
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePincer cobalt complex-catalyzed Z-selective hydrosilylation of terminal alkynes†
Xiaoyong
Du
,
Wenjun
Hou
,
Yanlu
Zhang
and
Zheng
Huang
*
State Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, 345 Lingling Road, Shanghai, 200032, China. E-mail: huangzh@sioc.ac.cn; Fax: +86 5492 5533; Tel: +86 5492 5522
First published on 28th April 2017
Abstract
A phosphine-iminopyridine (PCNN) cobalt-catalyzed Z-selective hydrosilylation of terminal alkynes with Ph2SiH2 has been developed for the synthesis of (Z)-β-vinylsilanes with high regio- and stereoselectivity and wide functional group tolerance. Furthermore, the Co-catalyzed hydrosilylations of unsymmetrical arylalkyl disubstituted internal alkynes afford syn-addition products with unique regioselectivity: the silyl group is added to the alkyl-substituted carbon, instead of the aryl-substituted carbon. The (Z)-β-vinylsilane products are further applied to Pd-catalyzed Hiyama–Denmark cross-couplings for stereoselective synthesis of (Z)-disubstituted alkenes.
Introduction
Transition metal-catalyzed hydrosilylation of alkynes with hydrosilanes is one of the most atom-economical approaches to access vinylsilanes,1 which are valuable intermediates in organic synthesis.2 The hydrosilylation of a terminal alkyne can generate three possible products, (E)-β-, (Z)-β-, and α-vinylsilanes (Scheme 1a), and thus controlling the regio- and stereoselectivity is a key issue during the H–Si addition process. The selective synthesis of (Z)-β-vinylsilanes is considered to be more challenging than the formation of the thermodynamically more stable (E)-β-isomers. Indeed, many transition metal-catalyzed hydrosilylation reactions selectively form (E)-β-vinylsilanes.3 Nevertheless, a number of catalysts using noble-metals, such as Rh,4 Ir4g,5 and Ru,6 have been developed for the (Z)-β-selective hydrosilylation of terminal alkynes.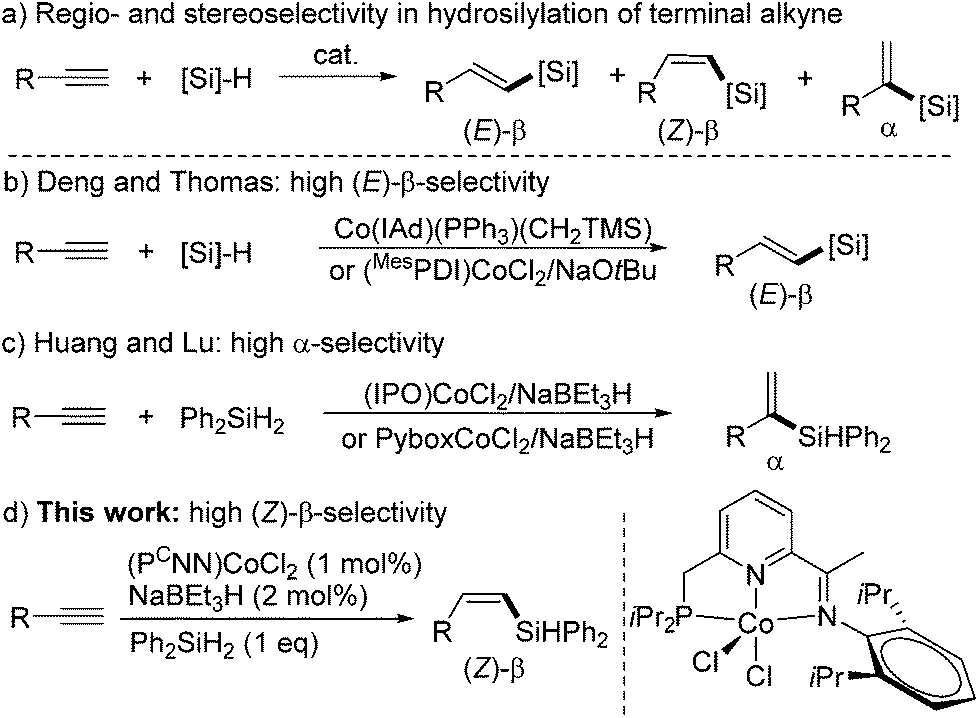 | ||
| Scheme 1 Cobalt-catalyzed hydrosilylation of terminal alkynes. | ||
Over the past decade, the low abundance, high cost, and the environmental concerns associated with the noble metals have spurred extensive studies toward developing earth-abundant base-metal alternatives.7 So far, Fe,8 Co,9 and Ni10 complexes have been proven effective for alkyne hydrosilylation, but only bis(imino)pyridine iron complexes gave high (Z)-β-selectivity in the hydrosilylations of two terminal alkyne substrates.8c,d Very recently, several groups reported that cobalt complexes could catalyze the hydrosilylation of terminal alkynes with high α- or (E)-β-selectivity. In 2014, Deng developed a carbene-ligated Co(I) complex and applied it to the hydrosilylation of alkynes with high (E)-β-selectivities (Scheme 1b).11 Thomas described a combination of the (MesPDI)CoCl2 complex of bis(imino)pyridine and NaOtBu for (E)-β-selective hydrosilylation with 1-hexyne with PhSiH3 (Scheme 1b).12 In 2016, Lu13 and our group14 independently reported Co complexes of iminopyridine-oxazoline (IPO) or Pybox ligands for the α-selective hydrosilylation of terminal aryl alkynes with Ph2SiH2 (Scheme 1c). However, the use of cobalt complexes for the hydrosilylation of terminal alkynes with (Z)-β-selectivity is rare.15 During the submission of this manuscript, Ge reported a bis(imino)pyridine cobalt-catalyzed Z-selective hydrosilylation of terminal alkynes with PhSiH3.16 Herein, we describe a highly Z-selective hydrosilylation of terminal alkynes with Ph2SiH2 using a (PCNN)CoCl2/NaBEt3H catalyst system (Scheme 1a), which has been previously developed by our group for the Markovnikov hydrosilylation of terminal alkenes.17 Moreover, the (PCNN)Co catalyst effects the hydrosilylation of internal alkynes, and intriguingly, the reactions of arylalkyl disubstituted alkynes produce vinylsilanes containing the silyl unit at the alkyl-substituted olefinic carbon, rather than the aryl-substituted one. The (Z)-β-vinylsilane products can undergo Pd-catalyzed Hiyama–Denmark cross-couplings to form (Z)-disubstituted alkenes.
Results and discussion
We commenced our studies by examining the PCNN ligand-supported Fe and Co catalysts for the hydrosilylation of a simple aliphatic alkyne, 1-hexyne (3a), with Ph2SiH2 as the silicon source. The results are summarized in Table 1. Using NaBEt3H (2 mol%) as the activator, both tBu- and iPr-substituted Fe complexes, (tBuPCNNiPr)FeCl2 (1a, 1 mol%) and (iPrPCNNiPr)FeCl2 (1b, 1 mol%), were inactive for the hydrosilylation (entries 1 and 2). However, 1-hexyne (1.2 equiv.) reacted with Ph2SiH2 to give (Z)-β-vinylsilane 4a and (E)-β-vinylsilane 5a when the Co analogues were employed. Using the most sterically hindered complex 2a (1 mol%) bearing a tBu-substituted phosphino group and iPr substituents at the 2,6-positions of the N-aryl ring, the hydrosilylation in THF at 25 °C gave 13% 4a and 18% 5a after 24 h (entry 3). Reducing the steric hindrance of the PCNN ligands resulted in a significant improvement of the activity and selectivity for the desired (Z)-β-product. The reactions using complexes (iPrPCNNiPr)CoCl22b and (iPrPCNNMe)CoCl22c containing iPr-substituted phosphino groups formed 4a in 85% and 83% yield; α-vinylsilane was not observed in both cases (entries 4 and 5).|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Cat. | Silane | Solvent | 3a/[Si] | Yield [%] | |
| 4a | 5a | |||||
| a Reaction conditions: 3a (0.72–0.9 mmol) and silane (0.6 mol) in THF (1 mL) or under neat conditions at 25 °C. The yields were determined by 1H NMR spectroscopy. b Without precat. c Without NaHBEt3. | ||||||
| 1 | 1a | Ph2SiH2 | THF | 1.2 | 0 | 0 |
| 2 | 1b | Ph2SiH2 | THF | 1.2 | 0 | 0 |
| 3 | 2a | Ph2SiH2 | THF | 1.2 | 13 | 18 |
| 4 | 2b | Ph2SiH2 | THF | 1.2 | 85 | 3 |
| 5 | 2c | Ph2SiH2 | THF | 1.2 | 83 | 3 |
| 6 | 2b | Ph2SiH2 | None | 1.5 | 95 | 1 |
| 7b | 2b | Ph2SiH2 | None | 1.5 | 0 | 0 |
| 8c | 2b | Ph2SiH2 | None | 1.5 | 0 | 0 |
| 9 | 2b | (EtO)3SiH | None | 1.5 | <5 | <5 |
| 10 | 2b | MD′M | None | 1.5 | 0 | 0 |
| 11 | 2b | Et3SiH | None | 1.5 | 0 | 0 |
| 12 | 2b | PhMe2SiH | None | 1.5 | 0 | 0 |
| 13 | 2b | PhSiH3 | None | 1.5 | 50 | 45 |
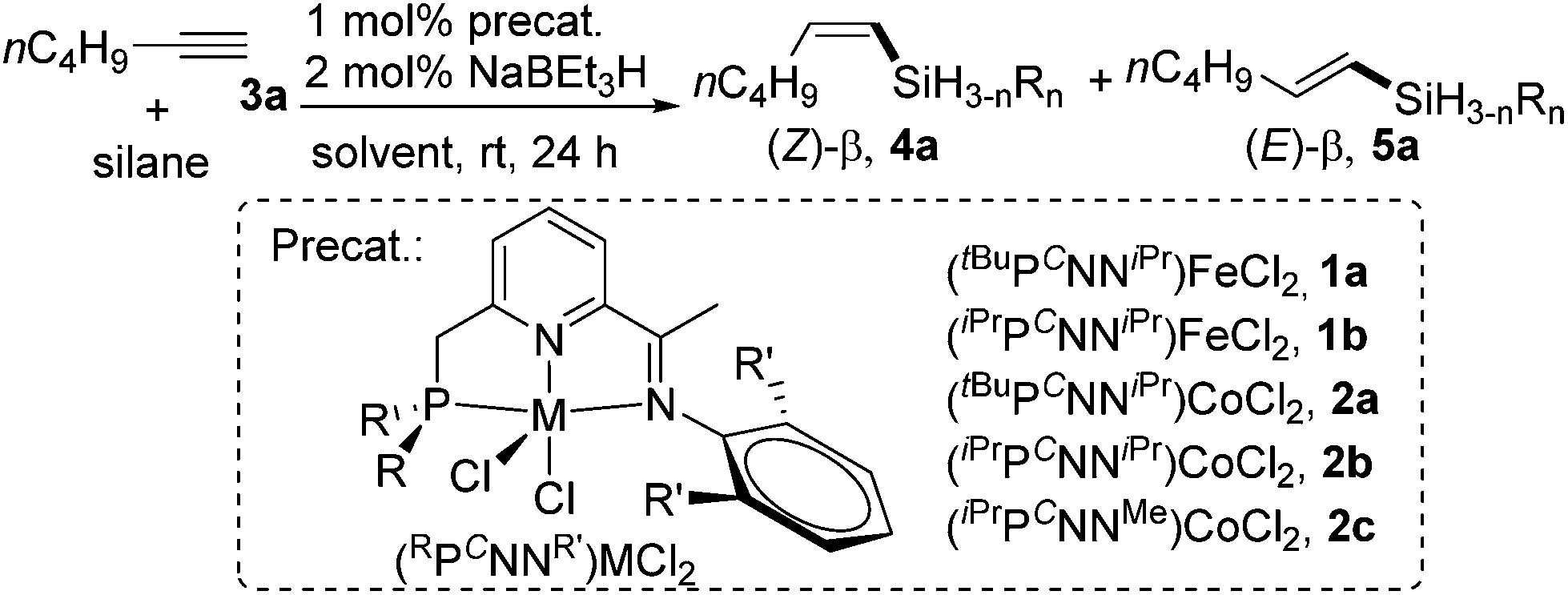
Further optimizations were conducted to improve the selectivity for cis-vinylsilane. The best Z-selectivity was obtained when the reaction occurred under solvent-free conditions using a 1.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of 3a:Ph2SiH2 in the presence of 1 mol% of 2b and 2 mol% of NaHBEt3; the run at 25 °C for 24 h produced (Z)-vinylsilane 4a in 95% yield with the Z/E stereoselectivity of 95:1 (entry 6). A control experiment without the precatalyst 2b, but with NaBEt3H, gave no hydrosilylation product (entry 7), and another control without NaHBEt3 indicated that the addition of the activator was necessary for the catalysis (entry 8). Finally, we explored the catalytic performance of this Co catalyst for hydrosilylations with primary and tertiary hydrosilanes. Tertiary silanes (EtO)3SiH, MD′M (1,1,1,3,5,5,5-heptamethyltrisiloxane), Et3SiH and PhMe2SiH were unreactive under the reaction conditions (entries 9–12). Although the reaction with PhSiH3 gave the anti-Markovnikov products in high yield (95% totally), the stereoselectivity was low (Z/E = ∼1:1) (entry 13).
:1 ratio of 3a:Ph2SiH2 in the presence of 1 mol% of 2b and 2 mol% of NaHBEt3; the run at 25 °C for 24 h produced (Z)-vinylsilane 4a in 95% yield with the Z/E stereoselectivity of 95:1 (entry 6). A control experiment without the precatalyst 2b, but with NaBEt3H, gave no hydrosilylation product (entry 7), and another control without NaHBEt3 indicated that the addition of the activator was necessary for the catalysis (entry 8). Finally, we explored the catalytic performance of this Co catalyst for hydrosilylations with primary and tertiary hydrosilanes. Tertiary silanes (EtO)3SiH, MD′M (1,1,1,3,5,5,5-heptamethyltrisiloxane), Et3SiH and PhMe2SiH were unreactive under the reaction conditions (entries 9–12). Although the reaction with PhSiH3 gave the anti-Markovnikov products in high yield (95% totally), the stereoselectivity was low (Z/E = ∼1:1) (entry 13).
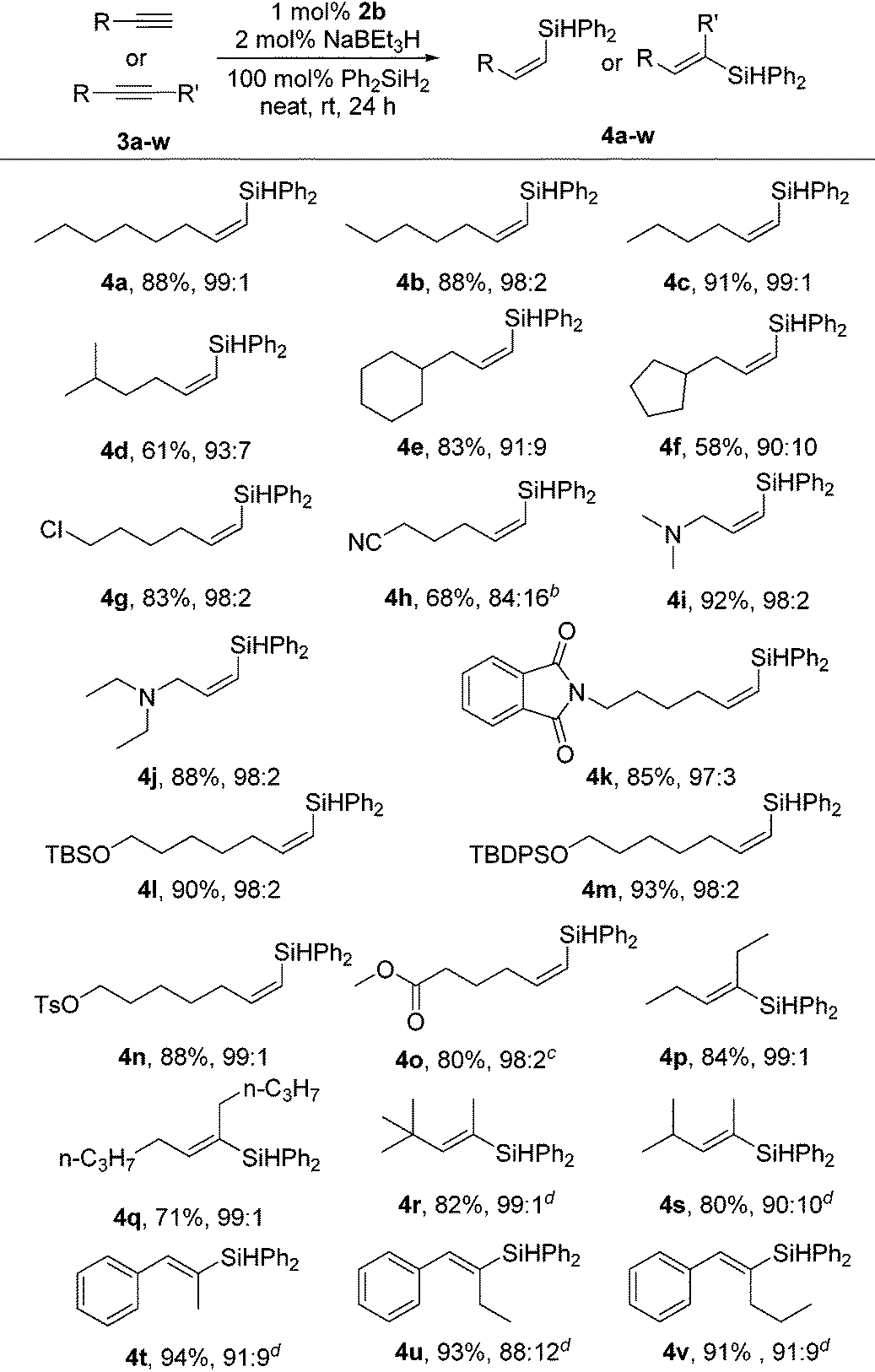
To evaluate the scope of the Co-catalyzed alkyne hydrosilylation, the reactions with a variety of functionalized terminal alkynes and a number of internal alkynes were carried out using 2b as the precatalyst. The results are summarized in Table 2. All reactions with Ph2SiH2 proceeded to completion within 24 h at room temperature under solvent-free conditions. For terminal alkyne substrates, most reactions were highly selective for the formation of the (Z)-β-vinylsilane products (Z/E > 95:5). Linear terminal alkynes gave the corresponding products 4a–d in moderate-to-high isolated yield with excellent selectivity. 3-Cyclohexyl-1-propyne 3e and 3-cyclopentyl-1-propyne 3f underwent hydrosilylations in useful yields and high selectivities. The catalyst is compatible with terminal alkynes bearing various functional groups, including chloride (4g), nitrile (4h), tertiary amine (4i, 4j), amide (4k) and even ester (4o). Aliphatic alkynes containing protecting groups, such as silylether (4l, 4m) and tosylate (4n, 88%), afforded the desired (Z)-β-vinylsilanes in high yields and high selectivities. Aryl terminal alkynes are unreactive under the catalytic conditions, as demonstrated by the full recovery of the alkyne in the reaction of phenylacetylene with Ph2SiH2.
a
|
a Reaction conditions: Ph2SiH2 (0.6 mmol) and alkyne (0.9 mmol) at 25 °C. Yield of isolated products. Z/E ratios of isolated products were determined by 1H NMR spectroscopy.
b With 1.2 mmol 3h. A 84:16 Z-β/α ratio was determined by 1H NMR spectroscopy.
c With 2 mol% 2b and 4 mol% NaBEt3H.
d Regioselectivities determined by 1H NMR spectroscopy.
|
|---|
|
Internal alkynes underwent the hydrosilylation reactions under the catalytic conditions to form the products in good-to-high yields. The hydrosilylation of symmetric dialkyl-substituted alkynes (3p, 3q) occurred to form the syn-addition products in high yields (4p, 4q). Next, we turned our attention to the more challenging unsymmetrical internal alkynes. The unsymmetrical dialkyl-substituted alkynes with different steric properties, such as 1-tert-butyl-1-propyne (3r) and 4-methyl-pent-2-yne (3s), were suitable for the selective hydrosilylation, producing the syn-addition products 4r and 4s with the incorporation of the silyl group into the less sterically demanding sp-hybridised carbon. The reaction of arylalkyl disubstituted alkynes (3t–v) gave the products in 91–94% yields (4t–v). Noteworthily, the processes exhibited good regioselectivities, furnishing tri-substituted olefins with the Ph2HSi unit selectively added to the alkyl-substituted olefinic carbon, which is in contrast to previous reports on Co-catalyzed hydrosilylations of arylalkyl disubstituted alkynes that yielded vinylsilanes with the Si atom adjacent to the aryl group.9e,11,13
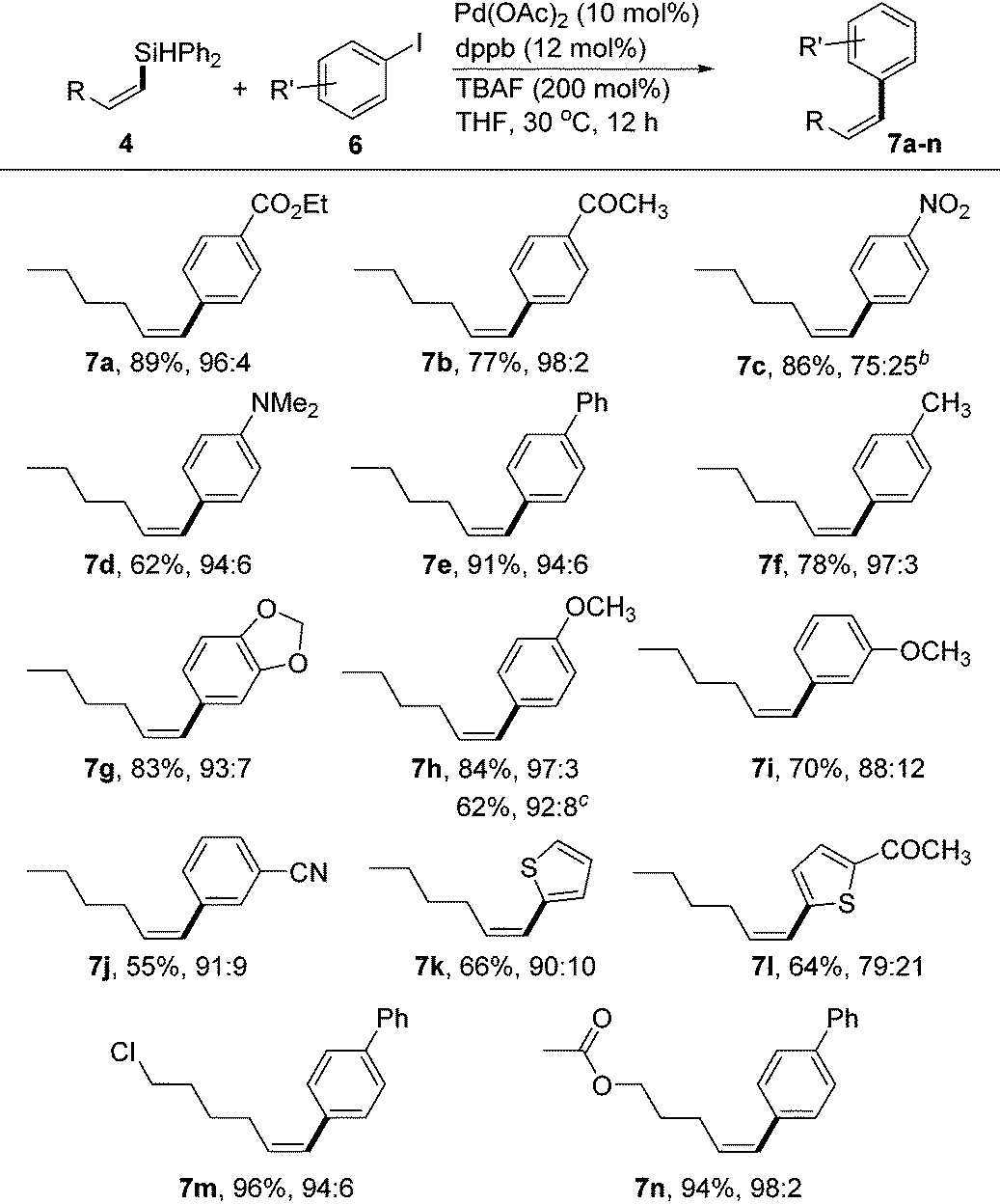
Due to their high stability, non-toxicity, and ease of handling, vinylsilanes have become valuable intermediates for Pd-catalyzed Hiyama–Denmark cross-couplings.18 However, the silane reagents used in the coupling reactions often require heteroatom substituent(s) (e.g., O or F) to make the Si atom feasible toward activation with a base or fluoride source.19 To this end, it is of interest to develop conditions for cross-couplings of our (Z)-vinylsilane products bearing the Ph2HSi moiety.20 After a screening of various mono- and bisphosphine ligands (see the ESI†), we identified the Pd catalyst generated from Pd(OAc)2 (10 mol%) and 1,4-bis(diphenylphosphino)butane (dppb, 12 mol%) was effective for stereoselective couplings of (Z)-vinylsilane (4) with aryl iodides using TBAF as the activator (2 equiv.). Most reactions occurred at 30 °C to form the cis-1,2-disubstituted alkenes in high yields and high stereoselectivities (Table 3). The electrophiles 6 bearing both electron-donating and -withdrawing groups were suitable coupling partners. Functionalized groups, such as ester (7a), ketone (7b), tertiary amine (7d) and nitrile (7j) could be tolerated. The reaction of 6c containing a nitro group provided the coupling product 7c in high yield, albeit with relatively low Z-selectivity (75:25). Aryl iodide 6i with a meta-MeO group gave the product with somewhat decreased selectivity (7i, 88:12) compared to the reaction with 4-iodoanisole (7h, 97:3). Aryl bromide is suitable for the coupling reaction as demonstrated by the formation of the desired product 7h in useful yield (62%) with 4-bromoanisole as the reagent. Heteroaromatic iodide, such as 2-iodothiophene and 2-acetyl-5-iodothiophene, gave the corresponding products 7k and 7l in 66 and 64% isolated yields, respectively. Functional Z-vinylsilanes such as 4g and 4o reacted with 4-iodo-1,1′-biphenyl under standard conditions and gave the corresponding Z-alkenes 7m and 7n in excellent yields and selectivities.
A mechanism accounting for the formation of (Z)-β-vinylsilane is presented in Scheme 2. On the basis of the precedents of Co-catalyzed hydrosilylation processes,11,16,17,21 we propose that the catalytic cycle involves a low-valent Co(I) silyl intermediate. The insertion of the coordinated terminal alkyne into the Co–Si bond forms a vinyl Co intermediate, (Z)-l-silyl-l-alken-2-yl-cobalt (C). Due to the significant steric repulsion between the bulky silyl group and the metal moiety, complex C undergoes isomerization to form a sterically less demanding complex F, (E)-l-silyl-l-alken-2-yl-cobalt, through the Ojima–Crabtree type rearrangement.4h,5gE then reacts with Ph2SiH2 to form (Z)-vinylsilane and regenerate the Co(I) silyl species A.
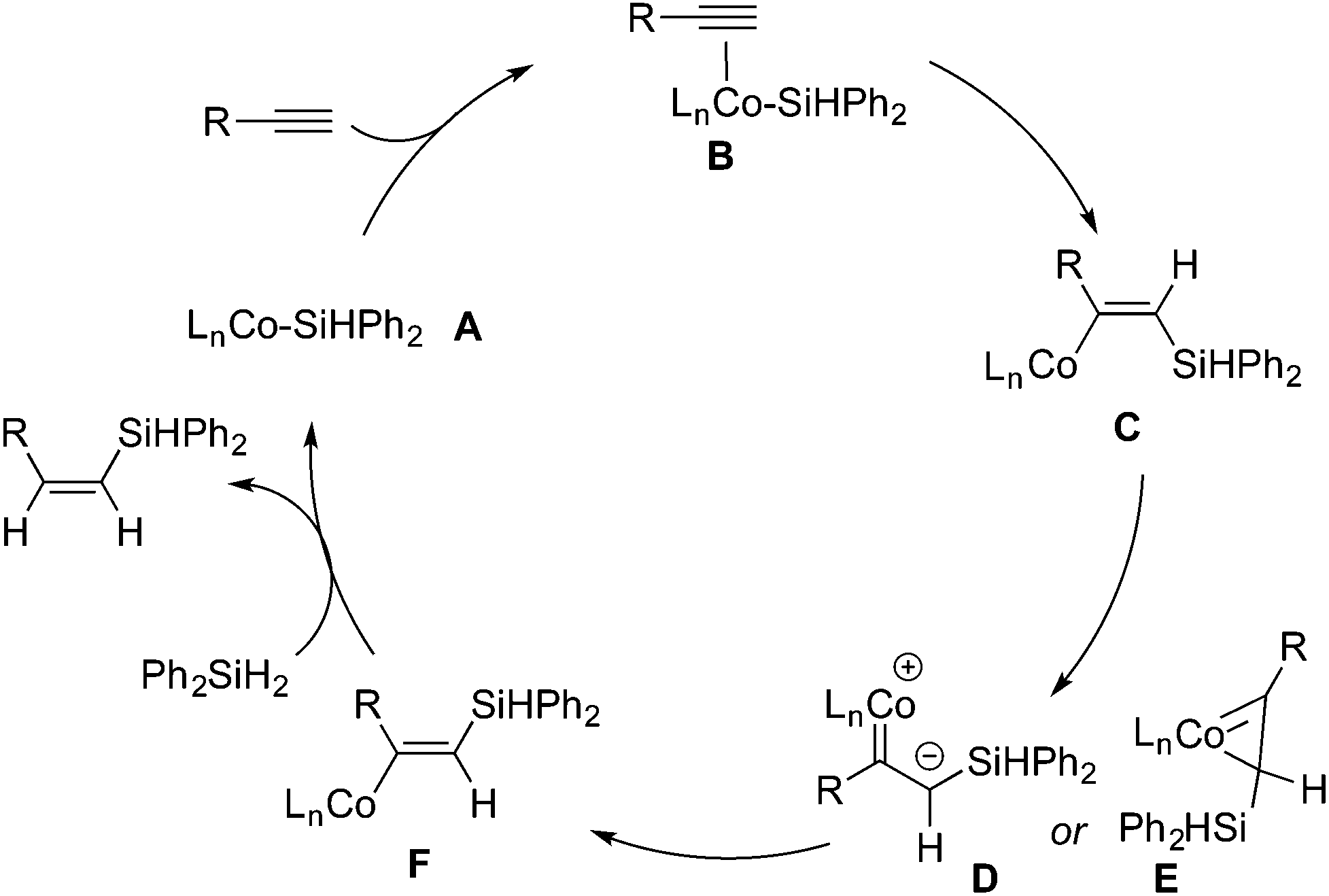 | ||
| Scheme 2 Proposed mechanism involving the Ojima–Crabtree type rearrangement for Co-catalyzed (Z)-selective hydrosilylation of terminal alkynes. | ||
Conclusions
In conclusion, we have developed a highly efficient cobalt-catalyzed regio- and stereoselective hydrosilylation of terminal alkynes with Ph2SiH2 to generate (Z)-β-vinylsilanes with high functional group tolerance. The silyl groups introduced into the vinylsilane products offer flexible synthetic manipulations as demonstrated by the Hiyama–Denmark cross-couplings with aryl halides, allowing a convenient, stereoselective synthesis of Cis-disubstituted alkenes.Acknowledgements
We gratefully acknowledge the financial support from the National Natural Science Foundation of China (No. 21272255, 21121062).Notes and references
- (a) H. Brunner, Angew. Chem., Int. Ed., 2004, 43, 2749 CrossRef CAS PubMed; (b) B. M. Trost and Z. T. Ball, Synthesis, 2005, 853 CrossRef CAS; (c) S. J. Clarson, Silicon, 2009, 1, 57 CrossRef CAS; (d) D. S. W. Lim and E. A. Anderson, Synthesis, 2012, 983 CAS.
- (a) T. H. Chan and I. Fleming, Synthesis, 1979, 761 CrossRef CAS; (b) T. A. Blumenkopf and L. E. Overman, Chem. Rev., 1986, 86, 857 CrossRef CAS; (c) E. Langkopf and D. Schinzer, Chem. Rev., 1995, 95, 1375 CrossRef CAS; (d) I. Fleming, A. Barbero and D. Walter, Chem. Rev., 1997, 97, 2063 CrossRef CAS PubMed.
- (a) H. Aneetha, W. Wu and J. G. Verkade, Organometallics, 2005, 24, 2590 CrossRef CAS; (b) G. De Bo, G. Berthon-Gelloz, B. Tinant and I. E. Markó, Organometallics, 2006, 25, 1881 CrossRef CAS; (c) Y. Nakao, H. Imanaka, J. Chen, A. Yada and T. Hiyama, J. Organomet. Chem., 2007, 692, 585 CrossRef CAS; (d) G. Berthon-Gelloz, J.-M. Schumers, G. De Bo and I. E. Markó, J. Org. Chem., 2008, 73, 4190 CrossRef CAS PubMed; (e) A. Hamze, O. Provot, J.-D. Brion and M. Alami, J. Organomet. Chem., 2008, 693, 2789 CrossRef CAS; (f) M. Blug, X.-F. Le Goff, N. Mézailles and P. Le Floch, Organometallics, 2009, 28, 2360 CrossRef CAS; (g) R. Cano, M. Yus and D. J. Ramón, ACS Catal., 2012, 2, 1070 CrossRef CAS; (h) K. Igawa, D. Yoshihiro, N. Ichikawa, N. Kokan and K. Tomooka, Angew. Chem., Int. Ed., 2012, 51, 12745 CrossRef CAS PubMed; (i) S. Dierick, E. Vercruysse, G. Berthon-Gelloz and I. E. Markó, Chem. – Eur.J., 2015, 21, 17073 CrossRef CAS PubMed; (j) D. Munz, C. Allolio, D. Meyer, M. Micksch, L. Roessner and T. Strassner, J. Organomet. Chem., 2015, 794, 330 CrossRef CAS; (k) L. Ortega-Moreno, R. Peloso, C. Maya, A. Suarez and E. Carmona, Chem. Commun., 2015, 51, 17008 RSC.
- (a) M. Atsunori, T. Eisuke, K. Hiroshi, H. Kazunori, N. Yasushi and H. Tamejiro, Chem. Lett., 1998, 27, 443 CrossRef; (b) J. W. Faller and D. G. D'Alliessi, Organometallics, 2002, 21, 1743 CrossRef CAS; (c) A. Mori, E. Takahisa, Y. Yamamura, T. Kato, A. P. Mudalige, H. Kajiro, K. Hirabayashi, Y. Nishihara and T. Hiyama, Organometallics, 2004, 23, 1755 CrossRef CAS; (d) M. V. Jiménez, J. J. Pérez-Torrente, M. I. Bartolomé, V. Gierz, F. J. Lahoz and L. A. Oro, Organometallics, 2008, 27, 224 CrossRef; (e) J. L. McBee, J. Escalada and T. D. Tilley, J. Am. Chem. Soc., 2009, 131, 12703 CrossRef CAS PubMed; (f) M. Iglesias, M. Perez-Nicolas, P. J. S. Miguel, V. Polo, F. J. Fernandez-Alvarez, J. J. Perez-Torrente and L. A. Oro, Chem. Commun., 2012, 48, 9480 RSC; (g) M. Iglesias, P. J. Sanz Miguel, V. Polo, F. J. Fernández-Alvarez, J. J. Pérez-Torrente and L. A. Oro, Chem. – Eur.J., 2013, 19, 17559 CrossRef CAS PubMed; (h) I. Ojima, N. Clos, R. J. Donovan and P. Ingallina, Organometallics, 1990, 9, 3127 CrossRef CAS.
- (a) C.-H. Jun and R. H. Crabtree, J. Organomet. Chem., 1993, 447, 177 CrossRef CAS; (b) M. Viciano, E. Mas-Marzá, M. Sanaú and E. Peris, Organometallics, 2006, 25, 3063 CrossRef CAS; (c) M. Yoshihiro, I. Eigo and I. Masahiko, Chem. Lett., 2006, 35, 836 CrossRef; (d) V. S. Sridevi, W. Y. Fan and W. K. Leong, Organometallics, 2007, 26, 1157 CrossRef CAS; (e) A. Zanardi, E. Peris and J. A. Mata, New J. Chem., 2008, 32, 120 RSC; (f) J. J. Pérez-Torrente, D. H. Nguyen, M. V. Jiménez, F. J. Modrego, R. Puerta-Oteo, D. Gómez-Bautista, M. Iglesias and L. A. Oro, Organometallics, 2016, 35, 2410 CrossRef; (g) R. S. Tanke and R. H. Crabtree, J. Am. Chem. Soc., 1990, 112, 7984 CrossRef CAS.
- (a) M. A. Esteruelas, J. Herrero and L. A. Oro, Organometallics, 1993, 12, 2377 CrossRef CAS; (b) H. Katayama, K. Taniguchi, M. Kobayashi, T. Sagawa, T. Minami and F. Ozawa, J. Organomet. Chem., 2002, 645, 192 CrossRef CAS; (c) C. S. Arico and L. R. Cox, Org. Biomol. Chem., 2004, 2, 2558 RSC; (d) S. V. Maifeld, M. N. Tran and D. Lee, Tetrahedron Lett., 2005, 46, 105 CrossRef CAS; (e) C. Menozzi, P. I. Dalko and J. Cossy, J. Org. Chem., 2005, 70, 10717 CrossRef CAS PubMed; (f) M. Nagao, K. Asano, K. Umeda, H. Katayama and F. Ozawa, J. Org. Chem., 2005, 70, 10511 CrossRef CAS PubMed; (g) R. Gao, D. R. Pahls, T. R. Cundari and C. S. Yi, Organometallics, 2014, 33, 6937 CrossRef CAS; (h) C. Conifer, C. Gunanathan, T. Rinesch, M. Hölscher and W. Leitner, Eur. J. Inorg. Chem., 2015, 333 CrossRef CAS; (i) Y. Na and S. Chang, Org. Lett., 2000, 2, 1887 CrossRef CAS PubMed.
- (a) K. Junge, K. Schroder and M. Beller, Chem. Commun., 2011, 47, 4849 RSC; (b) B. D. Sherry and A. Fürstner, Acc. Chem. Res., 2008, 41, 1500 CrossRef CAS PubMed; (c) I. Bauer and H.-J. Knölker, Chem. Rev., 2015, 115, 3170 CrossRef CAS PubMed; (d) M. D. Greenhalgh, A. S. Jones and S. P. Thomas, ChemCatChem, 2015, 7, 190 CrossRef CAS; (e) J. Sun and L. Deng, ACS Catal., 2016, 6, 290 CrossRef CAS; (f) X. Du and Z. Huang, ACS Catal., 2017, 1227 CrossRef CAS.
- (a) S. C. Bart, E. Lobkovsky and P. J. Chirik, J. Am. Chem. Soc., 2004, 126, 13794 CrossRef CAS PubMed; (b) C. Belger and B. Plietker, Chem. Commun., 2012, 48, 5419 RSC; (c) M. D. Greenhalgh, D. J. Frank and S. P. Thomas, Adv. Synth. Catal., 2014, 356, 584 CrossRef CAS; (d) A. J. Challinor, M. Calin, G. S. Nichol, N. B. Carter and S. P. Thomas, Adv. Synth. Catal., 2016, 358, 2404 CrossRef CAS.
- (a) M. Isobe, R. Nishizawa, T. Nishikawa and K. Yoza, Tetrahedron Lett., 1999, 40, 6927 CrossRef CAS; (b) L. Yong, K. Kirleis and H. Butenschön, Adv. Synth. Catal., 2006, 348, 833 CrossRef CAS; (c) T. Konno, K.-i. Taku, S. Yamada, K. Moriyasu and T. Ishihara, Org. Biomol. Chem., 2009, 7, 1167 RSC; (d) K.-H. Huang and M. Isobe, Eur. J. Org. Chem., 2014, 4733 CrossRef CAS; (e) A. Rivera-Hernández, B. J. Fallon, S. Ventre, C. Simon, M.-H. Tremblay, G. Gontard, E. Derat, M. Amatore, C. Aubert and M. Petit, Org. Lett., 2016, 18, 4242 CrossRef PubMed.
- (a) T. Bartik, G. Nagy, P. Kvintovics and B. Happ, J. Organomet. Chem., 1993, 453, 29 CrossRef CAS; (b) A. Tillack, S. Pulst, W. Baumann, H. Baudisch, K. Kortus and U. Rosenthal, J. Organomet. Chem., 1997, 532, 117 CrossRef CAS; (c) M. R. Chaulagain, G. M. Mahandru and J. Montgomery, Tetrahedron, 2006, 62, 7560 CrossRef CAS; (d) J. Berding, J. A. van Paridon, V. H. S. van Rixel and E. Bouwman, Eur. J. Inorg. Chem., 2011, 2450 CrossRef CAS.
- Z. Mo, J. Xiao, Y. Gao and L. Deng, J. Am. Chem. Soc., 2014, 136, 17414 CrossRef CAS PubMed.
- J. H. Docherty, J. Peng, A. P. Dominey and S. P. Thomas, Nat. Chem., 2017 DOI:10.1038/nchem.2697.
- (a) J. Guo and Z. Lu, Angew. Chem., Int. Ed., 2016, 55, 10835 CrossRef CAS PubMed; (b) J. Guo, X. Shen and Z. Lu, Angew. Chem., Int. Ed., 2017, 56, 615 CrossRef CAS PubMed.
- Z. Zuo, J. Yang and Z. Huang, Angew. Chem., Int. Ed., 2016, 55, 10839 CrossRef CAS PubMed.
- Note that (Z)-β-selective hydroboration of terminal alkynes has been previously reported by Chirik et al. See: J. V. Obligacion, J. M. Neely, A. N. Yazdani, I. Pappas and P. J. Chirik, J. Am. Chem. Soc., 2015, 137, 5855 CrossRef CAS PubMed.
- W. J. Teo, C. Wang, Y. W. Tan and S. Ge, Angew. Chem., Int. Ed., 2017, 56, 4328 CrossRef CAS PubMed.
- X. Du, Y. Zhang, D. Peng and Z. Huang, Angew. Chem., Int. Ed., 2016, 55, 6671 CrossRef CAS PubMed.
- (a) S. E. Denmark and J. H. C. Liu, Angew. Chem., Int. Ed., 2010, 49, 2978 CrossRef CAS PubMed; (b) Y. Nakao and T. Hiyama, Chem. Soc. Rev., 2011, 40, 4893 RSC; (c) H. F. Sore, W. R. J. D. Galloway and D. R. Spring, Chem. Soc. Rev., 2012, 41, 1845 RSC; (d) S. E. Denmark and A. Ambrosi, Org. Process Res. Dev., 2015, 19, 982 CrossRef CAS PubMed; (e) T. Komiyama, Y. Minami and T. Hiyama, ACS Catal., 2017, 7, 631 CrossRef CAS.
- C. Cheng, E. M. Simmons and J. F. Hartwig, Angew. Chem., Int. Ed., 2013, 52, 8984 CrossRef CAS PubMed.
- Lu reported cross-couplings of two diphenyl-substituted α-vinylsilanes with aryl iodide using Pd2(dba)3 as the catalyst. See ref. 13.
- C. C. H. Atienza, T. Diao, K. J. Weller, S. A. Nye, K. M. Lewis, J. G. P. Delis, J. L. Boyer, A. K. Roy and P. J. Chirik, J. Am. Chem. Soc., 2014, 136, 12108 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7qo00250e |
| This journal is © the Partner Organisations 2017 |