Bisisoindigo: using a ring-fusion approach to extend the conjugation length of isoindigo†
Received
18th September 2015
, Accepted 26th November 2015
First published on 26th November 2015
Abstract
The synthesis of bisisoindigo, a near IR absorbing electron acceptor in which two isoindigo units are ring-fused along the 6 and 7 positions, is reported. An electron-deficient bisisatin was first synthesized using the Martinet isatin synthesis, after which two concurrent aldol condensations with 1-alkyl-2-oxindole yielded the ring-fused isoindigo dimer. The ring fusion significantly extends the conjugation length of the molecule relative to the isoindigo parent compound. The bisisoindigo sub-unit was then coupled to 2,2′-bithiophene, yielding a new organic semiconductor with a donor–acceptor structure. Both bisisoindigo and the donor–acceptor compound absorb light well into the near IR; the donor–acceptor compound features an absorption edge of almost 1000 nm in the solid state. Both compounds were employed in prototype organic photovoltaic devices alongside common electron donors and acceptors.
Introduction
In the last several years, there have been significant advances in the field of organic semiconductor design that have produced substantial increases in the efficiency of organic photovoltaic (OPV) devices and the performance of organic field effect transistors (OFETs). One of the chief reasons behind these advances has been the rise of new organic semiconductors characterized by their use of alternating electron donor and acceptor units.1 Juxtaposition of these units leads to materials with substantially reduced band gaps and good charge carrier mobilities. Common electron donors are often thiophene based (e.g., benzodithiophene,2,3 thieno[3,2-b]thiophene4–6), due to the electron-rich nature of the thiophene ring. In contrast, electron-withdrawing imide and amide groups are prevalent in common acceptor units such as diketopyrollopyrrole and isoindigo.
Isoindigo was first incorporated into organic semiconductors by Mei et al.,7 and has since become ubiquitous throughout the field of organic electronics as an electron accepting unit in both polymers and small molecules.8–15 Many studies have varied the identity of donor groups and alkyl chains, but relatively few variations of isoindigo itself have been reported.16–21 Among the few reported modifications to isoindigo are the 7-haloisoindigos (both chloro and fluoro derivatives),16,17,22 and thienoisoindigo, where the benzene ring is replaced with thiophene.18–20 In recent work, the conjugation length of thienoisoindigo has been extended by the fusion of additional aromatic rings to the molecular structure. Both thienothiophene and benzothienothiophene have been used in this fashion to extend the planarity and conjugation length of isoindigos in organic electronics.23,24
More recently, the concept of using crossed aldol reactions to form expanded isoindigo units has been reported. The reaction of an isatin with benzodifurandione has been shown to produce an expanded, lactone-containing isoindigo system,25,26 and the same type of crossed aldol reaction between benzodifurandione and a thienoisatin yields the thienoisoindigo analogue.27 A second example of an expanded isoindigo system is prepared from the condensation of 2,2′-bithiophene-5,5′-dicarboxaldehyde with an isatin to form an isoindigo-like motif containing an interior bithiophene group.28 Most recently, two groups independently synthesized an expanded isoindigo structure in which one phenyl ring bears two lactam moieties oriented para- to one another.29,30 Our work has been inspired by the idea of exploiting crossed aldol chemistry to expand the isoindigo core unit, as well as using ring fusion to further extend the conjugation length of the system.
Herein, we report the synthesis of bisisoindigo, an expanded isoindigo structure that consists of two isoindigo moieties fused across the 6 and 7 positions. The bisisoindigo building block features a highly extended conjugation length, having an optical band gap well into the near infrared. The dibromobisisoindigo derivative was coupled to a 2,2′-bithiophene moiety to create an organic semiconductor with a donor–acceptor motif. Thin films of this material absorb across the entire visible region, with an absorption edge of nearly 1000 nm. Both bisisoindigo and the donor–acceptor compound were incorporated into the active layers of OPVs. While device performance was modest, both bisisoindigo-based compounds were able to function as electron donors. Bisisoindigo is an exciting new electron-deficient building block in the synthesis of organic semiconductors, and it is anticipated that through judicious choice of both donor sub-unit and alkyl chain length, it will lead to a number of new, high-performance organic semiconductors.
Experimental
Materials and characterization
Prior to use DMF and toluene were dried over activated 3 Å molecular sieves and stored under N2. Pd(PPh3)4 was stored in an inert atmosphere N2 glove box when not in use. All other solvents and reagents were used as received. NMR spectra were obtained using a Bruker Avance 500 MHz spectrometer. UV/vis spectroscopy measurements were performed in CHCl3 or as thin films on glass substrates using a Cary 6000 UV/vis spectrophotometer. AFM measurements were performed using a Dimensions Hybrid Nanoscope system (Veeco Metrology Group). Cyclic voltammetry was carried out in 0.05 mol L−1 tetrabutylammonium hexafluorophosphate dissolved in dry, degassed CH2Cl2. The working electrode was glassy carbon, the counter electrode was a Pt wire, and the reference electrode was a Ag wire. Voltammograms were referenced to an internal Fc/Fc+ standard. Mass spectra were acquired on a JEOL AccuToF 4G GCv mass spectrometer with an EiFi field desorption ionization source. Melting points were measured using a DigiMelt MPA160 (Stanford Research Systems) melting point analyser. Elemental analysis was carried out on a 2400 CHN Elemental analyser (Perkin Elmer).
Synthesis of bisisatin (2).
1,5-Diaminonaphthalene (2.01 g, 12.7 mmol) was dissolved in 20 mL of glacial acetic acid and heated to reflux. To the resulting purple solution was added a solution of diethylketomalonate (8.0 mL, 52 mmol) in glacial acetic acid (23 mL) dropwise over 0.75 hours. The resulting red-brown suspension was heated at reflux for 18 hours. The acetic acid was removed in vacuo and the resulting red solid was dissolved in 1 mol L−1 NaOH to a final solution pH of between 11 and 12. The resulting dark brown solution was heated at reflux with sparging air for 5 hours. The solution was then poured onto ice and acidified to pH 0 with aqueous 6 mol L−1 HCl. The resulting red-purple solid was collected by suction filtration, washed with H2O and dried in vacuo to yield the crude product as a black-purple solid (3.28 g, 97%). The crude product was determined to be approximately 75% pure by 1H NMR spectroscopy; however, due to its extremely low solubility, the product was carried forward without further purification. 1H NMR (500 MHz, DMSO-d6, δ): 11.78 (s, 2H), 7.79 (d, J = 8.3 Hz, 2H), 7.61 (d, J = 8.3 Hz, 2H).
Synthesis of N,N′-bis(2-ethylhexyl)bisisatin (3).
In an oven dried Schlenk flask, crude 2 (2.42 g, 9.08 mmol) and freshly dried K2CO3 (3.16 g, 22.9 mmol) were dissolved in 50 mL of dry DMF. The reaction mixture was heated to 70 °C, 2-ethylhexyliodide (8.60 g, 35.8 mmol) was added via syringe, and the temperature was increased to 100 °C for a further 5.5 hours. The reaction mixture was poured over 200 mL H2O and acidified to pH 7 with aqueous 1 mol L−1 HCl. The aqueous layer was extracted with CH2Cl2. The organic layers were dried over MgSO4 and concentrated to yield the crude product as a viscous black oil. The crude product was purified by column chromatography on silica gel (eluent: 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 EtOAc:hexanes) followed by recrystallization from 1:3 EtOAc:hexanes. The resulting blue solid was isolated by suction filtration and dried in vacuo. Total yield 3: 984 mg (30% yield, assuming 75% pure 1). Mp 175 °C (decomp.). 1H NMR (500 MHz, CDCl3, δ): 8.00 (d, J = 8.7 Hz, 2H), 7.67 (d, J = 8.6 Hz, 2H), 4.11–4.24 (m, 5H), 1.85–1.91 (m, 2H), 1.23–1.45 (m, 16H), 0.93 (t, J = 7.5 Hz, 6H), 0.87 (t, J = 6.6 Hz, 6H). 13C NMR (125 MHz, CDCl3, δ): 182.7, 159.3, 152.3, 127.4, 120.1, 120.0, 116.4, 47.2, 38.7, 30.1, 28.4, 28.3, 23.5, 23.0, 14.0, 10.35, 10.33. HRMS (m/z): (M˙+) calc. (C30H38N2O4): 490.28316 found: 490.28261. Anal. calcd for C30H38N2O4: C, 73.44; H, 7.81; N, 5.71; found: C, 73.43; H, 7.70; N, 5.64.
:3 EtOAc:hexanes) followed by recrystallization from 1:3 EtOAc:hexanes. The resulting blue solid was isolated by suction filtration and dried in vacuo. Total yield 3: 984 mg (30% yield, assuming 75% pure 1). Mp 175 °C (decomp.). 1H NMR (500 MHz, CDCl3, δ): 8.00 (d, J = 8.7 Hz, 2H), 7.67 (d, J = 8.6 Hz, 2H), 4.11–4.24 (m, 5H), 1.85–1.91 (m, 2H), 1.23–1.45 (m, 16H), 0.93 (t, J = 7.5 Hz, 6H), 0.87 (t, J = 6.6 Hz, 6H). 13C NMR (125 MHz, CDCl3, δ): 182.7, 159.3, 152.3, 127.4, 120.1, 120.0, 116.4, 47.2, 38.7, 30.1, 28.4, 28.3, 23.5, 23.0, 14.0, 10.35, 10.33. HRMS (m/z): (M˙+) calc. (C30H38N2O4): 490.28316 found: 490.28261. Anal. calcd for C30H38N2O4: C, 73.44; H, 7.81; N, 5.71; found: C, 73.43; H, 7.70; N, 5.64.
Synthesis of bisisoindigo (6).
Compounds 3 (399 mg, 0.812 mmol) and 4 (478 mg, 1.95 mmol) were dissolved in 15 mL of glacial acetic acid. Concentrated HCl (100 μL) was added and the reaction mixture was heated at reflux for 14 hours. After 14 hours the reaction mixture was poured over a mixture of ice and brine, and extracted with CH2Cl2. The combined organic layers were washed with brine, dried over MgSO4, and concentrated to yield the crude product as a black solid. The crude product was purified by column chromatography (silica gel, eluent: 60:40 CH2Cl2:hexanes) and concentrated in vacuo to yield a dark brown-black solid (347 mg, 45%). Mp 250–252 °C. 1H NMR (500 MHz, CD2Cl2, δ): 9.06 (d, J = 7.9 Hz, 2H), 9.00 (d, J = 9.3 Hz, 2H), 7.87 (d, J = 9.36, 2H), 7.37 (t, J = 8.2, 2H), 7.03 (t, J = 7.4 Hz, 2H), 6.82 (d, J = 7.8 Hz), 4.20–4.35 (m, 4H), 3.62–3.73 (m, 4H), 1.98–2.07 (m, 2H), 1.84–1.92 (m, 2H), 1.22–1.48 (m, 37H), 0.84–0.99 (m, 26H). 13C NMR (125 MHz, CDCl3, δ): 169.9, 168.1, 145.4, 143.2, 134.0, 132.9, 132.4, 129.2, 124.3, 123.4, 122.3, 122.1, 120.1, 117.2, 108.4, 47.0, 44.3, 38.5, 37.7, 30.8, 30.2, 28.8, 28.3, 24.1, 23.5, 23.1, 14.1, 14.0, 10.7, 10.5. HRMS (m/z): (M˙+) calc. (C62H80N4O4): 944.61795 found: 944.62061. Anal. calcd for C62H80N4O4: C, 78.77; H, 8.53; N, 5.93; found: C, 77.79; H, 8.60; N, 5.78.
Synthesis of dibromo-bisisoindigo (7).
Compound 3 (462.6 mg, 0.9433 mmol) and 5 (827.6 mg, 2.55 mmol) were dissolved in 20 mL of glacial acetic acid. Concentrated HCl (100 μL) was added and the reaction mixture was heated at reflux for 18 hours. The reaction mixture was poured over H2O and extracted with CH2Cl2. The combined organic layers were washed with brine, dried over MgSO4, and concentrated to yield the crude product. The crude product was purified by column chromatography (silica gel, eluent: 70:30 CH2Cl2:hexanes, gradient to 10:1 CH2Cl2:MeOH), concentrated in vacuo to yield a brown-black solid, and washed with acetone (235 mg, 23%). Mp 237–252 °C (decomp.). 1H NMR (500 MHz, CDCl3, δ): 8.95 (dd, J1 = 2.4 Hz, J2 = 9.0 Hz, 4H), 7.84 (d, J = 9.4 Hz, 2H), 7.17 (d, J = 8.6 Hz, 2H), 6.90 (s, 2H), 4.18–4.30 (m, 4H), 3.58–3.71 (m, 4H), 1.95–2.04 (m, 2H), 1.80–1.90 (m, 2H), 1.21–1.47 (m, 34H), 0.83–0.98 (m, 25H). 13C NMR (125 MHz, CDCl3, δ): 169.9, 168.0, 143.4, 132.6, 132.6, 130.3, 126.9, 125.2, 120.8, 117.3, 111.7, 47.0, 44.4, 38.5, 37.5, 30.6, 30.2, 28.6, 28.3, 24.0, 23.5, 23.1, 14.1, 14.0, 10.7, 10.5, 7.8. HRMS (m/z) (M˙+) calc. (C62H78Br2N2O4): 1102.43693 found: 1102.43715. Anal. calcd for C62H78Br2N2O4: C, 67.51; H, 7.13; N, 5.08; found: C, 67.15; H, 7.45; N, 5.91.
Synthesis of bis-bithiophene-bisisoindigo (8).
Compound 7 (145 mg, 0.132 mmol), Pd(PPh3)4 (11.1 mg, 9.6 μmol), and 5′-hexyl-5-tributylstannyl-2,2′-bithiophene (315 mg, 0.584 mmol) were added sequentially to 15 mL dry, degassed toluene. The reaction mixture was heated to reflux for 15 hours. The reaction mixture was concentrated in vacuo, purified by column chromatography (silica gel, eluent 70:30 toluene:hexanes, gradient to 3% methanol:toluene), and the product recrystallized from CH2Cl2/acetone to yield a blue-black solid (151 mg, 80%). Mp 237–245 °C. 1H NMR (500 MHz, CD2Cl2, δ): 9.02 (d, J = 8.4 Hz, 2H), 8.90 (d, J = 9.2 Hz, 2H), 7.78 (d, J = 9.6 Hz, 2H), 7.31 (d, J = 3.8 Hz, 2H), 7.21 (d, J = 8.7 Hz, 2H) 7.05 (q, J = 5.8 Hz, 4H), 6.71 (d, J = 3.6 Hz), 4.19–4.34 (m, 5H), 3.60–3.78 (m, 5H), 2.80 (t, J = 7.6 Hz, 3H), 1.97–2.04 (m, 3H), 1.82–1.91 (m, 2H), 1.65–1.73 (m, 4H), 1.53–1.63 (m, 6H), 1.21–1.49 (m, 39H), 0.82–1.00 (m, 24H). 13C NMR (125 MHz, CDCl3, δ): 169.8, 168.3, 146.2, 145.8, 142.6, 141.8, 138.9, 137.8, 134.5, 132.5, 130.6, 129.9, 125.2, 125.0, 124.1, 124.0, 123.8, 122.9, 121.1, 119.9, 118.5, 117.0, 104.5, 46.9, 44.0, 38.5, 37.8, 31.6, 31.56, 30.9, 30.3, 30.2, 29.7, 28.0, 28.8, 28.8, 28.4, 28.3, 24.2, 23.5, 23.2, 23.2, 23.1, 22.6, 14.2, 14.1, 10.9, 10.6, 10.6, 10.5. HRMS (m/z) (M˙+) calc. (C90H112N4O4S4): 1440.75664 found: 1440.75420. Anal. calcd for C90H112N4O4S4: C, 74.96; H, 7.83; N, 3.88; found: C, 74.78; H, 8.49; N, 3.29.
Results and discussion
Synthesis
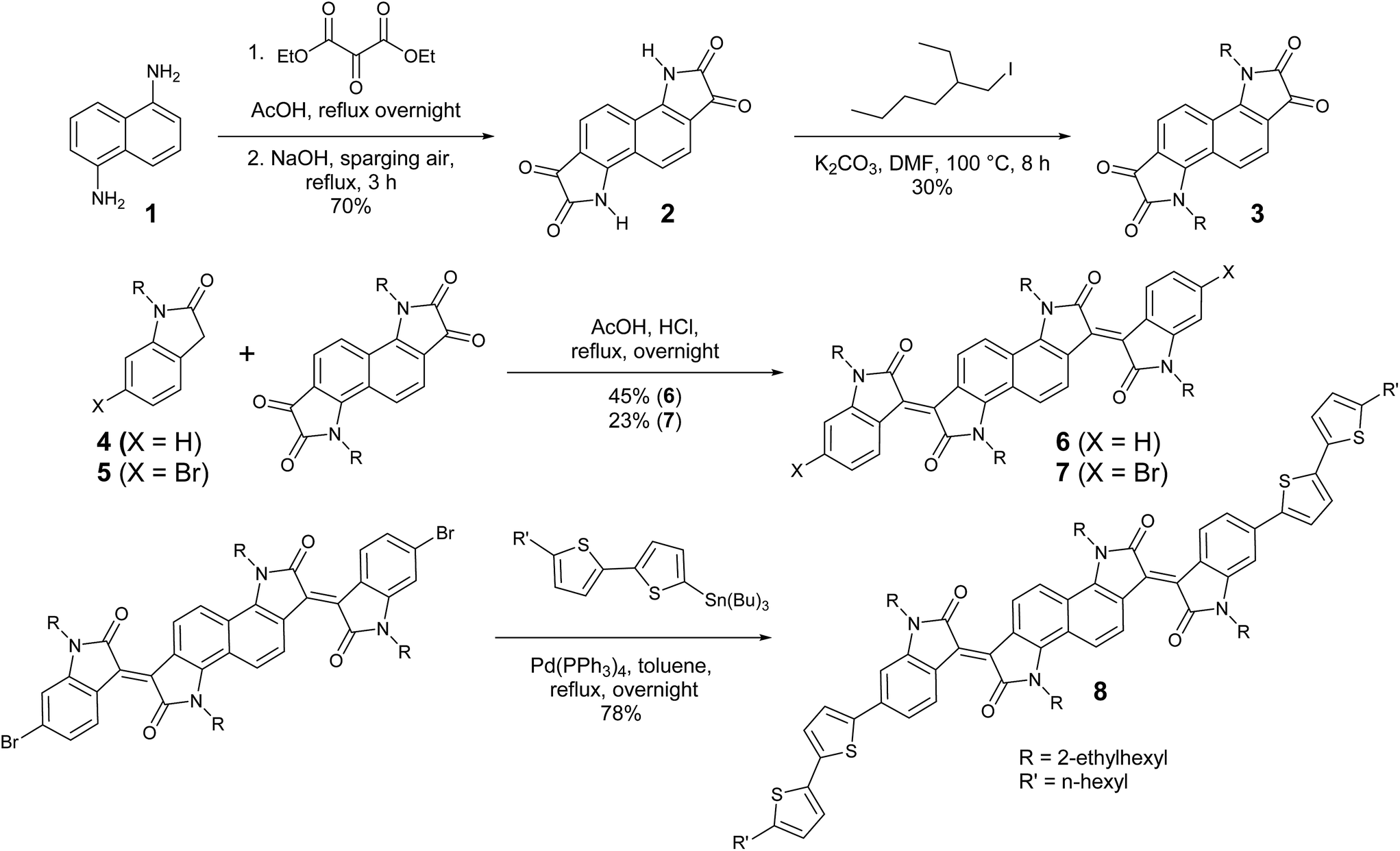
The preparation of bisisoindigo (Scheme 1) presented several distinct synthetic challenges. Isatins are often synthesized by the Sandmeyer isatin synthesis.31 Attempts to perform the Sandmeyer isatin synthesis using 1,5-diaminonaphthalene (1) as described by Kossmehl and Manecke32 yielded the desired bisisatin (2) as a minor product and aminobenzoisatin as the major product (Scheme S1 ESI†). The ring-closing step of the Sandmeyer isatin synthesis requires the electrophilic aromatic substitution of a hydroxyimine group onto a benzene ring.33 When attempting to perform this reaction twice on a single ring system, the formation of the first isatin withdraws much of the electron density from the ring, preventing the second ring closure. Therefore, in order to synthesize bisisatin (2), the Martinet isatin synthesis (Scheme S1 ESI†) was employed.34 In this reaction, the ring closing step produces a 3-hydroxy-2-oxindole intermediate. This group is much less electron withdrawing than an isatin, and therefore the aromatic ring is not deactivated towards the second ring closure. Bubbling air is sufficient to oxidize both 3-hydroxy-2-oxindole groups to the corresponding isatins, producing 2 as the major product. The electron deficient nature of 2 stabilizes the anion formed by deprotonation of the amide, making it a poor nucleophile, and alkyl iodides were required to alkylate the heterocyclic N-atom to form 3. An acid-catalysed double crossed aldol reaction of 3 and the appropriate 2-oxindole derivatives (4 or 5) was then used to complete the synthesis of 6 and 7. The dibrominated derivative was subsequently used in a Stille cross-coupling reaction with 5-tributylstannyl-5′-hexyl-2,2′-bithiophene to form a donor–acceptor–donor molecule, 8. Significant colour changes were noted with each subsequent extension of the conjugation; 3 is a blue solid, 6 is a brown-black solid, and 8 is a dark blue-black solid.
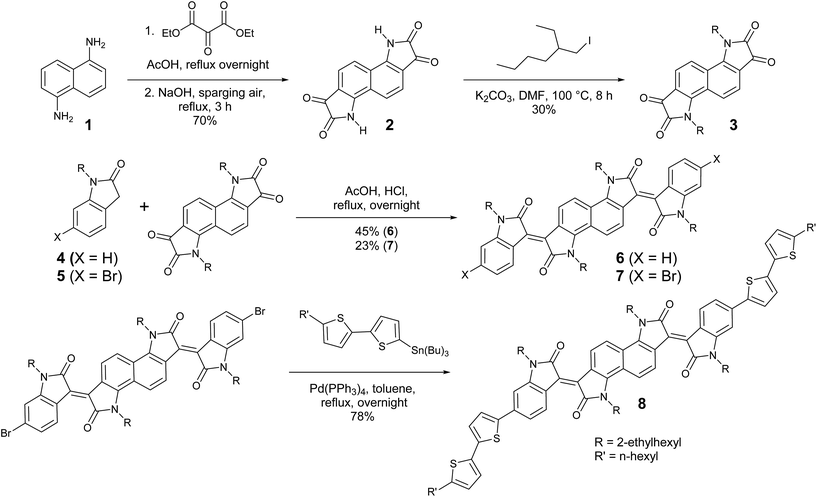 |
| | Scheme 1 Synthesis of bisisoindigo (6) and bis(5′-hexyl-2,2′-bithienyl)-bisisoindigo (8). | |
Energy levels and absorption spectra of 6 and 8
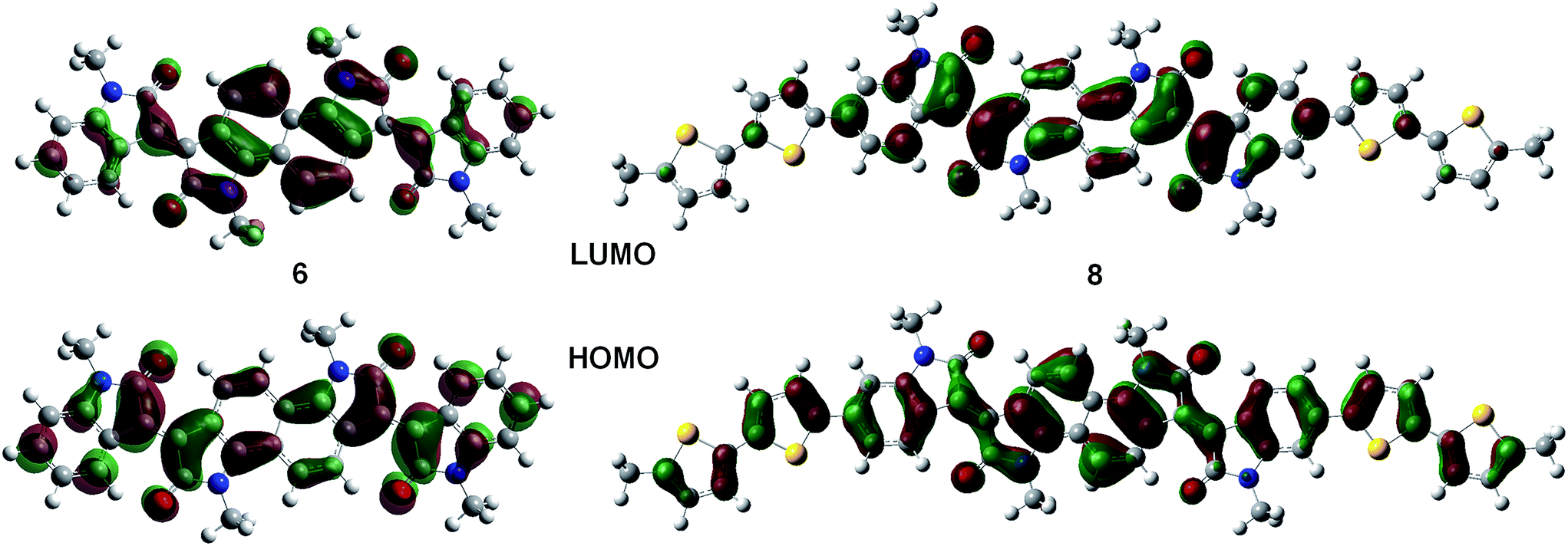
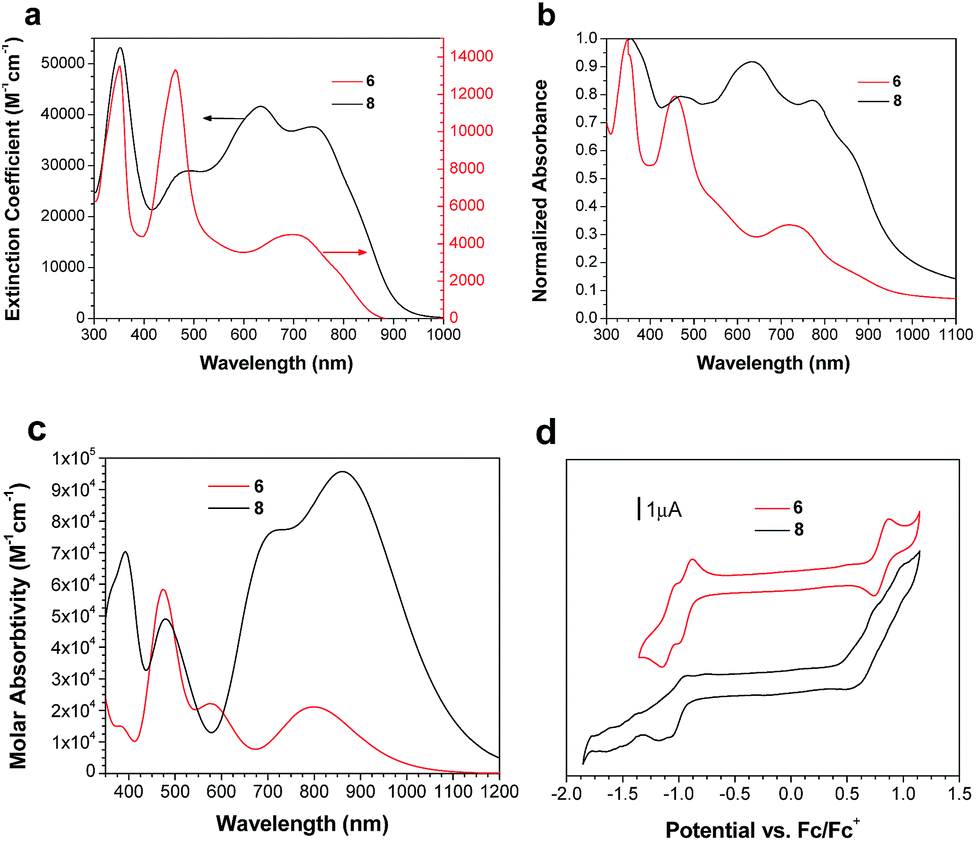
Solution and solid state UV/vis spectroscopy was performed on 6 and 8 (Fig. 2a and b). Using time dependent density functional theory (TDDFT) calculations (Fig. 1 and 2c), the lowest energy peak in the spectra of both 6 and 8 can be assigned to the HOMO-to-LUMO transition; the additional peak at 625 nm in the spectrum of 8 is assigned to a transition from a bithiophene-based π-orbital to the LUMO. As a result of the extended conjugation in bisisoindigo, both molecules absorb well into the near infrared. The effect of the donor bithiophene group is immediately apparent when the spectra of 6 and 8 are compared. While the HOMO-to-LUMO transition in 6 has a relatively low oscillator strength, the HOMO-to-LUMO transition in 8 is much more intense. This increase in oscillator strength is also predicted by the TDDFT calculations. Calculation of the frontier orbital geometries of both 6 and 8 (Fig. 1) reveals a higher degree of orbital localization in the LUMO of the donor–acceptor compound. While both the HOMO and LUMO of 6 are delocalized across the entire molecule, the LUMO of 8 is isolated almost entirely on the bisisoindigo core. This localization leads to an increase in the transition dipole moment of the HOMO-to-LUMO transition, greatly increasing its extinction coefficient. The large extinction coefficient and small band gap (Table 1) of 8 are ideal characteristics for application in organic photovoltaic devices. Cyclic voltammetry (Fig. 2d) was used to determine the absolute energy levels of 6 and 8 (Table 1). Consistent with the addition of electron donating bithiophene groups, the HOMO of 8 lies 0.2 eV higher in energy than that of 6; however, the energy of the LUMO is unchanged, providing further evidence that the bithiophene groups do not appreciably contribute to the LUMO. Compound 6 exhibits a quasi-reversible oxidation wave, as well as two independent quasi-reversible reduction peaks. Conversely, neither the oxidation nor reduction of 8 was reversible. Upon oxidation and reduction a solid film was noted on the surface of the glassy carbon electrode. The onset of oxidation and reduction were used to estimate the HOMO and LUMO levels of 6 and 8, and are listed in Table 1.
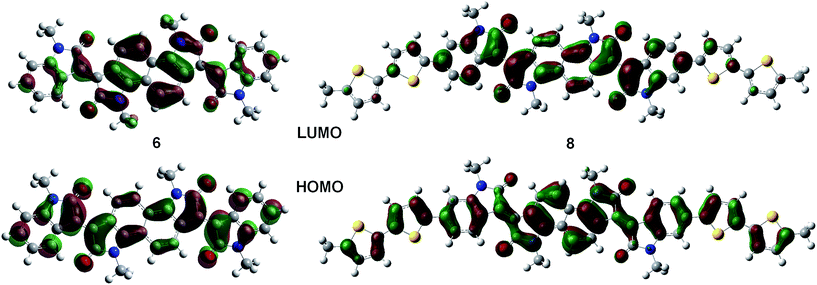 |
| | Fig. 1 Frontier molecular orbitals of 6 and 8. | |
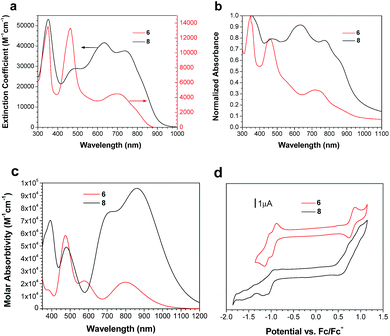 |
| | Fig. 2 UV/vis absorption spectra of 6 and 8 in (a) CHCl3 and (b) as a thin film on glass. (c) theoretical UV/vis spectra of 6 and 8. (d) Cyclic voltammograms of 6 and 8 in 0.05 mol L−1 (NBu4)PF6 in CH2Cl2. | |
Table 1 Calculated and experimental frontier orbital energy levels and band gaps of 6 and 8
| Compound |
E
HOMO
(eV) |
E
LUMO
(eV) |
E
Gap
(eV) |
E
Gap
(eV) |
E
Gap
(eV) |
|
From onset of oxidation, Fc/Fc+ referenced as 5.1 eV relative to vacuum.
From onset of reduction.
E
HOMO − ELUMO, as determined from voltammograms.
Calculated from TDDFT results (see ESI†).
Calculated from the absorption onset.
|
|
6
|
5.8 |
4.2 |
1.6 |
1.55 |
1.44 |
|
8
|
5.6 |
4.2 |
1.4 |
1.42 |
1.27 |
Performance in OPV devices
The cyclic voltammetry results indicated that the frontier orbital energies of both 6 and 8 lay in between those of OPV donor materials such as PTB7-Th,3 and fullerene-based acceptors such as phenyl-C71-butyric acid methyl ester (PC71BM). This suggested that 6 and 8 could be used as either the electron donor or the electron acceptor in an OPV device. Therefore, 6 and 8 were tested in both roles: as the electron acceptor (blended with PTB7-Th), and as the electron donor (blended with PC71BM). OPVs were first fabricated using an active layer blend of PTB7-Th:6 (Table 2, Fig. S1 and S2 ESI†). Device performance was modest, but improved greatly with the addition of 1,8-diiodooctane (DIO). Atomic force microscopy (AFM) images of PTB7-Th:6 blends (Fig. S3 ESI†) suggests that the addition of DIO decreases the domain size from ∼1 μm to <100 nm. Unfortunately, even upon addition of DIO, some large crystallites were still visible. However, although the overall efficiency was low, the devices did nonetheless display very high Voc values (up to 1.0 V). OPVs fabricated with PTB7-Th:8 active layers also performed relatively poorly. This is likely because 8 was insufficiently soluble in chlorobenzene and o-dichlorobenzene, the ideal solvents for processing PTB7-Th. The films cast from chloroform exhibited poor film morphologies and large domain sizes, which greatly reduced device performance (Fig. S1 and S4 ESI†).
Table 2 Champion photovoltaic device performance of ITO/PEDOT:PSS/active layer/LiF/Al bulk heterojunction solar cells
| Active layer |
V
oc (V) |
J
sc (mA cm−2) |
Fill factor (%) |
Efficiency (%) |
| PTB7-Th:6 |
1.0 |
0.33 |
31 |
0.10 |
|
6:PC71BM |
0.68 |
0.92 |
45 |
0.28 |
| PTB7-Th:8 |
0.60 |
0.030 |
27 |
0.0048 |
|
8:PC71BM |
0.59 |
1.28 |
30 |
0.23 |
Although poor film morphology is likely the cause of the poor photovoltaic performance, one alternative is a lack of energetic driving force for electron transfer at the donor/acceptor interface. Electron transfer between the donor (PTB7-Th) and acceptor (6/8) was therefore evaluated using solution phase fluorescence spectroscopy (Fig. S6–S8 ESI†); a decrease in donor fluorescence in the presence of 6 or 8 is indicative of electron transfer. In order to extract quantitative information regarding fluorescence quenching, a Stern–Volmer analysis was performed (Fig. S8 ESI†). The fluorescence quenching of PTB7-Th yielded Stern–Volmer constants of (6 ± 1) × 104 mol−1 L when quenched by 6, and (1.1 ± 0.2) × 104 mol−1 L when quenched by 8 (Table S5 ESI†). These values are similar to the Stern–Volmer quenching constant obtained when P3HT emission is quenched by PC61BM (1.6 × 104 mol−1 L).35 This suggests that there is a sufficiently large energy level offset to split the exciton at the donor/acceptor interface, and that either the morphology or the electron mobility limit the performance of these devices.
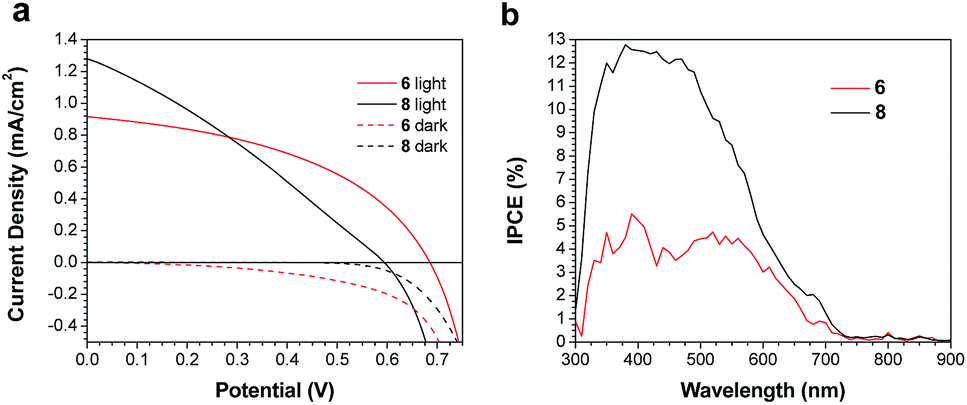
Both 6 and 8 were then evaluated as electron donors alongside PC71BM (Table 2, Fig. 3). DIO loadings between 0% and 3% were investigated (see ESI†) and the champion device using 6:PC71BM and 1% DIO produced an efficiency of 0.28%. The current densities produced by these cells were 3× greater than those of the PTB7-Th containing cells. Similarly, devices with an active layer of 8:PC71BM performed much better than the analogous PTB7-Th:8 devices. However, these devices suffered from a much lower fill factor than those using 6:PC71BM active layers (Fig. 3a). This is likely due to the poor blend morphology, which again results from the use of chloroform as the casting solvent. Optical microscopy revealed cracks in the 8:PC71BM films, which would be expected to lead to microshorts and carrier recombination, decreasing the fill factor. The larger extinction coefficient of 8 relative to 6 is reflected in a higher incident photon-to-current efficiency (IPCE) (Fig. 3b). As in the PTB7-Th based OPVs, the addition of DIO was required to optimize the performance of the 6/8:PC71BM devices; however, instead of seeing a decrease in domain size, the formation of larger domains is observed (Fig. 4). The addition of 1% DIO to 6:PC71BM blends greatly increased the device performance, while a 3% loading led to domains that were too large, leading to a decrease in device efficiency.
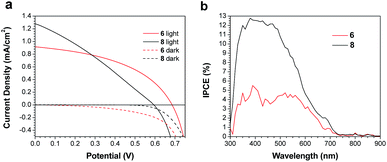 |
| | Fig. 3 (a) J–V curves for champion devices, and (b) IPCE spectra for devices based on active layers of 6 or 8 and PC71BM. | |
 |
| | Fig. 4 10 × 10 μm AFM images of active layer blends cast from 20 mg mL−1 solutions; (a–c) 6:PC71BM active layers with (a) 0%, (b) 1%, (c) 3% DIO (v/v); (d–f) 8:PC71BM active layers with (d) 0%, (e) 1%, (f) 3% DIO (v/v). | |
Conclusions
In conclusion, we have reported the synthesis of a unique electron acceptor sub-unit incorporating two isoindigo units fused along the 6 and 7 positions (6). This acceptor sub-unit was then coupled to 2,2′-bithiophene donor groups to form a donor–acceptor compound (8). Both 6 and 8 absorb light well into the near infrared, with 8 exhibiting a large extinction coefficient due to a higher degree of intramolecular charge transfer character in the HOMO-to-LUMO transition. 6 and 8 were incorporated into OPV active layers (paired with either PTB7-Th or PC71BM). Although the power conversion efficiencies were modest, bisisoindigo (6) features the deep energy levels desired in an electron acceptor, and 8 absorbs strongly throughout the visible and into the near infrared, demonstrating the utility of the bisisoindigo building block. The incorporation of 6 into new donor–acceptor compounds (both polymers and small molecules) may therefore yield a variety of new, high-performance organic semiconductors.
Acknowledgements
The Natural Science and Engineering Research Council of Canada (NSERC) and the University of Saskatchewan are acknowledged for financial support. T. L. K. is a Canada Research Chair in Photovoltaics. This research was undertaken in part, thanks to funding from the Canada Research Chairs Program. N. M. R. and P. C. B. thank NSERC for scholarship funding; P. C. B. also thanks the Government of Saskatchewan for the Queen Elizabeth II Centennial Aboriginal Scholarship.
Notes and references
- A. J. Heeger, Chem. Soc. Rev., 2010, 39, 2354–2371 RSC.
- C. Y. Mei, L. Liang, F. G. Zhao, J. T. Wang, L. F. Yu, Y. X. Li and W. S. Li, Macromolecules, 2013, 46, 7920–7931 CrossRef CAS.
- S. H. Liao, H. J. Jhuo, Y. S. Cheng and S. A. Chen, Adv. Mater., 2013, 25, 4766–4771 CrossRef CAS PubMed.
- I. McCulloch, M. Heeney, M. L. Chabinyc, D. DeLongchamp, R. J. Kline, M. Cölle, W. Duffy, D. Fischer, D. Gundlach, B. Hamadani, R. Hamilton, L. Richter, A. Salleo, M. Shkunov, D. Sparrowe, S. Tierney and W. Zhang, Adv. Mater., 2009, 21, 1091–1109 CrossRef CAS.
- J. C. Bijleveld, R. A. M. Verstrijden, M. M. Wienk and R. A. J. Janssen, J. Mater. Chem., 2011, 21, 9224–9231 RSC.
- J. A. Schneider, H. Black, H. P. Lin and D. F. Perepichka, ChemPhysChem, 2015, 16, 1173–1178 CrossRef CAS PubMed.
- J. Mei, K. R. Graham, R. Stalder and J. R. Reynolds, Org. Lett., 2010, 12, 660–663 CrossRef CAS PubMed.
- E. Wang, W. Mammo and M. R. Andersson, Adv. Mater., 2014, 26, 1801–1826 CrossRef CAS PubMed.
- R. Stalder, J. Mei, K. R. Graham, L. A. Estrada and J. R. Reynolds, Chem. Mater., 2013, 26, 664–678 CrossRef.
- C. C. Ho, C. A. Chen, C. Y. Chang, S. B. Darling and W. F. Su, J. Mater. Chem. A, 2014, 2, 8026–8032 CAS.
- Z. Wang, J. Zhao, Y. Li and Q. Peng, Polym. Chem., 2014, 5, 4984–4992 RSC.
- E. D. Głowacki, G. Voss and N. S. Sariciftci, Adv. Mater., 2013, 25, 6783–6800 CrossRef PubMed.
- X. Guo, A. Facchetti and T. J. Marks, Chem. Rev., 2014, 114, 8943–9021 CrossRef CAS PubMed.
- R. Stalder, J. Mei, J. Subbiah, C. Grand, L. A. Estrada, F. So and J. R. Reynolds, Macromolecules, 2011, 44, 6303–6310 CrossRef CAS.
- F. Grenier, P. Berrouard, J. R. Pouliot, H. R. Tseng, A. J. Heeger and M. Leclerc, Polym. Chem., 2013, 4, 1836–1841 RSC.
- T. Lei, J. H. Dou, Z. J. Ma, C. H. Yao, C. J. Liu, J. Y. Wang and J. Pei, J. Am. Chem. Soc., 2012, 134, 20025–20028 CrossRef CAS PubMed.
- T. Lei, J. H. Dou, Z. J. Ma, C. J. Liu, J. Y. Wang and J. Pei, Chem. Sci., 2013, 4, 2447–2452 RSC.
- Y. Koizumi, M. Ide, A. Saeki, C. Vijayakumar, B. Balan, M. Kawamoto and S. Seki, Polym. Chem., 2013, 4, 484–494 RSC.
- G. K. Dutta, A. R. Han, J. Lee, Y. Kim, J. H. Oh and C. Yang, Adv. Funct. Mater., 2013, 23, 5317–5325 CrossRef CAS.
- G. Kim, S. J. Kang, G. K. Dutta, Y. K. Han, T. J. Shin, Y. Y. Noh and C. Yang, J. Am. Chem. Soc., 2014, 136, 9477–9483 CrossRef CAS PubMed.
- N. M. Randell, A. F. Douglas and T. L. Kelly, J. Mater. Chem. A, 2014, 2, 1085–1092 CAS.
- Y. Deng, J. Liu, J. Wang, L. Liu, W. Li, H. Tian, X. Zhang, Z. Xie, Y. Geng and F. Wang, Adv. Mater., 2014, 26, 471–476 CrossRef CAS PubMed.
- I. Meager, M. Nikolka, B. C. Schroeder, C. B. Nielsen, M. Planells, H. Bronstein, J. W. Rumer, D. I. James, R. S. Ashraf, A. Sadhanala, P. Hayoz, J. C. Flores, H. Sirringhaus and I. McCulloch, Adv. Funct. Mater., 2014, 24, 7109–7115 CAS.
- W. Yue, R. S. Ashraf, C. B. Nielsen, E. Collado-Fregoso, M. R. Niazi, S. A. Yousaf, M. Kirkus, H. Y. Chen, A. Amassian, J. R. Durrant and I. McCulloch, Adv. Mater., 2015, 27, 4702–4707 CrossRef CAS PubMed.
- T. Lei, J. H. Dou, X. Y. Cao, J. Y. Wang and J. Pei, Adv. Mater., 2013, 25, 6589–6593 CrossRef CAS PubMed.
- X. Zhou, N. Ai, Z. H. Guo, F. D. Zhuang, Y. S. Jiang, J. Y. Wang and J. Pei, Chem. Mater., 2015, 27, 1815–1820 CrossRef CAS.
- G. Zhang, Z. Ye, P. Li, J. Guo, Q. Wang, L. Tang, H. Lu and L. Qiu, Polym. Chem., 2015, 6, 3970–3978 RSC.
- S. Li, Z. Yuan, J. Yuan, P. Deng, Q. Zhang and B. Sun, J. Mater. Chem. A, 2014, 2, 5427–5433 CAS.
- J. Pei, Y. Cao, J. S. Yuan, X. Zhou, X. Y. Wang, F. D. Zhuang and J. Y. Wang, Chem. Commun., 2015, 51, 10514–10516 RSC.
- Y. He, C. Guo, B. Sun, J. Quinn and Y. Li, Chem. Commun., 2015, 51, 8093–8096 RSC.
- J. F. D. Silva, S. J. Garden and A. C. Pinto, J. Braz. Chem. Soc., 2001, 12, 273–324 CrossRef.
- G. Kossmehl and G. Manecke, Makromol. Chem., 1975, 176, 333–340 CrossRef CAS.
- B. V. Silva, F. A. Violante, A. C. Pinto and L. S. Santos, Rapid Commun. Mass Spectrom., 2011, 25, 423–428 CrossRef CAS PubMed.
- K. C. Rice, B. J. Boone, A. B. Rubin and T. J. Rauls, J. Med. Chem., 1976, 19, 887–892 CrossRef CAS PubMed.
- W. Senevirathna and G. Sauvé, J. Mater. Chem. C, 2013, 1, 6684–6694 RSC.
Footnote |
| † Electronic supplementary information including detailed synthetic procedures, photovoltaic characterization, fluorescence quenching, atomic force microscopy and NMR spectra available online. See DOI: 10.1039/c5ta07511d |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence