
Non-stabilized enolates – versatile nucleophiles in transition metal-catalysed allylic alkylations†
Uli
Kazmaier
Institute of Organic Chemistry, Saarland University, P.O. Box 151150, 66041 Saarbrücken, Germany. E-mail: u.kazmaier@mx.uni-saarland.de
First published on 23rd August 2016
Abstract
While in the early days of palladium-catalyzed allylic alkylations mainly stabilized carbanions, such as malonates, have been used as standard nucleophiles, during the last 10–15 years also non stabilized enolates became more and more popular. Efficient protocols have been developed allowing their highly regio- as well as stereoselective allylations. In addition, catalysts based on other transition metals enlarge the synthetic potential of these valuable transformations, since each metal (complex) shows its own, typical reaction behaviour. This review will cover the new developments in this fast developing field.
 Uli Kazmaier | Uli Kazmaier studied chemistry at the University of Stuttgart where he obtained his PhD working with U. Schmidt. Afterwards he joined the research groups of M. T. Reetz (Marburg) and B. M. Trost (Stanford) as a postdoc. In 1992, he moved to Heidelberg starting his own scientific work at the Institute of Organic Chemistry. In 2000 he received a Novartis Chemistry Lectureship and in 2001 an offer of a full professorship at Saarland University. His current research interest is focused on new organometallic reagents and reactions especially for amino acid and peptide synthesis, and their application to natural product synthesis. |
Introduction
Organometallic chemistry is definitely one of the most powerful tools in modern organic synthesis. Besides stoichiometric reactions, especially transition metal-catalysed reactions play a dominant role, and a wide range of transition metal-catalysed cross-coupling reactions has been developed during the last decades, while the allylic alkylations became a major player in this field.1 In 1965, J. Tsuji discovered that π-allyl palladium complexes could be reacted with C-nucleophiles in a stoichiometric fashion. Later on, catalytic and enantioselective versions were developed mainly by B. M. Trost et al. While in the early years, the π-allyl chemistry was clearly dominated by the palladium complexes, in the meanwhile a wide range of other transition metals, such as Ir, Rh, Ru and others made their way into the limelight.2 Each of these transition metals has its own characteristics and reaction behaviour, enlarging the potential of allylic alkylations significantly. Although, in the beginning mainly stabilized carbanions such as malonates or β-ketoesters [pKa < 25 (DMSO)] have been used as nucleophiles besides hetero-nucleophiles like amines, in the meanwhile protocols have been developed allowing also the application of non-stabilized enolates, such as those of aldehydes and ketones or of carboxylic acid derivatives. This review will focus on allylic alkylations of such non-stabilized enolates.Allylic alkylations of ketone enolates
Allylation of metal enolates
In 1980 Trost and Keinan reported for the first time on allylic alkylations of ketone enolates.3 Simple enolates, for example obtained from acetophenone gave rise to disubstituted products preferentially, while the desired monoallylated derivative was obtained only as the side product. The problem arises from an equilibrium between the enolates of the product and the starting material. Shortly thereafter, Fiaud and Malleron reported the use of sterically demanding allylic substrates, such as cyclohexenyl acetate, providing the monoalkylated products in acceptable yields.4 A significant improvement was the switch to tributyltin enolates (1) which showed a clean monoalkylation with high regioselectivity (rs) (Scheme 1). Alkylation occurred at the steric less hindered position of the π-allyl-complex, and the E-configured product was obtained preferentially independent of the olefin geometry of the starting allylic acetate.5 If the tin enolate was generated in situ from the corresponding ketone, the regioselectivity of the enolate allylation depended on the reactants and the reaction conditions. Reactions with reactive allylic substrates led only to the 2,6-disubstituted product (kinetic enolate), while sluggish substrates gave rise to 2,2-disubstituted derivatives, the products resulting from the more stable tin enolate. Obviously, here an equilibrium of the tin enolates occurred faster than the allylation.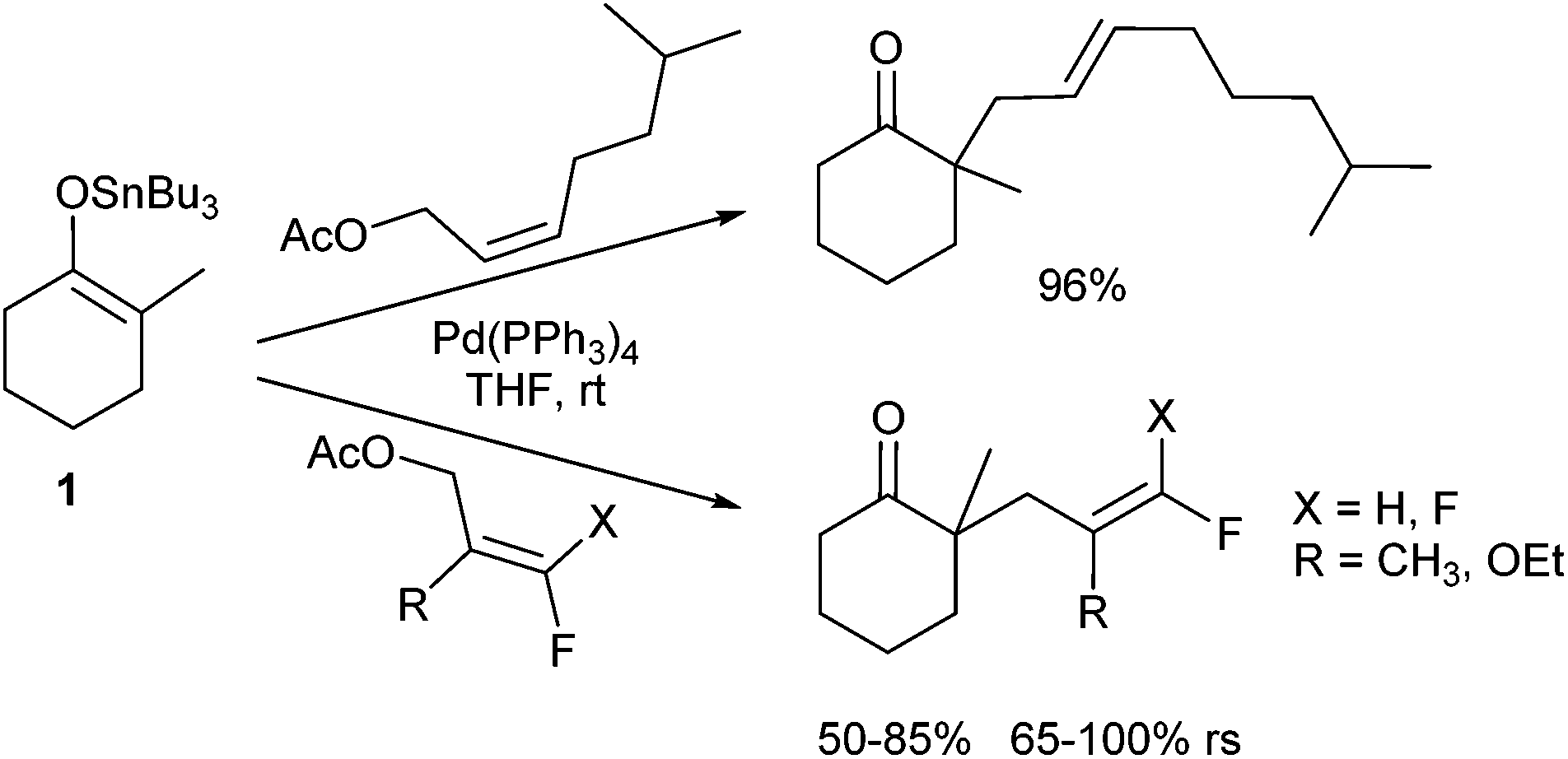 | ||
| Scheme 1 Allylic alkylation of tin enolates 1. | ||
The influence of substituents in the allylic substrate on the regioselectivity (rs) of the reaction was investigated by Shi et al.6 They used several fluoro- and difluoro-substituted allyl acetates and found a preferential attack of tin enolate 1 at the terminus remote from the fluorine (Scheme 1). The fluorinated acetates showed also a diminished reactivity in comparison to their non-fluorinated counterparts.
Negishi and John investigated the effect of the counter cation on the outcome of the reaction.7 They observed, that allylation of potassium enoxyborates proceeded well and various allylic halides and acetates could be used. Very similar results were obtained with the corresponding zinc enolates, while a wide range of other counter ions (ClMg+, Me2Al+, Cp2ClTi+, Me3Si+) gave unsatisfying results or did not show any reaction at all.
If chiral allylic substrates are used, the reactions proceed with overall net retention (Scheme 2). This can be explained by a double inversion, a mechanism which is typical for stabilized nucleophiles, and which is in sharp contrast to findings with aryl and vinyl metal compounds, which react under inversion (via transmetallation).8 If functionalized allylic substrates are used, the nucleophilic attack occurs regioselectively at the position distal to the electron withdrawing group.9 This approach is suitable to control the stereogenic centre in the allyl moiety, but not at the α-position of the ketone, which is formed as an almost 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 diastereomeric mixture.
:1 diastereomeric mixture.
 | ||
| Scheme 2 Stereoselective allylic alkylation of ketone enolates. | ||
The configuration at the α-position can be controlled by using chiral ketones, while the selectivity depends on a successful shielding of one face of the enolate. Riant et al. reported the regio- and diastereoselective allylation of ketone enolates generated in situ from chiral silyl enol ethers, which proceeded in good yields but only moderate stereoselectivity (Scheme 3). The selectivity could be increased by using additional chiral phosphine ligands.10
 | ||
| Scheme 3 Diastereoselective allylic alkylation of ketone enolates. | ||
Much better results were observed if only one face of the enolate could react, for example in intramolecular reactions. Williams et al. reported synthetic studies toward quinine using a regio- and stereoselective intramolecular ketone allylation as a key step (Scheme 4).11
 | ||
| Scheme 4 Intramolecular diastereoselective allylic alkylation of silyl enol ether 2. | ||
In this case, a tin enolate was formed in situ from silyl enol ether 2 and two stereogenic centres are created with excellent stereoselectivity. The α-allylated ketone was directly reduced to the corresponding alcohol to avoid epimerization and elimination.
Cook et al. used a similar strategy for the synthesis of closely related alkaloid structures using a palladium-catalysed domino cyclization to construct the core structure of Neosarpargine (Scheme 5).12 In this case, an N-allylation is followed by an intramolecular enolate allylation. Best results were obtained with the corresponding Zn-enolate.
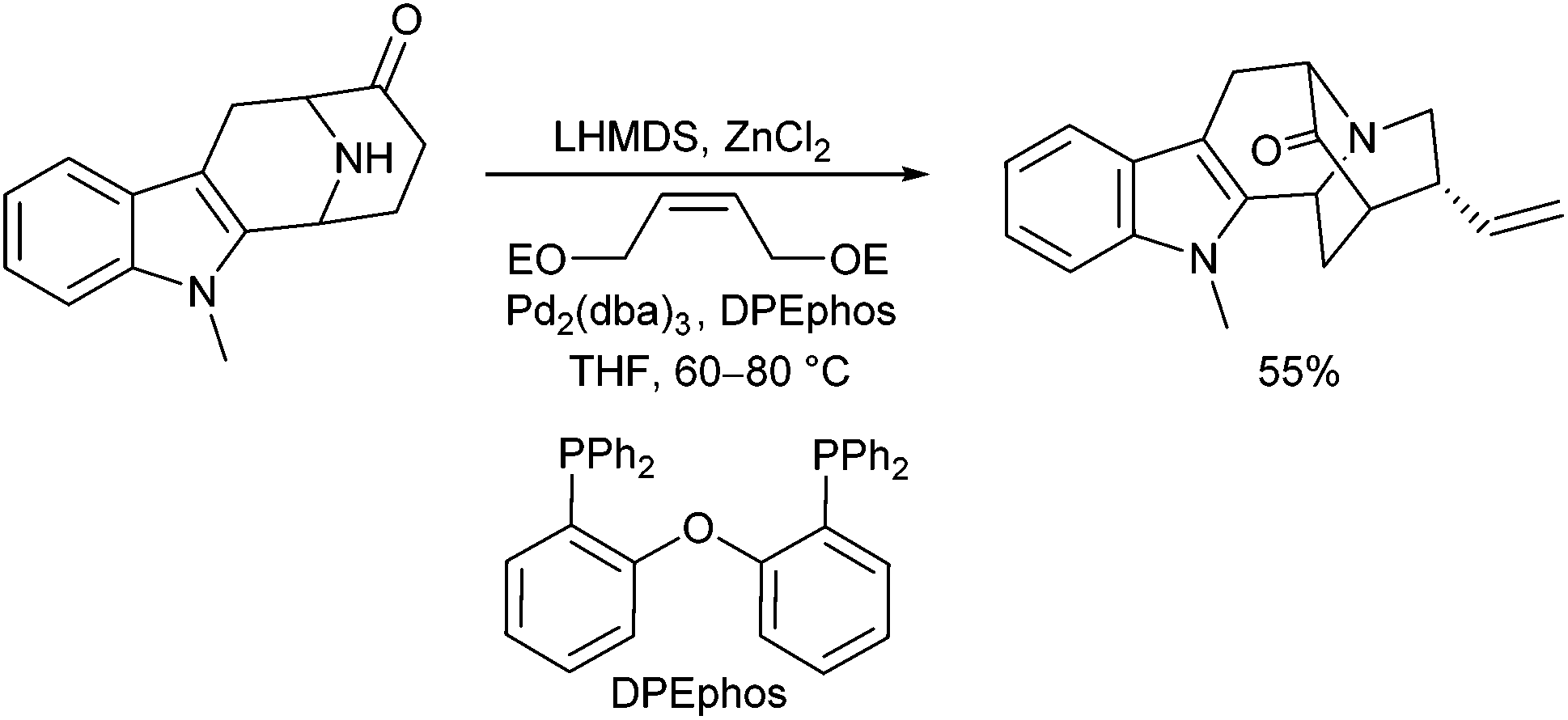 | ||
| Scheme 5 Domino N-allylation/intramolecular enolate allylation. | ||
Very recently, Kazmaier et al. reported a completely different approach for acyclic stereocontrol using chelated chiral α-aminoketone enolates.13 If aminoketones such as 3 were treated with an excess of base in the presence of ZnCl2 probably the sterically least hindered enolate A was formed, where the side chain shields one face of the enolate (Scheme 6). Deprotonation of the amide N–H prevents an epimerization of the stereogenic centre. Attack of the Pd–allyl complex occurs from the sterically least hindered face of the enolate giving the allylation product with excellent yield and selectivity. If chiral allylic substrates were used, also a stereogenic centre at the β-position could be introduced under substrate control.
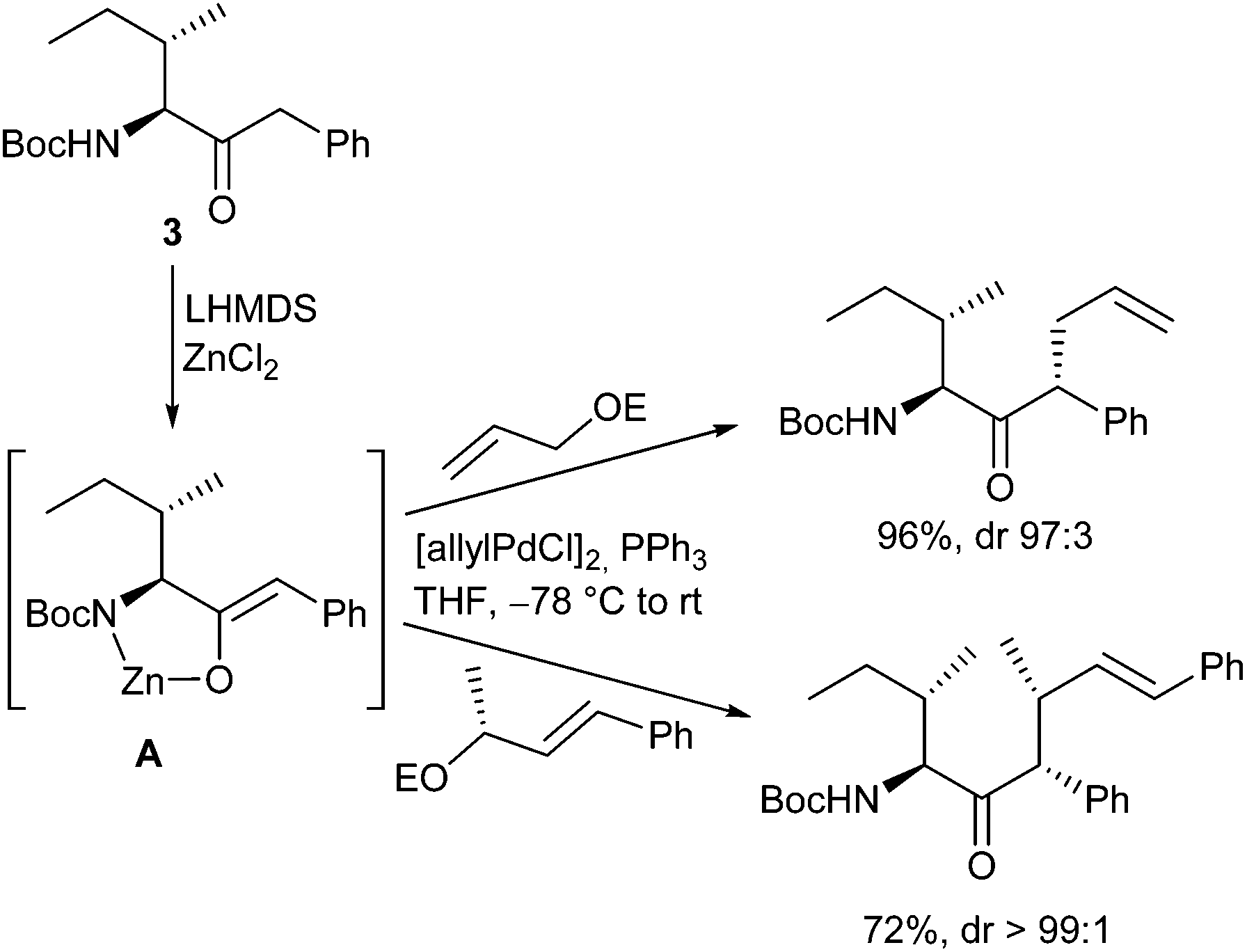 | ||
| Scheme 6 Stereoselective α-aminoketone allylations. | ||
During the last three decades, asymmetric catalysed allylic alkylations became more and more important and therefore, it is not surprising that this elegant technique found also some applications in the allylation of ketones. The control of the stereochemical outcome by using chiral ligands is not a trivial issue since bond-making and bond-breaking events occur outside the coordination sphere of the metal. Thus, they must transmit their stereochemical information through space. Discrimination of enantiotopic faces in the nucleophile is especially difficult as the nucleophile is segregated from the chiral environment by the π-allyl moiety.
In 1999 Trost and Schroeder reported the first asymmetric allylic alkylation of tin enolates obtained from 2-substituted cyclohexanone derivatives (Scheme 7).14 They developed chiral bidentate ligands (L1), derived e.g. from trans-diaminocyclohexane (L1a), which gave good results not only with stabilized enolates15 but also tin enolates of ketones. The tin enolates were found to be superior to boron enolates and best results were obtained by using an excess of LDA. Other cyclic ketones gave comparable results.16
 | ||
| Scheme 7 Enantioselective ketone allylation. | ||
This nice protocol was also applied in the total synthesis of natural products, such as Hamigeran B (Scheme 8).17 In this case, the enantiomeric ligand ent-L1a was used to introduce the stereogenic centre in the 5-membered ring. It should be mentioned that high ee's were only obtained in the presence of an excess t-butanol.
 | ||
| Scheme 8 Enantioselective ketone allylation. | ||
In 2001 Hou and Dai et al. reported related investigations, also using methyl tetralone, with a very similar chiral ligand.18 They used phosphinocarboxylic acids derived from ferrocene (L2) instead of phosphinobenzoic acid and obtained similar results as reported by Trost. They described also the enantioselective allylation of acyclic ketones later on (Scheme 9).19 In this case, the yield and selectivity was depended on the counter ion of the base, while LHMDS was found to be the base of choice. Good results were obtained with α-alkoxy ketones, especially in the presence of Lewis acids such as AgBr. The excellent results were explained by the formation of a chelated (Z)-enolate formed as an intermediate.
 | ||
| Scheme 9 Enantioselective α-alkoxy ketone allylation. | ||
In 2007, Paquin et al. described the enantioselective allylation of fluorinated silyl enol ethers using triphenyldifluorosilicate (TBAT) to activate the silyl ether in situ (Scheme 10).20 Although the enolate formed was structurally closely related to the previous system, only traces of product could be obtained in the presence of the Trost ligand (L1a). In this case, best results were obtained with phosphineoxazoline L3a in toluene.
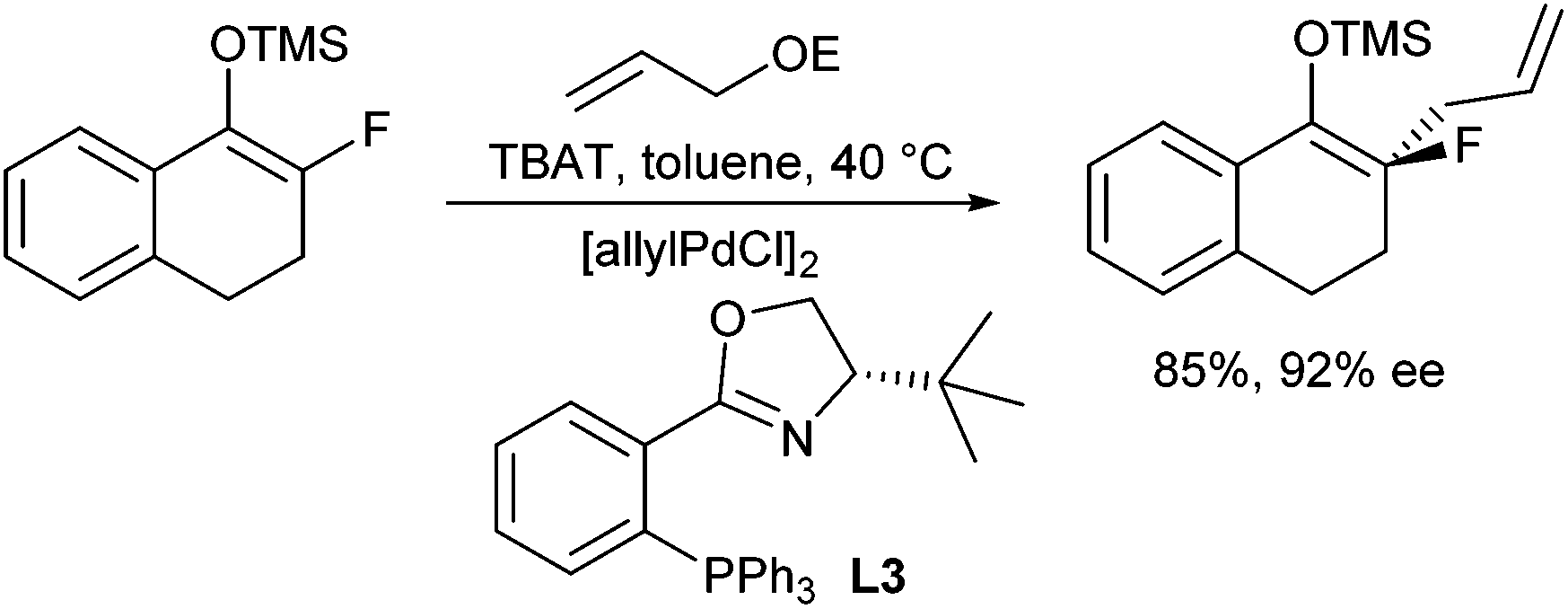 | ||
| Scheme 10 Enantioselective allylation of silyl enolate. | ||
Interesting investigations were carried out by Braun et al. not only on the enantiocontrol but also on the diastereoselective outcome of allylation reactions in the presence of achiral and chiral ligands.21 Enolates of cyclohexanone and propiophenone derivatives were used as nucleophiles and 1,3-diphenyl allyl acetate as allylation agent (Scheme 11). Best diastereoselectivities were obtained with lithium and magnesium enolates in the presence of ferrocenyl-derived ligand L4, both with cyclic and aromatic ketones. While cyclohexanone gave rise to the syn-product preferentially, the anti-product was obtained from aromatic ketone. Surprisingly, the enolate geometry in the latter case had no significant influence on the stereoselective outcome of the reaction. The (E)- as well as the corresponding (Z)-enolate gave the same major diastereomer, albeit the (E)-isomer gave the better selectivity. It was suggested that different transition states are involved in the allylation step.22 In case of cyclohexanone, also excellent enantioselectivities were obtained with (R)-Binap (L5) as a ligand, especially if the magnesium enolate was used.
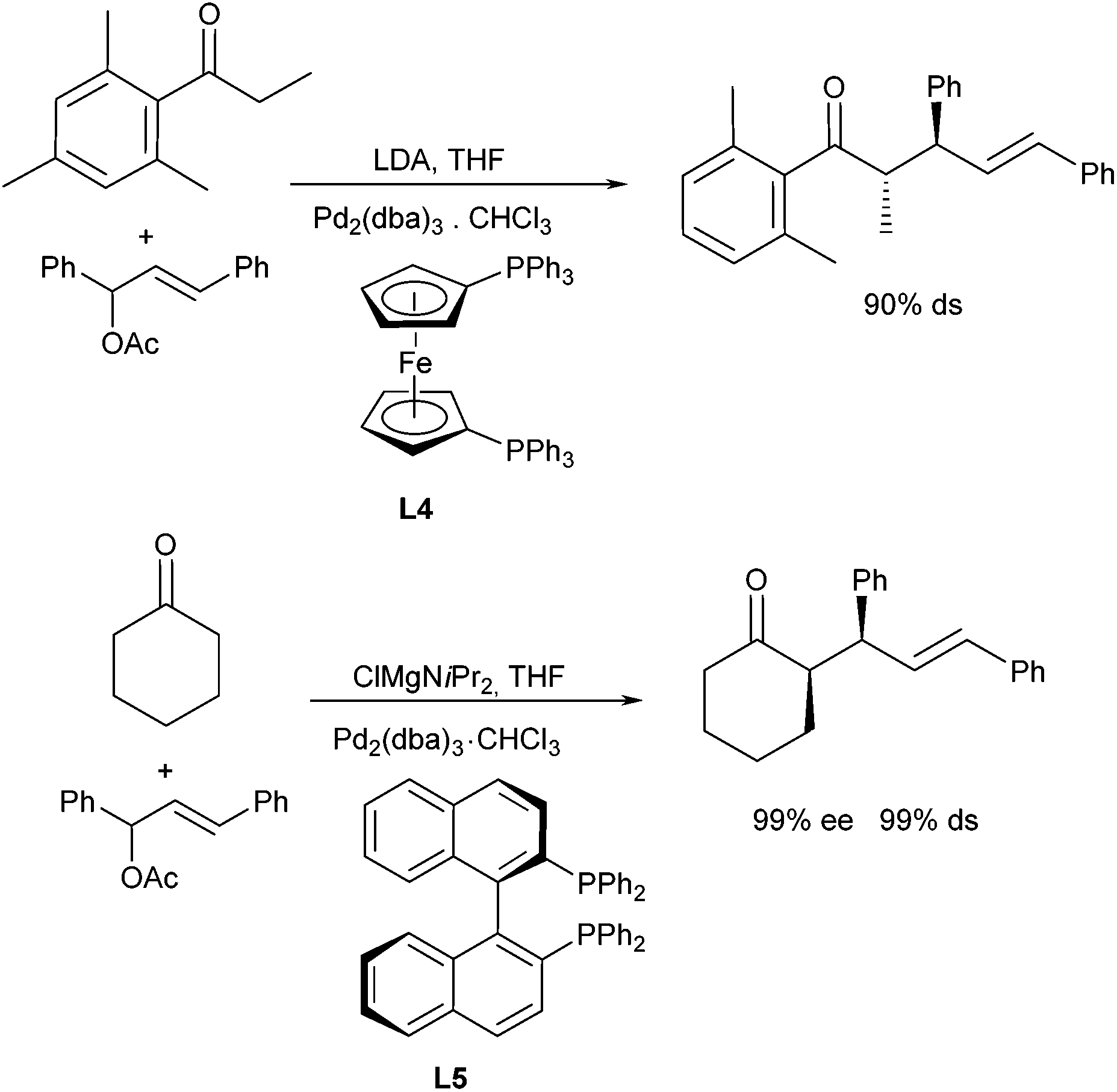 | ||
| Scheme 11 Enantio- and diastereoselective ketone allylation. | ||
Similar anti-selectivities were observed by Hou et al. in allylations of propiophenone and derivatives using a ferrocene-based oxazoline ligand L6a (Scheme 12). Surprisingly, cinnamyl acetates gave the branched product selectively with this ligand, while the regio- as well as the stereoselectivity depended on the solvent and some additives used.
 | ||
| Scheme 12 Enantio- and diastereoselective ketone allylation. | ||
Dimethoxyethane was found to be superior to THF and in the presence of LiCl the branched product was formed almost exclusively in a highly selective fashion.23 Similar results were also obtained with acylsilanes.24
The same type of ligand (L6b) was also used in the kinetic resolution of quinolone derivatives via allylic alkylation (Scheme 13).25 Allyl phosphates were found to be superior to the corresponding acetates and carbonates, and excellent selectivities were obtained at −50 °C, where the trans isomer was formed exclusively.
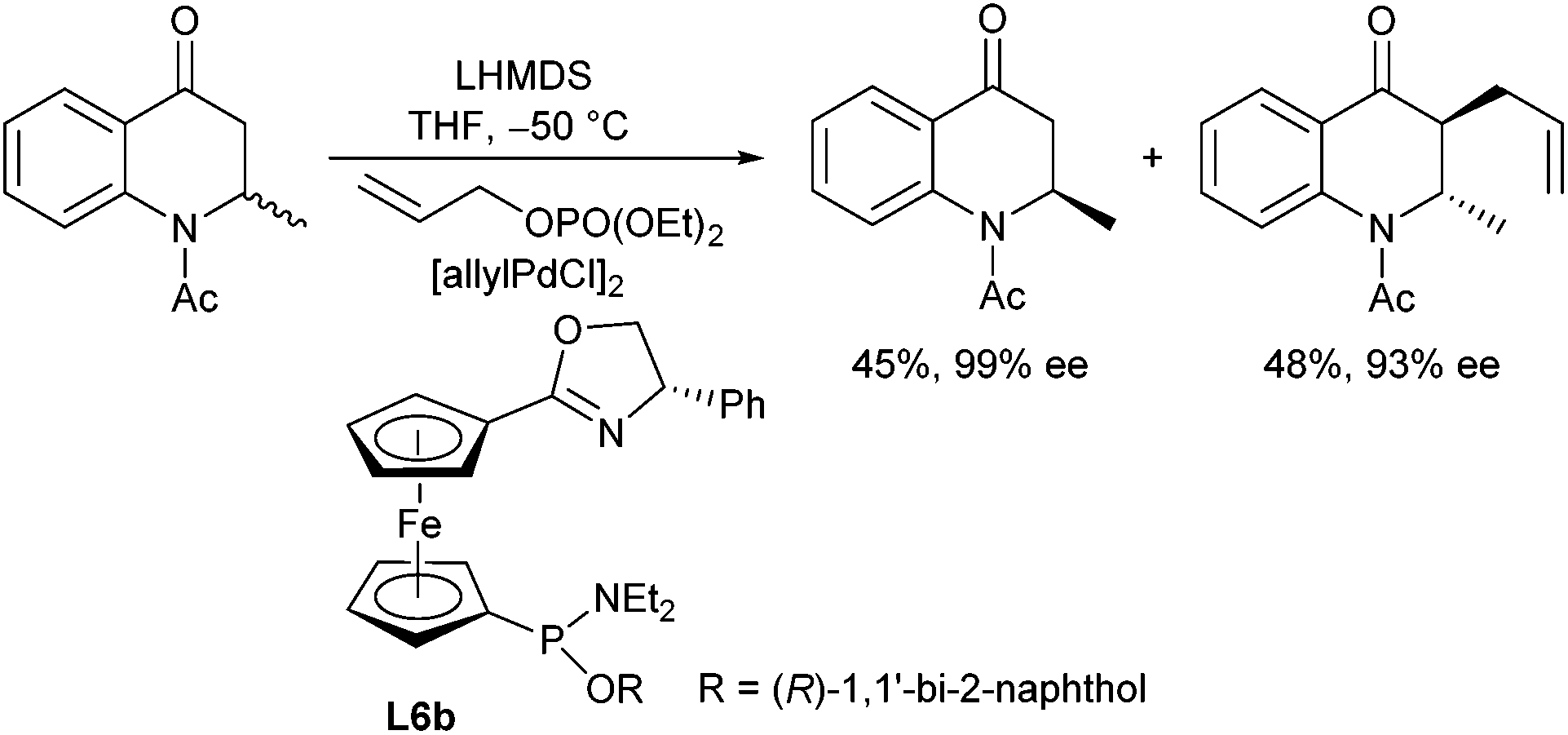 | ||
| Scheme 13 Kinetic resolution of quinolone derivative. | ||
The protocol could also be transformed to intramolecular allylations giving rise to trans-substituted cyclopentanone derivatives (Scheme 14).26 While LHMDS, which is often used as base gave not the expected cyclization product, but the O-allylation product, good yields and selectivities were obtained with K3PO4 as a base, especially in the presence of TBAF in alcoholic solvents.
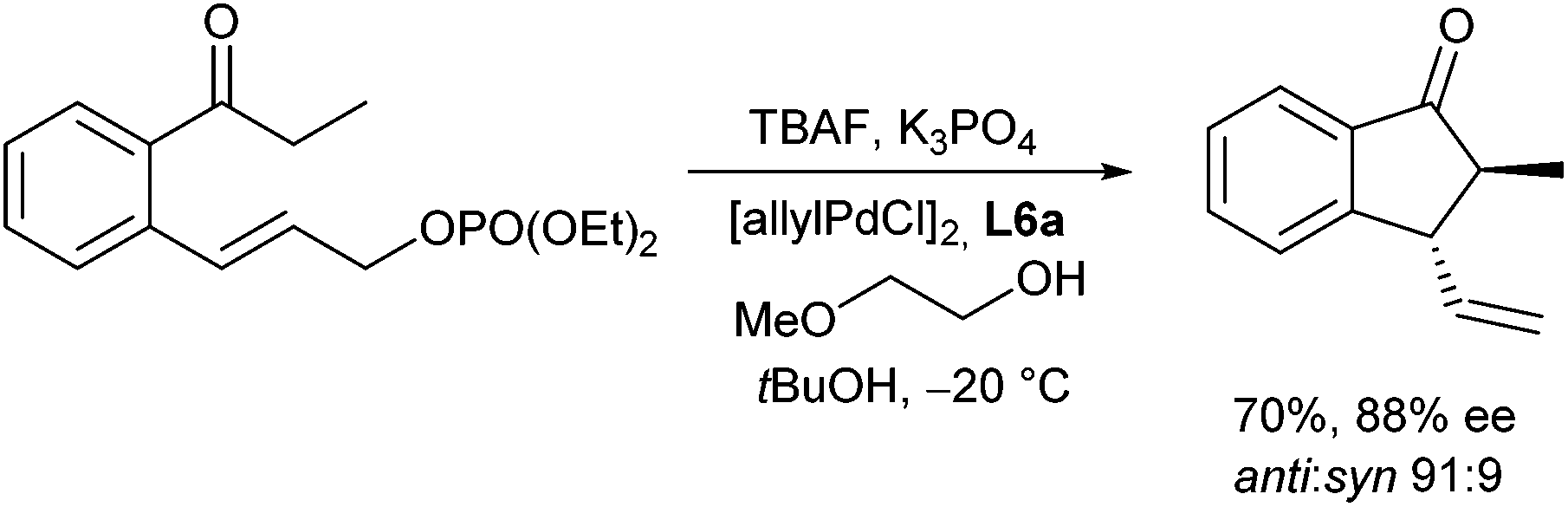 | ||
| Scheme 14 Asymmetric intramolecular allylation. | ||
The allylic alkylation of ketone enolates is not limited to Pd-catalysed reactions but can also be carried out with other transition metal catalysts. Graening and Hartwig showed that an unusual combination of two different metal fluorides activate trimethylsilyl enol ethers toward iridium-catalysed asymmetric allylic substitution.27 Reactions of allylic carbonates with silyl enol ethers and caesium fluoride alone as activator led to cleavage of the silyl enol ether and the caesium enolate reacted with unremarkable regio- and enantioselectivities, especially in the presence of phosphoramidite ligands (L7a) (Scheme 15). The same reaction, but with zinc fluoride alone, gave only low conversion. However, reactions of allylic carbonates with silyl enol ethers and a combination of these two metal fluorides provided homoallylic ketones with a β-stereogenic centre with high regio- and enantioselectivity. Reactions of aromatic ketones occurred in high yield, while reactions of aliphatic ketones occurred with high selectivities but in modest yields.
 | ||
| Scheme 15 Iridium-catalysed enantioselective allylation of silyl enolates. | ||
α,β-Unsaturated methyl ketones can also be converted into the corresponding silylenol ethers, which can be subjected to allylic alkylations. In this case, KF/18-K-6 was found to be better suitable than the CsF/ZnF2-combination.28 Cyclic ketones gave best results when they were converted into the corresponding barium enolates.29
Rhodium-catalysed allylic alkylations of ketone enolates were reported by Evans et al.30 Especially good results were obtained with copper enolates, especially with α-alkoxy ketones (Scheme 16).31 Using Wilkinson's catalyst, high yields and excellent regio- and stereoselectivities were obtained. In case enantiomerically pure allyl substrates were used, also a perfect chirality transfer was observed. The good selectivities obtained are explained by the formation of a chelated Cu-enolate with well-defined enolate geometry.
 | ||
| Scheme 16 Rh-Catalyzed α-alkoxy ketone allylation. | ||
In addition, an enantioselective version was reported using chiral monodentate phosphite ligands (L8).32 Interestingly, while K-enolates provided only racemic allylation products, the corresponding Li-enolates gave high ee's, especially with allyl benzoates (Scheme 17). Probably, also here chelated enolates are responsible for the good selectivities.
 | ||
| Scheme 17 Enantioselective Rh-catalyzed α-alkoxy ketone allylation. | ||
Very recently, Kanbayashi and Onitsuka et al. reported an asymmetric allylic alkylation of silyl enolates with allyl chlorides (Scheme 18).33 As a catalyst, a planar–chiral ruthenium complex K1 was used. In the presence of NaHCO3, the reaction proceeded under mild conditions giving the branched product regioselectively and with good enantioselectivity. Interestingly, functional groups such as aldehydes were tolerated. The Cp–Ru complex plays an important role in activating both, the allyl chloride and the silyl enolate.
 | ||
| Scheme 18 Ruthenium-catalysed enantioselective allylation of silyl enolates. | ||
In 2011, Jarugumilli and Cook described an iron-catalysed allylic alkylation of zinc enolates34 generated in situ via copper-catalysed Michael addition (Scheme 19).35 Diiron nonacarbonyl in combination with PPh3 provided the allylation products in modest to good yields and diastereoselectivity at room temperature. While PPh3 was the most effective of the investigated ligands, the exact nature of the catalytic active species is unknown.
 | ||
| Scheme 19 Iron-catalysed allylation of in situ formed zinc enolates. | ||
Allylation of ketones via enamine intermediates
Enamines can also serve as good nucleophiles in Pd-catalysed allylic alkylations. This approach avoids the use of strong bases to generate the non-stabilized enolates. Zhang et al. reported the allylation in the presence of chiral metallocene-based phosphinoxazoline ligands (L10), giving excellent ee's (Scheme 20).36 The enamines can be generated in situ from the corresponding ketone and a secondary amine such as pyrrolidine.37 It should be mentioned, that these reactions are not limited to activated allylic substrates such as acetates and carbonates, but can also be carried out with allylic alcohols.38 | ||
| Scheme 20 Enantio- and diastereoselective ketone allylation. | ||
A chiral Pd/proline-catalysed version was reported by Breit et al. in 2009.39 In this case, the choice of a large-bite angle ligand, such as Xantphos (L11) was essential for good yields (Scheme 21). Proline is also essential, since pyrrolidine gave no conversion. Unfortunately, L-proline provided only access to racemic allylation products.
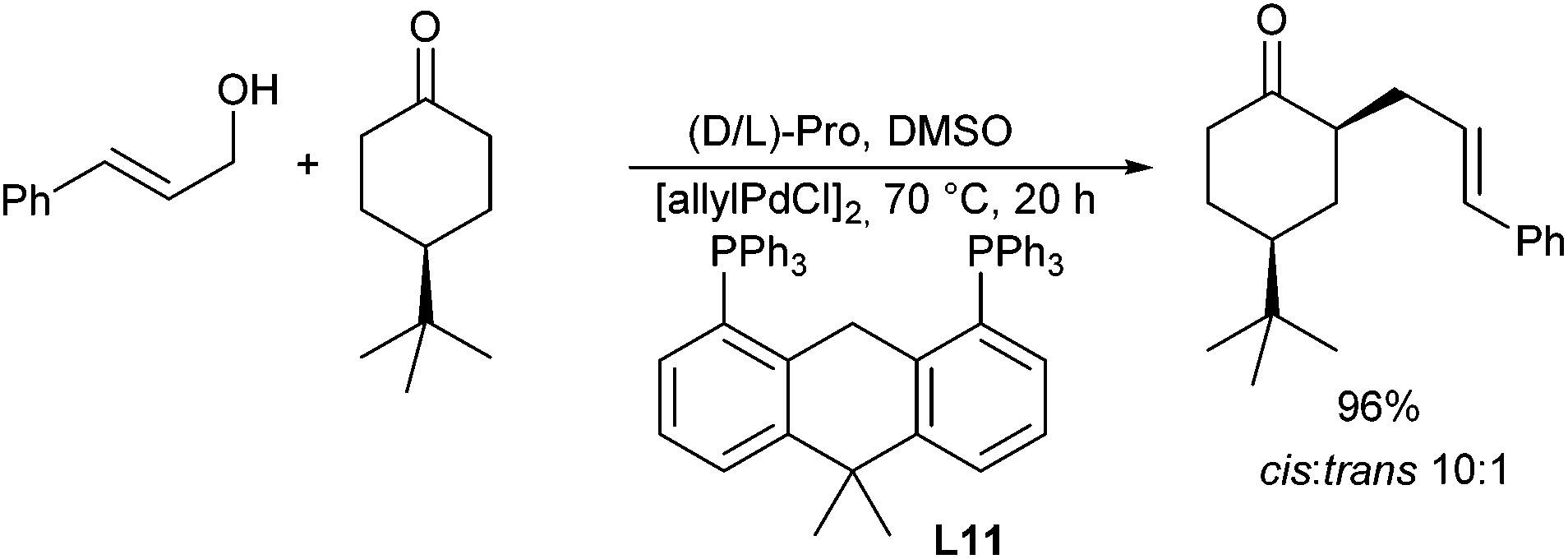 | ||
| Scheme 21 Diastereoselective ketone allylation. | ||
Hartwig et al. described the first Ir-catalysed enantioselective allylation of enamines in the presence of phosphoramidite ligands (Scheme 22).40 Allylation occurred at the sterically higher substituted position of the allyl substrate with excellent ee's.
 | ||
| Scheme 22 Enantioselective Ir-catalysed enamine allylation. | ||
Decarboxylative allylic alkylations
In the last years especially decarboxylative allylations using vinyl allyl carbonates41 or allyl β-oxo carboxylates42 became very popular. The protocol was developed by Tsuji et al. in the early 80s,43 and is nowadays mainly influenced by the work Trost,44 Tunge45 and Stoltz.46 The new developments are covered in a set of excellent recent reviews,47 and therefore the discussion here will focus only on the principle.In most cases, quaternary stereogenic centres are created, especially if chiral ligands are used, probably to avoid an epimerization of the stereogenic centres under the reaction conditions used.
With both types of substrates high yields and enantioselectivities were obtained, e.g. with Trost ligands (L1) or phosphinoxazolines, such as L3a (Scheme 23).
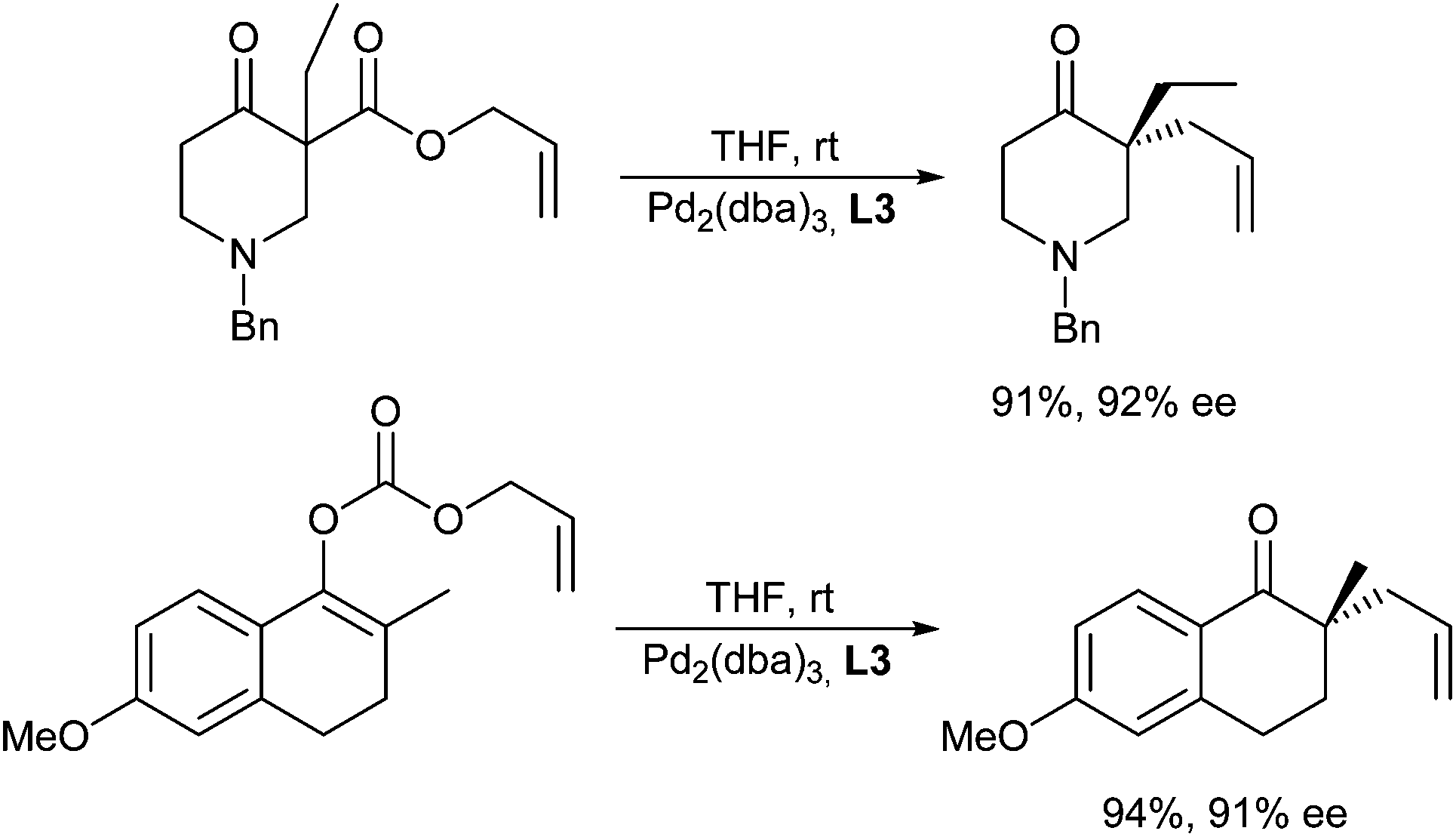 | ||
| Scheme 23 Decarboxylative allylic alkylations. | ||
If chiral allylic esters are used, the reactions proceed with net retention, as with stabilized nucleophiles.48 This protocol found widespread application in the synthesis of natural products containing quaternary stereogenic centres.49 Detailed mechanistic studies by Stoltz et al. indicate that these alkylation reactions occur through an inner-sphere mechanism involving binding of the prochiral enolate nucleophile, formed in the decarboxylation step, directly to the palladium centre (B) (Scheme 24).50
 | ||
| Scheme 24 Mechanism of decarboxylative allylic alkylations. | ||
In 2007, an interesting strategy for the generation of the enolate was demonstrated by Schulz and Blechert.51 The enolate as well as the π-allyl palladium complex were formed in a decarboxylative de Mayo ring expansion reaction52 and could be trapped in an asymmetric allylic alkylation. This allowed the preparation of enantioenriched substituted cycloheptane-1,4-dione and cyclooctane-1,5-dione derivatives (Scheme 25).
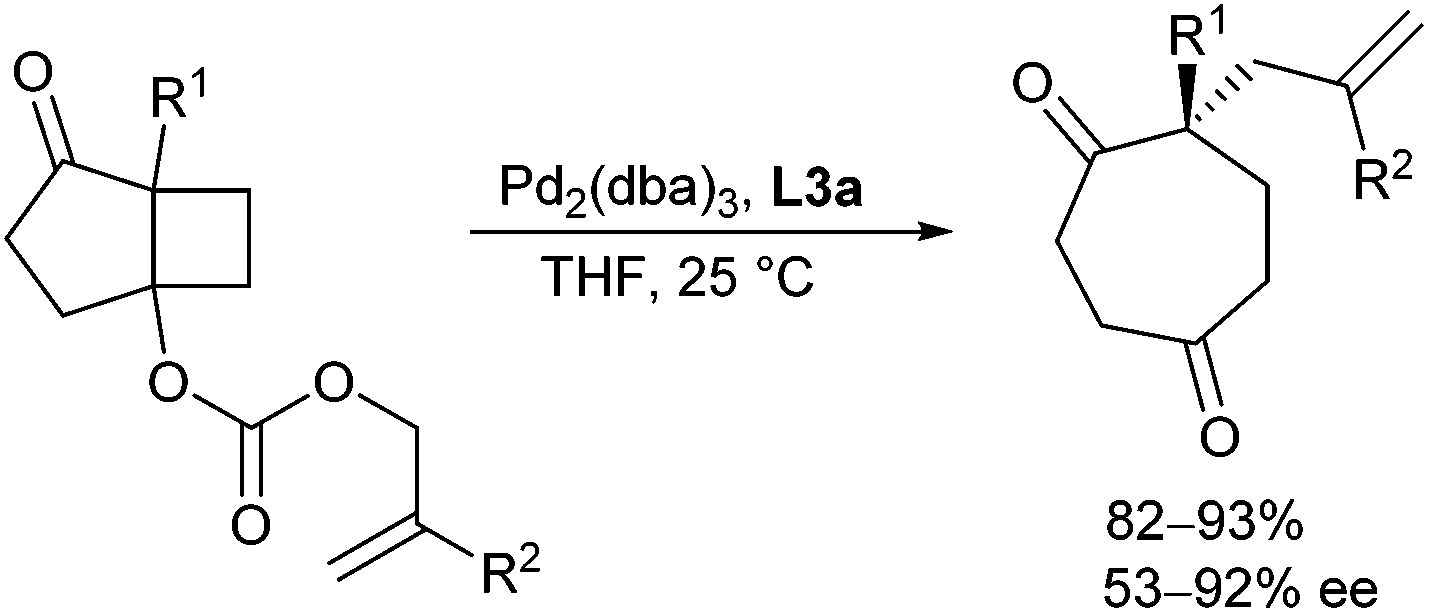 | ||
| Scheme 25 Decarboxylative de Mayo ring expansion/allylation. | ||
Allylic alkylations of aldehyde enolates
In principle, enolizable aldehydes can be subjected to allylic alkylation under the same conditions as described for ketones. Nevertheless, only a few examples are reported in the literature so far. Most protocols proceed via in situ formed enamines. In 2006, Córdova et al. reported the direct catalytic α-allylation of aldehydes by a combination of transition metal and organocatalysis.53 Secondary amines were used to generate an enamine, while best results were obtained with pyrrolidine. By using proline and derivatives thereof, the reaction could be performed in a highly enantioselective fashion.54In situ reduction gave rise to the corresponding primary alcohols with excellent ee (Scheme 26).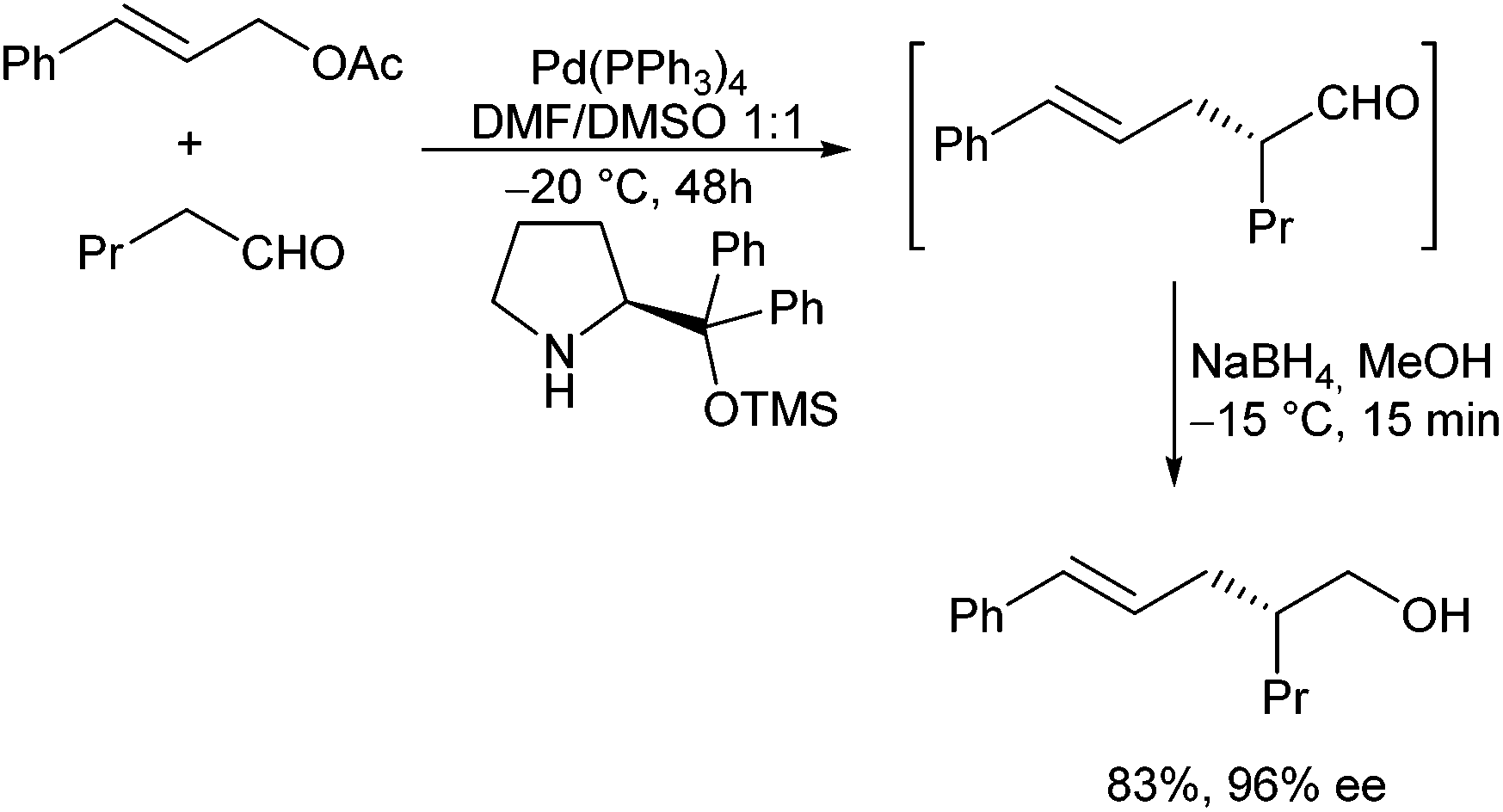 | ||
| Scheme 26 Enantioselective aldehyde allylation. | ||
Breit et al. could show, that in the presence of xantphos (L11) the corresponding allyl alcohols could directly be used in the allylation, without prior activation.39 Instead of proline (derivatives) also other chiral amino acids can be used, but in general the ee's are lower.55 Alternatively, the stereogenic outcome of the reaction can also be controlled by using chiral ligands such as metallocene-based ligand L12. The β-stereogenic centre could be introduced with good ee, but the simple diastereoselectivity was low in this case (Scheme 27).56
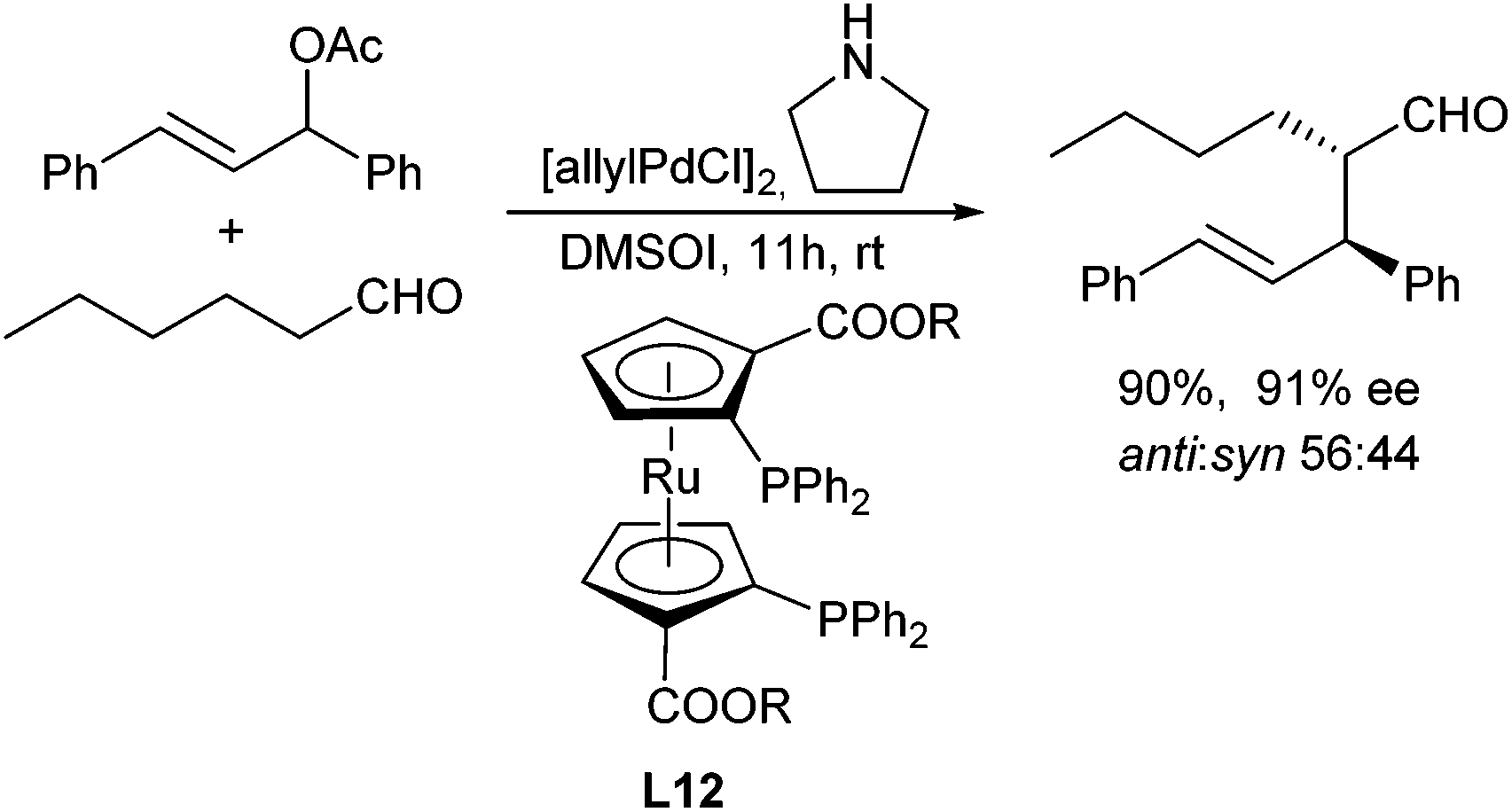 | ||
| Scheme 27 Enantio- and diastereoselective aldehyde allylation. | ||
Probably, the most straightforward approach is the direct allylation of aldehydes with unactivated allyl alcohols. Tamaru et al. reported such a direct α-allylation in the presence of (over)stoichiometric amounts of BEt3 as a Lewis acid to activate the alcohol.57 List et al. reported that Brønsted acids can be used as well for this purpose.58 In combination with enamine catalysis, chiral phosphoric acid derivatives, such as Trip, could be used to control the stereochemical outcome of the reaction (Scheme 28).59
 | ||
| Scheme 28 Chiral Brønsted acid catalysed asymmetric allylic alkylation. | ||
Trost et al. could show that the decarboxylative allylation approach, developed for ketone allylations, can also be applied to protected α-tertiary hydroxy aldehydes.60 Independent of the enol carbonate used (4 or 5) the desired α-allylated aldehyde was obtained in high yield and ee, especially in the presence of ligand L1b (Scheme 29). While the formation of the aldehyde is obvious in case of 4, the regioisomer 5 should give access to the corresponding ketone. The formation of the same product from the two different enol carbonates indicates that the Pd-enolates C and D formed in situ are in an equilibrium, and that obviously the aldehyde enolate C is the preferred one, leading to the observed product.
 | ||
| Scheme 29 Decarboxylative allylic alkylation of aldehydes. | ||
Allylic alkylations of ester enolates
Allylation of metal enolates
Quite different can be the situation if ester enolates are used as nucleophiles. First examples were reported by Hegedus et al. in 1980.61 The yields obtained were generally low, and it was assumed, that non-stabilized carbanions attack the metal in preference to the allyl group (inner sphere mechanism), resulting in reduction of the complex rather than alkylation.62 However, this reduction can be suppressed completely by addition of HMPA. For example, treatment of π-allyl palladium chloride (stoichiometric amount) with the enolate of methyl cyclohexanecarboxylate under standard conditions (PPh3, THF) gave the expected product in a very low yield, while running the reaction in the presence of HMPA and triethylamine (replacing the PPh3) gave rise not to the “expected” allylation product, but to a cyclopropane derivative in good yield (Scheme 30). Obviously, the carbanion attacked the central carbon of the π-allyl complex. This is in sharp contrast to the attack of stabilized nucleophiles and the observations made with ketone enolates. Interestingly CO has a positive effect on the yield of the reaction, although it is not incorporated.63 | ||
| Scheme 30 Allylic alkylation of ester enolates. | ||
Interestingly, α-allylated products were obtained, if ester enolates were reacted with vinyl epoxides, although the yields varied depending on the vinyl epoxide used (Scheme 31).64 As usual, nucleophilic attack occurred at the sterically less hindered position yielding an E/Z-isomeric product mixture. Similar results were obtained with silyl ketene acetals in the presence of bidentate phosphine ligands.65
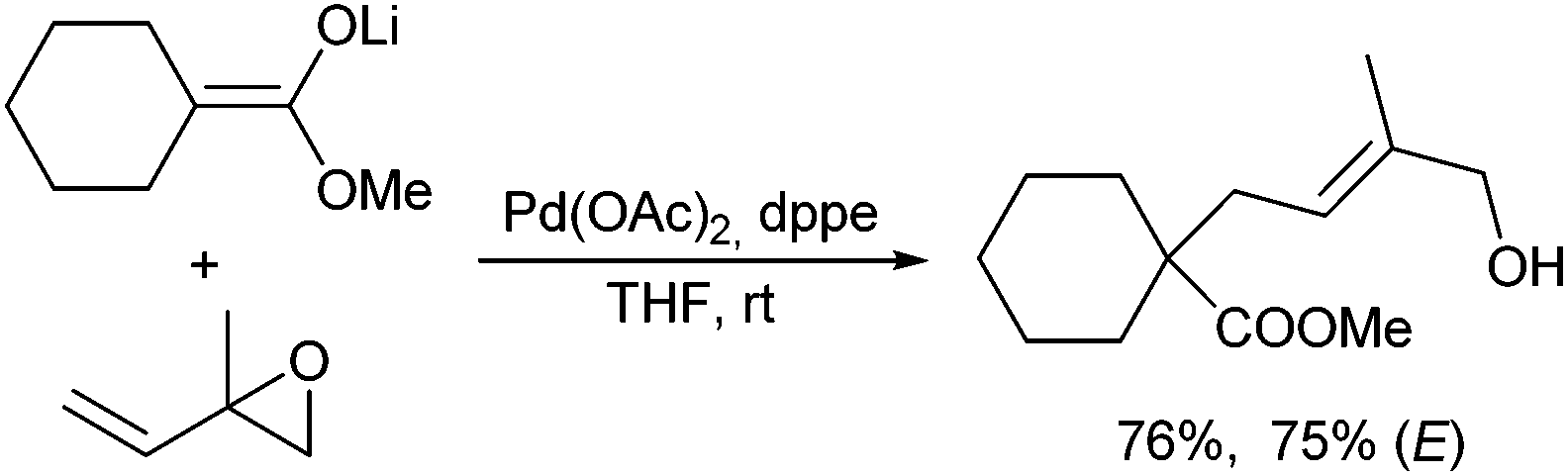 | ||
| Scheme 31 Allylic alkylation of ester enolates with vinyl epoxides. | ||
In 2007 Kawatsura et al. reported the allylic alkylation of a series of stabilized carbanions in the presence of 2-(diphenylphosphino)benzoic acid (dppb).66 In general good regio- and diastereoselectivities were obtained with substituted β-ketoesters, but simple aryl substituted esters gave only moderate diastereoselectivities. Interestingly, 2-pyridyl acetic esters could be used not only for the generation of tertiary stereogenic centres, but also quaternary ones (Scheme 32). In contrast, reactions with 2-phenyl propanoates failed. Reactions of the less acidic aliphatic esters were not reported.
 | ||
| Scheme 32 Allylic alkylation of 2-pyridyl acetic esters. | ||
Braun and Thiel described the allylic alkylation of preformed lactone enolates.67 They showed that high regio- and diastereoselectivities could be observed in the presence of additives, such as LiCl (Scheme 33). Excellent ee's were obtained in the presence of Binap (L5) as chiral ligand. Five-membered lactones gave better yields compared to six-membered rings, while the latter gave better selectivities. If chiral lactones were used, the problem of double stereodifferentiation arose. The stereochemical outcome of the reaction was mainly controlled by the chiral ligand and not by the stereogenic centre in the lactone. DFT calculations were performed to investigate the mechanism and the selectivity of the reaction.68
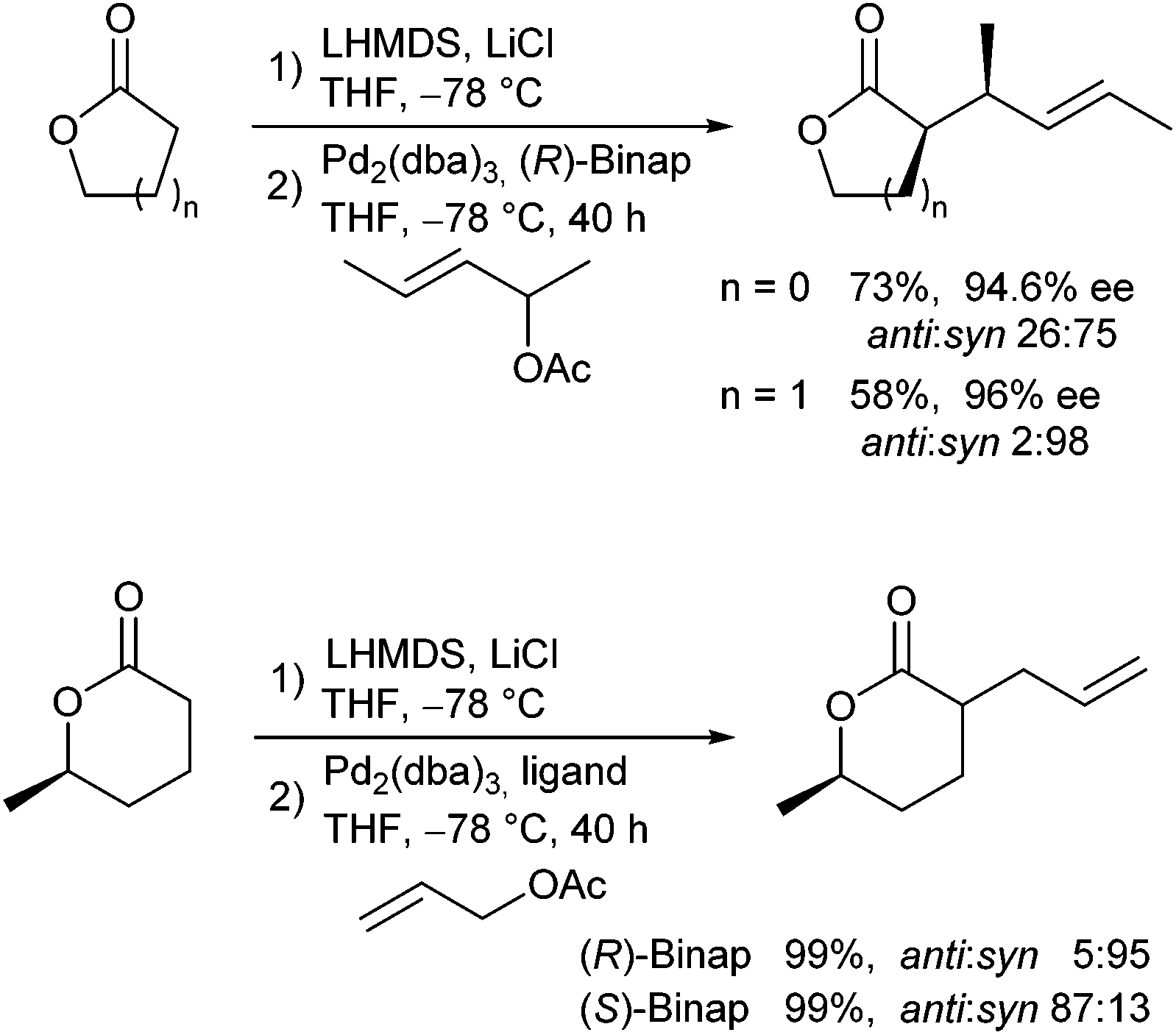 | ||
| Scheme 33 Diastereoselective allylic alkylation of lactones. | ||
Decarboxylative allylic alkylations
In principle, the decarboxylative allylation approach used widely for the stereoselective synthesis of α-allylated ketones can also be applied for the stereoselective generation of quaternary α-allylated esters. In this case, α,α-disubstituted allyl malonates can be used, which unfortunately are less reactive than the corresponding β-ketoesters, and therefore require harsher reaction conditions (DMF, 120 °C).69 In 2007 Ohta et al. reported that α-arylated diallyl malonates are much more reactive, undergoing the decarboxylative allylation already at room temperature in a wide range of solvents (Scheme 34).70 α-Quaternary benzylic esters were obtained in excellent yield in the presence of various types of P-ligands except phosphites.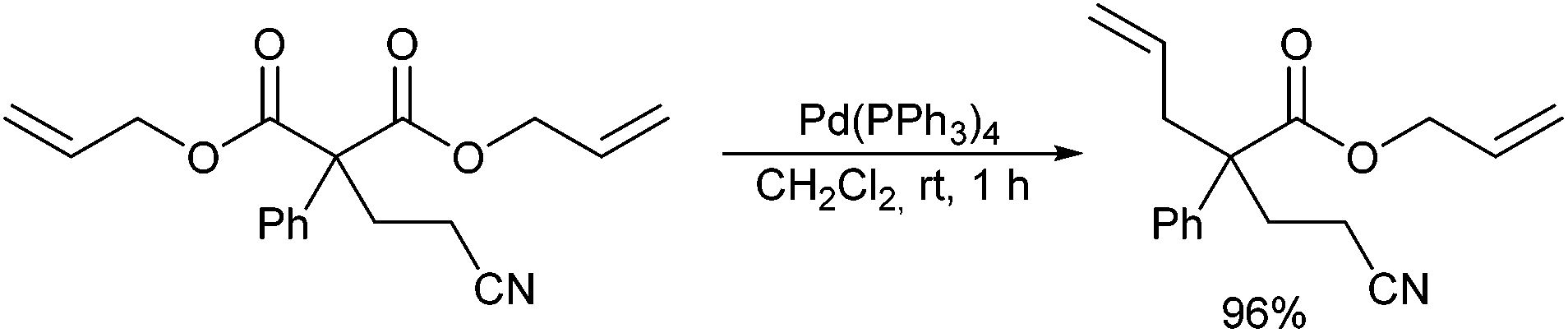 | ||
| Scheme 34 Decarboxylative allylation of esters. | ||
Deacylative allylic alkylations
Grenning and Tunge reported an alternative approach generating acyl substituted ester and lactone enolates via retro-Claisen condensation.45b In this deacylative allylation readily available allylic alcohols could be used without prior activation (Scheme 35). | ||
| Scheme 35 Deacylative allylation of esters and lactones. | ||
Allylation of chelated enolates
In 1999, Kazmaier and Zumpe introduced a new type of nucleophile, chelated glycine ester enolates.71 Chelation causes a marked enhancement of the thermal stability, without having any negative influence on the reactivity of these enolates.72 In addition, coordination of the deprotonated enolate toward a chelating metal prevents a coordination to the Pd-catalyst, a general problem of non-stabilized enolates. Due to the fixed enolate geometry conversions of these enolates proceed with a high degree of stereoselectivity in most cases. This is not only true for aldol reactions,73 Michael additions74 or chelate-enolate Claisen rearrangements,75 but also for transition metal catalysed allylic alkylations.76 Best results, with respect to yield and selectivity, were obtained with TFA- (trifluoroacetyl)-protected glycine tert-butyl esters. Besides allylic carbonates, the corresponding acetates or benzoates could be used as well, but with the more reactive carbonates, the yields and selectivities were usually better. Because of the high reactivity of the chelated enolates (E), the allylations already take place under very mild conditions at −78° C, what has an extremely positive effect on the selectivity of the reactions. With secondary allylic substrates, anti-configured β-branched amino acid derivatives were obtained in high yield and diastereoselectivity (Scheme 36). | ||
| Scheme 36 Diastereoselective allylic alkylation of chelated glycine ester enolates E. | ||
This approach allowed the introduction of a wide range of also functionalized side chains,77e.g. redox active ferrocene units,78 fluorescence labels79 or vinylstannanes, which could be subjected to further modifications via cross coupling reactions (Scheme 37).80
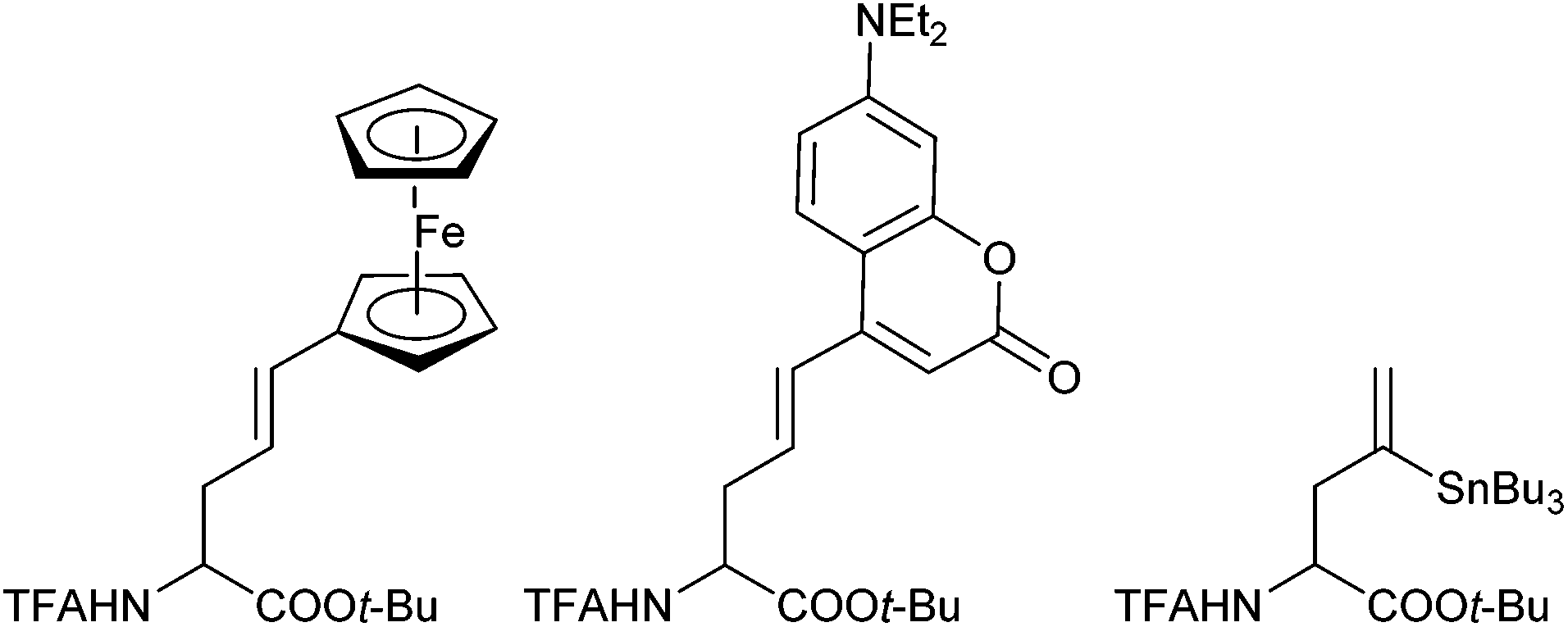 | ||
| Scheme 37 Applications of chelated enolate allylations. | ||
Vinyl epoxides are also excellent substrates for Pd-catalysed allylic alkylations of these enolates, allowing the introduction of hydroxylated allylic side chains (Scheme 38).81 Nucleophilic attack onto the formed π-allyl complex occurred regioselectively at the distal position. The E-configured product was formed preferentially, depending on the substitution pattern and the reaction conditions used.
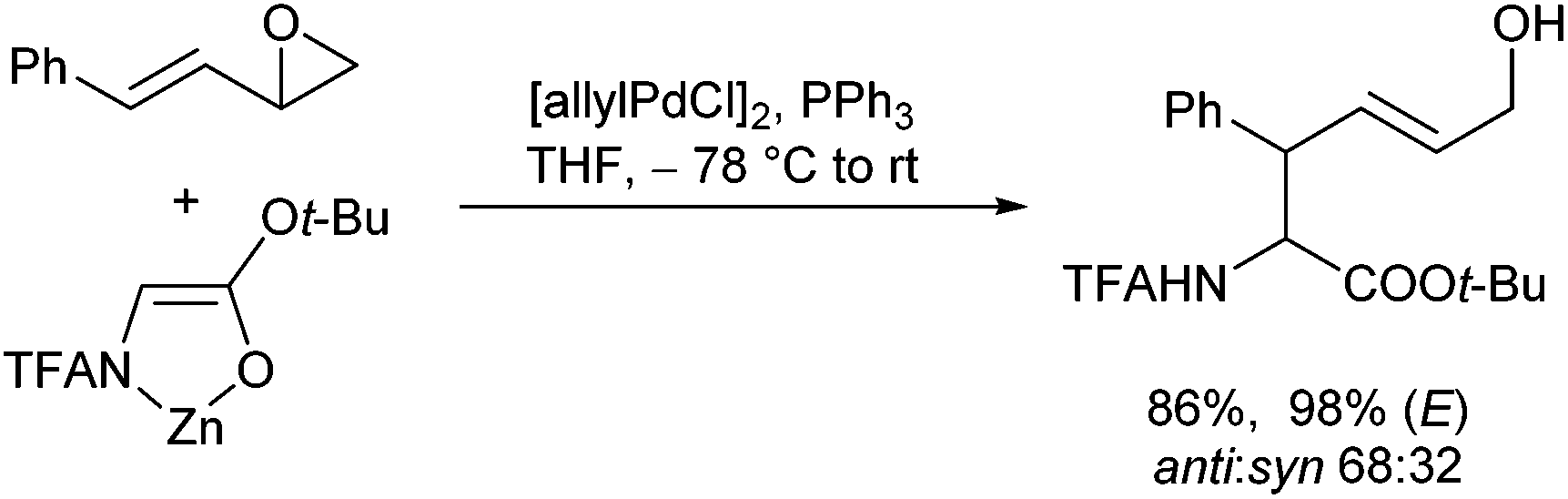 | ||
| Scheme 38 Allylic alkylations of chelated enolates using vinyl epoxides. | ||
In addition to amino acid ester enolates, deprotonated, chelated tert-butyl glycolates were found to be also suitable nucleophiles for Pd-catalysed allylic alkylations, generating γ,δ-unsaturated α-hydroxy acid derivatives (Scheme 39).82 This protocol is suitable for the introduction of different side chains, including stannylated ones. These could be subjected to further cross coupling reactions, giving access to a wide range of structural motifs.
 | ||
| Scheme 39 Allylic alkylations of chelated glycolates. | ||
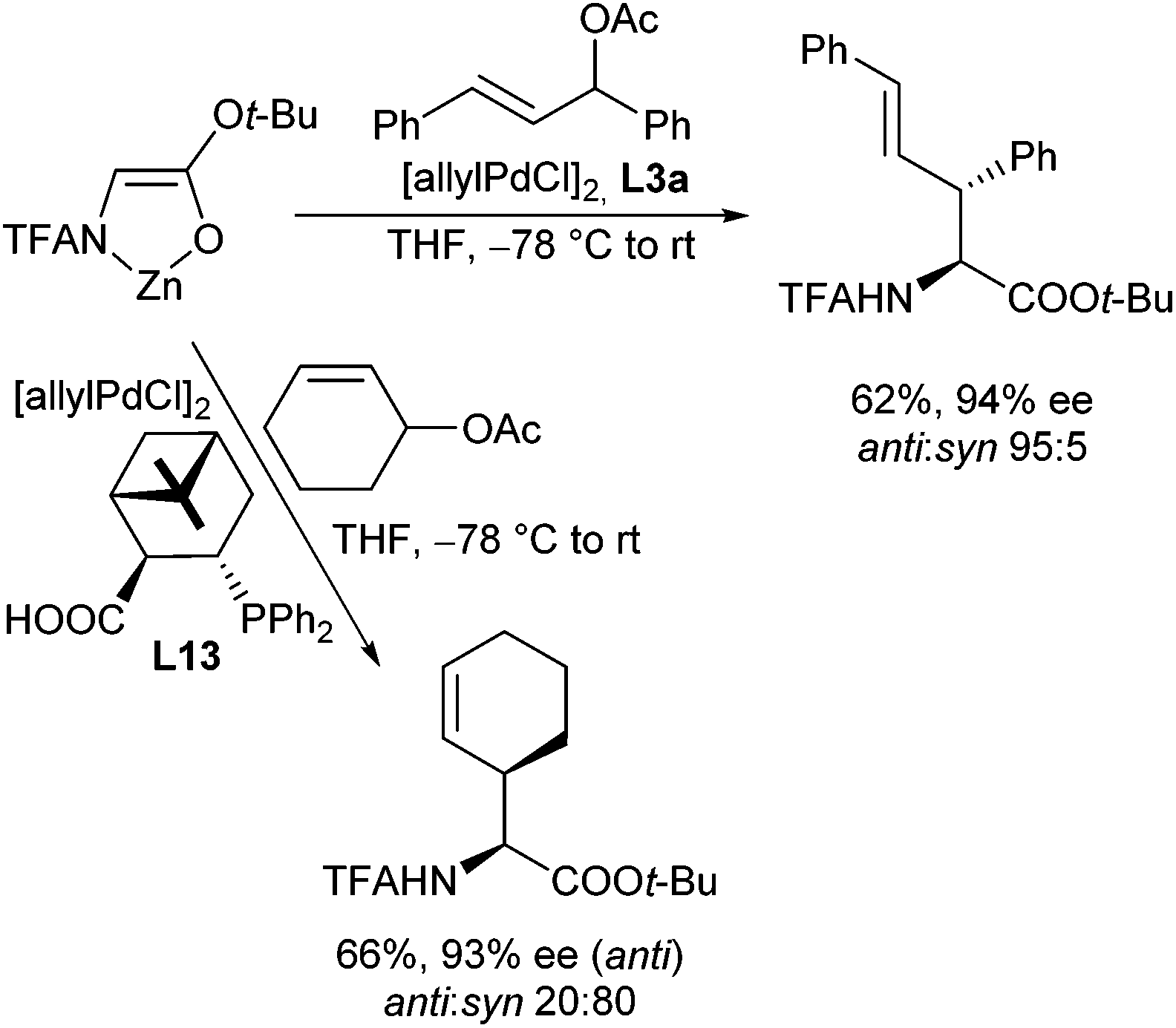 | ||
| Scheme 40 Enantio- and diastereoselective allylic alkylations of chelates enolates. | ||
On the other hand, if allyl derivatives with different substituents are used, unsymmetrical π-allyl-palladium-complexes are formed. Attack of nucleophiles on these complexes can provide mixtures of regioisomers, depending on the substitution pattern of the allyl moiety.85 Although this is a serious issue, these substrates also have a big advantage: if chiral allyl substrates are used, the stereogenic information of the allyl substrate can be transferred into the product. Based on the reaction mechanism, the stereochemical control in the allyl fragment is not a problem (double inversion), in contrast to chiral centres formed in the ‘nucleophile moiety’. The allylic substitutions with chiral allyl substrates such as 6 proceeded cleanly and with good yields (Scheme 41). The chirality could be transferred almost completely with good diastereoselectivities, and the only regioisomers obtained were those with the double bond in conjugation to the phenyl ring.
 | ||
| Scheme 41 Allylic alkylations using chiral allyl substrates. | ||
Interestingly, the diastereoselectivities obtained with acetates were a little worse in comparison to those obtained with carbonates, what can be explained by the higher reactivity of the carbonates. While these react already at −78 °C (or even lower), the corresponding acetates require temperatures around −60 °C for ionization. This allowed the first time to use these C-nucleophiles in isomerization-free allylations.
 | ||
| Scheme 42 π–σ–π-Isomerization of π-allyl-palladium complexes. | ||
Since the palladium-catalysed allylic substitution with chelated enolates already proceeds at −78 °C, this isomerization processes can be suppressed almost completely. For example, the reaction with the (Z)-carbonate 7 yielded the desired (Z)-substitution product almost exclusively (Z/E > 99/1) and with excellent stereoselectivity (Scheme 43).88 In contrast, the corresponding acetate furnished a (E/Z)-mixture in a very low yield. The selectivities were markedly worse than those obtained with the carbonate 7. This clearly indicated, that with the acetate both processes, direct allylic substitution and isomerization, compete with each other, while the carbonate react isomerization-free.
 | ||
| Scheme 43 Isomerization-free allylic alkylations. | ||
Terminal π-allyl-complexes show an even higher tendency towards isomerization than 1,3-disubstituted derivatives. But even with these substrates, an almost perfect retention of the olefin geometry was observed. In case chiral substrates (8) were used, significant chiral induction from the allyl substrate to the α-position of the new formed amino acid was observed.89
If the π–σ–π-isomerization can be suppressed like in this case, the question arises: What would happen to (Z)-allylic substrates with two identical substituents at the allyl termini? In principle, there are two different reaction pathways, because the anti/syn π-allyl complex formed as an intermediate has two different allylic positions (Scheme 44).90 Nucleophilic attack at a (anti-position) would provide a product with (E)-configured double bond, whereas attack at b (syn-position) would lead to a (Z)-double bond, and the product ratio should depend on the relative reactivity of these two positions.
 | ||
| Scheme 44 Nucleophilic attack onto anti/syn π-allyl complexes. | ||
This question is so far of interest, since symmetrically substituted allyl derivatives are normally used in asymmetric catalysed reactions, where achiral syn–syn complexes are formed even from enantiomerically pure allyl substrates. However, if one can suppress the isomerization during the reaction of (Z)-configured substrates, and if one of the two allylic positions is more reactive than the other one, it should be possible to generate optically active compounds also with these substrates. An indeed, allylation with chiral (Z)-allyl carbonate 9 (98% ee) proceeded cleanly with high regio- and diastereoselectivity and almost perfect chirality transfer (Scheme 45). This is a clear indication, that the anti-position is much more reactive than the syn-position,91 and that also here the allylation was faster than the isomerization. Otherwise, one would obtain an (almost) racemic product. The significantly higher reactivity of the anti-position was also used for regioselective allylations using allylic substrates bearing different substituents (10, 11).92
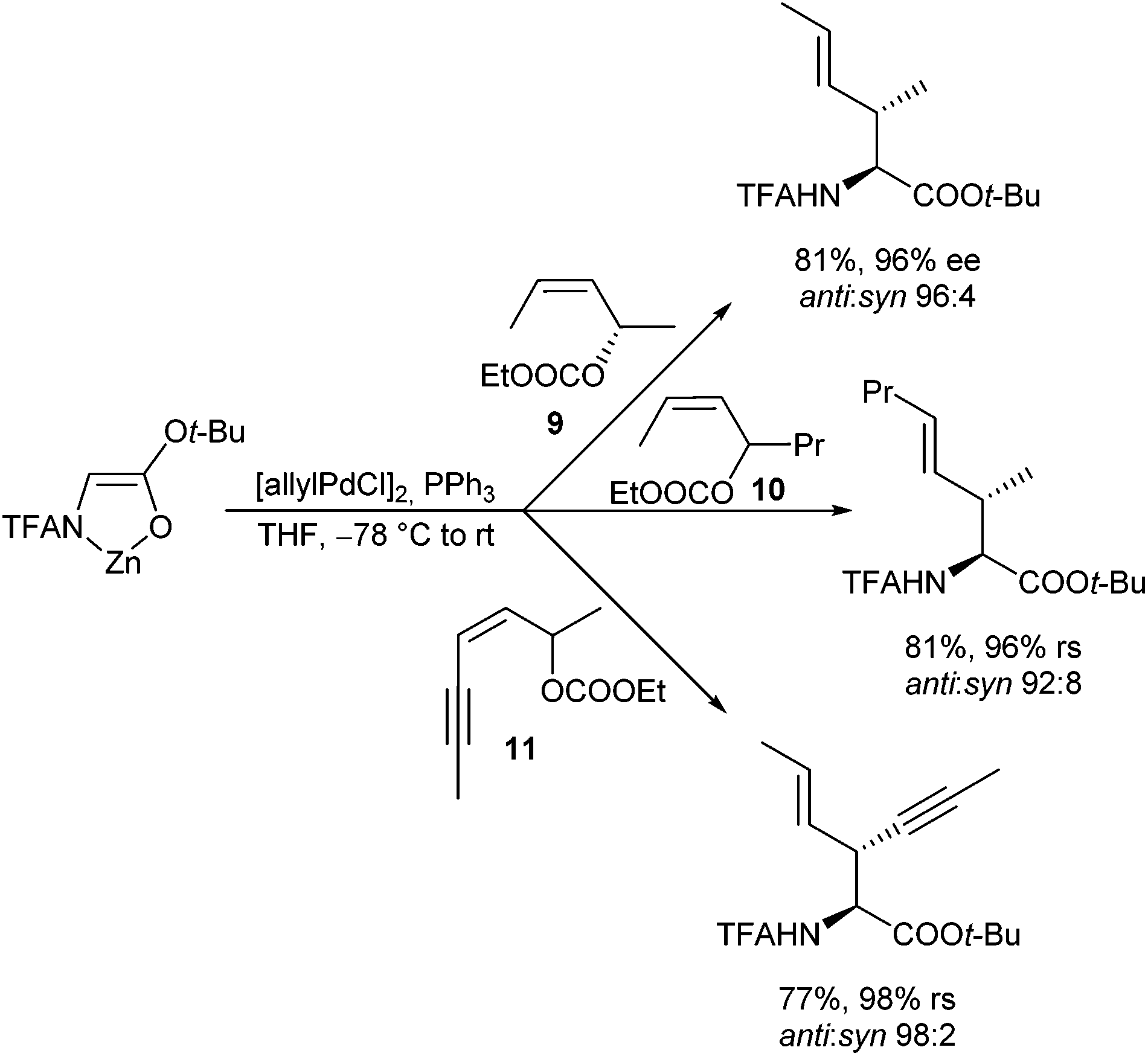 | ||
| Scheme 45 Regioselective allylic alkylations. | ||
If steric hindrance is not an issue, the expected (E)-configured products were formed with excellent selectivity, while with sterically demanding substituents at the double bond, isomerization became faster than the allylation, and mixtures of regioisomers were formed. It should be mentioned, that if the substituent at the (Z)-double bond is an electron withdrawing group, Michael additions of the chelated enolates74 become competitive to the allylic alkylations.93
In most cases an isomerization of the π-allyl intermediate is an undesired process, leading to side products or a loss of chiral information, especially if terminal π-allyl complexes are involved. On the other hand, a fast isomerization can also be used to control the stereochemical outcome of the reaction.94 For example, all isomeric allyl carbonates 12 gave rise to the same allylation product with the same regio- and stereoselectivity, independent of the position and configuration of the double bond and leaving group (Scheme 46). The primary formed terminal π-allyl-Pd-complexes isomerize to the most stable complex F, with the Pd opposite to the sterically demanding O-protecting group. Highly “diastereoselective” attack of the enolate at the terminal position from the “opposite face” provides the desired product with an excellent degree of 1,5 induction. This approach found application in the synthesis of Boletin.95 The opposite stereoisomer could be obtained by replacing the sterically demanding O-protecting group by a coordinating group such as dppb.96
 | ||
| Scheme 46 Diastereoselective allylic alkylation taking advantage of π–σ–π-isomerization. | ||
 | ||
| Scheme 47 Stereoselective modifications of dipeptides. | ||
This can be explained by the formation of a metal peptide enolate complex, where the peptide coordinates three or even more times towards the metal. In such an enolate complex, one face of the enolate is shielded by the side chain of the adjacent amino acid, generating the “opposite” configuration.
Since this approach is not only applicable to a wide range of different, also functionalized peptides, but also to rather complex allylic substrates, it allows an easy synthesis of peptide libraries.
Even higher is the flexibility if stannylated allyl carbonates98 are used in the allylation step.99 In most cases, the desired metallated peptide were obtained as single diastereomer, and could be subjected to a wide range of cross coupling reactions (Scheme 48).100 The products were obtained in very high yield and without epimerization of the stereogenic centres.
 | ||
| Scheme 48 Generation of peptide libraries from stannylated peptides. | ||
This approach allowed also the introduction of heterocyclic side chains, such as functionalized tryptophans.101 In principle with only two to three steps large libraries of rather divergent peptides can be generated, what makes this approach interesting for the synthesis of drug like molecules and investigations of structure–activity relationships.
The application of chelated enolates as nucleophiles is not limited to Pd-catalysed reactions, but can also be applied to other transition-metal complexes. In 2006, Stolz and Kazmaier reported highly regio- and stereoselective rhodium-catalysed allylations of these enolates.102 In contrast to the Pd-catalysed reactions, the Rh-catalysed process proceeded (under the same reaction conditions) not via a “symmetrical” π-allyl complex, but via a σ/π-allyl complex,103 explaining the regio- and stereoselectivities observed. Sterically unhindered secondary terminal allylic substrates react with excellent regio- and stereoretention, while the best results were obtained with allyl phosphates and sterically demanding phosphite ligands (Scheme 49).104 With sterically demanding allylic substrates, not the expected branched product was formed but the linear one. Interestingly, in this case a high chirality transfer was observed, clearly indicating that isomerization processes do not play a role.
 | ||
| Scheme 49 Rh-Catalysed allylic alkylations of chelated enolates. | ||
The high regioretention allowed also a chemoselective allylation using allyl substrates with competitive allyl moieties (13).105 Wilkinson's catalyst was found to be highly sensitive to steric hindrance, and therefore terminal allyl substrates could selectively be reacted in the presence of substituted ones (Scheme 50). The same selectivity was also observed with dienyl substrates (14).
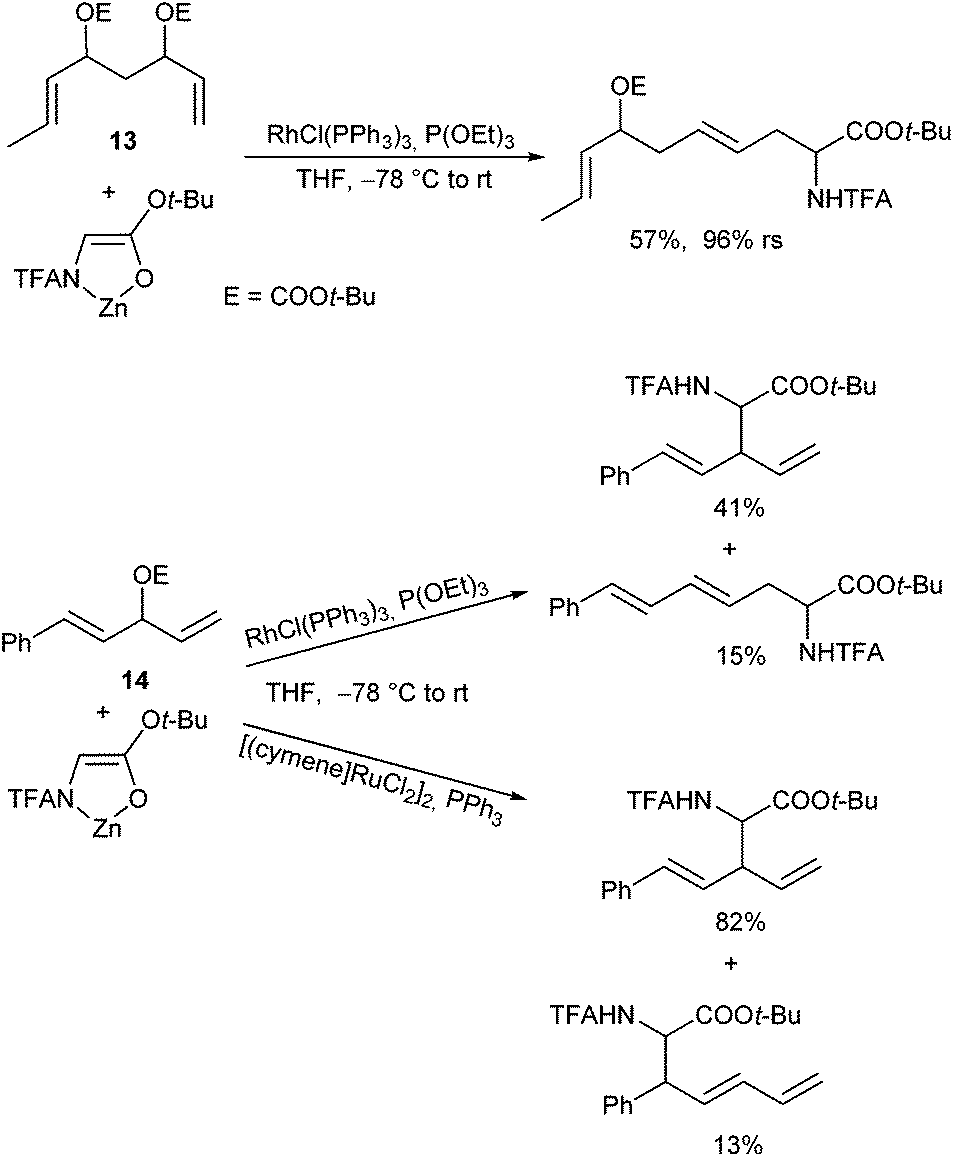 | ||
| Scheme 50 Chemoselective Rh- and Ru-catalysed allylic alkylations. | ||
Interestingly, complementary selectivities were observed with Ru-catalysts, where the sterically more hindered allyl moiety reacted preferentially.106 In comparison, under Pd-catalysed conditions mixtures of all possible substitution products are formed, clearly indicating that both allyl moieties undergo ionization.107
Best results so far were obtained with [(cymene)RuCl2]2,108 a catalyst that shows excellent regioretention with branched and (Z)-configured substrates, reacting without any isomerization (Scheme 51).109 [Cp*Ru(NCCH3)]PF6, a catalyst widely used by Trost110 and Bruneau111 gave different results, showing a high degree of isomerization.
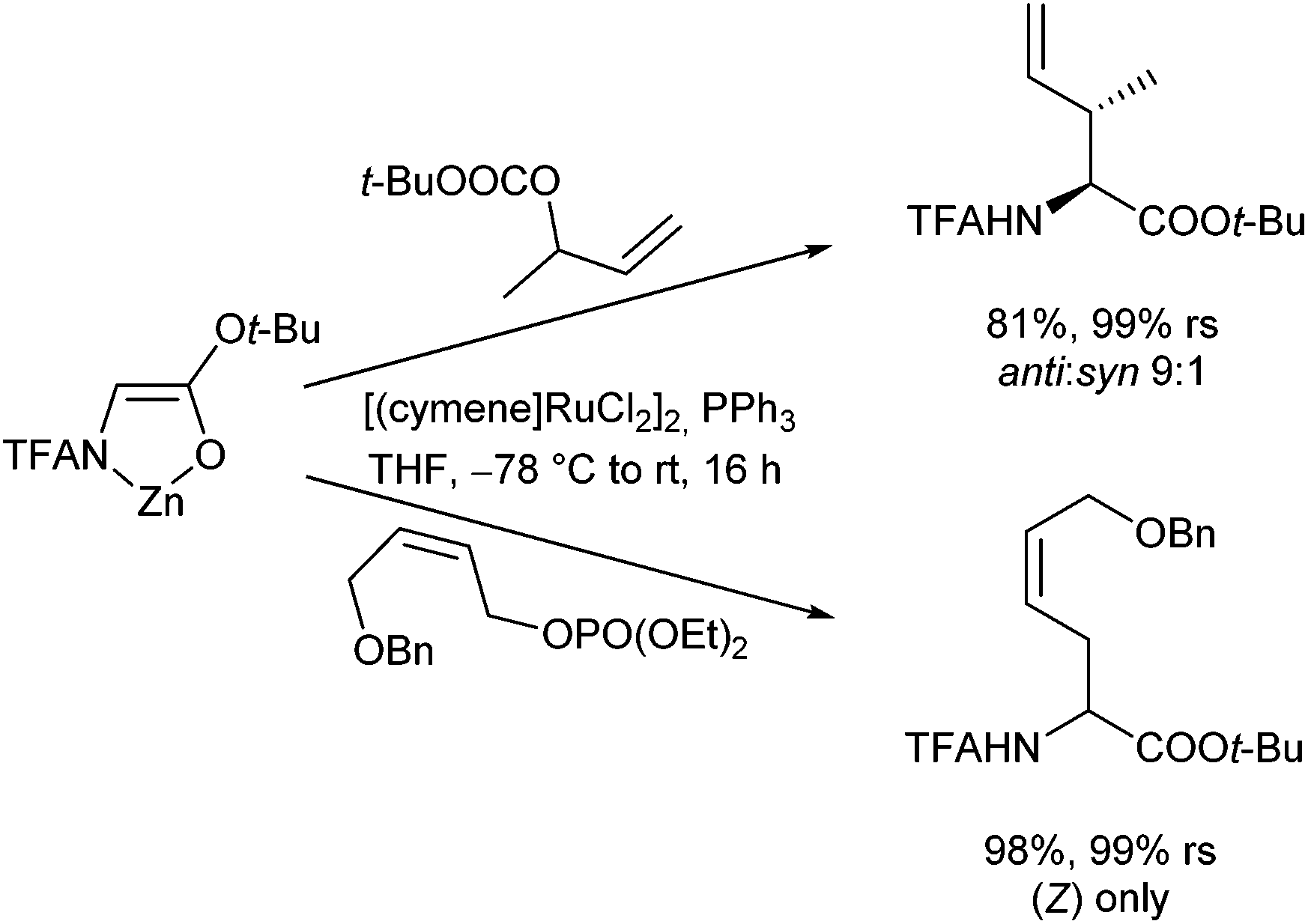 | ||
| Scheme 51 Regioselective Ru-catalysed allylic alkylations. | ||
In addition to the generally used allyl acetates and carbonates also 4-vinyl-substituted 1,3-dioxolan-2-ones were found to be good substrates for Ru-catalysed allylic alkylations (Scheme 52).112Cis-1,3-dioxolan-2-ones were more reactive than the corresponding trans-isomers.
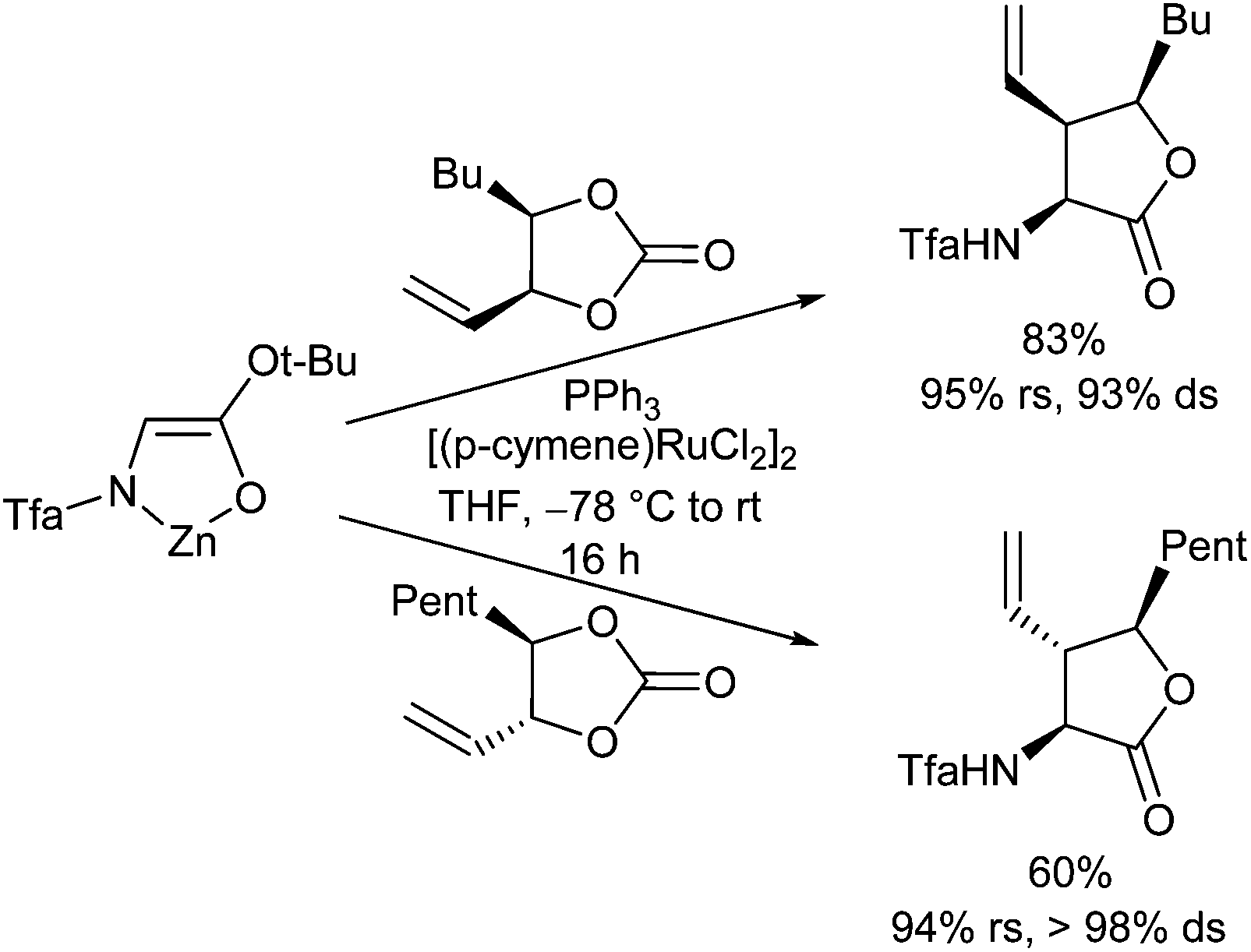 | ||
| Scheme 52 Regio- and stereoselective Ru-catalysed allylic alkylations using vinyl dioxolanones. | ||
The attack occurred preferentially with regioretention at the position of the leaving group with perfect chirality transfer. Therefore, this protocol is a good complement to the Pd-catalysed processes, which gave only linear products with this type of substrates. Under standard reaction conditions, when the reaction mixture is warmed up to room temperature overnight, the γ-hydroxyester formed primarily underwent cyclization toward the corresponding γ-lactone.
Allylic alkylations of carboxylic acids
Esters are by far the most common used enolate precursors, and allylic alkylations of other carboxylic acid derivatives are rather seldom. Only one report by Braun et al. described the application of double deprotonated carboxylic acids as nucleophiles in Pd-catalysed allylic alkylations (Scheme 53).113 Best results were obtained in the presence of LiCl, which was assumed to increase the reactivity of the enolate via disaggregation.114 The protocol could be applied to a wide range of carboxylic acids, also those with sterically demanding substituents, and was also suitable for the generation of quaternary α-centres. High ee's were obtained in the presence of Binap (L5) as chiral ligand. | ||
| Scheme 53 Stereoselective allylic alkylation of cyrboxylic acids. | ||
Allylic alkylations of carboxylic acid amides and imides
Besides esters, secondary carboxylic amides and imides are especially suitable for enolate formation and several applications as nucleophiles have been reported during the last years, although their number cannot compete with the ester enolates.One of the first examples in this direction was reported by Trost and Frederiksen using α-arylated oxindoles as nucleophiles.115 Weak bases such as NEt3 or K2CO3 could be used as bases to generate the “enolate”, which has to be considered as a stabilized one. Therefore, this type of enolates will not be discussed here.
Hou et al. reported the first allylic alkylations of acetic amides in 2008.116 The outcome of the reaction strongly depended on the substituents on the amide nitrogen. By far the best results were obtained with N,N-diphenyl amides. In the presence of ferrocene-based oxazoline ligands L6c high yields and ee's could be obtained (Scheme 54).
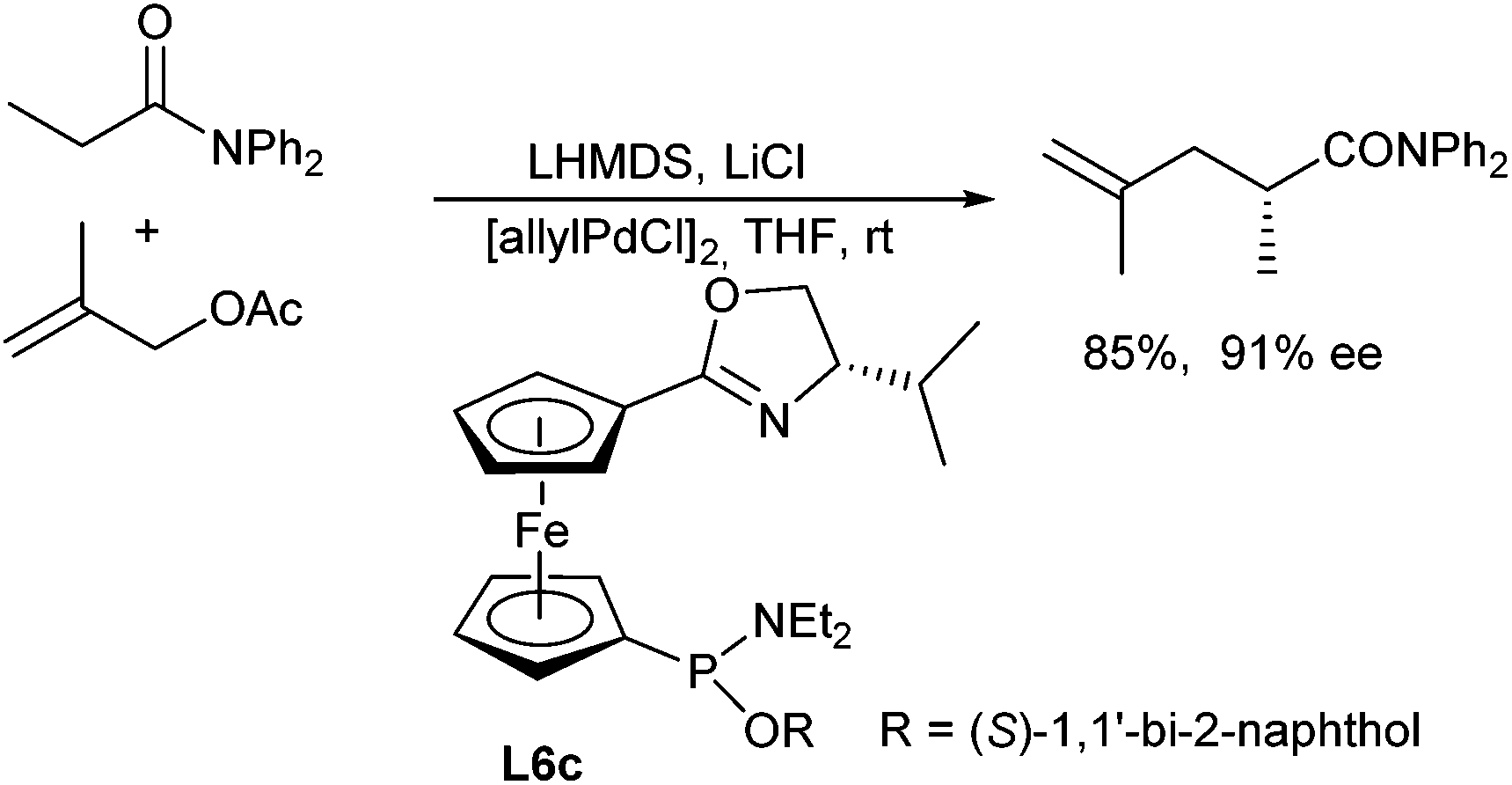 | ||
| Scheme 54 Enantioselective allylic alkylation of amides. | ||
Deacylative allylic alkylations
The decarboxylative allylation approach widely used for the allylic alkylation of in situ generated ketone enolates could also be used to generate amide or imide enolates. Trost et al. reported the decarboxylative allylation of N-acyloxazolinones, which can be considered as ester enolate equivalents.117 High yields and ee's could be obtained in the presence of Trost-ligand L1c (Scheme 55). | ||
| Scheme 55 Decarboxylative allylation of N-acyloxazolinone. | ||
The same approach was used by Stoltz et al. for the allylation of lactams and cyclic β-imidoesters.118 In general, they used allyl malonate derivatives as allylic substrates and PHOX ligands L3, while the best results were obtained with ligand L3b (Scheme 56). This protocol could be used for the allylation of a wide range of (heterocyclic) lactams.119
 | ||
| Scheme 56 Decarboxylative allylation of lactams. | ||
Allylation of chelated enolates
In 2011 Datta and Kazmaier could show, that chelated enolates of peptide amides can also be subjected to Pd-catalysed allylic alkylations.120 The stereochemical outcome of the reaction was controlled by the stereogenic centres in the peptide chain, in analogy to the corresponding peptide esters. This approach is especially interesting, because it allows the allylation of proline- or N-alkylated amino acid-containing peptides (Scheme 57).121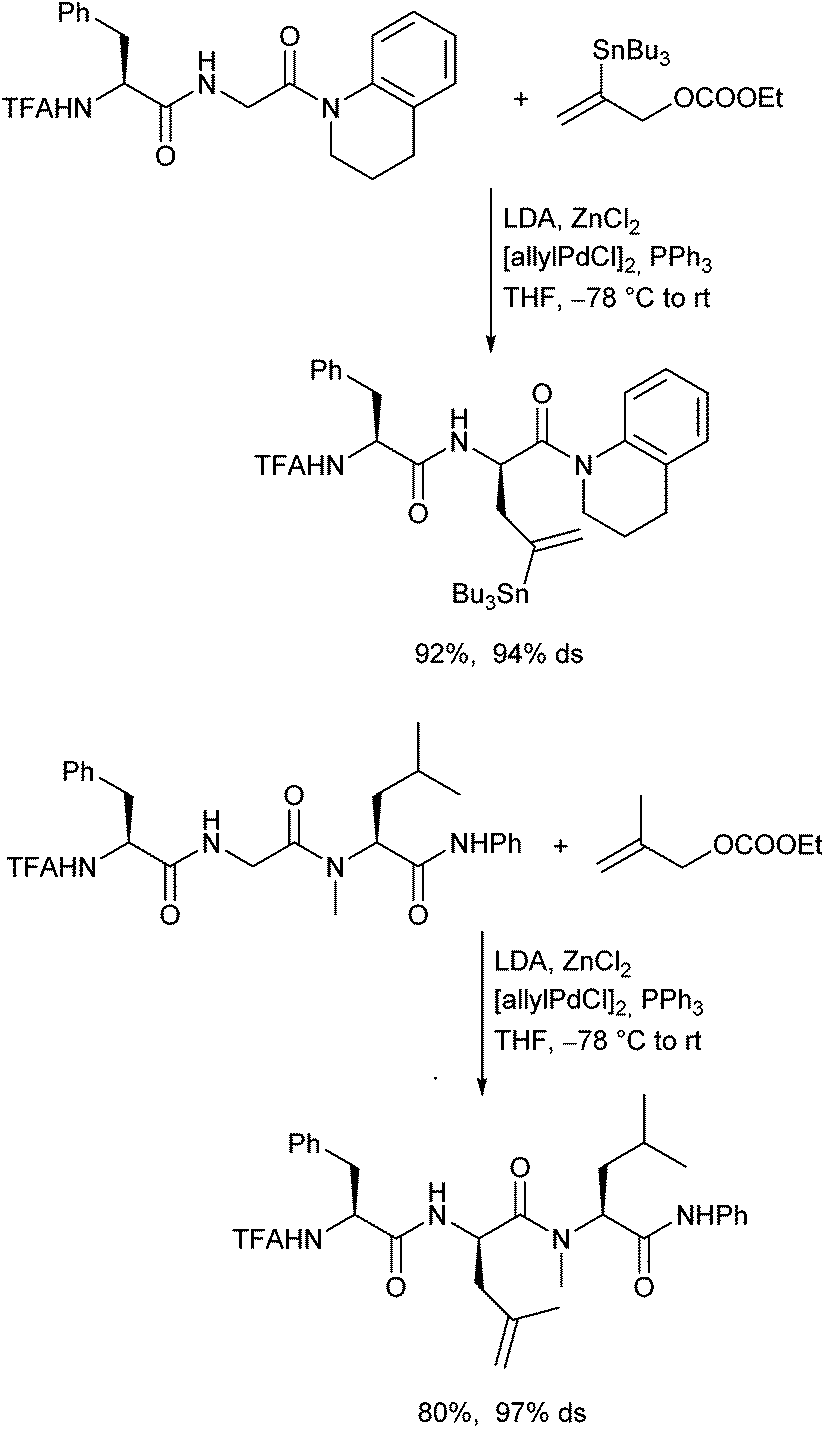 | ||
| Scheme 57 Pd-Catalysed allylic alkylation of peptide amides. | ||
The ruthenium-catalysed allylic alkylation is also here an interesting alternative to the palladium-catalysed process, since it can provide products, which are not accessible under Pd-catalysis.122 Chiral terminal allylic substrates could be reacted with perfect stereo- and regioretention, and (Z)-configured allylic substrates could be converted isomerization-free (Scheme 58). This allowed highly stereoselective modifications of peptides also in the middle of a peptide chain. The configuration at the α-position of the new generated α-amino acid can be controlled by the peptide and at the β-position by using chiral allylic substrates.
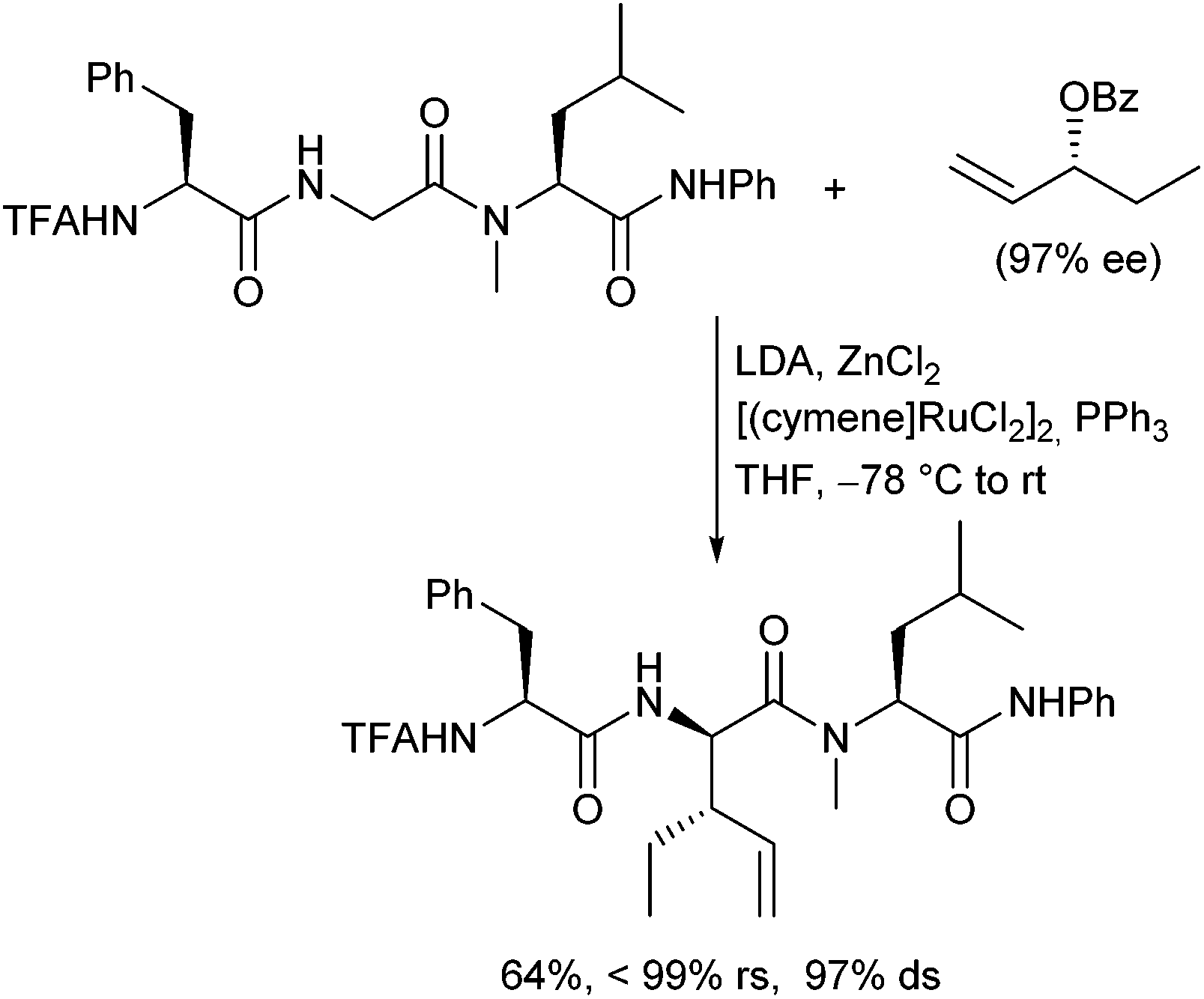 | ||
| Scheme 58 Ru-Catalysed allylic alkylation of peptide amides. | ||
In principle, N-protected amino acid allyl amides with an allylic leaving group can be used as substrates in palladium-catalysed allylic alkylation. While intermolecular allylations proceed with excellent yield under standard conditions for enolate reactions, the intramolecular version was not a trivial issue.123N-protected glycine amides preferentially formed piperidinones via N-allylation, but the corresponding sarcosine derivatives provided γ-lactams in acceptable to good yields in dichloromethane, especially when the corresponding titanium enolates were used (Scheme 59).
 | ||
| Scheme 59 Intramolecular allylic alkylation of sarcosine amides. | ||
Allylic alkylations of nitriles
In analogy to esters and secondary amides, nitriles can also be converted into their corresponding enolates, which should be suitable for allylic alkylation. But so far, only one report by Grenning and Tunge et al. describes Pd-catalysed nitrile allylations using the deacylation protocol.45b In the presence of NaH, quaternary α-arylated cyanoacetone derivatives and cyanoacetates could be allylated using allyl alcohol as allylating agent (Scheme 60). It is proposed that the alcoholat formed initiates a retro Claisen condensation generating the nitrile enolate in situ.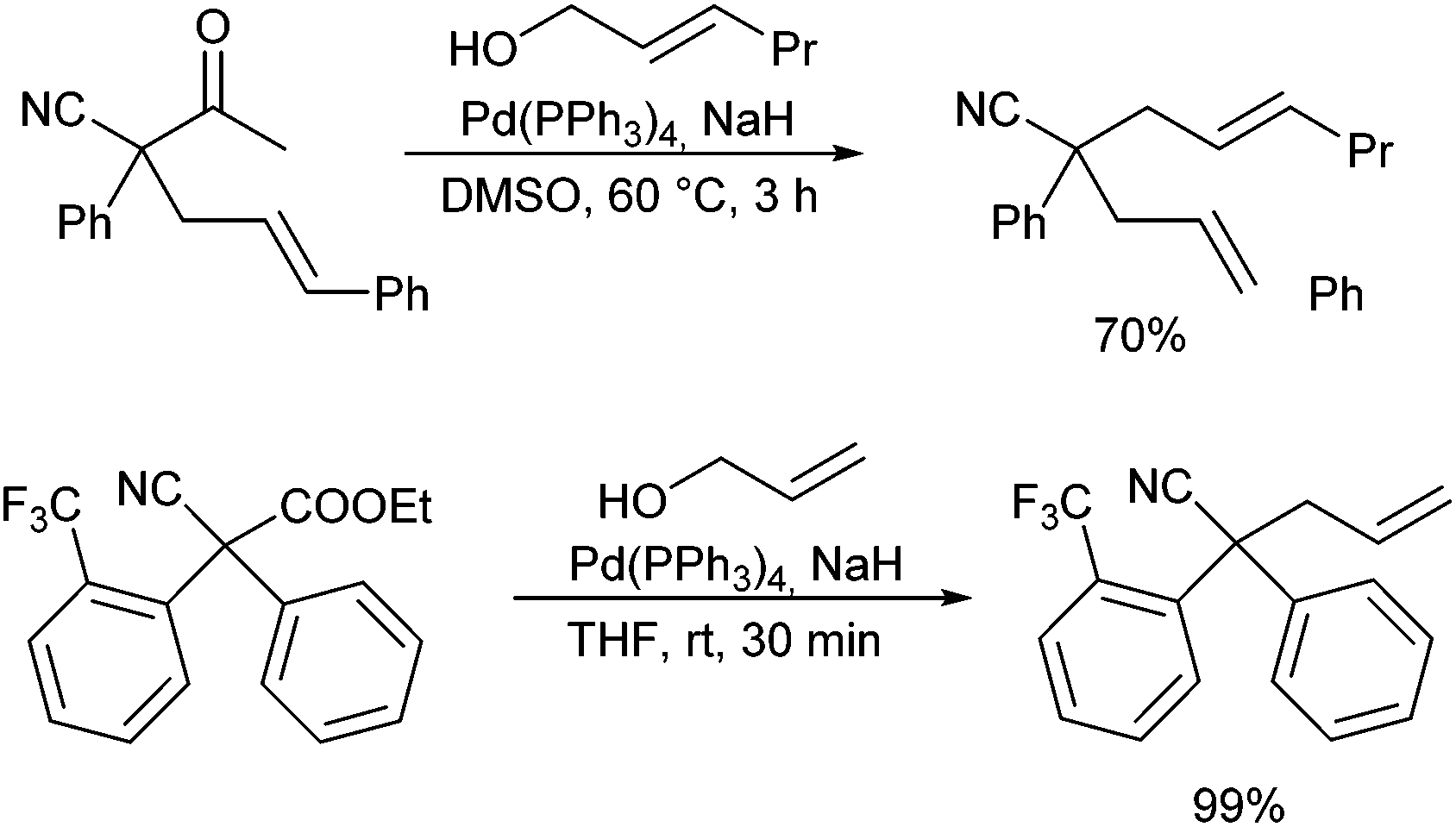 | ||
| Scheme 60 Deacylative allylation of nitriles. | ||
Evans and Oliver reported the Rh-catalysed allylic alkylation of O-silylated cyanohydrins derived from (substituted) benzaldehydes (Scheme 61).124 The deprotonated cyanohydrins function as acyl anion equivalents. Treatment of the allylation product with TBAF resulted in a cleavage of the protected cyanohydrin toward the corresponding ketone, which was stereoselectively allylated, because the allylation step proceeds with high regio- and stereoretention. The allyl carbonate could also be generated in situ in a one-pot protocol.
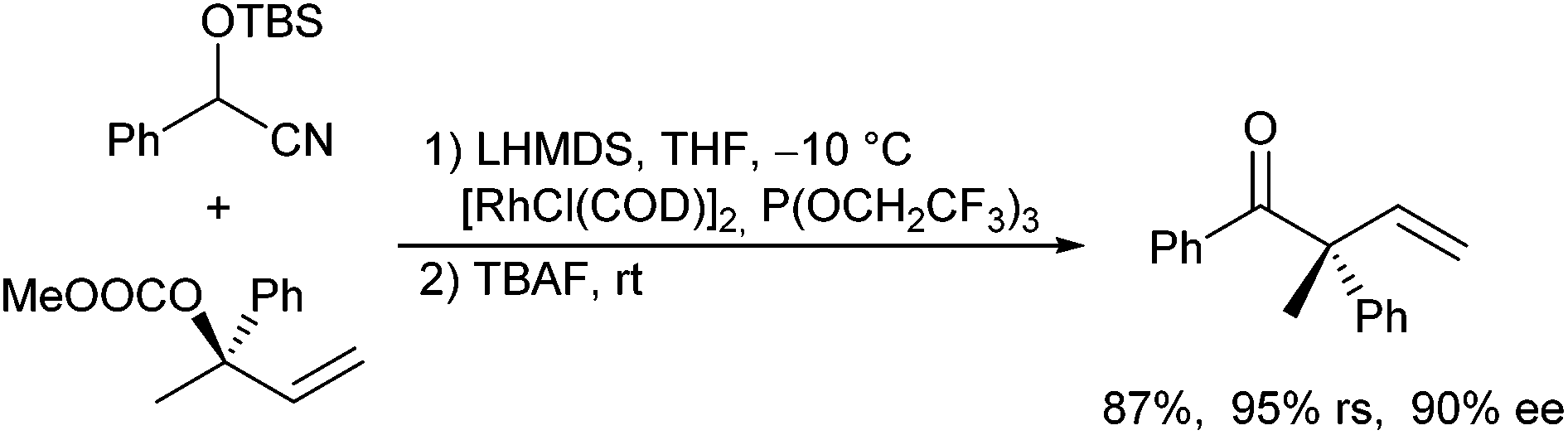 | ||
| Scheme 61 Rh-Catalysed allylic alkylation of O-silylated cyanohydrins. | ||
Conclusions
In conclusion, one can say that the transition metal-catalysed allylic alkylation of non-stabilized enolates made great progress in the last 10–15 years. Although sporadic examples were already reported in the 1980s, the development of new strategies, such as the decarboxylative allylations or the use of chelated enolates brought new impulses and enlarged the synthetic potential of this interesting reaction significantly. Also the use of other transition metals besides palladium opens up new perspectives.Acknowledgements
Financial support from the Deutsche Forschungsgemeinschaft was gratefully acknowledged. I also want to thank my excellent and very enthusiastic coworkers for their important contributions, and last but not least, I want to thank Prof. B. M. Trost for giving me the chance to work with him.References
- (a) B. M. Trost and M. L. Crawley, Chem. Rev., 2003, 103, 2921–2943 CrossRef CAS PubMed; (b) Z. Lu and S. Ma, Angew. Chem., 2008, 120, 264–303 ( Angew. Chem., Int. Ed. , 2008 , 47 , 258–297 ) CrossRef; (c) G. Helmchen, U. Kazmaier and S. Förster, in Catalytic Asymmetric Synthesis, ed. I. Ojima, Wiley-VCH, Weinheim, 2010, pp. 497–641 Search PubMed; (d) Topics in Organometallic Chemistry – Transition Metal Catalyzed Enantioselective Allylic Substitution in Organic Synthesis, ed. U. Kazmaier, Springer, Heidelberg, 2011 Search PubMed; (e) A. Bayer and U. Kazmaier, in Metal-Catalyzed Cross-Coupling Reactions and More, ed. A. De Meijere, S. Bräse and M. Oestreich, Wiley-VCH, Weinheim, 2014, pp. 926–994, and references cited therein Search PubMed.
- Reviews: (a) P. A. Evans and D. K. Leahy, Chemtracts, 2003, 16, 567–578 CAS; (b) B. L. Ashfeld, K. A. Miller, A. J. Smith, K. Tran and S. F. Martin, J. Org. Chem., 2007, 72, 9018–9031 CrossRef CAS PubMed; (c) G. Helmchen, A. Dahnz, P. Dübon, M. Schelwies and R. Weihofen, Chem. Commun., 2007, 675–691 RSC; (d) J. F. Hartwig and L. M. Stanley, Acc. Chem. Res., 2010, 43, 1461–1475 CrossRef CAS PubMed; (e) P. Tosatti, A. Nelson and S. P. Marsden, Org. Biomol. Chem., 2012, 10, 3147–3163 RSC; (f) B. M. Trost, Org. Process Res. Dev., 2012, 16, 185–194 CrossRef CAS PubMed; (g) C.-X. Zhuo, C. Zheng and S.-L. You, Acc. Chem. Res., 2014, 47, 2558–2573 CrossRef CAS PubMed.
- B. M. Trost and E. Keinan, Tetrahedron Lett., 1980, 21, 2591–2594 CrossRef CAS.
- J. C. Fiaud and J.-L. Malleron, J. Chem. Soc., Chem. Commun., 1981, 1159–1160 RSC.
- B. M. Trost and T. R. Verhoeven, J. Org. Chem., 1976, 41, 3215–3216 CrossRef CAS.
- G.-Q. Shi, X.-H. Huang and F.-J. Zhang, Tetrahedron Lett., 1995, 36, 6305–6308 CrossRef CAS.
- E.-I. Negishi and R. A. John, J. Org. Chem., 1983, 48, 4098–4102 CrossRef CAS.
- (a) L. V. Dunkerton and A. J. Sevino, J. Org. Chem., 1982, 47, 2812–2814 CrossRef CAS; (b) H. Matsushita and E.-I. Negishi, J. Chem. Soc., Chem. Commun., 1982, 160–161 RSC.
- W. Kinouchi, R. Saeki, H. Kawashima and Y. Kobayashi, Tetrahedron Lett., 2015, 56, 2265–2268 CrossRef CAS.
- F. Nahra, Y. Macé, D. Lambin and O. Riant, Angew. Chem., 2013, 125, 3290–3294 ( Angew. Chem., Int. Ed. , 2013 , 52 , 3208–3212 ) CrossRef.
- D. M. Johns, M. Mori and R. M. Williams, Org. Lett., 2006, 8, 4051–4054 CrossRef CAS PubMed.
- X. Liao, S. Huang, H. Zhou, D. Parrish and J. M. Cook, Org. Lett., 2007, 9, 1469–1471 CrossRef CAS PubMed.
- K. Huwig, K. Schultz and U. Kazmaier, Angew. Chem., 2015, 127, 9248–9251 ( Angew. Chem., Int. Ed. , 2015 , 54 , 9120–9123 ) CrossRef.
- B. M. Trost and G. M. Schroeder, J. Am. Chem. Soc., 1999, 121, 6759–6760 CrossRef CAS.
- B. M. Trost, R. Radinor and E.-M. Grenzer, J. Am. Chem. Soc., 1997, 119, 7879–7880 CrossRef CAS.
- (a) B. M. Trost and G. M. Schroeder, Chem. – Eur. J., 2005, 11, 174–184 CrossRef PubMed; (b) B. M. Trost, G. M. Schroeder and J. Kristensen, Angew. Chem., 2002, 114, 3642–3645 ( Angew. Chem., Int. Ed. , 2002 , 41 , 3492–3495 ) CrossRef.
- B. M. Trost, C. Pissot-Soldermann, I. Chen and G. M. Schroeder, J. Am. Chem. Soc., 2004, 126, 4480–4481 CrossRef CAS PubMed.
- S.-L. You, X.-L. Hou, L.-X. Dai and X.-Z. Zhu, Org. Lett., 2001, 3, 149–151 CrossRef CAS PubMed.
- X.-X. Yan, C.-G. Liang, Y. Zhang, W. Hong, B.-X. Cao, L.-X. Dai and X.-L. Hou, Angew. Chem., 2005, 117, 6702–6704 ( Angew. Chem., Int. Ed. , 2005 , 44 , 6544–6546 ) CrossRef.
- E. Belanger, K. Cantin, O. Messe, M. Tremblay and J.-F. Paquin, J. Am. Chem. Soc., 2007, 129, 1034–1035 CrossRef CAS PubMed.
- (a) M. Braun, F. Laicher and T. Meier, Angew. Chem., 2000, 112, 3637–3639 ( Angew. Chem., Int. Ed. , 2000 , 39 , 3494–3496 ) CrossRef; (b) M. Braun and T. Meier, Synlett, 2005, 2968–2972 CrossRef CAS.
- M. Braun, T. Meier, F. Laicher, P. Meletis and M. Fidan, Adv. Synth. Catal., 2008, 350, 303–314 CrossRef CAS.
- W.-H. Zheng, B.-H. Zheng, Y. Zhang and X.-L. Hou, J. Am. Chem. Soc., 2007, 129, 7718–7719 CrossRef CAS PubMed.
- J.-P. Chen, C.-H. Ding, W. Liu, X.-L. Hou and L.-X. Dai, J. Am. Chem. Soc., 2010, 132, 15493–15495 CrossRef CAS PubMed.
- B.-L. Lei, C.-H. Ding, X.-F. Yang, X.-L. Wan and X.-L. Hou, J. Am. Chem. Soc., 2009, 131, 18250–18251 CrossRef CAS PubMed.
- X.-H. Li, B.-H. Zheng, C.-H. Ding and X.-L. Hou, Org. Lett., 2013, 15, 6086–6089 CrossRef CAS PubMed.
- T. Graening and J. F. Hartwig, J. Am. Chem. Soc., 2005, 127, 17192–17193 CrossRef CAS PubMed.
- M. Chen and J. F. Hartwig, Angew. Chem., 2014, 126, 8835–8839 ( Angew. Chem., Int. Ed. , 2014 , 53 , 8691–8695 ) CrossRef.
- W. Chen, M. Chen and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 15825–15828 CrossRef CAS PubMed.
- P. A. Evans and D. K. Leahy, J. Am. Chem. Soc., 2003, 125, 8974–8975 CrossRef CAS PubMed.
- P. A. Evans and M. J. Lawler, J. Am. Chem. Soc., 2004, 126, 8642–8643 CrossRef CAS PubMed.
- P. A. Evans, E. A. Clizbe, M. J. Lawler and S. Oliver, Chem. Sci., 2012, 3, 1835–1838 RSC.
- N. Kanbayashi, A. Yamazawa, K. Takii, T. Okamura and K. Onitsuka, Adv. Synth. Catal., 2016, 358, 555–560 CrossRef CAS.
- (a) G. K. Jarugumilli and S. P. Cook, Org. Lett., 2011, 13, 1904–1907 CrossRef CAS PubMed; (b) G. K. Jarugumilli, C. Zhu and S. P. Cook, Eur. J. Org. Chem., 2012, 1712–1715 CrossRef CAS.
- R. Naasz, L. A. Arnold, M. Pineschi, E. Keller and B. L. Feringa, J. Am. Chem. Soc., 1999, 121, 1104–1105 CrossRef CAS.
- D. Liu, F. Xie and W. Zhang, Tetrahedron Lett., 2007, 48, 7591–7594 CrossRef CAS.
- X. Zhao, D. Liu, F. Xie, Y. Liu and W. Zhang, Org. Biomol. Chem., 2011, 9, 1871–1875 CAS.
- (a) S. Yasuda, N. Kumagai and M. Shibasaki, Heterocycles, 2012, 86, 745–757 CrossRef CAS; (b) X. Huo, G. Yang, D. Liu, Y. Liu, I. D. Gridnev and W. Zhang, Angew. Chem., 2014, 126, 6894–6898 ( Angew. Chem., Int. Ed. , 2014 , 53 , 6776–6780 ) CrossRef.
- I. Usui, S. Schmidt and B. Breit, Org. Lett., 2009, 11, 1453–1456 CrossRef CAS PubMed.
- D. J. Weix and J. F. Hartwig, J. Am. Chem. Soc., 2007, 129, 7720–7721 CrossRef CAS PubMed.
- (a) E. Bélanger, C. Houzé, N. Guimond, K. Cantin and J.-F. Paquin, Chem. Commun., 2008, 3251–3253 RSC; (b) B. M. Trost, J. Xu and T. Schmidt, J. Am. Chem. Soc., 2009, 131, 18343–18357 CrossRef CAS PubMed; (c) J. Fournier, O. Lozano, C. Menozzi, S. Arseniyadis and J. Cossy, Angew. Chem., 2013, 125, 1295–1299 ( Angew. Chem., Int. Ed. , 2013 , 52 , 1257–1261 ) CrossRef; (d) S. Arseniyadis, J. Fournier, S. Thangavelu, O. Lozano, S. Prevost, A. Archambeau, C. Menozzi and J. Cossy, Synlett, 2013, 2350–2364 CrossRef CAS.
- (a) T. Tsuda, Y. Chujo, S. Nishi, K. Tawara and T. Saegusa, J. Am. Chem. Soc., 1980, 102, 6381–6384 CrossRef CAS; (b) T. Tanaka, N. Okamura, K. Bannai, A. Hazato, S. Sugiura, K. Tomimori, K. Manabe and S. Kurozumi, Tetrahedron, 1986, 42, 6747–6758 CrossRef CAS; (c) C. J. Gartshore and D. W. Lupton, Angew. Chem., 2013, 125, 4207–4210 ( Angew. Chem., Int. Ed. , 2013 , 52 , 4113–4116 ) CrossRef; (d) Z. Li, S. Zhang, S. Wu, X. Shen, L. Zou, F. Wang, X. Li, F. Peng, H. Zhang and Z. Shao, Angew. Chem., 2013, 125, 4211–4214 ( Angew. Chem., Int. Ed. , 2013 , 52 , 4117–4121 ) CrossRef.
- (a) I. Shimizu, T. Yamada and J. Tsuji, Tetrahedron Lett., 1980, 21, 3199–3202 CrossRef CAS; (b) J. Tsuji, Pure Appl. Chem., 1982, 54, 197–206 CrossRef CAS; (c) J. Tsuji, I. Minami and I. Shimizu, Tetrahedron Lett., 1983, 24, 1793–1796 CrossRef CAS; (d) J. Tsuji, T. Yamada, I. Minami, M. Yuhara, M. Nisar and I. Shimizu, J. Org. Chem., 1987, 52, 2988–2995 CrossRef CAS; (e) J. Tsuji, Pure Appl. Chem., 1999, 71, 1539–1547 CrossRef CAS.
- (a) B. M. Trost and J. Xu, J. Am. Chem. Soc., 2005, 127, 2846–2847 CrossRef CAS PubMed; (b) B. M. Trost and J. Xu, J. Am. Chem. Soc., 2005, 127, 17180–17181 CrossRef CAS PubMed; (c) B. M. Trost, R. N. Bream and J. Xu, Angew. Chem., Int. Ed., 2006, 45, 3109–3112 CrossRef CAS PubMed; (d) B. M. Trost, J. Xu and M. Reichle, J. Am. Chem. Soc., 2007, 129, 292–283 CrossRef PubMed; (e) B. M. Trost, J. Xu and T. Schmidt, J. Am. Chem. Soc., 2008, 130, 11852–11853 CrossRef CAS PubMed; (f) B. M. Trost, J. Xu and T. Schmidt, J. Am. Chem. Soc., 2009, 131, 18343–18357 CrossRef CAS PubMed; (g) B. M. Trost, K. Lehr, D. J. Michaelis, J. Xu and A. K. Buckl, J. Am. Chem. Soc., 2010, 132, 8915–8917 CrossRef CAS PubMed; (h) B. M. Trost, B. Schäffner, M. Osipov and D. A. A. Wilton, Angew. Chem., 2011, 123, 3610–3613 ( Angew. Chem., Int. Ed. , 2011 , 50 , 3548–3551 ) CrossRef; (i) B. M. Trost, R. Koller and B. Schäffner, Angew. Chem., 2012, 124, 8415–8418 ( Angew. Chem., Int. Ed. , 2012 , 51 , 8290–8293 ) CrossRef; (j) B. M. Trost, D. J. Michaelis, J. Charpentier and J. Xu, Angew. Chem., 2012, 124, 208–212 ( Angew. Chem., Int. Ed. , 2012 , 51 , 204–208 ) CrossRef.
- (a) E. C. Burger and J. A. Tunge, Org. Lett., 2004, 6, 4113–4115 CrossRef CAS PubMed; (b) A. J. Grenning and J. A. Tunge, J. Am. Chem. Soc., 2011, 133, 14785–14794 CrossRef CAS PubMed.
- (a) N. H. Sherden, D. C. Behenna, S. C. Virgil and B. M. Stoltz, Angew. Chem., 2009, 121, 6972–6975 ( Angew. Chem., Int. Ed. , 2009 , 48 , 6840–6843 ) CrossRef; (b) J. Streuff, E. White, S. C. Virgil and B. M. Stoltz, Nat. Chem., 2010, 192–196 CrossRef CAS PubMed; (c) J. J. Day, R. M. McFadden, S. C. Virgil, H. Kolding, J. L. Alleva and B. M. Stoltz, Angew. Chem., Int. Ed., 2011, 50, 6814–6818 CrossRef CAS PubMed; (d) C. M. Reeves, C. Eidamshaus, J. Kim and B. M. Stoltz, Angew. Chem., 2013, 125, 6850–6853 ( Angew. Chem., Int. Ed. , 2013 , 52 , 6718–6721 ) CrossRef.
- Reviews: (a) J. A. Tunge and E. C. Burger, Eur. J. Org. Chem., 2005, 1715–1726 CrossRef CAS; (b) J. T. Mohr and B. M. Stoltz, Chem. – Asian J., 2007, 2, 1476–1491 CrossRef CAS PubMed; (c) A. Y. Hong and B. M. Stoltz, Eur. J. Org. Chem., 2013, 2745–2759 CrossRef CAS PubMed and references cited therein.
- (a) J. C. Fiaud and L. Aribi-Zouioueche, Tetrahedron Lett., 1982, 23, 5279–5282 CrossRef CAS; (b) J.-E. Bäckvall, R. E. Nordbert and J. Vågberg, Tetrahedron Lett., 1983, 24, 411–412 CrossRef.
- Y. Liu, S.-J. Han, W.-B. Liu and B. M. Stoltz, Acc. Chem. Res., 2015, 48, 740–751 CrossRef CAS PubMed and references cited therein.
- (a) J. A. Keith, D. C. Behenna, J. T. Mohr, S. Ma, S. C. Marinescu, J. Oxgaard, B. M. Stoltz and W. A. Goddard III, J. Am. Chem. Soc., 2007, 129, 11876–11877 CrossRef CAS PubMed; (b) D. C. Behenna, J. T. Mohr, N. H. Sherden, S. C. Marinescu, A. M. Harned, K. Tani, M. Seto, S. Ma, Z. Novák, M. R. Krout, R. M. McFadden, J. L. Roizen, J. A. Enquist Jr., D. E. White, S. R. Levine, K. V. Petrova, A. Iwashita, S. C. Virgil and B. M. Stoltz, Chem. – Eur. J., 2011, 17, 14199–14223 CrossRef CAS PubMed.
- S. R. Schulz and S. Blechert, Angew. Chem., Int. Ed., 2007, 46, 3966–3970 CrossRef CAS PubMed.
- P. de Mayo, H. Takeshita and A. B. M. Sattar, in Proceedings of the Chemical Society, The Royal Society of Chemistry, 1962, p. 119 Search PubMed.
- I. Ibrahem and A. Córdova, Angew. Chem., 2006, 118, 1986–1990 ( Angew. Chem., Int. Ed. , 2006 , 45 , 1952–1956 ) CrossRef.
- S. Afewerki, I. Ibrahem, J. Rydfjord, P. Breistein and A. Córdova, Chem. – Eur. J., 2012, 18, 2972–2977 CrossRef CAS PubMed.
- A. M. Yoshida, E. Masaki, T. Terumine and S. Hara, Synthesis, 2014, 1367–1373 CrossRef.
- X. Zhao, D. Liu, F. Xie, Y. Liu and W. Zhang, Org. Biomol. Chem., 2011, 9, 1871–1875 CAS.
- M. Kimura, Y. Horino, R. Mukai, S. Tanaka and Y. Tamaru, J. Am. Chem. Soc., 2001, 123, 10401–10402 CrossRef CAS PubMed.
- G. Jiang and B. List, Adv. Synth. Catal., 2011, 353, 1667–1670 CrossRef CAS.
- S. Mukherjee and B. List, J. Am. Chem. Soc., 2007, 129, 11336–11337 CrossRef CAS PubMed.
- B. M. Trost, J. Xu and M. Reichle, J. Am. Chem. Soc., 2007, 129, 282–283 CrossRef CAS PubMed.
- (a) L. S. Hegedus, W. H. Darlington and C. E. Russell, J. Org. Chem., 1980, 45, 5193–5196 CrossRef CAS; (b) L. S. Hegedus, R. E. Williams, M. A. McGuire and T. Hayashi, J. Am. Chem. Soc., 1980, 102, 4973–4979 CrossRef CAS.
- (a) S. Murahashi, M. Yamamura and N. Mita, J. Org. Chem., 1977, 42, 2870–2874 CrossRef CAS; (b) S. Murahashi, Y. Tamba, M. Yamamura and N. Yoshimura, J. Org. Chem., 1978, 43, 4099–4106 CrossRef CAS.
- H. M. R. Hoffmann, A. R. Otte and A. Wilde, Angew. Chem., 1992, 104, 224–225 ( Angew. Chem., Int. Ed. , 1992 , 31 , 234–236 ) CrossRef CAS.
- (a) M. Rychlet Elliott, A.-L. Dhimane and M. Malacria, J. Am. Chem. Soc., 1997, 119, 3427–3428 CrossRef; (b) M. R. Elliott, A.-L. Dhimane and M. Malacria, Tetrahedron Lett., 1998, 39, 8849–8852 CrossRef CAS.
- C. Carfagna, L. Mariani, A. Musco, G. Sallese and R. Santi, J. Org. Chem., 1991, 56, 3924–3927 CrossRef CAS.
- M. Kawatsura, D. Ikeda, Y. Komatsu, K. Mitani, T. Tanaka and J. Uenishi, Tetrahedron, 2007, 63, 8815–8824 CrossRef CAS.
- P. Meletis, M. Patil, W. Thiel, W. Frank and M. Braun, Chem. – Eur. J., 2011, 17, 11243–11249 CrossRef CAS PubMed.
- M. Patil and W. Thiel, Chem. – Eur. J., 2012, 18, 10408–10418 CrossRef CAS PubMed.
- J. Tsuji, T. Yamada, I. Minami, M. Yuhara, M. Nisar and I. Shimizu, J. Org. Chem., 1987, 52, 2988–2995 CrossRef CAS.
- D. Imao, A. Itoi, A. Yamazaki, M. Shirakura, R. Ohtoshi, K. Ogata, Y. Ohmori, T. Ohta and Y. Ito, J. Org. Chem., 2007, 72, 1652–1658 CrossRef CAS PubMed.
- U. Kazmaier and F. L. Zumpe, Angew. Chem., 1999, 111, 1572–1574 ( Angew. Chem., Int. Ed., Engl. , 1999 , 38 , 1468–1470 ) CrossRef.
- (a) U. Kazmaier, Liebigs Ann./Recl., 1997, 285–295 CrossRef CAS; (b) U. Kazmaier, in Bioorganic Chemistry, Wiley-VCH, Weinheim, 1999, pp. 201–206 Search PubMed; (c) U. Kazmaier, in Frontiers in Asymmetric Synthesis and Application of alpha-Amino Acids, ed. V. A. Soloshonok and K. Izawa, ACS Books, Washington, 2009, pp. 157–176 Search PubMed.
- (a) R. Grandel and U. Kazmaier, Tetrahedron Lett., 1997, 38, 8009–8012 CrossRef CAS; (b) R. Grandel, U. Kazmaier and F. Rominger, J. Org. Chem., 1998, 63, 4524–4528 CrossRef CAS.
- (a) M. Pohlman and U. Kazmaier, Org. Lett., 2003, 5, 2631–2633 CrossRef CAS PubMed; (b) B. Mendler and U. Kazmaier, Org. Lett., 2005, 7, 1715–1718 CrossRef CAS PubMed; (c) B. Mendler, U. Kazmaier, V. Huch and M. Veith, Org. Lett., 2005, 7, 2643–2646 CrossRef CAS PubMed.
- (a) U. Kazmaier and S. Maier, Tetrahedron, 1996, 52, 941–954 CrossRef CAS; (b) U. Kazmaier, J. Org. Chem., 1996, 61, 3694–3699 CrossRef CAS PubMed; (c) U. Kazmaier and C. Schneider, Tetrahedron Lett., 1998, 39, 817–818 CrossRef CAS.
- (a) U. Kazmaier, S. Maier and F. L. Zumpe, Synlett, 2000, 1523–1535 CAS; (b) U. Kazmaier, Curr. Org. Chem., 2003, 317–328 CrossRef CAS; (c) M. Bauer and U. Kazmaier, Recent Res. Dev. Org. Chem., 2005, 9, 49–69 CrossRef CAS.
- U. Kazmaier, A. Bayer and J. Deska, Synthesis, 2013, 1462–1468 CrossRef CAS.
- J. Gorges, A. Ullrich and U. Kazmaier, Eur. J. Org. Chem., 2013, 4372–4378 CrossRef CAS.
- L. Schmidt, T. Doroshenko, P. Barbie, A. Grueter, G. Jung and U. Kazmaier, Synthesis, 2016 DOI:10.1055/s-0035-1561628.
- U. Kazmaier, D. Schauß, S. Raddatz and M. Pohlman, Chem. – Eur. J., 2001, 7, 456–464 CrossRef CAS.
- S. Thies and U. Kazmaier, Synlett, 2010, 137–141 CAS.
- A. Kiefer, D. Gawas and U. Kazmaier, Eur. J. Org. Chem., 2015, 5810–5816 CrossRef CAS.
- T. D. Weiss, G. Helmchen and U. Kazmaier, Chem. Commun., 2002, 1270–1271 RSC.
- Review: G. Helmchen, J. Organomet. Chem., 1999, 576, 203–214 CrossRef CAS.
- B. Goldfuss and U. Kazmaier, Tetrahedron, 2000, 56, 6493–6496 CrossRef CAS.
- G. Consiglio and R. M. Waymouth, Chem. Rev., 1989, 89, 257–276 CrossRef CAS.
- P. Corradini, G. Maglio, A. Musco and G. Paiaro, J. Chem. Soc., Chem. Commun., 1966, 618–619 RSC.
- (a) U. Kazmaier and F. L. Zumpe, Angew. Chem., 2000, 112, 805–807 ( Angew. Chem., Int. Ed., Engl. , 2000 , 39 , 802–804 ) CrossRef; (b) U. Kazmaier and F. L. Zumpe, Eur. J. Org. Chem., 2001, 4067–4076 CrossRef CAS.
- K. Krämer and U. Kazmaier, J. Org. Chem., 2006, 71, 8950–8953 CrossRef PubMed.
- U. Kazmaier, D. Stolz, K. Krämer and F. Zumpe, Chem. – Eur. J., 2008, 14, 1322–1329 CrossRef CAS PubMed.
- (a) B. Åkermark, S. Hansson and A. Vitagliano, J. Am. Chem. Soc., 1990, 112, 4587–4588 CrossRef; (b) M. Sjögren, S. Hansson, P.-O. Norrby, B. Åkermark, M. E. Cucciolito and A. Vitagliano, Organometallics, 1992, 11, 3954–3964 CrossRef.
- U. Kazmaier and M. Pohlman, Synlett, 2004, 623–626 CrossRef CAS.
- M. Pohlman, U. Kazmaier and T. Lindner, J. Org. Chem., 2004, 69, 6909–6912 CrossRef CAS PubMed.
- (a) U. Kazmaier and T. Lindner, Angew. Chem., 2005, 117, 3368–3371 ( Angew. Chem., Int. Ed. , 2005 , 44 , 3303–3306 ) CrossRef; (b) T. Lindner and U. Kazmaier, Adv. Synth. Catal., 2005, 1687–1695 CrossRef CAS.
- D. Gawas and U. Kazmaier, Org. Biomol. Chem., 2010, 8, 457–462 CAS.
- B. Breit, P. Demel and C. Studte, Angew. Chem., 2004, 116, 3874–3877 ( Angew. Chem., Int. Ed. , 2004 , 43 , 3786–3789 ) CrossRef.
- (a) U. Kazmaier, J. Deska and A. Watzke, Angew. Chem., 2006, 118, 4973–4976 ( Angew. Chem., Int. Ed. , 2006 , 45 , 4855–4858 ) CrossRef; (b) J. Deska and U. Kazmaier, Chem. – Eur. J., 2007, 13, 6204–6211 CrossRef CAS PubMed; (c) J. Deska and U. Kazmaier, Curr. Org. Chem., 2008, 12, 355–385 CrossRef CAS.
- (a) U. Kazmaier, D. Schauß and M. Pohlman, Org. Lett., 1999, 1, 1017–1019 CrossRef CAS; (b) U. Kazmaier, M. Pohlman and D. Schauß, Eur. J. Org. Chem., 2000, 2761–2766 CrossRef CAS.
- J. Deska and U. Kazmaier, Angew. Chem., 2007, 119, 4654–4657 ( Angew. Chem., Int. Ed. , 2007 , 46 , 4570–4573 ) CrossRef.
- (a) G. T. Crisp and P. T. Glink, Tetrahedron Lett., 1992, 33, 4649–4652 CrossRef CAS; (b) G. T. Crisp and P. T. Glink, Tetrahedron, 1994, 50, 3213–3234 CrossRef CAS.
- (a) L. Junk and U. Kazmaier, Org. Biomol. Chem., 2016, 14, 2916–2923 RSC; (b) L. Junk and U. Kazmaier, Synlett, 2016, 1531–1536 CAS.
- (a) U. Kazmaier and D. Stolz, Angew. Chem., 2006, 118, 3143–3146 ( Angew. Chem., Int. Ed. , 2006 , 45 , 3072–3075 ) CrossRef; (b) D. Stolz and U. Kazmaier, Synthesis, 2008, 2288–2292 CAS.
- I. Minami, I. Shimizu and J. Tsuji, J. Organomet. Chem., 1985, 296, 269–280 CrossRef CAS.
- (a) P. A. Evans and J. D. Nelson, Tetrahedron Lett., 1998, 39, 1725–1728 CrossRef CAS; (b) P. A. Evans and L. J. Kennedy, J. Am. Chem. Soc., 2001, 123, 1234–1235 CrossRef CAS PubMed.
- S. Hähn and U. Kazmaier, Eur. J. Org. Chem., 2011, 4931–4939 Search PubMed.
- (a) A. Bayer and U. Kazmaier, Org. Lett., 2010, 12, 4960–4963 CrossRef CAS PubMed; (b) A. Bayer and U. Kazmaier, Chem. – Eur. J., 2014, 20, 10484–10491 CrossRef CAS PubMed.
- (a) S. Basak and U. Kazmaier, Org. Lett., 2008, 10, 501–504 CrossRef CAS PubMed; (b) S. Basak and U. Kazmaier, Eur. J. Org. Chem., 2008, 4169–4177 CrossRef.
- M. Kawatsura, F. Ata, S. Hayase and T. Itoh, Chem. Commun., 2007, 4283–4285 RSC.
- P. Servatius and U. Kazmaier, Synlett, 2015, 2001–2005 CAS.
- B. M. Trost, P. L. Fraisse and Z. T. Ball, Angew. Chem., 2002, 114, 1101–1103 ( Angew. Chem., Int. Ed. , 2002 , 41 , 1059–1061 ) CrossRef.
- (a) C. Bruneau, J. L. Renaud and B. Demerseman, Chem. – Eur. J., 2006, 12, 5178–5187 CrossRef CAS PubMed; (b) C. Bruneau, J.-L. Renaud and B. Demerseman, Pure Appl. Chem., 2008, 80, 861–871 CrossRef CAS; (c) H.-J. Zhang, B. Demerseman, L. Toupet, Z. Xi and C. Bruneau, Adv. Synth. Catal., 2008, 350, 1601–1609 CrossRef CAS.
- A. Bayer and U. Kazmaier, J. Org. Chem., 2014, 79, 8498–8504 CrossRef CAS PubMed.
- M. Braun, P. Meletis and R. Visse, Adv. Synth. Catal., 2011, 353, 3380–3384 CrossRef CAS.
- D. Seebach, Angew. Chem., 1988, 100, 1685–1715 ( Angew. Chem., Int. Ed., Engl. , 1988 , 27 , 1624–1654 ) CrossRef CAS.
- B. M. Trost and M. U. Frederiksen, Angew. Chem., 2005, 117, 312–314 ( Angew. Chem., Int. Ed. , 2005 , 44 , 308–310 ) CrossRef.
- K. Zhang, Q. Peng, X.-L. Hou and Y. D. Wu, Angew. Chem., 2008, 120, 1765–1768 ( Angew. Chem., Int. Ed., Engl. , 2008 , 47 , 1741–1744 ) CrossRef.
- B. M. Trost, D. J. Michaelis, J. Carpentier and J. Xu, Angew. Chem., 2012, 124, 208–212 ( Angew. Chem., Int. Ed. , 2012 , 51 , 204–208 ) CrossRef.
- (a) N. B. Bennett, D. C. Duquette, J. Kim, W.-B. Liu, A. N. Marziale, D. C. Behenna, S. C. Virgil and B. M. Stoltz, Chem. – Eur. J., 2013, 19, 4414–4418 CrossRef CAS PubMed; (b) A. N. Marziale, D. C. Duquette, R. A. Craig II, K.-E. Kim, M. Liniger, Y. Numajiri and B. M. Stoltz, Adv. Synth. Catal., 2015, 357, 2238–2245 CrossRef CAS PubMed.
- (a) Y. Numajiri, G. Jiménez-Osés, B. Wang, K. W. Houk and B. M. Stoltz, Org. Lett., 2015, 17, 1082–1085 CrossRef CAS PubMed; (b) Y. Kita, Y. Numajiri, N. Okamoto and B. M. Stoltz, Tetrahedron, 2015, 71, 6349–6353 CrossRef CAS PubMed.
- S. Datta and U. Kazmaier, Org. Biomol. Chem., 2011, 9, 872–880 CAS.
- S. Datta, A. Bayer and U. Kazmaier, Org. Biomol. Chem., 2012, 10, 8268–8275 CAS.
- A. Bayer and U. Kazmaier, J. Org. Chem., 2014, 79, 8491–8497 CrossRef CAS PubMed.
- S. Thies and U. Kazmaier, Eur. J. Org. Chem., 2014, 1695–1707 CrossRef CAS.
- P. A. Evans and S. Oliver, Org. Lett., 2013, 15, 5626–5629 CrossRef CAS PubMed.
Footnote |
| † Dedicated to Prof. B. M. Trost on the occasion of his 75th birthday. |
| This journal is © the Partner Organisations 2016 |