Trienamine catalysis with linear deconjugated 3,5-dienones†
Peng-Qiao
Chen
a,
You-Cai
Xiao
a,
Cai-Zhen
Yue
a and
Ying-Chun
Chen
*ab
aKey Laboratory of Drug-Targeting and Drug Delivery System of the Education Ministry, West China School of Pharmacy, Sichuan University, Chengdu 610041, China. E-mail: ycchen@scu.edu.cn; Fax: +86 28 85502609
bCollege of Pharmacy, Third Military Medical University, Chongqing 400038, China
First published on 1st April 2014
Abstract
An array of deconjugated linear 3,5-dienones with substantial substitutions have been successfully used in β,ε-regioselective Diels–Alder cycloadditions with 3-olefinic oxindole-based dienophiles via trienamine catalysis of cinchona-based primary amines, efficiently producing spirocyclic oxindole architectures with dense and diverse substitutions in high stereoselectivity (up to 99% ee, >19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 d.r.).
:1 d.r.).
Asymmetric organocatalysis has been established as a powerful strategy for the development of stereoselective reactions of saturated or unsaturated carbonyl compounds via diverse catalytic modes, such as enamine, dienamine, iminium, or vinylogous iminium catalysis, leading to C–C or C–X bond formations at α, β, γ, or δ-sites.1 Recently, trienamine catalysis has been reported as a new activation mode for 2,4-dienal substrates, furnishing more remote β,ε-regioselective Diels–Alder cycloaddition reactions with an array of electron-deficient dienophiles to construct multifunctional six-membered carbo- or heterocycles, generally in excellent stereoselectivity.2 However, the similar catalytic strategy could not be simply switched to analogous 2,4-dienone substances, and only those with δ,δ-disubstituted and α′-aryl or styryl patterns could participate in the desired β,ε-regioselective cycloadditions via trienamine catalysis.3
In fact, almost no reaction occurred for the combination of linear 2,4-dienone 1 with an α′-enolizable methyl group and diverse dienophiles in the presence of a primary amine catalyst, probably because the cross-conjugated trienamine intermediate I would be generated, as outlined in Scheme 1. Very recently, we developed a new strategy, in which the previously positioned δ,ε-C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond could perform as an inducing group for the formation of linear trienamines from interrupted cyclic 2,5-dienones bearing an α′-CH group.4 Inspired by such observation, we envisaged that similar inducing effect would be applicable to the deconjugated linear 3,5-dienone 2a, rendering the formation of the requisite extended trienamine intermediate and facilitating the later β,ε-regioselective cycloaddition process.
C bond could perform as an inducing group for the formation of linear trienamines from interrupted cyclic 2,5-dienones bearing an α′-CH group.4 Inspired by such observation, we envisaged that similar inducing effect would be applicable to the deconjugated linear 3,5-dienone 2a, rendering the formation of the requisite extended trienamine intermediate and facilitating the later β,ε-regioselective cycloaddition process.
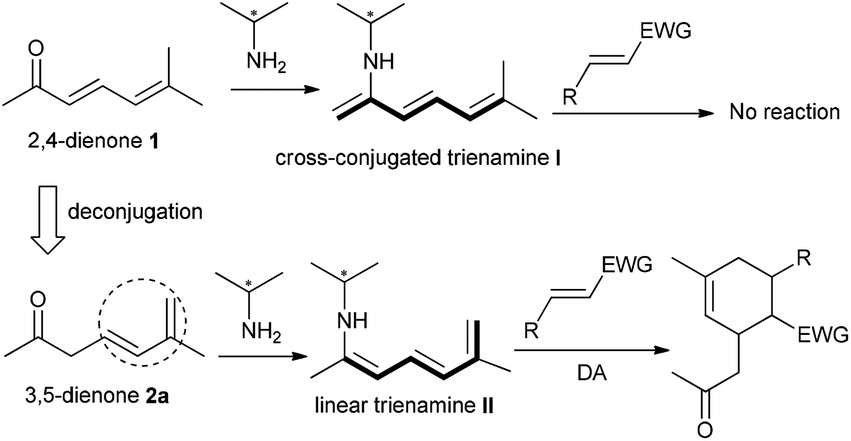 | ||
| Scheme 1 A deconjugation strategy for linear trienamine catalysis with 3,5-dienone substrate. | ||
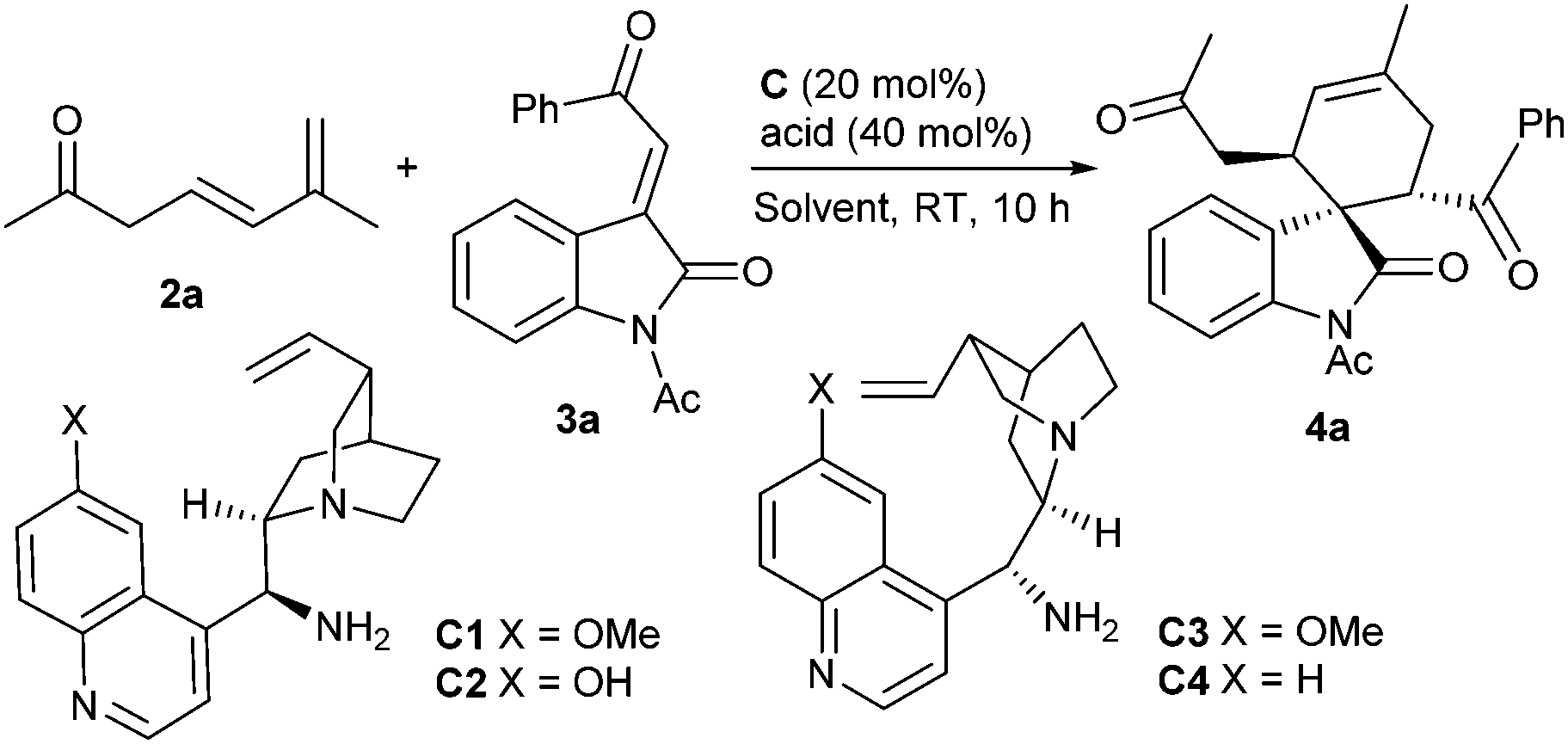
Based on the above-mentioned considerations, the initial investigation was set up for the model reaction of 6-methylhepta-4,6-dien-2-one 2a, which was effectively obtained by the deconjugation of 6-methylhepta-3,5-dien-2-one 1,5 with 3-olefinic oxindole 3a,6 in the presence of 9-amino-9-deoxyepiquinine (C1, 20 mol%) and salicylic acid (SA, 40 mol%).7 To our gratification, the desired Diels–Alder reaction occurred smoothly in toluene at room temperature, and the spirocyclic oxindole 4a was produced in remarkable stereoselectivity and with a good yield (Table 1, entry 1).8 It should be pointed out that the cycloaddition reaction did not occur in the absence of a primary amine catalyst, indicating that the HOMO-activation of the β,γ,δ,ε-diene moiety by the formation of a conjugated enamine intermediate is necessary. Consequently, other catalytic parameters were briefly screened. Amine C2 with a free OH group provided a decreased ee value and lower yield (entry 2). In addition, both 9-amino-9-deoxyepiquinidine (C3) and 9-amino-9-deoxyepicinchonine (C4) delivered the product with an opposite configuration, also in high stereoselectivity (entries 3 and 4). The cycloaddition could take place in an array of solvents, though diminished enantioselectivity was generally observed (entries 5–10). Lower enantiocontrol was also produced when benzoic acid (BA) or o-fluorobenzoic acid (OFBA) was used (entries 11 and 12). In addition, the reaction proceeded smoothly with 10 mol% of the catalyst, while the enantiocontrol was slightly reduced (entry 13). Similar inferior data were observed on a larger scale under the optimized conditions (entry 14).
|
|
|||||
|---|---|---|---|---|---|
| Entry | 1 | Solvent | Acid | Yieldb (%) | eec (%) |
|
a Unless noted otherwise, reactions were performed with 3,5-dienone 2a (0.2 mmol), 3-olefinic oxindole 3a (0.1 mmol), amine C (20 mol%) and acid (40 mol%) in a solvent (1.0 mL) at rt in 10 h.
b Yield of the isolated product.
c Determined by HPLC analysis on a chiral column; d.r. > 19:1 by 1H NMR analysis.
d With 10 mol% of C1 and 20 mol% SA for 18 h.
e At 1.0 mmol scale for 12 h.
|
|||||
| 1 | C1 | Toluene | SA | 91 | 96 |
| 2 | C2 | Toluene | SA | 78 | 87 |
| 3 | C3 | Toluene | SA | 86 | −94 |
| 4 | C4 | Toluene | SA | 86 | −90 |
| 5 | C1 | CHCl3 | SA | 84 | 92 |
| 6 | C1 | DCE | SA | 86 | 87 |
| 7 | C1 | DCM | SA | 86 | 89 |
| 8 | C1 | THF | SA | 91 | 90 |
| 9 | C1 | Dioxane | SA | 84 | 92 |
| 10 | C1 | MeCN | SA | 78 | 67 |
| 11 | C1 | Toluene | BA | 84 | 87 |
| 12 | C1 | Toluene | OFBA | 89 | 89 |
| 13d | C1 | Toluene | SA | 86 | 87 |
| 14e | C1 | Toluene | SA | 89 | 87 |
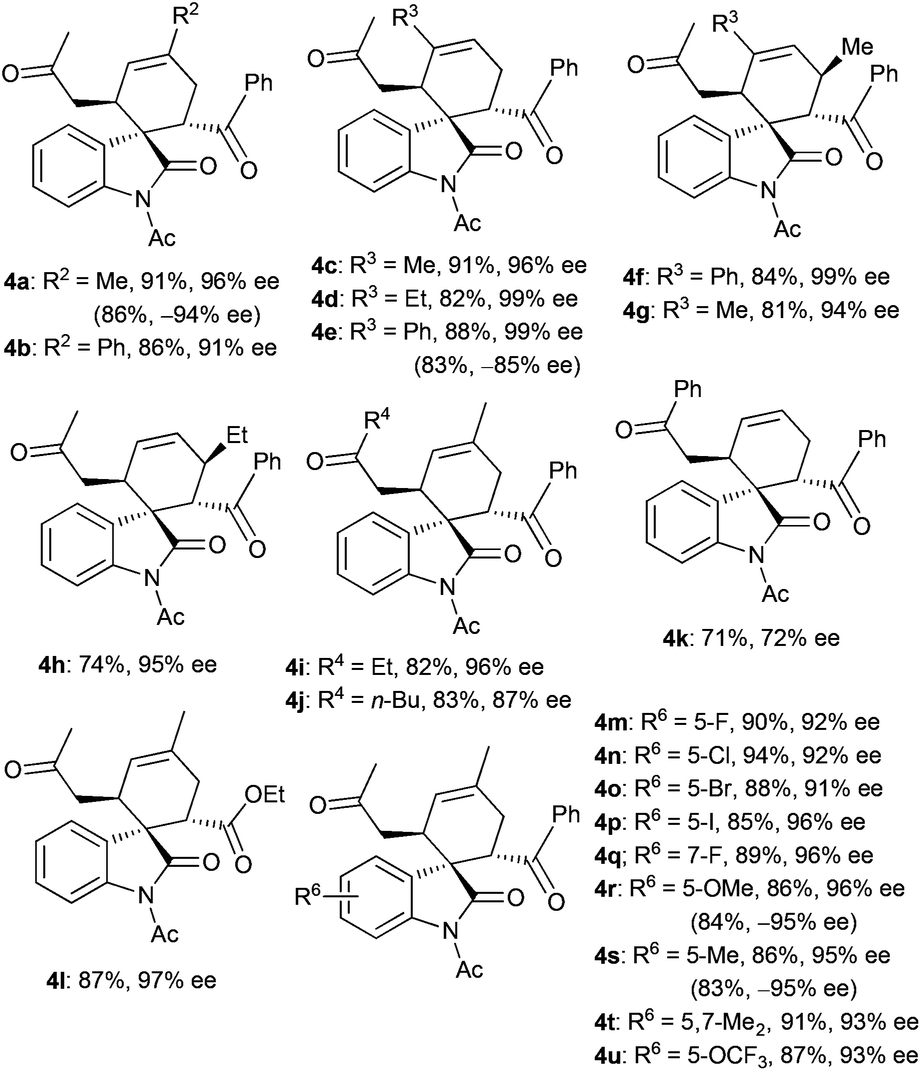
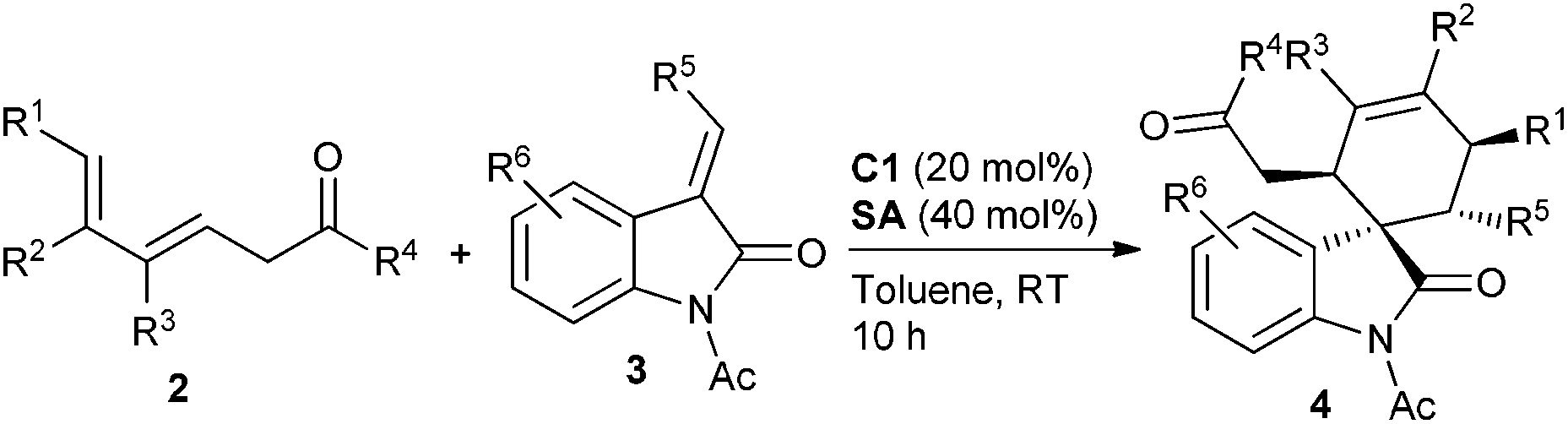
With the optimal reaction conditions in hand, we then investigated a variety of 3,5-dienones and 3-olefinic oxindoles catalyzed by amine C1 (20 mol%) and SA (40 mol%) in toluene at room temperature. The results are summarized in Table 2. At first, an array of deconjugated 3,5-dienones 2 with diverse substituted patterns was investigated in the reaction with dienophile 3a. Both aliphatic and aromatic substituents could be well compatible in the γ- or δ-position of 3,5-dienones 2, delivering the corresponding cycloadducts 4a–4e without apparent influence on the reaction outcome. It was gratifying that γ,ε-disubstituted 3,5-dienones exhibited good reactivity, and products 4f and 4g were obtained in good yields with excellent diastereo- and enantioselectivity. When a single ethyl group was introduced at the ε-position of a 3,5-dienone, the desired cycloadduct 4h was obtained in a moderate yield but still with outstanding enantioselectivity. Notably, other 3,5-dienones carrying a larger α′-alkyl group could also be successfully employed to obtain cycloadducts 4i and 4j in good yields. A 3,5-dienone with an α′-phenyl group without substitution on the backbone still exhibited acceptable reactivity, though modest yield and enantioselectivity were attained for product 4k. Unfortunately, a simple hepta-4,6-dien-2-one showed much lower reactivity, and a very poor yield with bad stereoselectivity was observed. Moreover, the results seemed reasonable when the benzoyl group was replaced by an ethoxycarbonyl group, and the cycloadduct 4l was produced in high yield and with prominent enantioselectivity, but 3-olefinic oxindole with a β-phenyl group (R5 = Ph) showed no reactivity. On the other hand, a spectrum of oxindole-based dienophiles 3 with diverse electron-withdrawing or -donating substituents was tested in the reaction with 6-methylhepta-4,6-dien-2-one 2a. Pleasingly, the corresponding cycloadducts 4m–4u were generally obtained with similar good results. A few substrates were further explored by the catalysis of amine C3, generally affording the cycloadducts with an opposite configuration in excellent enantioselectivity (see more data in parentheses). It should be pointed out that remarkable diastereoselectivity (d.r. > 19:1) was universally achieved in the illustrated reactions.
|
|
|---|
|
a Unless noted otherwise, reactions were performed with 3,5-dienones 2 (0.2 mmol), dienophiles 3 (0.1 mmol), amine C1 (20 mol%) and SA (40 mol%) in toluene (1.0 mL) at rt for 10 h.
b Isolated yield.
c ee was determined by chiral HPLC analysis; d.r. >19:1.
d The absolute configuration of 4i was determined by X-ray analysis, see ESI. Other products were assigned by analogy.
e Data in parentheses were obtained with amine C3.
|
|
To further expand the utility of this strategy, more electron-deficient dienophiles were explored in the reactions with 3,5-dienone 2a under the similar catalytic conditions. It was found that a good ee value with a high yield could be obtained for cycloadduct 6 using maleimide 5 with an N-aryl group (Scheme 2). However, attempts to improve the stereocontrol at a lower temperature resulted in no success. On the other hand, other potential dienophiles, such as β-nitrostyrene, were further investigated, but generally failed to give the desired cycloadducts, and decomposition or other transformations of 3,5-dienone 2a were observed when harsher conditions were applied.9 Therefore, more research remains to be conducted to expand the application of the current catalytic protocol.
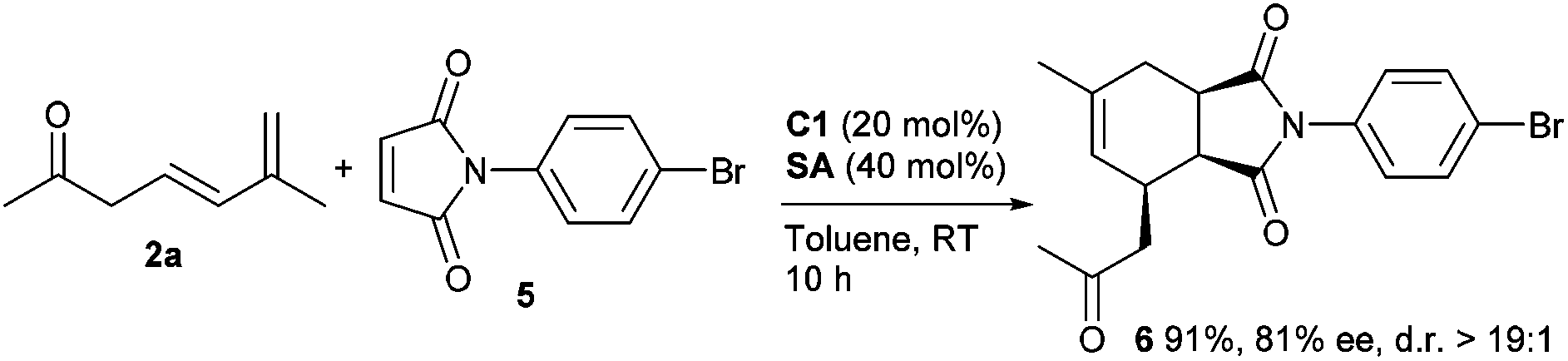 | ||
| Scheme 2 Asymmetric Diels–Alder reaction of maleimide. | ||
In conclusion, we have successfully accomplished the trienamine catalysis of linear polyunsaturated ketones with substantial substitutions using deconjugated 3,5-dienones as the substrates, which was previously not feasible with conjugated 2,4-dienone analogues. Highly stereoselective and β,ε-regioselective Diels–Alder cycloadditions were developed with 3-olefinic oxindole-based dienophiles under the catalysis of readily available cinchona-based primary amines, efficiently producing a spectrum of spirocyclic oxindole architectures with dense and diverse substitutions. Currently, more investigation is being conducted to develop asymmetric reactions with polyunsaturated carbonyl substances via aminocatalysis in this laboratory.
We are grateful for the financial support from the National Natural Science Foundation of China (21125206, 21372160 and 21321061) and the National Basic Research Program of China (973 program, 2010CB833300).
Notes and references
- For selected reviews on aminocatalysis, see: (a) S. Mukherjee, J. W. Yang, S. Hoffmann and B. List, Chem. Rev., 2007, 107, 5471 CrossRef CAS PubMed; (b) A. Erkkilä, I. Majander and P. M. Pihko, Chem. Rev., 2007, 107, 5416 CrossRef PubMed; (c) D. B. Ramachary and Y. V. Reddy, Eur. J. Org. Chem., 2012, 865 CrossRef CAS; (d) X. Yu and W. Wang, Org. Biomol. Chem., 2008, 6, 2037 RSC; (e) P. Melchiorre, Angew. Chem., Int. Ed., 2009, 48, 1360 CrossRef CAS PubMed; (f) C. Grondal, M. Jeanty and D. Enders, Nat. Chem., 2010, 2, 167 CrossRef CAS PubMed; (g) K. L. Jensen, G. Dickmeiss, H. Jiang, Ł. Albrecht and K. A. Jørgensen, Acc. Chem. Res., 2012, 45, 248 CrossRef CAS PubMed; (h) O. V. Serdyuk, C. M. Heckel and S. B. Tsogoeva, Org. Biomol. Chem., 2013, 11, 7051 RSC; (i) Ł. Albrecht, H. Jiang and K. A. Jørgensen, Chem. – Eur. J., 2014, 20, 358 CrossRef PubMed.
- (a) Z.-J. Jia, H. Jiang, J.-L. Li, B. Gschwend, Q.-Z. Li, X. Yin, J. Grouleff, Y.-C. Chen and K. A. Jørgensen, J. Am. Chem. Soc., 2011, 133, 5053 CrossRef CAS PubMed; (b) Z.-J. Jia, Q. Zhou, Q.-Q. Zhou, P.-Q. Chen and Y.-C. Chen, Angew. Chem., Int. Ed., 2011, 50, 8638 CrossRef CAS PubMed; (c) H. Jiang, B. Gschwend, Ł. Albrecht, S. G. Hansen and K. A. Jørgensen, Chem. – Eur. J., 2011, 17, 9032 CrossRef CAS PubMed; (d) Y. Liu, M. Nappi, E. Arceo, S. Vera and P. Melchiorre, J. Am. Chem. Soc., 2011, 133, 15212 CrossRef CAS PubMed; (e) K. S. Halskov, T. K. Johansen, R. L. Davis, M. Steurer, F. Jensen and K. A. Jørgensen, J. Am. Chem. Soc., 2012, 134, 12943 CrossRef CAS PubMed; (f) Ł. Albrecht, F. C. Acosta, A. Fraile, A. Albrecht, J. Christensen and K. A. Jørgensen, Angew. Chem., Int. Ed., 2012, 51, 9088 CrossRef PubMed; (g) C. Ma, Z.-J. Jia, J.-X. Liu, Q.-Q. Zhou, L. Dong and Y.-C. Chen, Angew. Chem., Int. Ed., 2013, 52, 948 CrossRef CAS PubMed; (h) K. Zhu, H. Huang, W. Wu, Y. Wei and J. Ye, Chem. Commun., 2013, 49, 2157 RSC; (i) A. Dieckmann, M. Breugst and K. N. Houk, J. Am. Chem. Soc., 2013, 135, 3237 CrossRef CAS PubMed; (j) Z.-J. Jia, K. Jiang, Q.-Q. Zhou, L. Dong and Y.-C. Chen, Chem. Commun., 2013, 49, 5892 RSC; (k) S.-J. Zhang, J. Zhang, Q.-Q. Zhou, L. Dong and Y.-C. Chen, Org. Lett., 2013, 15, 968 CrossRef CAS PubMed; (l) Ł. Albrecht, C. V. Gómez, C. B. Jacobsen and K. A. Jørgensen, Org. Lett., 2013, 15, 3010 CrossRef PubMed; (m) X. Li, M.-H. Lin, Y. Han, F. Wang and J.-P. Cheng, Org. Lett., 2014, 16, 114 CrossRef CAS PubMed; (n) C. Ma, J. Gu, B. Teng, Q.-Q. Zhou, R. Li and Y.-C. Chen, Org. Lett., 2013, 15, 6206 CrossRef CAS PubMed. For reviews, see: (o) E. Arceo and P. Melchiorre, Angew. Chem., Int. Ed., 2012, 51, 5290 CrossRef CAS PubMed; (p) J.-L. Li, T.-Y. Liu and Y.-C. Chen, Acc. Chem. Res., 2012, 45, 1491 CrossRef CAS PubMed; (q) I. Kumar, P. Ramaraju and N. A. Mir, Org. Biomol. Chem., 2013, 11, 709 RSC; (r) H. Jiang, Ł. Albrecht and K. A. Jørgensen, Chem. Sci., 2013, 4, 2287 RSC; (s) I. D. Jurberg, I. Chatterjee, R. Tannert and P. Melchiorre, Chem. Commun., 2013, 49, 4869 RSC.
- X.-F. Xiong, Q. Zhou, J. Gu, L. Dong, T.-Y. Liu and Y.-C. Chen, Angew. Chem., Int. Ed., 2012, 51, 4401 CrossRef CAS PubMed.
- (a) X. Feng, Z. Zhou, C. Ma, X. Yin, R. Li, L. Dong and Y.-C. Chen, Angew. Chem., Int. Ed., 2013, 52, 14173 CrossRef CAS PubMed; (b) For an example with 2,5-dienals, see: L. Prieto, G. Talavera, U. Uria, E. Reyes, J. L. Vicario and L. Carrillo, Chem. – Eur. J., 2014, 20, 2145 CrossRef CAS PubMed.
- N. Ito and Y. Yamagami, Jpn. Kokai Tokkyo Koho., 2002241337, 28 August, 2002 Search PubMed.
- N-Boc olefinic oxindole exhibited lower reactivity. For other applications of related acceptors, see: (a) G. Bencivenni, L.-Y. Wu, A. Mazzanti, B. Giannichi, F. Pesciaioli, M.-P. Song, G. Bartoli and P. Melchiorre, Angew. Chem., Int. Ed., 2009, 48, 7200 CrossRef CAS PubMed; (b) K. Jiang, Z.-J. Jia, S. Chen, L. Wu and Y.-C. Chen, Chem. – Eur. J., 2010, 16, 2852 CrossRef CAS PubMed; (c) A. Voituriez, N. Pinto, M. Neel, P. Retailleau and A. Marinetti, Chem. – Eur. J., 2010, 16, 12541 CrossRef CAS PubMed; (d) X.-C. Zhang, S.-H. Cao, Y. Wei and M. Shi, Chem. Commun., 2011, 47, 1548 RSC; (e) G. Li, T. Liang, L. Wojtas and J. C. Antilla, Angew. Chem., Int. Ed., 2013, 52, 4628 CrossRef CAS PubMed.
- For reviews of cinchona-based primary aminocatalysis, see: (a) P. Melchiorre, Angew. Chem., Int. Ed., 2012, 51, 9748 CrossRef CAS PubMed; (b) L. Jiang and Y.-C. Chen, Catal. Sci. Technol., 2011, 1, 354 RSC.
- (a) For a review on catalytic synthesis of chiral spirocyclic compounds, see: R. Rios, Chem. Soc. Rev., 2012, 41, 1060 RSC. For selected examples of catalytic asymmetric construction of spirocyclic 2-oxindoles, see: (b) B. M. Trost, N. Cramer and S. M. Silverman, J. Am. Chem. Soc., 2007, 129, 12396 CrossRef CAS PubMed; (c) D. Hojo, K. Noguchi, M. Hirano and K. Tanaka, Angew. Chem., Int. Ed., 2008, 47, 5820 CrossRef CAS PubMed; (d) X.-H. Chen, Q. Wei, S.-W. Luo, H. Xiao and L.-Z. Gong, J. Am. Chem. Soc., 2009, 131, 13819 CrossRef CAS PubMed; (e) K. Jiang, Z.-J. Jia, X. Yin, L. Wu and Y.-C. Chen, Org. Lett., 2010, 12, 2766 CrossRef CAS PubMed; (f) A. P. Antonchick, C. Gerding-Reimers, M. Catarinella, M. Schürmann, H. Preut, S. Ziegler, D. Rauh and H. Waldmann, Nat. Chem., 2010, 2, 735 CrossRef CAS PubMed; (g) X. Jiang, Y. Cao, Y. Wang, L. Liu, F. Shen and R. Wang, J. Am. Chem. Soc., 2010, 132, 15328 CrossRef CAS PubMed; (h) W.-B. Chen, Z.-J. Wu, Q.-L. Pei, L.-F. Cun, X.-M. Zhang and W.-C. Yuan, Org. Lett., 2010, 12, 3132 CrossRef CAS PubMed; (i) F. Zhong, X. Han, Y. Wang and Y. Lu, Angew. Chem., Int. Ed., 2011, 50, 7837 CrossRef CAS PubMed; (j) L.-T. Shen, W.-Q. Jia and S. Ye, Angew. Chem., Int. Ed., 2013, 52, 585 CrossRef CAS PubMed; (k) F. Manoni and S. J. Connon, Angew. Chem., Int. Ed., 2014, 53, 2628 CrossRef CAS PubMed.
- For more details, see the ESI.†.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, structural proofs, the CIF file of the enantiopure product 4i. CCDC 992018. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4qo00079j |
| This journal is © the Partner Organisations 2014 |