
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructure–performance relationships of Cr/Ti/SiO2 catalysts modified with TEAl for oligomerisation of ethylene: tuning the selectivity towards 1-hexene
D.
Cicmil
a,
I. K.
van Ravenhorst
a,
J.
Meeuwissen
b,
A.
Vantomme
b and
B. M.
Weckhuysen
*a
aInorganic Chemistry and Catalysis, Debye Institute for Nanomaterials Science, Utrecht University, Universiteitsweg 99, 3584 CG Utrecht, The Netherlands. E-mail: b.m.weckhuysen@uu.nl
bTotal Research and Technology Feluy, Zone Industrielle C, B-7181 Seneffe, Belgium
First published on 18th November 2015
Abstract
A study on the influence of H2 and the degree of titanation of a Cr/SiO2 Phillips polymerisation catalyst on the selective oligomerisation of ethylene induced by pre-contacting the catalyst with triethylaluminium (TEAl) is presented. Ethylene oligomerisation reactions were performed at 373 K and 1 bar, inside a quartz reactor of a specially designed operando experimental setup, which allowed examination of the catalysts by UV-vis-NIR diffuse reflectance spectroscopy, while the gas phase was continuously monitored by on-line mass spectroscopy and subsequently analysed by gas chromatography. With this combination of techniques, it was possible to acquire detailed insight into the different distributions of produced oligomers, which were highly dependent on the catalyst structure. The addition of small amounts of Ti significantly changes the electronic environment of Cr oligomerisation sites by the formation of Cr–O–Ti–O–Si linkages, favouring β-H transfer and increasing the selectivity towards butene at the expense of 1-hexene. Moreover, we show that ethylene oligo-/polymerisation reactions follow at least two different pathways, i.e. metallacycle for olefinic species with a broken Schulz-Flory distribution, and Cossee–Arlman for other hydrocarbon species.
Introduction
Phillips-type Cr/SiO2 polymerisation catalysts are responsible for the commercial production of more than one third of all polyethylene sold worldwide.1–4 This has attracted a great deal of research attention both in academia and chemical industries since their discovery in the early 1950s by J. P. Hogan and R. L. Banks at Phillips Petroleum Company.5,6 Despite the numerous research efforts on the oxidation states of the catalyst,7–11 the molecular structure of the active sites,2,12–16 and the mechanism of ethylene polymerisation with Phillips-type catalysts,17–23 however, these key scientific questions still require further attention.No univocal consensus has been yet achieved in both academia and chemical industries, especially in studies with polymerisation conditions similar to industrial ones, as many of the research studies involved model systems, temperatures and pressures, which were quite different from the conditions utilised in industrial systems. One of the reasons is the very high sensitivity of the Cr/SiO2 ethylene polymerisation catalyst to trace amounts of catalytic poisons, i.e. H2O, O2 and small oxygenated organic compounds. Furthermore, the fast rate of polymerisation and high productivity of the catalysts make the study of the initial stages of ethylene polymerisation very challenging.
Certain adjustments of the catalyst furthermore complicate the picture. Cr/SiO2 ethylene polymerisation catalysts can often be modified by incorporation of titania, yielding a Cr/Ti/SiO2 catalyst with increased activity and capability of producing polyethylene with a lower molecular weight and a broader molecular weight distribution than the pristine catalyst. Titanation can be performed in two manners, either by co-precipitation with silica gel, when titania becomes highly dispersed in the bulk catalyst material,24 or by reaction of a titanium compound with the hydroxyl groups of the support, therefore coating the silica surface with a layer of titania.25–28 Consequently, both methods include the formation of Cr–O–Ti–O–Si linkages,29 which change the electronic environment of the active sites and influence the mechanism of ethylene polymerisation.
One of the unique aspects of Phillips-type Cr/SiO2 catalysts is that they do not require activation by a metal alkyl co-catalyst as compared to Ziegler–Natta and metallocene polymerisation catalysts and therefore these compounds are often excluded, even at industrial production sites.30–32 However, when metal alkyls are used in combination with Phillips-type Cr/SiO2 catalysts, the development of the polymerisation rate is commonly accelerated and the induction time is decreased. It is believed that these effects are caused by a more facile reduction of the Cr6+ species of the activated catalyst to Cr species in a lower oxidation state, which are thought to be responsible for the polymerisation of ethylene.1 Most of the authors attribute the polymerisation-active sites to Cr2+ species of different coordinative unsaturations19,33–35 although other species of higher valences, i.e. Cr3+, Cr4+ and Cr5+, should still be taken into consideration,8,10,36–38 especially in view of the recent work of the group of Copéret.20,21,39 Secondly, metal alkyl co-catalysts can improve the polymerisation rate by alkylation of Cr sites and provide chain initiation similarly to Ziegler–Natta catalysts. However, with Phillips-type catalysts, this chain initiation by a co-catalyst is not necessary. Thirdly, as highly reactive compounds, metal alkyls can remove the poisons still present in the gas feed or in the system, such as the ethylene oxidation products formed during the initiation stage that might remain adsorbed on the Cr sites. Depending on the type of co-catalyst, there is an optimal amount expressed as the metal-to-Cr ratio, which gives the highest catalytic activity. Besides the improvement of the activity and decrease of the induction time, further increase of the co-catalyst amount can deteriorate the polymerisation rate or even destroy the catalyst due to attack on Cr–support bonds. Some of the metal alkyls can act as chain transfer agents, exchanging the alkyl groups with Cr polymerisation sites, and therefore affect the properties of the produced polyethylene, although less than in Ziegler–Natta and metallocene catalysis. Co-catalysts can also influence the polymerisation sites by reacting with Cr or neighbouring groups and directly modifying the polymerisation activity and the type of polyethylene that is produced.
One of the most interesting facts regarding co-catalysts and their interaction with the Cr sites is that the addition of metal alkyl compounds to the reactor containing a Phillips-type catalyst changes the amount of short-chain branching of the produced polyethylene, even if no α-olefin co-monomers were added to the system. This short-chain branching of the polyethylene chain originates from the co-polymerisation of ethylene with the in situ produced ethylene α-oligomers on modified polymerisation active sites.40,41In situ branching attracts high interest in the industrial world due to the possibility of reducing or even eliminating the separate feed of α-olefin co-monomers, their transportation, storage and purification, therefore simplifying manufacturing procedures and production costs of MDPE and LLDPE. The recent drop in ethylene price of more than $500 per metric ton from January 2014 to January 201542 decreases the production cost of polyethylene and increases the economic importance of in situ ethylene oligomerisation. Therefore, an accurate description and finding the means to directly influence in situ ethylene oligomerisation, i.e. selectivity, olefin distribution and their yield, proves to be one of the hottest topics in the ethylene polymerisation industry.
Although a lot of progress has been made in the last few decades in resolving these issues by applying advanced characterisation techniques,18,40,43–46 no unifying picture has yet been achieved. Here, we present an operando UV-vis-NIR diffuse reflectance study (DRS) of a Cr/Ti/SiO2 catalyst in order to elucidate the TEAl-induced in situ ethylene oligomerisation properties.
Experimental
Catalyst preparation
The catalyst samples were provided by Total Research and Technology Feluy, Belgium. Catalyst preparation includes the treatment of two commercially available Cr/SiO2 pre-catalysts containing either a higher or a lower Cr loading, with the following properties: a pre-catalyst with ~1.0 wt% Cr loading, a surface area of 500 m2 g−1, a pore volume of 1.5 cm3 g−1 and a D50 particle size diameter of 72 μm, and a pre-catalyst with ~0.5 wt% Cr loading, a surface area of 318 m2 g−1, a pore volume of 1.55 cm3 g−1 and a D50 particle size diameter of 47 μm. The pale white silica pre-catalyst was placed in a fluidised bed reactor under N2 flow and slowly heated to 543 K for dehydration. While still under N2 flow, in the case of the titanated samples, the pre-catalyst material was surface-titanated, according to the procedure described by Debras et al., to either 2 wt% or 4 wt% of the total Ti loading using titanium isopropoxide (99.999% trace metals basis, Sigma-Aldrich) which was added dropwise to the fluidised bed.25 After flushing under N2 for 45 min, the gas flow was changed to dry air and the temperature was slowly increased to the desired activation temperature. During the 6 h oxidation step, the colour of the prepared Cr/SiO2 and Cr/Ti/SiO2 catalysts changed to intense yellow/orange due to the formation of Cr6+ species. After cooling down to RT and switching back to N2 flow, the activated Cr/SiO2 and Cr/Ti/SiO2 catalysts could be obtained under inert N2 atmosphere and transferred to an Ar glove box for storage and further use. An overview of the catalysts' properties and activation temperatures is given in Table 1.| Sample name | Act. temp. (K) | Cr loading (wt%) | Ti loading (wt%) | Surface area (m2 g−1) | Pore volume (cm3 g−1) | O → Cr6+ CT (cm−1) | ||
|---|---|---|---|---|---|---|---|---|
| S | 1048 | 0 | 0 | 246 | 1.39 | n.a. | ||
| TS | 1048 | 0 | 4.7 | 254 | 1.51 | n.a. | ||
| CS | 1048 | 0.52 | 0 | 296 | 1.30 | 38![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 500 |
29500 |
21900 |
| CTS1 | 1048 | 0.62 | 2.2 | 293 | 1.44 | 38100 |
28700 |
21700 |
| CTS2 | 1048 | 0.56 | 3.9 | 277 | 1.39 | 37500 |
27800 |
21600 |
| CTS3 | 923 | 0.56 | 4.0 | 289 | 1.34 | 37200 |
27800 |
21600 |
| CTS4 | 823 | 0.55 | 3.8 | 294 | 1.35 | 36500 |
27500 |
21600 |
| CTS5 | 1048 | 0.98 | 4.7 | 367 | 1.13 | 36500 |
27500 |
21700 |
Characterisation of the Cr/SiO2 ethylene polymerisation catalysts
 | ||
| Fig. 1 UV-vis-NIR diffuse reflectance spectroscopy, mass spectrometry and gas chromatography setup developed for the testing of solid catalysts and products obtained in gas phase in reactions at 1 bar and temperatures in the range from RT to 473 K. The operando setup includes changeable gas reactant sources, mass flow controller setup (green), septum for the injection of the co-catalyst, 4 cm3 air-tight quartz reactor (red), Varian Cary 500 spectrophotometer (blue), OmniStar mass spectrometer (orange) and gas sampling bag for the GC(-MS) analysis of gas phase (purple). All lines and the reactor are traced and heated to the desired reaction temperature monitored by a couple of thermocouples. | ||
:Cr ratio of 2:1) triethylaluminium co-catalyst (TEAl) in heptanes (~94 wt% TEAl, with ~6 wt% predominately tri-n-butylaluminium and less than 0.1 wt% triisobutylaluminium residue, Acros Organics), which was injected through a silicon septum into a N2 stream of 10 cm3 min−1. After the stabilisation of the spectra, a gas reaction mixture consisting of either 45 vol% N2, 45 vol% C2H4 and 10 vol% H2 or 50 vol% N2 and 50 vol% C2H4 was fed into the cell for a total flow of 10 cm3 min−1. The gases were provided by Linde Gas with the following purities N2 (99.999%), H2 (99.999%) and C2H4 (99.95%). Modification of the catalyst with TEAl and polymerisation reaction were simultaneously monitored with operando UV-vis-NIR DRS, while the gaseous products were analysed on-line with MS and collected for GC analysis.
000 cm−1, with 17 cm−1 spectral resolution and 33 ms data point scan time in the UV-vis region (12500–45000 cm−1) and 7.1 cm−1 spectral resolution and 33 ms data point scan time in the NIR region (4000–12500 cm−1). The UV-vis-NIR DRS spectra were corrected for the detector/grating and light source changeover steps at 12500 cm−1 and 28570 cm−1, respectively, and smoothed in the UV-vis region using a FFT filter with a 0.00147 cut-off frequency. The spectra were repeatedly taken every 120 s.
Results
Integration of UV-vis-NIR diffuse reflectance spectroscopy, mass spectrometry and gas chromatography: an experimental example
For the sake of clarity, the results obtained by the integration of multiple analytical techniques within the specially designed setup, i.e. UV-vis-NIR diffuse reflectance spectroscopy (DRS), mass spectrometry (MS) and gas chromatography (GC) (Fig. 1), will be first explained for the ethylene polymerisation reaction using the CS catalyst (Table 1), after which the differences between the catalyst structures and reaction conditions will be discussed in the following sections.Fig. 2A shows the on-line MS analysis and time evolution of several fragments of the compounds of interest during the pre-run, catalyst modification with TEAl, ethylene oligo-/polymerisation reaction and post-run. The UV-vis-NIR DRS spectra were repeatedly collected during the TEAl-modification stage and polymerisation reaction, while the gas phase was sampled for the GC analysis during the reaction stage. Numerous mass fragments were monitored although only the evolution of the most relevant fragments was presented, based on the GC analysis shown in Fig. 2B.† The injection of TEAl solution in heptanes into the N2 stream results in a sudden increase of the detector current and saturation of the signal due to the high concentration of the solvents. After 17 min of flushing off the solvent, the gas feed was changed to the reaction mixture consisting of 45 vol% N2, 45 vol% C2H4 and 10 vol% H2. The change in the gas composition is testified by a high rise of the signal with m/z values of 2 and 27 due to H2 and C2H4 reactants, respectively. After the start of the reaction, the signals belonging to the fragments of the in situ produced 1-hexene slowly rise and remain constant until the feed of the reaction mixture was switched off. Furthermore, the TEAl-modified CS catalyst is able to oligomerise ethylene to 1-butene, although in smaller amounts than 1-hexene. At ~30 min of the reaction, a sudden increase of the signals suggests a temporary production of the oligomers with carbon numbers higher than 6. After 60 min of reaction, the gas feed was switched back to N2. The signals due to C2H4 and H2 immediately drop due to the abrupt change and continue to decline slowly together with the signals belonging to the oligomerisation products.
 | ||
| Fig. 2 A) On-line MS analysis of the gas phase during the modification of the CS Phillips-type Cr/SiO2 catalyst with the TEAl co-catalyst and subsequent oligo-/polymerisation of ethylene using the in-house designed operando setup. UV-vis-NIR DRS repeated measurements were started at t = 14 min. At t = 16 min, TEAl was injected into the system via evaporation into the N2 stream. After 17 min, the gas feed was changed to the reaction mixture consisting of 45% N2, 45% C2H4 and 10% H2, at a total flow rate of 10 cm3 min−1 and the gas phase was sampled for the GC analysis. After 60 min of reaction, the gas feed was changed back to N2 and UV-vis-NIR DRS and GC sampling stopped. Assignment of the m/z is as follows: 2 H2; 27 ethylene, 1-butene, 1-hexene, hexane, TEAl, butane, heptanes; 43 1-butene, butane, heptanes, octane; 41 1-butene, 1-hexene, hexane, butane, heptanes; 56 1-hexene, hexane, 1-butene; 42, 55, 69, 84 1-hexene, 1-octene; 57 hexane, TEAl, heptanes; 71, 85 TEAl, octane; 71, 85, 114 TEAl; 86 hexane and 112 1-octene. B) GC analysis of the gas phase collected during oligo-/polymerisation of ethylene after the modification of the catalyst with TEAl. | ||
The origin of the fragments detected during the on-line MS analysis could be identified and confirmed by the GC-MS and GC analysis of the sampled gas phase (Fig. 2B). In this particular case when the CS catalyst was used, the main oligomer product detected was 1-hexene. Moreover, a small amount of the in situ produced saturated and unsaturated ethylene dimers and tetramers was identified. The GC and MS results unambiguously confirm the ethylene oligomerisation properties of the tested catalyst material.
Furthermore, the UV-vis-NIR DRS spectrometer integrated within the setup offered monitoring of the catalyst material during the course of the reaction. Analysis of the UV-vis part is suitable for inspecting the electronic spectra of supported transition metal ions, which allows determination of the oxidation number, coordination state and symmetry of a transition metal ion by studying d–d and charge transfer (CT) transitions.47,48 Moreover, the NIR part offers the possibility of examining vibrational properties i.e. combination bands and overtones of the vibrational modes of the materials and reaction products. The expected electronic and vibrational bands due to the silica-supported Cr and Ti species (if present inside the catalyst),14,16,49,50 and polymer51 produced therewith are summarised in Table 2.
| Assignment |
![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) (cm−1) (cm−1) |
|---|---|
| σ → σ* C–C | ~67000 |
π → π* C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C C |
52000–59000 |
| O → Ti4+ CT | 37500–40000 |
| O → Cr6+ CT | 37600–39600 |
| O → Cr6+ CT | 27000–31000 |
| O → Cr6+ CT | 21000–22000 |
| Cr3+Oh d–d | ~16000 |
| Cr2+Oh d–d | ~10000 |
| 3νas(CH2/CH3) | 8423; 8233 |
| 2ν(SiOH) | 7325 |
| 2ν(TiOH) | 7270 |
| 2νas/s(CH2/CH3) + δ(CH2/CH3) | 7189; 7059; 6966; 6482 |
| 2νas/s(CH2/CH3) | 5777; 5668 |
| ν(H2O) + δ(H2O) | 5260 |
| ν(SiOH) + δ(SiOH) | 4525 |
| ν as/s(CH2/CH3) + δ(CH2/CH3) | 4325; 4250 |
| ν as(CH2/CH3) + w(CH2/CH3) | 4185 |
During the pre-treatment of the CS catalyst with TEAl (Fig. 3A and B), the UV-vis-NIR DRS spectra of the catalyst show a small intensity increase of the bands at 16000 cm−1 and 10000 cm−1 due to the 4A2g → 4T2g and 5Eg → 5T2g d–d transitions of the formed Cr3+Oh and Cr2+Oh species. Due to the small amount of modified surface sites, the reduction is more evident after the subtraction of the fresh catalyst spectrum from the final spectrum of the sample after TEAl modification, as shown in Fig. 4. The negative bands at 21000–22000 cm−1, 27000–31000 cm−1 and 37000–40000 cm−1 originating from the decrease of the CT bands of Cr6+ species are accompanied with the positive bands of the reduced Cr3+Oh and Cr2+Oh species. The addition of the co-catalyst can be clearly noted in the NIR region of the time-resolved spectra (Fig. 3B). The intensity increase of the bands at 4000–4400 cm−1 is due to the combination modes of stretching and deformation vibrations of CH2/CH3 groups originating from the adsorbed alkyl groups of the TEAl and heptane solvents. At higher wavenumbers, the first overtone of the fundamental symmetric and asymmetric CH2/CH3 stretching vibration appears in the 5600–5900 cm−1 region. Furthermore, the first stretching vibration overtone and deformation band can be observed in the 6900–7200 cm−1 region. In the first overtone OH stretching region, consumption of the silanol groups at 7325 cm−1 occurs due to the interaction with adsorbed TEAl and heptanes. After flushing with N2, the OH functionality of the catalysts is mostly restored, giving almost identical spectra to those obtained before the treatment with TEAl, although a broadening and red shift of the associated absorption band can be noted.
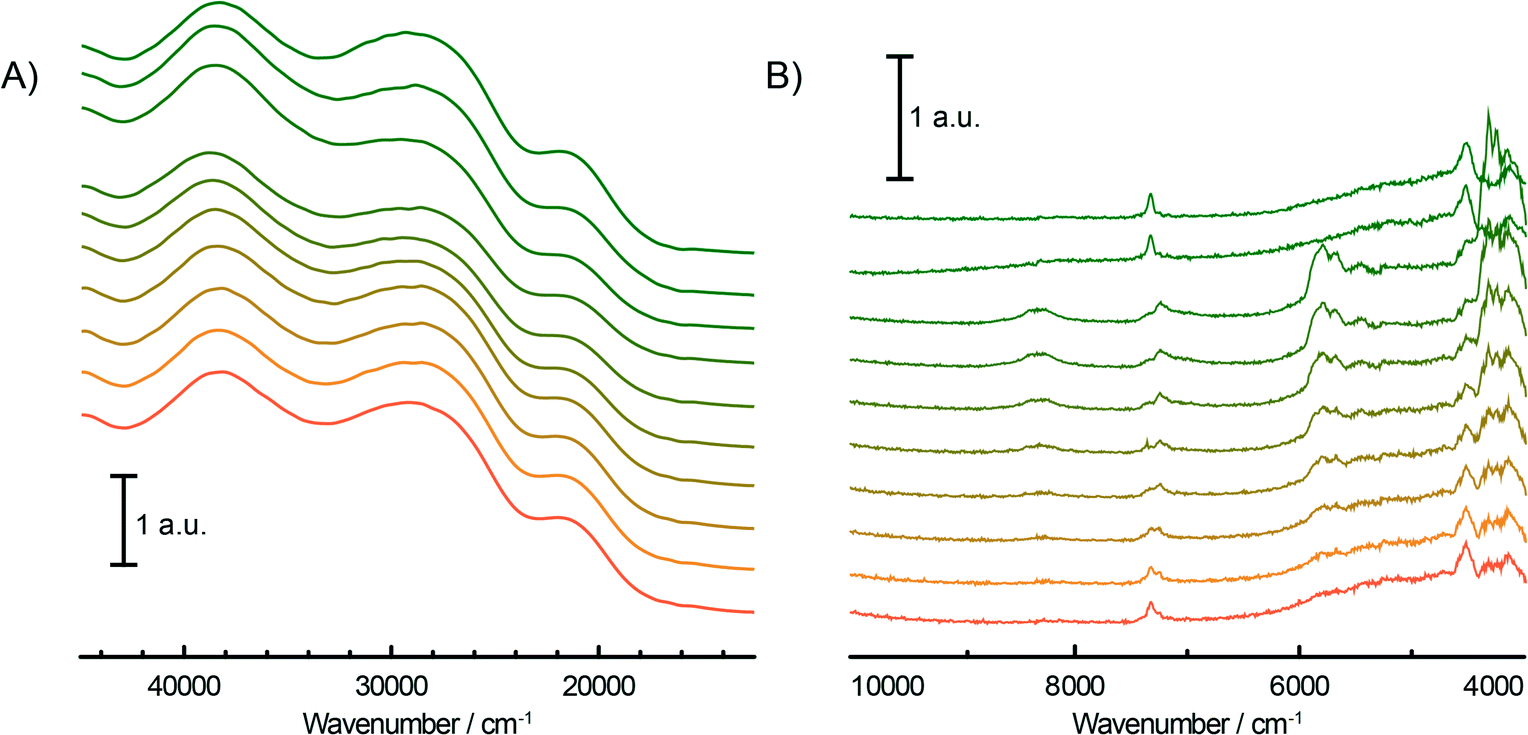 | ||
| Fig. 3 Time evolution (from green to orange) of the operando UV-vis-NIR DRS spectra of the Cr/SiO2 Phillips-type catalyst (CS sample) during the addition of TEAl in the UV-vis (A) and the NIR (B) region. | ||
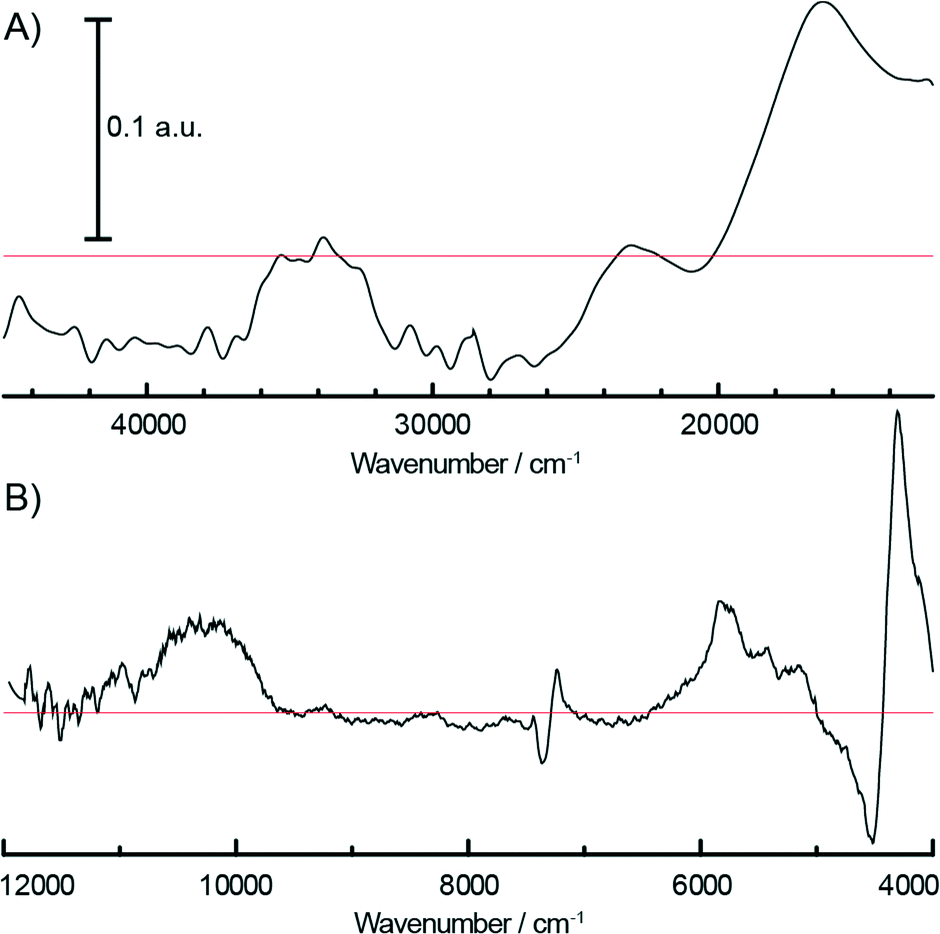 | ||
| Fig. 4 Difference spectrum of the catalyst modified with TEAl and the fresh Cr/SiO2 Phillips-type catalyst in the UV-vis (A) and the vis-NIR (B) region showing the reduction of some of the Cr6+ species as testified by the negative O → Cr6+ charge transfer bands and positive bands of the formed Cr3+ and Cr2+ species at approximately 16000 and 10000 cm−1, respectively. Zero is presented with the red line. | ||
At this stage, ethylene polymerisation reaction with the CS catalyst was started by changing the N2 flow in the quartz reactor to the gas reactant mixture consisting of 45 vol% ethylene, 45 vol% N2 and 10 vol% H2. Fig. 5A shows the time evolution of the UV-vis-NIR DRS spectra in the UV-vis region during the in situ polymerisation of ethylene using the Cr/SiO2 (CS) catalyst. During the reaction, the intensities of the O → Cr6+ CT bands belonging to mono- and dichromates are decreasing due to the reduction of Cr6+ species by ethylene with a simultaneous increase of the d–d transition bands of Cr3+Oh and Cr2+Oh species at ~16000 cm−1 and ~10000 cm−1, respectively. Consumption of surface silanol groups is evidenced in the NIR region (Fig. 5B) by the disappearance of the OH stretching overtone and combination bands at ~7325 cm−1 and 4535 cm−1. On the other hand, the formation of the polymerisation product can be monitored in situ by the growing bands originating from the CH2 and CH3 of the growing polymer chain and possible shorter oligomers. These bands evolve in the regions of 4000–4400 cm−1, 5600–5900 cm−1 and 6900–7200 cm−1 with the assignation given in Table 2.
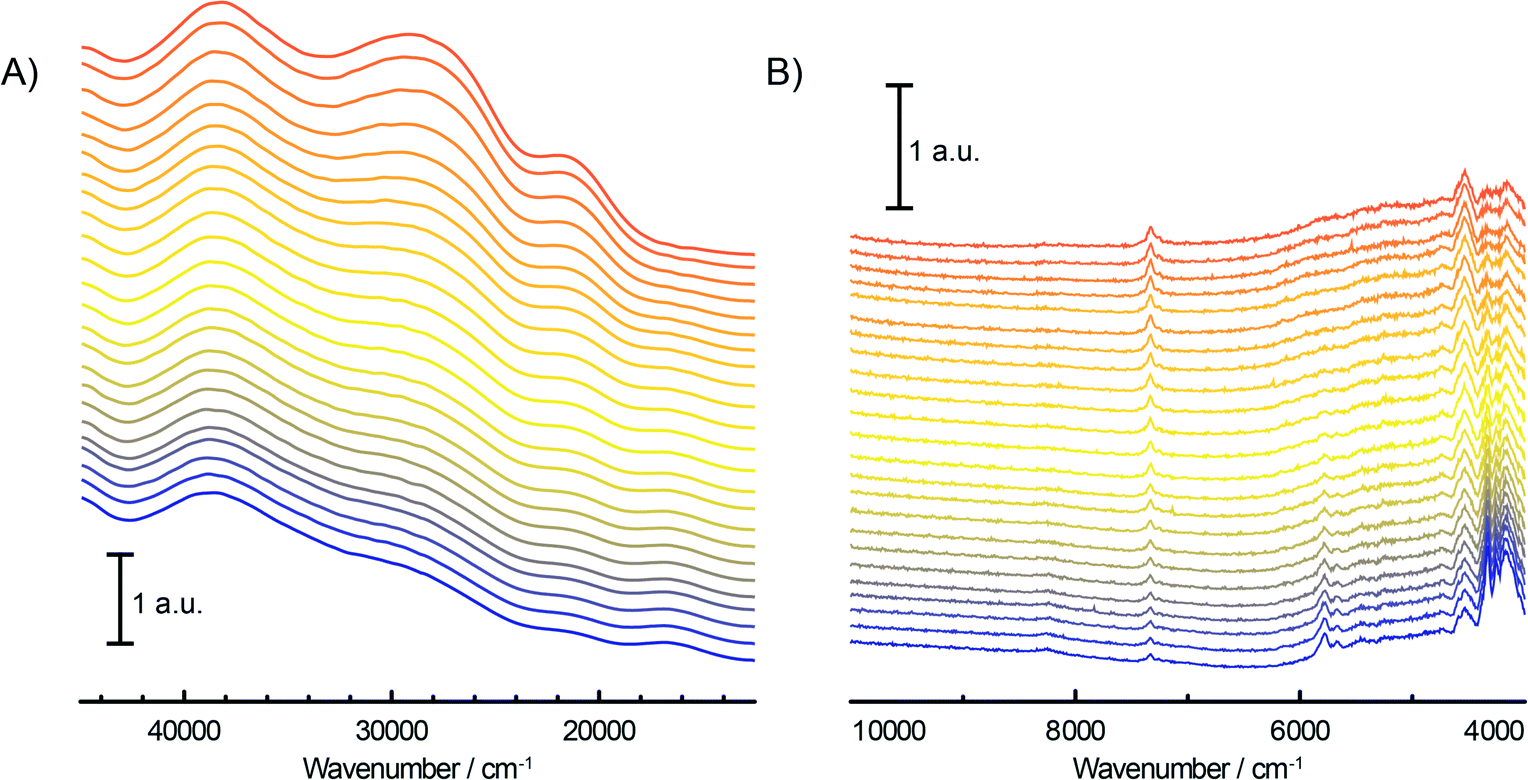 | ||
| Fig. 5 Time evolution (from orange to blue) of the operando UV-vis-NIR DRS spectra of the fresh Cr/SiO2 Phillips-type catalyst (CS sample) during the oligo-/polymerisation of ethylene, in the UV-vis (A) and the NIR (B) region. | ||
From the on-line MS and operando UV-vis-NIR DRS data, we can identify two stages of the catalyst reaction. During the first stage, right from the start of the ethylene feed, the catalyst is being reduced. No bands due to the polymer product can yet be seen in the NIR region of the spectra. On the other hand, it is possible to observe a rise of the MS signal belonging to the fragments of the produced ethylene oligomers. In this stage, ethylene oligomerisation sites were being created and ethylene oligomerised mainly to 1-hexene. The second stage starts later and includes the production of heavier oligomers and polyethylene, as confirmed by the growing NIR bands of the CH2 and CH3 groups. It must be noted that the oligomerisation activity of the catalyst is still preserved during the later stage.
UV-vis-NIR diffuse reflectance spectroscopy
In this study we have investigated the influence of Ti and H2 on the oligomerisation properties of a TEAl-modified Phillips-type ethylene polymerisation catalyst and therefore tested the catalysts with different amounts of titanation (Table 1) and a different content of the gas reactant mixture. As was shown in the previous section, the UV-vis-NIR DRS spectra of the fresh CS catalyst show a highly dehydroxylated catalyst surface, evidenced by a sharp 2ν(SiOH) stretching overtone at ~7325 cm−1 and a combination ν(SiOH) + δ(SiOH) band at ~4525 cm−1. The addition of Ti during the preparation of the catalyst materials introduces a 2ν(TiOH) overtone band at 7270 cm−1 as a shoulder of the silanol stretching band, with the intensity increasing with increasing Ti loading. This group increases the acidity of the catalyst surface and appears at lower wavenumbers as expected due to the increased reduced mass of the functional group. Another characteristic of the fresh catalysts is the presence of three O → Cr6+ CT bands of mono- and polychromate species appearing in the UV-vis region. The exact positions of these absorption bands (Table 1) are dependent on the catalyst architecture since any changes in the electronic environment influence the energy levels of Cr. Titanation and an increased amount of Ti from CS to CTS2 cause a shift of the maxima of the UV-vis bands towards lower energy. This is triggered by the formation of Si–O–Ti–O–Cr bonds where Si is replaced with the less electronegative Ti atom, causing a slight shift of the electron cloud towards the O closest to Cr, populating its energy levels and narrowing the O → Cr6+ CT energy gap, therefore decreasing the energy required for the charge transfer to occur.52Prior to ethylene polymerisation, all of the tested catalysts were pre-contacted with TEAl. Some of the Cr6+ sites were reduced to Cr3+ and Cr2+ species, as testified by a small intensity decrease of the O → Cr6+ CT bands and the appearance of the 4A2g → 4T2g d–d transition band of Cr3+Oh and the 5Eg → 5T2g d–d transition band of Cr2+Oh. Furthermore, TEAl also reacts with the hydroxyl groups of the support, as shown by the broadening and lowering of the intensity of the silanol and titanol group stretching first overtone. However, these changes in the UV-vis-NIR DRS spectra are not substantial, suggesting that TEAl modifies only some of the Cr sites, which were still enough to produce ethylene oligomers.
Fig. 6 shows the evolution of the UV-vis-NIR DRS spectra during ethylene polymerisation using the CS and CTS2 catalysts, with and without H2, after the modification with TEAl. During ethylene polymerisation, Cr6+ mono- and dichromate species become significantly reduced by the ethylene reactant to Oh coordinated Cr3+ and Cr2+ species.9 Some of these new-formed sites may not only be the active ethylene polymerisation species, but also the products of deactivation of polymerisation sites as a slight shift of the 16000 cm−1 band to higher energies indicates a possible formation of crystalline Cr2O3 on the catalyst surface. The influence of titanation on the polymerisation of ethylene can be clearly seen in the time evolution of the operando UV-vis-NIR DRS spectra. By changing the electronic and spatial environments around Cr sites, Ti gives rise to a new type of Cr6+ species, which prove to be more reducible than the Cr6+ species of the non-titanated catalyst. This effect is evidenced by a more rapid intensity decrease of the O → Cr6+ CT bands in the case of CTS2 compared to CS. Consequently, easier reducibility causes a faster development of the polymerisation rate, which can be seen by the growth rate of the overtone and combination vibrational bands of the CHx groups of the growing polyethylene. In the presence of H2 (Fig. 6B and D), the reduction of Cr is even more pronounced due to the reducing properties of H2.
 | ||
| Fig. 6 UV-vis part of the operando UV-vis-NIR DRS spectra of CTS2 and CS Phillips catalysts during polymerisation of ethylene without H2 (A and C) and with H2 (B and D), respectively, after pre-reacting the catalyst with the TEAl co-catalyst. NIR part of the operando UV-vis-NIR DRS spectra of CTS2 and CS Phillips catalysts during polymerisation of ethylene without H2 (E and G) and with H2 (F and H) after pre-reacting the catalyst with the TEAl co-catalyst. Time evolution of the spectra is indicated by colour transition from orange to blue with 2 minutes between each successive spectrum. | ||
The UV-vis absorption bands of the oligo/polymerisation products could not be measured, since they appear outside of the detectable range of the UV-vis-NIR DRS spectrophotometer. Absorption, due to the σ → σ* transition of saturated hydrocarbons, is expected at ~67000 cm−1. Additional unsaturation of the C bond, in the case of alkene species, introduces an additional π → π* transition band in the 52000–59000 cm−1 range.53
Gas chromatography analysis of the reaction products
Although operando UV-vis-NIR DRS is a spectroscopic technique useful to study both the catalyst and the product during the polymerisation reaction, it cannot discriminate between different types of products due to the overlap of NIR bands appearing at the same wavenumbers. Fig. 7–9 show the GC analysis results of the gas phase sampled during the polymerisation of ethylene using several different catalysts, with varying Ti loading (0, ~2 and ~4 wt% Ti), Cr loading (0, ~0.5 and ~1 wt% Cr) and activation temperature (823, 923 and 1048 K), as summarised in Table 1. Each catalyst was tested both with and without 10 vol% H2, while the ratio of ethylene and N2 was kept at 1:1.
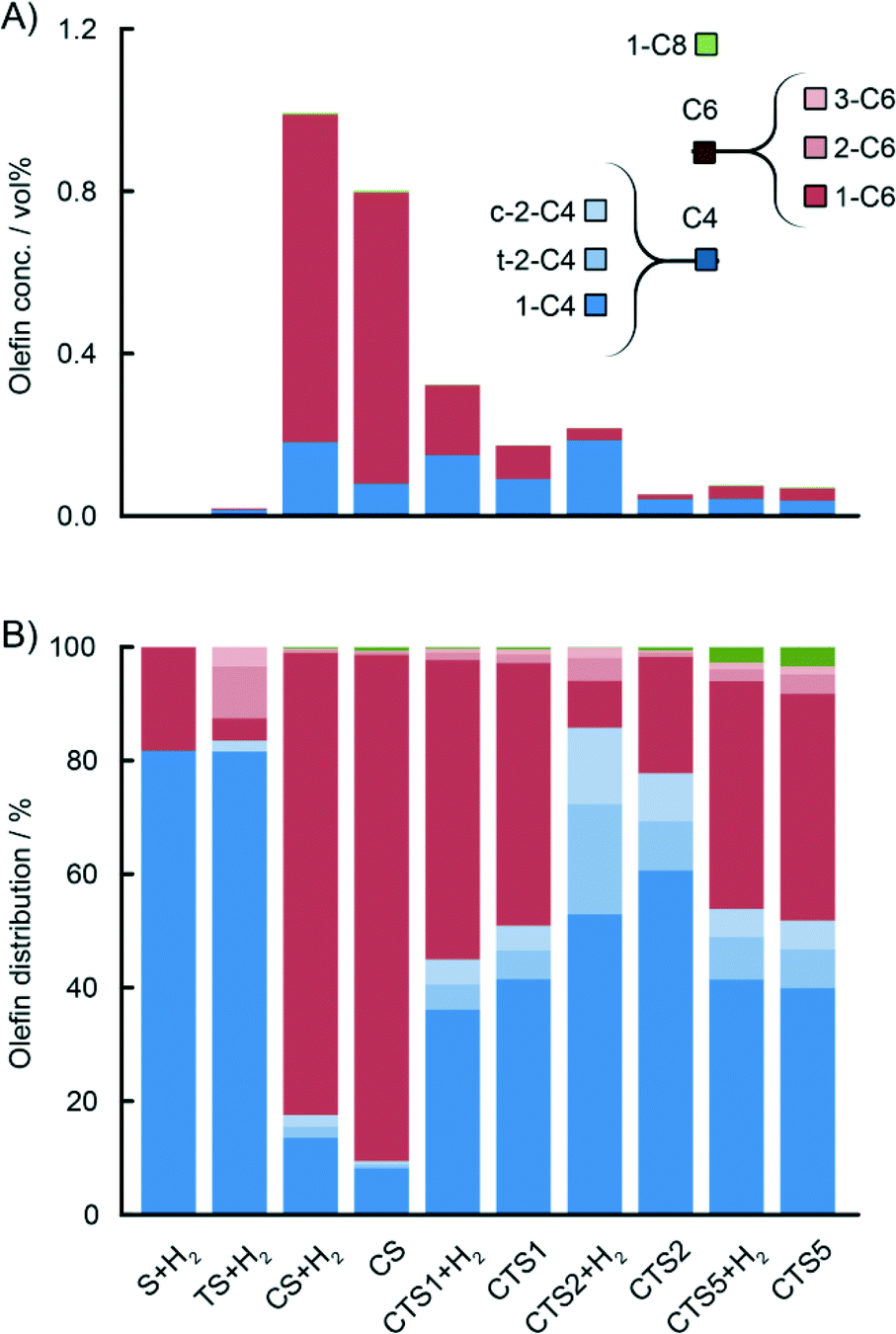 | ||
| Fig. 7 GC analysis of the gas phase during ethylene polymerisation with various Phillips-type catalysts showing different gas phase concentrations (A) of the in situ produced C4 (blue), C6 (red) and C8 (green) olefins depending on the catalyst composition. Olefin distribution normalised to 100% (B) reveals that the titanation of the Phillips-type catalysts has a detrimental effect on ethylene oligomerisation and selectivity towards industrially important 1-hexene fraction. | ||
 | ||
| Fig. 8 GC analysis of the gas phase during ethylene polymerisation with various Phillips-type catalysts showing different gas phase concentrations (A) of the in situ produced C4 (blue), C6 (red) and C8 (green) paraffins depending on the catalyst composition. Paraffin distribution normalised to 100% (B) reveals that the titanation of the Phillips-type catalysts does not change significantly the ratio of produced oligomers. | ||
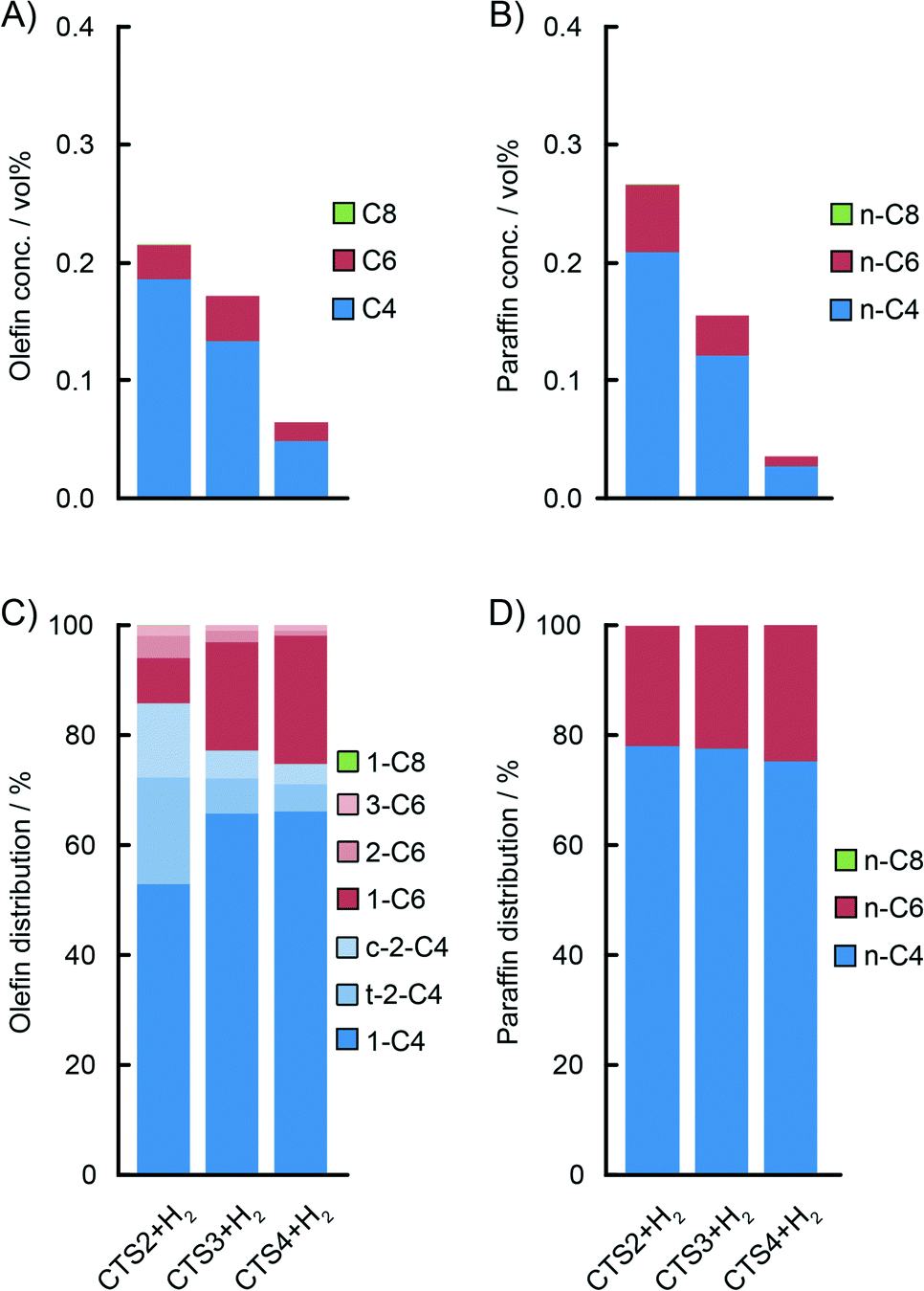 | ||
| Fig. 9 GC analysis of the gas phase during ethylene polymerisation with CTS2, CTS3 and CTS4 Cr/Ti/SiO2 Phillips-type catalysts activated at 1048 K, 923 K and 823 K, respectively. Decrease of the activation temperature lowers the yields of the in situ produced C4 (blue) and C6 (red) olefins (A) and paraffins (C). A lower activation temperature slightly shifts the distribution of in situ produced olefins towards α-olefins, particularly 1-hexene (B), having practically no influence on the distribution of paraffins (D). | ||
For the sake of excluding any ethylene oligomerisation activity of the silica carrier or the titania on the silica carrier,‡ two additional materials without any Cr were tested, i.e. wide-pore silica support (S) and 4 wt% titanated silica (TS), both activated using the same procedure as the actual catalyst. During ethylene polymerisations using these materials with H2 and the TEAl co-catalyst, no substantial ethylene oligomerisation was reported (Fig. 7A and 8A).
For the actual Phillips-type catalysts, it was observed that the concentration of the produced ethylene oligomers greatly depends on the composition of the catalyst. The highest yield of all oligomers was obtained with the CS catalyst containing no Ti, and it decreases with increasing Ti loading. Detailed analysis of the amounts of olefinic and paraffinic fractions shows that the yield of olefins significantly drops with increasing Ti loading, while the yield of paraffins is slightly increased. The addition of H2 to the reaction mixture caused an increase in the overall oligomer yield, with a higher influence on the production of saturated oligomers. Therefore, H2 might be involved in the termination of polymer chain growth at the early stages, increasing the concentration of saturated oligomers in the gas phase.
Fig. 7B and 8B show a detailed distribution of the in situ produced olefinic and paraffinic hydrocarbons, respectively, normalised to 100%. Firstly, a clear dependence of the ratio of butenes, hexenes and octenes can be observed, depending on the amount of Ti present in the catalyst. The CS system, without any Ti, exhibited the highest selectivity towards ethylene trimerisation, predominantly producing 1-hexene. The addition of Ti caused a further decrease in the overall ethylene trimerisation selectivity as can be noted for the CTS1 catalyst with ~2 wt% Ti. The most pronounced Ti effect is observed for the catalyst with the highest Ti loading of ~ 4 wt% where a strong inhibition of ethylene trimerisation sites diminished the amount of hexenes compared to the higher yield of butenes and octenes. In the reaction with the CTS5 catalyst, which contains double the amount of Cr loading compared to the other examined catalysts, the distribution of olefins matches the distribution produced with the CTS1 catalyst of a similar Cr-to-Ti ratio. The distribution of paraffins shows that the relative ratio of the in situ produced n-butane, n-hexane and n-octane is independent of the Ti loading in the catalyst and that the differences can mainly be attributed to the presence of H2 in the reactant gas mixture. Overall, a higher Ti loading decreases the olefin-to-paraffin ratio within the ethylene oligomers of all chain lengths.
A higher activation temperature of the catalyst showed the promoting effect on the in situ ethylene oligomerisation as can be seen by the increasing amount of produced oligomers starting from CTS4, over CTS3 to the CTS2 catalyst, activated in dry air at 823 K, 923 K and 1048 K, respectively (Fig. 9). A higher activation temperature improves the dehydroxylation of the catalyst surface, liberating the coordination sphere of Cr producing more reactive Cr sites, which increase the gas phase oligomer yield. A rise in the activation temperature also shifts the olefin distribution towards butenes and promotes the isomerisation and production of linear β- and γ-olefins.
Fig. 10 shows the total distribution of hydrocarbon oligomers, split into olefin and paraffin contributions, which can be further presented as Schulz–Flory (SF) diagrams.† A clear 1-hexene spike can be observed with the CS catalyst with no Ti. An increase of the Ti loading significantly decreases the 1-hexene selectivity. If the products follow the SF distribution, caused by the linear-insertion mechanism, a straight curve can be expected with the slope, which represents the chain-growth probability α, determined by the rates of chain growth and termination. The distribution of the in situ produced paraffins exhibits an exponential decay with the increase of carbon number, which is reflected in linear SF curves with α values given in Table 3. Addition of 10 vol% H2 to the reactant mixture increases α and therefore decreases the selectivity to shorter paraffin oligomers, i.e. n-butane. On the other hand, the olefinic oligomers do not show a linear trend in the SF plot in the case of the CS catalyst containing no Ti. With increasing Ti loading, due to the decrease in 1-hexene selectivity, the curves become more linear.
 | ||
| Fig. 10 The increase of Ti loading in the Phillips-type catalyst (from light to dark blue) shifts the selectivity towards C4 at the expense of C6 products in the experiments with (□) and without (Δ) hydrogen. A different trend in the distribution of olefinic species (A) compared to paraffinic species (B) is also clearly visible in the Schulz–Flory plots. While paraffinic species show linear curves independent of the Ti loading (D), olefin species exhibit a discrepancy from linearity with increasing Ti loading (C), indicating an oligomerisation mechanism, which does not follow SF distribution. | ||
| Catalyst name | α, no H2 | α, with H2 |
|---|---|---|
| CS | 0.30 | 0.45 |
| CTS1 | 0.33 | 0.37 |
| CTS2 | 0.27 | 0.51 |
| CTS5 | 0.38 | 0.46 |
Similar trends can be observed with the CTS5 catalyst with the increased Cr loading of ~1 wt% (Fig. 11). A higher Cr loading, while keeping the amount of Ti constant, increases the number of Ti-free Cr sites, which are able to produce selectively ethylene trimers, as illustrated by the non-exponential decay of olefins and the presence of 1-hexene spike. This catalyst sample still contained Ti, which explains the higher amount of ethylene dimers. Nevertheless, the increase in Cr loading appeared to be beneficial for the promotion of ethylene trimerisation.
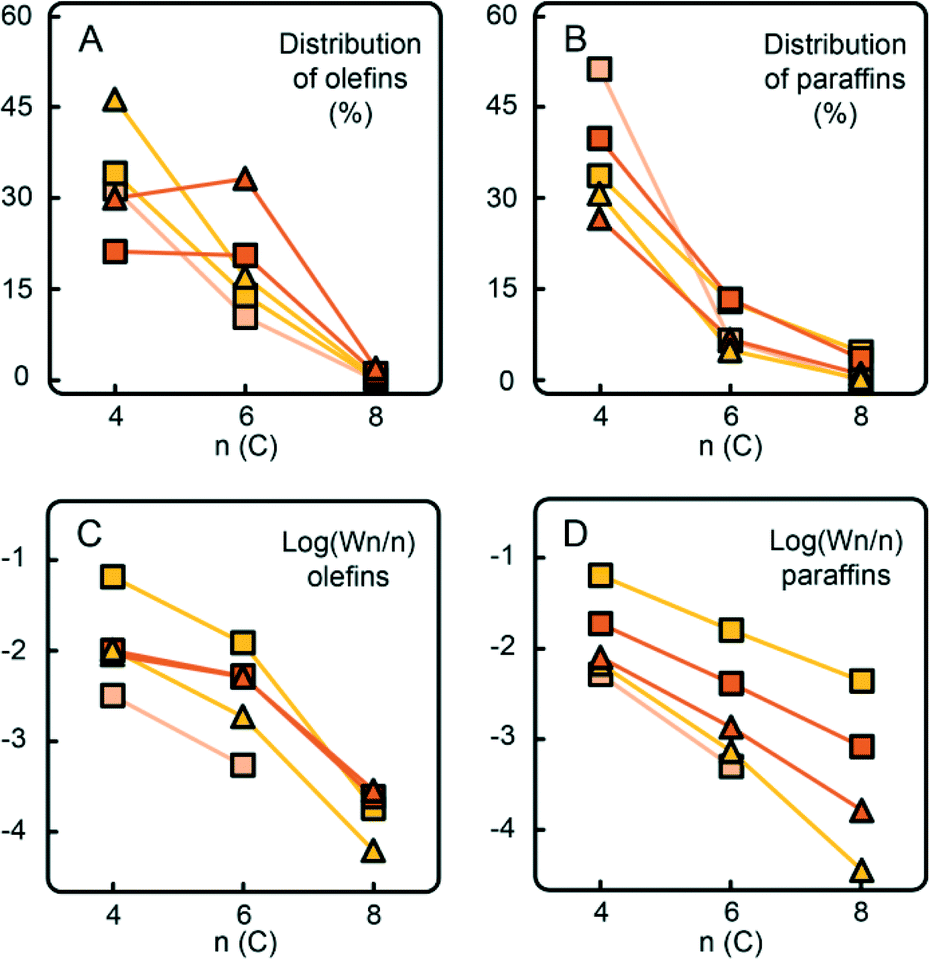 | ||
| Fig. 11 The increase of Cr loading in the Phillips-type catalyst (from light yellow to orange) increases the selectivity towards C6 products in the experiments with (□) and without (Δ) hydrogen. A different trend in the distribution of olefinic species (A) compared to paraffinic species (B), also visible in the Schulz–Flory plots (C and D), causes a discrepancy from linearity with increasing Cr loading. A higher amount of Cr, while keeping the Ti loading constant, increases the amount of ethylene trimerisation sites, which produce higher amounts of 1-hexene. | ||
Discussion
The analyses of the gas phase collected during the ethylene polymerisation show that the titanation of the Cr/SiO2 Phillips-type catalyst significantly reduces the yield of ethylene oligomers and shifts the selectivity towards butene fractions. A lower Ti-to-Cr loading ratio appeared to be beneficial for the promotion of the trimerisation of ethylene, as the number of Cr sites, which are not inhibited by Ti, is increased and therefore able to selectively produce more ethylene trimers, indicating a change in the oligomerisation pathway.Currently, the most widely accepted proposed mechanism of ethylene polymerisation is the Cossee–Arlman mechanism, which explains the chain propagation by linear insertion of the ethylene monomer into an existing alkyl group or polymer, bonded to a Cr active site. The termination of the chain occurs via β-H elimination leaving a vinyl end group in the released hydrocarbon product. As a result of the mentioned ethylene insertion mechanism, the products are expected to follow a statistical hydrocarbon distribution, known as the Schulz–Flory (SF) distribution, with only even-membered hydrocarbon chains due to the presence of ethylene as the only monomer. However, the high 1-hexene spike produced with the non-titanated catalyst can be attributed to the complex structure of the catalyst, with different types of active sites and a complex mechanism of oligomerisation, which cannot be explained solely by the Cossee–Arlman mechanism of statistical chain growth.
From the results of our study, it is found that two types of distinct oligomerisation mechanisms occur simultaneously with the extent between each other depending on the precise catalyst compositions and hence the electronic environment of the different active sites. Besides the Cossee–Arlman mechanism of linear insertion, in the case of alkane oligomers and polyethylene, alkene species are predominantly being produced via a metallacyclic mechanism (Scheme 1). Two ethylene molecules are able to coordinate to the vacant coordination sphere of the Cr active site and form a chromacyclopentane ring. Two processes can occur at this stage, β-H transfer and release of 1-butene, or coordination and insertion of another ethylene monomer to form a chromacycloheptane ring. This process can continue even further and form bigger chromacyclic rings, where the release of olefin via β-H transfer occurs when the probability of insertion of a new monomer becomes lower than the probability of β-H transfer. The stability of the chromacyclic ring is highly influenced by the structure of Cr active sites. As revealed in the operando UV-vis-NIR DRS spectra, titanation of the catalyst changes the structure of the Cr oligomerisation sites by forming Si–O–Ti–O–Cr linkages, as testified by the red shifts of O → Cr6+ CT bands, and introduces free surface titanol groups, which increase the acidity of the catalyst surface. Consequently, an increasing Ti loading decreases the stability of the 5-membered chromacyclopentane ring, favouring β-H transfer and release of 1-butene and therefore reducing the selectivity towards 1-hexene. Alternatively, given the DFT studies of Robinson et al.54 and the work of Emrich et al.,55 the high 1-butene selectivity in the case of the titanated catalysts can be explained by the Cossee–Arlman mechanism over the oligomerisation sites with a very low α, while 1-hexene is still being produced over trimerisation sites via the metallacyclic mechanism.
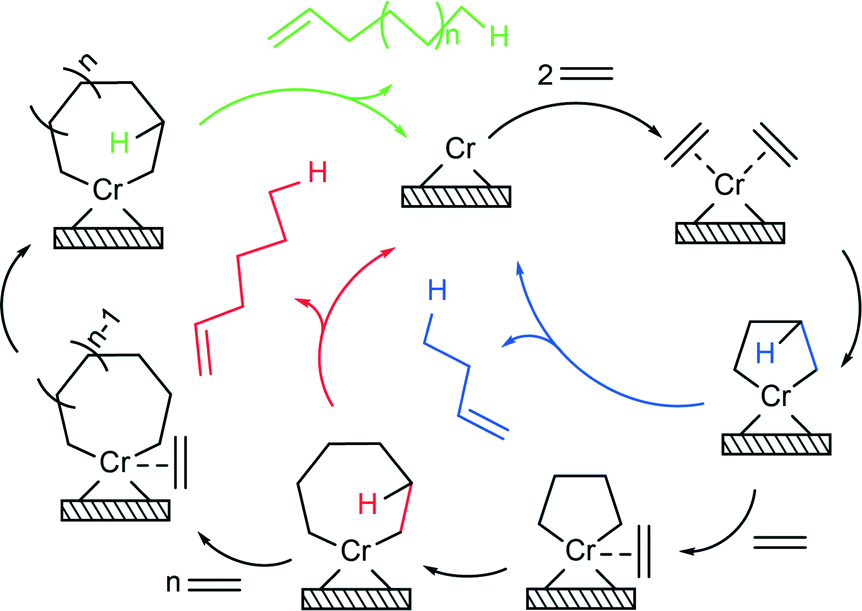 | ||
| Scheme 1 Metallacyclic mechanism for selective ethylene oligomerisation. | ||
Ethylene trimerisation catalysts have been reported for chromium-based homogeneous systems, basically consisting of multidentate ligands reacted with chromium salts and activated with aluminium alkyl co-catalyst, some of them being commercialised.56–59 The isotopic labelling and regiochemistry studies of Agapie et al.60,61 on such catalytic systems strongly support the metallacyclic mechanism of the selective ethylene trimerisation and tetramerisation, which might occur through Cr+/Cr3+ and Cr2+/Cr4+ redox cycles as shown by XAS and EPR.62–64 Similarly, the metallacyclic mechanism was proposed for the silica-supported chromium catalyst discussed in this paper, although it has to be noted that a heterogeneous catalyst is more difficult to define due to the heterogeneity of different surface chromium sites. The advantage of the catalyst material examined in this study is its multifunctionality, i.e. the selective ethylene oligomerisation capabilities of some of the chromium sites and co-/polymerisation activity of other sites, within a single Phillips catalyst.
Conclusions
The oligomerisation properties of a TEAl-modified Phillips-type ethylene polymerisation catalyst were tested in a specially developed operando setup allowing the relation of data gathered by UV-vis-NIR diffuse reflectance spectroscopy, mass spectrometry and gas chromatography techniques. This combination of methods allowed a real-time study of working catalysts and reaction products. The incorporation of Ti in the Cr/SiO2 Phillips-type catalysts by the formation of Cr–O–Ti–O–Si linkages alters the electronic environment of Cr ions and changes the oligomerisation capabilities of the catalyst. Consequently, different distributions of the in situ produced ethylene oligomers were revealed, highly dependent on the architecture of the catalyst. Paraffinic oligomers show an exponential decay explained by the Cossee–Arlman linear insertion mechanism. On the contrary, olefinic oligomers in the case of the non-titanated Cr/SiO2 catalyst exhibit a much more complex distribution, with a higher selectivity towards 1-hexene than what is expected from the linear insertion mechanism. High ethylene trimerisation activity is a consequence of the metallacyclic oligomerisation mechanism. The addition of Ti modifies the trimerisation Cr sites and creates new oligomerisation sites, which favour β-H transfer in the chromacyclopentane intermediate and release of the 1-butene product, therefore decreasing the selectivity towards 1-hexene. These results open new possibilities for both academic and industrial research of the Phillips polymerisation system, as they show that by a proper selection of the catalyst structure it is possible to change the distribution of the in situ produced oligomers and therefore change the amount of α-olefins, which are able to co-polymerise with ethylene to form the desired grade of polyethylene.Acknowledgements
This research was funded by Total Research and Technology Feluy and Total SA. G. van Hauwermeiren and J. Decrom (Total Research and Technology, Feluy) are acknowledged for catalyst preparations. A. Mens (Utrecht University) is acknowledged for the construction of the experimental setup.Notes and references
- M. P. McDaniel, Adv. Catal., 2010, 53, 123–606 CAS.
- E. Groppo, C. Lamberti, S. Bordiga, G. Spoto and A. Zecchina, Chem. Rev., 2005, 105, 115–184 CrossRef CAS PubMed.
- B. M. Weckhuysen and R. A. Schoonheydt, Catal. Today, 1999, 51, 215–221 CrossRef CAS.
- R. Cheng, Z. Liu, L. Zhong, X. He, P. Qiu, M. Terano, M. S. Eisen, S. L. Scott and B. Liu, Adv. Polym. Sci., 2013, 257, 135–202 CrossRef CAS.
- J. P. Hogan and R. L. Banks, US Pat., 2825721, 1958 Search PubMed.
- J. P. Hogan and R. L. Banks, US Pat., 2951816, 1960 Search PubMed.
- R. Merryfield, M. P. McDaniel and G. Parks, J. Catal., 1982, 77, 348–359 CrossRef CAS.
- B. Liu, P. Šindelář, Y. Fang, K. Hasebe and M. Terano, J. Mol. Catal. A: Chem., 2005, 238, 142–150 CrossRef CAS.
- B. M. Weckhuysen, L. M. De Ridder and R. A. Schoonheydt, J. Phys. Chem., 1993, 97, 4756–4763 CrossRef CAS.
- B. M. Weckhuysen, L. M. De Ridder, P. J. Grobet and R. A. Schoonheydt, J. Phys. Chem., 1995, 99, 320–326 CrossRef CAS.
- B. M. Weckhuysen, A. A. Verberckmoes, A. R. de Baets and R. A. Schoonheydt, J. Catal., 1997, 166, 160–171 CrossRef CAS.
- L. Zhong, M.-Y. Lee, Z. Liu, Y.-J. Wanglee, B. Liu and S. L. Scott, J. Catal., 2012, 293, 1–12 CrossRef CAS.
- C. A. Demmelmaier, R. E. White, J. A. van Bokhoven and S. L. Scott, J. Catal., 2009, 262, 44–56 CrossRef CAS.
- A. Zecchina, E. Groppo, A. Damin and C. Prestipino, in Topics in Organometallic Chemistry, ed. C. Copéret and B. Chaudret, Springer, Berlin, 2005, vol. 16, pp. 1–35 Search PubMed.
- B. M. Weckhuysen, A. A. Verberckmoes, A. L. Buttiens and R. A. Schoonheydt, J. Phys. Chem., 1994, 98, 579–584 CrossRef CAS.
- B. M. Weckhuysen, I. E. Wachs and R. A. Schoonheydt, Chem. Rev., 1996, 96, 3327–3350 CrossRef CAS PubMed.
- D. S. McGuinness, N. W. Davies, J. Horne and I. Ivanov, Organometallics, 2010, 29, 6111–6116 CrossRef CAS.
- E. Groppo, C. Lamberti, S. Bordiga, G. Spoto and A. Zecchina, J. Catal., 2006, 240, 172–181 CrossRef CAS.
- E. Groppo, J. Estephane, C. Lamberti, G. Spoto and A. Zecchina, Catal. Today, 2007, 126, 228–234 CrossRef CAS.
- M. P. Conley, M. F. Delley, G. Siddiqi, G. Lapadula, S. Norsic, V. Monteil, O. V. Safonova and C. Copéret, Angew. Chem., Int. Ed., 2014, 53, 1872–1876 CrossRef PubMed.
- M. F. Delley, M. P. Conley and C. Copéret, Catal. Lett., 2014, 144, 805–808 CrossRef CAS.
- S. L. Scott and J. Amor Nait Ajjou, Chem. Eng. Sci., 2001, 56, 4155–4168 CrossRef CAS.
- A. Fong, Y. Yuan, S. L. Ivry, S. L. Scott and B. Peters, ACS Catal., 2015, 5, 3360–3374 CrossRef CAS.
- R. E. Dietz, US Pat., 3887494, 1975 Search PubMed.
- G. Debras and J.-P. Dath, EP Pat., 0882743B1, 1998 Search PubMed.
- T. J. Pullukat, D. Plaines and M. Shida, US Pat., 3780011, 1973 Search PubMed.
- J. P. Hogan and D. R. Witt, US Pat., 3622521, 1971 Search PubMed.
- B. Horvath, US Pat., 3625864, 1971 Search PubMed.
- T. J. Pullukat, R. E. Hoff and M. Shida, J. Polym. Sci., Polym. Chem. Ed., 1980, 18, 2857–2866 CrossRef CAS.
- L. L. Böhm, Angew. Chem., Int. Ed., 2003, 42, 5010–5030 CrossRef PubMed.
- G. Wilke, Angew. Chem., Int. Ed., 2003, 42, 5000–5008 CrossRef CAS PubMed.
- H. H. Brintzinger, D. Fischer, R. Mülhaupt, B. Rieger and R. M. Waymouth, Angew. Chem., Int. Ed. Engl., 1995, 34, 1143–1170 CrossRef CAS.
- D. L. Myers and J. H. Lunsford, J. Catal., 1986, 99, 140–148 CrossRef CAS.
- B. M. Weckhuysen, H. J. Spooren and R. A. Schoonheydt, Zeolites, 1994, 14, 450–457 CrossRef CAS.
- G. Ghiotti, E. Garrone and A. Zecchina, J. Mol. Catal., 1988, 46, 61–77 CrossRef.
- D. D. Beck and J. H. Lunsford, J. Catal., 1981, 68, 121–131 CrossRef CAS.
- B. M. Weckhuysen, R. A. Schoonheydt, F. E. Mabbs and D. Collison, J. Chem. Soc., Faraday Trans., 1996, 92, 2431–2436 RSC.
- J.-M. Jehng, I. E. Wachs, B. M. Weckhuysen and R. A. Schoonheydt, J. Chem. Soc., Faraday Trans., 1995, 91, 953–961 RSC.
- M. F. Delley, F. Núñez-Zarur, M. P. Conley, A. Comas-Vives, G. Siddiqi, S. Norsic, V. Monteil, O. V. Safonova and C. Copéret, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 11624–11629 CrossRef CAS PubMed.
- D. Cicmil, J. Meeuwissen, A. Vantomme, J. Wang, I. K. Ravenhorst, H. E. van der Bij, A. Muñoz-Murillo and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2015, 54, 13073–13079 CrossRef CAS PubMed.
- W. Xia, B. Liu, Y. Fang, K. Hasebe and M. Terano, J. Mol. Catal. A: Chem., 2006, 256, 301–308 CrossRef CAS.
- Platts Global Ethylene Price Index, can be found under http://www.platts.com/news-feature/2015/petrochemicals/pgpi/ethylene, 2015 Search PubMed.
- E. Groppo, K. Seenivasan and C. Barzan, Catal. Sci. Technol., 2013, 3, 858–878 CAS.
- C. Lamberti, A. Zecchina, E. Groppo and S. Bordiga, Chem. Soc. Rev., 2010, 39, 4951–5001 RSC.
- A. Damin, F. Bonino, S. Bordiga, E. Groppo, C. Lamberti and A. Zecchina, ChemPhysChem, 2006, 7, 342–344 CrossRef CAS PubMed.
- W. Xia, B. Liu, Y. Fang, T. Fujitani, T. Taniike and M. Terano, Appl. Catal., A, 2010, 389, 186–194 CrossRef CAS.
- F. C. Jentoft, Adv. Catal., 2009, 52, 129–211 CAS.
- R. A. Schoonheydt, Chem. Soc. Rev., 2010, 39, 5051–5066 RSC.
- B. M. Weckhuysen and R. A. Schoonheydt, Catal. Today, 1999, 51, 223–232 CrossRef CAS.
- S. Petit, J. Madejova, A. Decarreau and F. Martin, Clays Clay Miner., 1999, 47, 103–108 CAS.
- M. Watari, Appl. Spectrosc. Rev., 2013, 49, 462–491 CrossRef.
- C. E. Housecroft and A. G. Sharpe, Inorganic Chemistry, Pearson Education Limited, Harlow, 2nd edn, 2005, ch. 20, pp. 555–588 Search PubMed.
- P. S. Kalsi, Spectroscopy of Organic Compounds, New Age International (P) Ltd., Publishers, New Delhi, 6th edn, 2007, ch. 2, pp. 11–14 Search PubMed.
- R. Robinson, D. S. Mcguinness and B. F. Yates, ACS Catal., 2013, 3, 3006–3015 CrossRef CAS.
- R. Emrich, O. Heinemann, P. W. Jolly, C. Krüger and G. P. J. Verhovnik, Organometallics, 1997, 16, 1511–1513 CrossRef CAS.
- W. K. Reagen, EP Pat., 0417477A2, 1990 Search PubMed.
- J. R. Briggs, US Pat., 4668838, 1987 Search PubMed.
- F.-J. Wu, US Pat., 5550305, 1996 Search PubMed.
- J. T. Dixon, P. Wasserscheid, D. S. McGuinness, F. M. Hess, H. Maumela, D. H. Morgan and A. Bollmann, EP Pat., 2075242A1, 2007 Search PubMed.
- T. Agapie, S. J. Schofer, J. A. Labinger and J. E. Bercaw, J. Am. Chem. Soc., 2004, 126, 1304–1305 CrossRef CAS PubMed.
- T. Agapie, J. A. Labinger and J. E. Bercaw, J. Am. Chem. Soc., 2007, 129, 14281–14295 CrossRef CAS PubMed.
- L. H. Do, J. A. Labinger and J. E. Bercaw, ACS Catal., 2013, 3, 4–7 Search PubMed.
- I. Y. Skobelev, V. N. Panchenko, O. Y. Lyakin, K. P. Bryliakov, V. A. Zakharov and E. P. Talsi, Organometallics, 2010, 29, 2943–2950 CrossRef CAS.
- J. Rabeah, M. Bauer, W. Baumann, A. E. C. Mcconnell, W. F. Gabrielli, P. B. Webb, D. Selent and A. Bru, ACS Catal., 2013, 3, 95–102 CrossRef CAS.
- P. D. Smith, D. D. Klendworth and M. P. McDaniel, J. Catal., 1987, 105, 187–198 CrossRef CAS.
Footnotes |
| † The concentration of the oligomers in the gas phase may be altered by the consumption of the oligomers in the polymer. However, we assume that the oligomer consumption by polymer formation is negligible due to the small amount of PE formed and the low reactivity of α-olefins with the Phillips-type catalyst. For example, Cr/SiO2 incorporates only 1.85 mol% of 1-hexene at a constant concentration in the reactor.1 |
| ‡ Supported Ti alkoxides on silica reduced by a metal alkyl co-catalyst are reported as active catalysts for ethylene dimerisation.65 In the case of our catalysts, we assume that virtually all alkoxide groups have been burnt off during the calcination step. |
| This journal is © The Royal Society of Chemistry 2016 |