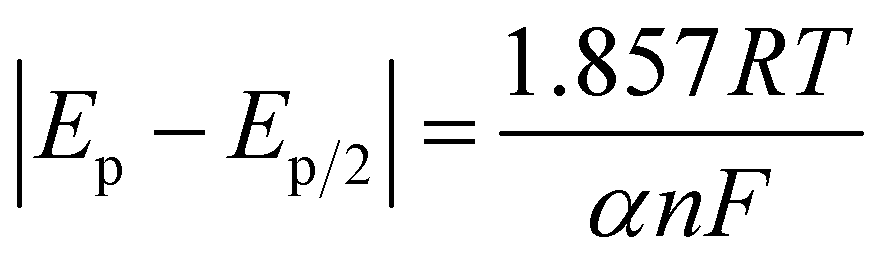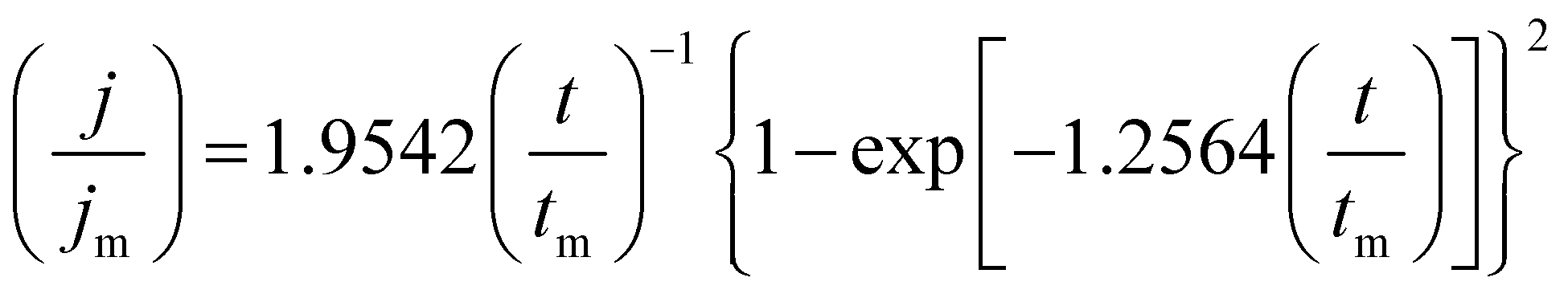DOI:
10.1039/C4RA06855F
(Paper)
RSC Adv., 2014,
4, 38012-38026
Electrodeposit copper from alkaline cyanide-free baths containing 5,5′-dimethylhydantoin and citrate as complexing agents†
Received
9th July 2014
, Accepted 31st July 2014
First published on 4th August 2014
Abstract
An alkaline cyanide-free bath containing 5,5′-dimethylhydantoin (DMH) and citrate, as complexing agents, was investigated and developed for copper electrodeposition on carbon steel substrate. The cathodic electrodeposition process was studied by electrochemical measurements including cyclic voltammetry, chronoamperometry, chronopotentiometry, and cathodic polarization on platinum and glass carbon disk electrodes. Copper layers were characterized with SEM for surface morphology, XRD for crystal structure, and three qualitative methods for adhesion evaluation. Based on the cyclic voltammetry analysis with various switching potentials the discharge processes were proven to have the typical two steps for solutions containing either DMH, or citrate, or both as complexing agents. The discharge processes of both copper–DMH and copper–citrate complexes are irreversible according to cyclic voltammetry with various scan rates, which combined with sample current voltammetry generates the kinetic parameters. Diffusion coefficients were calculated from chronoamperometry according to Conttrell equation. The nucleation and crystal growth processes on the glass carbon electrode, in these three solutions, show an agreement with the progressive nucleation process of SH models. Molecular dynamic simulations reveal that the plane of the heterocycle of all the four DMH molecules simulated is parallel to the Fe (111) surface and the interaction energy between various molecules and Cu (111) as well as Fe (111) surface were compared. DMH molecules adsorbed on the carbon steel electrode desorb at the initial stage of electrodeposition, which was determined from potential vs. time curves. The concentrations of DMH and K2CO3 as well as the value of pH have a crucial effect on the surface morphology, preferable crystal orientation, and the adhesion of copper layers electroplated on the carbon steel substrate. The electrodeposited copper layers had good adhesion with the carbon steel substrate and the average grain size was about 30 nm. The optimum bath is composed of 0.1 M CuSO4, 0.2 M DMH, 0.3 M citrate, and 0.3 M K2CO3 at pH 9–10.5 and 50 °C.
1 Introduction
Alkaline cyanide baths are still used to electroplate copper and have several applications for the metal-finishing industry.1 In recent decades many different alternative electrolytes, such as solutions containing amine–ammonia, chloride, citrate, EDTA, glycerolate, phosphate, pyrophosphate, tartrate, triethanolamine,1 sorbitol,2,3 tartrate,4 and glycine,5,6 have been proposed and investigated around the world to replace cyanide baths for its environmental concerns. Because of economic, chemical, environmental or some other limitations, the majority of these electrolyte systems, however, cannot entirely replace cyanide baths.
Our research group has developed cyanide-free baths for gold and silver electroplating using 5,5′-dimethylhydantoin (DMH) as one of the complexing agents.7,8 From previous studies, we discovered that the features of complexation and adsorption of DMH were the two key factors that influenced the cathodic deposition processes of gold and silver. In addition, the nature of gold, silver, and copper is similar to some degree. To our knowledge, two patents,9,10 whose statements are very vague, have claimed to use DMH as an optional agent for copper electroplating but no related literatures or detailed studies have been found. Therefore, we are interested to investigate solutions containing DMH from an electrochemical point of view and to develop a potentially robust bath.
To obtain copper layers with good adhesion on a carbon steel substrate, a key point is to prevent the formation of a copper immersion coating on the steel substrate. However, this is not the only factor decreasing the adhesion, except for poor cleaning and pretreatment.11 As the steel substrate easily oxidizes in air and water, the thin passive film that forms before the electrolytic cell is powered on also plays a significant role in decreasing the adhesion between copper electrodeposits and the steel substrate.11–13 Hydantoin and its derivatives that are commonly used as drugs can coordinate with several metal ions, including Au, Ag, Co, Ni, Cu, and Zn ions.14,15 The formation of the copper–DMH complex may prevent the immersion coating. By forming an adsorbed layer on the metal surface, some organic molecules containing polar groups, such as organic N, amine, S, and OH groups, can inhibit the corrosion of iron in acid.16 Hydantoin also has a polar group (N-heterocyclic) and some hydantoin derivatives have been confirmed to be capable of inhibiting the corrosion of iron in acid solution.17,18 Thus, we anticipated that DMH might also be adsorbed on the carbon steel surface, which could influence the electrodeposition process so that the adhesion between electrodeposits and the steel substrate would be enhanced. Molecular dynamic simulation can predict and investigate the adsorption behavior and the interaction energy of organic molecules on metal surfaces.19,20 Although by using a solution with DMH as the only complexing agent, the immersion coating could be prevented efficiently at a high concentration ratio of DMH to copper ions, the current efficiency (CE) in our experiments was too low to be acceptable. For this reason, we chose a second complexing agent, citrate, which could coordinate stably with cupric ions and has been extensively studied.21–24
In the present work, we investigated the cathodic discharge processes and kinetic features of solutions in the presence of either DMH, or citrate, or both as complexing agents using cyclic voltammetry and sample current voltammetry. Chronopotentiometry and chronoamperometry were employed to explore the initial process of electrodeposition, including the nucleation, growth and desorption of molecular DMH, whose adsorption behavior on a steel substrate was revealed by molecular dynamic simulations. Cathodic polarization was demonstrated by potentiodynamic cathodic polarization curves on a platinum rotating disk electrode. The CE of the baths, surface morphology, crystal structure, and the adhesion of copper layers electrodeposited on a carbon steel substrate were also discussed.
2 Experimental
2.1 Preparing baths and electrochemical measurements
All the reagents were of analytical grade. Distilled water was used throughout the studies. The baths composed of CuSO4·5H2O as the main salt, DMH and citrate (potassium citrate) as complexing agents, and K2CO3 as the buffer. Sulphuric acid and sodium hydroxide solutions were used to adjust the pH of the baths. The composition of the main solutions in the experiments is presented in Table 1. The pH was 9 and the temperature was 50 °C, if not mentioned specifically.
Table 1 The composition of the main solutions used in this work
| Solution no. |
Concentration (M) |
| CuSO4·5H2O |
DMH |
Citrate |
K2CO3 |
| Sp1 |
0.1 |
0.0 |
0.6 |
0.3 |
| Sp2 |
0.1 |
0.2 |
0.6 |
0.3 |
| Sp3 |
0.1 |
0.5 |
0.6 |
0.3 |
| Sp4 |
0.1 |
0.5 |
0.0 |
0.3 |
| Sp5 |
0.1 |
0.5 |
0.3 |
0.3 |
| S1 |
0.1 |
0.0 |
0.3 |
0.3 |
| S2 |
0.1 |
0.2 |
0.3 |
0.3 |
| S3 |
0.1 |
0.4 |
0.3 |
0.3 |
| S4 |
0.1 |
0.2 |
0.1 |
0.3 |
| S5 |
0.1 |
0.2 |
0.5 |
0.3 |
| S6 |
0.1 |
0.2 |
0.3 |
0.0 |
| S7 |
0.1 |
0.2 |
0.3 |
0.5 |
All the electrochemical measurements were conducted in a typical three-electrode cell connected to a potentiostat/galvanostat. A glass carbon (GC), a platinum (Pt) electrode (φ = 3 mm) and a carbon steel disk (φ = 5 mm) embedded in Teflon were used as the working electrodes for cyclic voltammetry, chronoamperometry, and chronopotentiometry measurements. A glass carbon and a platinum rotating disk electrode (RDE, φ = 5 mm) were employed as the working electrodes for polarization curves and CE measurements. The carbon steel electrode was polished to a mirror finish immediately prior to the measurements. A platinum plate was employed as the counter electrode and a saturated calomel electrode (SCE) as the reference electrode for all the electrochemical experiments.
The deposition/stripping method based on constant-current chronopotentiometry25–27 was employed to estimate the current efficiency. First, copper electrodeposition was performed at constant cathodic current density for 10 minutes. Then, stripping was carried out at constant anodic current density to oxidize the electrodeposits in the electrolyte containing 300 g L−1 NH4NO3. The potential was monitored as a function of time when stripping (One example of the potential vs. time curves obtained by oxidizing the copper deposits is shown in Fig. S1 of the ESI†). The time at which the potential increased to 1.0 V vs. SCE was chosen as the end point of the oxidization process for all the experiments. The CE was calculated by eqn (1).
| |
 | (1) |
where
ia and
ic are the anodic oxidation and the cathodic reduction currents, respectively.
ta and
tc represent the time used for oxidation and reduction, respectively. Here, the CE of the oxidation process was assumed to be 100%.
A cell consisting of a copper sheet (5 × 5 cm) as the anode and a carbon steel sheet (3 × 3 cm) as the cathode was employed for electroplating experiments. All the electroplating experiments were performed at galvanostatic conditions with relatively strong mechanical agitation (around 200 rpm with a blade agitator).
2.2 Molecular dynamic simulation
Molecular dynamic simulations were performed in simulation boxes with periodic boundary conditions using Materials Studio (from Accelrys Inc). The boxes consisted of a surface (cleaved along the (111) plane for ferrum and along the (111) plane for copper) and a liquid phase. The liquid phase contained water molecules with a density of 1 g cm−3 and several organic molecules in every box. All the molecular dynamic simulations were carried out at 323 K, NVT ensemble, and COPMASS force field with a time step of 1 fs and simulation time of 500 ps. The interaction energy Einteraction between the metal surface and one organic molecule was calculated by eqn (2):| |
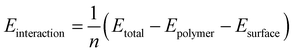 | (2) |
where Etotal is the total energy of the metal crystal together with the adsorbed organic molecules, Esurface and Epolymer represent the total energy of the metal surface and free organic ions, respectively, and n is the number of organic molecules simulated in the box. The binding energy value is the negative value of the interaction energy.
2.3 Characterizing copper layers
An environmental scanning electron microscope (SEM, Quanta 200F) was employed to observe the surface morphology and cross-sections of the copper layers. Crystal structure was revealed by X-ray diffraction (XRD, PANalytical X'Pert PRO).
It is very difficult to quantitatively measure adhesion and a “yes or no” test procedure is still used in many cases even today.12 The bend test, the cross-hatch test, and the thermal shock test can be utilized to evaluate the adhesion of electroplated copper layers on a carbon steel substrate.12 In this work, the sample sheet was bent to and fro with an angle of ±90° until it was broken into two parts for the bend test. A harden steel engraver with an angle of 30 degrees was used to cross-hatch a mesh pattern of 1 to 3 mm in length for the cross-hatch test. For the thermal shock test samples were heated in an oven to 250 °C and then suddenly immersed into cool water (25 °C). The carbon steel substrate was polished to a mirror finish, then degreased, and activated in a weak acid solution before electrodeposition. After electroplating copper in the investigated baths for 30 minutes, electrodeposition for another 30 minutes was performed in a typical sulfate bath to make the copper layers sufficiently thick. Then, the adhesion was evaluated with the three methods mentioned above.
3 Results and discussion
Three types of electrochemical measurements including cyclic voltammetry, chronoamperometry, and chronopotentiometry were employed to analyse the cathodic discharge process of copper in three solutions, Sp4 containing only DMH, Sp1 containing only citrate, and Sp3 with both DMH and citrate as complexing agents. Cyclic voltammetry measurements with various switching potentials and various scan rates were performed to investigate the reactions and their kinetic features. Sample current voltammetry with a simple analysis method, proposed by Michael V. Mirkin and Allen J. Bard,28 was used to obtain the kinetic parameters of the Pt or GC electrode in the solution containing only DMH or only citrate as the complexing agent. The diffusion coefficient was calculated by fitting the current vs. time curves according to the Conttrell equation. Nucleation was compared with the relatively simple and most frequently used nucleation models proposed by Scharifker and Hills.29,30
3.1 Cyclic voltammetry for the discharge processes
3.1.1 Various switching potentials. Cyclic voltammograms of the Pt electrode from 1.0 V to various switching potentials (−0.40, −0.60, −0.80, −1.00, −1.10, −1.15, −1.30, and −1.50 V) vs. SCE in the Sp4 solution containing only DMH as complexing agent are shown in Fig. 1. The entire potential scan was initially swept away from the rest potential toward the negative direction at a scan rate of 50 mV s−1. In the range from 1.0 V to −1.5 V in Fig. 1(a), three cathodic peaks (C1 (−0.20 V), C2 (−0.85 V), C3 (−1.24 V)), and five anodic peaks (A1 (−0.27 V), A2 (−0.17 V), A3 (−0.04 V), A4 (0.23 V), A5 (0.70 V)) can be observed. The C1 peak shifts negatively in the 2nd cycle compared with the 1st cycle, which is attributed to the copper deposited on the Pt electrode, which is, however, not entirely stripped in the 1st cycle because copper covering the Pt electrode was macroscopically observed only after five cycles. When the switching potential is −0.40 V, −0.60 V, and −0.80 V, the curves have only one cathodic peak C1 and only one anodic peak A4 in Fig. 1(b). The anodic A1 appears when the scan is switched to −1.00 V, and the anodic A2 appears when the scan is switched to −1.10 V. A3 appears in the curves with the switching potential of −1.15 V. The curve with the switching potential of −1.30 V has all the five anodic peaks.
 |
| | Fig. 1 Cyclic voltammograms of the Pt electrode in the Sp4 solution containing only DMH as complexing agent. (a) Ranging from 1.0 V to −1.5 V vs. SCE and (b) ranging from 1.0 V to various switching potentials vs. SCE. The scan rate is 50 mV s−1. The blue dash line signed by “Empty with DMH” in (a) was obtained from the background solution composed of 0.2 M DMH and 0.3 M K2CO3 at pH = 9 and 50 °C. | |
We assume that the main copper–DMH species is [Cu(DMH−)3]− in the Sp4 solution, because the three copper–DMH complexes are [Cu(DMH−)]+, [Cu(DMH−)2], and [Cu(DMH−)3]−, for which the formation constants (log10(β)) are 4.3, 8.2, and 12.1, respectively14 and the pH of the Sp4 solution was 9. Although no report provides definite data regarding the complexation between Cu+ and DMH has been found, except for one patent,10 here we assume that the coordination of Cu+ and DMH forms [Cu(DMH−)]. According to all the results above, we can conclude that the main copper–DMH species is discharged in two steps, even though the voltammograms have three cathodic peaks. The first cathodic peak C1 refers to the first discharge step from Cu2+ to Cu+ as can be seen in eqn (3). Corresponding to C1, the anodic peak A4 is attributed to the oxidation of Cu+. The second cathodic peak C2 is related to the second discharge step from Cu+ to Cu0 as shown in eqn (4), and the anodic peak A1 is attributed to the oxidation of Cu0. Unlike other related report of literatures,31 a sharp cathodic peak, C3, appears after the second one. This behaviour may be related to a catalytic reaction. The small amount of copper electrodeposited at C2 accelerates the discharge of Cu+. The second cathodic peak C2 is more like a platform, indicating that the diffusion of Cu+ did not reach a limit after peak C2. Moreover, the significantly larger area of the anodic peaks of the curves displaying the C3 peak implies that much more copper is deposited at the C3 peak. The anodic peaks A2, A3, and A5, which are not easy to define, may be attributed to the production of hydroxides or oxides that may be adsorbed or forming soluble species.32
| | |
[Cu(DMH−)3]− + e− → [Cu(DMH−)] + 2DMH−
| (3) |
| | |
[Cu(DMH−)] + e− → Cu0 + DMH−
| (4) |
Cyclic voltammograms of the GC electrode from 1.0 V to various switching potentials (−1.00, −1.30, −1.35, −1.40, and −1.5 V) vs. SCE in the Sp4 solution containing only DMH as complexing agent are shown in Fig. 2. The entire potential scan started in the negative direction from the rest potential at a scan rate of 50 mV s−1. The voltammogram with the potential range from 1.0 V to −1.5 V vs. SCE in Fig. 2(a) has two cathodic peaks (C1 (−0.30 V), C2 (−1.45 V)), and four anodic peaks (A1 (−0.30 V), A2 (−0.20 V), A3 (−0.02 V), A4 (0.15 V)). Unlike the voltammogram obtained on the Pt electrode, the C1 peak does not shift in the 2nd cycle comparing with the 1st cycle. However, the initial point of the second peak C2 shifts positively, which is also attributed to the deposited copper that is not completely stripped in the 1st cycle. The voltammogram has a large peak separation and crossover on the cathodic branches, indicating the presence of nucleation and growth processes,29 whereas the voltammogram on the Pt in Fig. 1 does not have this feature. When the switching potentials are −1.00 V and −1.30 V, the curves have only one cathodic peak C1 and only one anodic peak A4 in Fig. 2(b). The anodic A1 and A2 appear when the scan is switched to −1.35 V. The anodic A3 appears when the scan is switched to −1.40, and at the same time the crossover related to the nucleation also appears.
 |
| | Fig. 2 Cyclic voltammograms of the GC electrode in the Sp4 solution containing only DMH as complexing agent. (a) Ranging from 1.0 V to −1.5 V vs. SCE and (b) ranging from 1.0 V to various switching potentials vs. SCE. The scan rate is 50 mV s−1. | |
From these results, it appears that the discharge process of the main copper–DMH species on the GC electrode also has two steps but is different from that on the Pt electrode. The cathodic peak C1 and the anodic peak A4 refer to the first step, Cu2+ to Cu+, as shown in eqn (3). The cathodic peak C2 and the anodic peaks A1 and A2 are attributed to the second step, Cu+ to Cu0, as can be seen in eqn (4). Similar to the condition on the Pt electrode, the anodic peak A3 may be attributed to the production of hydroxides or oxides.
Cyclic voltammograms of the GC electrode from 1.0 V to various switching potentials (−0.60, −0.93, −1.35, and −1.5 V) vs. SCE in the Sp1 solution containing only citrate as the complexing agent are shown in Fig. 3. The entire potential scan started in the negative direction from the rest potential at a scan rate of 50 mV s−1. The voltammogram with the potential range from 1.0 V to −1.5 V vs. SCE in Fig. 3(a) has one big cathodic peak (C3 (−1.20 V)), four small cathodic peaks (C1 (−0.925 V), C2 (−0.99 V), C4 (−1.33 V), C5 (−1.60 V)) and four anodic peaks (A1 (−0.285 V), A2 (−0.09 V), A3 (−0.00 V)). No peak shift can be observed in the 2nd cycle compared with the 1st cycle. The A2 peak can be observed in all the three curves with the switching potentials of −0.60, −0.93, −1.35 V, in the zoom view of Fig. 3(b). The peaks A1 and A3 cannot be observed clearly in the curves with the switching potentials of −0.60, −0.93 V, thus it is not easy to determine whether the two curves have the two peaks. The curve with the switching potential of −1.35 V has all the three anodic peaks, which can be observed clearly in Fig. 3(b).
 |
| | Fig. 3 Cyclic voltammograms of the GC electrode in the Sp1 solution containing only citrate as complexing agent. (a) Ranging from 1.0 V to −1.5 V vs. SCE and (b) ranging from 1.0 V to various switching potentials vs. SCE. The scan rate is 50 mV s−1. | |
The cathodic peaks C1, C3, C4, and C5 are very small and difficult to define, therefore, here we only discuss the predominant cathodic peak C2. According to the reports of Rode, S., et al.,22 the predominant copper–citrate species is the [Cu2(Cit2H−2)]4− dimer with the formation constant (log10(β)) of 5.1, and the discharge process of [Cu2(Cit2H−2)]4− has two steps with an adsorbed blocking intermediate, [Cu2(Cit2H−2)]6−ads. Considering the discussion of the results in Fig. 1 and 2, it can be inferred that the anodic peaks A2 and A3 in Fig. 3(b) refer to the oxidation of Cu+ and the peak A1 to the oxidation of Cu0. In the voltammogram with the switching potential of −0.60 V, although no obvious cathodic peak can be observed, the anodic peak proves that the first discharge step exists as eqn (5). Therefore, the discharge of [Cu2(Cit2H−2)]4− in our experiments also has the two steps. The cathodic peak C2 and the anodic peak A1 are attributed to the reaction demonstrated by eqn (6). In addition, the cyclic voltammetry at various scan rates, combined with the sample current voltammetry, confirms that the discharge in this work is in agreement with the mechanism proposed by Rode, S., et al.
| | |
[Cu2(Cit2H−2)]4− + 2e− → [Cu2(Cit2H−2)]6−ads
| (5) |
| | |
[Cu2(Cit2H−2)]6−ads + 2e− + 2H2O → 2Cu + 2Cit3− + 2OH−
| (6) |
Cyclic voltammograms of the GC electrode from 1.0 V to −1.25 and −1.5 V vs. SCE in the Sp3 solution containing both DMH and citrate as complexing agents is shown in Fig. 4. The entire potential scan started in the negative direction from the rest potential at a scan rate of 50 mV s−1. Two cathodic peaks (C1 (−0.90 V) and C2 (−1.40 V)), four anodic peaks (A1 (−0.26 V), A2 (−0.15 V), A3 (−0.05), and A4 (−0.17)), and a nucleation crossover are observed in the voltammogram ranging from 1.0 V to −1.5 V, whereas only one cathodic peak C1 and two anodic peaks exist in the voltammogram ranging from 1.0 V to −1.25 V. These results indicate that the predominant cupric complexes also discharge in two steps with C1, A3, and A4, referring to Cu2+ to Cu+ and C2 and A1, A2 referring to Cu+ to Cu0, respectively. This is in agreement with the discharge processes of cupric complexes in solution containing either DMH or citrate as complexing agent because the feature of the voltammogram in Fig. 3 is very similar to the synthesis of the ones in Fig. 1(a) and 2(a). In addition, Peak C1 shifts negatively and peak C2 is smoother in the 2nd cycle compared with that in the 1st cycle in Fig. 3, which can also be observed in the curves obtained from the former two solutions. This results from the fact that the surface of the electrode is changed by the potential scan in the 1st cycle.
 |
| | Fig. 4 Cyclic voltammograms of the GC electrode in the Sp3 solution containing both DMH and citrate as complexing agents in the range of 1.0 V to −1.25 V and −1.5 V vs. SCE. The scan rate is 50 mV s−1. | |
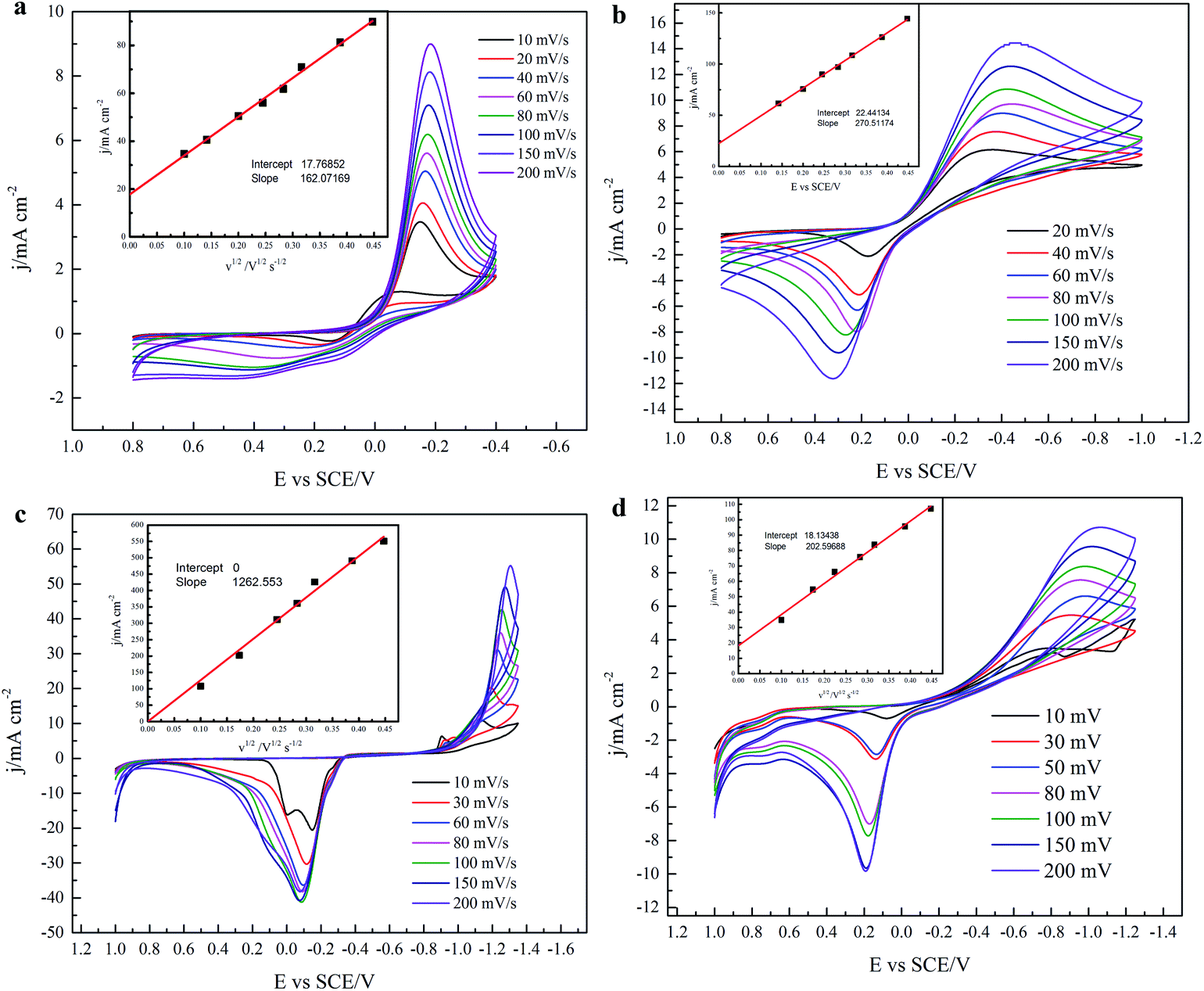
3.1.2 Various scan rates. Voltammetry measurements at various scan rates were conducted in the three solutions, Sp4, Sp1, and Sp3. The results are presented in Fig. 5. To maintain consistency of the electrode surface in the experiments of this part, the electrodes were pretreated by voltammetry until no peak shift occurred in the last two cycles before every voltammetry measurement was performed. Fig. 5(a) and (b) show the voltammograms at various scan rates of the Pt and GC electrode, respectively, in the Sp4 solution containing only DMH as complexing agent. Fig. 5(c) and (d) show the voltammograms of the GC electrode in Sp1 and Sp3 solutions, respectively. The potential scan in Fig. 5(a) and (b) is performed in the range in which only the first discharge step occur, whereas the potential scan in Fig. 5(c) and (d) is carried out in the range of the second step discharge. All the voltammograms in the four figures feature the irreversibility reaction: the relationship of the peak current density (ip) and the square root of the scan rate (v1/2) is linear, and the peak potential Ep shifts negatively with the increase in the scan rate. However, among the four plots of ip vs. v1/2 only the one in Fig. 5(c) has a zero intercept, and the diffusion coefficient calculated from voltammetry according to eqn (7) is not in agreement with the results from chronoamperometry (see Fig. 3). The transfer coefficients (α) listed in Fig. 3 were also calculated from cyclic voltammetry according to eqn (8) (detailed data are listed in Table S1 of the ESI†). The α coefficient calculated from the cyclic voltammetry of the Pt electrode in the Sp4 solution containing only DMH as the complexing agent is in agreement with the value from the sample current voltammetry. The α coefficient calculated from the cyclic voltammetry of the GC electrode in the Sp1 solution containing only citrate as the complexing agent is also very close to the value obtained from sample current voltammetry, when the number of the transferred electrons n is 2, which confirms the validity of eqn (5). These results allow us to speculate that the reaction of the copper–DMH complex on the GC electrode is much more intricate than that presented in eqn (3), considering that the α coefficient calculated from the cyclic voltammetry of the GC electrode in the Sp4 solution is half of the value obtained from sample current voltammetry when n = 1. Incidentally, no significant data was obtained from the polarization curves on a GC rotating disk electrode with a slow scan rate of 1 mV s−1. The electrodeposition process on the GC electrode appears to be controlled by the charge transfer process in the Sp4 and Sp3 solutions, and it exhibits features of the diffusion control in the Sp1 solution (see Fig. S2–S5 of the ESI†). The data from the Sp1 solution was used to calculate the diffusion coefficient based on the 0.01 cm2 s−1 kinematic viscosity according to the Koutecký–Levich equation, which, however, differs largely from the one obtained from cyclic voltammetry. The value, 61.12 × 10−7 cm2 s−1, obtained from the rotating disk electrode is higher than that reported, 21.30 × 10−7 cm2 s−1.33| |
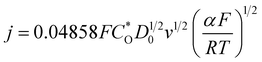 | (7) |
| |
 | (8) |
where j is the current density, F the Faraday Constant, C*O the mole concentration of the electroactive species, assumed to be 0.1 M in this work, D0 the diffusion coefficient of the electroactive species, v is the scan rate, R is the gas constant, T is the thermodynamic temperature, Ep is the peak potential, and Ep/2 is the potential, where the current is at half the peak value.
 |
| | Fig. 5 Cyclic voltammograms at various scan rates. (a) The Pt electrode in the Sp4 solution containing only DMH as complexing agent, (b) the GC electrode in the Sp4 solution containing only DMH as complexing agent, (c) the GC electrode in the Sp1 solution containing only citrate as complexing agent, (d) the GC electrode in the Sp3 solution containing both DMH and citrate as complexing agents. The insets are jp vs. v1/2 lines fitted from the peak of cathodic current density at various scan rates. | |
3.2 Chronoamperometry
3.2.1 Sample current voltammetry for kinetic parameters. Chronoamperometry was used to obtain the sample current voltammograms (the original current density vs. time curves are presented in Fig. S6–S8 of the ESI†). The sample current voltammogram of the Pt electrode in the Sp4 solution is shown in Fig. 6(a) (and the ones of the GC electrode in the Sp4 and Sp1 solutions are presented in Fig. S9 and S10 of the ESI†). The kinetic parameters based on the database proposed by Mirkin, M. V. and A. J. Bard28 are presented in Table 2. Simple linear interpolation and the temperature coefficient, 298/T, for E1/4 − E1/2 and E1/2 − E3/4 have been employed in the analysis. Fig. 6(b) is the Tafel curve based on the data of the sample current voltammogram in Fig. 6(a). The α coefficient calculated from the Tafel curve is very close to the ones from the database, which confirms the validity of the analysis proposed by Mirkin, M. V. and A. J. Bard. However, the calculated conditional (formal) potential E0′ may be invalid because it is smaller than the rest of the potential of the systems. The standard rate constant k0 was calculated by eqn (9).28| |
 | (9) |
where λ is a dimensionless rate constant, and t is the time at which the current density is used for the sample current voltammogram.
 |
| | Fig. 6 (a) The sample current voltammogram of the Pt electrode in the Sp4 solution containing only DMH as complexing agent. (b) The E vs. ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) j curves fitted from the data of the sample current voltammetry. j curves fitted from the data of the sample current voltammetry. | |
Table 2 Kinetic parameters calculated from sample current voltammetry based on the database proposed by Mirkin, M. V. and A. J. Barda
| |
E1/4 − E1/2 (mV) |
E1/2 − E3/4 (mV) |
λ |
k0 (cm s−1) |
nΔE0′ (mV) |
E0′ (mV) |
α |
αTafel |
αCV |
| DCu2+ (10−7 cm2 s−1): 3.54 for Pt electrode in Sp4, 3.61 for GC electrode in Sp4, and 61.12 for GC electrode in Sp1. αTafel was obtained from the data of sample current voltammetry according to Tafel equation. αCV was calculated from the voltammetry in Fig. 5 according to eqn (8), of which the number of transfer electron n = 1 for Pt electrode in Sp4, n = 1 for Pt electrode in Sp4, n = 2 for GC electrode in Sp1. |
| Pt in Sp4 |
42 |
43 |
0.17 |
1.04 × 10−5 |
72 |
−0.02 |
0.65 |
0.61 |
0.64 |
| GC in Sp4 |
67 |
66 |
0.20 |
1.23 × 10−5 |
106 |
0.01 |
0.41 |
0.41 |
0.20 |
| GC in Sp1 |
84 |
81 |
0.20 |
5.10 × 10−5 |
128 |
−0.48 |
0.34 |
0.30 |
0.34 |
Obviously, the Pt and GC electrodes in the Sp4 solution containing only DMH as the complexing agent have different α values, indicating different discharge mechanisms on the two electrodes. This is in agreement with the results of the cyclic voltammetry because the cyclic voltammograms obtained on the two electrodes have different shapes.
3.2.2 Contrell equation fitting for the diffusion coefficient. The plots of j vs. t−1/2 of the Pt and GC electrodes in the Sp4 solution containing only DMH as the complexing agent is shown in Fig. 7. The current vs. time curves with potential stepping from 0.2 V to −0.16 V vs. SCE was used to fit the plots of j vs. t−1/2, while eqn (10) and (11) are the two equations derived from the intercepts and slopes of the two plots. Theoretic curves based on eqn (10) and (11) match very well with the curves measured, as shown in the zoom view of Fig. 7. The diffusion coefficients, calculated from the two plots according to Conttrell equation,34,35 are very similar and are listed in Table 3. No significant data were obtained from the current vs. time curves of the GC electrode in the Sp1 solution because the nucleation feature appeared early at a relative small step potential (see Fig. S8 of the ESI†).| | |
j = 19.063 + 32.417t1/2
| (10) |
| | |
j = 20.839 + 32.727t1/2
| (11) |
 |
| | Fig. 7 The j vs. t1/2 line fitted according to the Conttrell equation from the j vs. time curves obtained from the Sp4 solution containing only DMH as complexing agent. (a) On the Pt electrode and (b) on the GC electrode with the potential changed from 0.2 V to −0.16 vs. SCE. | |
Table 3 Diffusion coefficients calculated from cyclic voltammetry and chronoamperometry
| DCu2+ (10−7 cm2 s−1) |
Pt in Sp4 |
GC in Sp4 |
GC in Sp1 |
| Cyclic voltammetry |
1.59 |
7.06 |
181.60 |
| Chronoamperometry |
3.54 |
3.61 |
— |
| RDE |
— |
— |
61.12 |
3.2.3 Nucleation modelling. Potentiostatic current transients presented in Fig. 8 were recorded in the potential ranges of the electrochemical deposition of copper on the GC electrode from solutions Sp4 (Fig. 8(a)), Sp1 (Fig. 8(b)), and Sp3 (Fig. 8(c)). The potential range used is in agreement with the results of the previous cyclic voltammetry. All the current transients have the following characteristic features: an abrupt drop followed by an increase in the cathodic current density up to a maximum, jmax at tmax, which is related with the crystal nucleation and growth processes, then a decaying in the current density after tmax, converging to the limiting current density corresponding to the linear diffusion of the electroactive species to a planar electrode, and a bigger jmax and a shorter tmax with a more negative step potential.
 |
| | Fig. 8 The chronoamperometry at various step potentials and the corresponding dimensionless experimental curves with their theoretical curves for instantaneous and progressive nucleation processes of the GC electrode in different solutions. (a) and (A) In the Sp4 solution containing only DMH as complexing agent, (b) and (B) in the Sp1 solution containing only citrate as complexing agent, (c) and (C) in the Sp3 solution with both DMH and citrate as complexing agents. | |
The SH nucleation models classify the nucleation and crystal growth in three dimensions (3D-dc) controlled by the diffusion of two types: the instantaneous process (eqn (12)) with simultaneously activated active sites followed by a slow growth of nuclei, and the progressive process (eqn (13)) with continually formed active sites followed by a fast growth of nuclei.36,37 The comparison of the experimental curves to the dimensionless theoretical curves of the three solutions are presented in Fig. 8(A)–(C). At all the step potentials in this work, the curves (−1.300 to −1.388 V) in Fig. 8(A) from the Sp4 solution containing only DMH as the complexing agent and the curves (−1.25 to −1.35 V) in Fig. 8(C) from the Sp3 solution containing both DMH and citrate as complexing agents show a good agreement with the progressive nucleation process before tmax, while departing downward after tmax, becoming stronger with a more negative step potential. According to Floate, S., et al.,37 the concomitant hydrogen evolution or the rapid replenishment of electroactive species through hemi-spherical diffusion to growth centres may contribute to the departure. The former exerts a greater effect with a more negative step potential while the latter has more influence with a less negative step potential. No departure is observed in the curves (−1.300 to −1.388 V) in Fig. 8(B) from the Sp1 solution containing only citrate as the complexing agent, which also exhibits an agreement with the progressive nucleation process. It can be concluded that DMH have a stronger influence on the nucleation and crystal growth process of copper on the GC electrode in the Sp3 solution containing both DMH and citrate as complexing agents in the experimental composition.
| |
 | (12) |
| |
 | (13) |
3.3. The adsorption of DMH molecules on a steel substrate
3.3.1 Molecular dynamic simulation. Molecular dynamic simulations were conducted to reveal the adsorption behaviours of DMH and citric acid molecules on the Fe (111) and Cu (111) surfaces. All the simulations were executed for 500 ps, in which time the computed systems reached a steady state (as shown by the energy and potential vs. time curves in supplementary materials). The results are shown in Fig. 9 and the interaction energy Einteraction values between the organics and the metal surface are listed in Table 4. All the DMH molecules simulated, which were placed randomly in the boxes before computing, got close to the surfaces of Cu (111) and Fe (111) at the end of the simulation. Two of the four DMH simulated molecules have the planes of their heterocycle parallel to the Cu (111) surface, and the other two have the planes of their heterocycle perpendicular to the Cu (111) surface. Moreover, the planes of the heterocycle of all the four simulated molecules are parallel to the Fe (111) surface. The simulated adsorption behaviors for DMH− (DMH molecule lacking one proton), citric acid molecule (Cit), Cit− (citric acid molecule lacking three protons), and hydrogen cyanide molecule on Fe (111) and Cu (111) surfaces are not presented in the paper (see Fig. S14–S23 of ESI†). According to Table 4, both DMH and DMH− have a stronger adsorption on the Fe (111) surface than that on the Cu (111) surface and both Cit and Cit− also have a stronger adsorption on the Fe (111) surface than that on the Cu (111) surface. In fact, Cit and Cit− did not get close to the surface of Cu (111) at the end of the simulation and the Ebinding between Cit− and Cu (111) surface was negative. In addition, neutral molecules, either DMH or Cit, have a larger Ebinding than molecules with negative charges on both the Fe (111) and Cu (111) surfaces.
 |
| | Fig. 9 Front and top views of the simulated behaviors of DMH on Cu (111) (A and a) and Fe (111) (B and b) surfaces. | |
Table 4 Interaction energy between the organic molecules and metal surfaces involved in the molecular dynamic simulation
| |
Etotal (kJ mol−1) |
Esurface (kJ mol−1) |
Epolymer (kJ mol−1) |
Ebinding (kJ mol−1) |
| DMH on Fe (111) |
259862399.10 |
259865287.90 |
−1554.32 |
333.64 |
| DMH− on Fe (111) |
255802131.80 |
255802937.10 |
231.43 |
259.16 |
| Cit on Fe (111) |
259863609.30 |
259865356.90 |
−1013.50 |
183.53 |
| Cit− on Fe (111) |
259867179.60 |
259865287.90 |
1744.84 |
−36.70 |
| HCN on Fe (111) |
259865055.30 |
259865356.90 |
42.18 |
42.97 |
| DMH on Cu (111) |
2078040.71 |
2080417.59 |
−1554.00 |
205.72 |
| DMH− on Cu (111) |
2080252.76 |
2080417.59 |
266.67 |
107.88 |
| Cit on Cu (111) |
2079465.50 |
2080417.59 |
−923.67 |
7.10 |
| Cit− on Cu (111) |
2083831.67 |
2080417.59 |
3294.77 |
−29.83 |
| HCN on Cu (111) |
2080139.38 |
2080417.59 |
43.83 |
40.25 |
3.3.2 Chronopotentiometry. Chronopotentiometry was used to investigate the initial stage of the copper electrodeposition process on the carbon steel substrate in S2. The potential vs. time curves obtained from S2 at various current densities and from background solutions with and without DMH are shown in Fig. 10. The background solution is composed of 0.0 or 0.2 M DMH, 0.3 M citrate, and 0.3 M K2CO3. These curves (10, 15, 20, 25 mA cm−2) are generally characterized by five stages: (1) an abrupt potential drop which can be attributed to the ohmic potential drop, (2) a slow decrease of the potential which can be attributed to the charging of the double-layer capacitance, (3) a negative potential peak, (4) another slow decrease of the potential which can be attributed to the decrease of the reactant concentration at the interface caused by the decay of the diffusion process, and lastly (5) a potential platform on which copper electrodeposition and hydrogen evolution occur simultaneously. However, the curves obtained at 5 mA cm−2 do not have the fifth stage and end at a potential platform on which only the discharge of copper complexes occurs. The results indicate that there is a strong hydrogen evolution when copper is electrodeposited galvanostatically at values above 10 mA cm−2.
 |
| | Fig. 10 Potential vs. time curves of a carbon steel electrode as a function of step-current in S2 and cupric ions free solutions. Curve (a) was obtained from the background solution consisting of 0.3 M citrate, and 0.3 M K2CO3. Curve (b) from the background solution consisting of 0.2 M DMH, 0.3 M citrate, and 0.3 M K2CO3. All solutions were at pH 9 and 50 °C. | |
The negative potential peak, as observed clearly from the inset in Fig. 10, in the third stage may be attributed to the reduction of the passive film on the surface of the steel electrode13,38,39 or to the desorption of DMH molecules adsorbed on the steel electrode. The latter assumption is confirmed by the potential curves obtained from background solutions without and with DMH (curve (a) and (b) Fig. 5) because the potential vs. time curve (b) from the background solution with DMH also has a negative potential peak initially, although the peak value is lower than that of the curves from the solution containing cupric ions. However, no negative potential peak can be observed in the curve (a) obtained from the background solution without DMH.
3.4 Cathodic polarization curves
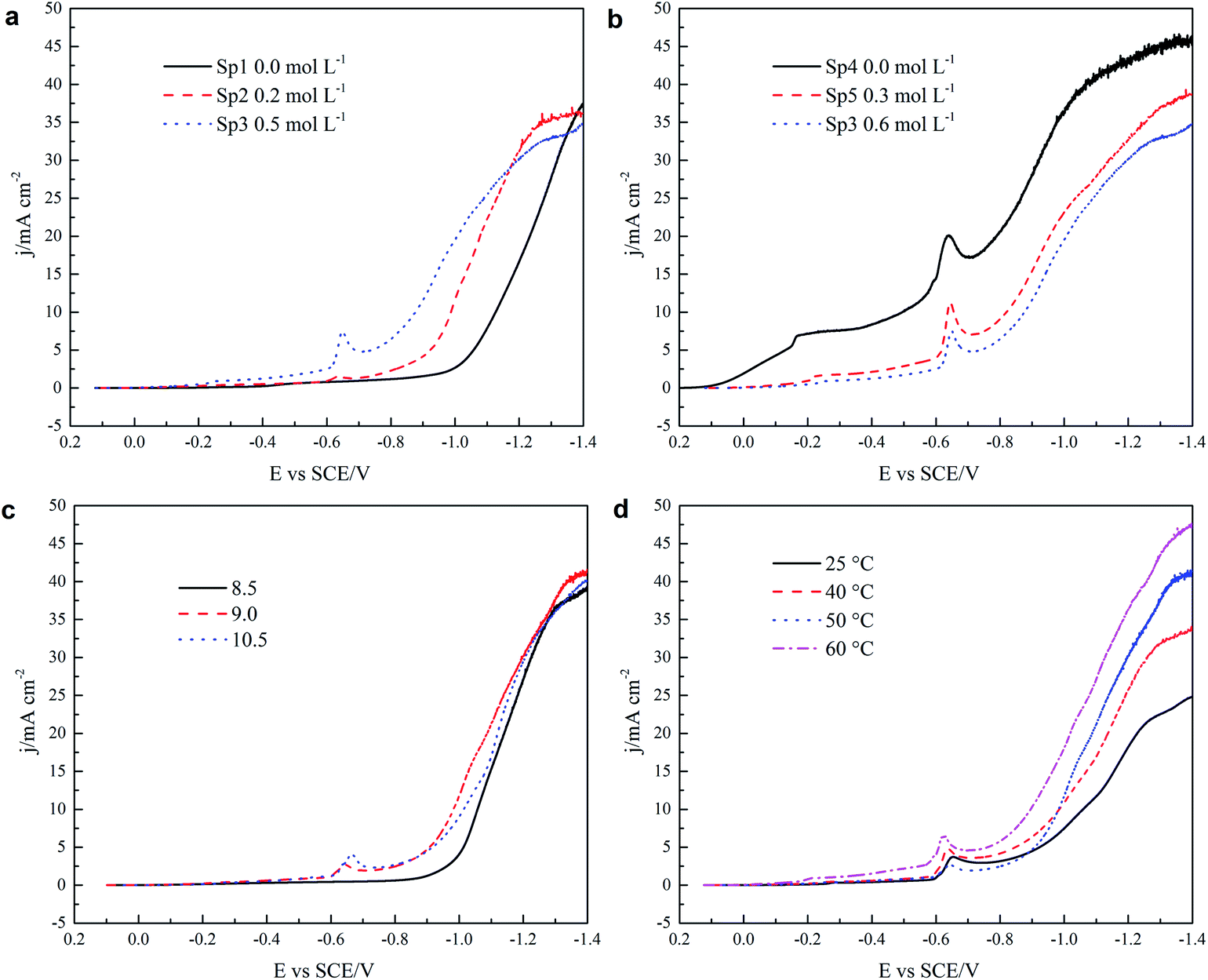
The cathodic polarization curves with the Pt RDE as the working electrode at 300 rpm were obtained from alkaline DMH/citrate baths with various compositions and conditions. The scan rate for all the curves was 1 mV s−1. Cathodic polarization curves for copper electrodeposition with the effect of adding increasing amounts of DMH and citrate are shown in Fig. 11(a) and (b), respectively. Two reduction waves and a peak around −0.65 V can be observed in the curves obtained from solutions containing DMH. The peak current density of the first wave, associated with the discharge of copper–DMH complex according to the cyclic voltammograms, increases with increasing DMH concentration, while it decreases when increasing the citrate concentration.
 |
| | Fig. 11 Cathodic polarization curves as a function of (a) citrate, (b) DMH, (c) pH, and (d) temperature with the Pt RDE as the working electrode at 300 rpm and a scan rate of 1 mV s−1. (a) and (b) were obtained from S1, S2, S3, S4, S5 at pH 9 and 50 °C, (c) at various pH values from the S2 solution at 50 °C, and (d) at various temperatures from the S2 solution at pH 9. | |
Fig. 11(a) shows how the polarization is influenced by increasing the amount of DMH in solutions (Sp2, Sp3). In the solution (Sp1) in the absence of DMH there is no significant current before about −0.95 V, indicating a big overpotential. The current density increases linearly between −0.95 to −1.35 V with increasing cathodic potential and then reaches the limiting current plateau. However, the extent of the cathodic polarization falls with an increase in the DMH concentration. According to literature reports,14,22 the stability constants (log10(β)) of copper–DMH and copper–citrate complexes from our experimental conditions were very close. Therefore, on adding DMH into solutions, there was a competition between DMH and citrate to coordinate with cupric ions because the stability constants of the two complexes were very similar. These results demonstrate that it is easier to discharge the copper–DMH complex than the copper–citrate complex. It can also be observed that increasing the DMH concentration decreases the limiting current density, indicating that the copper–DMH complex diffused slower than the copper–citrate complex, which is in agreement with previous results.
The effect of the citrate concentration (Sp3, Sp4, Sp5) on cathodic polarization is given in Fig. 11(b). Both the cathodic polarization and the limiting current density decrease significantly with the addition of citrate. This results from a more difficult discharge as more copper–citrate complexes formed. Fig. 11(c), with data from the S2 solution at 50 °C, shows that the overpotential decreases slightly when the pH is increased from 8.5 to 9 but no significant difference is observed in the limiting current density at various pH values (8.5, 9, 10.5). The influence of temperature on cathodic polarization measured in the S2 solution at pH 9 is shown in Fig. 11(d). The polarization decreases and the limiting current density increases with the increment in temperature from 25 to 60 °C. This trend can be attributed to the effect of temperature on the diffusion coefficient, which increases with increasing temperature.
3.5 Current efficiency
CE as a function of current density at 40 and 50 °C is presented in Fig. 12. The investigated bath had a CE of above 70% at 50 °C, which was similar to the CE of classical cyanide baths.1 The CE of the S2 solution in Fig. 12(a) falls when increasing the current density from 5 to 15 mA cm−2, then remains stable from 15 to 25 mA cm−2, and finally tends to fall again after 25 mA cm−2. This trend is expected due to the simultaneous hydrogen evolution reaction, which is enhanced by an increasing current density and confirmed by the set of potential vs. time curves in Fig. 10. Comparing curve (a) at 50 °C with (b) at 40 °C in Fig. 12 shows that the CE increases with increasing temperature because of the influence of temperature on the diffusion coefficient. This result is in good agreement to the data in Fig. 11.
 |
| | Fig. 12 CE as a function of current density from various solutions and conditions with pH 9. (a) S2 at 50 °C, (b) S2 at 40 °C, (c) 0.1 M Cu2+, 0.4 M DMH, and 0.3 M K2CO3 at 50 °C and pH 9. | |
3.6 Characteristics of copper electrodeposits
3.6.1 Morphology. The surface morphology of the copper layers electrodeposited for 30 min at 15 mA cm−2 from solutions with the various concentrations of DMH, citrate, and K2CO3 and at various pH values was observed by using SEM and the images are given in Fig. 13 (more images are presented in Fig. S11 and S12,† and the thickness of the layers calculated from cross-sectional SEM images like Fig. S15† are listed in Table S2 of ESI†). The surface of the copper layer obtained from S1 that did not contain DMH is dark and rough, in Fig. 13(1), and clusters with cracks in between can be observed. This is because the citrate concentration is very low to reduce free cupric ions by coordination to an adequately low level. With the addition of DMH into the bath (S2), the cracks between the clusters disappear and the layer becomes smoother with small islands dispersing on the surface in Fig. 13(2). The small islands, which can also be observed on the other layers, may be ascribed to the discharge of species, which are easily reduced. The layer is denser and smoother but too thin (2.50 μm) when the DMH concentration is 0.4 M.
 |
| | Fig. 13 SEM images illustrating the morphology of copper layers electrodeposited from various solutions at pH 9 and 50 °C: (1) S1, (2) S2, (3) S4, (4) S5, (5) S6, and (6) S7, and from S2 with various values of the pH at 50 °C: (7) pH 8.5, and (8) pH 10.5. | |
From the images of Fig. 13(3) and (4), when increasing the citrate concentration the copper layer is first compact with numerous small islands on the surface, and then has some dents that may be attributed to hydrogen evolution, and lastly becomes very smooth. Moreover, the thickness of the layer increases at first and then decreases on further addition of citrate, which is related to the coordinating environment in the solution. The K2CO3 concentration also has a significant effect on the surface morphology in Fig. 13(5) and (6). The layer becomes smoother with the addition of K2CO3, compared with that obtained from S6 without K2CO3. When the K2CO3 concentration is 0.5 M, the dents become holes and the islands are more disperse, which is attributed to strong hydrogen evolution. Furthermore, the thickness decreases with the increase of the K2CO3 concentration. These results indicate that K2CO3 also influences the coordination environment and the electrodeposition process. On the other hand, the layer becomes smoother and the thickness increases with increasing pH. The amount of islands is small but some dents can be observed at pH 9. In addition, the electroplated copper layer, from the bath containing only DMH as the complexing agent, is not present because to prevent the immersing coating of copper, adequate amount of DMH had to be used but no copper layer can be obtained in a large current density range (5–40 mA cm−2) even at a DMH concentration of 0.4 M.
3.6.2 Crystal structure. XRD was used to reveal the structure and calculate the average crystal sizes of the electroplated copper layers from different baths and at different pH values. The main peaks in the spectra match very well with the pattern of a standard copper powder sample (PDF#4-0836) (see Fig. S14 of ESI†), indicating that the copper layer has a crystalline cubic structure. The average crystal size calculated from the intensity of peaks (a) and (b) by using Scherrer equation is about 30 nm (see Table S5 of ESI†),40 which is nearly 70% smaller than those (91 nm) reported for copper electrodeposits obtained from an additive-free acid copper sulfate bath.41 The copper layer deposited from the solution containing only citrate as complexing agent prefers the (111) orientation of the crystals (Fig. 14(a)). With the addition of DMH into the solution, the (111) orientation of the crystals turns to a preferable (200) orientation. This result is in agreement with Fig. 14(b), in which the peak of (111) orientation becomes reversely stronger than that of the (200) orientation, with an increase in the citrate concentration from 0.1 M to 0.5 M. The behaviors of crystal orientation when changing solution composition confirm the competitive relationship between DMH and citrate in coordination with cupric ions because more citrates can form more copper–citrate complexes, which dominate the discharge and produce more crystals with the (111) orientation. Although not changed, the preferable (200) crystal orientation becomes stronger with the increase in the K2CO3 concentration, while becoming weaker with increasing pH. This indicates that more K2CO3 and lower pH enhance the discharge of copper–DMH.
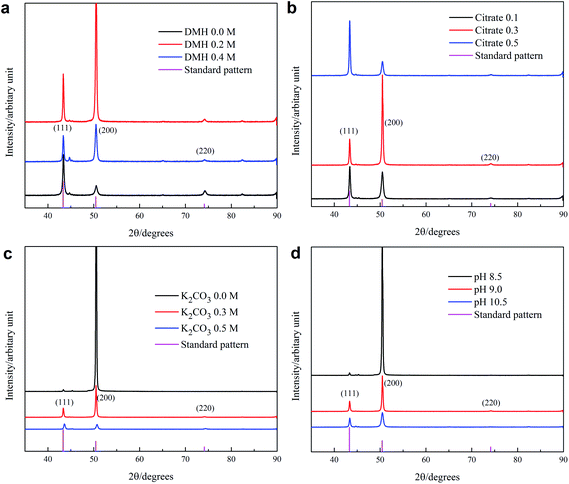 |
| | Fig. 14 XRD patterns of the copper layers corresponding to the images in Fig. S11.† The copper layers were electroplated from various solutions at pH 9 and 50 °C: (1) S1, (2) (5) and (8) S2, (3) S3, (4) S4, (6) S5, (7) S6, and (9) S7, and from S2 with various values of the pH at 50 °C: (10) pH 8.5, (11) pH 9, and (12) pH 10.5. | |
3.6.3 Adhesion on steel substrate. The influence of the concentrations of DMH, citrate, and K2CO3 and the value of pH on the adhesion of electrodeposited copper layers on a carbon steel substrate was investigated with three qualitative methods and the results are listed in Table 5. The copper layers obtained from solutions without DMH did not pass the bend test. This may result from the immersion coating of copper on the steel substrate because the citrate concentration was very low to prevent the contact replacement reaction by coordinating with cupric ions. The adhesion was good in the investigated range (0.1 to 0.5 M) of the citrate concentration. In the solution with high concentration of K2CO3 (0.5 M), the adhesion was also not good enough to pass the bend test, which may be attributed to the strong hydrogen evolution as demonstrated in Fig. 13(6).42,43 The copper layers obtained at pH 8.5 detached from the substrate during the bend test because copper ions could not coordinate with DMH and citrate very well at low pH conditions. In addition, the cross-sectional SEM image of the copper layer electroplated from S2 at 15 mA cm−2 is given in Fig. 15 (see more in Fig. S12 of ESI†). No slit can be observed at the joint area, which further confirms the good adhesion of the copper layer on the carbon steel substrate.
Table 5 The adhesion of the copper layers electrodeposited from various solutions
| DMH (M) |
Citrate (M) |
K2CO3 (M) |
pH |
| 0.0 |
No |
0.1 |
Yes |
0.1 |
Yes |
8.5 |
No |
| 0.2 |
Yes |
0.3 |
Yes |
0.3 |
Yes |
9 |
Yes |
| 0.4 |
Yes |
0.5 |
Yes |
0.5 |
No |
10.5 |
Yes |
 |
| | Fig. 15 Cross-sectional SEM image of the copper layer electrodeposited from S2 at 15 mA cm−2 for 30 min. | |
4 Conclusions
We have used cyclic voltammetry, chronoamperometry, chronopotentiometry, and cathodic polarization to study the electrodeposition of copper in solutions containing either DMH, or citrate, or both as complexing agents. Molecular dynamic simulation reveals the adsorption behavior and the interaction energy of DMH, citric acid, and HCN molecules on Cu (111) and Fe (111) surfaces. Copper layers were characterized by SEM for surface morphology, XRD for crystal structure, and three qualitative methods for adhesion evaluation.
Cyclic voltammetry studies at various switching potentials and at various scan rates show that the discharge process on the Pt or GC electrode in the solutions containing either DMH, or citrate, or both as complexing agents has two steps and is irreversible. Kinetic parameters are obtained easily from the sample current voltammograms with the results in agreement with the ones from cyclic voltammetry. The different transfer coefficients α obtained from the Pt and GC electrodes in the solution containing only DMH as complexing agent indicate different discharge mechanism, which is also confirmed by cyclic voltammetry because cyclic voltammograms of the two systems have two different shapes.
The analysis of chronoamperometry on the Pt and GC electrodes in solutions containing only DMH as complexing agent gives two almost identical results, 3.54 × 10−7 and 3.61 × 10−7 cm2 s−1. At the same time, the theoretic current density vs. time curves with fitted results based on the Conttrell equation match very well the experimental curves. All the three systems containing either DMH, or citrate, or both as complexing agents have the progressive nucleation process of SH, according to the comparison of the experimental curves with the dimensionless theoretical curves. Moreover, DMH has a stronger influence on the nucleation and crystal growth process of copper on the GC electrode in the Sp3 solution containing both DMH and citrate as complexing agents in the experimental composition.
In addition, DMH molecules adsorb on the Fe (111) surface with the plane of the heterocycle parallel to the Fe (111) surface revealed by molecular dynamic simulation. DMH and citric acid, whether with or without negative charges, adsorb stronger on Fe (111) than on Cu (111). DMH molecules show larger interaction energy than citric acid molecules on both Fe (111) and Cu (111). The chronopotentiometry study shows that DMH molecules adsorbed on a carbon steel electrode desorb at the initial stage of the electrodeposition. This confirms that DMH acts not only as the complexing agent, but also as the inhibitor by adsorbing on the substrate surface, which can prevent the passivation of the steel substrate.
The concentration of DMH and K2CO3 and the value of pH have a crucial effect on the adhesion, the morphology, and the preferable crystal orientation of the copper layers electroplated on a carbon steel substrate. The baths have a current efficiency higher than 80% at 10 mA cm−2 according to the deposition/stripping method based on constant-current chronopotentiometry.
Finally, the optimum bath composition and conditions for copper electroplating on a carbon steel substrate are as follows: 0.1 M CuSO4, 0.2 M DMH, 0.3 M citrate, and 0.3 M K2CO3, pH = 9–10, at 50 °C, current density range from 5 to 25 mA cm−2 under strong agitation. Copper layers with average grain size of about 30 nm and good adhesion can be electroplated on a carbon steel substrate from the baths. As the study in this paper only focuses on the electrodeposition mechanism, more work on specific technologies and applications is required in further studies. We suggest the following studies: longer-term deposition, thicker deposit, bath ageing studies, Hull cell studies to determine acceptable electroplating ranges of current density for various operating conditions and bath compositions, controlled hydrodynamic studies using rotating disc and cylinder electrodes, and Haring–Blum throwing power studies.
Acknowledgements
The authors are grateful for financial support from the State Key Laboratory of Urban Water Resource and Environment (Harbin Institute of Technology) (2012DX03).
References
- F. A. Lowenheim, Modern Electroplating, J. Wiley & Sons, New York, USA, 5th edn, 2010 Search PubMed.
- Z. A. Hamid and A. A. Aal, Surf. Coat. Technol., 2009, 203, 1360–1365 CrossRef PubMed.
- L. L. Barbosa, M. R. H. de Almeida, R. M. Carlos, M. Yonashiro, G. M. Oliveira and I. A. Carlos, Surf. Coat. Technol., 2005, 192, 145–153 CrossRef CAS PubMed.
- C. N. Tharamani, B. N. Maruthi and S. M. Mayanna, Trans. Inst. Met. Finish., 2002, 80, 37–39 CAS.
- J. C. Ballesteros, E. Chainet, P. Ozil, Y. Meas and G. Trejo, Int. J. Electrochem. Sci., 2011, 6, 1597–1616 CAS.
- J. C. Ballesteros, E. Chainet, P. Ozil, G. Trejo and Y. Meas, J. Electroanal. Chem., 2010, 645, 94–102 CrossRef CAS PubMed.
- Y. Xiaowei, A. Maozhong, Z. Yunwang and Z. Lin, Electrochim. Acta, 2011, 58, 516–522 CrossRef PubMed.
- A. M. Liu, X. F. Ren, M. Z. An, J. Q. Zhang, P. X. Yang, B. Wang, Y. M. Zhu and C. Wang, Sci. Rep., 2014, 4, 3837 Search PubMed.
- S. Schäfer, US Pat., US20110180415 A1, 2011.
- W. R. Brasch, US Pat., US5750018 A, 1998.
- A. Gerasimenko, M. Kirvoruchko and V. Korzhavina, Prot. Met., 2000, 6, 1597–1616 Search PubMed.
- N. Kanani, Electroplating: Basic Principles, Processes and Practice, Elsevier Science, 2004 Search PubMed.
- S. B. Feng, S. B. Shang, X. Bao, L. T. Feng, J. W. Zhang and Z. H. Li, Acta Phys.-Chim. Sin., 2005, 21, 463–467 CAS.
- G. Z. Pavlovich and R. G. Luthy, Water Res., 1988, 22, 327–336 CrossRef CAS.
- K. Oyaizu, Y. Ohtani, A. Shiozawa, K. Sugawara, T. Saito and M. Yuasa, Inorg. Chem., 2005, 44, 6915–6917 CrossRef CAS PubMed.
- R. W. Revie, Corrosion and Corrosion Control, Wiley, 2008 Search PubMed.
- L. H. Madkour, A. M. Hassanein, M. M. Ghoneim and S. A. Eid, Monatsh. Chem., 2001, 132, 245–258 CrossRef CAS.
- A. O. Yüce and G. Kardaş, Corros. Sci., 2012, 58, 86–94 CrossRef PubMed.
- C. Wang, M. An, P. Yang and J. Zhang, Electrochem. Commun., 2012, 18, 104–107 CrossRef CAS PubMed.
- S. Monti, C. Li and V. Carravetta, J. Phys. Chem. C, 2013, 117, 5221–5228 CAS.
- F. I. Lizama-Tzec, L. Canché-Canul and G. Oskam, Electrochim. Acta, 2011, 56, 9391–9396 CrossRef CAS PubMed.
- S. Rode, C. Henninot, C. C. Vallieres and M. Matlosz, J. Electrochem. Soc., 2004, 151, C405–C411 CrossRef CAS PubMed.
- T. A. Green, A. E. Russell and S. Roy, J. Electrochem. Soc., 1998, 145, 875–881 CrossRef CAS PubMed.
- E. Beltowska-Lehman and P. Ozga, Electrochim. Acta, 1998, 43, 617–629 CrossRef CAS.
- F. Vangaever and J. Vanhumbeeck, US Pat., US4595462 A, 1986.
- P. Bos and E. V. Dalen, J. Electroanal. Chem. Interfacial Electrochem., 1968, 17, 21–30 CrossRef CAS.
- A. Plankova, K. M. Matar, P. Rauko and L. Novotny, J. Pharm. Pharm. Sci., 2010, 13, 391–399 CAS.
- M. V. Mirkin and A. J. Bard, Anal. Chem., 1992, 64, 2293–2302 CrossRef CAS.
- B. Scharifker and G. Hills, Electrochim. Acta, 1983, 28, 879–889 CrossRef CAS.
- M. E. Hyde and R. G. Compton, J. Electroanal. Chem., 2003, 549, 1–12 CrossRef CAS.
- R. Drissi-Daoudi, A. Irhzo and A. Darchen, J. Appl. Electrochem., 2003, 33, 339–343 CrossRef CAS.
- A. Ramos, M. Miranda-Hernandez and I. Gonzalez, J. Electrochem. Soc., 2001, 148, C315–C321 CrossRef CAS PubMed.
- R. Y. Ying, P. K. Ng, Z. Mao and R. E. White, J. Electrochem. Soc., 1988, 135, 2964–2971 CrossRef CAS PubMed.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, Wiley, 2000 Search PubMed.
- H. A. Laitinen and I. M. Kolthoff, J. Am. Chem. Soc., 1939, 61, 3344–3349 CrossRef CAS.
- D. Grujicic and B. Pesic, Electrochim. Acta, 2002, 47, 2901–2912 CrossRef CAS.
- S. Floate, M. Hyde and R. G. Compton, J. Electroanal. Chem., 2002, 523, 49–63 CrossRef CAS.
- K. Ogura and H. Wada, Electrochim. Acta, 1980, 25, 913–917 CrossRef CAS.
- A. J. Davenport, J. A. Bardwell and C. M. Vitus, J. Electrochem. Soc., 1995, 142, 721–724 CrossRef CAS PubMed.
- S. Ghosh and S. Roy, Surf. Coat. Technol., 2014, 238, 165–173 CrossRef CAS PubMed.
- A. Ibañez and E. Fatás, Surf. Coat. Technol., 2005, 191, 7–16 CrossRef PubMed.
- J. Gui and T. M. Devine, Corros. Sci., 1995, 37, 1177–1189 CrossRef CAS.
- J. Banaś, U. Lelek-Borkowska, B. Mazurkiewicz and W. Solarski, Electrochim. Acta, 2007, 52, 5704–5714 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra06855f |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.