profile compared to RGO (Fig. 8e). The onset of weight loss (∼12 wt%) observed at about 350 °C is attributed to the thermal removal of organic functional groups and the weight loss is greater than that caused by the departure of only labile oxygen functionalities in this range of temperature. Two relevant fragments m/z = 64 (SO2) and m/z = 78 (C6H12) are detected between 250 and 550 °C and correlate with the mass loss (Fig. 8e and f). The maximum of the SO2 and C6H12 peaks is around 350–400 °C, confirming the chemical bonding between graphene sheets and aryl groups.81 The signal with m/z 44, attributed to CO2, showed a different profile compared to unmodified RGO, with a maximum at 600 °C. The grafting of sulfophenyl groups on RGO surface is confirmed by elemental analysis (ESI, Table S2†). Their mass loading was calculated to be 12 wt% from the data in Table S2,† by considering the presence of a sulfur atom per grafted group.
4 Conclusion
Reduced graphene oxide (RGO) was synthesized via thermal reduction of GO under argon/hydrogen between 200 and 800 °C. Thermogravimetric analysis coupled with mass spectra analysis was firstly used to examine oxygen functionalities of GO and secondly to confirm the presence of sulfophenyl groups on the surface of chemically modified RGO. The immobilization of organic molecules on the graphene sheets was demonstrated by TEM, FTIR, XPS and nitrogen gas adsorption. Electronic conductivity measurements and Raman spectroscopy confirmed the covalent bonds between the graphene sheets and the organic molecules. The optical band gap of RGO was found to decrease following grafting of sulfophenyl groups. Finally, sulfophenyl and other aryl modified-graphene have a wide variety of potential applications. Similarly to modified carbons, SRGO and more general aryl-modified graphene could be employed in proton exchange membrane fuel cells, electrochemical capacitors, batteries, inks for printing, sensors as well as automotive and biomedical coatings.105,106
Conflict of interest
The authors declare no competing financial interest.
Acknowledgements
This work was financially supported by the Natural Sciences and Engineering Research Council of Canada (NSERC) by a Discovery Grant to DB. The research infrastructure of NanoQAM was used during this work.
References
- Z.-S. Wu, K. Parvez, X. Feng and K. Müllen, Nat. Commun., 2013, 4, 2487–3487 CrossRef.
- J. H. Seol, I. Jo, A. L. Moore, L. Lindsay, Z. H. Aitken, M. T. Pettes, X. Li, Z. Yao, R. Huang, D. Broido, N. Mingo, R. S. Ruoff and L. Shi, Science, 2010, 328, 213–236 CrossRef CAS.
- S. Stankovich, D. A. Dikin, G. H. B. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282–286 CrossRef CAS.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- X. Zhou, Y. Li, G. Ma, Q. Ma and Z. Lei, J. Alloys Compd., 2016, 685, 216–221 CrossRef CAS.
- Q. Li, Y. Chen, J. He, F. Fu, J. Lin and W. Zhang, J. Alloys Compd., 2016, 685, 294–299 CrossRef CAS.
- D. Wei, L. Grande, V. Chundi, R. White, C. Bower, P. Andrew and T. Ryhänen, Chem. Commun., 2012, 48, 1239–1241 RSC.
- R. Raccichini, A. Varzi, S. Passerini and B. Scrosati, Nat. Mater., 2015, 14, 271–279 CrossRef CAS.
- G. Pognon, T. Brousse and D. Bélanger, Carbon, 2011, 49, 1340–1348 CrossRef CAS.
- A. Ambrosi, C. K. Chua, B. Khezri, Z. Sofer, R. D. Webster and M. Pumera, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 12899–12904 CrossRef CAS PubMed.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- S. Park, J. An, J. R. Potts, A. Velamakanni, S. Murali and R. S. Ruoff, Carbon, 2011, 49, 3019–3023 CrossRef CAS.
- J. R. Lomeda, C. D. Doyle, D. V. Kosynkin, W.-F. Hwang and J. M. Tour, J. Am. Chem. Soc., 2008, 130, 16201–16206 CrossRef CAS.
- T. Kuila, A. K. Mishra, P. Khanra, N. H. Kim and J. H. Lee, Nanoscale, 2013, 5, 52–71 RSC.
- H. J. Shin, K. K. Kim, A. Benayad, S. M. Yoon, H. K. Park, I. S. Jung, M. H. Jin, H. K. Jeong, J. M. Kim, J. Y. Choi and Y. H. Lee, Adv. Funct. Mater., 2009, 19, 1987–1992 CrossRef CAS.
- S. Pei, J. Zhao, J. Du, W. Ren and H. M. Cheng, Carbon, 2010, 48, 4466–4474 CrossRef CAS.
- A. K. Das, M. Srivastav, R. K. Layek, M. E. Uddin, D. Jung, N. H. Kim and J. H. Lee, J. Mater. Chem. A, 2014, 2, 1332–1340 RSC.
- R. S. Park and S. Ruoff, Nat. Nanotechnol., 2009, 4, 217–224 CrossRef.
- J. Park, S. B. Jo, Y. J. Yu, Y. Kim, J. W. Yang, W. H. Lee, H. H. Kim, B. H. Hong, P. Kim, K. Cho and K. S. Kim, Adv. Mater., 2012, 24, 407–411 CrossRef CAS.
- B. D. Ossonon and D. Bélanger, Carbon, 2017, 111, 83–93 CrossRef CAS.
- S. Umrao, S. Abraham, F. Theil, S. Pandey, V. Ciobota, P. K. Shukla, C. J. Rupp, S. Chakraborty, R. Ahuja, J. Popp, B. Dietzek and A. Srivastava, RSC Adv., 2014, 4, 59890–59901 RSC.
- L. Song, L. Ci, H. Lu, P. B. Sorokin, C. Jin, J. Ni, A. G. Kvashnin, D. G. Kvashnin, J. Lou, B. I. Yakobson and P. M. Ajayan, Nano Lett., 2010, 10, 3209–3215 CrossRef CAS.
- M. Saranya, R. Ramachandran, P. Kollu, S. K. Jeong and A. N. Grace, RSC Adv., 2015, 5, 15831–15840 RSC.
- Y. N. Singhbabu, K. K. Sahu, D. Dadhich, A. K. Pramanick, T. Mishra and R. K. Sahu, J. Mater. Chem. C, 2013, 1, 958–966 RSC.
- X. Li, H. Zhu, J. Wei, K. Wang, E. Xu, Z. Li and D. Wu, Appl. Phys. A: Mater. Sci. Process., 2009, 97, 341–344 CrossRef CAS.
- J. Landers, G. Y. Gor and A. V. Neimark, Colloids Surf., A, 2013, 437, 3–32 CrossRef CAS.
- D. S. Yu, T. Kuila, N. H. Kim, P. Khanra and J. H. Lee, Carbon, 2013, 54, 310–322 CrossRef CAS.
- G. Wang, B. Wang, J. Park, Y. Wang, B. Sun and J. Yao, Carbon, 2009, 47, 3242–3246 CrossRef CAS.
- Y. Zhu, M. D. Stoller, W. Cai, A. Velamakanni, R. D. Piner, D. Chen and R. S. Ruoff, ACS Nano, 2010, 4, 1227–1233 CrossRef CAS PubMed.
- Y. Wang, X. Tong, X. Guo, Y. Wang, G. Jin and X. Guo, Nanotechnology, 2013, 24, 1–5 CAS.
- S. Umrao, S. Abraham, F. Theil, S. Pandey, V. Ciobota, P. K. Shukla, C. J. Rupp, S. Chakraborty, R. Ahuja, J. Popp, B. Dietzek and A. Srivastava, RSC Adv., 2014, 4, 59890–59901 RSC.
- A. Mathkar, D. Tozier, P. Cox, P. Ong, C. Galande, K. Balakrishnan, A. Leela Mohana Reddy and P. M. Ajayan, J. Phys. Chem. Lett., 2012, 3, 986–991 CrossRef CAS.
- J. M. Englert, C. Dotzer, G. Yang, M. Schmid, C. Papp, J. M. Gottfried, H.-P. Steinrück, E. Spiecker, F. Hauke and A. Hirsch, Nat. Chem., 2011, 3, 279–286 CrossRef CAS.
- K. T. Shruthi, M. S. Kumar, A. Pratap and N. Chandrasekaran, RSC Adv., 2015, 5, 93423–93432 RSC.
- Y. Gao, D. Ma, C. Wang, J. Guan and X. Bao, Chem. Commun., 2011, 47, 2432–2434 RSC.
- S. Brunauer, L. S. Deming, W. E. Deming and E. Teller, J. Am. Chem. Soc., 1940, 62, 1723–1732 CrossRef CAS.
- A. Gambou-Bosca and D. Belanger, J. Electrochem. Soc., 2015, 162, A5115–A5123 CrossRef CAS.
- K. S. W. Sing and R. T. Williams, Adsorpt. Sci. Technol., 2004, 22, 773–782 CrossRef CAS.
- T. Ohkubo, J. Miyawaki, K. Kaneko, R. Ryoo and N. A. Seaton, J. Phys. Chem. B, 2002, 106, 6523–6528 CrossRef CAS.
- R. F. Bruinsma, P. G. De Gennes, J. B. Freund and D. Levine, Nature, 2004, 427, 523–527 CrossRef.
- M. D. Stoller, S. Park, Y. Zhu, J. An and R. S. Ruoff, Nano Lett., 2008, 8, 3498–3502 CrossRef CAS.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- C. Moreno-Castilla, M. A. Ferro-Garcia, J. P. Joly, I. Bautista-Toledo, F. Carrasco-Marín and J. Rivera-Utrilla, Langmuir, 1995, 11, 4386–4392 CrossRef CAS.
- M. Toupin and D. Bélanger, Langmuir, 2008, 24, 1910–1917 CrossRef CAS.
- A. Le Comte, D. Chhin, A. Gagnon, R. Retoux, T. Brousse and D. Bélanger, J. Mater. Chem. A, 2015, 3, 6146–6156 RSC.
- H. Li, Y. Wang, Y. Shi, J. Li, L. H. Yanga and H. Ying, RSC Adv., 2013, 35, 14839–15492 Search PubMed.
- D. D. Jiang, Q. Yao, M. A. McKinney and C. A. Wilkie, Polym. Degrad. Stab., 1999, 63, 423–434 CrossRef CAS.
- J. Lu, Y. Li, S. Li and S. P. Jiang, Sci. Rep., 2016, 6, 21530–21542 CrossRef CAS.
- D. H. Johnston and D. F. Shriver, Inorg. Chem., 1993, 32, 1045–1047 CrossRef CAS.
- Z. Xia, F. Leonardi, M. Gobbi, Y. Liu, V. Bellani, A. Liscio, A. Kovtun, R. Li, X. Feng, E. Orgiu, P. Samorì, E. Treossi and V. Palermo, ACS Nano, 2016, 10, 7125–7134 CrossRef CAS.
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 14095–14107 CrossRef CAS.
- A. C. Ferrari and D. M. Basko, Nat. Nanotechnol., 2013, 8, 235–246 CrossRef CAS.
- A. C. Ferrari, J. C. Meyer, V. Scardaci, C. Casiraghi, M. Lazzeri, F. Mauri, S. Piscanec, D. Jiang, K. S. Novoselov, S. Roth and A. K. Geim, Phys. Rev. Lett., 2006, 97, 187401–187405 CrossRef CAS.
- W. Gao, M. Majumder, L. B. Alemany, T. N. Narayanan, M. A. Ibarra, B. K. Pradhan and P. M. Ajayan, ACS Appl. Mater. Interfaces, 2011, 3, 1821–1826 CrossRef CAS.
- M. Fang, K. Wang, H. Lu, Y. Yang and S. Nutt, J. Mater. Chem., 2009, 19, 7098–7105 RSC.
- M. A. Pimenta, G. Dresselhaus, M. S. Dresselhaus, L. G. Cançado, A. Jorio and R. Saito, Phys. Chem. Chem. Phys., 2007, 9, 1276–1291 RSC.
- W. Chen and L. Yan, Nanoscale, 2010, 2, 559–563 RSC.
- Y. Wang, D. C. Alsmeyer and R. L. Mccreery, Carbon, 1990, 2, 557–563 CAS.
- K. N. Kudin, B. Ozbas, H. C. Schniepp, R. K. Prud’homme, I. A. Aksay and R. Car, Nano Lett., 2008, 8, 36–41 CrossRef CAS PubMed.
- G. Wei, M. Yan, R. Dong, D. Wang, X. Zhou, J. Chen and J. Hao, Chem.–Eur. J., 2012, 18, 14708–14716 CrossRef CAS.
- H. Lim, J. S. Lee, H. J. Shin, H. S. Shin and H. C. Choi, Langmuir, 2010, 26, 12278–12284 CrossRef CAS.
- M. S. Dresselhaus, A. Jorio, M. Hofmann, G. Dresselhaus and R. Saito, Nano Lett., 2010, 10, 751–758 CrossRef CAS.
- A. Ganguly, S. Sharma, P. Papakonstantinou and J. Hamilton, J. Phys. Chem., 2011, 115, 17009–17019 CAS.
- A. Guermoune, T. Chari, F. Popescu, S. S. Sabri, J. Guillemette, H. S. Skulason, T. Szkopek and M. Siaj, Carbon, 2011, 49, 4204–4210 CrossRef CAS.
- Z. Jin, T. P. McNicholas, C. J. Shih, Q. H. Wang, G. L. C. Paulus, A. J. Hilmer, S. Shimizu and M. S. Strano, Chem. Mater., 2011, 23, 3362–3370 CrossRef CAS.
- Y. I. Zhang, L. Zhang and C. Zhou, Acc. Chem. Res., 2013, 46, 2329–2339 CrossRef CAS.
- K. Parvez, S. Yang, X. Feng and K. Müllen, Synth. Met., 2015, 210, 123–132 CrossRef CAS.
- S. Pei and H. M. Cheng, Carbon, 2012, 50, 3210–3228 CrossRef CAS.
- C. Singh, A. K. Mishra and A. Paul, J. Mater. Chem. A, 2015, 3, 18557–18563 RSC.
- J. Wang, T. Zhou, H. Deng, F. Chen, K. Wang, Q. Zhang and Q. Fu, Colloids Surf., B, 2013, 101, 171–176 CrossRef CAS.
- J. Guan, X. Chen, T. Wei, F. Liu, S. Wang, Q. Yang, Y. Lu and S. Yang, J. Mater. Chem. A, 2015, 3, 4139–4146 RSC.
- G. Gao, D. Liu, S. Tang, C. Huang, M. He, Y. Guo, X. Sun and B. Gao, Sci. Rep., 2016, 6, 1–8 CrossRef.
- X. Fan, W. Peng, Y. Li, X. Li, S. Wang, G. Zhang and F. Zhang, Adv. Mater., 2008, 20, 4490–4493 CrossRef CAS.
- C.-Y. Su, A.-Y. Lu, Y. Xu, F.-R. Chen, A. N. Khlobystov and L.-J. Li, ACS Nano, 2011, 5, 2332–2339 CrossRef CAS PubMed.
- S. Baranton and D. Bélanger, J. Phys. Chem. B, 2005, 109, 24406–24410 CrossRef.
- E. Lebègue, T. Brousse, J. Gaubicher and C. Cougnon, Electrochim. Acta, 2013, 88, 680–687 CrossRef.
- F. Hauquier, N. Debou, S. Palacin and B. Jousselme, J. Electroanal. Chem., 2012, 677–680, 127–132 CrossRef CAS.
- J. Lyskawa, A. Grondein and D. Bélanger, Carbon, 2010, 48, 1271–1278 CrossRef CAS.
- D. B. Ossonon, M. Weissmann and D. Bélanger, Electrochim. Acta, 2014, 122, 210–217 CrossRef.
- M. Toupin and D. Bélangeer, J. Phys. Chem. C, 2007, 111, 5394–5401 CrossRef CAS.
- L. Madec, K. A. Seid, J.-C. Badot, B. Humbert, P. Moreau, O. Dubrunfaut, B. Lestriez, D. Guyomard and J. Gaubicher, Phys. Chem. Chem. Phys., 2014, 16, 22745–22753 RSC.
- C. Saby, B. Ortiz, G. Y. Champagne and D. Bélanger, Langmuir, 1997, 7463, 6805–6813 CrossRef.
- T. S. Sreeprasad, S. M. Maliyekkal, K. P. Lisha and T. Pradeep, J. Hazard. Mater., 2011, 186, 921–931 CrossRef CAS.
- C. Mattevi, G. Eda, S. Agnoli, S. Miller, K. A. Mkhoyan, O. Celik, D. Mastrogiovanni, C. Cranozzi, E. Carfunkel and M. Chhowalla, Adv. Funct. Mater., 2009, 19, 2577–2583 CrossRef CAS.
- S. Park, K.-S. Lee, G. Bozoklu, W. Cai, S. T. Nguyen and R. S. Ruoff, ACS Nano, 2008, 2, 572–578 CrossRef CAS.
- K. Hu, X. Xie, T. Szkopek and M. Cerruti, Chem. Mater., 2016, 28, 1756–1768 CrossRef CAS.
- S. Eigler, C. Dotzer, A. Hirsch, M. Enzelberger and P. Müller, Chem. Mater., 2012, 24, 1276–1282 CrossRef CAS.
- C. Hontoria-Lucas, A. J. López-Peinado, J. de D. López-González, M. L. Rojas-Cervantes and R. M. Martín-Aranda, Carbon, 1995, 33, 1585–1592 CrossRef CAS.
- A. Sinitskii, A. Dimiev, D. A. Corley, A. A. Fursina, D. V. Kosynkin and J. M. Tour, ACS Nano, 2010, 4, 1949–1954 CrossRef CAS.
- A. Mesnage, X. Lefe, P. Je, G. Deniau and S. Palacin, Langmuir, 2012, 28, 11767–11778 CrossRef CAS.
- J. Marwan, T. Addou and D. Bélanger, Chem. Mater., 2005, 17, 2395–2403 CrossRef CAS.
- A. L. Gui, G. Liu, M. Chockalingam, G. Le Saux, J. B. Harper and J. J. Gooding, Electroanalysis, 2010, 22, 1283–1289 CrossRef CAS.
- A. Y. Romanchuk, A. S. Slesarev, S. N. Kalmykov, D. V. Kosynkin and J. M. Tour, Phys. Chem. Chem. Phys., 2013, 15, 2321–2327 RSC.
- H. Dai, Z. Xu and X. Yang, J. Phys. Chem. C, 2016, 120, 22585–22596 CrossRef CAS.
- V. Shubina, L. Gaillet, S. Ababou-Girard, V. Gaudefroy, T. Chaussadent, F. Farças, T. Meylheuc, C. Dagbert and J. Creus, Appl. Surf. Sci., 2015, 351, 1174–1183 CrossRef CAS.
- A. Ganguly, S. Sharma, P. Papakonstantinou and J. Hamilton, J. Phys. Chem. C, 2011, 115, 17009–17019 CrossRef CAS.
- Y. Ishii, D. Yang, A. Velamakanni, S. J. An and M. Stoller, Science, 2008, 321, 1815–1818 CrossRef.
- T. Szabo, O. Berkesi, P. Forgo, K. Josepovits, Y. Sanakis, D. Petridis and I. Dékany, Chem. Mater., 2006, 18, 2740–2749 CrossRef CAS.
- G. Gunasekaran and L. R. Chauhan, Electrochim. Acta, 2004, 49, 4387–4395 CrossRef CAS.
- Y. Xiong, Y. Xie, Z. Li and C. Wu, Chem.–Eur. J., 2003, 9, 1645–1651 CrossRef CAS.
- A. Buchsteiner, A. Lerf and J. Pieper, J. Phys. Chem. B, 2006, 110, 22328–22338 CrossRef CAS.
- L. B. Casabianca, M. A. Shaibat, W. W. Cai, S. Park, R. Piner, R. S. Ruoff and Y. Ishii, J. Am. Chem. Soc., 2010, 132, 5672–5676 CrossRef CAS.
- C. Zhao, H. Gao, C. Chen and H. Wu, J. Mater. Chem. A, 2015, 3, 18360–18364 RSC.
- B. Desalegn, T. Brousse and D. Bélanger, Carbon, 2015, 92, 362–381 CrossRef.
- D. Bélanger and J. Pinson, Chem. Soc. Rev., 2011, 40, 3995–4048 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra28311j |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*