
Low-valent cobalt-catalyzed C–H allylation†
Xuefeng
Cong‡
a,
Shengxian
Zhai‡
b and
Xiaoming
Zeng
*a
aCenter for Organic Chemistry, Frontier Institute of Science and Technology, Xi'an Jiaotong University, Xi'an 710054, P. R. China. E-mail: zengxiaoming@mail.xjtu.edu.cn
bThe State Key Laboratory of Applied Organic Chemistry, College of Chemistry and Chemical Engineering, Lanzhou University, Lanzhou, Gansu 730000, P. R. China
First published on 18th March 2016
Abstract
An efficient allylation of C–H bonds promoted by a simple cobalt salt combined with a Grignard reagent has been developed. This reaction provides an alternative protocol to the build-up of fundamentally interesting allylated aromatics at room temperature. Mechanistically different from the Cp*Co(III)-promoted transformation, a low-valent cobalt complex that was formed in situ by the reduction with a Grignard reagent can be considered as the catalytically active species, allowing the C–H activation to proceed effectively followed by straightforward coupling with an allylic electrophile to give the product.
 Xiaoming Zeng | Xiaoming Zeng received his B.Sc. degree at Sichuan Normal University, 2002, and his Ph.D. degree at Sichuan University, 2009 (with Professor Meiming Luo). He spent two years as a joint Ph.D. student in Professor Guy Bertrand's laboratory at the University of California at Riverside, funded by the China Scholarship Council (2007–2009). As a JSPS postdoctoral fellow, he pursued synthetic chemistry with Professor Eiichi Nakamura at the University of Tokyo. He started his independent research group at Xi'an Jiaotong University as a Principal Investigator in January 2012. His research interests focus on developing new carbenes and their application in transition metal catalysis. |
Transition metal-catalyzed C–H functionalization is one of the most powerful and useful tools in modern organic chemistry.1 Recent progression in the area typically focuses on developing cost-effective strategies with earth-abundant, inexpensive first-row metal catalysts.2 Among these, the use of group 9 metal cobalt in uncovering new synthetic procedures has attracted much attention of chemists recently.3 Ascribing to their diverse transformations of allyl groups by numerous classic methods,4 the exploration of efficient methods to introduce allyl scaffolds via C–H functionalization has attracted broad interest,5 which allows the straightforward synthesis of allyl-bearing structural motifs in a step-economic manner. Such an approach was initially achieved by the use of ruthenium catalysts.6 More recently, Ackermann, Matsunaga and Kanai demonstrated that a complex of Cp*Co(III) demonstrates high reactivity in the promotion of allylation of C–H bonds with the assistance of a 2-pyrimidyl (pym) directing group (Scheme 1a).7 Glorius remarkably expanded the scope with diverse directing group-containing substrates.8 A Cp*Co(III)-promoted cleavage of the C–H bond by a mechanism of concerted metalation–deprotonation (CMD) was thought to be involved in these transformations as a key elementary step.7a,8b,9
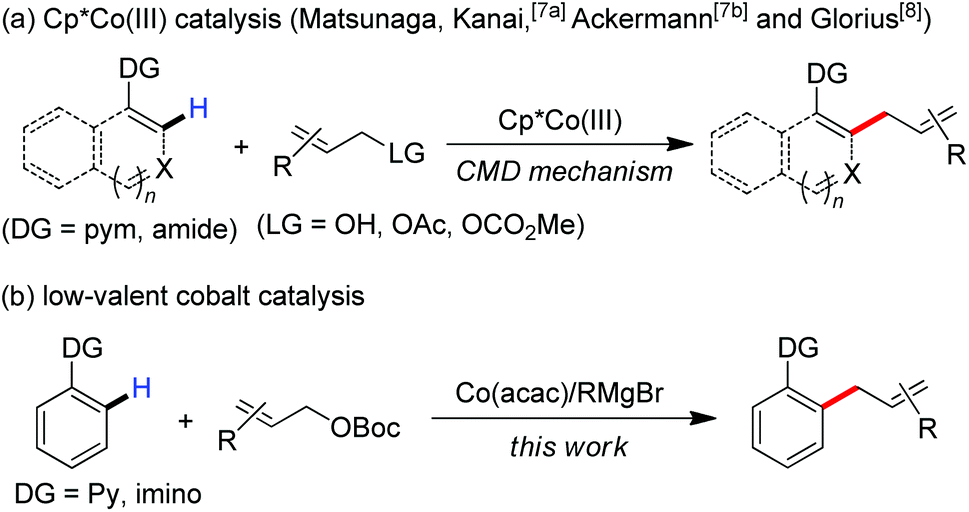 | ||
| Scheme 1 Cobalt-catalyzed C–H allylation reactions. | ||
Inspired by recent advances in low-valent cobalt-catalyzed C–H functionalization,10,11 we hypothesized that a low-valent complex, which can be generated in situ by reducing the cobalt(III) salt with a Grignard reagent, may enable the cleavage of inert C–H bonds with the assistance of a chelation auxiliary,12 and then followed by treating with an allyl electrophile to provide access to the allylated derivatives. With our effort to disclose the unique ability of low-valent metal catalysts in the promotion of the direct functionalization of inert chemical bonds,13 herein, we report that a combination of a simple cobalt(III) salt and a Grignard reagent allows the directed allylation of C–H bonds to proceed effectively at room temperature, leading to the formation of linear allylated aromatics in high regioselectivity (Scheme 1b).
We started our experiment by studying the effect of the leaving group (LG) of allyl electrophiles on the C–H allylation (Table 1). With the assistance of pyridyl auxiliary, the reaction of C–H bonds with cinnamyl alcohol did not proceed with a mixed Co(acac)3 and TMSCH2MgCl (entry 1). Variation of the allylic partner to 3-methoxy-1-phenylpropene allowed the formation of the linear compound 3a in 23% yield (entry 2). To our delight, the use of tert-butyl-substituted cinnamyl carbonate largely improves the transformation to produce 3a in 70% yield (entry 5). Other allyl electrophiles including cinnamyl acetate and diethyl phosphate resulted in low conversions (entries 3 and 4). Cobalt(II) salts such as Co(acac)2, CoCl2, CoBr2 and Co(OAc)2 gave an inferior performance in the allylation (entries 6–9), while replacement of TMSCH2MgCl by a phenyl or methyl Grignard reagent led to a low yield of 3a (entries 10 and 11). Organomagnesium bearing an isopropyl- or tert-butyl group cannot promote the reaction, indicating an important role of a Grignard reagent in generating a catalytically active cobalt species (entries 12 and 13). Note that increasing the amount of allyl carbonate slightly improves the conversion, leading to 3a in 86% yield combined with the formation of a diallylated compound in less than 10% yield via sequential allylation of two ortho-C–H bonds (entry 16).
|
|
||||
|---|---|---|---|---|
| Entry | Cobalt salt | LG (leaving group) | RMgX | Yieldb (3a) |
| a Reaction conditions: 1a (0.2 mmol), 2 (0.3 mmol), cobalt salt (0.02 mmol), RMgX (0.4 mmol), 25 °C, 12 h. b Isolated yield. c The yield was determined by GC analysis. d Not detected by GC-MS and 1H NMR analyses. e 2 (0.4 mmol) was employed. | ||||
| 1 | Co(acac)3 | OH | TMSCH2MgCl | nd |
| 2 | Co(acac)3 | OMe | TMSCH2MgCl | 23% |
| 3 | Co(acac)3 | OAc | TMSCH2MgCl | 67% |
| 4 | Co(acac)3 | OP(OEt)2 | TMSCH2MgCl | 30% |
| 5 | Co(acac)3 | OBoc | TMSCH2MgCl | 70% |
| 6 | Co(acac)2 | OBoc | TMSCH2MgCl | 62% |
| 7 | CoCl2 | OBoc | TMSCH2MgCl | 56% |
| 8 | CoBr2 | OBoc | TMSCH2MgCl | 50% |
| 9 | Co(OAc)2 | OBoc | TMSCH2MgCl | 9% |
| 10 | Co(acac)3 | OBoc | PhMgBr | <10%c |
| 11 | Co(acac)3 | OBoc | MeMgCl | <10%c |
| 12 | Co(acac)3 | OBoc | iPrMgCl | ndd |
| 13 | Co(acac)3 | OBoc | n BuMgBr | ndd |
| 14 | Co(acac)3 | OBoc | t BuMgBr | Tracec |
| 15 | Co(acac)3 | OBoc | CyMgBr | Tracec |
| 16e | Co(acac)3 | OBoc | TMSCH2MgCl | 86% |
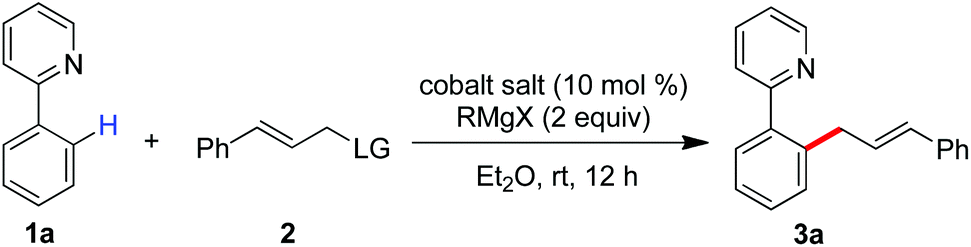
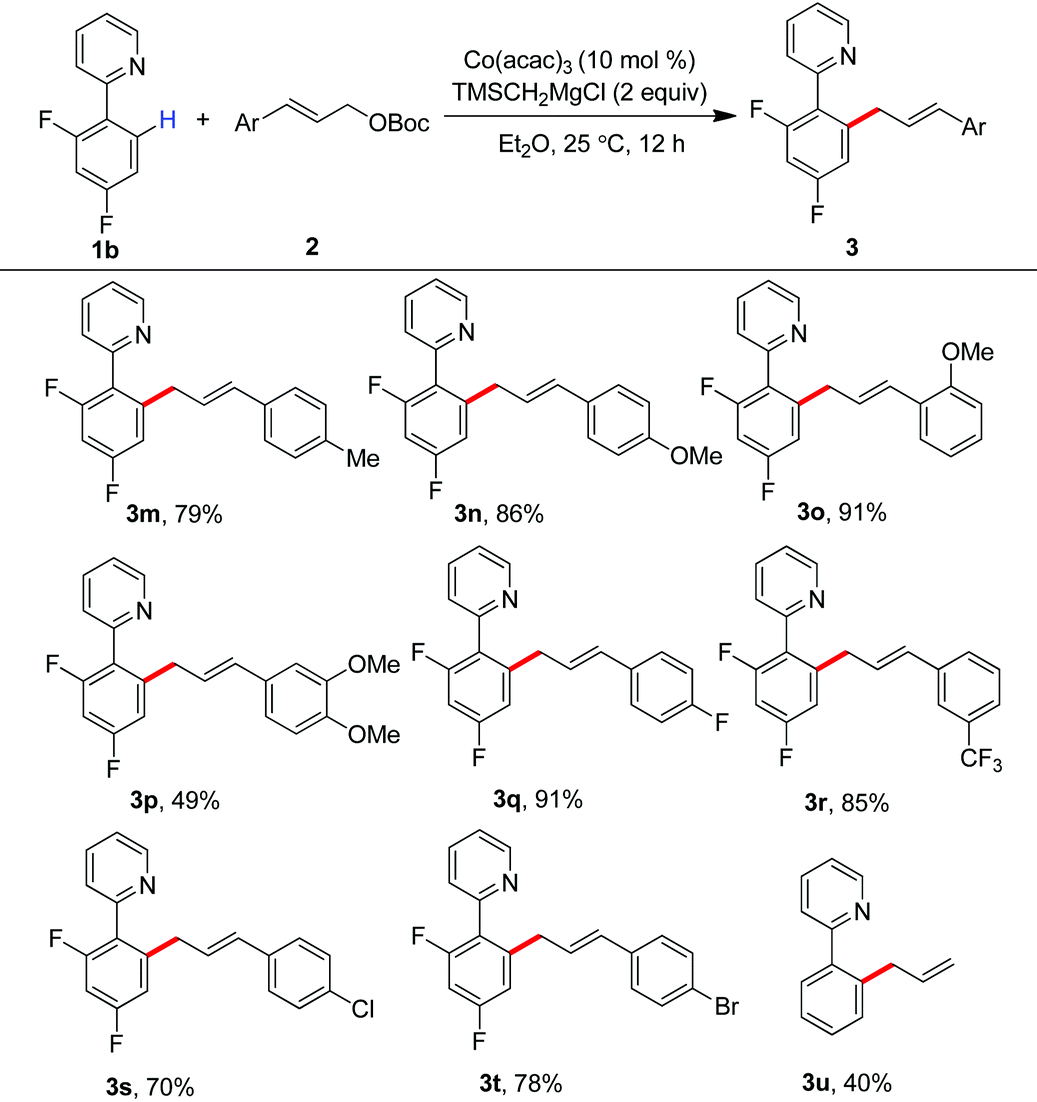
Having the optimal reaction conditions, we next examined the substrate scope. As shown in Table 2, the incorporation of a methyl group at the para and ortho positions of the aromatic ring has no influence on the allylation, giving the desired products in good yields (3b and 3c). Interestingly, with the introduction of the methyl group at the meta site of the aromatic ring, the formation of mixed regioisomeric products 3d and 3d′ was observed via cleavage of two different ortho-C–H bonds. Probably due to the secondary directing effect, the allylation preferably occurred at a position of steric hindrance adjacent to the methoxy group (3f).11c,g,i,14 An analogous result was obtained by the replacement with a fluoro substituent (3h). Note that a chloride substituent can be well compatible with the catalytic system (3k). Altering the directing group to 2-methyl-pyridyl has no effect on the conversion (3l). In particular, the pmp-substituted imino group was proved to be an effective auxiliary, allowing the reaction to occur effectively at room temperature (Scheme 2). The related ortho-allylated aromatic ketones were accessible in good yields (5a–5d). The formation of 2,3-disubstituted products was observed using meta-methoxy- or fluoride-containing aryl imines (5e and 5f).
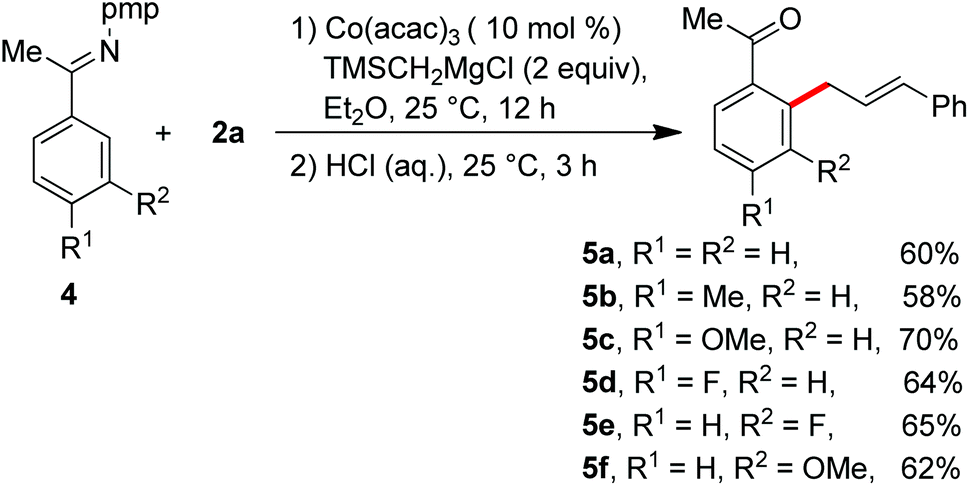 | ||
| Scheme 2 Imino-assisted allylation of C–H bonds for the preparation of ortho-allylated aromatic ketones. | ||
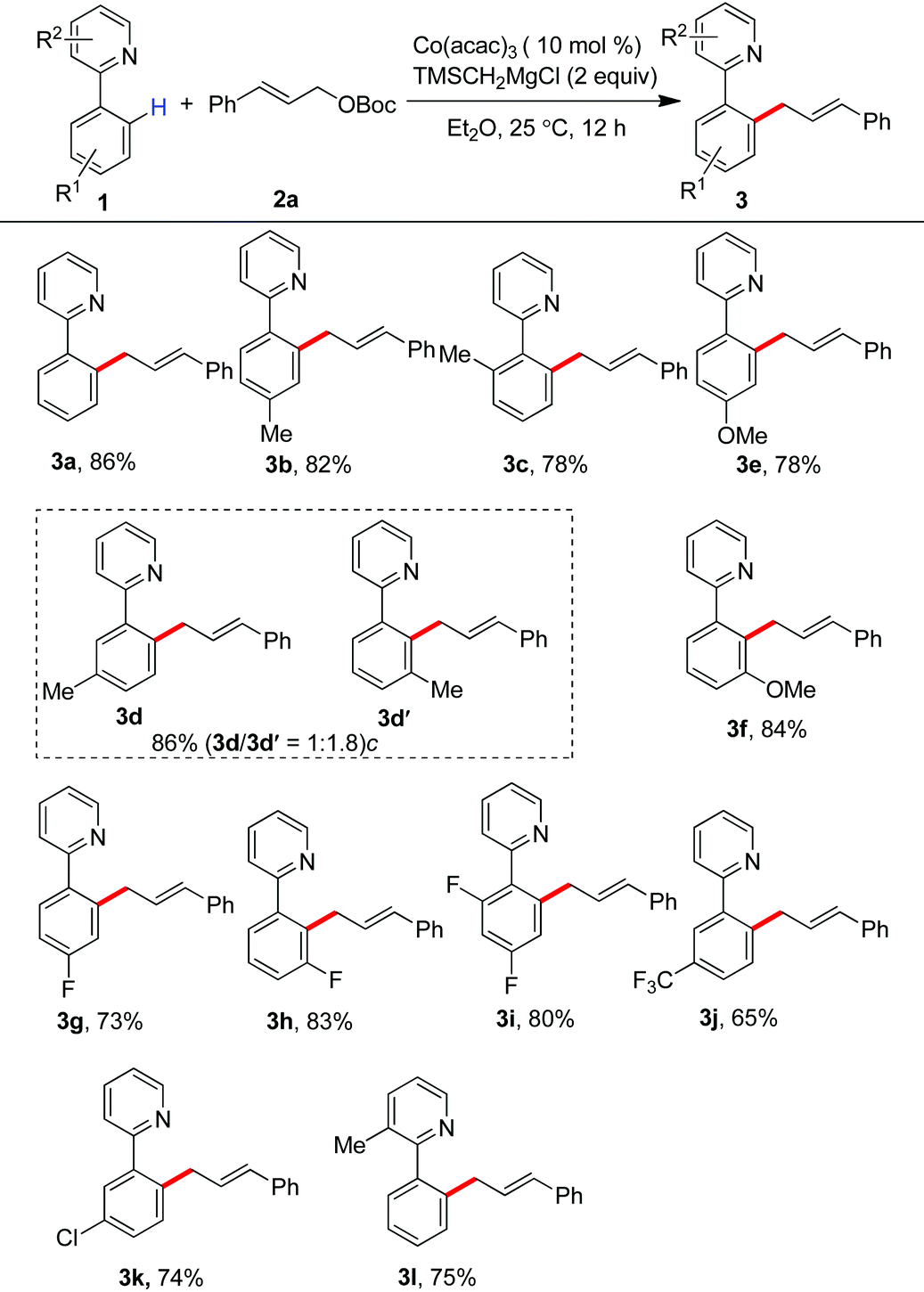
Encouraged by these results, we then investigated the scope of allyl partners by treating them with 2-(2,4-difluorophenyl)pyridine (Table 3). The installation of methyl, methoxy, fluoride and trifluoromethyl groups onto the phenyl scaffold of cinnamyl carbonates did not impact on the allylation. The production of the products 3m–3r in good to excellent yields was observed in these cases. Particularly interesting was that synthetically useful functionalities such as chloride and bromide can be well retained in the C–H allylation, providing an entry for late-stage functionalization (3s and 3t). It was found that the reaction with parent allyl carbonate also took place smoothly (3u).
After the realization of the C–H allylation with a simple salt of cobalt(III) acetylacetonate and a Grignard reagent, a preliminary mechanistic study for understanding the reaction pathway was next carried out. Starting from Z-cinnamyl carbonate, the reaction gave rise to a single E-selective product 3a in 89% yield (Scheme 3a). This indicates that the formation of a π-(allyl)cobalt intermediate can be considered in the catalytic cycle. Meanwhile, the same product was formed in complete γ-regioselectivity by the use of branched tert-butyl(1-phenylallyl)carbonate 2b, suggesting that the regioselectivity of the reaction was heavily influenced by the steric environment around the allyl electrophile, and the allylation proceeded preferentially at the position of less steric hindrance (Scheme 3b). Moreover, the intermolecular kinetic isotopic experiment (KIE) showed that a relatively high value of 4.0 was obtained, implying that the cleavage of ortho-C–H bonds may occur irreversibly in the catalytic cycle (Scheme 3c).11d,k
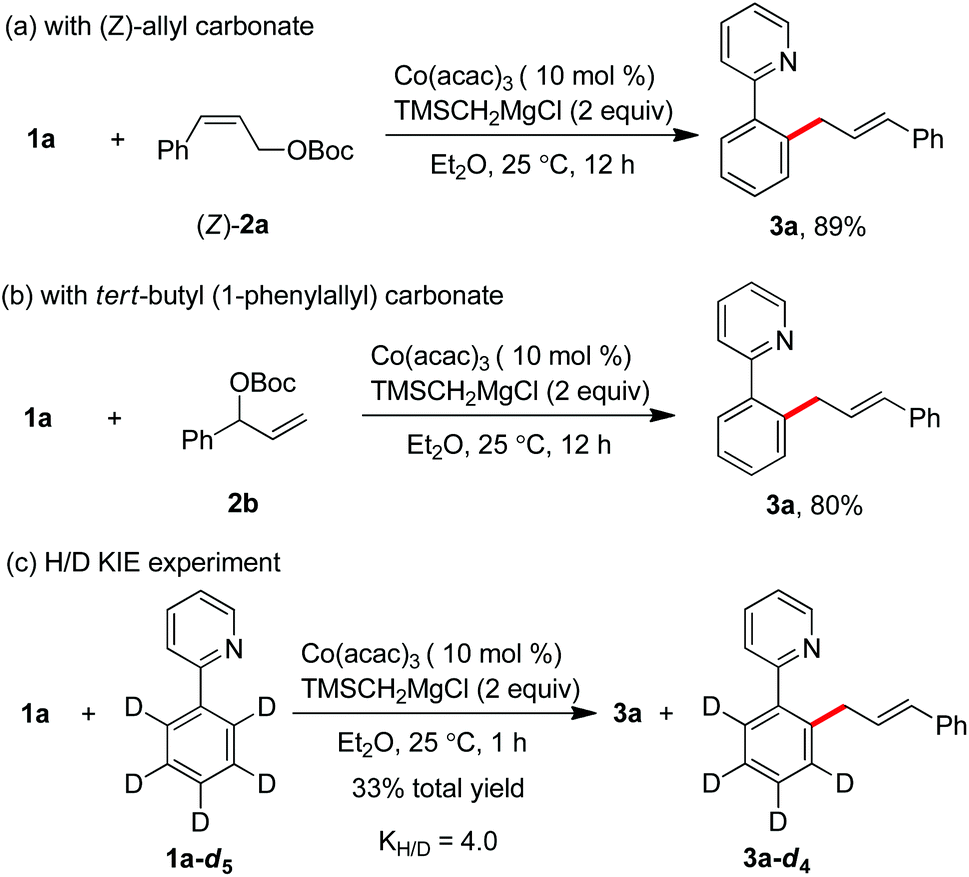 | ||
| Scheme 3 Mechanistic studies. | ||
Based on these preliminary results, we proposed that a low-valent cobalt species A can be formed in situ by the treatment of the cobalt(III) salt with a Grignard reagent, which allows insertion into the ortho-C–H bond with the assistance of a directing group to give a cyclometalated complex B (Scheme 4). An oxidative addition may proceed by the treatment with allyl carbonate to form π-(allyl)cobalt intermediate C,15 which allows undergoing a reductive elimination from the orientation of sterically favourable to give complex D. Subsequently a transmetalation with a Grignard reagent followed by the dissociation of the product may proceed leading to the product and regeneration of active cobalt species. On the other hand, an alternative pathway involving a single-electron transfer (SET) mechanism10e with a radical-type recombination cannot be ruled out for the formation of the corresponding π-(allyl)cobalt complex.
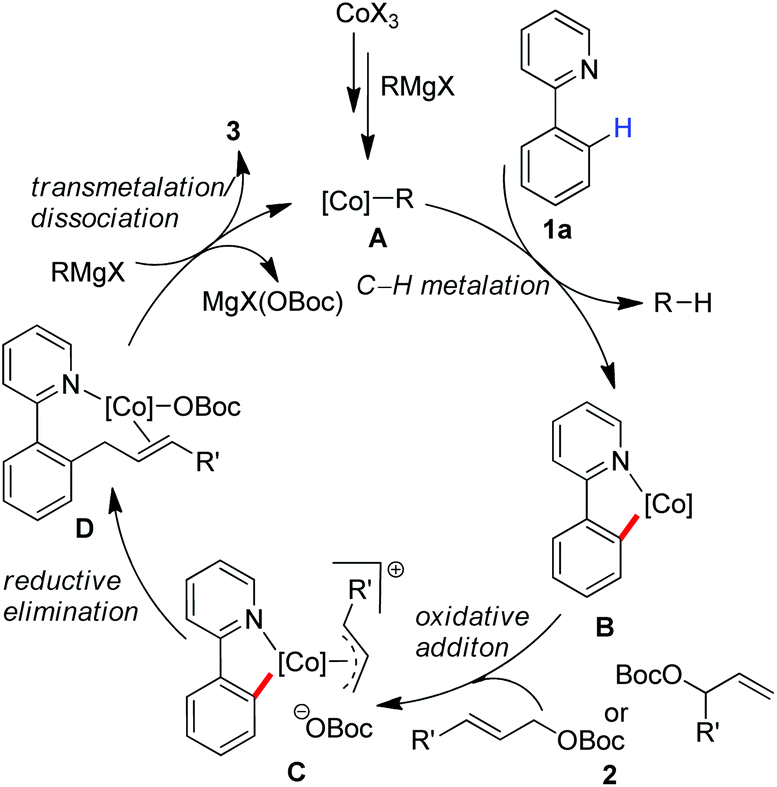 | ||
| Scheme 4 Proposed mechanism for the cobalt-catalyzed C–H allylation. | ||
Conclusions
We have developed a regioselective allylation reaction of C–H bonds by the use of a combination of a cobalt(III) salt and a Grignard reagent. This protocol allows the preparation of linear allylated compounds at room temperature, and provides an alternative strategy to Cp*Co(III)-promoted C–H allylation with a different reaction pathway. Further exploring the unique ability of first-row metal catalysts such as cobalt in the promotion of the direct functionalization of inert chemical bonds is ongoing.Acknowledgements
We thank the National Natural Science Foundation of China [no. 21202128, 21572175] for generous financial support.Notes and references
- For selected reviews on C–H activation, see: (a) J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369 CrossRef CAS PubMed; (b) B.-J. Li and Z.-J. Shi, Chem. Soc. Rev., 2012, 41, 5588 RSC; (c) K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788 CrossRef CAS PubMed; (d) C.-L. Sun, B.-J. Li and Z.-J. Shi, Chem. Rev., 2011, 111, 1293 CrossRef CAS PubMed; (e) S. H. Cho, J. Y. Kim, J. Kwak and S. Chang, Chem. Soc. Rev., 2011, 40, 5068 RSC; (f) R. Jazzar, J. Hitce, A. Renaudat, J. SofackKreutzer and O. Baudoin, Chem. – Eur. J., 2010, 16, 2654 CrossRef CAS PubMed; (g) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed; (h) R. Giri, B.-F. Shi, K. M. Engle, N. Maugel and J.-Q. Yu, Chem. Soc. Rev., 2009, 38, 3242 RSC; (i) N. Kuhl, M. N. Hopkinson, J. Wencel-Delord and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 10236 CrossRef CAS PubMed; (j) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215 CrossRef CAS PubMed; (k) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624 CrossRef CAS PubMed; (l) O. Daugulis, H.-Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074 CrossRef CAS PubMed; (m) P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879 CrossRef CAS PubMed; (n) M. Lautens and P. Thansandote, Chem. – Eur. J., 2009, 15, 5874 CrossRef PubMed; (o) K. M. Engle and J.-Q. Yu, J. Org. Chem., 2013, 78, 8927 CrossRef CAS PubMed.
- For selected reviews on first-row transition-metal-promoted C–H functionalization: (a) B. Su, Z.-C. Cao and Z.-J. Shi, Acc. Chem. Res., 2015, 48, 886 CrossRef CAS PubMed; (b) E. Nakamura, T. Hatakeyama, S. Ito, K. Ishizuka, L. Ilies and M. Nakamura, Org. React., 2014, 83, 1 CAS; (c) J. Yamaguchi, K. Muto and K. Itami, Eur. J. Org. Chem., 2013, 19 CrossRef CAS; (d) N. Yoshikai, Synlett, 2011, 1047 CrossRef CAS; (e) Y. Nakao, Chem. Rec., 2011, 11, 242 CrossRef CAS PubMed; (f) E. Nakamura and N. Yoshikai, J. Org. Chem., 2010, 75, 6061 CrossRef CAS PubMed; (g) A. Kulkarni and O. Daugulis, Synthesis, 2009, 4087 CAS.
- For selected examples on Co(iii)-catalyzed C–H activation: (a) D. Zhao, J. H. Kim, L. Stegemann, C. A. Strassert and F. Glorius, Angew. Chem., Int. Ed., 2015, 54, 4508 CrossRef CAS PubMed; (b) J. Li and L. Ackermann, Angew. Chem., Int. Ed., 2015, 54, 3635 CrossRef CAS PubMed; (c) T. Yoshino, H. Ikemoto, S. Matsunaga and M. Kanai, Angew. Chem., Int. Ed., 2013, 52, 2207 CrossRef CAS PubMed; (d) J. R. Hummel and J. A. Ellman, J. Am. Chem. Soc., 2015, 137, 490 CrossRef CAS PubMed; (e) L. Grigorjeva and O. Daugulis, Angew. Chem., Int. Ed., 2014, 53, 10209 CrossRef CAS PubMed; (f) L.-B. Zhang, X.-Q. Hao, S.-K. Zhang, Z.-J. Liu, X.-X. Zheng, J.-F. Gong, J.-L. Niu and M.-P. Song, Angew. Chem., Int. Ed., 2015, 54, 272 CrossRef CAS PubMed.
- For selected book chapters, see: (a) Comprehensive Organic Functional Group Transformations, ed. A. R. Katritzky, O. Meth-Cohn and C. W. Rees, Elsevier Science, New York, 1995 Search PubMed; (b) Comprehensive Asymmetric Catalysis, ed. E. N. Jacobsen, A. Pfaltz and H. Yamamoto, Springer, New York, 1999 Search PubMed; (c) Supplement A: The Chemistry of Double-Bonded Functional Groups, ed. S. Patai, The Chemistry of Functional Groups Series, Parts 1 and 2, Wiley, New York, 1997, vol. 1 and 2 Search PubMed.
- For selected recent examples, see: (a) H. Wang, N. Schröder and F. Glorius, Angew. Chem., Int. Ed., 2013, 52, 5386 CrossRef CAS PubMed; (b) S. Asako, L. Ilies and E. Nakamura, J. Am. Chem. Soc., 2013, 135, 17755 CrossRef CAS PubMed; (c) T. Yao, K. Hirano, T. Satoh and M. Miura, Angew. Chem., Int. Ed., 2011, 50, 2990 CrossRef CAS PubMed; (d) Y. Makida, H. Ohmiya and M. Sawamura, Angew. Chem., Int. Ed., 2012, 51, 4122 CrossRef CAS PubMed; (e) S. Fan, F. Chen and X. Zhang, Angew. Chem., Int. Ed., 2011, 50, 5918 CrossRef CAS PubMed; (f) Y.-B. Yu, S. Fan and X. Zhang, Chem. – Eur. J., 2012, 18, 14643 CrossRef CAS PubMed; (g) X. Cong, Y. Li, Y. Wei and X. Zeng, Org. Lett., 2014, 16, 3926 CrossRef CAS PubMed; (h) C.-H. Ying and W.-L. Duan, Org. Chem. Front., 2014, 1, 546 RSC.
- S. Oi, Y. Tanaka and Y. Inoue, Organometallics, 2006, 25, 4773 CrossRef CAS.
- (a) Y. Suzuki, B. Sun, K. Sakata, T. Yoshino, S. Matsunaga and M. Kanai, Angew. Chem., Int. Ed., 2015, 54, 9944 CrossRef CAS PubMed; (b) M. Moselage, N. Sauermann, J. Koeller, W. Liu, D. Gelman and L. Ackermann, Synlett, 2015, 1596 CAS.
- (a) D.-G. Yu, T. Gensch, F. de Azambuja, S. Vásquez-Céspedes and F. Glorius, J. Am. Chem. Soc., 2014, 136, 17722 CrossRef CAS PubMed; (b) T. Gensch, S. Vásquez-Céspedes, D.-G. Yu and F. Glorius, Org. Lett., 2015, 17, 3714 CrossRef CAS PubMed.
- (a) D. Lapointe and K. Fagnou, Chem. Lett., 2010, 39, 1118 CrossRef; (b) L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef CAS PubMed.
- For recent reviews of low-valent cobalt-catalyzed C–H functionalization: (a) K. Gao and N. Yoshikai, Acc. Chem. Res., 2014, 47, 1208 CrossRef CAS PubMed; (b) L. Ackermann, J. Org. Chem., 2014, 79, 8948 CrossRef CAS PubMed; (c) N. Yoshikai, Bull. Chem. Soc. Jpn., 2014, 87, 843 CrossRef CAS; (d) N. Yoshikai, J. Synth. Org. Chem., Jpn., 2014, 72, 1198 CrossRef CAS; (e) M. Moselage, J. Li and L. Ackermann, ACS Catal., 2015, 6, 498 CrossRef.
- For selected examples of low-valent cobalt catalysis, see: (a) Q. Chen, L. Ilies and E. Nakamura, J. Am. Chem. Soc., 2011, 133, 428 CrossRef CAS PubMed; (b) L. Ilies, Q. Chen, X. Zeng and E. Nakamura, J. Am. Chem. Soc., 2011, 133, 5221 CrossRef CAS PubMed; (c) K. Gao and N. Yoshikai, J. Am. Chem. Soc., 2013, 135, 9279 CrossRef CAS PubMed; (d) K. Gao, P.-S. Lee, T. Fujita and N. Yoshikai, J. Am. Chem. Soc., 2010, 132, 12249 CrossRef CAS PubMed; (e) J. Li and L. Ackermann, Chem. – Eur. J., 2015, 21, 5718 CrossRef CAS PubMed; (f) B. Punji, W. Song, G. A. Shevchenko and L. Ackermann, Chem. – Eur. J., 2013, 19, 10605 CrossRef CAS PubMed; (g) W. Song and L. Ackermann, Angew. Chem., Int. Ed., 2012, 51, 8251 CrossRef CAS PubMed; (h) B. Li, Z.-H. Wu, Y.-F. Gu, C.-L. Sun, B.-Q. Wang and Z.-J. Shi, Angew. Chem., Int. Ed., 2011, 50, 1109 CrossRef CAS PubMed; (i) K. Gao, R. Paira and N. Yoshikai, Adv. Synth. Catal., 2014, 356, 1486 CrossRef CAS; (j) Z. Ding and N. Yoshikai, Angew. Chem., Int. Ed., 2013, 52, 8574 CrossRef CAS PubMed; (k) P.-S. Lee, T. Fujita and N. Yoshikai, J. Am. Chem. Soc., 2011, 133, 17283 CrossRef CAS PubMed; (l) B. J. Fallon, E. Derat, M. Amatore, C. Aubert, F. Chemla, F. Ferreira, A. Perez-Luna and M. Petit, J. Am. Chem. Soc., 2015, 137, 2448 CrossRef CAS PubMed.
- For selected pioneering examples of low-valent cobalt-catalyzed C–H functionalization, see: (a) S. Murahashi, J. Am. Chem. Soc., 1955, 77, 6403 CrossRef CAS; (b) G. Halbritter, F. Knoch, A. Wolski and H. Kisch, Angew. Chem., Int. Ed. Engl., 1994, 33, 1603 CrossRef; (c) P. Lenges and M. Brookhart, J. Am. Chem. Soc., 1997, 119, 3165 CrossRef; (d) A. D. Bolig and M. Brookhart, J. Am. Chem. Soc., 2007, 129, 14544 CrossRef CAS PubMed.
- X. Cong, H. Tang and X. Zeng, J. Am. Chem. Soc., 2015, 137, 14367 CrossRef CAS PubMed.
- (a) R. F. Heck and D. S. Breslow, J. Am. Chem. Soc., 1960, 82, 750 CrossRef CAS; (b) W. McClellan, E. L. Muetterties, B. W. Howk, H. H. Hoehn and H. N. Cripps, J. Am. Chem. Soc., 1961, 83, 1601 CrossRef CAS.
- (a) R. F. Heck and D. S. Breslow, J. Am. Chem. Soc., 1960, 82, 750 CrossRef CAS; (b) W. McClellan, E. L. Muetterties, B. W. Howk, H. H. Hoehn and H. N. Cripps, J. Am. Chem. Soc., 1961, 83, 1601 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Characterization data for the allylated products and kinetic isotope effect. See DOI: 10.1039/c6qo00011h |
| ‡ These authors contributed equally. |
| This journal is © the Partner Organisations 2016 |