
Biogenic synthesis of ZnO–Ag nano custard apples for efficient photocatalytic degradation of methylene blue by sunlight irradiation†
S. Kaviya and
Edamana Prasad*
Department of Chemistry, Indian Institute of Technology Madras, Chennai-600 036, TN, India. E-mail: pre@iitm.ac.in; Fax: +91-44-2257-4202; Tel: +91-44-2257-4232
First published on 29th January 2015
Abstract
Photocatalytic degradation of pollutants will be attractive if the degradation process is under direct sunlight in the presence of a cost effective catalyst. We have synthesized a novel, monodispersed ZnO–Ag nano custard apple (NCA) using pomegranate peel extract as reducing and stabilizing agent. The ZnO–Ag NCA was characterized by various spectroscopic and microscopic methods. The photocatalytic performance of ZnO–Ag NCA was evaluated using the degradation of methylene blue dye in the presence of direct sunlight. It was observed that NCA show ∼40% and 24% enhanced photocatalytic activity compared to commercial ZnO nanoparticles and TiO2 (P25). Moreover, the performance of the photocatalyst NCA has been examined in the presence of oxidative reagents such as peroxomonosulfate (PMS), peroxodisulfate (PDS) and hydrogen peroxide (H2O2). The mineralization data of methylene blue, performed by total organic carbon (TOC) analysis, revealed that 95% and 70% mineralization was achieved in 3 h using a NCA–PMS combination and NCA, respectively. The catalyst did not lose its catalytic activity for up to five cycles. Overall, this system is relatively inexpensive, reproducible, extremely stable and efficient for complete degradation of methylene blue in aqueous solution.
Introduction
Industrial effluents, especially dyes, cause serious problems for environmental protection.1 Most of the dyes produced by the paper, textile and other industries are difficult to decompose due to the presence of aromatic dyes with relatively stable chemical structures.2 Methylene blue (MB) is one of the frequently used dyes for paper-making, textile, cosmetic, and pharmaceutical purposes.3 General treatments of such wastewater include adsorption,4 reverse osmosis,5 and/or chemical coagulations.6 In all of the above methods, further treatments are required to recycle the material and the associated costs are relatively high. The efficient oxidation reaction of contaminants by photocatalysts under irradiation processes provides an alternative way due to its easy operation and non-toxic end products.7 The photocatalytic degradation of pollutants under direct sunlight is a better option due to the renewable nature of the energy source. Semiconductor photocatalysts have been a potential field for treating various water pollutants.8 In particular, ZnO has attracted much attention with respect to the degradation of various pollutants due to its high photo sensitivity, stability, larger excitonic binding energy (60 meV), electron affinity (4.2 eV) and wide band gap (3.37 eV). The main drawback of ZnO is the rapid recombination of photoexcited electrons and holes which reduce its photocatalytic efficiency.9 The combination of semiconductor substrate and metal nanoparticles has been reported to give improved photocatalytic activity by trapping the photoinduced charge carriers, thereby improving the charge transfer processes.10,11 Moreover, silver nanoparticles (NPs) have been shown to have a beneficial influence on the photocatalytic activity of nano crystalline semiconductor due to their localized surface plasmon resonance (LSPR).12,13Reports have shown that the photocatalytic efficiency of ZnO nanocomposites can be significantly affected by morphologies.14,15 Various synthetic strategies have been developed to modify the photocatalytic performance of ZnO–Ag photo catalyst by different morphologies such as spherical,16 core–shell,17 rod,18 plate,19 wire20 and flower.21 In this article, we report a novel morphology of ZnO–Ag catalyst, achieved via one-pot, inexpensive, template free, rapid biosynthesis. The shape of the nano-cluster is identical with that of a custard apple and hence we name the monodispersed ZnO–Ag catalyst as a Nano Custard Apple (NCA). To the best of our knowledge, this is the first successful synthesis of monodispersed ZnO–Ag nano custard apple (NCA). While researchers have been using various synthetic methods to prepare ZnO nanoparticles (NPs) such as flame reactor,7 wet-phase,7 laser ablation,22 hydrothermal,23 seed layer,24 sol–gel,25 sputtering,26 chemical vapour deposition (CVD),27 molecular beam epitaxy (MBE),28 we have adopted a greener approach in this work where pomegranate peal extract is used as reducing and stabilizing agent for the nanosystem. Biological approach29–31 using plant or plant fractions for synthesizing nano-structures are attractive over other synthetic methodologies7,22–28 due to their simple, cost effective and eco-friendly nature. The NCA has been utilized for photocatalytic degradation of methylene blue (MB) under sunlight irradiation. The effect of the photocatalytic efficiency of ZnO–Ag NCA in presence of other oxidants such as peroxomonosulfate (PMS), peroxodisulfate (PDS) and hydrogen peroxide (H2O2) has been examined. We have also investigated the complete mineralization of MB and reusability of the catalyst.
Experimental
Materials
All the chemicals were purchased from Sigma-Aldrich (USA) and used as received without further purification. Doubly distilled water was used for the experiments. Fresh and ripened pomegranate was used for the experiment.Preparation of the extract
Pomegranate peels were removed and washed with distilled water before and after peeling. About 2 g of peels were weighted and transferred into a clean beaker containing 100 mL of distilled water and boiled for 5 min. The extract was filtered using Whatman filter paper no. 40 and stored in the refrigerator for further use.Synthesis of ZnO–Ag nanostructure
Zinc acetate solution was prepared by dissolving 3 g of Zn(OAc)2·2H2O into 30 mL of distilled water. To synthesis silver loaded ZnO nanoparticles, an aqueous solution of AgNO3 were prepared with different wt% ranging between 0.2 to 2.0 wt%. About 1 mL of AgNO3 solution was added into zinc acetate solution. Next, 3 mL of pomegranate peel extract was added drop wise into the well dissolved precursor solution under vigorous shaking using glass rod. The solution was kept under sunlight for 15 min. The formed precipitate was collected from the reaction vessel simply by decanting the upper solution. The precipitate was washed twice with distilled water to remove the excess capping agent and unreacted Zn and Ag ions from the solution. The sample was kept in the oven at 120 °C for 2 h. The as-synthesized compound was calcined at 300 °C in air atmosphere.Characterization
Diffused reflectance spectra (DRS) of the samples were recorded on a Shimadzu UV-vis spectrophotometer (UV-2550) fitted with ISR-2200 DRS accessory. Solid-state photoluminescence (PL) spectra were recorded on Shimadzu RF-5301 spectroflurophotometer. Fluoromax-4 (HORIBA Jobin Yuvin) spectrofluorometer was used for photoluminescence measurement of liquid sample. Bruker D8 advance powder X-day diffractometer with Cu Kα radiation was used for X-ray diffraction analysis. X-ray photoelectron spectroscopy (XPS) measurements were conducted to analyze elemental and chemical state of elements in the sample using Al Kα radiation as the source (Omicron model). FT-Raman spectra have been recorded from Bruker RFS 27 spectrometer. The size and morphology of ZnO–Ag NPs were examined using transmission electron microscopy (TEM) (Philips Tecnai 12). Energy dispersive X-ray spectroscopy (EDAX) was performed by TEM, equipped with an EDAX attachment. Scanning electron microscopy (SEM) images were taken by FEI Quanta FEG 200. The photocatalytic degradation of methylene blue (MB) was followed spectrophotometrically using UV-vis absorption spectroscopy (UV-3100 Hitachi). Total organic carbon content (TOC) of the reaction mixture was determined by total organic carbon analyzer (Shimadzu TOC-VCPH model). We measured the intensity of sunlight (measured between 11.00 am to 2.00 pm in the month of April to June in TN, India) using digital Lux meter (Metravi 1332).Evaluation of photocatalytic activity
Photocatalytic activity of ZnO–Ag nanostructures towards the degradation of methylene blue (MB) as a model dye was performed under sunlight. The intensity of observed sun light radiation was 95 mW cm−1. A known amount of photocatalysts were added into 150 mL of desired concentration of MB solution and stirred for about 30 min in dark for reaching adsorption–desorption equilibrium prior to the sunlight irradiation. The concentration of MB in the bulk solution at this condition (measured from the absorbance of MB at its λmax = 663 nm) was taken as the initial concentration (C0). 3 mL of aliquots were withdrawn after a certain period of irradiation of the suspension and the catalyst was removed by filtration using a 0.2 μL PVDF syringe filter and then the concentration of MB in the filtrate was determined using UV-vis spectroscopy. The concentration obtained at certain irradiation time (t) is denoted as Ct. Triplicate experiments were conducted at optimized conditions and the mean values of the results are presented along with the standard deviations. The catalyst is removed from the solution, washed in distilled water, and dried at 100 °C for 2 h for recycling process. We followed the similar reaction conditions for testing the photocatalytic performance of ZnO–Ag NCA with oxidants such as PMS, PDS, H2O2, commercial TiO2 (P25) and ZnO NPs.Results and discussion
Characterization of ZnO–Ag nanoparticles
The crystalline structure of ZnO–Ag nano particle was investigated by XRD analysis (Fig. 1). The peaks at 2θ = 31.7, 34.4, 36.2, 47.5, 56.6 and 62.8 can be indexed to (100), (002), (101), (102), (110) and (103) diffractions of ZnO wurtzite structure (JCPDS card no. 36-1451). The extra peaks at 2θ = 38.04, 44.30 and 64.38 can be indexed as (111), (200) and (220) planes of face centred cubic (fcc) nature of silver NPs (JCPDS card no. 4-0783). We observed the gradual increment in the intensity of these diffraction peaks (2θ = 38.04, 44.30 and 64.38) as the amount of silver (0.2–2.0 wt%) loading is increased. The peaks from AgNP are relatively weak since the amount of silver is less compared to that of ZnO. XRD spectra did not show any additional peaks indicating that the sample is free from impurities. It should be noted that silver NPs are obtained in the as-produced material without the presence of any separate reducing agent. This suggests that the phytochemicals such as punicalagin and punicalin in the aqueous pomegranate peel extract act as the reducing agent.32–34 | ||
| Fig. 1 XRD pattern of ZnO–Ag nanoparticles with different loading of Ag (0.2–2.0 wt%) calcined at 300 °C. | ||
The size and morphology of ZnO–Ag NPs are studied by SEM and TEM analysis. The SEM images of NPs formed at different loading of Ag (wt%) are shown in Fig. 2 and S1.† The morphology of ZnO–Ag NP formed in presence of 0.2 wt% of Ag indicates the tendency of the system to form custard apple kind of structures (Fig. S1a†). However, the particles formed at 0.6 wt% loading of Ag have clearly shown custard-apple morphology with the diameter of about ∼100 nm and the assemblies are monodispersed in nature (Fig. 2a and b). TEM image of ZnO–Ag (0.6 wt%) NPs also show similar morphology (Fig. 2c). The high resolution TEM image displayed the distance of two adjacent lattice fringe spacing in ZnO and AgNPs and the values were about 0.24 and 0.23 nm, respectively (Fig. 2d and S2†). It also confirms the well dispersed nature of AgNP on ZnO surface in ZnO–Ag NCA. The lattice fringe distance corresponds to (101) and (111) planes in the ZnO and Ag crystal, respectively.35 As the concentration of Ag is increased from 1.0 to 2 wt%, the NCA structure is collapsed and irregular morphologies were obtained (Fig. S1b–d†). This suggests that excess amount of silver leads to the aggregation of particles. While it is known that calcination temperature (300 °C) has some influence on the shape of the particles, the above results strongly suggest that silver play a major contribution towards the size, shape and morphology of ZnO–Ag NP formation. The presence of Ag on ZnO–Ag nano clustered apple was further confirmed by EDAX analysis (Fig. S3†). The elemental mapping obtained from EDS analysis shows that Zn, O and Ag are distributed in the ZnO–Ag NCA (Fig. S4†).
 | ||
| Fig. 2 (a and b) SEM, (c) TEM and (d) HR-TEM image of ZnO–Ag (0.6 wt%) nano-custard apple. | ||
DRS peaks of silver loaded (0.2 to 2.0 wt%) ZnO are shown in Fig. 3. The pure commercial ZnO NPs exhibits absorption only in UV region indicating its incapability of light harvesting at visible region. The spectra of ZnO–Ag NPs contain both ZnO peak around 350 nm and the surface plasmon resonance peak of silver NPs around 430 nm. This also confirms the formation of AgNPs in the reaction mixture during the synthesis. While increasing the concentration of Ag from 0.2 wt% to 0.6 wt%, the peak intensity of ZnO is decreased and enhancement in the SPR peak (420 nm) of AgNPs has been observed. The peak at 420 nm indicates the formation of well dispersed spherical AgNPs in NCA. The SPR peak position was relatively stable between 1.4 wt% and 2.0 wt% of Ag, indicating that the surface saturation of ZnO NPs by AgNPs. The SPR peak is broadened (400–750 nm) as the concentration of Ag is further increased (1 wt% to 2 wt%). This indicates the increment in the particle size of AgNPs on ZnO, due to higher order of surface deposition of AgNPs.29 The band gap of ZnO–Ag NCA (3.0 eV) calculated by Tauc's plot and the value is significantly lower than pure ZnO (Fig. S5†). The result clearly indicates that the absorption properties of ZnO were altered by the formation of ZnO–Ag NCA.
 | ||
| Fig. 3 (a) DRS spectra of ZnO–Ag NPs (0.2–0.6 wt%). | ||
Photocatalytic property of the NPs highly depends on the PL intensity and recombination rate of the excited electrons and holes.36 Fig. S6† shows the room temperature photo luminescent (PL) spectra (excitation at 325 nm) recorded from the bare ZnO and ZnO–Ag nano particles. Each spectrum consists of two bands, one is in the UV region (383 nm) and another is in a visible region (550 nm).37 The peak at 383 nm is attributed to the near-band edge (NBE) emission of ZnO NP, which is mainly due to the recombination of electron–hole in ZnO.37 A broad green emission band centred at 550 nm is due to recombination of photogenerated hole and the singly ionized-charge oxygen vacancy.38 The reduced intensity at NBE of ZnO–Ag (0.6 wt%) NCA indicates strong exchange interaction of electrons between Ag and ZnO.37 Conversely, the PL intensity at 550 nm is decreased as the concentration of silver is increased to 0.6 wt% of Ag, presumably because of dispersion of AgNPs on ZnO reaches a maximum at the particular Ag concentration. The decreased emission intensity at NBE indicates a lower electron–hole recombination rate and hence a longer life time of the photogenerated carries caused by AgNPs.36 The results taken together revealed that, charge transfer from ZnO to Ag is rapid in the case of ZnO–Ag NCA, at 0.6 wt% of Ag.
To investigate the molecular vibration modes in ZnO–Ag nano custard apple, FT-Raman measurement was carried out at room temperature by exciting the sample with a laser source λ = 1064 nm. Fig. S7† shows the Raman spectrum of ZnO–Ag nano custard apple. A pure hexagonal wurtzite phase of ZnO in NCA shows Raman peaks at 583.1, 435.3, 382.0, 331.6 and 99 cm−1 which are corresponding to the E1 (LO-longitudinal optical), E2 (high), A1 (TO-transverse optical), A1 and E2 (low) vibrational modes.39,40 ZnO–Ag nano custard apple shows dominant Raman peaks at 436 and 99 cm−1 which correspond to E2 (high) and E2 (low), respectively.41 The second order Raman peak at 333 cm−1 was originated from the zone boundary phonons.41 The less intense peak at 580 cm−1 is associated with the oxygen vacancies and surface defects caused by the addition of Ag on ZnO nanostructure. This is also consistent with the PL spectra at 550 nm (Fig. S6†). Further, the presence of Ag on ZnO–Ag nano custard apple is confirmed by observing the peaks at 1036, 1102, 1348 and 1583 cm−1 which are assigned to Ag nanoparticles.42,43
The surface chemical composition and chemical states of elements present in ZnO–Ag NCA was investigated by XPS measurement. Fig. S8† shows the XPS spectra of ZnO–Ag NCA which indicates the presence of Zn, O, and Ag. The strong peak at 1021.8 eV is ascribed to Zn 2p3/2 and peak at 1044.2 eV for Zn 2p½, indicating the oxidation state of Zn as +2.44 The binding energy of O1s peak (528.5 to 530.14 eV) was attributed to lattice oxygen present in ZnO, whereas, the peak at 531.57 eV was assigned to surface hydroxyl group in ZnO–Ag NCA. The presence of surface hydroxyl group facilitates the trapping of photoinduced electron and hole, leading to enhanced photocatalytic degradation process.44,45 Ag 3d spectrum of AgNP in ZnO–Ag NCA contains two peaks at 368.3 and 374.1 eV which are assigned to Ag 3d5/2 and Ag 3d3/2, respectively.45
Photocatalytic degradation of methylene blue
The photocatalytic degradation of MB was evaluated with biosynthesized ZnO–AgNPs (0.2 to 2 wt%), commercial ZnO NPs and TiO2 (P25) under sunlight irradiation. The changes in the absorption intensity of MB (663 nm) was monitored by UV-vis spectroscopy. The photocatalytic degradation process complies with first-order kinetics, C = C0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−kt), Where C is the MB concentration after irradiation, C0 is the initial MB concentration, t is the irradiation time and constant k represents the reaction rate.46 Fig. 4 depicts the plot of Ct/C0 against irradiation time. We observed no degradation for MB in the absence of catalyst under sunlight irradiation. Conversely, the intensity of MB is gradually decreased in presence of catalysts with time (Fig. 4). The data suggests that, the photocatalytic activity of the catalyst are in the following order ZnO < ZnO–Ag (2.0 wt%) < ZnO–Ag (1.4 wt%) < TiO2 (P25) < ZnO–Ag (0.2 wt%) < ZnO–Ag (1.0 wt%) < ZnO–Ag (0.6 wt%). A significant decrease in the absorption of MB was noticed only in the case of ZnO–Ag nano clustered apple (Fig. 4). The decolorization of MB ([catalyst] = 0.5 g L−1, [MB] = 0.02 mM) was occurred in 60 min in the presence of ZnO–Ag NCA under sun light irradiation (Fig. S9†).
exp(−kt), Where C is the MB concentration after irradiation, C0 is the initial MB concentration, t is the irradiation time and constant k represents the reaction rate.46 Fig. 4 depicts the plot of Ct/C0 against irradiation time. We observed no degradation for MB in the absence of catalyst under sunlight irradiation. Conversely, the intensity of MB is gradually decreased in presence of catalysts with time (Fig. 4). The data suggests that, the photocatalytic activity of the catalyst are in the following order ZnO < ZnO–Ag (2.0 wt%) < ZnO–Ag (1.4 wt%) < TiO2 (P25) < ZnO–Ag (0.2 wt%) < ZnO–Ag (1.0 wt%) < ZnO–Ag (0.6 wt%). A significant decrease in the absorption of MB was noticed only in the case of ZnO–Ag nano clustered apple (Fig. 4). The decolorization of MB ([catalyst] = 0.5 g L−1, [MB] = 0.02 mM) was occurred in 60 min in the presence of ZnO–Ag NCA under sun light irradiation (Fig. S9†).
 | ||
| Fig. 4 Photocatalytic degradation of methylene blue in the presence of different photocatalysts ([catalyst] = 0.5 g L−1, [MB] = 0.02 mM). | ||
The dye degradation rate is highly depended on the morphology and crystallinity of the sample.47 The enhanced photocatalytic activity of biosynthesized ZnO–Ag NPs is due to the formation of nano heterojunctions compared to ZnO NPs and TiO2 (P25).48 We observed a better catalytic activity for ZnO–Ag NCA, presumably due to the uniform morphology and optimum loading of Ag, which results in the strong electronic interaction between Ag and ZnO NPs which retards the recombination of charge carriers (Fig. S6†). Since the rate of charge recombination is less, an electric field at the ZnO–Ag nano-junction will be built in, upon sunlight irradiation.49 The presence of AgNPs in the catalyst acts as nano-antenna for light trapping due to the characteristic localized surface plasmon resonance (LSPR) of AgNPs which is converted into locally excited electric field as a result of photo excitation of LSPR.48 While shining sunlight into ZnO–Ag NCA the electron in the valance band of ZnO will transfer into the conduction band. ZnO in ZnO–Ag NCA will allow the charge transfer from ZnO to AgNPs due to LSPR and accelerates the separation of photogenerated electron–hole pairs. The holes in the valence band will react with water and generate ˙OH radicals and electron in the conduction band of AgNPs will produce superoxide anion radicals (O2˙−) during the photocatalytic degradation process. The formation of these active radicals results in the efficient photocatalytic degradation of MB under direct sunlight.
Alternately, sunlight can excite the adsorbed methylene blue on the surface of the catalyst, which injects electrons into the conduction band of ZnO nano particles.50 The electrons on the ZnO surface can be trapped by the AgNPs and thereby prevent the recombination process. Further, the electrons are consumed by the dissolved oxygen in the solution to generate reactive oxidative species to degrade the dye molecules.45,51 The overall mechanistic picture of the photocatalytic degradation of MB in presence of ZnO–Ag NCA under sunlight irradiation is shown schematically in Fig. 5. The various reactions involved in the process can be summarized as follows.45,52,53
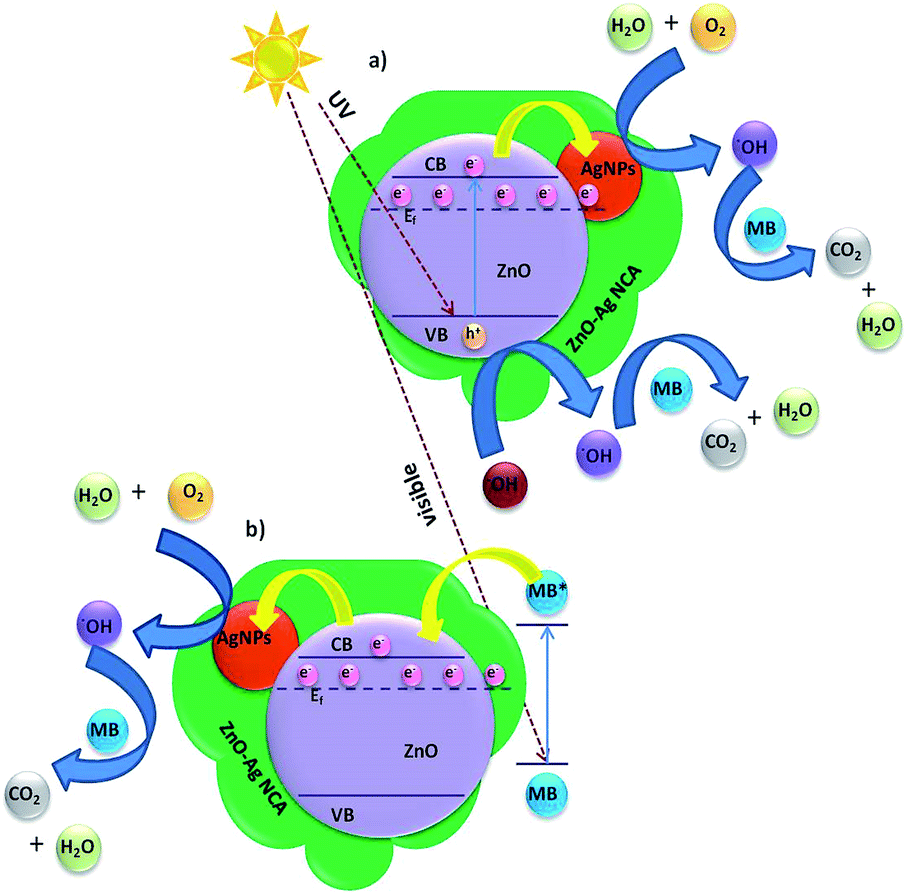 | ||
| Fig. 5 Proposed mechanism for the electron transfer events occurred in ZnO–Ag NCA during the degradation of MB upon sunlight irradiation: (a) band gap excitation and (b) sensitization. | ||
The amount of catalyst required for the photocatalytic reaction is important since it can strongly influence the photocatalytic degradation of MB. As we found ZnO–Ag NCA shows a better photocatalytic activity towards the degradation of MB, we optimized the amount of catalyst required for a better degradation rate of MB. We varied the catalyst amount from 0.1 to 0.6 g L−1 and found that the degradation rate of MB was increased up to 0.5 g L−1 of ZnO–Ag NCA (Fig. 6). The higher amount of the catalyst (0.5 g L−1) offers more active surface area for the photocatalytic degradation of MB. A decrease in the degradation rate of ZnO–Ag NCA at 0.6 g L−1 is presumably due to emergence of light scattering by excess amount of catalyst present in the reaction medium.50
 | ||
| Fig. 6 Photocatalytic degradation of methylene blue at various concentration of ZnO–Ag NCA under sunlight irradiation ([catalyst] = 0.5 g L−1, [MB] = 0.02 mM). | ||
The generation of ˙OH during the photocatalytic reaction in the presence of catalyst was monitored using terephthalic acid (TA) as a probe molecule. The fluorescent intensity of TA is directly proportional to the amount of ˙OH production during the photocatalytic reaction due to the formation of hydroxy terephthalic acid.49 Fig. 7a shows the fluorescent intensity of hydroxy terephthalic acid formed in presence of different catalyst during the photocatalytic reaction under sunlight irradiation for 60 min. The results revealed that there is no formation of ˙OH in the dark reaction condition in presence of ZnO–Ag NCA. The fluorescent intensity is enhanced with time in presence of catalyst upon sunlight irradiation. We observed that ZnO–Ag NCA generates significantly higher amount of ˙OH than other catalyst. We have monitored the PL intensity of terephthalic acid in presence of ZnO–Ag NCA with different irradiation time (Fig. S10†). The maximum intensity was obtained at 60 min of sunlight irradiation. The results indicates that plasmonic effect of well dispersed and smaller sized AgNPs in ZnO–Ag NCA could lead efficient trapping of higher number of photogenerated charge carriers and generate more number of ˙OH during the reaction.
 | ||
| Fig. 7 (a) Fluorescence spectra of terephthalic acid (excited at 315 nm) in the presence of different photocatalysts under sunlight irradiation ([TA] = 0.5 mM) and (b) photocatalytic degradation of MB in presence of ZnO–Ag NCA with various oxidants under sunlight irradiation. | ||
We investigated the effects of oxidant such as peroxomonosulfate (PMS), peroxodisulfate (PDS) and hydrogen peroxide (H2O2) on the photocatalytic degradation of MB in presence of ZnO–Ag NCA under sunlight irradiation. We observed that the photocatalytic degradation of MB by ZnO–Ag NCA is higher in presence of PMS than PDS or H2O2 (Fig. 7b). An efficient charge separation at the interface of ZnO/Ag in ZnO–Ag NCA, leads higher surface tapped electrons. PMS oxidise faster and simultaneously produce ˙OH and SO4˙− during the reaction.54 Hence the overall rate of degradation of MB was increased in ZnO–Ag NCA with PMS combination.
The total organic carbon (TOC) analysis was performed to evaluate the mineralization efficiency of ZnO–Ag NCA in presence and absence of other oxidants (Fig. 8a). Generally, complete mineralization of MB takes place in two steps. The initial step involves the ring opening of MB, which is followed by subsequent oxidation of the fragments. Hence, mineralization will take more time than decolorization of dye molecule in presence of photo catalyst. The results revealed that, in the absence of oxidants, the mineralization rate of ZnO–Ag NCA is significantly low. The higher rate of mineralization was observed in the case of ZnO–Ag NCA with PMS. The complete mineralization of MB in presence of ZnO–Ag NCA with PMS under sunlight irradiation was taken place in 3 h.
 | ||
| Fig. 8 (a) Mineralization of MB in presence of ZnO–Ag NCA with various oxidants under sunlight irradiation and (b) photocatalytic degradation of MB using recycled ZnO–Ag NCA under sun light irradiation. | ||
Finally, the reusability of biogenic ZnO–Ag NCA during the photo catalytic degradation of MB was investigated (Fig. 8b). The results indicate that, even five consecutive cycles the NCA did not lose the photocatalytic activity towards the degradation of MB. It is noteworthy that the NCA based photo catalyst was stable under sunlight irradiation. The catalytic activity is decreased after the use of fifth cycle. The reason for this observation is the leaching of AgNPs from the surface of ZnO which can be seen from the XRD pattern (Fig. S11a†). Corroborating to this, the corresponding SEM images (Fig. S11b†) of the sample showed the collapsed structure of ZnO–Ag NCA.
Conclusions
We have shown a very simple but efficient method for the synthesis of ZnO–Ag NCA via a greener route and used the system for photocatalytic degradation of methylene blue under direct sunlight irradiation. The uniform morphology of ZnO–Ag NCA and well dispersed AgNPs on ZnO lead higher charge separation which leads to better photocatalytic activity towards the degradation of the dye. The degradation rate is significantly enhanced with the addition of peroxomonosulfate (PMS) into NCA and the complete mineralization of MB was occurred in 3 h. The catalyst is highly stable and its reusability is tested up to 5 cycles. ZnO–Ag NCA might be used as an excellent candidate for the degradation for other organic pollutant.Acknowledgements
S. Kaviya thanks for the INSPIRE fellowship from DST, Govt. of India. We thank the financial support from DST Nano Mission {Ref. no. SR/NM/MS-115/2010 (G)}. We thank SAIF, IITM for SEM analysis.Notes and references
- E. Bizani, K. Fytianos, I. Poulios and V. Tsiridis, J. Hazard. Mater., 2006, 136, 85–94 CrossRef CAS PubMed.
- T. Robinson, G. M. Mullan, R. Marchant and P. Nigam, Bioresour. Technol., 2001, 77, 247–255 CrossRef CAS.
- K. Rastogi, J. N. Sahu, B. C. Meikap and M. N. Biswas, J. Hazard. Mater., 2008, 158, 531–540 CrossRef CAS PubMed.
- D. Ozer, G. Dursun and A. Ozer, J. Hazard. Mater., 2007, 144, 171–179 CrossRef PubMed.
- T. Liu, M. Takeshi and S. Sourirajan, Ind. Eng. Chem. Res., 1983, 22, 77–85 CrossRef.
- P. Niranjan and J. Karthikeyan, Indian J. Environ. Prot., 1992, 12, 599–603 CAS.
- M. J. Height, S. E. Pratsinis, O. Mekasuwandumrong and P. Praserthdam, Appl. Catal., B, 2006, 63, 305–312 CrossRef CAS PubMed.
- L. B. Khalil, W. E. Mourad and M. W. Rophael, Appl. Catal., B, 1998, 17, 267–273 CrossRef CAS.
- H. Lu, S. Wang, L. Zhao, J. Li, B. Dong and Z. Xu, J. Mater. Chem., 2011, 21, 4228–4234 RSC.
- K. Zhu, N. R. Neale, A. Miedaner and A. J. Frank, Nano Lett., 2007, 7, 69–74 CrossRef CAS PubMed.
- Y. Dong, C. Feng, P. Jiang, G. Wang, K. Li and H. Miao, RSC Adv., 2014, 4, 7340–7346 RSC.
- B. Subash, B. Krishnakumar, M. Swaminathan and M. Shanthi, Langmuir, 2013, 29, 939–949 CrossRef CAS PubMed.
- C. Mondal, J. Pal, M. Ganguly, A. K. Sinha, J. Jana and T. Pal, New J. Chem., 2014, 38, 2999–3005 RSC.
- A. M. Ali, E. A. C. Emanuelsson and D. A. Patterson, Appl. Catal., B, 2010, 97, 168–181 CrossRef CAS PubMed.
- L. Xu, Y. L. Hu, C. Pelligra, C. H. Chen, L. Jin, H. Huang, S. Sithambaram, M. Aindow, R. Joesten and S. L. Suib, Chem. Mater., 2009, 21, 2875–2885 CrossRef CAS.
- R. J. V. Michael, B. Sambandam, T. Muthukumar, M. J. Umapathy and P. T. Manoharan, Phys. Chem. Chem. Phys., 2014, 16, 8541–8555 RSC.
- M. E. Aguirre, H. B. Rodriguez, E. S. Roman, A. Feldhoff and M. A. Grela, J. Phys. Chem. C, 2011, 115, 24967–24974 CAS.
- X. He, H. Wang, Z. Li, D. Chen and Q. Zhang, Phys. Chem. Chem. Phys., 2014, 16, 14706–14712 RSC.
- R. Zamiri, A. Rebelo, G. Zamiri, A. Adnani, A. Kuashal, M. S. Belsley and J. M. F. Ferreira, RSC Adv., 2014, 4, 20902–20908 RSC.
- I. C. Chen, Y. C. M. Liou, J. Yang and T. Y. Shieh, J. Raman Spectrosc., 2011, 42, 339–344 CrossRef CAS.
- X. Yin, W. Que, D. Fei, F. Shen and Q. Guo, J. Alloys Compd., 2012, 524, 13–21 CrossRef CAS PubMed.
- T. J. Whang, M. T. Hsieh and H. H. Chen, Appl. Surf. Sci., 2012, 258, 2796–2801 CrossRef CAS PubMed.
- H. Silva, C. M. Pedrero, C. Magen, D. A. P. Tanaka and A. Mendes, RSC Adv., 2014, 4, 31166–31176 RSC.
- J. Song and S. Lim, J. Phys. Chem. C, 2007, 111, 596–600 CAS.
- M. Yang, G. Pang, J. Li, L. Jiang, D. Liang and S. Feng, J. Phys. Chem. C, 2007, 111, 17213–17220 CAS.
- Z. G. Yu, P. Wu and H. Gong, Appl. Phys. Lett., 2006, 88, 132114–132116 CrossRef PubMed.
- J. H. Park, S. J. Jang, S. S. Kim and B. T. Lee, Appl. Phys. Lett., 2006, 89, 121108–121110 CrossRef PubMed.
- P. Fons, H. Tampo, A. V. Kolobov, M. Ohkubo, S. Niki, J. Tominaga, R. Carboni and S. Friedrich, Phys. Rev. Lett., 2006, 96, 045504–045507 CrossRef.
- S. Kaviya and E. Prasad, ACS Sustainable Chem. Eng., 2014, 2, 699–705 CrossRef CAS.
- P. Mukherjee, A. Ahmad, D. Mandal, S. Senapati, S. R. Sainkar, M. I. Khan, R. Parishcha, P. V. Ajaykumar, M. Alam, R. Kumar and M. Sastry, Nano Lett., 2001, 1, 515–519 CrossRef CAS.
- S. Kaviya, J. Santhanalakshmi, B. Viswanathan, J. Muthumary and K. Srinivasan, Spectrochim. Acta, Part A, 2011, 79, 594–598 CrossRef CAS PubMed.
- M. Cam and Y. Hisil, Food Chem., 2010, 123, 878–885 CrossRef CAS PubMed.
- T. Ismail, P. Sestili and S. Akhter, J. Ethnopharmacol., 2012, 143, 397–405 CrossRef CAS PubMed.
- S. Kaviya and E. Prasad, Anal. Methods, 2014, 7, 168–174 RSC.
- J. M. Song, Z. Zhang, J. J. Ni, H. L. Niu, C. J. Mao, S. Y. Zhang and Y. H. Shen, CrystEngComm, 2014, 16, 2652–2659 RSC.
- M. M. Khan, S. A. Ansari, J. A. Lee and M. H. Cho, Nanoscale, 2013, 5, 4427–4435 RSC.
- B. Panigrahy, M. Aslam and D. Bahadur, J. Phys. Chem. C, 2010, 114, 11758–117563 CAS.
- K. Vanheusden, W. L. Warren, C. H. Seager, D. R. Tallant, J. A. Voit and B. E. Gnade, J. Appl. Phys., 1996, 79, 7983–7990 CrossRef CAS PubMed.
- V. G. Pol, J. M. C. Moreno and P. Thiyagarajan, Langmuir, 2008, 24, 13640–13645 CrossRef CAS PubMed.
- H. Li, Y. Huang, Q. Zhang, Y. Qiao, Y. Gu, J. Liu and Y. Zhang, Nanoscale, 2011, 3, 654–660 RSC.
- M. Khan, C. Wei, M. Chen, J. Tao, N. Huang, Z. Qi and L. Li, J. Alloys Compd., 2014, 612, 306–314 CrossRef CAS PubMed.
- G. Naja, P. Bouvrette, S. Hrapovic and J. H. Luong, Analyst, 2007, 132, 679–686 RSC.
- A. Ahern and R. Garrell, Anal. Chem., 1987, 59, 2813–2816 CrossRef CAS.
- S. A. Ansari, M. M. Khan, M. O. Ansari, J. Lee and M. H. Cho, J. Phys. Chem. C, 2013, 117, 27023–27030 CAS.
- B. Subash, B. Krishnakumar, M. Swaminathan and M. Shanthi, Langmuir, 2013, 2, 939–949 CrossRef PubMed.
- N. V. Kaneva, G. G. Yordanov and C. D. Dushkin, React. Kinet. Catal. Lett., 2009, 98, 259–263 CrossRef CAS PubMed.
- J. H. Zeng, B. B. Jin and Y. F. Wang, Chem. Phys. Lett., 2009, 472, 90–95 CrossRef CAS PubMed.
- S. Ma, J. Xue, Y. Zhou and Z. Zhang, J. Mater. Chem. A, 2014, 2, 7272–7280 CAS.
- J. A. Scholl, A. L. Koh and J. A. Dionne, Nature, 2012, 483, 421–427 CrossRef CAS PubMed.
- C. Nasr, K. Vinodgopal, L. Fisher, S. Hotchandani, A. K. Chattopadhyay and P. V. Kamat, J. Phys. Chem., 1996, 100, 8436–8442 CrossRef CAS.
- G. Z. Xiong, L. L. Zhang, Z. J. Ma and S. X. Zhao, Chem. Commum., 2010, 46, 6099–6101 RSC.
- N. Pugazhenthiran, S. Murugesan and S. Anandan, J. Hazard. Mater., 2013, 263, 541–549 CrossRef CAS PubMed.
- C. Mondal, J. Pal, M. Ganguly, A. K. Sinha, J. Jana and T. Pal, New J. Chem., 2014, 38, 2999–3005 RSC.
- P. S. S. Kumar, R. Sivakumar, S. Anandan, J. Madhavan, P. Maruthamuthu and M. Ashokkumar, Water Res., 2008, 42, 4878–4884 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: See DOI: 10.1039/c4ra15293j |
| This journal is © The Royal Society of Chemistry 2015 |