
Cu(II)-catalyzed C6-selective C–H thiolation of 2-pyridones using air as the oxidant†
Panfeng
Peng
ab,
Jiang
Wang
b,
Chunpu
Li
b,
Wei
Zhu
b,
Hualiang
Jiang
*ab and
Hong
Liu
*b
aSchool of Pharmaceutical Engineering, Shenyang Pharmaceutical University, 103 Wen Hua Road, Liaoning, Shenyang 110016, China. E-mail: hljiang@simm.ac.cn
bCAS Key Laboratory of Receptor Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, 555 Zu Chong Zhi Road, Zhangjiang Hi-Tech Park, Shanghai 201203, People's Republic of China. E-mail: hliu@simm.ac.cn
First published on 10th June 2016
Abstract
The Cu(II)-catalyzed and chelate-directed C6-selective C–H thiolation of 2-pyridones with disulfides was developed to provide aryl and alkyl thioethers. This transformation uses a catalytic amount of Cu(OAc)2 and molecular oxygen in air as an oxidant, no cocatalysts or metallic oxidants are required. The reaction accommodated both electronic and steric factors at the C3–C5 positions of 2-pyridones, which is efficient for the C6 thiolation of a broad range of 2-pyridones with up to 93% yield.
The 6-substituted-2-pyridone structural motif is a prevalent heterocyclic core structure in many biologically active compounds (Fig. 1).1 Therefore, many researchers have developed synthetic methods for functional pyridones. Generally, they are prepared by the SN2 reaction between thiols or metal thiolates and organic halides.1d,f Transition metal-mediated cross-coupling of halogenated pyridones with organometallic reagents has gradually become a practical strategy.2 Disadvantages of these classic transformations include the unpleasant smell, low substrate scope, and the prefunctionalization of substrates. For the past decades, chemical approaches based upon direct C–H bond functionalization have allowed the formation of C–C and C-heteroatom bonds through more effective synthetic routes with high atom economy.3 Although, studies of transition metal-mediated C–H functionalization of comparably reactive C3 and C5 positions of 2-pyridones have been reported, only few examples of transition metal-mediated C6-selective C–H functionalization exist in the literature.4 Hiyama et al. discovered C6-selective alkenylation and alkylation of 2-pyridones by employing Ni(0)/Lewis-acid catalysts.4g,4h Recently, Miura et al. developed a Cu(II)-mediated C6-selective dehydrogenative heteroarylation of 2-pyridones with 1,3-azoles (Scheme 1a).4i Cramer et al. demonstrated the synthesis of 1,6-annulated 2-pyridones by Ni(0)-catalyzed intramolecular cyclization.4j However, to the best of our knowledge, the formation of C–S bond via transition metal-catalyzed C6-selective C–H functionalization of 2-pyridones has not been reported.
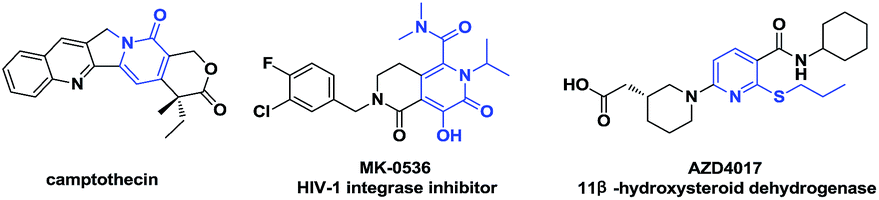 | ||
| Fig. 1 Biologically active molecules with 6-substituted-2-pyridone scaffolds. | ||
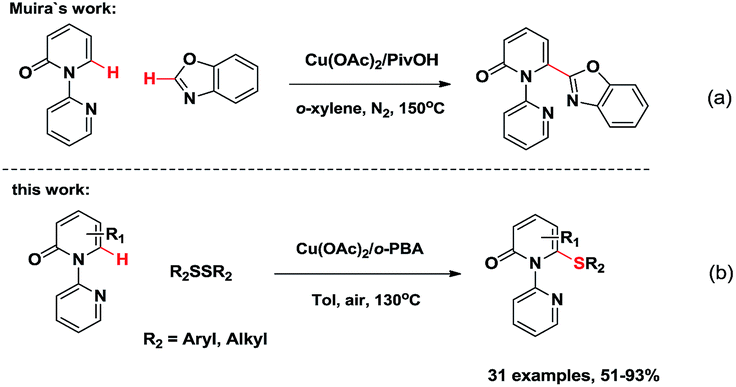 | ||
| Scheme 1 Cu(II)-mediated thiolation of C(sp2)–H bond. | ||
Although significant advances have been made in the formation of C–C or C-heteroatom bonds through direct C–H functionalization, reports of C–S bond formation through direct C–H thiolation remain scarce. This is presumably due to the deactivation of the transition metals by strong coordination to the sulfur atom.5 Yu et al. reported the first direct thiolation of 2-phenylpyridine via a Cu(II)-mediated C–H functionalization process.6 Elegant studies have demonstrated the high potential of Cu catalysts in the thiolation of arenes C(sp2)–H bonds.7 However, they are largely restricted to the using of stoichiometric Cu catalysts. Recently, Pd,8 Rh9 and Ni10 catalysts have been reported for the direct thiolation of arenes to produce diverse aryl sulfides. Nevertheless, many methods relied on the use of expensive transition metal catalysts and stoichiometric Cu(II)11 or Ag(I) salts as oxidants. Therefore, the development of a strategy for the synthesis of diverse C(sp2)–S using more sustainable and efficient catalytic system would be highly desirable.
Among the various transition metal catalysts, copper has attracted increasing attention owing to its low-cost, earth-abundance, and environmentally-benign.12 Herein, we report the Cu(II)-catalyzed C6-selective C–H thiolation of 2-pyridones for the synthesis of the functionalized 6-sulfenylation-2-pyridones in moderate to excellent yields (Scheme 1b). The directing group was readily removed, thus leading to 2-pyridone derivatives with a free N–H group. The reaction occurred without using any noble metals. Moreover, atmospheric oxygen is a suitable oxidant for the catalytic turnover of the Cu salt.
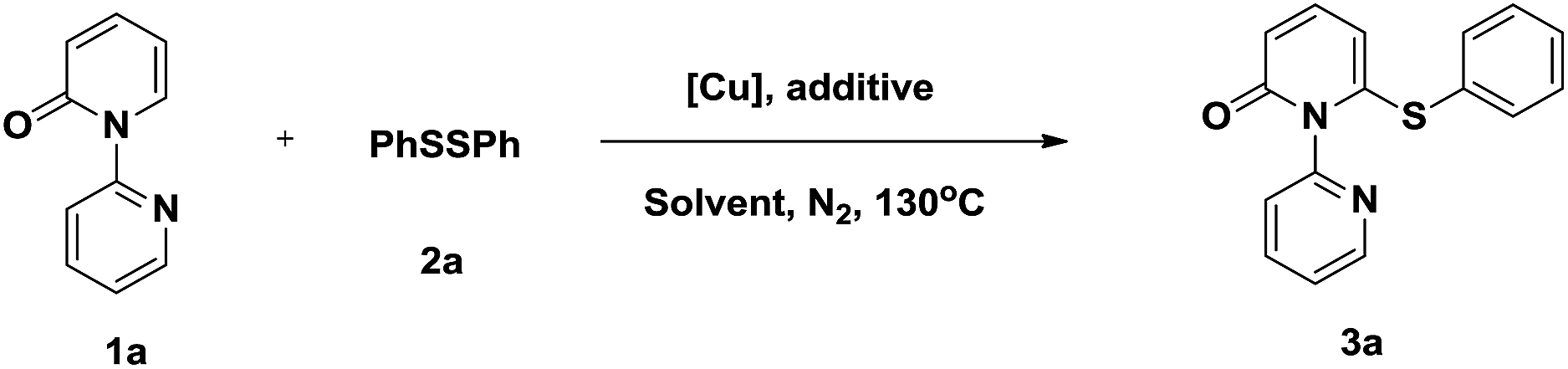
To verify the proposed strategy, we discovered that the reaction of 1-(2-pyridyl)-2-pyridone (1a) with 1.0 equiv. of diphenyl disulfide (2a) and 1.0 equiv. of Cu(OAc)2 in toluene under N2 at 130 °C for 20 h gave the desired thiolated product 3a in 81% yield (Table 1, entry 1). The structure of thiolated product 3a was confirmed by single-crystal X-ray analysis (Fig. S1 in the ESI†).12 The use of other Cu(II) catalysts, including Cu(OTf)2, CuBr2, and CuF2, led to poor results (entries 2–4). The optimization of various solvents showed toluene was optimal, other solvents, such as DCE, DME, t-BuOH, and DMF, were also gave in acceptable yields (entries 5–8). The aerobic oxidation of Cu(I) to Cu(II) is a facile process allowing catalytic turnover and has been employed in numerous reactions.13 We wander our reaction could proceeded when catalytic Cu(OAc)2 was employed. Firstly, we found the reaction yield (73%) was almost maintained when 0.8 equiv. Cu(OAc)2 was used (entry 9). Inspired by this result, further studies were conducted and displayed the reactions gave in 61% and 43% yields, respectively, when 0.5 and 0.2 equiv. Cu(OAc)2 were used (entries 10 and 11). Further optimization showed the reaction yield recovered to 71% when performed under air atmosphere (entries 12 and 13). The efficiency of the reaction was also significantly affected by the carboxylic acid additives. Notably, 2-biphenylcarboxylic acid (o-PBA), a sterically bulky benzoic acid, proved to be the most effective additive, affording the desired product 3a in 88% yield (entry 17). Other organic acids were less efficient (entries 14–16).
|
|
||||
|---|---|---|---|---|
| Entry | Cu salt (equiv.) | Solvent | Additive | Yieldb (%) |
| a Reaction conditions: 1a (0.2 mmol), 2a (0.2 mmol), Cu(II) salt, solvent (2.0 mL), at 130 °C for 20 h under Ar protection. b Isolated yield. c Under O2 atmosphere. d Under air atmosphere. PivOH = 2,2,2-trimethylacetic acid, 1-AdCOOH = 1-adamantanecarboxylic acid, o-NBA = 2-nitrobenzoic acid, o-PBA = 2-biphenylcarboxylic acid. | ||||
| 1 | Cu(OAc)2 (1.0) | Toluene | — | 81% |
| 2 | Cu(OTf)2 (1.0) | Toluene | — | 0% |
| 3 | CuBr2 (1.0) | Toluene | — | 51% |
| 4 | CuF2 (1.0) | Toluene | — | <5% |
| 5 | Cu(OAc)2 (1.0) | DCE | — | 73% |
| 6 | Cu(OAc)2 (1.0) | DME | — | 61% |
| 7 | Cu(OAc)2 (1.0) | t-BuOH | — | 48% |
| 8 | Cu(OAc)2 (1.0) | DMF | — | 75% |
| 9 | Cu(OAc)2 (0.8) | Toluene | — | 73% |
| 10 | Cu(OAc)2 (0.5) | Toluene | — | 61% |
| 11 | Cu(OAc)2 (0.2) | Toluene | — | 43% |
| 12c | Cu(OAc)2 (0.2) | Toluene | — | 52% |
| 13d | Cu(OAc)2 (0.2) | Toluene | — | 71% |
| 14d | Cu(OAc)2 (0.2) | Toluene | PivOH | 76% |
| 15d | Cu(OAc)2 (0.2) | Toluene | 1-AdCOOH | 81% |
| 16d | Cu(OAc)2 (0.2) | Toluene | o-NBA | 79% |
| 17d | Cu(OAc)2 (0.2) | Toluene | o-PBA | 88% |
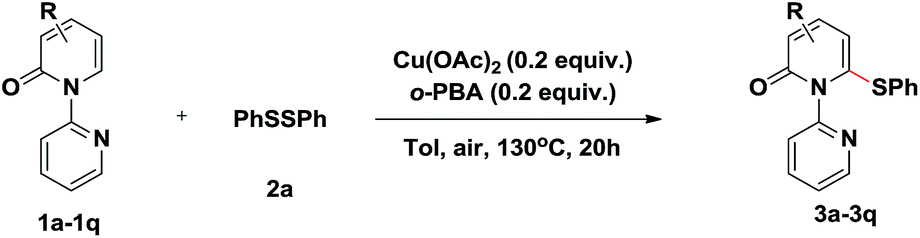
With the optimized conditions identified, a variety of 2-pyridones were investigated (Table 2). Satisfyingly, the C3 position of 2-pyridones bears either electron-donating or -withdrawing groups (3b–3f). It should be mentioned that strong electron-withdrawing 3-trifluoromethyl substituted 1b gave corresponding product up to 93% yield. Similarly, the C4 position of 2-pyridones accommodated both electron-donating and -withdrawing groups and proceeded smoothly to providing the thiolation products in excellent yields (3g–3j). Moreover, a fused 2-isoquinolinone 1k also provided product 3k in 86% yield. Remarkably, in spite of steric factors, substituents at the C5 position were feasible. 5-Fluoro substituted 2-pyridone 1l gave thiolation product 3l in 86% yield under the optimal conditions. Other electron-donating and -withdrawing substituents, such as chloro (1m), bromo (1n), methyl (1o) and trifluoromethyl (1p) worked efficiently to provide the desired products in high yields when using 0.5 equiv. Cu(OAc)2 and 0.5 equiv. o-PBA (3m–3p).

We then evaluated the scope of various disulfides (Table 3). Generally, the disulfides reacted with both electronic and steric variations under the optimal conditions. Disulfide containing ortho-substitution was well tolerated though the presence of steric hindrance (4a). Electronically diverse disulfides were efficient coupling partners, although a preference for electron-rich disulfides was observed (4b–4f). Importantly, halogen substituents in the diaryl disulfides were tolerated and delivered the desired products in good yields (4g–4i), which allowed for the further modification. Besides the aromatic disulfides, the heteroaromatic disulfides, such as 2-methyl-3-furyl disulfide and 2-pyridinyl disulfide showed acceptable reactivity to give the corresponding sulfides when using 0.5 equiv. Cu(OAc)2 and 0.5 equiv. o-PBA (4j–4k). Notably, less reactive dialkyl disulfides, even dibenzyl disulfide, also reacted in moderate yields (4l–4o).
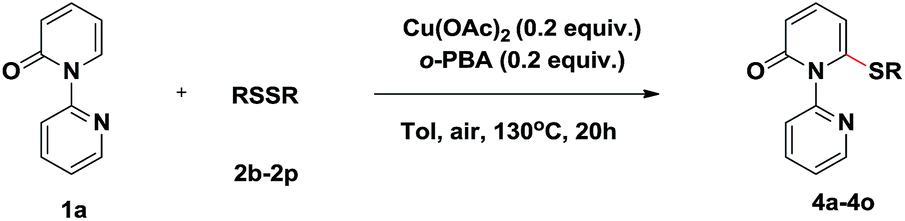
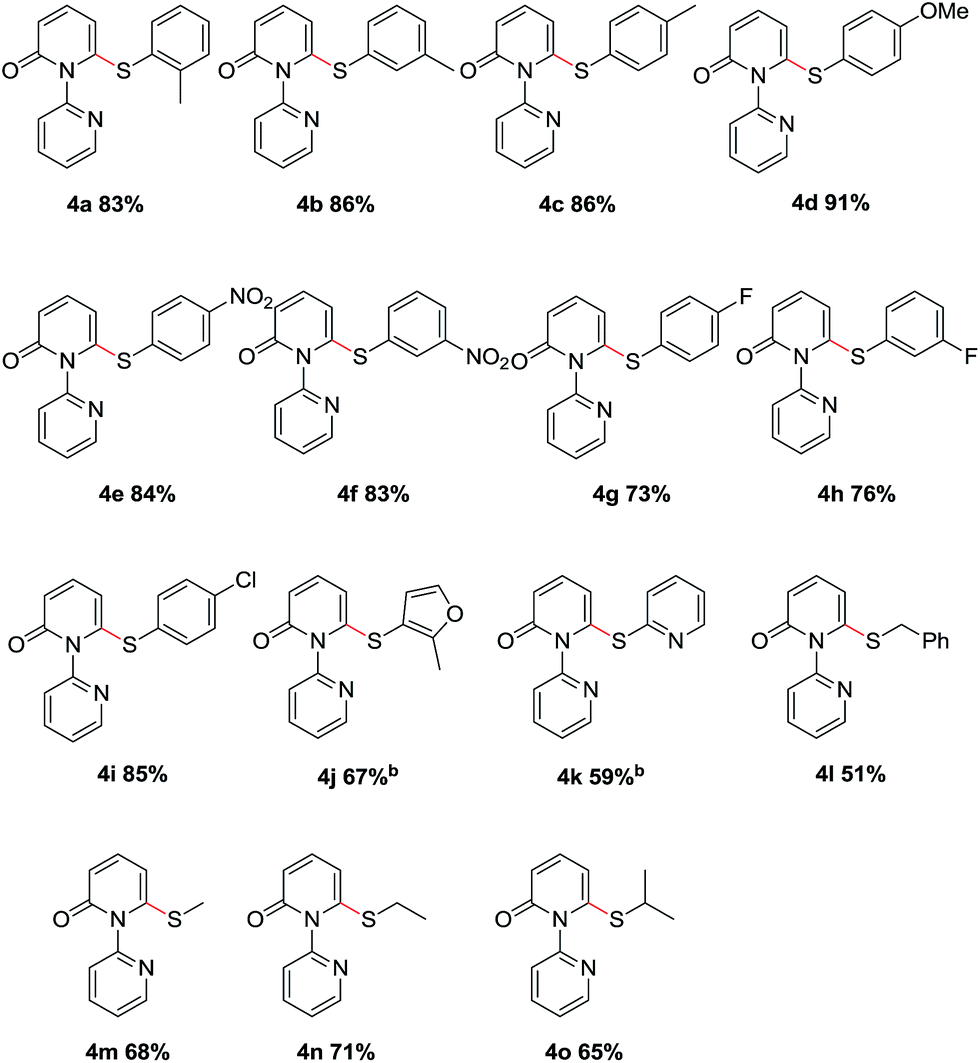
To demonstrate the applicability of this thiolation reaction in the synthesis of sulfone derivatives which show a wide range of biological activities, further transformation was explored (Scheme 2). Treatment of product 3a with 3-chloroperoxybenzoic acid (m-CPBA) at room temperature gave the corresponding sulfone 5a in 75% yield. Furthermore the directing group could be removed efficiently by the “quaternization–hydride reduction” strategy.4i,14 The corresponding 6-phenylthio-2-pyridone 6a was easily obtained from product 3a through a mild, two-step sequence in 66% yield (Scheme 2).
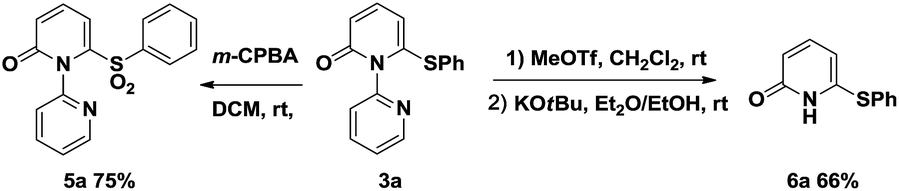 | ||
| Scheme 2 Synthetic transformations of 3a. | ||
To shed light on the possible pathway of this thiolation reaction, a series of controlled experiments were performed. First, radical scavenger experiments were conducted (Scheme 3a). Two separate reactions were carried out by the addition of a radical inhibitor (2,2,6,6-tetramethylpiperidin-1-yl)oxidanyl (TEMPO) or an electron-transfer scavenger 2,6-di-tert-butyl-4-methylphenol (BHT). These reactions produced the thiolated product 3a without significantly affecting the efficiency, indicating that the single electron transfer (SET) process is not involved in this transformation.
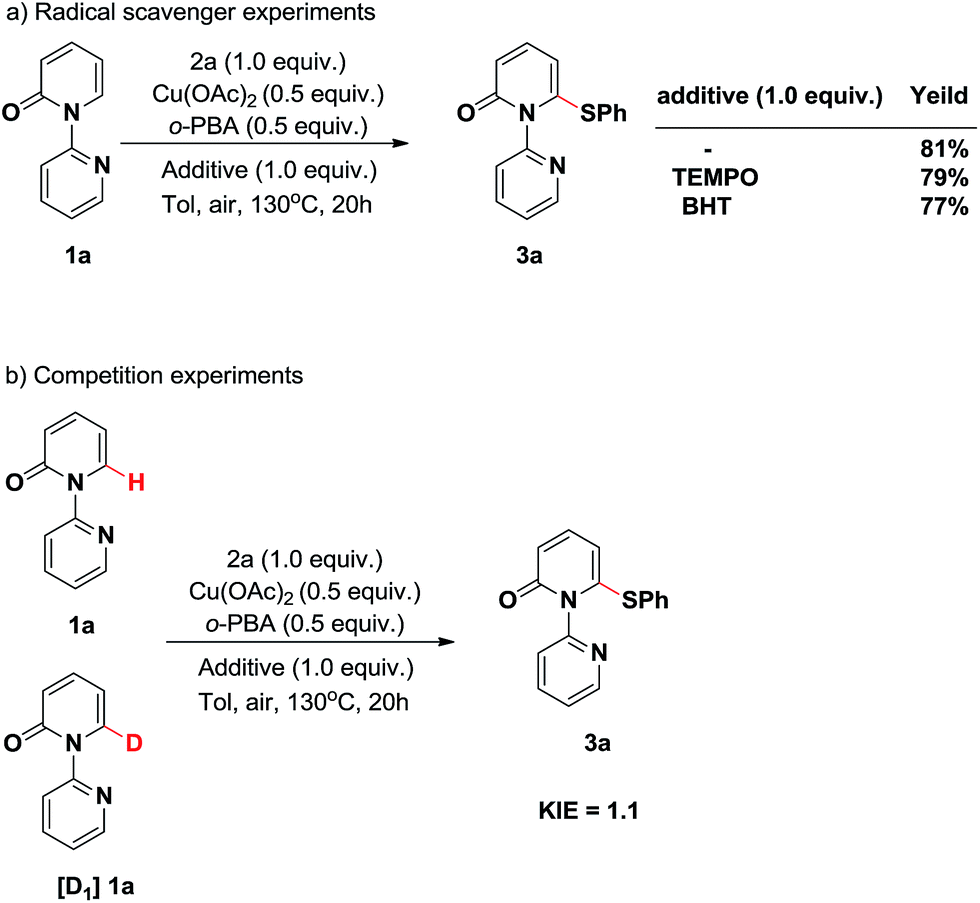 | ||
| Scheme 3 Proposed reaction mechanism. | ||
Subsequently, to gain some mechanistic insight into the C–H cleavage step, we carried out the deuterium-labeling experiments (Scheme 3b). No kinetic isotope effect (KIE) was observed when 1a and [D1]1a were employed under the reaction conditions in the initial stage of competition experiments (KIE = 1.1, Table S1 in the ESI†), which suggested that C–H bond cleavage is not the rate-determining step.4j
Although the mechanism of the reaction remains unclear at this moment, on the basis of the above results and previously reported studies, two relevant observations may be discussed. First, Cu(II)-catalyzed and mediated arene C–H bond activation and transformations through arylcopper(II) and arylcopper(III) complexes has been well-defined recently.16 Thus, aryl-Cu(III) species can undergo a nucleophilic-addition-type reaction with disulfides to afford the desired product.7b,8d,9 However, we could not rule out another pathway, it is known disulfides can oxidize Cu(I) to Cu(III) and stabilize high-valent Cu(III), followed by reductive elimination to give the desired product.7b,15 Moreover, deuterium-labeling experiments implied that oxidative addition of disulfides is slower than cleavage of the C–H bond and seems to be the rate-determining step.8a,8d,16d Further efforts are essential to clarify the detailed mechanism.
Conclusions
We have developed Cu(II)-catalyzed C6-selective C–H thiolation of 2-pyridones with aryl and alkyl disulfides. This transformation uses Cu(OAc)2 as a catalyst, o-PBA as an additive, and O2 in air as an oxidant. Catalytic turnover of the Cu salt and removable directing group is a significant practical advantage. This reaction showed a remarkably broad substrate scope and good functional group tolerance, providing an efficient and economic protocol for the synthesis of 6-thiolation-2-pyridones.Notes and references
- (a) I. M. Lagoja, Chem. Biodiversity, 2005, 2, 1–50 CrossRef CAS; (b) M. E. Wall, M. C. Wani, C. E. Cook and K. H. Palmer, J. Am. Chem. Soc., 1966, 88, 3888–3890 CrossRef CAS; (c) F. Surup, O. Wagner, J. von Frieling, M. Schleicher, S. Oess, P. Müller and S. Grond, J. Org. Chem., 2007, 72, 5085–5090 CrossRef CAS; (d) G. Manfroni, F. Meschini, M. L. Barreca, P. Leyssen, A. Samuele, N. Iraci, S. Sabatini, S. Massari, G. Maga, J. Neyts and V. Cecchetti, Bioorg. Med. Chem., 2012, 20, 866–876 CrossRef CAS; (e) M. Metifiot, B. Johnson, S. Smith, X. Z. Zhao, C. Marchand, T. Burke, S. Hughes and Y. Pommier, Antimicrob. Agents Chemother., 2011, 55, 5127–5233 CrossRef CAS; (f) J. S. Scott, F. W. Goldberg and A. V. Turnbull, J. Med. Chem., 2014, 57, 4466–4486 CrossRef CAS.
- (a) L. A. Hasvold, W. Wang, S. L. Gwaltney, T. W. Rockway, L. T. G. Nelson, R. A. Mantei, S. A. Fakhoury, G. M. Sullivan, Q. Li, N.-H. Lin, L. Wang, H. Zhang, J. Cohen, W.-Z. Gu, K. Marsh, J. Bauch, S. Rosenberg and H. L. Sham, Bioorg. Med. Chem. Lett., 2003, 13, 4001–4005 CrossRef CAS; (b) C. Bengtsson and F. Almqvist, J. Org. Chem., 2010, 75, 972 CrossRef CAS; (c) M. D. Hill and M. Movassaghi, Chem.–Eur. J., 2008, 14, 6836–6844 CrossRef CAS.
- For selected reviews, see: (a) D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174–238 CrossRef CAS; (b) L. C. Lewis, R. G. Bergman and J. A. Ellman, Acc. Chem. Res., 2008, 41, 1013–1025 CrossRef; (c) O. Daugulis, H.-Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074–1086 CrossRef CAS; (d) X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094–5115 CrossRef CAS; (e) L. Ackermann, R. Vicente and A. R. Kapdi, Angew. Chem., Int. Ed., 2009, 48, 9792–9826 CrossRef CAS; (f) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147–1169 CrossRef CAS; (g) C.-L. Sun, B.-J. Li and Z.-J. Shi, Chem. Commun., 2010, 46, 677–685 RSC; (h) C. Liu, H. Zhang, W. Sui and A. Lei, Chem. Rev., 2011, 111, 1780–1824 CrossRef CAS; (i) G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11726–11743 CrossRef CAS.
- (a) A. Nakatani, K. Hirano, T. Satoh and M. Miura, Chem.–Eur. J., 2013, 19, 7691–7695 CrossRef CAS; (b) A. Nakatani, K. Hirano, T. Satoh and M. Miura, J. Org. Chem., 2014, 79, 1377–1385 CrossRef CAS; (c) E. A. Anagnostaki, A. D. Fotiadou, V. Demertzidou and A. L. Zografos, Chem. Commun., 2014, 50, 6879–6882 RSC; (d) A. Modak, S. Rana and D. Maiti, J. Org. Chem., 2015, 80, 296–303 CrossRef CAS; (e) T. Itahara and F. Ouseto, Synthesis, 1984, 488–489 CrossRef CAS; (f) Y. Chen, F. Wang, A. Jia and X. Li, Chem. Sci., 2012, 3, 3231–3236 RSC; (g) Y. Nakao, H. Idei, K. S. Kanyiva and T. Hiyama, J. Am. Chem. Soc., 2009, 131, 15996–15997 CrossRef CAS; (h) R. Tamura, Y. Yamada, Y. Nakao and T. Hiyama, Angew. Chem., Int. Ed., 2012, 51, 5679–5682 CrossRef CAS; (i) R. Odani, K. Hirano, T. Satoh and M. Miura, Angew. Chem., Int. Ed., 2014, 53, 10784–10788 CrossRef CAS; (j) P. A. Donets and N. Cramer, Angew. Chem., Int. Ed., 2015, 54, 633–637 CAS.
- A. Correa, M. Carril and C. Bolm, Angew. Chem., Int. Ed., 2008, 47, 2880–2883 CrossRef CAS.
- X. Chen, X. S. Hao, C. E. Goodhue and J.-Q. Yu, J. Am. Chem. Soc., 2006, 128, 6790–6791 CrossRef CAS PubMed.
- (a) L. Chu, X. Yue and F.-L. Qing, Org. Lett., 2010, 12, 1644–1647 CrossRef CAS; (b) L. D. Tran, I. Popov and O. Daugulis, J. Am. Chem. Soc., 2012, 134, 18237–18240 CrossRef CAS.
- (a) M. Iwasaki, M. Lyanaga, Y. Tsuchiya, Y. Nishimura, W. Li, Z. Li and Y. Nishihara, Chem.–Eur. J., 2014, 20, 2459–2462 CrossRef CAS; (b) M. Iwasaki, W. Kaneshika, Y. Tsuchiya, K. Nakajima and Y. Nishihara, J. Org. Chem., 2014, 79, 11330–11338 CrossRef CAS PubMed; (c) P. Sharma, S. Rohilla and N. Jain, J. Org. Chem., 2015, 80, 4116–4122 CrossRef CAS; (d) S. Vásquez-Céspedes, A. Ferry, L. Candish and F. Glorius, Angew. Chem., Int. Ed., 2015, 54, 5772–5776 CrossRef.
- Y. Yang, W. Hou, L. Qin, J. Du, H. Feng, B. Zhou and Y. Li, Chem.–Eur. J., 2014, 20, 416–420 CrossRef CAS.
- (a) K. Yang, Y. Wang, X. Chen, A. A. Kadi, H.-K. Fun, H. Sun, Y. Zhang and H. Lu, Chem. Commun., 2015, 51, 3582–3585 RSC; (b) S.-Y. Yan, Y.-J. Liu, B. Liu, Y.-H. Liu and B.-F. Shi, Chem. Commun., 2015, 51, 4069–4072 RSC; (c) C. Lin, D. Li, B. Wang, J. Yao and Y. Zhang, Org. Lett., 2015, 17, 1328–1331 CrossRef CAS; (d) S.-Y. Yan, Y.-J. Liu, B. Liu, Y.-H. Liu, Z.-Z. Zhang and B.-F. Shi, Chem. Commun., 2015, 51, 7341–7344 RSC; (e) X. Ye, J. L. Petersen and X. Shi, Chem. Commun., 2015, 51, 7863–7886 RSC; (f) C. Lin, W. Yu, J. Yao, B. Wang, Z. Liu and Y. Zhang, Org. Lett., 2015, 17, 1340–1343 CrossRef CAS; (g) X. Wang, R. Qiu, C. Yan, V. P. Reddy, L. Zhu, X. Xu and S.-F. Yin, Org. Lett., 2015, 17, 1970–1973 CrossRef CAS.
- L. Zhu, X. Cao, R. Qiu, T. Iwasaki, V. P. Reddy, X. Xu, S.-F. Yin and N. Kambe, RSC Adv., 2015, 5, 39358–39365 RSC.
- (a) A. E. Wendlandt, A. M. Suess and S. S. Stahl, Angew. Chem., Int. Ed., 2011, 50, 11062–11087 CrossRef CAS; (b) C. Zhang, C. H. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3464–3484 RSC; (c) S. E. Allen, R. R. Walvoord, R. P. Salinas and M. C. Kozlowski, Chem. Rev., 2013, 113, 6234–6458 CrossRef CAS.
- Copies of crystallographic data (CCDC 1439142†).
- V. Smout, A. Peschiulli, S. Verbeeck, E. A. Mitchell, W. Herrebout, P. Bultinck, C. M. L. Vande Velde, D. Berthelot, L. Meerpoel and B. U. W. Maes, J. Org. Chem., 2013, 78, 9803–9814 CrossRef CAS.
- (a) P. T. Beurskens, J. A. Cras and J. Steggerda, J. Inorg. Chem., 1968, 7, 810–813 CrossRef CAS; (b) M. A. Willert-Porada, D. J. Burton and N. C. Baenziger, J. Chem. Soc., Chem. Commun., 1989, 1633–1634 RSC.
- (a) B. Yao, D. X. Wang, Z. T. Huang and M. X. Wang, Chem. Commun., 2009, 2899–2901 RSC; (b) X. Ribas, C. Calle, A. Poater, A. Casitas, L. Gomez, R. Xifra, T. Parella, J. Benet-Buchholz, A. Schweiger, G. Mitrikas, M. Sola, A. Llobet and T. D. Stack, J. Am. Chem. Soc., 2010, 132, 12299–12306 CrossRef CAS; (c) A. Casitas and X. Ribas, Chem. Sci., 2013, 4, 2301–2318 RSC; (d) H. Zhang, B. Yao, L. Zhao, D. X. Wang, B. Q. Xu and M. X. Wang, J. Am. Chem. Soc., 2014, 136, 6326–6332 CrossRef CAS; (e) B. Yao, Y. Liu, L. Zhao, D. X. Wang and M. X. Wang, J. Org. Chem., 2014, 79, 11139–11145 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and additional spectra. CCDC 1439142. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra04388g |
| This journal is © The Royal Society of Chemistry 2016 |