
Synthesis of hybrid silica nanoparticles grafted with thermoresponsive poly(ethylene glycol) methyl ether methacrylate via AGET-ATRP†
Zhiping Du*ab,
Xiaofeng Sunb,
Xiumei Taib,
Guoyong Wangb and
Xiaoying Liub
aInstitute of Resources and Environment Engineering, Shanxi University, No. 92 Wucheng Road, Taiyuan 030006, PR China. E-mail: duzhiping@sxu.edu.cn; Fax: +86 0351 4040802; Tel: +86 0351 4084691 Tel: +86 0351 2027036
bChina Research Institute of Daily Chemical Industry, No. 34 Wenyuan Road, Taiyuan 030001, PR China
First published on 27th January 2015
Abstract
Thermosensitive SiO2 nanoparticles were prepared with graft-on poly(ethylene glycol) methyl ether methacrylate (POEGMA, Mn ∼ 300 or 500 g mol−1) via atom transfer radical polymerization with activators generated through electron transfer (AGET ATRP). CuBr2/PMDETA and ascorbic acid (AA) were employed as the catalyst and reducing agent respectively in the fabrication process. The structure and surface composition of our hybrid materials were analyzed using Fourier Transform Infrared (FTIR) spectroscopy and X-ray Photoelectron Spectroscopy (XPS) in detail. The results of Thermogravimetric Analysis (TGA) show that AGET-ATRP could provide a higher surface grafting density to get more pronounced thermosensitive properties. The morphology of our hybrid SiO2 nanoparticles was characterized by Transmission Electron Microscopy (TEM) and Scanning Electron Microscopy (SEM). measurements of turbidity, Dynamic Light Scattering (DLS), and phase transfer were conducted to investigate the thermal responses of the hybrid material. In addition, Pickering emulsions were prepared using such hybrid SiO2-POEGMA as an emulsifier. Moreover, its fast de-emulsification above a lower critical solution temperature (LCST) may be exploited for the efficient separation and recycling of catalysts in future.
Introduction
Silica/polymer hybrid nanoparticles have attracted much attention due to their fascinating optical, electronic, magnetic, biological, pharmacological, and catalytic properties.1–7 Typically, hybrid nanoparticles incorporate polymer shells which are the principal determinants of the chemical properties of nanoparticles and their responsiveness to external stimuli.8–11Silica/polymer hybrid nanoparticles can be prepared by efficient controlled polymerization techniques, such as atom transfer radical polymerization (ATRP), which can incorporate organic polymers with a precise degree of substitution, composition, and functionality into both planar surfaces,12–14 and colloidal/particulate substrates.15–19 Patten and colleagues20,21 demonstrated that styrene and methyl methacrylate could be controllably polymerized from silica nanoparticles through surface-initiated ATRP.
However, the initiator system of ATRP is generally toxic and expensive and the catalyst such as CuCl is unstable under air and easily oxidated.22,23 To overcome these drawbacks, Wang and Matyjaszewski discovered reverse ATRP, in which the initiation step involves the active radical combining a halogen atom with the catalyst to form a dormant halide species and a reduced transition metal activator.24,25 However, to reach the ATRP equilibrium, a conventional radical initiator such as 2,2-azobisisobutyronitrile (AIBN) is typically added to generate radicals that reduce Cu(II) to Cu(I) and generate the halogen-containing initiator in situ. In reverse ATRP, the amount of catalyst cannot be independently reduced and should be comparable to the radical initiator because the added Cu(II) complex is the only source of transferable atoms.26
Fortunately, a new method to achieve the formation of an active catalyst, a procedure for preparing an “activator generated by electron transfer” for ATRP (AGET ATRP), was developed by Krzysztof Matyjaszewski.27 This method resolved these issues by using electron transfer instead of employing a conventional radical initiator. A reducing agent, typically Sn(EH)2, ascorbic acid (AA) or phenol, is used to generate the activator through reduction of the Cu(II) complex without the involvement of organic radicals that can initiate new chains.26,28,29 Therefore, AGET ATRP has the advantages of normal ATRP and reverse ATRP alongside additional benefits such as a high rate of conversion of the monomer in a well controlled manner.30–32 Zhu and colleagues synthesised a series of versatile magnetic nanoparticles with graft-on functional polymers employing iron-catalyzed AGET ATRP.33–36
To date, perhaps the most extensively studied thermosensitive hybrid nanoparticles have been coated with poly(N-isopropylacrylamide) (PNIPAM) brushes, which exhibit LCST at ∼32 °C, close to body temperature.37–40 Wang and colleagues41 synthesised SiO2 nanoparticles grafted with PNIPAM shells by surface-initiated ATRP in aqueous solution, using CuCl/bpy as a catalytic system. Despite its widespread popularity in materials science, PNIPAM has narrow and low LCST, which limits its practical applications. So, the pursuit of tunable LCST is one of major objectives of thermosensitive hybrid nanoparticles.42–47 Thermosensitive poly(ethylene glycol) methyl ether methacrylate was recently proposed as an attractive alternative to PNIPAM.46,48–52 In fact, the balance between hydrophobic and hydrophilic moieties in the molecular structure of the amphiphilicity polymers is the key-parameter that determines LCST.53 The side-chains (i.e., EO unit) of poly(ethylene glycol) methyl ether methacrylate are hydrophilic group by forming stabilizing H-bonds with water.54,55 The introduction of hydrophilic group, such as poly(ethylene glycol, EO unit) moieties, into the temperature-responsive polymers might raise the LCST, whereas that of a hydrophobic group might lower the LCST.49,51,56 Therefore, the LCST can be finely tuned through modification of the length of the oligo(ethylene glycol) side chain.43,53,57 In addition, POEGMA is well-known to be an uncharged, nontoxic, protein-resistant, and biocompatible polymer and has been widely applied in biomedical applications.58,59
The efficient segregation and recycling of catalysts is one of major obstacles for sustainable and green chemistry.60–65 Pickering emulsions using nanoparticles as emulsifiers are emerging as an attractive platform for designing efficient catalytic systems by providing a large oil–water reaction interface.66–72 Yang67 successfully developed a novel method for in situ separation and recycling of submicrometer-sized solid catalysts by tuning the pH value based on Pickering-emulsion inversion.
In this paper we describe the synthesis of thermosensitive poly(ethylene glycol) methyl ether methacrylate (POEGMA, Mn ∼ 300 or 500 g mol−1) brushes on silica nanoparticles using AGET ATRP and study the thermal properties of this hybrid material through measurements of turbidity, phase transfer, and dynamic light scattering (DLS). Interestingly, the LCST can be finely tuned through modification of the length of the oligo(ethylene oxide) side chain. Then, Pickering emulsions prepared using hybrid SiO2-POEGMA were investigated for the efficient separation and recycling of catalysts in future. The general scheme for the preparation of bare SiO2 nanoparticles (SiO2), amino-functionalized (SiO2-NH2), 2-bromoisobutyrate-functionalized (SiO2-Br), and thermoresponsive POEGMA grafted SiO2 nanoparticles (SiO2-POEGMA) are shown in Scheme 1.
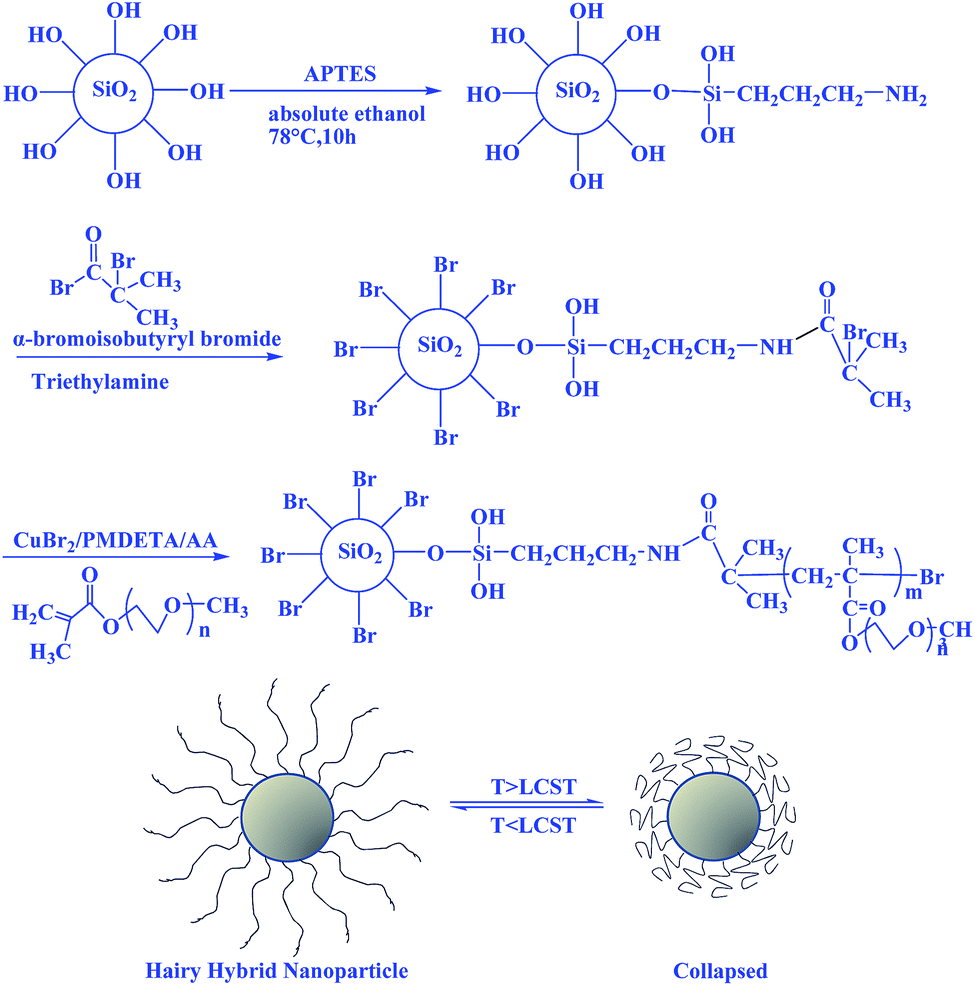 | ||
| Scheme 1 Schematic illustration of the synthesis of hybrid silica nanoparticles coated with thermoresponsive poly(ethylene glycol) methyl ether methacrylate (POEGMA) brushes via AGET-ATRP. The densely grafted POEGMA brushes exhibit thermosensitive swelling/collapse phase transitions. | ||
Experimental
Materials
Tetraethoxysilane (TEOS), CuBr2, CH2Cl2, 2-bromoisobutyryl bromide, 3-aminopropyltriethoxysilane (APTES), N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA), and ascorbic acid (AA) were obtained from Aladdin Reagent Co. Poly(ethylene glycol) methyl ether methacrylate (POEGMA300 and POEGMA500) were purchased from Sigma-Aldrich and used without further purification. Absolute alcohol and ammonia were of analytical grade from Beijing Chemical works. Toluene, mineral oil, ethyl acetate and triethylamine were purchased from Tianjin Fengchuan Chemical Reagent Co., Ltd. Water used in the experiments was triply distilled by a quartz water purification system. Its conductivity was lower than 1.8 μS cm−1, as measured by a DDSJ-308A type conductivity instrument in our laboratory.Synthesis of SiO2 and SiO2-NH2
Nano-SiO2 particles were prepared in absolute ethanol according to the Stöber method.73,74 TEOS (9 mL) and ethanol (300 mL) were added into a three-necked round-bottomed flask at ambient temperature, and ammonium hydroxide (25% (w/w) in water, 30 mL) then added to the system through a dropping funnel while the flask was stirred vigorously. The solution was incubated for eight hours to obtain a bare nano-SiO2 particle suspension (solution A). APTES (9 mL), ethanol (15 mL) and ammonium hydroxide (25% (w/w) in water, 3 mL) were added into a round-bottomed flask, and then injected dropwise into solution A while the flask was stirred vigorously. The reaction mixture was heated to reflux temperature (78 °C) for 10 h. The solution was then cooled to room temperature and a powdery white product obtained. The powder was filtered and washed with ethanol. The sample was then dried in a vacuum oven at 60 °C for 12 h.Synthesis of SiO2-Br
SiO2-NH2 (2.50 g) was dispersed in 150 mL of anhydrous toluene by ultrasonication. After cooling to 0 °C, triethylamine (4 mL) and 2-bromoisobutyryl bromide (3 mL) were added dropwise. The solution was stirred at 0 °C for 6 h and then at room temperature for 12 h. The nanoparticles were purified and isolated following similar procedures to those described for the synthesis of SiO2-NH2. The final product was dried in a vacuum oven at 60 °C for 12 h.Synthesis of SiO2-POEGMA via AGET-ATRP
SiO2-Br (1.50 g) was dispersed in 10 mL of CH2Cl2 via ultrasonication. Then, CuBr2 (0.1671 g) and PMDETA (0.7793 g) were added to a stirred round-bottomed flask at room temperature to form a homogeneous solution. POEGMA500 (3.0 g) was then added and, once the solution became well mixed and had returned to room temperature, 30 mL of deionized water was added. The CH2Cl2 was slowly evaporated from the mixture through purging with nitrogen for 30 min. Ascorbic acid (0.04 mmol) was injected dropwise into the deoxygenated aqueous solution at 70 °C. The SiO2-POEGMA500 nanoparticles were recovered by filtration, washed with ethanol several times and then extracted 12 h using a Soxhlet extractor. The collected sample was dried in a vacuum oven at 60 °C for 12 h. The SiO2-POEGMA300 nanoparticles were prepared by similar steps.Characterization
Fourier transform infrared spectroscopy (FTIR) spectra were obtained using a Bruker V70 instrument by incorporating hybrid particles into KBr pellets.X-ray photoelectron spectroscopy (XPS) was recorded with a PHI quantum ESCA micro-probe system (THERMO VG ESCALAB250), using the Al Kα line as the radiation source at an energy of 1486.6 eV, 16 mA × 12 kV. The fitting XPS curves were analyzed with multipeak 6.0 A software.
Thermogravimetric analysis (TGA) was carried out using a thermal analyzer (HTC-3) under a nitrogen flow (10 mg of the sample was heated in steps of 10 °C min−1 in an Al2O3 pan for analysis).
Weight- and number-average molecular weights, Mw and Mn, respectively, were determined using a Waters-greeze gel permeation chromatography (GPC) with polystyrene as the standard for molecular weight calibration. GPC analyses were run in THF at 20 °C with an RI-detector.
1H NMR spectra were obtained in CDCl3 at room temperature using a Bruker DRX-300 instrument and tetramethylsilane as an internal standard.
The morphologies of pure silica and the polymer grafted silica nanoparticles were characterized with a JEM-1100 TEM (JEOL, Tokyo, Japan) at 100 kV. They were dispersed in ethanol in an ultrasonic bath for 15 min, deposited on a copper grid, and the grid then dried at room temperature.
Scanning electron microscopy (SEM) images were acquired on a LEO 1525 scanning electron microscope. The SEM samples were prepared by adding a few drops of a dilute suspension onto a clean silicon wafer and allowing the solvent to evaporate in air.
DLS measurements were conducted with a Zeta Plus Particle Size Analysis instrument (Brookhaven, USA) at a scattering angle of 90°. The light source was a Spectra-physics 127 helium neon laser (633 nm, power 35 mW). The sample was held at each temperature for 15 min to ensure that it had reached equilibrium. The optical transmittance of aqueous solutions of hybrid nanoparticles was measured at a wavelength of 560 nm on a Unico UV/vis 2802PCS spectrophotometer using a thermostatically controlled bath.
Emulsion droplets were observed by microscope (Leica DMI 6000) at 10 × 10 magnification.
Results and discussion
FTIR spectra
Infrared spectroscopy was employed to verify that molecules had been successfully attached to the particle surface.75 The FTIR spectra of bare SiO2, SiO2-Br and SiO2-POEGMA500 are shown in Fig. 1. For bare SiO2 (Fig. 1A), absorption bands characteristic of tetrahedral silicate structures occur at 1100 cm−1 (Si–O stretching) and 465 cm−1 (Si–O bending). Si–OH bending can be observed at 945 cm−1 and Si–O–Si bending at 800 cm−1. After amidation, a doublet appears in the vicinity of 1380 cm−1, characteristic absorptions due to the deformation of the two isopropyl methyl groups in 2-bromoisobutyrate residues.76 (Fig. 1B and insert diagram.) For SiO2-POEGMA500, the absorption band at 2980 cm−1 is ascribed to the –C–H antisymmetric stretching vibration, and the absorption band at 2850 cm−1 is the –CH2 stretching vibration. The absorption band at 1730 cm−1 belongs to –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching (Fig. 1C). The presence of these bands confirm that the SiO2-POEGMA500 has been prepared successfully. Similar FTIR spectra were noticed for SiO2-POEGMA300 (data not shown).
O stretching (Fig. 1C). The presence of these bands confirm that the SiO2-POEGMA500 has been prepared successfully. Similar FTIR spectra were noticed for SiO2-POEGMA300 (data not shown).
 | ||
| Fig. 1 FTIR spectra of (A) bare SiO2, (B) SiO2-Br, and (C) SiO2-POEGMA500. | ||
Surface composition analysis by XPS
XPS was used to determine the composition of samples and identify the valence states of various species.77 As shown in Fig. 2A, the POEGMA500's C1s spectra could be fitted with three peak components attributable to: hydrocarbon (CxHy: 284.71 eV), carbon adjacent to oxygen (C–O: 286.02 eV), and carbonyl (O–CO: 288.64 eV). For O1s spectra of bare SiO2 (Fig. 2B), there is only one peak at 532.67 eV (O atom in Si–O bond), while a further peak with a binding energy of 533.66 eV (O atom in CO bond) appears in O1s spectra of SiO2-POEGMA500 (Fig. 2C). From a wide scan of the bare SiO2 and SiO2-POEGMA500 (Fig. S1 in the ESI†), we could see that the carbon content of SiO2-POEGMA500 increased and silicon content decreased in comparison to bare SiO2. The specific elemental composition of bare SiO2 and SiO2-POEGMA500 is shown in Tables S1 and S2 of ESI† respectively. The above results can be considered clear evidence of our success in preparing SiO2-POEGMA500. Similar XPS spectra were got for SiO2-POEGMA300 (data not shown).
 | ||
| Fig. 2 XPS spectra of (A) C1s of SiO2-POEGMA500, (B) O1s of the bare SiO2, (C) O1s of SiO2-POEGMA500. | ||
Thermogravimetric analysis
Thermogravimetric analysis (TGA), which was performed under a nitrogen flow at a heating rate of 10 °C min−1, indicated that the weight retention at 420 °C was 93.5% for bare SiO2 and 92.2% for SiO2-NH2 nanoparticles (Fig. 3A and B). The weight loss of bare particles might be due to the associated water loss and continued condensation reaction.78 The mass retention of SiO2-POEGMA500 was far less than of SiO2-Br (Fig. 3C and D), showing that POEGMA500 was grafted onto SiO2 by high-density.79 We got alike mass retention for SiO2-POEGMA300 (data not shown). The molecular weights (Mn = 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 234 and Mw = 38237) of the polymer POEGMA500 resulting from AGET ATRP conducted by GPC further verified that POEGMA500 could be well polymerized by AGET ATRP.80 A typical 1H NMR spectrum of the polymer POEGMA500 was shown in Fig. S2 in the ESI.†
234 and Mw = 38237) of the polymer POEGMA500 resulting from AGET ATRP conducted by GPC further verified that POEGMA500 could be well polymerized by AGET ATRP.80 A typical 1H NMR spectrum of the polymer POEGMA500 was shown in Fig. S2 in the ESI.†
 | ||
| Fig. 3 Thermogravimetric analysis (TGA) of (A) bare SiO2, (B) SiO2-NH2, (C) SiO2-Br, and (D) SiO2-POEGMA500. | ||
Morphology analysis
The morphology (size, shape and distribution) of the nanoparticles was observed by transmission electron microscopy (TEM) and scanning electron microscopy (SEM). TEM images of bare SiO2, SiO2-NH2, SiO2-Br and SiO2-POEGMA500 composites are showed in Fig. 4. As can be seen from the images, bare SiO2 exhibits an average external diameter about 327 nm and nothing can be seen around the nanoparticles (Fig. 4A). After grafted with POEGMA500, the observed diameter of SiO2-POEGMA500 became larger and clear core/shell structures were formed (Fig. 4D). This implied that the polymer chains had been linked to SiO2. | ||
| Fig. 4 TEM images of (A) bare SiO2 (327 nm), (B) SiO2-NH2 (337 nm), (C) SiO2-Br (345 nm), and (D) SiO2-POEGMA500 (357 nm). | ||
SEM images of the colloidal silica particles before and after the surface initiated polymerization of POEGMA500 were depicted in Fig. 5. The bare SiO2 morphology was spherical and monodisperse, as expected (Fig. 5A). As can be seen in Fig. 5B, each silica particle is coated with a relatively thick outer layer of POEGMA500 chains compared with bare SiO2, consistent with the observations of C. Perruchot.81 Similar TEM and SEM images of SiO2-POEGMA300 were noticed (data not shown).
 | ||
| Fig. 5 SEM images of (A) bare SiO2, (B) SiO2-POEGMA500. | ||
Turbidity measurements
Fig. 6 shows the temperature dependence of optical transmittance at 560 nm obtained for aqueous solutions of SiO2-POEGMA and bare SiO2. For SiO2-POEGMA500 (Fig. 6B), no appreciable changes in the optical transmittance were observed within the range 25–85 °C. In this temperature range, the outer zone of POEGMA brushes remained hydrophilic and aggregation between particles did not occur. Above 85 °C, however, transmittance increased abruptly from 2% to 70%, implying that the hydrogen bonds had broken and that aggregation of hybrid nanoparticles occurred in the temperature range 85–94 °C. For SiO2-POEGMA300 (Fig. 6C), transmittance increased abruptly in the temperature range 58–68 °C. Liu8 predicted that this type of collision will contribute to the aggregation between different hybrid nanoparticles since the PNIPAM corona becomes “sticky” above the critical phase transition temperature. However, no noticeable variations were recognized for bare SiO2 at all temperatures trialed. Illustrative images of bare SiO2 and SiO2-POEGMA500 before and after heating are shown in Fig. 7. | ||
| Fig. 6 Temperature-dependent optical transmittance at 560 nm obtained for 5.0 × 10−2 g mL−1 aqueous solution of SiO2-POEGMA300 (red line), SiO2-POEGMA500 (blue line), bare SiO2 (black line). | ||
 | ||
| Fig. 7 Illustrative images of bare SiO2 and SiO2-POEGMA500 before and after heating: bare SiO2: (A) 25 °C, (B) 90 °C; SiO2-POEGMA500: (C) 25 °C, (D) 90 °C. | ||
DLS analysis
The temperature-dependent size and size distribution of SiO2-POEGMA300 and SiO2-POEGMA500 in aqueous solutions were measured by dynamic light scattering (DLS). Aqueous dispersions of SiO2-POEGMA500 at a concentration of 0.005 g mL−1 were gradually heated from 25 °C to 95 °C. As shown in Fig. 8A, when the temperature dropped below 75 °C, no significant changes in the average hydrodynamic diameter were observed with increasing temperature. The average hydrodynamic size decreased over a broad temperature range from 75 °C (366.5 nm) to 95 °C (183.2 nm), a much wider range than the LCST of free POEGMA in water (90 °C), consistent with the observations by Kizhakkedathu and colleagues of PNIPAm brushes on PS latex particles.82 While, as shown in Fig. 8B the average hydrodynamic size decreased from 60 °C (352.9 nm) to 67 °C (219.8 nm) for SiO2-POEGMA300 (LCST ∼ 65 °C). | ||
| Fig. 8 Average hydrodynamic diameters of the SiO2-POEGMA300 and SiO2-POEGMA500 in the different temperature measured by dynamic light scattering. | ||
Owing to the relatively high graft densities (see Fig. 3D) of POEGMA chains on the nanoparticles, the interactions between neighboring polymer chains may play a role in the transition. It was predicted by Zhulina and colleagues that spherical polymer brushes should undergo a transition in which the collapse of polymer brushes would begin in the outermost layers.83 Inter-chain interactions make the collapse of the grafted polymers weaker than that of free polymer chains in a dilute solution as the inter-chain contacts reduce water–monomer interactions. Zhao and colleagues84 proposed that the broader transitions of polymer brushes on the nanoparticles might be also related to different degrees of freedom of segments along the grafted polymer chains.
Fig. 9 shows the particle size distribution of SiO2-POEGMA500 at 55 °C and 92 °C. It can be seen that the distribution of the particle size was monodisperse, in agreement with the results of TEM and SEM.
 | ||
| Fig. 9 The particle size distribution of the SiO2-POEGMA500 measured by dynamic light scattering at 55 °C and 92 °C, respectively. | ||
Phase transfer triggered by temperature
Bare SiO2 (0.05 g) and SiO2-POEGMA500 (0.05 g) were dispersed in 4 mL of deionized water by ultrasonication, respectively. Ethyl acetate (4 mL) was added into the bottle and emulsified for 2 min. Illustrative images showing phase transfer before and after heating are recorded in Fig. 10. Bare SiO2 precipitated at all temperatures trialed (Fig. 10A and B). SiO2-POEGMA500 remained in the aqueous layer at 25 °C, however, they then transferred into the oil phase above 90 °C (Fig. 10C and D), which is similar to the observations of Zhao and colleagues.85 For SiO2-POEGMA300, the phase transfer temperature was about 65 °C. A similar conclusion was got when the oil phase was changed to toluene (Fig. 10E and F) or mineral oil (Fig. 10G and H). | ||
| Fig. 10 Phase transfer triggered by temperature of bare SiO2 and SiO2-POEGMA500. | ||
Pickering-emulsion preparation
SiO2-POEGMA500 (0.2 g) was dispersed in 4 mL of deionized water using a vortex mixer (Fisher Scientific Analogue Vortex Mixer, 120 V at 3000 rpm) for 2 min. Toluene (4 mL) was added into the bottle and emulsified for 2 min and emulsions were formed. As can be seen in Fig. 11A, SiO2-POEGMA500 was well distributed in the upper layer with good stability against sedimentation (at least two weeks). Microscopic images confirmed that the upper layer was a Pickering emulsion phase; spherical droplets can clearly be observed in Fig. 11C. Interestingly, de-emulsification was observed when the Pickering emulsion was heated above 90 °C (Fig. 11B) and emulsion droplets were damaged (Fig. 11D). Intriguingly, a Pickering emulsion could be rapidly reformed by cooling down the sample to allow re-emulsification, and emulsion droplets were still observed (Fig. S3 ESI†). Similar phenomenon was observed for Pickering-emulsion prepared by SiO2-POEGMA300. This emulsification–demulsification inversion related to the temperature-responsive surface chemistry could in future be exploited for separation and recycling of catalysts.67,86,87 | ||
| Fig. 11 Photographs and light micrographs of the emulsification–demulsification inversion of the Pickering emulsion. | ||
Conclusions
Narrow-disperse thermoresponsive hybrid silica nanoparticles grafted with POEGMA (Mn ∼ 300 or 500 g mol−1) brushes were synthesized by AGET ATRP from SiO2-Br nanoparticles. The experimental results confirmed that the SiO2-POEGMA was prepared successfully. Optical transmittance measurement, dynamic light scatter (DLS) and phase transfer indicated that thermo-induced phase transitions of SiO2-POEGMA300 were around 65 °C, while thermo-induced phase transitions of SiO2-POEGMA500 was around 90 °C. Pickering emulsions prepared by hybrid SiO2-POEGMA possessed the ability to undergo emulsification–demulsification inversion in response to temperature, which may have potential applications in biomaterials, sensors, drug delivery, stimuli-responsive materials, and catalyst separation and recycling.Acknowledgements
We gratefully acknowledge financial support from the National Science & Technology Pillar Program during the Twelfth Five-year Plan Period (Grant no. 2014BAE03B03), Natural Science Found of Shanxi Province (Grant no. 2014011014-1), China National Petroleum & gas Corporation science and technology development project “Nano intelligent chemical flooding agent” (2011A-1001), Shanxi Scholarship Council of China (no. 2013-151), Shanxi Natural Science Foundation (no. 2012011046-3) and Shanxi Province Graduate Outstanding Innovation Project (no. 0113127).Notes and references
- J. Feng and H. Zhang, Chem. Soc. Rev., 2013, 42, 387 RSC.
- E. Hutter and J. H. Fendler, Adv. Mater., 2004, 16, 1685 CrossRef CAS.
- X. Sun, X. Xin, N. Tang, L. Guo, L. Wang and G. Xu, J. Phys. Chem. B, 2014, 118, 824 CrossRef CAS PubMed.
- B. Daglar, E. Ozgur, M. Corman, L. Uzun and G. Demirel, RSC Adv., 2014, 4, 48639 RSC.
- G. L. Li, Z. Zheng, H. Möhwald and D. G. Shchukin, ACS Nano, 2013, 7, 2470 CrossRef CAS PubMed.
- X. Sun, Z. Du, E. Li, X. Xin, N. Tang, L. Wang and J. Yuan, Colloids Surf., A, 2014, 457, 345 CrossRef CAS PubMed.
- Z. Du, E. Li, G. Wang and F. Cheng, RSC Adv., 2014, 4, 4836 RSC.
- T. Wu, Y. Zhang, X. Wang and S. Liu, Chem. Mater., 2008, 20, 101 CrossRef CAS.
- Y. Hu, H. An, X. Liu, J. Yin, H. Wang, H. Zhang and L. Wang, Dalton Trans., 2014, 43, 2488 RSC.
- Q. Fang, Q. Cheng, H. Xu and S. Xuan, Dalton Trans., 2014, 43, 2588 RSC.
- M. Cargnello, N. L. Wieder, P. Canton, T. Montini, G. Giambastiani, A. Benedetti, R. J. Gorte and P. Fornasiero, Chem. Mater., 2011, 23, 3961 CrossRef CAS.
- D. M. Jones, A. A. Brown and W. T. Huck, Langmuir, 2002, 18, 1265 CrossRef CAS.
- V. L. Osborne, D. M. Jones and W. T. Huck, Chem. Commun., 2002, 1838 RSC.
- W. Huang, G. L. Baker and M. L. Bruening, Angew. Chem., 2001, 113, 1558 CrossRef.
- J. N. Kizhakkedathu and D. E. Brooks, Macromolecules, 2003, 36, 591 CrossRef CAS.
- P. von Natzmer, D. Bontempo and N. Tirelli, Chem. Commun., 2003, 1600 RSC.
- W. Huang, J.-B. Kim, M. L. Bruening and G. L. Baker, Macromolecules, 2002, 35, 1175 CrossRef CAS.
- J. Pyun and K. Matyjaszewski, Chem. Mater., 2001, 13, 3436 CrossRef CAS.
- Q. Li, L. Zhang, Z. Zhang, N. Zhou, Z. Cheng and X. Zhu, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2006 CrossRef CAS.
- T. Von Werne and T. E. Patten, J. Am. Chem. Soc., 1999, 121, 7409 CrossRef CAS.
- T. von Werne and T. E. Patten, J. Am. Chem. Soc., 2001, 123, 7497 CrossRef CAS PubMed.
- G. Wang, X. Zhu, Z. Cheng and J. Zhu, Eur. Polym. J., 2003, 39, 2161 CrossRef.
- D.-Q. Qin, S.-H. Qin and K.-Y. Qiu, Macromolecules, 2000, 33, 6987 CrossRef CAS.
- P. Li and K.-Y. Qiu, Polymer, 2002, 43, 3019 CrossRef CAS.
- J.-S. Wang and K. Matyjaszewski, Macromolecules, 1995, 28, 7572 CrossRef CAS.
- K. Min, H. Gao and K. Matyjaszewski, J. Am. Chem. Soc., 2005, 127, 3825 CrossRef CAS PubMed.
- W. Jakubowski and K. Matyjaszewski, Macromolecules, 2005, 38, 4139 CrossRef CAS.
- J. Pietrasik, H. Dong and K. Matyjaszewski, Macromolecules, 2006, 39, 6384 CrossRef CAS.
- Z. Du, X. Sun, X. Tai, G. Wang and X. Liu, Appl. Surf. Sci., 2014, 329, 234 CrossRef PubMed.
- T. B. Mai, T. N. Tran, M. R. Islam, J. M. Park and K. T. Lim, J. Mater. Sci., 2014, 49, 1519 CrossRef CAS.
- S. Aleksanian, Y. Wen, N. Chan and J. K. Oh, RSC Adv., 2014, 4, 3713 RSC.
- H. Zhao, X. Kang and L. Liu, Macromolecules, 2005, 38, 10619 CrossRef CAS.
- W. He, L. Cheng, L. Zhang, Z. Liu, Z. Cheng and X. Zhu, Polym. Chem., 2014, 5, 638 RSC.
- J. Liu, W. He, L. Zhang, Z. Zhang, J. Zhu, L. Yuan, H. Chen, Z. Cheng and X. Zhu, Langmuir, 2011, 27, 12684 CrossRef CAS PubMed.
- W. He, L. Cheng, L. Zhang, Z. Liu, Z. Cheng and X. Zhu, ACS Appl. Mater. Interfaces, 2013, 5, 9663 CAS.
- W. He, L. Cheng, L. Zhang, X. Jiang, Z. Liu, Z. Cheng and X. Zhu, Nanotechnology, 2014, 25, 045602 CrossRef PubMed.
- H. Schild, Prog. Polym. Sci., 1992, 17, 163 CrossRef CAS.
- S. K. Saha, S. Das, P. Chowdhury and S. K. Saha, RSC Adv., 2014, 4, 14457 RSC.
- P. Liu, J. Liang, S. Chen and H. Zhang, RSC Adv., 2014, 4, 49028 RSC.
- G. Marcelo and M. Fernández-García, RSC Adv., 2014, 4, 11740 RSC.
- K. Zhang, J. Ma, B. Zhang, S. Zhao, Y. Li, Y. Xu, W. Yu and J. Wang, Mater. Lett., 2007, 61, 949 CrossRef CAS PubMed.
- N. Fechler, N. Badi, K. Schade, S. Pfeifer and J.-F. Lutz, Macromolecules, 2008, 42, 33 CrossRef.
- N. Badi and J.-F. Lutz, J. Controlled Release, 2009, 140, 224 CrossRef CAS PubMed.
- Y. Zou, D. E. Brooks and J. N. Kizhakkedathu, Macromolecules, 2008, 41, 5393 CrossRef CAS.
- S. Li, Y. Su, M. Dan and W. Zhang, Polym. Chem., 2014, 5, 1219 RSC.
- Y. Li, H. Guo, Y. Zhang, J. Zheng, J. Gan, X. Guan and M. Lu, RSC Adv., 2014, 4, 17768 RSC.
- E. Dashtimoghadam, H. Mirzadeh, F. A. Taromi and B. Nyström, RSC Adv., 2014, 4, 39386 RSC.
- Y. Li, H. Guo, J. Zheng, J. Gan, Y. Zhang, X. Guan, K. Wu and M. Lu, RSC Adv., 2014, 4, 54268 RSC.
- S. Han, M. Hagiwara and T. Ishizone, Macromolecules, 2003, 36, 8312 CrossRef CAS.
- M. M. Ali and H. D. Stöver, Macromolecules, 2004, 37, 5219 CrossRef CAS.
- H. Kitano, T. Hirabayashi, M. Gemmei-Ide and M. Kyogoku, Macromol. Chem. Phys., 2004, 205, 1651 CrossRef CAS.
- L. Wang, D. Su, L. Zeng, N. Liu, L. Jiang, X. Feng, K. Neoh and E. Kang, Dalton Trans., 2013, 42, 13642 RSC.
- J. F. Lutz, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3459 CrossRef CAS.
- K. Tasaki, J. Am. Chem. Soc., 1996, 118, 8459 CrossRef CAS.
- J. Israelachvili, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 8378 CrossRef CAS.
- M. Mertoglu, S. Garnier, A. Laschewsky, K. Skrabania and J. Storsberg, Polymer, 2005, 46, 7726 CrossRef CAS PubMed.
- N. Fechler, N. Badi, K. Schade, S. Pfeifer and J.-F. Lutz, Macromolecules, 2009, 42, 33 CrossRef CAS.
- W. Zhu, A. Nese and K. Matyjaszewski, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1942 CrossRef CAS.
- H. Dong, V. Mantha and K. Matyjaszewski, Chem. Mater., 2009, 21, 3965 CrossRef CAS.
- V. K. Dioumaev and R. M. Bullock, Nature, 2000, 424, 530 CrossRef PubMed.
- D. J. Cole-Hamilton, Science, 2003, 299, 1702 CrossRef CAS PubMed.
- M. Mokhadinyana, S. L. Desset, D. B. G. Williams and D. J. Cole-Hamilton, Angew. Chem., Int. Ed., 2012, 51, 1648 CrossRef CAS PubMed.
- P. Pollet, R. J. Hart, C. A. Eckert and C. L. Liotta, Acc. Chem. Res., 2010, 43, 1237 CrossRef CAS PubMed.
- F.-C. Zheng, Q.-W. Chen, L. Hu, N. Yan and X.-K. Kong, Dalton Trans., 2014, 43, 1220 RSC.
- X. Duan, Y. Liu, Q. Zhao, X. Wang and S. Li, RSC Adv., 2013, 3, 13748 RSC.
- S. Crossley, J. Faria, M. Shen and D. E. Resasco, Science, 2010, 327, 68 CrossRef CAS PubMed.
- H. Yang, T. Zhou and W. Zhang, Angew. Chem., 2013, 125, 7603 CrossRef.
- P. A. Zapata, J. Faria, M. P. Ruiz, R. E. Jentoft and D. E. Resasco, J. Am. Chem. Soc., 2012, 134, 8570 CrossRef CAS PubMed.
- L. Leclercq, A. Mouret, A. Proust, V. Schmitt, P. Bauduin, J. M. Aubry and V. Nardello-Rataj, Chem.–Eur. J., 2012, 18, 14352 CrossRef CAS PubMed.
- B. M. Mosby, A. Díaz and A. Clearfield, Dalton Trans., 2014, 43, 10328 RSC.
- O. V. Kharissova, B. I. Kharisov and E. G. de Casas Ortiz, RSC Adv., 2013, 3, 24812 RSC.
- J. Pan, W. Zhu, X. Dai, X. Yan, M. Gan, L. Li, H. Hang and Y. Yan, RSC Adv., 2014, 4, 4435 RSC.
- W. Stöber, A. Fink and E. Bohn, J. Colloid Interface Sci., 1968, 26, 62 CrossRef.
- A. Philipse and A. Vrij, J. Colloid Interface Sci., 1989, 128, 121 CrossRef CAS.
- G. A. Hudalla and W. L. Murphy, Langmuir, 2009, 25, 5737 CrossRef CAS PubMed.
- Y. V. Pan, R. A. Wesley, R. Luginbuhl, D. D. Denton and B. D. Ratner, Biomacromolecules, 2001, 2, 32 CrossRef CAS PubMed.
- F. Li and X. Li, Appl. Catal., A, 2002, 228, 15 CrossRef CAS.
- S. Blomberg, S. Ostberg, E. Harth, A. W. Bosman, B. Van Horn and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 1309 CrossRef CAS.
- E. Effati and B. Pourabbas, Powder Technol., 2013, 246, 473 CrossRef CAS PubMed.
- J. Pietrasik, C. M. Hui, W. Chaladaj, H. Dong, J. Choi, J. Jurczak, M. R. Bockstaller and K. Matyjaszewski, Macromol. Rapid Commun., 2011, 32, 295 CrossRef CAS PubMed.
- C. Perruchot, M. Khan, A. Kamitsi, S. v. Armes, T. Von Werne and T. Patten, Langmuir, 2001, 17, 4479 CrossRef CAS.
- J. N. Kizhakkedathu, R. Norris-Jones and D. E. Brooks, Macromolecules, 2004, 37, 734 CrossRef CAS.
- E. Zhulina, O. Borisov, V. Pryamitsyn and T. Birshtein, Macromolecules, 1991, 24, 140 CrossRef CAS.
- D. Li, G. L. Jones, J. R. Dunlap, F. Hua and B. Zhao, Langmuir, 2006, 22, 3344 CrossRef CAS PubMed.
- D. Li and B. Zhao, Langmuir, 2007, 23, 2208 CrossRef CAS PubMed.
- H. Liu, Z. Zhang, H. Yang, F. Cheng and Z. Du, ChemSusChem, 2014, 7, 1888 CrossRef CAS PubMed.
- W. Zhang, L. Fu and H. Yang, ChemSusChem, 2014, 7, 391 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra17013j |
| This journal is © The Royal Society of Chemistry 2015 |