
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Variable coordination modes and catalytic dehydrogenation of B-phenyl amine–boranes†
Amit
Kumar
,
Isobel K.
Priest
,
Thomas N.
Hooper
* and
Andrew S.
Weller
*
Department of Chemistry, University of Oxford, 12 Mansfield Road, Oxford, OX1 3TA, UK. E-mail: andrew.weller@chem.ox.ac.uk
First published on 24th February 2016
Abstract
The chemistry of N-substituted amine–boranes and their reactivity towards transition metal centres is well established but the chemistry of B-substituted amine–boranes is not. Here we present the coordination chemistry of H2PhB·NMe3 towards a range of Rh(I) fragments with different P–Rh–P ligand bite angles, {Rh(PiPr3)2}+, {Rh(PiBu3)2}+, {Rh(iPr2P(CH2)3PiPr2)}+, {Rh(Ph2P(CH2)nPPh2)}+ (n = 3, 5), as characterised by NMR spectroscopy and single-crystal X-ray diffraction. This reveals a difference in the coordination mode of the amine–borane, with large bite angle fragments favouring η2-coordination through a sigma-interaction with BH2, whereas fragments with small bite angles favour η6-coordination through the aryl group of the amine–borane. The catalytic dehydrocoupling of H2PhB·NMe2H is also explored, with the aminoborane HPhB![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NMe2 found to be the sole dehydrogenation product. Stoichiometric reactivity with H2PhB·NMe2H again showed small bite angle fragments to prefer η6-aryl coordination, while the larger bite angle {Rh(PiPr3)2}+ gave rapid dehydrogenation to form a mixture of the Rh(III) dihydride [Rh(PiPr3)2(H)2(η2-H2PhB·NMe2H)][BArF4] and the low coordinate aminoboryl complex [Rh(PiPr3)2(H)(BPhNMe2)][BArF4]. These results suggest that precatalysts which η6-bind arenes strongly should be avoided for the dehydrocoupling of amine–boranes bearing aryl substituents.
NMe2 found to be the sole dehydrogenation product. Stoichiometric reactivity with H2PhB·NMe2H again showed small bite angle fragments to prefer η6-aryl coordination, while the larger bite angle {Rh(PiPr3)2}+ gave rapid dehydrogenation to form a mixture of the Rh(III) dihydride [Rh(PiPr3)2(H)2(η2-H2PhB·NMe2H)][BArF4] and the low coordinate aminoboryl complex [Rh(PiPr3)2(H)(BPhNMe2)][BArF4]. These results suggest that precatalysts which η6-bind arenes strongly should be avoided for the dehydrocoupling of amine–boranes bearing aryl substituents.
Introduction
Amine–boranes, defined by the simplest example H3B·NH3, have been the subject of significant interest and research effort in the past decade with regard to their potential as molecular hydrogen storage materials (i.e. dehydrogenation)1 and as precursors to polyaminoboranes (i.e. dehydrocoupling).2 Much of this research has focussed on developing homo- and heterogeneous catalytic methodologies for dehydrogenation/dehydrocoupling that allow for control of kinetics and final product distributions.3N-Alkyl substituted amine–boranes, particularly those bearing methyl groups4 (although aryl substituents are also known5) have received the bulk of attention because of their thermal stability (N-alkyl especially as N-aryl undergo spontaneous dehydrocoupling5b) relative ease of synthesis, high weight% H, and as precursors to polyaminoboranes.4 The coordination chemistry, and subsequent reactivity, of such species is also well developed, often operating through 3 centre–2 electron (3c–2e) sigma M⋯H–B interactions.3,4,6 Developments in B-substituted analogues have, surprisingly, lagged behind; perhaps due to their more challenging synthesis,7 and potential instability due to weaker B–N bonds.7b,8The reactivity and coordination chemistry of B-alkyl (or heteroalkyl) substituted amine–boranes, particularly with respect to dehydrocoupling, has only recently attracted significant attention. Manners and co-workers reported the synthesis of B-substituted amine–boranes containing relatively exotic substituents [e.g. C6F5 or SR, Scheme 1(i)]9 that undergo dehydrogenation to form the corresponding aminoboranes. Liu and co-workers have developed a range of cyclic amine–boranes [selected examples shown in Scheme 1(ii)],10 that can be dehydrocoupled by transition metal catalysts to form discrete, well-characterised products.11 In some cases intermediate sigma-complexes can be isolated, e.g.AScheme 2.11f Liu and Manners have independently described the dehydrogenation of the B-methyl amine–boranes MeH2B·NMexH3−x [x = 0, 1, 2; Scheme 1(iii)] by catalytic and non-catalytic (thermal) routes.7
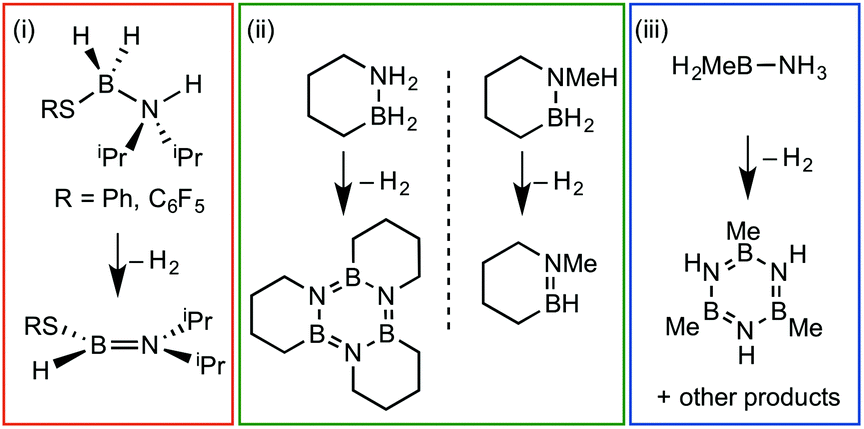 | ||
| Scheme 1 Selected examples of B-substituted amine–boranes and the products of dehydrogenation. | ||
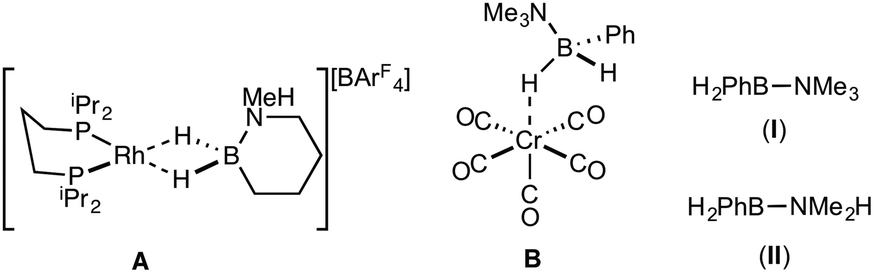 | ||
| Scheme 2 Coordination complexes of B-substituted amine–boranes, and B-aryl precursors used in this study. ArF = 3,5-(CF3)2C6H3. | ||
Reports of B-aryl amine–boranes are scarce. H2PhB·NMe3 (I)12 has been shown to form sigma-complexes with suitable group 6 and 7 metal fragments (e.g.B, Scheme 2).13 H2PhB·NMe2H (II) is known14 but its coordination chemistry or dehydrocoupling has not been reported. The B-phenyl amine–borane H2PhB·NH3 can be dehydrocoupled using [Pd(NCMe)4][BF4]2 to form a material tentatively identified as [PhBNHx]n, but insolubility prevented further characterisation.15
We report here a detailed study into the coordination chemistry of B-aryl substituted amine–boranes (I) and (II) with {Rh(L2)}+ fragments (L2 = (PR3)2 or chelating diphosphine) in which the steric and electronic (bite angle, β16) demands of the phosphine ligands are varied. Unlike B-alkyl (or N-alkyl) substituted amine–boranes, B-aryl analogues offer two potential binding motifs: either through the aryl (e.g. η6) or 3c–2e Rh⋯H–B interactions (e.g. η2), Scheme 3. The relative strength of amine–borane sigma binding with increasing bite angle has been commented upon before in [Rh(L2)(η2-H3B·NMe3)][BArF4] complexes, with larger L–Rh–L bite angles favouring tighter Rh⋯H2B interactions (as measured by NMR spectroscopy).17 Conversely, larger bite angles in the simple arene complexes [Rh(L2)(η6-C6H5F)][BArF4] result in weaker Rh⋯arene interactions, as measured by collision-induced dissociation in Electrospray Mass Spectrometry (ESI-MS) and solution equilibrium measurements.18 These trends presumably reflect the optimisation of bonding between the d8–Rh(I)–{ML2} fragment and either the B–H sigma donating orbitals13 or the π-arene orbitals,19 as modified by the L–Rh–L angle.20 This can be interpreted by the energy of the C2v-{ML2}+ LUMO that is of π-symmetry (b1) becoming lower in energy with increasing bite angle,17a,21 thus finding a worse match with the arene HOMO and a better one with the relatively low lying B–H σ-orbitals. In this contribution we demonstrate empirically that with B-aryl amine–boranes the L–Rh–L bite-angle dictates which mode of binding is observed (i.e. η6 or η2), present equilibrium thermochemical data on the relative binding strengths of each motif when the two binding modes are finely balanced, and show that dehydrocoupling of H2PhB·NMe2H forms an unusual example of a B-substituted acyclic aminoborane which undergoes subsequent B–H activation to form a B-substituted amino–boryl complex.
 | ||
| Scheme 3 Potential coordination modes of B-aryl amine–boranes with {Rh(L2)}+ fragments, and previous observations regarding bite angle and strength of binding of a generic arene and amine–borane fragments. | ||
Results and discussion
Synthesis of precursors
H2PhB·NMe3 (I)12 and H2PhB·NMe2H (II)14 have been reported, and their original syntheses comes from the reaction of diboranes (PhBH2)2 with NMe3 or NMe2H respectively. An alternative, expedient, synthesis of (I) and (II) is based on the methods of Hawthorne,22 Shimoi,13 and Liu.7a Li[PhBH3], prepared by reaction of phenylboronic acid with lithium aluminium hydride in diethyl ether,23 was combined with the appropriate ammonium salt, [NMe3H]Cl or [NMe2H2]Cl, to give H2PhB·NMe3 (I) and H2PhB·NMe2H (II) respectively, which were isolated as white solids in good yield. NMR spectroscopic data for (I) in CD2Cl2 are consistent those previously described13 [e.g. BH2: δ(1H) 2.37; δ(11B) −0.8, t, J(BH) 97 Hz], while as far as we are aware NMR data for (II) have not been previously reported; BH2: δ(1H) 2.34; δ(11B) −4.7, t, J(BH) 95 Hz. In contrast to N-aryl amine boranes, such as H3B·NPhH2,5b compounds (I) and (II) were found be stable towards thermal dehydrocoupling or B–N bond cleavage, remaining unchanged on heating (C6H5F, 80 °C, 12 h).Coordination chemistry of H2PhB·NMe3
Reaction of a stoichiometric amount of (I) with [Rh(L2)(η6-C6H5F)][BArF4] [L2 = (PiPr3)2,24 (PiBu3)2,25iPr2P(CH2)3PiPr2,18b Ph2P(CH2)3PPh2 and Ph2P(CH2)5PPh2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 17b] in 1,2-difluorobenzene solvent resulted in displacement of the fluorobenzene ligand and formation of new complexes in solution as determined by NMR spectroscopy. These fragments were chosen to probe changes in phosphine bite-angle, while keeping the electronic contribution from the phosphine substituent as constant as possible. For example PiBu3 and PiPr3 have different cone angles of 143° & 160° respectively but similar electronic properties;26 L–Rh–L bite angles can be varied in Ph2P(CH2)nPPh2 (n = 3 or 5); and mono-dentate versus chelating coordination modes can be probed with PiPr3 and iPr2P(CH2)3PiPr2. These fragments have also been used to form well-defined sigma amine–borane complexes with, for example, H3B·NR3 type ligands,11f,17,27 whose structures and solution NMR spectroscopic markers are well-established.
17b] in 1,2-difluorobenzene solvent resulted in displacement of the fluorobenzene ligand and formation of new complexes in solution as determined by NMR spectroscopy. These fragments were chosen to probe changes in phosphine bite-angle, while keeping the electronic contribution from the phosphine substituent as constant as possible. For example PiBu3 and PiPr3 have different cone angles of 143° & 160° respectively but similar electronic properties;26 L–Rh–L bite angles can be varied in Ph2P(CH2)nPPh2 (n = 3 or 5); and mono-dentate versus chelating coordination modes can be probed with PiPr3 and iPr2P(CH2)3PiPr2. These fragments have also been used to form well-defined sigma amine–borane complexes with, for example, H3B·NR3 type ligands,11f,17,27 whose structures and solution NMR spectroscopic markers are well-established.
The reaction of [Rh(PiPr3)2(η6-C6H5F)][BArF4] with (I) resulted in the immediate formation of a deep purple solution. Recrystallisation by addition of pentane gave blue crystalline material in 69% isolated yield, identified by NMR spectroscopy and single crystal X-ray diffraction as [Rh(PiPr3)2(η2-H2PhB·NMe3)][BArF4] (1) in which the amine–borane binds through two Rh⋯H–B 3c–2e interactions (Scheme 4). In the 11B{1H} NMR spectrum a single broad peak is observed at δ 34.8, with a characteristic downfield shift (35.6 ppm) of the borane resonance compared to free ligand (δ −0.8) that signals η2 Rh⋯H–B binding.27,28 In the 1H NMR spectrum the BH2 resonance is observed at δ −6.36 (2 H relative integral), an upfield shift of 8.73 ppm compared to free ligand. The aryl protons [δ 7.37, 3H; δ 7.25, 2H] are not significantly shifted from free ligand.13 In the 31P{1H} NMR spectrum a doublet is observed at δ 69.6 [J(PRh) = 176 Hz], shifted 14.1 ppm downfield from the starting material.24 In the solid state the complex crystallises with two cations (and two anions) in the asymmetric unit. An overlay of the independent cations (ESI) did not reveal any significant difference in amine–borane geometry but the PiPr3 ligands vary slightly in position and conformation [e.g. P(1)–Rh(1)–P(2) 106.19(3) Å, P(3)–Rh(2)–P(4) 101.86(3)°] which we attribute to crystal packing effects due to a rather flat potential energy surface as only one set of resonances could be observed in the 1H, 11B and 31P{1H} NMR spectra. This observation of different ligand conformation/bite angles of two independent molecules in the asymmetric unit has been noted in amine–borane complexes of H3B·NMe2BH2·NMe2H and [Me2NBH2]2 with the {Rh(PiBu3)2}+ fragment.27 In the solid-state short Rh⋯B distances [2.150(3) and 2.159(3) Å] are consistent with an η2-binding mode, by comparison to previously reported structures,17b,27,29 including sigma amine–borane complexes of closely related tBuCH2CH2BH2·NMe3.30 This distance in the structurally similar [Rh(PiPr3)2(η2-H3B·NMe3)][BArF4] is slightly shorter [2.1376(3) Å],17a perhaps as a result of the extra steric demand caused by B-substitution in 1. High quality X-ray diffraction data allowed the hydrogen atoms of the BH2 unit to be located in the difference map and refined freely, confirming the η2-coordination mode.
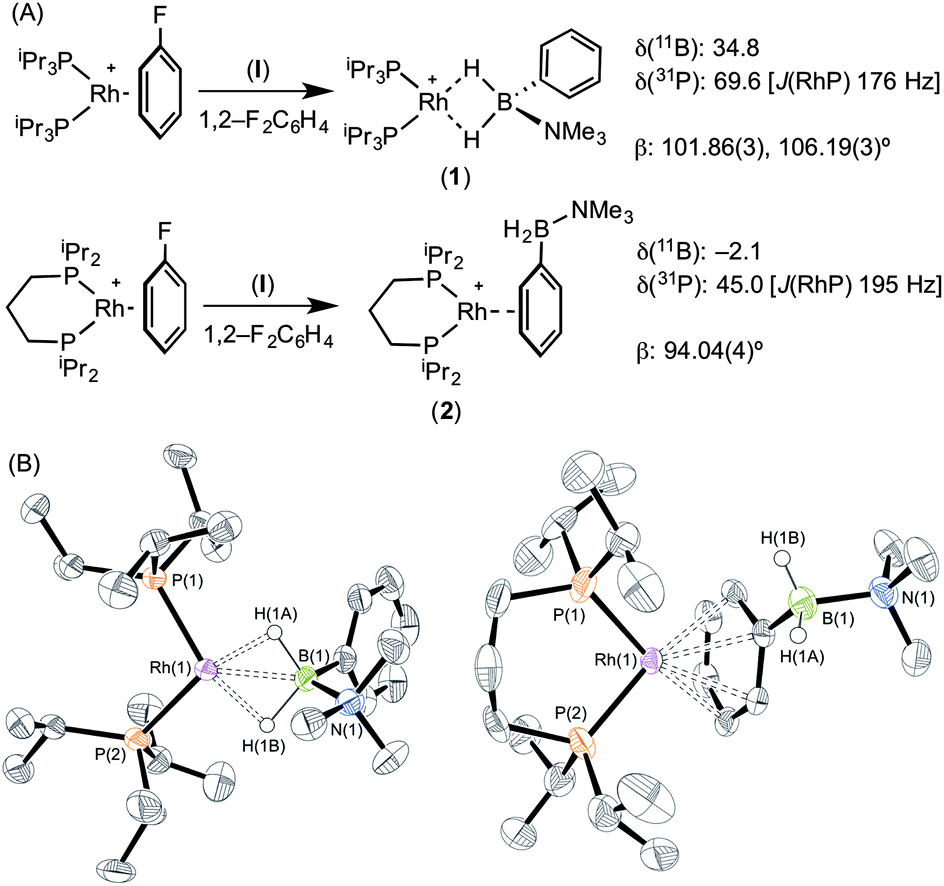 | ||
| Scheme 4 (A) Synthesis, selected NMR spectroscopic data and (B) molecular structures of [Rh(PiPr3)2(η2-H2PhB·NMe3)][BArF4] (1) and [Rh(iPr2P(CH2)3PiPr2)(η6-H2PhB·NMe3)][BArF4] (2). [BArF4]− anions and selected H atoms are omitted for clarity. Ellipsoids shown at 50% probability level. Selected bond lengths (Å) and angles (°) (1) [values are also given for the second cation in the asymmetric unit which is not shown in the figure]: Rh(1)–P(1) 2.2581(6), Rh(1)–P(2) 2.2748(7), Rh(1)–B(1) 2.150(3), B(1)–N(1) 1.623(4), Rh(2)–P(3) 2.2628(7), Rh(2)–P(4) 2.2655(7), Rh(2)–B(2) 2.159(3), B(2)–N(2) 1.629(4), P(1)–Rh(1)–P(2) 106.19(3), P(3)–Rh(2)–P(4) 101.86(3). (2) [Only major component of disorder shown]. Ellipsoids shown at 50% probability level. Selected bond lengths (Å) and angles (°): Rh(1)–P(1) 2.2340(9), Rh(1)–P(2) 2.2403(8), Rh(1)–Ph centroid 1.848, B(1)–N(1) 1.635(4), P(1)–Rh(1)–P(2) 94.04(4). | ||
Forcing the P–Rh–P bite angle to be significantly smaller, while keeping the electronic contribution of the P-substituents the same, is achieved by use of the chelating phosphine complex [Rh(iPr2P(CH2)3PiPr2)(η6-C6H5F)][BArF4]. Reaction of this with a stoichiometric amount of amine–borane (I) resulted in the formation of an orange solution, rather than the purple one observed for 1. X-ray diffraction quality crystals were obtained from a 1,2-difluorobenzene/pentane recrystallisation, from which a single crystal X-ray diffraction study demonstrated η6-binding of the arene, rather than Rh⋯H–B bonding: [Rh(iPr2P(CH2)3PiPr2)(η6-PhH2B·NMe3)][BArF4] (2) (Scheme 4B). The P–Rh–P bite angle [94.04(4)°] is significantly smaller than in η2-bound complex 1 [e.g. 101.86(3)°]. Consistent with this different binding mode, that does not involve the borane fragment, in the 11B{1H} NMR spectrum a single resonance is observed at δ −2.1 that is now only slightly shifted from free amine–borane (δ −0.8). In the 31P{1H} NMR spectrum a single species is observed [δ 45.0; J(RhP) = 195 Hz] a chemical shift that is barely changed when compared with [Rh(iPr2P(CH2)3PiPr2)(η6-C6H5F)][BArF4].18b No resonance was observed in the high-field region of the 1H NMR spectrum that would signal Rh⋯H2B interactions, but peaks at δ 6.93 [relative integral 1 H] and δ 6.31 [4 H, a 2 + 2 coincidence] demonstrate η6-binding through the phenyl moiety of (I).31 Thus a change in the bite angle from 101.83(3)° in (1) to 94.04(4)° in (2) is also reflected in a change in the coordination mode from η2 to η6.
This preference comes into fine balance when the mono-dentate phosphine PiBu3 is used, that has a cone angle of 143° and thus might be expected to have a smaller P–Rh–P bite angle than (1).17a Reaction of (I) with the precursor complex [Rh(PiBu3)2(η6-C6H5F)][BArF4] again led to formation of a purple solution. However, more complicated NMR data were observed than for either (1) or (2) which suggested the presence of two species in solution. In the 11B{1H} NMR spectrum (CD2Cl2) two peaks are observed at δ 29.5 and −2.1 in a ratio of 10:11 respectively. The peak observed at δ 29.5 suggested the formation of a sigma complex with a η2-Rh⋯H2B interaction, being shifted 30.3 ppm downfield compared to I, cf. complex (1). The higher field signal at δ −2.1 is only shifted 1.3 ppm upfield compared with free ligand suggesting an alternative coordination mode for the amine–borane, more like (2). In the 31P{1H} NMR spectrum two resonances are observed in the same ratio as measured in the 11B NMR spectrum, one at δ 34.1 [d, J(RhP) = 177 Hz] with a similar downfield shift and coupling constant to (1), consistent with sigma complex formulation; while a signal at δ 25.2 [d, J(RhP) = 202 Hz] suggests a binding mode as for (2). These data indicate both η2-Rh⋯H2B and η6-aryl bound complexes are present in solution. The 1H NMR spectrum is consistent with this description. In the high field region a broad resonance is observed at δ −5.06 (Rh⋯H2B) which integrates to 1.1 H relative to the [BArF4]– signals, and 2 singlets are observed at δ 2.76 and 2.50 corresponding to NMe3 protons in the different coordination modes of the amine–borane. In addition, resonances can be observed upfield of the aryl region indicative of η6-aryl coordination. These two complexes are formulated as [Rh(PiBu3)2(η2-H2PhB·NMe3)][BArF4], (η2-3), and [Rh(PiBu3)2(η6-PhH2B·NMe3)][BArF4], (η6-3), Scheme 5A.
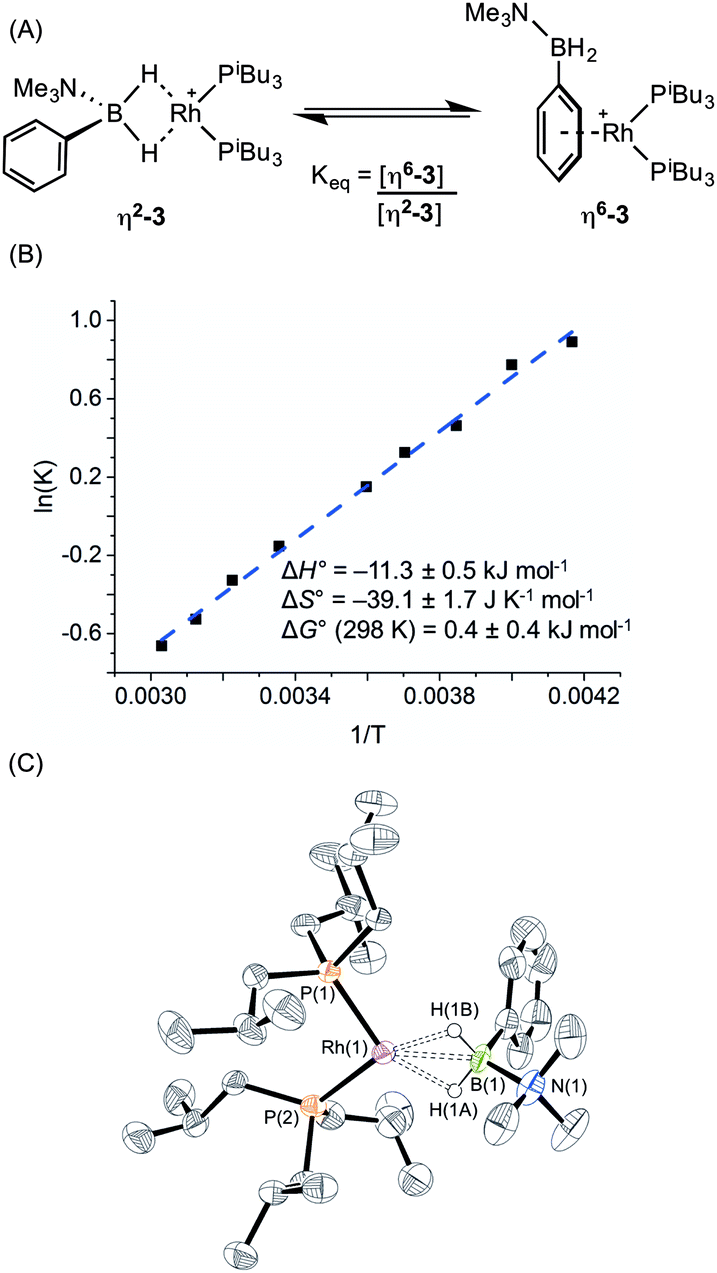 | ||
| Scheme 5 (A) Equilibrium between η2-3 and η6-3, (B) Van't Hoff plot and (C) molecular structure of [Rh(PiBu3)2(η2–H2PhB·NMe3)][BArF4] (η2-3). The second cation in asymmetric unit, [BArF4]− anions and selected H atoms are omitted for clarity. Only major component of disorder shown. Ellipsoids shown at 50% probability level. Selected bond lengths (Å) and angles (°) (values are also given for the second cation in the asymmetric unit which is not shown in the figure): Rh(1)–P(1) 2.2254(14), Rh(1)–P(2) 2.2436(14), Rh(1)–B(1) 2.153(6), B(1)–N(1) 1.605(8), Rh(2)–P(3) 2.2353(14), Rh(2)–P(4) 2.2289(14), Rh(2)–B(2) 2.172(6), B(2)–N(2) 1.617(9), P(1)–Rh(1)–P(2) 98.84(5), P(3)–Rh(2)–P(4) 95.14(5). | ||
A variable temperature NMR spectroscopy study was carried out to determine if exchange between these isomers was occurring in solution. At 298 K in 1,2-difluorobenzene, the concentrations of η2-3 and η6-3 were found to be approximately equal. Lowering the temperature to 240 K resulted in a relative increase in η6-3 while at higher temperature (330 K) (η2-3) was favoured, demonstrating the two isomers to be in dynamic equilibrium. The equilibrium constant at each temperature was calculated from integration of the 31P{1H} NMR spectra; and the resulting Van't Hoff plot (Scheme 5B) allowed for determination of the thermodynamic parameters for this exchange: ΔH° = −11.3 ± 0.5 kJ mol−1; ΔS° = −39.1 ± 1.7 J K−1 mol−1; ΔG(298 K) = 0.4 ± 0.4 kJ mol−1. Thus binding between the two modes is approximately thermoneutral. The negative enthalpy indicates η6-binding of the aryl group is stronger than the η2-binding through BH2 but this is moderated by the associated negative entropy, which is likely to be the result of loss of free rotation of the phenyl group upon η6-binding. The negative entropy also means that η2 Rh⋯H–B binding will become increasingly favoured at higher temperature. A similar entropy change (ΔS° = −16.3 ± 3.3 J K−1 mol−1) upon loss of phenyl group free rotation was observed in the epimerisation of 2-phenyl-c-4,c-6-dimethyl-1,3-dioxane;32 while for the anion exchange equilibrium between [1-closo-CB11H6Br6]− and [BArF4]− η6-binding through an aryl group of [BArF4]− was also shown to be enthalpically favoured but entropically disfavoured (ΔS° = −87.6 ± 0.8 J K−1 mol−1).33
Layering a 1,2-difluorobenzene solution of this mixture with pentane at −30 °C led to formation of purple crystals and an orange oil. Isolation of a crystal suitable for X-ray diffraction by mechanical separation allowed the solid-state structure of the purple material to be determined (Scheme 5C). As for complex (1), two cations are present in the asymmetric unit; an overlay of these independent structures (ESI) did not reveal significant differences in amine–borane binding and orientation, although some conformational differences and a difference in ligand bite angle was observed for the PiBu3 ligands. The structure shows a close interaction between the rhodium centre and the BH2 moiety in [Rh(PiBu3)2(η2-H2PhB·NMe3)][BArF4] (η2-3) with Rh⋯B distances of 2.153(6) and 2.172(6) Å consistent with η2-binding. Although the hydrogen atoms could not be located in the difference map and were placed in calculated positions, the metrical data are consistent with this description as well as the NMR data. The two P–Rh–P bite angles measured for each independent molecule, 95.14(5) and 98.84(5)°, are smaller than for (1), but larger than for (2), consistent with the equilibrium observed in solution. [Rh(PiBu3)2(η6-C6H5F)][BArF4], a model for η6-binding of (I), has a P–Rh–P angle of 94.14(4)°, placing the approximate tipping point between the two structural motifs as lying between 94 and 95°.
Bulk mechanical separation of the crystals from the oil for further analysis was not possible, but we propose the orange oil to be the η6-phenyl bound amine–borane complex [Rh(PiPr3)2(η6-PhH2B·NMe3)][BArF4] (η6-3). Dissolving the mixture of blue crystals and orange oil isolated gave a solution that showed the same NMR spectra as a freshly prepared sample.
To extend this study into the effect of bite angle [Rh(Ph2P(CH2)5PPh2)(η6-C6H5F)][BArF4] was used as a starting material, which has a flexible chelating phosphine with a 5-carbon backbone. This ligand has been shown to be able to access to a wide range of bite angles, and values of 93.98(4) to 117.3(1)° have been determined crystallographically.16b,34 Reaction of a stoichiometric amount of this starting material with (I) resulted in an orange solution with NMR data characteristic of an η6-aryl bound. Crystallisation from layering a dichloromethane solution with pentane allowed a single crystal X-ray diffraction study to be carried out and confirmed the η6-coordination mode in [Rh(Ph2P(CH2)5PPh2)(η6-PhH2B·NMe3)][BArF4] (4) (Fig. 1). The phosphine ligand bite angle was found to be only 92.21(4)°, the smallest observed crystallographically for this ligand but consistent with the observed binding mode. By contrast the corresponding H3B·NMe3 complex is η2-bound, [Rh(Ph2P(CH2)5PPh2)(η2-H3B·NMe3)][BArF4], and shows a P–Rh–P bite angle of 98.18(3)°. This shows that the observed bite angle for a flexible ligand such as Ph2P(CH2)5PPh2 is very dependent on the ancillary ligands. The analogous complex formed with the smaller bite angle aryl diphosphine, [Rh(Ph2P(CH2)3PPh2)(η6-PhH2B·NMe3)][BArF4] [(5), 87.955(15)°, ESI] also shows a η6-coordination mode. The data for the ligation of (I) is summarised in Fig. 2; in which a plot of Rh⋯B distance against bite angle shows that larger bite angles give η2-complexes, smaller bite angles result in η6-complexes, with a crossover point at approximately 95°. The Rh⋯B distance in the η2-binding mode appears to be rather insensitive to bite angle.
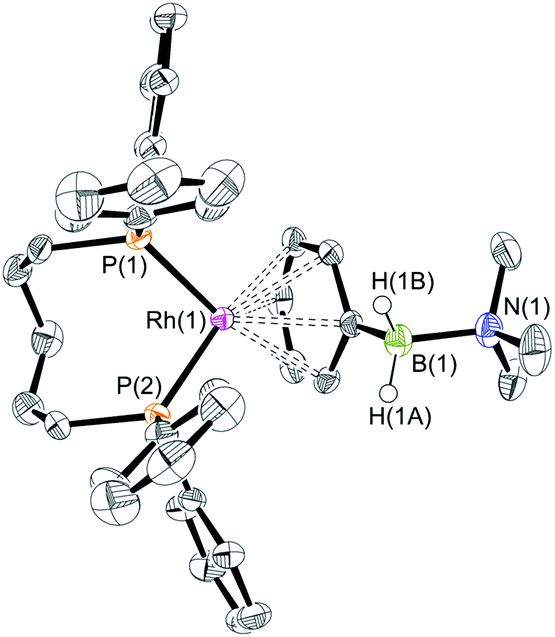 | ||
| Fig. 1 X-ray molecular structure of [Rh(Ph2P(CH2)5PPh2)(η6-PhH2B·NMe3)][BArF4] (4). [BArF4]− anion and selected H atoms omitted for clarity. Ellipsoids shown at 50% probability level. Selected bond lengths (Å) and angles (°): Rh(1)–P(1) 2.2416(8), Rh(1)–P(2) 2.2469(7), Rh(1)–Ph centroid 1.860, B(1)–N(1) 1.638(3); P(1)–Rh(1)–P(2) 92.21(4). | ||
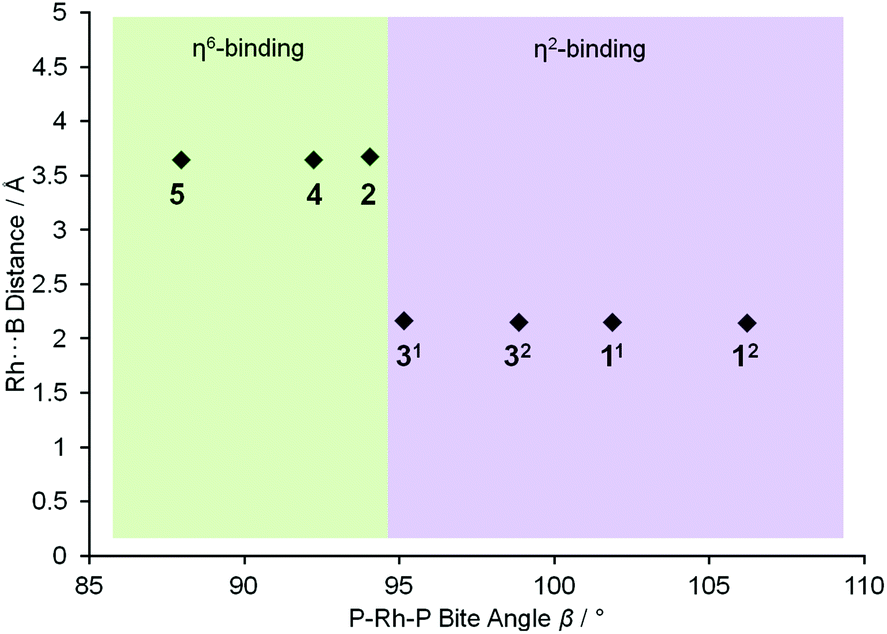 | ||
| Fig. 2 Plot of Rh⋯B distance against bite angle β showing different binding modes for complexes 1, 2, 3, 4 and 5. The values for independent cations in the asymmetric units of 1 and 3 are given separately. | ||
Catalytic dehydrogenation of H2PhB·NMe2H (II)
{Rh(P2)}+ fragments have been shown to catalyse the dehydrocoupling of secondary and primary amine–boranes;17,27 and the ligand bite angle has been shown to affect the rate of dehydrocoupling of H3B·NMe2H in particular.17 Although empirically it is found that the smaller bite angles promoted in larger turnover frequencies, the precise factors behind these differences are not yet fully delineated and likely involve a combination of relative accessibility of Rh(I)/Rh(III) oxidation states/ease of H2 loss/relative barriers to BH and NH activation all as modified by the bite angle.3,35 We therefore sought to probe the effect of the ligands on the dehydrocoupling of secondary amine–borane (II) by comparing two electronically similar precatalysts but with very different P–Rh–P angles: [Rh(PiPr3)2(η6-C6H5F)][BArF4] and [Rh(iPr2P(CH2)3PiPr2)(η6-C6H5F)][BArF4] which form η2 and η6-complexes with (I), i.e. (1) and (2) respectively.Reaction of 5 mol% of these two precatalysts with (II) in 1,2-difluorobenzene (25 °C, closed system) resulted in very slow3 consumption of (II) in both cases: less than 15% conversion in 5 hours (TOF less than 0.6 h−1). Although slow, dehydrogenation is also not fast with other amine–boranes using these systems.11f,17a The major 11B-containing product displayed a single resonance at δ 39.4 which split into a doublet [J(BH) = 123 Hz] in the 11B NMR spectrum. This was assigned as aminoborane HPhBNMe2 (6) from its characteristic 11B NMR chemical shift and the presence of a single B–H bond (Scheme 6). For example aminoboranes bearing two alkyl groups at nitrogen show similar 11B NMR chemical shift values (e.g. H2BNMe2, δ 37.5; H2BNEt2, δ 36.6; H2BNiPr2, δ 35.1);36 while the recently reported B-substituted HMeBNMe2, displays a doublet at δ 41.2 [J(BH) = 123 Hz] in the 11B NMR spectrum,7b and cyclo-HBNMeC4H8 is observed at δ 40.8 [J(BH) = 125 Hz].11f Heating to 77 °C in a sealed NMR tube resulted in the complete consumption of (II) in less than 1 hour for both catalysts. The product (>95% by 11B NMR spectroscopy) of dehydrocoupling was again found to be free aminoborane HPhBNMe2 from the in situ NMR spectrum. Unfortunately, due to its apparent instability and similar volatility to the 1,2-difluorobenzene solvent, separation and isolation of pure (6) was not possible due to decomposition upon vacuum distillation. Nevertheless NMR data are unambiguous, and as we show in situ generated (6) can also be used for onward reactivity. The formation of (6) is in contrast with the metal catalysed dehydrocoupling of H2MeB·NMe2H which forms cyclic B-dimethyl-N-tetramethyldiborazane,7b [Me2NBHMe]2, as well the aminoborane, HMeBNMe2. The B-phenyl group in (6) inhibits any appreciable dimerisation to the corresponding diborazane.
 | ||
| Scheme 6 Catalytic dehydrogenation of amine–borane (II) to form aminoborane (6) at 77 °C [cat.] = [Rh(PiPr3)2(η6-C6H5F)][BArF4] or [Rh(iPr2P(CH2)3PiPr2)(η6-C6H5F)][BArF4]. Inset shows 11B NMR spectrum after 15 minutes, [cat.] = [Rh(iPr2P(CH2)3PiPr2)(η6-C6H5F)][BArF4]. | ||
[Rh(Ph2P(CH2)3PPh2)(η6-C6H5F)][BArF4] has been shown to be an excellent catalyst for dehydrocoupling of H3B·NMe2H (0.2 mol%, open system, TOF ∼ 1250 h−1).17b However this was also a slow catalyst for dehydrocoupling of (II) at room temperature, with full conversion to (6) only observed after 23 hours at 5 mol% catalyst loading (25 °C, TOF ∼ 1 h−1). In order to probe the causes of the slow dehydrogenation of (II) with this catalyst, stoichiometric studies were performed.
Stoichiometric reactivity of H2PhB·NMe2H (II)
The presence of the phenyl group which provides a competitive (η6) site for amine–borane binding at the metal centre is a possible cause of the slow dehydrogenation of (II), as B–H activation at the metal centre requires the formation of a precursor sigma complex.2b,3 Preferential η6-coordination through the aryl ring makes this less likely.Addition of a slight excess of (II) (1.2 equiv.) to [Rh(iPr2P(CH2)3PiPr2)(η6-C6H5F)][BArF4] results in the formation of an η6-bound complex [Rh(iPr2P(CH2)3PiPr2)(η6-PhH2B·NMe2H)][BArF4] (7) alongside a small amount of dehydrogenation product (6). Complex (7) was characterised by NMR spectroscopy and single crystal X-ray diffraction (Fig. 3). The solid-state structure reveals a ligand bite angle of 94.27(4)° which is very similar to that in (2) [94.04(4)°] which also displayed an η6-coordination mode. In addition to the expected resonances in the NMR spectra an N–H resonance is observed in the 1H NMR spectrum at δ 3.55. A similar complex is formed on reaction of [Rh(Ph2P(CH2)3PPh2)(η6-C6H5F)][BArF4] with (II), as characterised by NMR spectroscopy: [Rh(Ph2P(CH2)3PPh2)(η6-PhH2B·NMe2H)][BArF4] (8). When three equivalents of (II) were combined with [Rh(iPr2P(CH2)3PiPr2)(η6-C6H5F)][BArF4] slow dehydrocoupling (hours) to form aminoborane (6) occurs, with η6-bound (7) observed at the end of catalysis.
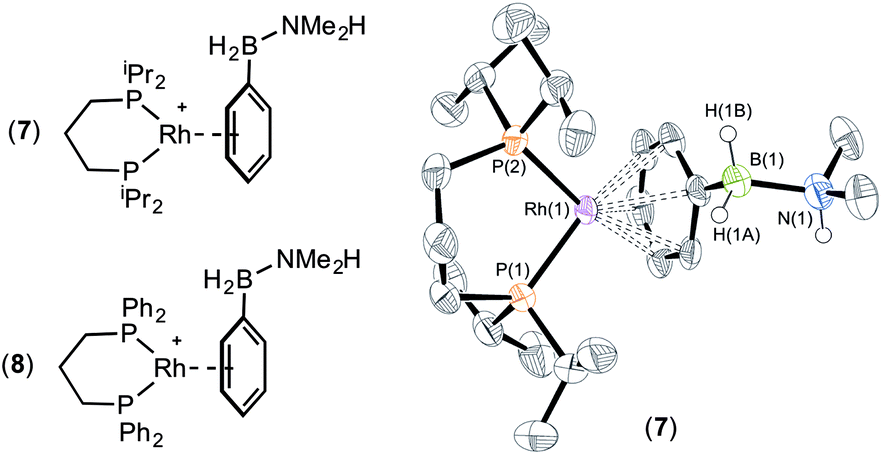 | ||
| Fig. 3 X-ray molecular structure of (7). [BArF4]− anion and selected H atoms omitted for clarity. Ellipsoids shown at 50% probability level. Selected bond lengths (Å) and angles (°): Rh(1)–P(1) 2.2441(11), Rh(1)–P(2) 2.2422(10), Rh(1)–Ph centroid 1.851, B(1)–N(1) 1.618(6); P(1)–Rh(1)–P(2) 94.27(4). | ||
That η6-coordination of ligand (II) in (7) and (8) is preferred to η2-binding suggests that this competitive binding mode contributes to the slow dehydrogenation rate under catalytic conditions for these chelating systems. However, that dehydrogenation does occur catalytically, albeit slowly, indicates that if an inner sphere mechanism is operating, access to the η2-coordination mode through BH2 is possible, but the equilibrium lies heavily in favour of η6-coordination. Complexes (7) and (8) do not dehydrogenate to any significant degree in the absence of exogenous amine–borane, and we, and others, have previously commented upon the role of B–H⋯H–N interactions in lowering barrier to dehydrocoupling.37 Given the η6 binding mode we cannot discount an outer-sphere mechanism in which π-coordination38 of the metal activates the amine–borane to alternative dehydrogenation pathways. However, the N–H resonance does not change significantly on coordination [δ 3.55 versus δ 3.52] suggesting only a minimal perturbation to this bond.
By using a metal fragment which can adopt a large ligand bite angle, {Rh(PiPr3)2}+, in which η2-coordination is favoured (i.e. complex 1) this effect of competitive aryl binding can potentially be avoided. Upon mixing equal amounts of [Rh(PiPr3)2(η6-C6H5F)][BArF4] and (II) in 1,2-F2C6H4 solvent a blue solution was immediately formed which rapidly decolourised (less than 5 min) to yield a very pale yellow solution. This blue colour likely results from the sigma-complex [Rh(PiPr3)2(η2-H2PhB·NMe2H)][BArF4], (9), although its short lifetime meant full characterisation was not possible. 31P{1H} NMR spectroscopy measured in situ after 2 minutes revealed three doublets [δ 69.3, J(RhP) = 174 Hz; δ 64.4 J(RhP) = 109 Hz; and δ 48.5, J(RhP) = 116 Hz] in an approximate ratio of 10:45:45 respectively. After 10 minutes the resonance at δ 69.3 had disappeared leaving the remaining two in a 1:1 ratio. We propose the doublet at δ 69.3 is therefore likely to result from (9). In the 11B{1H} NMR spectrum no signals for free (II) or (6) were observed. There was no further change after 24 hours. Recrystallisation at −26 °C resulted in the formation of two distinct crystalline products: colourless block- and pale yellow plate-type crystals that could be separated mechanically. Although relatively poor crystal quality compounded with significant disorder of the phosphine alkyl groups in both complexes prevented collection of high-quality data in single crystal X-ray diffraction experiments, the data were sufficient to identify the products as [Rh(PiPr3)2(H)2(η2-H2PhB·NMe2H)][BArF4] (10) and [Rh(PiPr3)2(H)(BPhNMe2)][BArF4] (11), Scheme 7 and Fig. 4.
 | ||
| Scheme 7 Complexes (10) and (11). [BArF4]− anions not shown. | ||
 | ||
| Fig. 4 X-ray molecular structures of [Rh(H)2(PiPr3)2(η2-H2PhB·NMe2H)][BArF4] (10), left, one independent cation from the asymmetric unit is shown; and [Rh(H)(PiPr3)2(BPhNMe2)][BArF4] (11), right. [BArF4]− anions, minor components of disorder and selected H atoms omitted for clarity. Ellipsoids shown at 50% probability level. Selected bond lengths (Å) and angles (°). (10) [Values are also given for the second cation in the asymmetric unit which is not shown in the figure]: Rh(1)–P(1) 2.336(2), Rh(1)–P(2) 2.330(2), Rh(1)–B(1) 2.274(9), B(1)–N(1) 1.650(11), Rh(2)–P(3) 2.3227(19), Rh(2)–P(4) 2.327(2), Rh(2)–B(2) 2.333(8), B(2)–N(2) 1.608(11), P(1)–Rh(1)–P(2) 158.35(8), P(3)–Rh(2)–P(4) 155.75(8). (11): Rh(1)–P(1) 2.3292(18), Rh(1)–P(2) 2.350(2), Rh(1)–B(1) 1.929(9), B(1)–C(1) 1.536(14), B(1)–N(1) 1.411(14), P(1)–Rh(1)–P(2) 157.55(7), P(1)–Rh(1)–B(1) 100.2(3), P(2)–Rh(1)–B(1) 100.6(3), Rh(1)–B(1)–N(1) 138.0(8), Rh(1)–B(1)–C(1) 98.3(7), N(1)–B(1)–C(1) 123.7(8). | ||
The solid-state structure of (10) contains two independent cations (and two anions) in the asymmetric unit with broadly similar metric parameters, but disorder is observed in the iPr groups of the phosphine ligand. The N–H and B–H hydrogen atoms were placed in calculated positions, the Rh–H hydride ligands could not be reliably placed and so were omitted, although their presence was confirmed by NMR spectroscopy and ESI-MS of pure, isolated material, vide infra. The structure, and NMR data, of (10) are similar to the closely related complex [Rh(PiPr3)2(H)2(η2-H3B·NMe3)][BArF4],17a with the PiPr3 ligands in trans orientation, cis Rh–H functionality and Rh–B distances of 2.274(9) and 2.333(8) Å respectively]. The 31P{1H} NMR spectrum (CD2Cl2) of isolated (10) confirmed this species as one of the products observed in the mixture, δ 64.7 [d, J(RhP) = 108 Hz]. Presumably the amine–borane in (10) is undergoing a fluxional process that makes the phosphine ligands equivalent at room temperatures, similar to that observed in related complexes39 in which an η2 to η1 change in coordination is accompanied by a rotation around the Rh⋯H–B bond. In the 1H NMR spectrum a broad resonance at δ −2.08 is assigned to the Rh⋯H2B interaction, and a sharper one at δ −19.05 (integral 2 H) assigned to Rh–H. The 11B{1H} NMR spectrum shows a signal at δ 8.0 assigned to the amine–borane. ESI-MS confirmed the formulation of the cation to be [Rh(PiPr3)2(H)2(η2-H2PhB·NMe2H)]+ (m/z = 560.32 (found), 560.32 (calculated)). Complex (10) forms by sequential B–H/N–H activation at a Rh(I) centre, to form a Rh(III) dihydride, which then coordinates another equivalent of (II) to liberate free (6). Such activation processes are well established.3,27
The second product isolated from the reaction mixture, [Rh(PiPr3)2(H)(BPhNMe2)][BArF4] (11), is more unusual. The solid-state structure shows a complex in which the phosphine ligands are arranged in a trans orientation, and a molecule of aminoborane (6) has undergone overall oxidative addition of the B–H bond at the {Rh(PiPr3)2}+ fragment to form a terminal hydride (located in the final difference map) and a direct Rh–B bond, i.e. an amino–boryl species.
Group 9 amino–boryl species have been isolated previously, coming from B–H activation, e.g. [Rh(IMes)2(H){B(H)NMe2}]+ [C, IMes = N,N-bis(2,4,6-trimethylphenyl)imidazol-2-ylidene]40 or [Rh(κ3-POP-Xantphos)(H)(B(H)NiPr2)(NCMe)][BArF4].41 The Rh–B distance in (11) [1.929(9) Å] is, within error, the same as that found in (C) [1.960(9) Å]. A relatively short B–N bond [1.411(14) Å, cf (C) 1.390(15) Å] suggests aminoborane character is retained, and the B–Caryl bond distance of 1.536(14) Å is consistent with a single bond. The angles around boron sum to 360°, demonstrating sp2 character, although the Rh–B(1)–C(1) angle of 98.3(7)° is much smaller than might be expected for such hybridisation. Overall these metrics point to an amino–boryl species, rather than an alternative borylene structure.42 There appears to be a vacant site that sits cis to Rh–H and Rh–B. There are no close25,43 Rh⋯C interactions from the iPr ligands that would point to an agostic interaction [shortest Rh⋯C 3.227 Å]. There is, however, a relatively short Rh⋯C distance to the ipso-phenyl carbon atom [2.634(8) Å] that, when combined with the compressed Rh–B–C angle, suggests a Rh⋯C(ipso) interaction. The next closest distance to the phenyl group is 3.042(9) Å (ortho carbon), longer than would be expected for a η2-arene type interaction. The geometry is reminiscent of the η2-benzyl complexes that interact via methylene and ipso carbon atoms.44 Including the Rh⋯C(ipso) interaction complex (11) can be described as a 16-electron Rh(III) species, and (11) is also related to the 16-electron Rh(PiPr3)2(Bcat)(H)Cl that comes from oxidative addition of HBcat to a Rh(I) precursor (cat = catechol).45 Interestingly, in the system when a chelating phosphine is used an η6-complex is isolated, Rh(iPr2PCH2CH2PiPr2){(η6-cat)Bcat}, paralleling the observations described herein.
In the 31P{1H} NMR spectrum of (11) a doublet is observed [δ 48.8, J(RhP) = 120 Hz]. The chemical shift of δ 50.1 for the aminoborane observed in the 11B{1H} NMR spectrum places the complex as a boryl, rather than a borylene42 [cf. aminoboryl (C) δ 50.1]. The 1H NMR spectrum displays the terminal Rh–H at δ −21.71 [doublet of triplets, J(RhH) = 64 Hz, J(PH) = 12 Hz], the unusually large Rh–H coupling constant31a indicates a hydride bound to low coordinate Rh(III) centre; e.g. (C) J(RhH) = 43 Hz.40a or [Rh(PtBu3)2(H)2][BArF4] J(RhH) = 59 Hz.25 Two distinct resonances are observed for the N–Me groups demonstrating a lack of rotation around the B–N bond on the NMR timescale, consistent with a BN multiple bond character. The phenyl region shows 3 signals in the ratio 1:3:1 (in addition to the [BArF4]− resonances), demonstrating that there is not free rotation around B(1)–C(1).
A plausible mechanism for the formation of complex (11) invokes dehydrogenation of (II) to form (10) and aminoborane (6), followed by much faster reaction of the latter with residual [Rh(PiPr3)2(η6-C6H5F)][BArF4] to form (11), overall in equal ratio to (10). When three equivalents of (II) were combined with [Rh(PiPr3)2(η6-C6H5F)][BArF4] the rapid (15 minutes) formation of only (10) was observed followed by the slow dehydrocoupling (hours) to form aminoborane (6), during which time (10) is observed as a resting state (Scheme 8). From this reaction mixture (10) could be isolated pure in good yield (57%) by layering with pentane and storage at −25 °C. A solution of pure (10) did not show any changes, suggesting (11) does not form from (10). Using aminoborane (6) generated catalytically (Scheme 6) reaction (overall oxidative addition) with [Rh(PiPr3)2(η6-C6H5F)][BArF4] is very rapid (on time of mixing) to form (11). In contrast there is no reaction of (6) with the Rh(I) sigma-complex (1) over 24 h, showing that aminoborane (6) will not displace amine–borane (I) under these conditions. A proposed mechanism is summarised in Scheme 9.
 | ||
| Scheme 8 Reactivity of (II) and (6). | ||
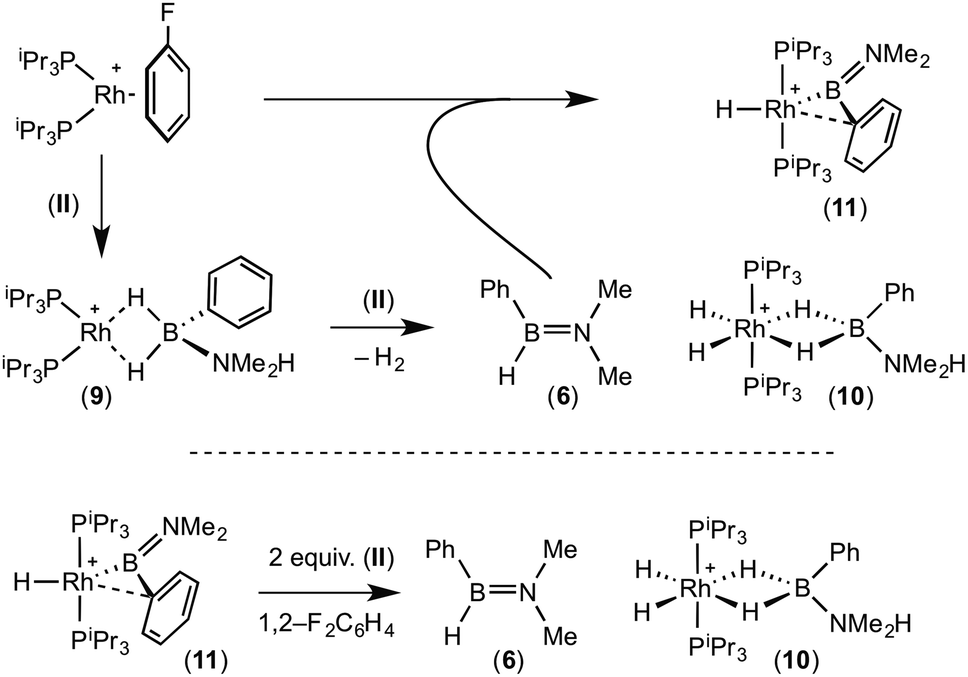 | ||
| Scheme 9 Proposed mechanism for the formation of (10) and (11), and reactivity of (11) with amine–borane (II). | ||
Complex (11) reacts with 2 equivalents of amine–borane (II) to give (10) and (6) on time of mixing. The current data do not discriminate between two possible mechanisms for this transformation. A sigma-bond metathesis/β-elimination of (11) with (II) to eliminate (6), or a reversible reductive elimination of (6) to give a {Rh(PiPr3)2}+ fragment which undergoes reaction with (II) as described (Scheme 8). Whatever the mechanism, complex (10) and aminoborane (6) are the ultimate products when excess amine–borane is present, consistent with their observation during catalysis. Pure complex (10) was found to be a slow catalyst for the dehydrogenation of (II) to form (6). Slow amine–borane dehydrogenation when catalysts sit in a Rh(III) dihydride resting state has been noted previously.35b
Conclusions
We have shown here that the bite angle in {Rh(P2)}+ type fragments can have a significant effect in determining whether Rh⋯H2B η2-sigma amine–borane complexes or Rh⋯arene η6 complexes are formed with B-substituted amine–boranes. Wider bite angles (i.e. monodentate phosphines) tend to favour η2 coordination modes, and relatively rapid B–H/N–H activation with a secondary B-substituted amine–borane to afford a Rh(III) dihydride complex and a B-substituted aminoborane. With constrained, chelating, phosphines η6 complexes can be isolated instead in which the amine–borane moiety is intact and dehydrogenation is slow. This difference in stoichiometric reactivity balances out the reported large differences in catalytic dehydrocoupling rate of {Rh(PR3)2(H)2}+ fragment (slow) versus {Rh(chelating phosphine)}+ (fast) with H3B·NMe2H, which does not bear aryl substituents. Thus, although {Rh(PiPr3)2(H)2}+ dehydrocouples H2PhB·NMe2H slowly, the η6 coordination mode observed with {Rh(iPr2P(CH2)3PiPr2)}+ means that B–H (and subsequent N–H) activation by an inner sphere coordination/activation mechanism are also slowed so that now both fragments operate at a similar rate. Such observations are potentially important in the design of systems that dehydropolymerise arene-substituted amine–boranes (i.e. BN polystyrene analogues) as rapid dehydrogenation to form putative aminoborane intermediates that then can undergo B–N bond forming process are likely central to any successful catalyst system. We thus suggest that systems that form strong adducts with arene π-systems are less likely to be good candidates for such transformations.Experimental
General experimental details
All manipulations, unless otherwise stated, were performed under an atmosphere of argon, using standard Schlenk and glove-box techniques. Glassware was oven dried at 130 °C overnight and flamed under vacuum prior to use. Dichloromethane, diethyl ether and pentane were dried using a Grubbs type solvent purification system (MBraun SPS-800) and degassed by successive freeze–pump–thaw cycles.46 CD2Cl2 and 1,2-F2C6H4 were distilled under vacuum from CaH2 and stored over 3 Å molecular sieves, 1,2-F2C6H4 was stirred over alumina for two hours prior to drying. NMR spectra were recorded on a Bruker AVD 500 MHz spectrometer at room temperature unless otherwise stated. For NMR spectra measured in situ in 1,2-F2C6H4, the spectrometer was pre-locked and pre-shimmed using a C6D6 (0.1 mL) and 1,2-F2C6H4 (0.3 mL) sample and 1H NMR spectra were referenced to the centre of the downfield solvent multiplet (δ 7.07). 31P and 11B NMR spectra were referenced against 85% H3PO4 (external) and Et2O·BF3 (external) respectively. Chemical shifts are quoted in ppm and coupling constants in Hz. ESI-MS were recorded on a Bruker MicrOTOF instrument. In all ESI-MS spectra there was a good fit to both the principal molecular ion and the overall isotopic distribution. Microanalyses were performed by Stephen Boyer at the London Metropolitan University.Metal precursor compounds [Rh(PiPr3)2(C6H5F)][BArF4],24 [Rh(PiBu3)2(C6H5F)][BArF4],25 [Rh(iPr2P(CH2)3PiPr2)(C6H5F)][BArF4],18b [Rh(Ph2P(CH2)3PPh2)(C6H5F)][BArF4]17b and [Rh(Ph2P(CH2)5PPh2)(C6H5F)][BArF4]17b were prepared by literature methods and all other starting materials were used as received.
PhH2B·NMe3 (I) and PhH2B·NMe2H (II)
Li[PhBH3] was prepared from PhB(OH)2 and Li[AlH4] as reported in the literature.23 In a typical synthesis, Li[PhBH3] (350 mg, 3.57 mmol) and the appropriate ammonium chloride salt, [NMe3H]Cl (291 mg, 3.57 mmol) or [NMe2H2]Cl (341 mg, 3.57 mmol) were added to a Schlenk flask and immediately dissolved in diethyl ether (20 mL). The mixture (a suspension of white solid) was stirred vigorously for 2 hours and evolution of hydrogen was observed. The mixture was evaporated to dryness in vacuo and pentane (100 ml) added and the mixture stirred vigorously. The solution was transferred by filter cannula to another Schlenk and the remaining white solid washed with pentane (2 × 10 mL). The combined fractions were evaporated to dryness in vacuo to yield the amine–borane PhH2B·NMe3 (309 mg, 58%) or PhH2B·NMe2H (340 mg, 64%) as white solids which were stored in the glove box.Both compounds have been reported previously although prepared by slightly different synthetic routes. The synthesis of PhH2B·NMe3 has been reported several times12,13,22,47 and NMR spectroscopy data have been reported in C6D6.13 Our data for PhH2B·NMe3 matched that previously reported. The synthesis of PhH2B·NMe2H has been reported by a different route14 but no NMR data was given and so is reported below for the first time.
Synthesis of metal complexes
Catalytic generation of aminoborane PhHB![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) NMe2 (6)
NMe2 (6)
The aminoborane PhHBNMe2 (II) was generated catalytically by heating a mixture of amine–borane PhH2B·NMe2H (0.9 mg, 0.0070 mmol) and [Rh(PiPr3)2(C6H5F)][BArF4] (0.5 mg, 0.00035 mmol, 5 mol%) in 1,2-F2C6H4 (0.35 mL) in a high pressure Young's tap NMR tube for 1 hour (77 °C). Attempts to isolate (6) from this mixture by vacuum distillation resulted in decomposition of the aminoborane to uncharacterised products however, we found this solution was sufficiently pure for further reaction. The NMR data reported was measured in situ in 1,2-F2C6H4 after complete catalytic conversion. PhHB![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) NMe2(6):1H NMR (500 MHz, 1,2-F2C6H4): δ 5.05 (broad q, JHB = 120 Hz, 1H, BH), 3.02 (s, 3H, NMe), 2.88 (s, 3H, NMe), phenyl resonances were obscured due to the solvent (1,2-F2C6H4) peaks. 11B{1H} NMR (160 MHz, 1,2-F2C6H4): δ 39.4 (s). 11B NMR (160 MHz, 1,2-F2C6H4): δ 39.4 (d, 1JBH = 123 Hz).
NMe2(6):1H NMR (500 MHz, 1,2-F2C6H4): δ 5.05 (broad q, JHB = 120 Hz, 1H, BH), 3.02 (s, 3H, NMe), 2.88 (s, 3H, NMe), phenyl resonances were obscured due to the solvent (1,2-F2C6H4) peaks. 11B{1H} NMR (160 MHz, 1,2-F2C6H4): δ 39.4 (s). 11B NMR (160 MHz, 1,2-F2C6H4): δ 39.4 (d, 1JBH = 123 Hz).
Reaction of aminoborane (6) with sigma-complex (1)
PhHBNMe2 (6) was generated catalytically as above using PhH2B·NMe2H (0.9 mg, 0.0070 mmol) and [Rh(PiPr3)2(C6H5F)][BArF4] (0.5 mg, 0.00035 mmol, 5 mol%). Clean conversion to (6) was checked by 11B NMR spectroscopy and this solution transferred via cannula to an NMR tube containing [Rh(PiPr3)2(η2-H2PhB·NMe3)][BArF4] (1) (10.0 mg, 0.0070 mmol). The reaction was mixed and no colour change was observed. Immediate NMR spectroscopy showed no reaction between (6) and (1) and no change in these spectra was observed after 24 h mixing of the solution by inversion at room temperature.
Reaction of aminoborane (6) with [Rh(PiPr3)2(C6H5F)][BArF4] – an alternative synthesis of (11)
PhHBNMe2 (6) was generated catalytically as above using PhH2B·NMe2H (1.8 mg, 0.0133 mmol) and [Rh(PiPr3)2(C6H5F)][BArF4] (0.9 mg, 0.00067 mmol, 5 mol%). Clean conversion to (6) was checked by 11B NMR spectroscopy and this solution transferred via cannula to an NMR tube containing [Rh(PiPr3)2(η6-C6H5F)][BArF4] (18.4 mg, 0.0133 mmol). The solution was mixed and immediate NMR spectroscopy showed almost quantitative conversion (>95%) to (11) with NMR spectra (measured in situ in 1,2-F2C6H4) matching those reported above. Crystallisation of this solution by layering with pentane and storage at −18 °C resulted in formation of crystals of (11).
Acknowledgements
The EPSRC (EP/J02127X) and the Rhodes Trust for funding.Notes and references
- (a) C. W. Hamilton, R. T. Baker, A. Staubitz and I. Manners, Chem. Soc. Rev., 2009, 38, 279–293 RSC; (b) A. Staubitz, A. P. M. Robertson and I. Manners, Chem. Rev., 2010, 110, 4079–4124 CrossRef CAS PubMed.
- (a) E. M. Leitao, T. Jurca and I. Manners, Nat. Chem., 2013, 5, 817–829 CrossRef CAS PubMed; (b) R. Waterman, Chem. Soc. Rev., 2013, 42, 5629–5641 RSC.
- H. C. Johnson, T. N. Hooper and A. S. Weller, in Synthesis and Application of Organoboron Compounds, Topics in Organometallic Chemistry, ed. E. Fernández and A. Whiting, Springer International Publishing, 2015, vol. 49, pp. 153–220 Search PubMed.
- A. Staubitz, A. P. M. Robertson, M. E. Sloan and I. Manners, Chem. Rev., 2010, 110, 4023–4078 CrossRef CAS PubMed.
- (a) C. A. Jaska, K. Temple, A. J. Lough and I. Manners, J. Am. Chem. Soc., 2003, 125, 9424–9434 CrossRef CAS PubMed; (b) H. Helten, A. P. M. Robertson, A. Staubitz, J. R. Vance, M. F. Haddow and I. Manners, Chem. – Eur. J., 2012, 18, 4665–4680 CrossRef CAS PubMed; (c) S.-K. Kim, S.-A. Hong, H.-J. Son, W.-S. Han, A. Michalak, S.-J. Hwang and S. O. Kang, Dalton Trans., 2015, 44, 7373–7381 RSC.
- (a) M. Shimoi, S.-i. Nagai, M. Ichikawa, Y. Kawano, K. Katoh, M. Uruichi and H. Ogino, J. Am. Chem. Soc., 1999, 121, 11704–11712 CrossRef CAS; (b) G. J. Kubas, Metal Dihydrogen and σ-Bond Complexes, Springer, New York, 2001 Search PubMed; (c) G. Alcaraz and S. Sabo-Etienne, Angew. Chem., Int. Ed., 2010, 49, 7170–7179 CrossRef CAS PubMed.
- (a) P. G. Campbell, J. S. A. Ishibashi, L. N. Zakharov and S.-Y. Liu, Aust. J. Chem., 2014, 67, 521–524 CrossRef CAS; (b) N. E. Stubbs, A. Schäfer, A. P. M. Robertson, E. M. Leitao, T. Jurca, H. A. Sparkes, C. H. Woodall, M. F. Haddow and I. Manners, Inorg. Chem., 2015, 54, 10878–10889 CrossRef CAS PubMed.
- (a) H. Anane, A. Jarid, A. Boutalib, I. Nebot-Gil and F. Tomás, J. Mol. Struct. (THEOCHEM), 1998, 455, 51–57 CrossRef CAS; (b) D. J. Grant, M. H. Matus, K. D. Anderson, D. M. Camaioni, S. R. Neufeldt, C. F. Lane and D. A. Dixon, J. Phys. Chem. A, 2009, 113, 6121–6132 CrossRef CAS PubMed.
- (a) A. P. M. Robertson, G. R. Whittell, A. Staubitz, K. Lee, A. J. Lough and I. Manners, Eur. J. Inorg. Chem., 2011, 2011, 5279–5287 CrossRef CAS; (b) A. P. M. Robertson, M. F. Haddow and I. Manners, Inorg. Chem., 2012, 51, 8254–8264 CrossRef CAS PubMed.
- P. G. Campbell, A. J. V. Marwitz and S.-Y. Liu, Angew. Chem., Int. Ed., 2012, 51, 6074–6092 CrossRef CAS PubMed.
- (a) P. G. Campbell, L. N. Zakharov, D. J. Grant, D. A. Dixon and S.-Y. Liu, J. Am. Chem. Soc., 2010, 132, 3289–3291 CrossRef CAS PubMed; (b) W. Luo, L. N. Zakharov and S.-Y. Liu, J. Am. Chem. Soc., 2011, 133, 13006–13009 CrossRef CAS PubMed; (c) W. Luo, P. G. Campbell, L. N. Zakharov and S.-Y. Liu, J. Am. Chem. Soc., 2011, 133, 19326–19329 CrossRef CAS PubMed; (d) W. Luo, D. Neiner, A. Karkamkar, K. Parab, E. B. Garner Iii, D. A. Dixon, D. Matson, T. Autrey and S.-Y. Liu, Dalton Trans., 2013, 42, 611–614 RSC; (e) G. Chen, L. N. Zakharov, M. E. Bowden, A. J. Karkamkar, S. M. Whittemore, E. B. Garner, T. C. Mikulas, D. A. Dixon, T. Autrey and S.-Y. Liu, J. Am. Chem. Soc., 2015, 137, 134–137 CrossRef CAS PubMed; (f) A. Kumar, J. S. A. Ishibashi, T. N. Hooper, T. C. Mikulas, D. A. Dixon, S.-Y. Liu and A. S. Weller, Chem. – Eur. J., 2016, 22, 310–322 CrossRef CAS PubMed.
- E. Wiberg, J. E. F. Evans and H. Noth, Z. Naturforsch., B: Anorg. Chem. Org. Chem. Biochem. Biophys. Biol., 1958, 13, 263–264 Search PubMed.
- Y. Kawano, K. Yamaguchi, S.-y. Miyake, T. Kakizawa and M. Shimoi, Chem. – Eur. J., 2007, 13, 6920–6931 CrossRef CAS PubMed.
- B. M. Mikhailov and V. A. Dorokhov, Russ. Chem. Bull., 1962, 11, 1138–1142 CrossRef.
- S.-K. Kim, W.-S. Han, T.-J. Kim, T.-Y. Kim, S. W. Nam, M. Mitoraj, Ł. Piekoś, A. Michalak, S.-J. Hwang and S. O. Kang, J. Am. Chem. Soc., 2010, 132, 9954–9955 CrossRef CAS PubMed.
- (a) P. Dierkes and P. W. N. M. van Leeuwen, J. Chem. Soc., Dalton Trans., 1999, 1519–1530 RSC; (b) D. Aguila, E. Escribano, S. Speed, D. Talancon, L. Yerman and S. Alvarez, Dalton Trans., 2009, 6610–6625 RSC.
- (a) A. B. Chaplin and A. S. Weller, Inorg. Chem., 2010, 49, 1111–1121 CrossRef CAS PubMed; (b) R. Dallanegra, A. P. M. Robertson, A. B. Chaplin, I. Manners and A. S. Weller, Chem. Commun., 2011, 47, 3763–3765 RSC.
- (a) S. D. Pike, I. Pernik, R. Theron, J. S. McIndoe and A. S. Weller, J. Organomet. Chem., 2015, 784, 75–83 CrossRef CAS; (b) I. Pernik, J. F. Hooper, A. B. Chaplin, A. S. Weller and M. C. Willis, ACS Catal., 2012, 2, 2779–2786 CrossRef CAS.
- J. Silvestre and T. A. Albright, J. Am. Chem. Soc., 1985, 107, 6829–6841 CrossRef CAS.
- T. A. Albright, J. K. Burdett and M. H. Whangbo, Orbital Interactions in Chemistry, 2nd edn, Wiley, New York, 2013 Search PubMed.
- M.-D. Su and S.-Y. Chu, Inorg. Chem., 1998, 37, 3400–3406 CrossRef CAS.
- M. F. Hawthorne, J. Am. Chem. Soc., 1958, 80, 4291–4293 CrossRef CAS.
- B. Singaram, T. E. Cole and H. C. Brown, Organometallics, 1984, 3, 774–777 CrossRef CAS.
- A. B. Chaplin, A. I. Poblador-Bahamonde, H. A. Sparkes, J. A. K. Howard, S. A. Macgregor and A. S. Weller, Chem. Commun., 2009, 244–246 RSC.
- T. M. Douglas, A. B. Chaplin and A. S. Weller, Organometallics, 2008, 27, 2918–2921 CrossRef CAS.
- C. A. Tolman, Chem. Rev., 1977, 77, 313–348 CrossRef CAS.
- T. M. Douglas, A. B. Chaplin, A. S. Weller, X. Yang and M. B. Hall, J. Am. Chem. Soc., 2009, 131, 15440–15456 CrossRef CAS PubMed.
- N. Merle, G. Koicok-Köhn, M. F. Mahon, C. G. Frost, G. D. Ruggerio, A. S. Weller and M. C. Willis, Dalton Trans., 2004, 3883–3892 RSC.
- G. Alcaraz, E. Clot, U. Helmstedt, L. Vendier and S. Sabo-Etienne, J. Am. Chem. Soc., 2007, 129, 8704–8705 CrossRef CAS PubMed.
- (a) H. C. Johnson, C. L. Mcmullin, S. D. Pike, S. A. Macgregor and A. S. Weller, Angew. Chem., Int. Ed., 2013, 52, 9776–9780 CrossRef CAS PubMed; (b) L. J. Sewell, A. B. Chaplin and A. S. Weller, Dalton Trans., 2011, 40, 7499–7501 RSC.
- (a) P. Pregosin, NMR in Organometallic Chemistry, Wiley-VCH, Weinheim, 2012 Search PubMed; (b) A. Woolf, A. B. Chaplin, J. E. McGrady, M. A. M. Alibadi, N. Rees, S. Draper, F. Murphy and A. S. Weller, Eur. J. Inorg. Chem., 2011, 2011, 1614–1625 CrossRef.
- W. F. Bailey, H. Connon, E. L. Eliel and K. B. Wiberg, J. Am. Chem. Soc., 1978, 100, 2202–2209 CrossRef CAS.
- T. M. Douglas, E. Molinos, S. K. Brayshaw and A. S. Weller, Organometallics, 2007, 26, 463–465 CrossRef CAS.
- (a) P. Herich, J. Kameníček, K. Kuča, M. Pohanka and M. Olšovský, Polyhedron, 2009, 28, 3565–3569 CrossRef CAS; (b) M. Schwach, H. D. Hausen and W. Kaim, Chem. – Eur. J., 1996, 2, 446–451 CrossRef CAS.
- (a) A. D. Wilson, A. J. M. Miller, D. L. DuBois, J. A. Labinger and J. E. Bercaw, Inorg. Chem., 2010, 49, 3918–3926 CrossRef CAS PubMed; (b) L. J. Sewell, G. C. Lloyd-Jones and A. S. Weller, J. Am. Chem. Soc., 2012, 134, 3598–3610 CrossRef CAS PubMed.
- (a) Y. Kawano, M. Uruichi, M. Shimoi, S. Taki, T. Kawaguchi, T. Kakizawa and H. Ogino, J. Am. Chem. Soc., 2009, 131, 14946–14957 CrossRef CAS PubMed; (b) L. Pasumansky, D. Haddenham, J. W. Clary, G. B. Fisher, C. T. Goralski and B. Singaram, J. Org. Chem., 2008, 73, 1898–1905 CrossRef CAS PubMed.
- (a) R. Dallanegra, A. B. Chaplin and A. S. Weller, Angew. Chem., Int. Ed., 2009, 48, 6875–6878 CrossRef CAS PubMed; (b) X. Chen, J.-C. Zhao and S. G. Shore, Acc. Chem. Res., 2013, 46, 2666–2675 CrossRef CAS PubMed; (c) V. S. Nguyen, M. H. Matus, D. J. Grant, M. T. Nguyen and D. A. Dixon, J. Phys. Chem. A, 2007, 111, 8844–8856 CrossRef CAS PubMed; (d) A. Kumar, H. C. Johnson, T. N. Hooper, A. S. Weller, A. G. Algarra and S. A. Macgregor, Chem. Sci., 2014, 5, 2546–2553 RSC.
- J. F. Hartwig, Organotransition Metal Chemistry, University Science Books, Sausalito, USA, 2010 Search PubMed.
- A. G. Algarra, L. J. Sewell, H. C. Johnson, S. A. Macgregor and A. S. Weller, Dalton Trans., 2014, 43, 11118–11128 RSC.
- (a) C. Y. Tang, N. Phillips, J. I. Bates, A. L. Thompson, M. J. Gutmann and S. Aldridge, Chem. Commun., 2012, 48, 8096 RSC; (b) M. O'Neill, D. A. Addy, I. Riddlestone, M. Kelly, N. Phillips and S. Aldridge, J. Am. Chem. Soc., 2011, 133, 11500–11503 CrossRef PubMed; (c) C. Y. Tang, A. L. Thompson and S. Aldridge, J. Am. Chem. Soc., 2010, 132, 10578–10591 CrossRef CAS PubMed.
- H. C. Johnson, E. M. Leitao, G. R. Whittell, I. Manners, G. C. Lloyd-Jones and A. S. Weller, J. Am. Chem. Soc., 2014, 136, 9078–9093 CrossRef CAS PubMed.
- H. Braunschweig, R. D. Dewhurst and V. H. Gessner, Chem. Soc. Rev., 2013, 42, 3197–3208 RSC.
- A. C. Cooper, E. Clot, J. C. Huffman, W. E. Streib, F. Maseras, O. Eisenstein and K. G. Caulton, J. Am. Chem. Soc., 1999, 121, 97–106 CrossRef CAS.
- N. H. Dryden, P. Legzdins, J. Trotter and V. C. Yee, Organometallics, 1991, 10, 2857–2870 CrossRef CAS.
- (a) S. A. Westcott, N. J. Taylor, T. B. Marder, R. T. Baker, N. J. Jones and J. C. Calabrese, J. Chem. Soc., Chem. Commun., 1991, 304–305 RSC; (b) W. H. Lam, S. Shimada, A. S. Batsanov, Z. Lin, T. B. Marder, J. A. Cowan, J. A. K. Howard, S. A. Mason and G. J. McIntyre, Organometallics, 2003, 22, 4557–4568 CrossRef CAS.
- A. B. Pangborn, M. A. Giardello, R. H. Grubbs, R. K. Rosen and F. J. Timmers, Organometallics, 1996, 15, 1518–1520 CrossRef CAS.
- J. J. Miller and F. A. Johnson, Inorg. Chem., 1970, 9, 69–74 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1445275–1445282. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt00197a |
| This journal is © The Royal Society of Chemistry 2016 |