
Stimuli-responsive peptide hydrogels for biomedical applications
Haoran
Zhou
a,
Yanhua
Zhu
a,
Bingbing
Yang
a,
Yehong
Huo
a,
Yuanyuan
Yin
 b,
Xuemei
Jiang
*a and
Wei
Ji
*a
b,
Xuemei
Jiang
*a and
Wei
Ji
*a
aKey Laboratory of Biorheological Science and Technology, Ministry of Education, College of Bioengineering, Chongqing University, Chongqing 400044, P. R. China. E-mail: jiangxuemei@cqu.edu.cn; weiji@cqu.edu.cn
bChongqing Key Laboratory of Oral Diseases and Biomedical Sciences, Chongqing Municipal Key Laboratory of Oral Biomedical Engineering of Higher Education, Stomatological Hospital of Chongqing Medical University, Chongqing 401147, P. R. China
First published on 20th January 2024
Abstract
Stimuli-responsive hydrogels can respond to external stimuli with a change in the network structure and thus have potential application in drug release, intelligent sensing, and scaffold construction. Peptides possess robust supramolecular self-assembly ability, enabling spontaneous formation of nanostructures through supramolecular interactions and subsequently hydrogels. Therefore, peptide-based stimuli-responsive hydrogels have been widely explored as smart soft materials for biomedical applications in the last decade. Herein, we present a review article on design strategies and research progress of peptide hydrogels as stimuli-responsive materials in the field of biomedicine. The latest design and development of peptide hydrogels with responsive behaviors to stimuli are first presented. The following part provides a systematic overview of the functions and applications of stimuli-responsive peptide hydrogels in tissue engineering, drug delivery, wound healing, antimicrobial treatment, 3D cell culture, biosensors, etc. Finally, the remaining challenges and future prospects of stimuli-responsive peptide hydrogels are proposed. It is believed that this review will contribute to the rational design and development of stimuli-responsive peptide hydrogels toward biomedical applications.
 Haoran Zhou | Haoran Zhou received his bachelor's degree in agriculture from Sichuan Agricultural University, China, in 2023. In September 2023, he was admitted to pursue a master's degree in biomedical engineering at Chongqing University. His research focuses on bio-inspired self-assembling biomaterials and piezoelectric biomaterials. |
 Xuemei Jiang | Xuemei Jiang is an associate professor at the Bioengineering College of Chongqing University. She obtained her PhD degree in biomedical engineering from Chongqing University (co-trained with the University of Copenhagen) in 2009, and then she joined the Bioengineering College of Chongqing University. Her main research interests include the analysis and characterization of biomaterials. |
 Wei Ji | Wei Ji is a professor at the College of Bioengineering, Chongqing University. He obtained his PhD degree in materials science and engineering from Shanghai Jiao Tong University in 2016, and then worked as a postdoc at Okinawa Institute of Science and Technology and Tel Aviv University. His research interests include peptide self-assembly, supramolecular hydrogels, piezoelectric biomaterials, and bio-inspired smart materials. |
1. Introduction
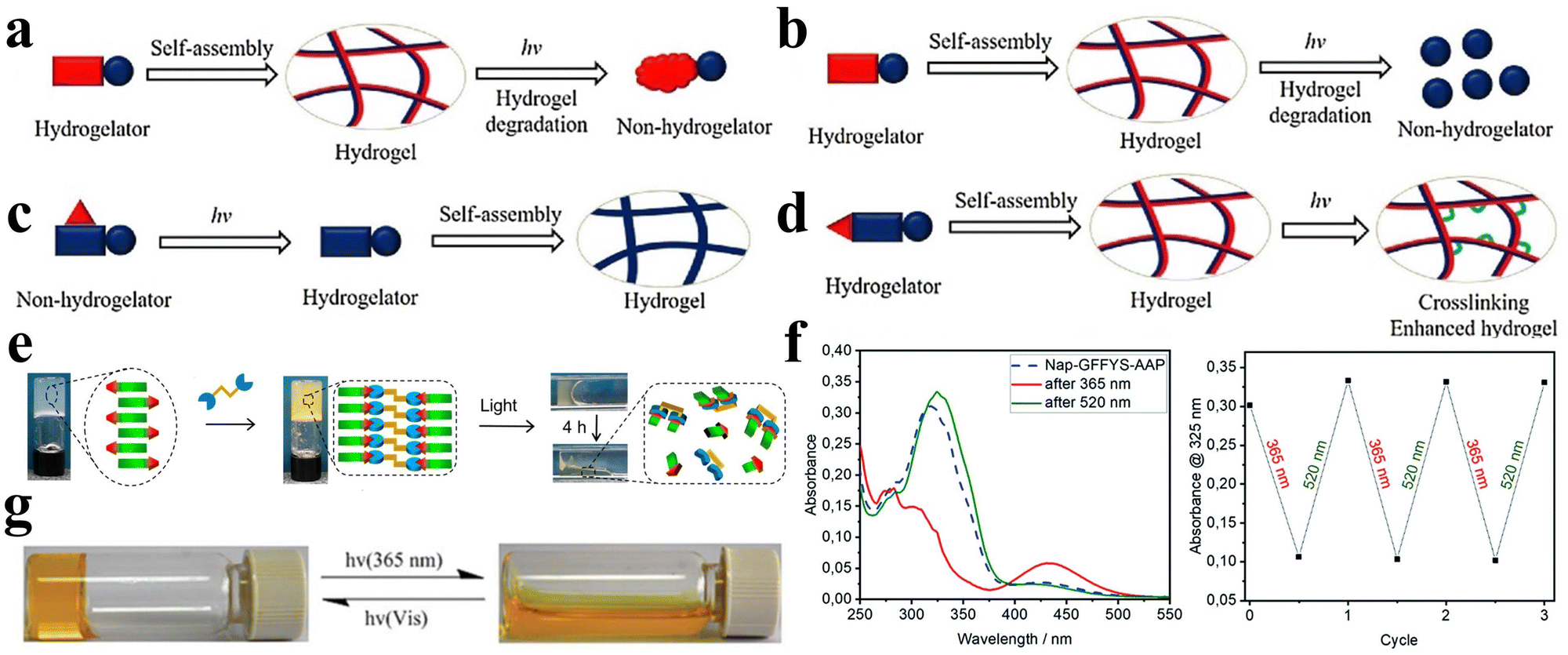
Hydrogels are soft substances with a 3D cross-linked polymer network, which have the exceptional ability to absorb and retain large amounts of water or other liquids without undergoing dissolution,1–3 which renders them closely akin to living tissues. This attribute bestows upon them great biocompatibility,3 rendering them suited for a diverse array of biomedical applications, spanning drug delivery, tissue engineering, anti-bacterial treatment, etc.4–9Stimuli-responsive hydrogels constitute an essential subclass of hydrogels. Within these systems, the polymer networks forming the hydrogel backbone incorporate stimuli-responsive units that enable the hydrogel to undergo controlled and reversible transitions between the gel and sol phases or between swelled and de-swelled states. Stimuli-responsive hydrogels represent an important class of functional materials that react to external stimuli such as temperature, pH, or light, thereby modulating their properties or behaviors correspondingly.10–14 Stimuli-responsive hydrogels can be constructed through weaker, reversible non-covalently crosslinked chains (e.g., hydrogen bonding, van der Waals forces, ionic bonding, or electrostatic interactions), and the stimuli can induce the formation or disappearance of these non-covalently crosslinked chains, resulting in a transition of the material between the solution state and the denser, more solid gel state. On the other hand, hydrogels are also capable of forming covalently crosslinked networks through stable chemical bonds (e.g., covalent bonds), which are capable of swelling or de-swelling when externally stimulated, but the overall network structure remains unchanged. Due to their intelligent responsiveness to environmental cues, these materials have sparked significant interest across various scientific domains and applications. For instance, specific physiological regions exhibit substantial pH fluctuations during normal function or in the presence of pathological states. Such variations within the human body can elicit either swelled or de-swelled responses in pH-responsive hydrogels, thereby facilitating the precise delivery of a spectrum of various therapeutic agents, encompassing proteins, small-molecule drugs, compounds, and genetic material, to anatomically precise locations.15 Temperature-responsive hydrogels adeptly exploit the thermal variance between external and internal milieu, effecting a transition from a soluble state to an insoluble one. This attribute facilitates their intracorporeal administration through injection, swiftly gelling under physiologic temperature conditions, thus serving as tissue regeneration scaffolds for treating various tissues and organs, including bone repair and damaged cardiac tissue repair.16
Frequently employed stimuli-responsive hydrogels encompass synthetic polymers such as polyethylene glycol (PEG),17 polyvinyl alcohol (PVA),18,19 or poly(2-hydroxyethyl methacrylate) (PHEMA),20,21 alongside naturally derived compounds such as agarose, alginate, chitosan, and gelatin.22–25 Although these conventional hydrogels are readily accessible, their limitations in terms of biocompatibility, biodegradability, tunability, and other features curtail their scope within the biomedical arena. Peptides, a category of short chains composed of amino acids, epitomize an optimal selection of materials for the construction of stimuli-responsive hydrogels due to their excellent, controllable self-assembly, and notable biological activity.26–30 Owing to their strong capability for supramolecular self-assembly through non-covalent interactions including hydrogen bonding, π–π stacking, hydrophobic effects, electrostatic forces, and van der Waals forces, peptides can self-assemble into nanostructures characterized by unique properties,31 which can form three-dimensional network-like structures to form peptide hydrogels.32–34 Furthermore, the inherent modifiability of peptides facilitates imparting stimulus-responsiveness to the resultant hydrogels. Peptide-based hydrogels can acquire varied properties through the incorporation of biologically functional groups into the peptide chain and by adjusting the composition of these hydrogels.35 Through modifying the amino acid sequence and controlling the self-assembly process, peptide-based hydrogels with specific stimulus responsiveness can be designed and synthesized, rendering them more intelligent in physiological environments and tailored to address distinct application scenarios. Therefore, peptide-based stimuli-responsive hydrogels introduce new dimensions and promising perspectives to the field of stimuli-responsive hydrogels.
Herein, we propose a review paper to summarize recent advancements in the design and applications of stimuli-responsive peptide hydrogels for biomedical purposes (Fig. 1). Firstly, this article gives a brief introduction to the synthesis, self-assembly mechanism, and properties intrinsic to peptide-based hydrogels. It aims to delineate the construction of stimuli-responsive peptide hydrogels, encompassing variables such as temperature, pH, light, enzymes, etc. Further, the review provides an overview of the diverse applications of stimuli-responsive peptide hydrogels in biomedical fields, spanning tissue engineering, drug delivery, wound healing, anti-bacterial, cell culture, and biosensors. Finally, it examines the combination of insights into current trends, remaining challenges, and future perspectives of stimuli-responsive peptide hydrogels in the realm of biomedical applications. We hope that this review will promote the advancement of innovative stimuli-responsive hydrogels, enriching the landscape of biomedical applications.
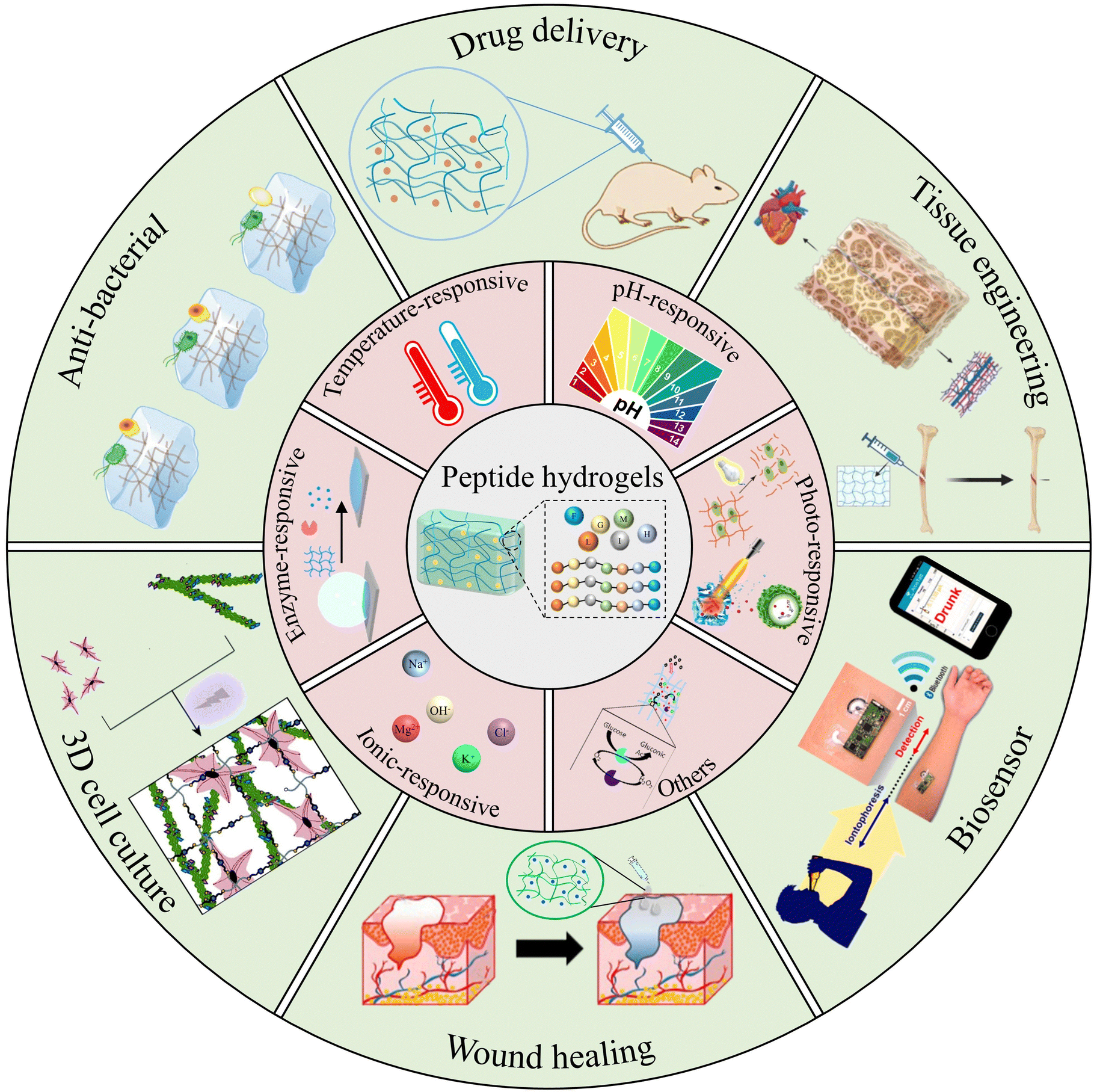 | ||
| Fig. 1 Peptide-based stimuli-responsive hydrogels and their various biomedical applications. Reproduced with permission from ref. 36–45. Photo-responsive:36 Copyright 2017, Elsevier B.V.37 Copyright 2019, Wiley-VCH GmbH. Enzyme-responsive:38 Copyright 2020, Springer Nature. Others:39 Copyright 2018, Royal Society of Chemistry. Drug delivery:40 Copyright 2019, Wiley-VCH GmbH. Tissue engineering:41 Copyright 2023, MDPI. Anti-bacterial:42 Copyright 2022, Royal Society of Chemistry. 3D cell culture:43 Copyright 2020, Royal Society of Chemistry. Wound healing:44 Copyright 2019, Elsevier B.V. Biosensor:45 Copyright 2016 American Chemical Society. | ||
2. Stimuli-responsive peptide hydrogels
Stimuli-responsive hydrogels constitute a distinct category of gel materials that can undergo reversible alterations in form or properties upon exposure to precise external stimuli.46 Peptides can respond to specific stimuli, such as temperature, pH, and ion concentration, by modulating the amino acid sequence. In recent years, peptide hydrogels have been extensively employed in the construction of intelligent hydrogel materials, leveraging their outstanding responsiveness to stimuli.2.1. Temperature-responsive
Temperature serves as one of the most prevalent stimuli triggering structural changes in peptide hydrogels. Through precise fine-tuning of the arrangement of peptide molecules, hydrogels can be engineered to shift between states of expansion and contraction within specific temperature windows. This section reviews the research progress in temperature-responsive peptide hydrogels in recent years. It focuses on the influence of temperature as a critical stimulus on the solution–gel transition, structure assembly, mechanical properties, and other aspects of peptide hydrogels and their regulatory mechanisms. Temperature-responsive hydrogels are divided into two main categories, namely, positively responsive hydrogels with upper critical solution temperature (UCST) and negatively responsive hydrogels with lower critical solution temperature (LCST).47 Positively responsive hydrogels exhibit miscibility with water when the temperature is above the UCST, with the molecules in a non-crosslinked state and the entire system presented as a solution phase, whereas when the temperature falls below the UCST, the polymer becomes increasingly hydrophobic and insoluble, and the molecules crosslink with each other to form a gel. The opposite holds true for negatively responsive hydrogels. Given that 37 °C represents the prevailing physiological temperature within the human body, numerous intrinsic control mechanisms are linked to this temperature. Consequently, temperature-responsive peptide hydrogels have consistently been the subject of profound scrutiny.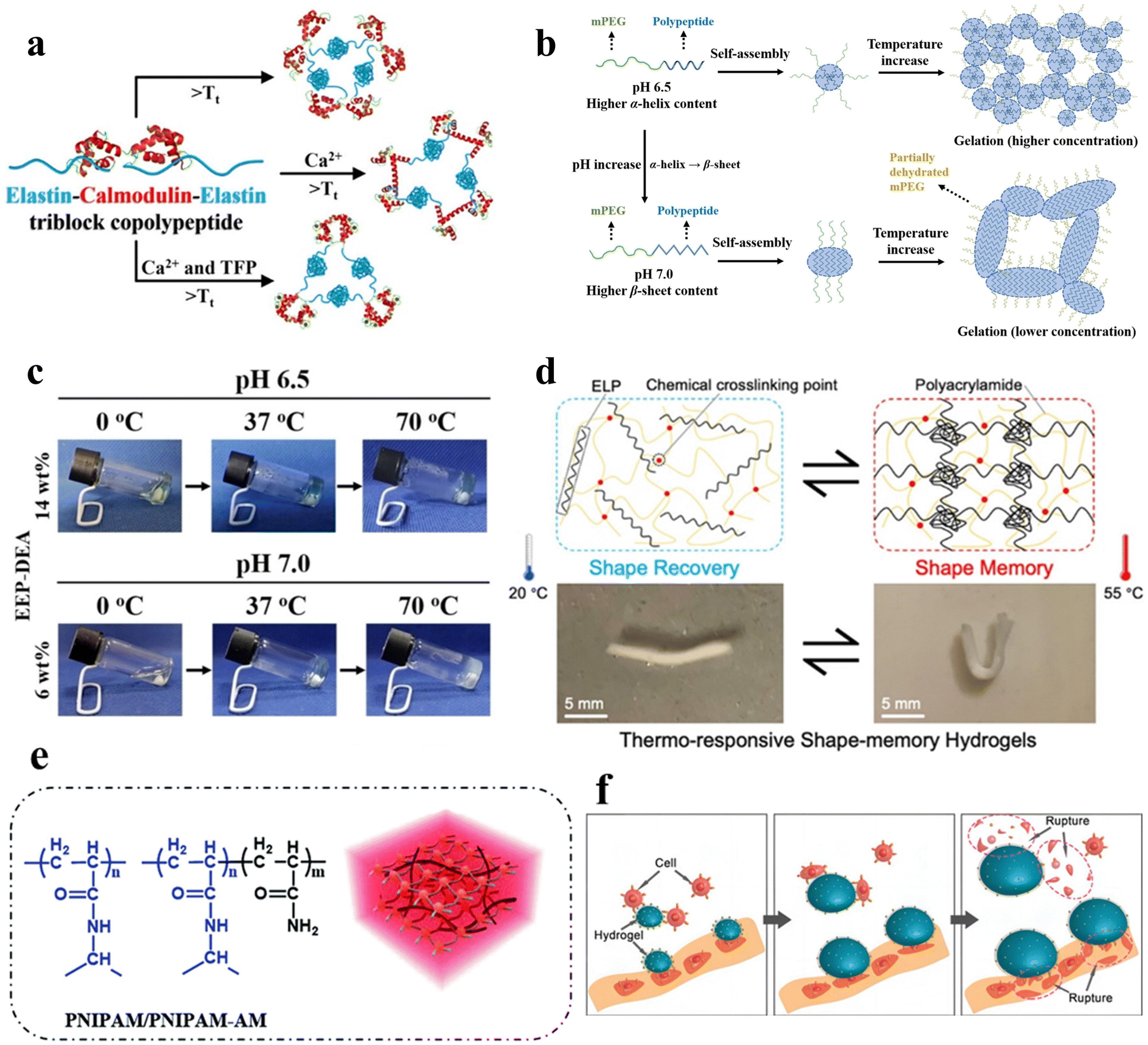 | ||
| Fig. 2 Thermo-responsive peptide-based hydrogels. (a) Schematic of the molecular design of the ECE triblock copolypeptides peptide, the temperature-triggered sol–gel transition of the ECE triblock copolymerization peptide, and the conformational alteration of CaM upon ligand binding.50 Copyright 2022 American Chemical Society. (b) Schematic illustration of the secondary structure transitions, self-assembly process, and thermo-induced gelation of EEP-diethylamine (DEA) aqueous solutions at pH 6.5 and 7.0, respectively. Left: Secondary structure of EEP-DEA at pH 6.5 and 7.0; middle: self-assembled micelles formed by EEP-DEA at pH 6.5 or 7.0; right: gelation of EEP-DEA induced by temperature increase. (c) Pictures of the sol–gel phase transition of EEP-DEA in PBS at pH 6.5 or 7.0.53 Copyright 2023 Wiley-VCH GmbH. (d) Schematic of the structural changes in the shape memory hydrogel ELP/PAM at high and low temperatures and photographs of the shape memory function.54 Copyright 2020 American Chemical Society. (e) Schematic illustration of the chemical structure and hydrogel network of PNIPAM/PNIPAM-acrylamide.56 Copyright 2018 Royal Society of Chemistry. (f) Mechanism of physical destruction of cancer cells by the RGD-PNIPAM hydrogel. Cancer cells adhere to the surface of the stimulus-responsive hydrogel; upon temperature stimulation, the hydrogel undergoes swelling, leading to the rupture of cancer cells.57 Copyright 2018 Springer Nature. | ||
Mizuguchi et al. utilized temperature-responsive elastin-like polypeptides (ELPs), polyaspartic acid (polyD) chains, helical peptides, and biofunctional peptides to engineer hydrogels.51 These hydrogels exhibited controllable sol–gel transitions and adjustable mechanical properties. With increasing temperature, elastin undergoes hydrophobic aggregation and forms non-covalent cross-linking points, which gels the solution. In the gel state, the four-arm structure of elastin forms a three-dimensional network through end-to-end connections, which gives the gel a particular mechanical strength. The mechanical properties can be adjusted by changing the temperature and protein concentration. polyD segments can adsorb troponin and pro-angiogenic factors through electrostatic interaction to achieve synergistic regulation of cellular behaviors. The temperature-responsive shape memory hydrogel material holds promising applications in the development of programmable intelligent biomedical materials.
Furthermore, polyisocyanopeptide (PIC) hydrogels represent a novel class of synthesized temperature-responsive non-covalently crosslinked hydrogels. They maintain a liquid state below 16 °C and seamlessly transition into a gel state at or above room temperature.52
In addition, Zhang et al. engineered shape-memory hydrogels, denoted as V250/PAM, based on a double-network polymer structure constructed through the utilization of physically cross-linked ELP in conjunction with chemically cross-linked polyacrylamide (PAM).54 These hydrogels were synthesized via ultraviolet (UV) irradiation at 20 °C. After molding them into a specific shape and subjecting them to a temperature of 55 °C while immersed in water, a “memory” phenomenon was induced within the hydrogels. Reverting the hydrogels to 20 °C allowed the initial shape to be reinstated, effectively demonstrating repeatability in the shape-memory capability (Fig. 2d).
Poly(N-isopropylacrylamide) (PNIPAM) hydrogels represent a widely used class of temperature-responsive non-covalently crosslinked hydrogels in aqueous environments (Fig. 2e). The polymer chain contains hydrophilic (amide) and hydrophobic (isopropyl) groups. At low temperatures, hydrogen bonding between the polymer chain and water molecules causes it to take on a ductile conformation. As the temperature increases, the entropy effect weakens the hydrogen bonding, and water molecules are expelled from the gel. The polymer chain exhibits a contracted spherical conformation, enabling the PNIPAM gel to undergo tightening and swelling phase transitions upon temperature changes. Significantly, PNIPAM's LCST closely approximates that of the human body, affording adjustable attributes that greatly enhance its potential across a diverse array of applications.55,56 Fang et al. developed a temperature-responsive non-covalently crosslinked hydrogel by encapsulating arginine–glycine–aspartic acid (RGD) peptides within PNIPAM, facilitating targeted adhesion of cancer cells.57 Through temperature modulation, the hydrogel undergoes swelling in response to stimuli, resulting in the rupture of adherent cancer cells (Fig. 2f).
Temperature-responsive peptide-based hydrogels demonstrate distinctive gelation behavior in response to temperature variations, rendering them invaluable for biomedical applications. The underlying mechanism involves the conformational changes of peptides within the hydrogel structure, responding to temperature alterations, and influencing the formation or dissolution of the gel network. Typically, these peptides exhibit a LCST, undergoing a phase transition between gel and solution. The preparation methods for temperature-responsive peptide-based hydrogels involve the self-assembly of peptides using various techniques, such as physical cross-linking and chemical cross-linking. Despite their promising features, temperature-responsive peptide-based hydrogels encounter certain limitations, including a restricted temperature responsiveness range, potential instability under specific conditions, and complex, costly synthesis. Furthermore, issues related to the speed of response to temperature changes and the adaptability of these hydrogels across diverse biomedical applications remain active areas of research and improvement. Through ongoing optimization and design of peptide structures, as well as the introduction of various functional molecules, researchers can develop temperature-responsive hydrogels that are more efficient, stable, and sensitive. This advancement holds great potential to propel the application of peptide-based hydrogels in biomedical fields such as targeted drug release.
2.2. pH-responsive
pH serves as a readily controllable and extensively investigated stimulus, with variations in pH within different regions and physiological states of the human body.47 Furthermore, pH influences both the amino and carboxyl groups inherent in the amino acid residues of peptide sequences.58 The construction of pH-responsive peptide hydrogels can achieve the sensing of different pH in vivo to control the assembly and release of peptide chains and control gel formation and dissolution. Therefore, the study of pH-responsive peptide hydrogels has attracted much attention. The main mechanisms affecting the pH responsiveness of peptide hydrogels include the charge effect and conformational effect. The charge effect uses the protonation/deprotonation of acidic amino acid side chains in the peptide chain to change the overall charge state. In contrast, the conformational effect controls the assembly by designing peptide chains with variable conformations. Understanding these mechanisms is essential for the design of pH-responsive peptide hydrogel systems. In this section, a comprehensive review of the current status of pH-responsive peptide hydrogels is presented to provide a reference for the rational design and development of highly pH-sensitive peptide-based smart gel materials.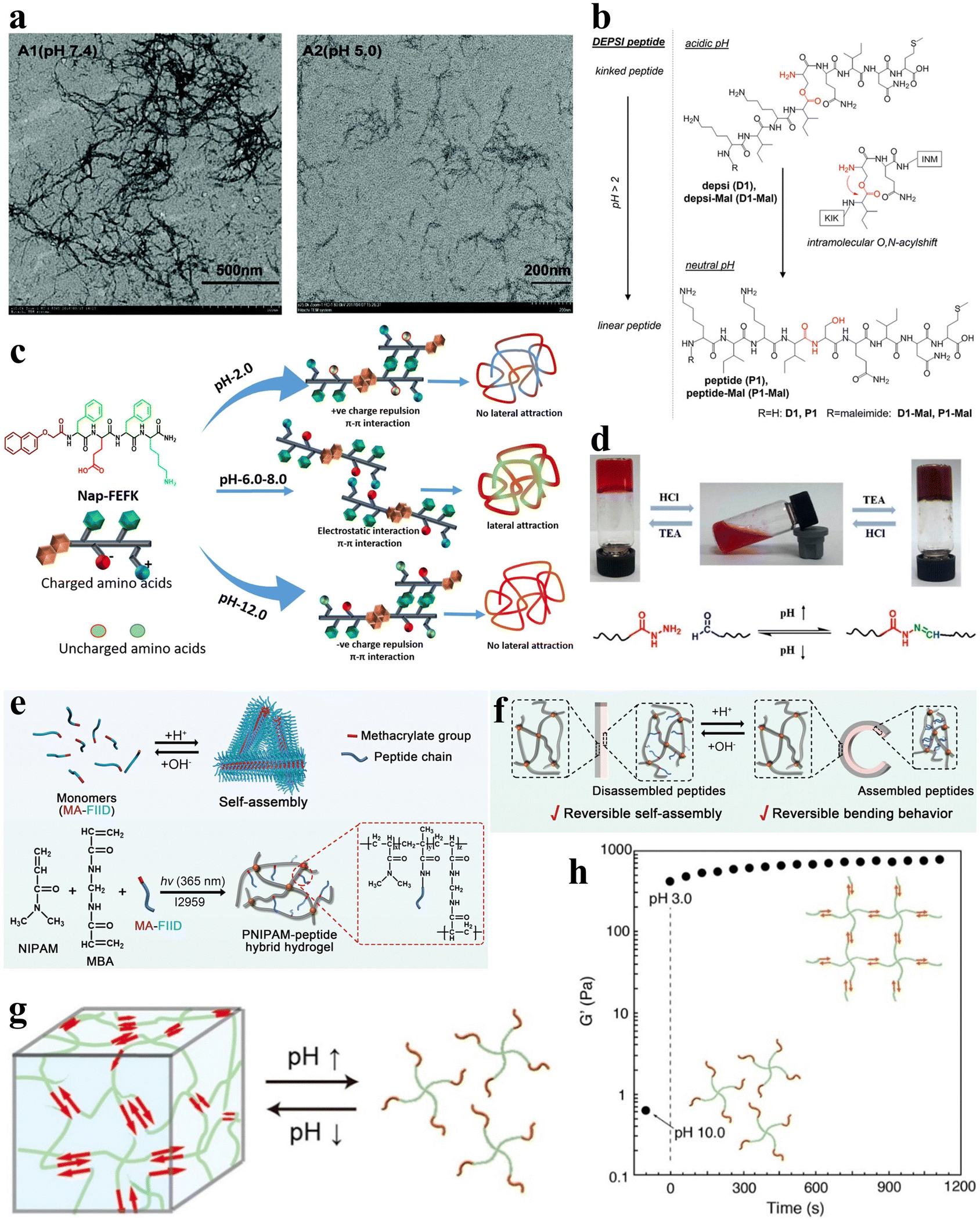 | ||
| Fig. 3 pH-responsive peptide-based hydrogels. (a) TEM images of negatively stained fibrils in aged fibril solutions of F-4 at pH 7.4 (left) and 5.0 (right).39 Copyright 2018 Royal Society of Chemistry. (b) pH-induced intramolecular O–N–acyl migration of the Depsi peptides yielding linear peptides.60 Copyright 2018 Wiley-VCH GmbH. (c) Chemical structure of NAP-FEFK and schematic of pH-controlled self-assembly behavior. pH controls the overall surface charge and interactions of peptides, leading to the formation of different self-assembled structures.65 Copyright 2020 American Chemical Society. (d) Schematic of pH-responsive sol–gel transition in hydrogels. The reversible sol–gel transition of hydrogels is achieved through the reversible breaking and reconstruction of acylhydrazone bonds in response to changes in pH levels.66 Copyright 2019 American Chemical Society. (e) Schematic of the preparation and network structure of PNIPAM/MA-FIID peptide hybrid hydrogels. (f) Schematic illustrating the reversible macroscopic shape deformation of a bilayer hydrogel in response to pH fluctuations.67 Copyright 2023 Wiley-VCH GmbH. (g) Schematic illustration of pH-responsive hydrogel formation through self-assembly of star polymers. (h) Time-scan curve of the G' value for the peptide-PEG hybrid star polymer hydrogel (2 wt%) showing rapid sol-to-gel transition as pH decreases from 10.0 to 3.0.68 Copyright 2022 American Chemical Society. | ||
Gačanin et al. synthesized non-covalently crosslinked hybrid hydrogels with unique rheological properties based on a Depsi peptide through pH-controlled structural transformation.60 Under physiological conditions, the Depsi peptide was in a curved conformation at low pH and could not self-assemble into peptide nanofibers (PNFs). As the pH increases to neutral, acyl migration occurs, transforming the ester bond within the peptide chain into a peptide bond, which turns the Depsi peptide into a linear one and allows it to self-assemble into β-sheeted PNFs. This structural transformation transforms the Depsi peptide from an unstable conformation into a β-sheeted fibrous structure, which is the key to forming the gel from this material. In other words, pH induces the conformational change of the Depsi peptide, which triggers the gelation (Fig. 3b). The self-assembly of oligopeptide sequences into nanoscale structures holds great potential for various applications.61–63 Clarke et al. systematically examined three distinct pentapeptide sequences characterized by variations in charge distributions.64 In an alkaline aqueous solution with a pH of 10, the peptides were dissolved and subsequently underwent gelation via non-covalent interactions upon neutralization with HCl to attain a neutral pH.
Incorporating pH-responsive peptides into hydrogels with diverse substrates proves to be an effective strategy for designing and synthesizing pH-responsive hydrogels. Kaur et al. utilized the classical non-gelling factor, N-terminal-conjugated aromatic group naphthoxyacetic acid (Nap), with an ion-complementary sequence FEFK, imparting pH-responsive gelation behavior, and the hydrogels were constructed through non-covalently crosslinked chains.65 Notably, distinct nanoscale gel structures are evident under different pH conditions. Extreme pH values (2.0 and 12.0) induce thinner fiber formation due to the charged state, while thicker fibers are apparent around physiological pH (Fig. 3c).
Xiang et al. developed a pH-responsive peptide, methylacrylated-FIID (MA-FIID), which was integrated into a PNIPAM framework (Fig. 3e).67 The FIID sequence can self-assemble at neutral pH by non-covalent interactions such as hydrophobic interaction, hydrogen bonding, and π–π stacking, and aspartic acid deprotonation when pH decreases weakens the aggregation effect and depolymerizes the gel. By harnessing the assembly of peptide molecules in varying pH environments, they successfully fabricated bilayer and heterogeneous hydrogel actuators. These hydrogel actuators enable controlled bending, flexing, and intricate deformations in response to pH stimulation (Fig. 3f). Koga et al. introduced a novel four-arm self-assembling peptide-PEG hybrid star polymer hydrogel with non-covalent crosslinks. The polymer demonstrated a rapid and highly responsive behavior to variations in pH, undergoing gelation within mere seconds following the pH alteration (Fig. 3g and h).68 Drawing inspiration from histidine-rich cross-linking domains present in marine mussel byssus proteins,69 Rammal et al. engineered a covalently crosslinked gel by combining the mussel-inspired peptide HRD-NGN (N-GHGGGHGGGHGGGHGGSASAAAHAACONH2) with PEG.70 This gel, crafted under alkaline pH conditions, forms a scaffold characterized by microporosity, self-healing properties, and elasticity. Remarkably, the aggregated granular hydrogels can be disassembled in the presence of a metal chelator or under acidic circumstances.
Furthermore, the utilization of covalent ion coordination/oxidative cross-linking emerged as a viable approach for synthesizing pH-responsive peptide hydrogels. Pham et al. employed enzymatic cross-linking or coordinated/oxidized cross-linking with Fe3+ ions, culminating in the successful synthesis of L-dopa-conjugated (GPLD) peptides, resulting in the formation of pH-responsive peptide hydrogels.71
pH-Responsive peptide-based hydrogels constitute a distinctive class of materials that undergo structural changes in response to variations in environmental pH. These pH-induced structural transformations prompt the peptide hydrogels to exhibit distinct functions in various physiological environments, such as the tumor microenvironment, characterized by a lower pH than normal tissues. Therefore, pH-responsive peptide-based hydrogels hold vast potential applications in drug delivery. The fundamental mechanism involves the ionization of amino acid residues within the peptide sequence, resulting in modifications to the charge of the hydrogel, and consequently, its conformational state. These alterations induce either gelation or dissolution, presenting a reversible and tunable response to pH fluctuations. The methods for crafting pH-responsive peptide-based hydrogels typically involve the design of peptides with pH-sensitive amino acid residues, such as histidine, or acidic residues like glutamic acid and aspartic acid. Including these residues confers pH sensitivity to the hydrogel, enabling it to adapt to changes in surrounding acidity or alkalinity. However, pH-responsive peptide-based hydrogels encounter certain limitations, including a confined pH range for optimal responsiveness, potential instability under extreme pH conditions, concerns regarding long-term stability, and challenges to achieving precise control over gelation properties. Additionally, issues such as biocompatibility, scalability of synthesis, and applicability in specific biomedical contexts remain active areas of investigation and refinement.
2.3. Ionic-responsive
Ions such as calcium, sodium, potassium, and chloride ions are abundant in nature and are widely distributed in biological systems. They can interact with specific peptide hydrogels, recognizing individual peptide sequences, thereby facilitating intermolecular cross-linking and promoting the self-assembly of peptides into structurally stable frameworks.58 Consequently, ionic-responsive peptide hydrogels hold significant value for extensive research.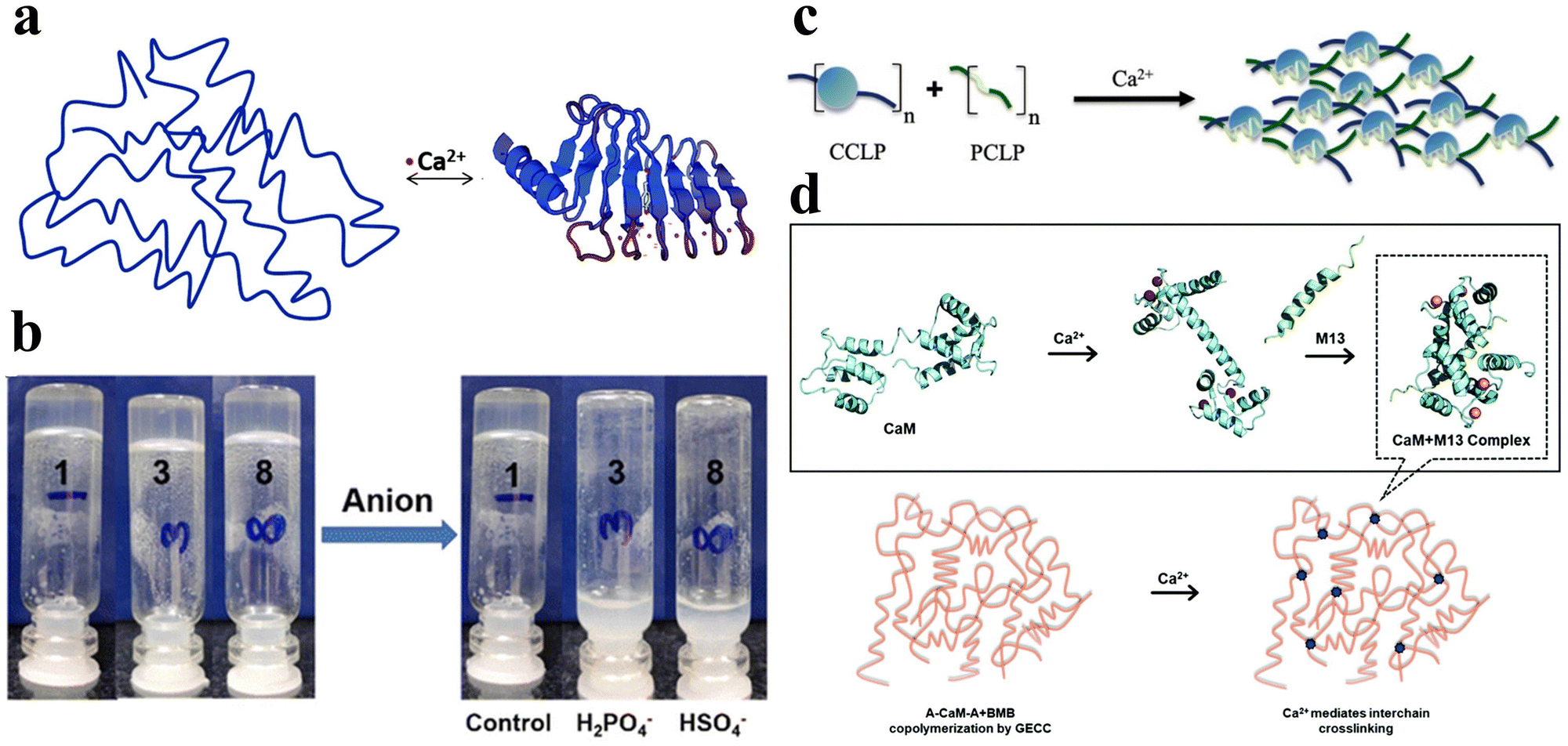 | ||
| Fig. 4 Ionic-responsive peptide-based hydrogels. (a) Schematic of the β-roll structure of RTX upon binding to Ca2+. Initially, the peptide sequence was disordered. Upon the addition of Ca2+, the RTX underwent a reversible conformational change to form the β-roll structure.72 Copyright 2018 Royal Society of Chemistry. (b) Effect of the addition of H2PO4− and HSO4− anions on supramolecular hydrogels formed by NapF-TREN.74 Copyright 2022 Royal Society of Chemistry. (c) Schematic of the CaM block polymer (blue circle) and M13 block polymer (green line) interacting in the presence of calcium ions to form a physically cross-linked hydrogel.75 Copyright 2020 American Chemical Society. (d) Schematic illustration of Ca2+-responsive protein polymers resulting from the SpyTag–ELP–CaM–ELP–SpyTag (A-CaM-A) and SpyCatcher–ELP–M13–ELP–SpyCatcher (BMB) reaction. In the absence of Ca2+, CaM adopts a closed conformation; upon the addition of Ca2+, CaM undergoes a conformational change into a dumbbell-like extended shape and wraps around the M13 domain.76 Copyright 2020 Royal Society of Chemistry. | ||
Tsutsumi et al. developed an amphiphilic peptide, E1Y9 (Ac-E-YEYKYEYKY-NH2), which self-assembles into nanofibers and forms a non-covalently crosslinked hydrogel in the presence of Ca2+.73 To expand its functionality, four derivatives of E1Y9 were designed, namely E1Y9-ALK (ALKRQGRTLYGF), E1Y9-DGR (DGRDSVAYG), E1Y9-PRG (PRGDSGYRGDS), and E1Y9-RGD (RGDS). These derivatives, along with E1Y9 itself, exhibit co-assembly behavior, forming a mesh-like nanofiber network that ultimately results in the formation of a responsive hydrogel upon interaction with Ca2+. Peptide hydrogels can be purposely designed to respond not only to metal cations but also to anions.
Peptide hydrogels can be designed to respond not only to metal cations but also to anions. In a separate study, Pradhan et al. designed an amphiphilic molecule comprising of phenylalanine and tris(aminoethyl)amine (TREN), NapF–TREN.74 The injectable hydrogel with non-covalent crosslinks formed by this molecule can be disrupted by monovalent anions such as H2PO4− and HSO4− (Fig. 4b).
Ion-responsive peptide-based hydrogels constitute a specialized class of materials designed to undergo structural changes in response to variations in ionic strength or the presence of specific ions in the surrounding environment. The fundamental principle involves the electrostatic interactions between charged amino acid residues within the peptide sequence and ions in the solution, inducing conformational changes that lead to either gelation or dissolution. This ion-induced modulation of hydrogel properties imparts reversible and stimuli-responsive behavior. The methods for constructing ion-responsive peptide-based hydrogels often entail the incorporation of ion-binding motifs or charged amino acid residues into the peptide sequence. These motifs may encompass metal-coordinating groups like histidine, cysteine, or other ion-binding moieties conferring sensitivity to specific ions. Despite their potential utility, ion-responsive peptide-based hydrogels encounter certain limitations, including dependence on specific ion types for responsiveness, potential interference from complex biological environments, challenges in achieving precise control over gelation kinetics, and concerns about the stability of the hydrogel under prolonged exposure to ions. Scalability for large-scale applications also remains the subject of ongoing research.
2.4. Photo-responsive
Light is a widely employed stimulus for precise control over the shape deformation of hydrogels. Peptides possess complex optical properties including photocatalysis, photothermal effects, and photodynamic effects.77 Photo-responsive peptide hydrogels have attracted considerable attention due to their dramatic volume changes induced by non-contact light irradiation, exhibiting high spatial and temporal resolution. Hydrogels can exploit various light-responsive mechanisms. These include photoisomerization/ionization reactions, which modify the gel network, as well as the incorporation of light-absorbing nanoparticles that transform light energy into thermal energy, inducing a phase transition.78 Additionally, precise regulation of the microscopic morphology of the gel can be achieved through the control of the formation and cleavage of non-covalent cross-links, and the utilization of photogenerated reactive species initiates photosensitized rupture of the gel network, enhancing the range of applications for these innovative materials.Various types of photoreactive peptide hydrogels can be synthesized by covalently conjugating diverse photosensitive moieties with peptides. These include photoisomerization hydrogels, which undergo structural changes upon light exposure, photocaging or photocleavage hydrogels that release or activate specific molecules upon light irradiation, and photo-cross-linking hydrogels that form stable networks in response to light (Fig. 5a–d).79 One such molecule capable of photoisomerization is 4,4′-azopyridine (APY), which contains azobenzene groups. Ji et al. co-assembled the dipeptide carboxybenzyl-protected diphenylalanine (ZFF) with APY to create a non-covalently crosslinked peptide hydrogel exhibiting photo-responsive characteristics.33 The photo-responsiveness of the ZFF/APY gel was examined by UV irradiation. Following 4 hours of UV irradiation, the ZFF/APY gel underwent collapse, whereas it remained stable when subjected to 4 hours of visible light irradiation (Fig. 5e). This is due to the fact that UV illumination induces a conformational change in bipyridine, which disrupts the co-assembled gel network and induces a gel-to-solution transition. This demonstrates that the ZFF/APY hydrogel displays reversible photo-induced volume changes in response to different wavelengths of light. The incorporation of the photoisomerizable APY conferred tunable photosensitivity and light-triggered actuation to the peptide hydrogel. This co-assembly approach enables the facile preparation of peptide hydrogels with stimuli-responsive properties. Nowak et al. reported a photo-responsive supramolecular hydrogel using an aromatic arylazopyrazole (AAP) modified pentapeptide gelator Nap-GFFYS and β-cyclodextrin vesicles (CDV).80 The chemical cross-linking resulting from the host–guest interaction of the AAP moiety and CDV makes the gel photo-responsive. Upon separate irradiation with UV and visible light, the host–guest interactions between the AAP moiety and the hydrophobic cavity of β-CD can be selectively deactivated, leading to a reversible reduction in the stiffness of the hydrogel (Fig. 5f). Li et al. proposed a novel approach to trigger the self-assembly of peptide hydrogels, serving as initiators and cross-linking agents for the resulting nanocomposite hydrogels to form supramolecular hydrogels.81 Carbon nanotubes (CNTs) were employed due to their photothermal behavior under near-infrared light (NIR), enabling the NIR-induced release of encapsulated contents. Diverse amyloid templates can be utilized as dynamic self-propagating templates to construct complex functional materials.82,83
 | ||
| Fig. 5 Photo-responsive peptide-based hydrogels. (a)–(d) Schematic illustrations of a (a) photo-isomerization hydrogel, (b) photo-cleavage hydrogel, (c) photo-cage hydrogel, and (d) photo-cross-linking hydrogel.79 Copyright 2018 Elsevier B.V. (e) Stimuli-responsive images of the ZFF/APY hydrogel under light irradiation. ZFF/APY gel collapsed by UV irradiation.33 Copyright 2021 American Chemical Society (f) UV/vis spectra and reversible photo isomerization of the E- and Z-isomers of Nap–GFFYS–AAP (0.1 mM) in 20 mM HEPES buffer (pH = 8.01).80 Copyright 2019 Royal Society of Chemistry. (g) Optical images and structural alterations during the photo-induced phase transition of azo–Gln–Phe–Ala hydrogels.86 Copyright 2011 Royal Society of Chemistry. | ||
Ji et al. selected the extensively studied diphenylalanine with Fmoc protection (FmocFF) as an ultrashort amyloid model and utilized 1,2-bis(4-pyridyl)ethylene (BPE) to co-assemble with FmocFF and prepared a non-covalently crosslinked photo-responsive hydrogel material.32 The co-assembled gel displayed no detectable fluorescence under 365 nm excitation prior to UV irradiation, whereas it exhibited significantly enhanced fluorescence after 1 h of UV irradiation.
Furthermore, azobenzene, a conventional conformational switch, was used to construct photo-responsive polymer hydrogels.84,85 Huang et al. investigated photo-responsive hydrogels based on azobenzene-substituted short peptides (Fig. 5g).86 Their study demonstrated that the judicious combination of azobenzene moieties and peptide sequences effectively established various non-covalently crosslinked photo-responsive supramolecular hydrogel systems with diverse functionalities and applications.
Photo-responsive peptide-based hydrogels belong to a distinct class of materials capable of undergoing structural or volume changes in response to light stimuli, providing control over their properties. The underlying principle involves integrating photoactive moieties, such as azobenzene or spiropyran, into the peptide sequence. Upon exposure to a specific wavelength of light, these moieties undergo reversible photoisomerization, inducing changes in the peptides and subsequently the gel–sol phases or swelled and de-swelled state transition of the hydrogel. The methods for constructing photo-responsive peptide-based hydrogels typically encompass the synthesis of peptides with covalently linked photoreactive groups. The choice of the photoactive moiety and the specific design of the peptide sequences are critical for achieving precise control over the photo-responsive behavior. However, current photo-responsive peptide-based hydrogel technology faces certain limitations. These include challenges related to achieving optimal spatial and temporal control of gelation, potential cytotoxicity associated with specific photoactive moieties, and the limited penetration depth of light in biological tissues.
2.5. Enzyme-responsive
Enzymes, as profoundly vital biomolecules, play indispensable roles in numerous metabolic processes and essential biological functions within the human body. Therefore, enzymes themselves offer potential as bio-stimulators. Simultaneously, through rational design, peptide hydrogels can directly interact with enzymes and exhibit responsive behaviors, thus possessing enzyme responsiveness. Owing to the remarkable substrate specificity of enzymes and their propensity for overexpression in certain tissues, enzyme-responsive peptide hydrogels find extensive biomedical applications.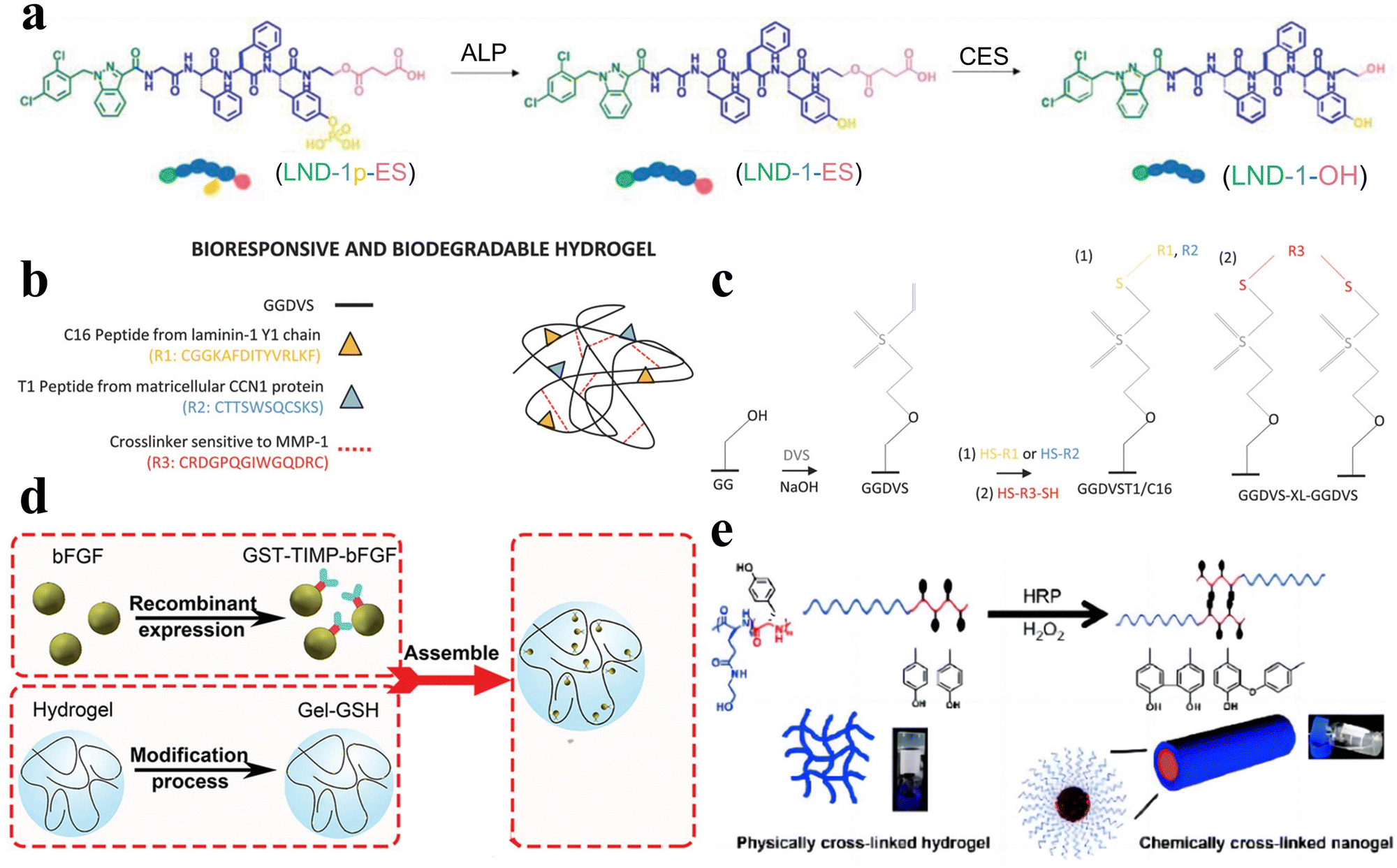 | ||
| Fig. 6 Enzyme-responsive peptide-based hydrogels. (a) Schematic illustrations of enzymatic transformations of LND-1p-ES. ALP first catalyzes the conversion of LND-1p-ES to LND-1-ES, and then CES further catalyzes the hydrolysis of the ester bond in LND-1-ES to produce the potent hydrogelator LND-1-OH.88 Copyright 2021 Wiley-VCH GmbH. (b) Schematic of GG biofunctionalisation and GGDVS hydrogel formation. (c) Schematic diagram of chemical modification of GG polymer chains by DVS and biofunctionalisation with cell adhesion peptide sequences.90 Copyright 2018 Wiley-VCH GmbH. (d) Schematic of MMP-responsive hydrogel preparation. GST-TIMP-bFGF was prepared by the recombinant protein expression method, and Gel-GSH was prepared by the chemical cross-linking method, and the two components were connected by the bond between GST and GSH.92 Copyright 2019 Wiley-VCH GmbH. (e) Schematic of the transformation of physically cross-linked hydrogels to chemically cross-linked nanogels by HRP.95 Copyright 2018 Science China Press. | ||
Peptide hydrogels that exhibit responsiveness to MMPs can also be deliberately tailored to respond to various other enzymes. For example, Hu et al. synthesized an unconventional amino acid, 2-nitroimidazol-1-ylalanine, through a Mitsunobu reaction involving 2-nitroimidazole and serine derivatives.93 They subsequently employed this amino acid in the development of a nitroreductase (NTR)-responsive peptide-based supramolecular hydrogel with non-covalent crosslinks. Zhou et al. developed an elastase-responsive hydrogel by covalently cross-linking a blend of carboxymethyl cellulose and PEG with an elastase-degradable peptide.94 Upon elastase introduction, the hydrogel transitioned from a gel to a sol state, resulting in the release of the encapsulated cargo.
Furthermore, Liu et al. engineered a series of nonionic peptide amphiphiles (PAs) using sequential ring-opening polymerization (ROP) of γ-benzyl-L-glutamic acid N-carboxyanhydride (BLG-NCA) and L-tyrosine N-carboxyanhydride (Tyr-NCA).95 These PAs self-assembled into non-covalently crosslinked peptide hydrogels. The incorporation of tyrosine residues imparted enzymatic responsiveness to the hydrogel. Under physiological conditions in the presence of horseradish peroxidase (HRP), the hydrogel transitioned from a hydrogel to a nanogel structure (Fig. 6e).
Enzyme-responsive peptide-based hydrogels constitute a specialized class of materials designed to undergo structural changes in the presence of specific enzymes, offering targeted and controlled responses in the biological environment. The fundamental principle involves incorporating enzyme-sensitive peptide sequences in the hydrogel structure, often susceptible to cleavage by proteases or other enzymes. Upon exposure to the target enzyme, these peptide sequences undergo enzymatic cleavage, inducing alterations in the conformation or degradation of the hydrogels. Methods for constructing enzyme-responsive peptide-based hydrogels typically revolve around the design and synthesis of peptide sequences featuring specific enzyme-sensitive motifs. The incorporation of peptide sequences serving as substrates for enzymes facilitates a controlled and triggered response. Despite their potential applications, enzyme-responsive peptide-based hydrogels encounter certain limitations. These include challenges in achieving precise control over the rate of enzymatic cleavage, potential interference from complex biological environments, and concerns related to the stability and specificity of the enzymatic response. Additionally, apprehensions regarding the potential immunogenicity or cytotoxicity of the peptide sequences and their degradation products may impact their biomedical applications.
2.6. Others
In addition to the aforementioned stimuli, essential biological signaling molecules, including glutathione (GSH), nitric oxide (NO), and hydrogen sulfide (H2S), can also function as external triggers, initiating the gel–sol phase transition in hydrogels (Fig. 7a).96 Aside from these bioactive molecules, common endogenous compounds like reactive oxygen species97,98 and glucose99 can also act as stimuli, inducing analogous responses in peptide hydrogels (Fig. 7b). The controlled application of an external magnetic field can be harnessed to selectively modulate the physical characteristics of the hydrogel and facilitate the transition from gel to sol phase (Fig. 7c).100,101 Likewise, oxidative-reductive stimuli offer a means to fine-tune various properties of peptide hydrogels (Fig. 7d and e).32,59,102,103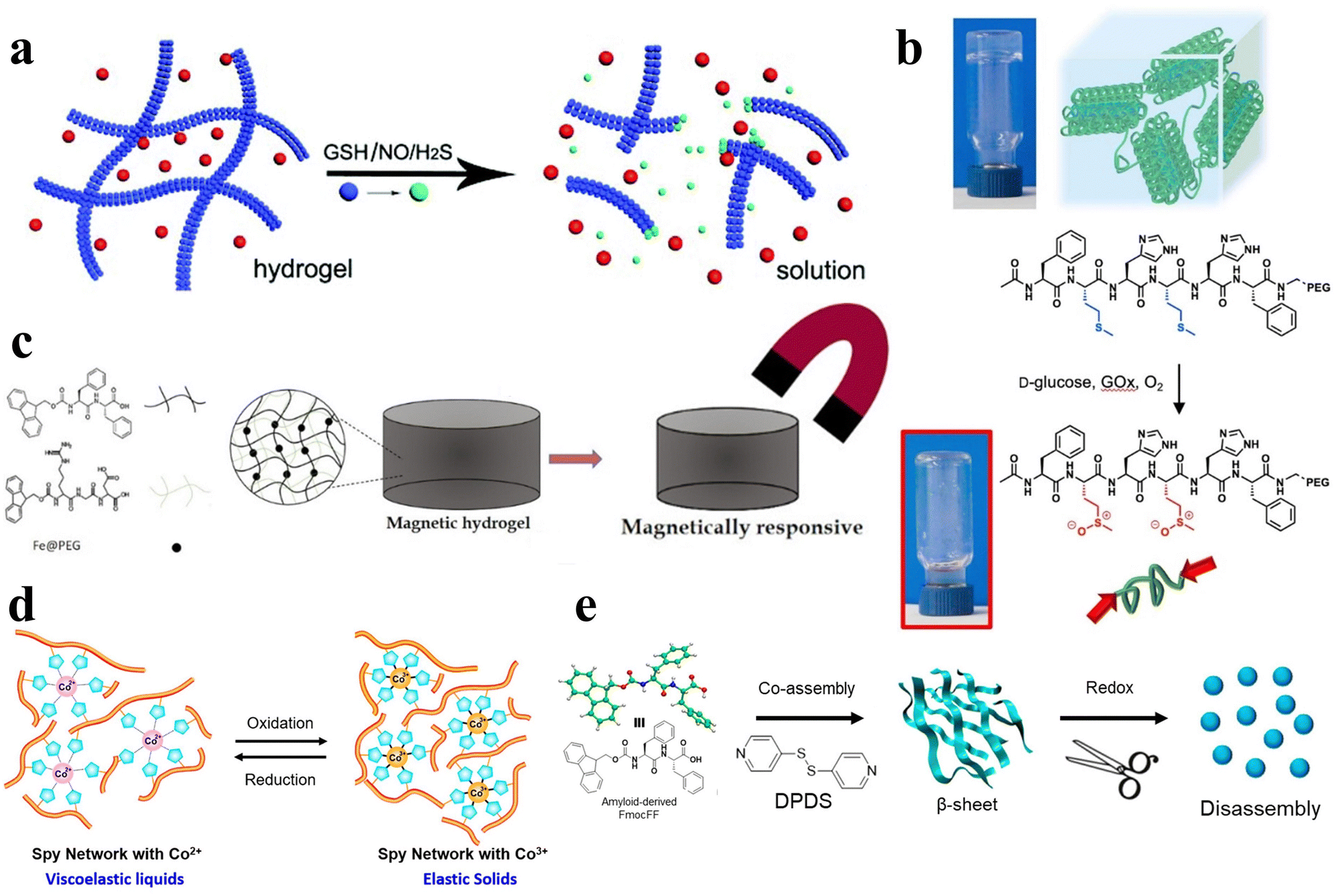 | ||
| Fig. 7 Various stimulus responsive peptide-based hydrogels. (a) Schematic of the gel–sol phase transition of the stimuli-responsive peptide hydrogel stimulated by GSH, NO, and H2S.96 Copyright 2018 Royal Society of Chemistry. (b) Schematic of oxidation-mediated decomposition of supramolecular hydrogels.97 Copyright 2018 Elsevier Ltd. (c) Schematic of the properties of short peptide supramolecular magnetic hydrogels.100 Copyright 2022 American Chemical Society. (d) Schematic of the reversible redox response of Spy network protein hydrogels.102 Copyright 2019 American Chemical Society. (e) Schematic of 4,4′-dipyridyl disulfide (DPDS) inducing a secondary structure transformation of FmocFF amyloid-like self-assembly into a redox-responsive hydrogel through co-assembly.32 Copyright 2020 American Chemical Society. | ||
Despite their potential applications, these hydrogels encounter specific limitations. Challenges encompass achieving high specificity in recognizing complex biological signals, ensuring both a controlled and reversible response, and addressing potential interference from other components within biological environments.
In summary, this section summarizes the effects of various external stimuli on the formation of peptide hydrogels that can respond to specific stimuli (e.g., temperature, pH, light, enzymes, and ionic concentration) by altering their composition or modulating their properties, such as swelling, degradation, or cross-linking. The mechanisms affecting gel assembly by external stimuli are also discussed in depth. These stimulus-responsive peptide hydrogels provide opportunities for the creation of “smarter” materials and provide a reference for the optimal design and innovative applications of stimulus-responsive peptide hydrogels in the future.
3. Biomedical applications of stimuli-responsive peptide hydrogels
Peptides are remarkably versatile biomaterials due to their ability to self-assemble, facile functionalization, responsiveness to stimuli, and multifunctionality.34,104,105 By designing the amino acid sequence, the chemical properties, secondary structure, and self-assembly behavior of peptide-based stimuli-responsive hydrogels can be controlled to achieve the desired properties.106 Moreover, stimuli-responsive peptide hydrogels have attracted research attention for a wide range of biotechnological and biomedical applications,47,107–109 including tissue engineering, drug delivery, wound healing, anti-bacterial therapy, 3D cell culture, and biosensors.58,107,108,110,111 In this context, we have systematically categorized various biomedical applications and presented instances of stimuli-responsive peptide hydrogels employed in these realms.3.1. Tissue engineering
Tissue engineering has emerged as a pivotal discipline in biomedical science and engineering over the past few decades. Tissue engineering aims to create artificial tissues with tissue compatibility, ultimately promoting the repair or replacement of tissues and organs.112–114 Stimuli-responsive peptide hydrogels offer adaptability and customization, with the ability to optimize their mechanical strength, response characteristics, and injectability via altering amino acid composition and sequence.115 This versatility allows them to meet the specific demands of diverse tissue engineering applications.116 Furthermore, hydrogels that incorporate bioactive peptide sequences can promote cell adhesion, further enhancing tissue regeneration. Therefore, stimuli-responsive peptide hydrogels represent highly promising materials for tissue engineering with vast application prospects.In the field of tissue engineering medicine, stimuli-responsive peptide hydrogels can be applied to blood vessel regeneration. RADA16-I (AcN-RADARADARADA-CONH2) belongs to a class of peptides capable of self-assembling into nanofiber scaffolds. In a related study, Liu et al. expanded the C-terminus of RADA16-I by incorporating three distinct short peptide sequences, leading to the development of three functionalized self-assembling non-covalently crosslinked peptide hydrogels (Fig. 8a).117 These hydrogels possess the potential to serve as 3D artificial microenvironments for controlling the behavior of human adipose-derived stem cells (hASCs) in vitro. Furthermore, Pal et al. developed a temperature-responsive, dual-cross-linked biocompatible hydrogel modified with a vascularization peptide (QK, a 15-amino acid peptide) for application in regenerative medicine, particularly tissue vascularization.118 Preliminary in vivo studies in mice demonstrated an increasing trend in blood vessel formation in the presence of the hydrogels that incorporated the QK peptide, along with well-defined capillaries and luminal structures within the area where the hydrogel was injected (Fig. 8b). Additionally, Mizuguchi et al. developed a temperature-responsive non-covalently crosslinked hydrogel as previously mentioned.51 The hydrogel was modified with bone sialoprotein-derived RGD (bRGD) as a bioactive peptide, to result in the hydrogel bRGD-coiled-coil unit bound ELP (bRGD-CUBE). This hydrogel demonstrated the capability of supporting the 3D cultivation of human umbilical vein endothelial cells (HUVECs) and enhanced their angiogenic activity. The results showed that the efficiency of blood vessel formation depends on the density of bRGD within the hydrogel, falling within a specific range (Fig. 8c). Therefore, bRGD-CUBE can be regarded as an advanced smart biomaterial with potential applications in tissue engineering.
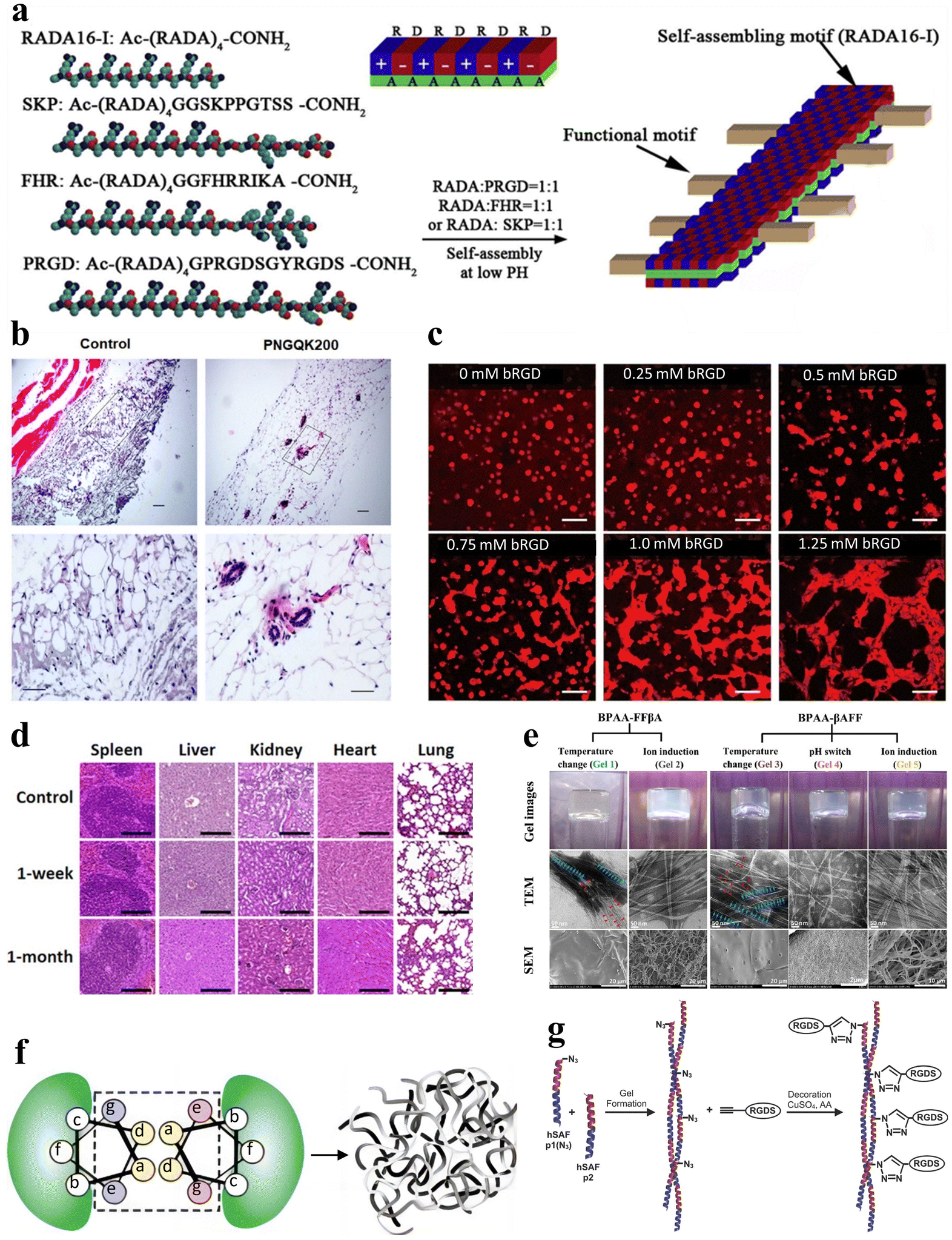 | ||
| Fig. 8 Stimuli-responsive peptide hydrogels for tissue engineering. (a) Schematic diagram of the self-assembly process of RADA16-I peptide nanofiber scaffolds. The side chains are distributed on both the polar and non-polar sides for self-assembly, with the functional peptide motifs extended outward from the β-sheet double ribbon structure.117 Copyright 2013 Elsevier Ltd. (b) Histological examination of the angiogenic properties in vivo for control and PNGQK200 materials.118 Copyright 2020 Elsevier Ltd. (c) HUVEC morphology of CUBE hydrogel cultures with varying concentrations of bRGD.51 Copyright 2020 American Chemical Society. (d) Histological analysis of five major organs (spleen, liver, kidney, heart, and lungs) in mice at 1 week and 1 month after in vivo transplantation of magnetic nanoparticles (MNP), FF + RGD or FF + RGD + MNP hydrogels.100 Copyright 2022 American Chemical Society. (e) Optical, TEM and SEM images of BPAA-FFβA-OH and BPAA-βAFF-OH hydrogels formed by the temperature switch/ion induction/pH switch method.119 Copyright 2021 Elsevier Ltd. (f) Schematic of the design principle of hSAF. Replacing select stronger interactions with more general weak interactions results in a nanofiber network cross-linked by non-covalent interactions, leading to the formation of a physical hydrogel.121 Copyright 2009, Springer Nature Limited. (g) Schematic of the click reaction of hSAFs-RGDS hydrogels. The N-terminally azido-modified hSAF-p1 gel undergoes a click reaction with alk-RGDS for decoration.122 Copyright 2014 Wiley-VCH GmbH. | ||
In addition to blood vessel regeneration, stimulus-responsive peptide hydrogels can also be applied to bone tissue engineering. For example, Mañas-Torres et al. delved into the application of biocompatible and biodegradable non-covalently crosslinked hydrogels comprising Fmoc-FF and Fmoc-RGD short peptides.100 These hydrogels were employed as 3D scaffolds to facilitate cellular growth. Notably, they exhibited the capacity to enhance cellular adhesion, proliferation, and differentiation in in vitro environments. Following in vivo transplantation for durations of 1 week and 1 month, all transplanted subjects showcased histological findings that lacked indications of inflammation, infection, necrosis, hemorrhage, or tumorigenesis (Fig. 8d).
Additionally, a noteworthy example is the PA, a peptide molecule featuring a hydrophilic peptide segment and a hydrophobic alkyl chain, enabling self-assembly into supramolecular nanostructures. Hartgerink et al. pioneered the use of pH-responsive PA self-assembly to fabricate nanofibrous scaffolds mimicking the extracellular matrix.1 This was achieved through PA self-assembly, covalent capture, and mineralization. The seminal work greatly enabled the use of PA and its derived stimulus-responsive peptide hydrogels as artificial extracellular matrices in tissue engineering. Inspired by this research, Li et al. synthesized 4-biphenylacetic acid (BPAA)-βAFF and BPAA-FFβA biphenyl-tripeptide non-covalently crosslinked supramolecular hydrogels with stimulus-responsive properties achieved through temperature switch, ion induction and pH switch, to mimic the extracellular matrix (ECM) scaffold (Fig. 8e).119 Both types of hydrogels effectively facilitated the adhesion and proliferation of L929 cells. The BPAA-βAFF hydrogel has been demonstrated in in vitro experiments to not only enhance chondrocyte proliferation and ECM secretion but also preserve a transparent cartilage phenotype. In summary, self-assembling peptides possess the capability of creating intricate nanofiber networks within hydrogels via ion-complementary assembly. This unique attribute allows them to effectively mimic the natural ECM, rendering them satisfactory substrates for tissue engineering applications.120
In another study, Tsutsumi et al. designed a series of hydrogels based on the E1Y9 peptide and its derivatives. In this study, the non-covalently crosslinked E1Y9/E1Y9-ALK mixed hydrogel exhibited the highest levels of cellular proliferation and differentiation activity compared with E1Y9/E1Y9-DGR, E1Y9/E1Y9-PRG, and E1Y9/E1Y9-RGD.73 The hydrogels demonstrated the effective induction of osteogenic differentiation when applied to MC3T3-E1 cells.
Some peptide hydrogels can also play a role in neural tissue engineering. Hydrogel self-assembling fibers (hSAFs) are self-assembling hydrogels based on standard linear peptides with a pure α-helical structure, as proposed by Banwell et al. (Fig. 8f).121 In their study, a comparative analysis was carried out between hSAFs and the extensively employed Matrigel matrix gel. This analysis showed that hSAFs, akin to Matrigel, facilitate the continual growth and differentiation of rat pheochromocytoma (PC12) cells into neurons throughout the cultivation process. Inspired by the research on hSAFs, Mehrban et al. introduced a method for synergistically integrating a cell-adhesion motif into a designed self-assembling peptide-based non-covalently crosslinked hydrogel system.122 Through the modification of hSAF gels with Arg–Gly–Asp–Ser (RGDS), a commonly used peptide in hydrogel synthesis that enhances cell adhesion and proliferation, they developed a hydrogel platform that amplifies PC12 cell growth and differentiation (Fig. 8g).
The application of stimuli-responsive peptide hydrogels in various tissue engineering fields, including blood vessel, bone, and neural tissue regeneration, has made research progress in recent years. Through the design and control of amino acid sequences, these peptides can self-assemble to form gel structures responsive to stimuli. The incorporated bioactive molecules enable the hydrogels to mimic the extracellular matrix. In vivo and in vitro evaluations have also demonstrated the good biocompatibility of the hydrogels. However, a more systematic investigation of the long-term in vivo stability and degradation rates of peptide hydrogels as tissue engineering scaffolds is still needed to achieve clinical translation, and the current biological safety assessment duration is insufficient. Meanwhile, injectable hydrogels possess the advantage of easy operation and align with the minimally invasive surgery rationale, making them suitable for tissue engineering applications. Therefore, more efforts should be devoted to designing injectable peptide hydrogels. These peptide-based intelligent materials have shown good application potential in tissue engineering, but further expansive evaluation and design optimization from multiple perspectives are still necessary to meet clinical demands. In the future, with the aid of computational methods to assist in the design and screening of peptide sequences, establishing tissue effect correlation models, and realizing rapid optimization and upgrading of material performance, it is expected to further promote the translation of these cutting-edge technology platforms into actual clinical applications.
3.2. Drug delivery
The precise delivery of therapeutic agents, including drugs, proteins, hormones, or genes, to specific targets within the body is a focus of extensive research and a significant challenge in the field of biomedicine.123,124 Materials for drug delivery should possess controllable drug loading and release, high encapsulation efficiency, and the capability of enhancing drug stability to prolong circulation time. Stimuli-responsive peptide hydrogels meet these criteria by enabling responsive controlled release, enhancing encapsulation via peptide networks, and safeguarding drug molecules from degradation. Owing to these advantageous features, stimuli-responsive peptide hydrogels exhibit great potential for drug delivery applications, and many related studies have been reported.Polypeptide hydrogels, loaded with immunomodulatory drugs, can effectively regulate the immune system by influencing gene expression and other mechanisms. This enables them to combat bacteria, viruses, and inflammation, providing a hopeful prospect for treating infectious diseases and immune-related conditions. The targeted delivery of specific drugs enabled by stimuli-responsive peptide hydrogels has shown a wide range of application potential in anti-bacterial and antiviral therapy. Firstly, Cao et al. prepared a reversible temperature-responsive sol–gel peptide hydrogel system by mixing a thermosensitive polymer, PNIPAM, into fibrillar nanostructures self-assembled from a short peptide I3K.125 This non-covalently crosslinked hydrogel system enables facile loading and delivery of the antibacterial peptide, G(IIKK)3I–NH2, with the release rate tunable through adjusting the concentration of short peptide I3K (Fig. 9a). On the other hand, Monroe et al. devised a novel strategy to create an antiviral prodrug hydrogel by incorporating tenofovir (TFV, a nucleotide reverse transcriptase inhibitor) into amphiphilic peptide sequences.126 The resulting TFV-PA3 hydrogel exhibited a nearly linear release of TFV over a month in vitro, and the prodrug conversion did not adversely affect the antiviral activity of TFV-Pas. Under reduced conditions, free TFV could be released to control viral replication, thereby delaying disease progression to a severe state.
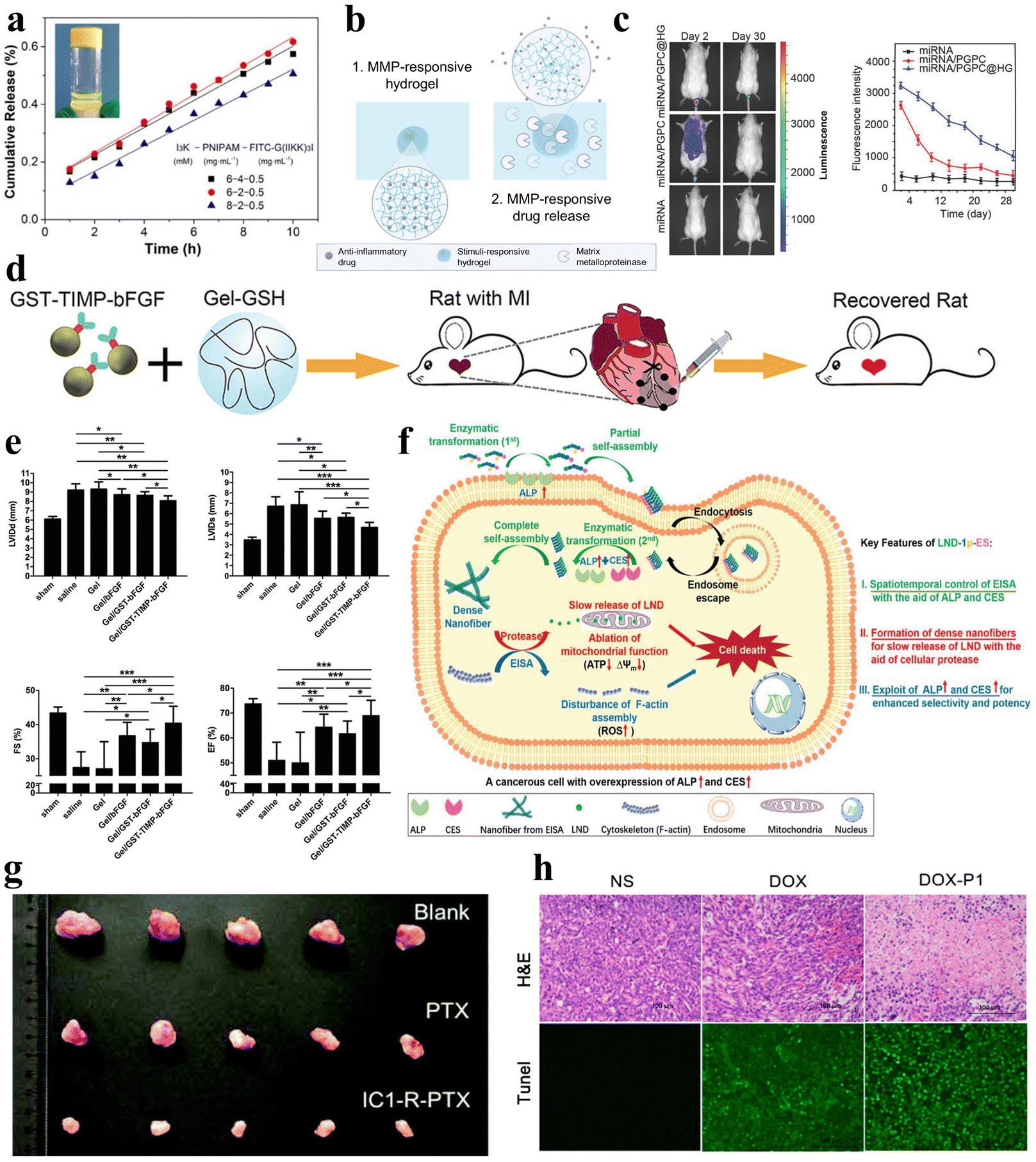 | ||
| Fig. 9 Stimuli-responsive peptide hydrogels for drug delivery. (a) Cumulative release of FITC-G(IIKK)3I-NH2 as a function of time from the I3K/PNIPAM/FITC-G(IIKK)3I-NH2 hybrid hydrogel, and images after 1 hour of release in 40 °C water.125 Copyright 2019 American Chemical Society. (b) Schematic of an MMP-responsive hydrogel encapsulating an anti-inflammatory drug that is released upon MMP stimulation.127 Copyright 2022 Elsevier Ltd. (c) Fluorescence images of Cy3-miRNA, Cy3-miRNA/poly(ethylene glycol)-GPLGVRG-poly{N′-[N-(2-aminoethyl)-2-aminoethyl]aspartamide}-cholesteryl (PGPC) and Cy3-miRNA/PGPC@hydrogels (HG) after injection into mice (left) and quantitative fluorescence intensity profiles of Cy3-miRNA (right). miRNA/PGPC@HG enables sustained release of encapsulated miRNA/PGPC polyplex micelles in MMP overexpressing intervertebral disc lesion tissues.91 Copyright 2018 Wiley-VCH GmbH. (d) Schematic diagram of MMP-responsive hydrogels injected into rat myocardial infarction wound sites for localized drug delivery. (e) Cardiac function measurements in mice after injection of various hydrogels: end-diastolic left ventricular (LV) internal diameter (upper left); end-systolic LV internal diameter (upper right); fractional shortening (lower left); and ejection fraction (lower right).92 Copyright 2019 Wiley-VCH GmbH. (f) Schematic diagram of LND-1p-ES intracellular enzyme-directed self-assembly for selective killing of cancer cells.88 Copyright 2021 Wiley-VCH GmbH. (g) Tumour images were obtained from mice injected for 10 days with saline, PTX, and IC1-R-PTX.59 Copyright Royal Society of Chemistry. (h) Hematoxylin and eosin (H&E) staining (top) and terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) assay (bottom) of tumour sections after different treatments within 7 days. Apoptotic cells showed stronger green fluorescence in the DOX-P1 hydrogel group.140 Copyright 2023 MDPI. | ||
Additionally, in a study, Noddeland et al. developed a non-covalently crosslinked hydrogel delivery system that is biocompatible and able to release anti-inflammatory drugs for treating inflammatory skin diseases.127 The hydrogel was formed by cross-linking peptide linkers, which are sensitive to MMPs, with PEG in order to deliver the anti-inflammatory Janus kinase inhibitor tofacitinib citrate (Fig. 9b). The researchers utilized the modified hydrogel in cellular investigations and demonstrated minimal cytotoxicity for fibroblasts and keratinocytes exposed to the hydrogel or its degradation products. Zhang et al. synthesized a non-covalently crosslinked supramolecular hydrogel combining graphene oxide (GO) and an L-phenylalanine derivative (LPFEG).128 This hydrogel can function as a photosensitive carrier for selective uptake of specific chiral drug enantiomers, L-dopa and S-naproxen sodium, and for the helical inversion induced by light conditions, leading to on-demand drug release. Moreover, the aforementioned hydrogel, formed by mixing the PEG gel precursor and MMP-cleavable peptide cross-linker, can be utilized for the treatment of intervertebral disc degeneration (IDD), utilizing overexpression of various MMPs to deliver miR-29a to nucleus pulposus cells in a sustained and biologically responsive manner to treat IDD (Fig. 9c).91 In a similar manner, the collagen-GSH non-covalently crosslinked hydrogel prepared by Fan et al. is responsive to MMPs.92 After targeted myocardial infarction, this hydrogel can selectively deliver drugs through upregulation of MMP-2/9, promoting angiogenesis and inhibiting cardiac remodeling (Fig. 9d and e).
In addition to the examples above, stimuli-responsive peptide hydrogels can serve as hormone delivery systems to effectively regulate physiological processes and maintain body homeostasis.99,129 For instance, Fu et al. developed a glucose-responsive insulin delivery system comprising non-covalently crosslinked pH-responsive peptide hydrogels, encapsulated insulin, and enzymes.39 Within this system, glucose undergoes catalytic conversion to gluconic acid, leading to localized acidification. This acidification subsequently degrades the pH-responsive peptide hydrogels and enables the controlled release of encapsulated insulin, effectively reducing blood glucose levels. Similarly, Bao et al. developed a non-covalently crosslinked pH-responsive hydrogel for the controlled release of insulin and bovine serum albumin (BSA).130 This hydrogel, composed of 4-arm PEG-block-poly L-glutamic acid (4a-PEG-PLG) copolymers, enables pH-regulated drug release without affecting the biological activity of insulin and BSA. Furthermore, Roth-Konforti et al. designed and synthesized 6-nitroveratryloxycarbonyl-diphenylalanine (Nvoc-FF) as a photo-responsive hydrogelator that self-assembles into a 3D, self-supporting, nanofibrous hydrogel.131 The non-covalently crosslinked hydrogel is capable of encapsulating insulin-fluorescein isothiocyanate (insulin-FITC) and undergoes cleavage upon UV irradiation, thereby facilitating the degradation of the hydrogel and the subsequent release of insulin-FITC. Turabee et al. designed a non-covalently crosslinked pH- and temperature-responsive peptide-based triblock copolymer hydrogel, poly[(2-(dibutylamino)ethyl-L-glutamate)-co-(γ-benzyl-L-glutamate)]-poly(ethylene glycol)-b-poly[(2-(dibutylamino)ethyl-L-glutamate)-co-(γ-benzyl-L-glutamate)] (PNLG-co-PBLG-b-PEG-b-PBLG-co-PNLG), as a therapeutic protein delivery platform.132 The peptide hydrogel is capable of forming stable polymer–protein complexes when combined with human growth hormone (hGH). The hGH-loaded peptide hydrogel can sustain therapeutic levels for one week, demonstrating sustained drug delivery properties and the potential for continuous hGH release. These studies collectively illuminate the extensive application domains of peptide hydrogels, rendering them a versatile biomedical carrier.
Herein, peptide hydrogels also reveal unique application potential in the field of cancer therapy. In the realm of cancer therapy, the utilization of peptide hydrogels presents promising prospects, due to their inherent attributes as carriers for anticancer drugs, facilitating targeted therapy.133–136 Wu et al. developed a novel self-assembling non-covalently crosslinked peptide derivative, designated as LND-1p-ES, that responds to three enzymes for efficient and targeted delivery of lornidamine to cancer cells (Fig. 9f).88 Significantly, LND-1p-ES serves as both a therapeutic agent and a carrier, effectively integrating the advantages of both components. Leveraging the overexpression of ALP, CES, and proteases in cancer cells, the LND-1p-ES hydrogel demonstrated amplified efficacy and selectivity against melanoma cells (A375) both in vitro and in vivo. These results validate the anti-tumor properties of this triple-enzyme-responsive peptide hydrogel. Zhu et al. designed a covalently crosslinked dual-responsive peptide hydrogel, which also has the ability to serve as a drug delivery material for the anticancer drug paclitaxel (PTX).59 This biocompatible PTX-loaded peptide hydrogel can be formed in vitro and then directly injected into the tumor site. Within the tumor microenvironment, the hydrogel network is disrupted by mild acidity and a high glutathione concentration, enabling sustained and controlled release of PTX to achieve an antitumor effect (Fig. 9g). Similarly, Yu et al. selected another peptide that can transport PTX, WMWVTNLRTD (WMW10), as both an assembling and therapeutic peptide.137 They synthesized a WMW10-PTX conjugate by linking PTX to WMW10 via disulfide bonds. This design enabled the controlled release of PTX in response to glutathione stimulation, leveraging the environment of cancer cells.
In another experiment, Tao et al. designed non-covalently crosslinked supramolecular hydrogels (E3FID and E3FNP) composed of flexible peptides and nonsteroidal anti-inflammatory drugs (NSAIDs) that were capable of encapsulating the anticancer drug docetaxel (DTX).138 The release of DTX from these hydrogels followed a non-Fickian diffusion pattern, indicating a sustained release profile. Testing against DU-145 and PC-3 cancer cell lines demonstrated that these DTX-loaded hydrogels exhibit anticancer effects, indicating their potential as carriers for anticancer drug delivery. Mei et al. engineered a co-assembled hydrogel incorporating the chemotherapeutic agent doxorubicin (DOX) and the RGD-derived peptide conjugate (1-RGDH).139 This hydrogel demonstrates pH-responsive release of DOX specifically in slightly acidic environments. Testing in non-small cell lung cancer cells (A549) showcased the anti-tumor potency of this targeted drug delivery system. Coincidentally, Zhu et al. designed and prepared a pH-responsive nonapeptide hydrogel, which can also achieve tumor-targeted delivery of DOX (Fig. 9h).140 In a similar study, Li et al. developed a temperature-responsive hydrogel composed of oligo (L-lysine) modified F127 (FL) that can deliver the anticancer drug 5-fluorouracil (5-FU).141
The working principle of stimuli-responsive peptide hydrogel drug delivery systems is to design peptide sequences that are sensitive to specific stimuli such as pH, temperature, reducing environment, etc. Under the stimulation of tumor microenvironments, the hydrogel can achieve a controlled release of drugs loaded inside. Stimuli-responsive peptide hydrogels demonstrate great application potential in drug delivery, but further research is needed to achieve actual clinical applications. Further optimization in drug loading, controlled release, in vivo stability, and enhanced response to physiological stimuli is necessary. It is possible to try to introduce multi-functional units such as targeting groups into the peptide hydrogel network to construct multi-functional intelligent delivery systems, and realize more efficient and precise targeted drug delivery and controlled release, making the drug delivery system more intelligent. In general, despite the broad application prospects of peptide hydrogels, more targeted optimization and improvement of each specific material is still needed to transform more stimuli-responsive peptide hydrogels into practical applications.
3.3. Wound healing
Wound healing is an intricate, multi-stage biological process involving hemostasis, inflammation, proliferation, and tissue remodeling, and the promotion of wound healing requires comprehensive and convenient therapeutic solutions.59,142 Peptide can be utilized for the modulation of gel adhesion, ensuring effective wound site coverage and expediting the wound healing process. Moreover, they can incorporate procoagulant peptide sequences to promote coagulation and hemostasis, along with antimicrobial peptide (AMP) sequences to mitigate wound infection. The remarkable attributes of peptide hydrogels have spurred extensive interest and research into their application as wound dressings.Wound infection remains a prevalent concern throughout the wound healing process, and peptide hydrogels as wound dressings can be loaded with antimicrobial agents applied to combat wound infection. Jeong et al. developed injectable non-covalently crosslinked supramolecular hydrogels by a straightforward amalgamation of cyclodextrin-modified hyaluronic acid (HA-CD) and adamantane-modified hyaluronic acid (Ad-HA).143 They employed cyclic peptide linkers with MMP and reactive oxygen species (ROS)-responsive cleavage sequences to tether AMP (Ad-HA-AMP), thus facilitating diabetic chronic wound healing (Fig. 10a). The hydrogel effectively regulated AMP release in vivo when wound infection was present, while the introduction of Ad-HA-AMP into non-infected wounds did not result in AMP release, thereby demonstrating a high level of safety. Likewise, Suo et al. developed a composite hydrogel (AMP-HA) by covalently cross-linking hyaluronic acid (HA) with another AMP, KK(SLKL)3KK, for wound healing.144 Both AMP and AMP-HA hydrogels exhibited broad-spectrum antibacterial activity in vitro and in vivo, and the AMP-HA hydrogel exhibited the highest healing rate compared to the control group and AMP alone (Fig. 10b). In another intriguing study, Veld et al. designed a PIC-based temperature-responsive hydrogel with non-covalent crosslinks, which can serve as an innovative wound dressing.52 Its ability to transition into a liquid state below 16 °C enables facile, painless application and removal. This property mitigates the risks of disturbing the wound healing process that can occur with conventional dressings requiring painful changes.
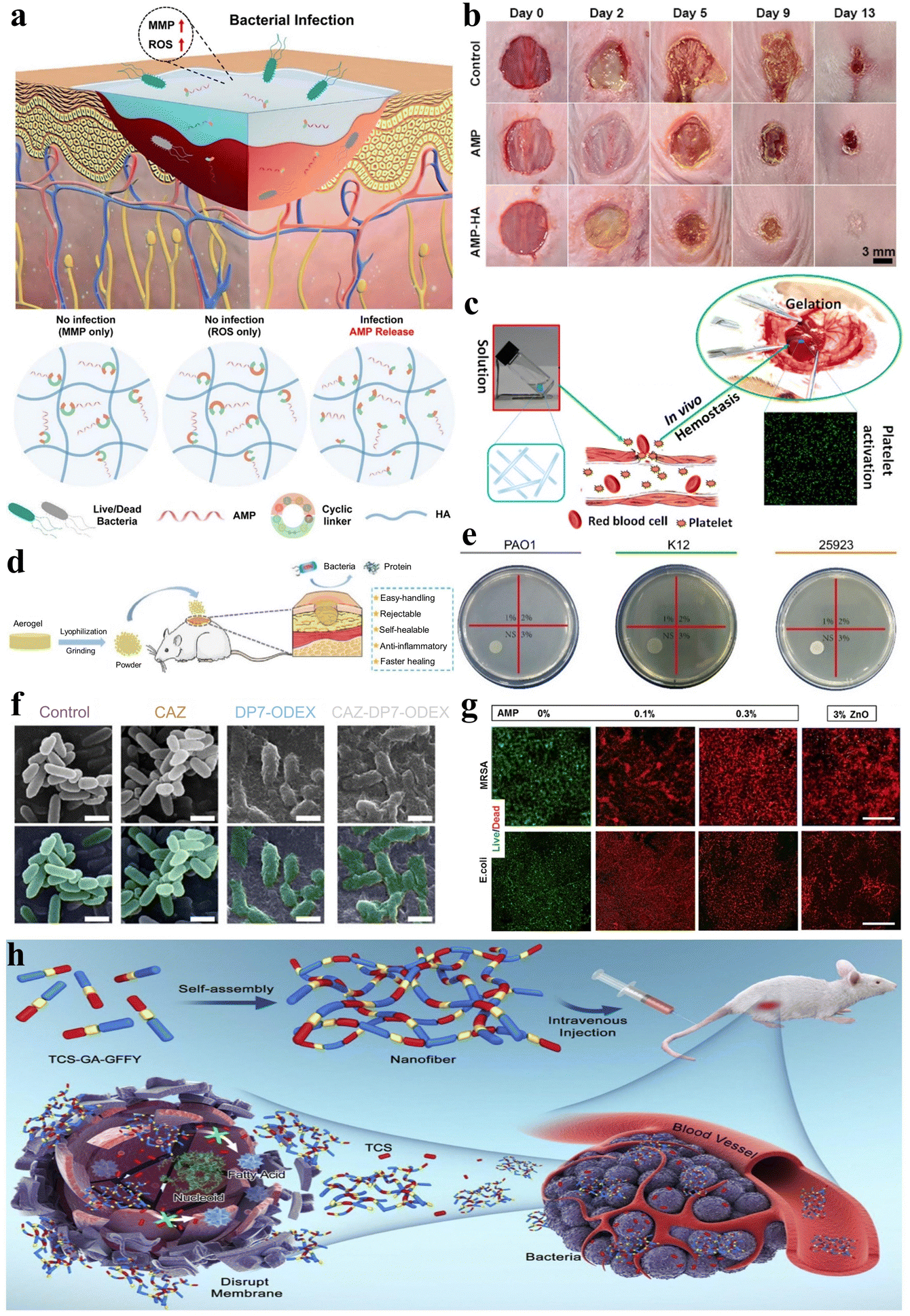 | ||
| Fig. 10 Stimuli-responsive peptide hydrogels for wound healing and anti-bacterial applications. (a) Schematic of an injectable hydrogel for controlled release of AMP to treat the diabetic chronic wound.143 Copyright 2023 American Chemical Society. (b) Images of S. aureus infected mice wounds under different treatments (control, AMP, and AMP-HA) on days 0, 2, 5, 9, and 13.144 Copyright 2021 American Chemical Society. (c) Schematic illustration of the accelerated hemostatic effect of I3QGK peptide solution by gelling blood and promoting platelet adhesion.145 Copyright 2016 American Chemical Society. (d) Schematic of the lyophilized CHX-loaded poly[3-dimethyl(methacryloyloxyethyl) ammonium propane sulfonate] (PDMAPS)/keratin hydrogel, pulverized into a powder for wound healing in mice.149 Copyright 2023 Elsevier B.V. (e) Photographs of Staphylococcus aureus (ATCC 25923), Escherichia coli (K12) and Pseudomonas aeruginosa (PAO1) following co-culture with LB agar and different levels of the DP7-ODEX hydrogel (1, 2, 3, wt% DP7) for 4 hours. (f) Biofilm inhibition ability of the CAZ solution (1/4 minimal inhibitory concentration (MIC)), DP7-ODEX hydrogel (3 wt% DP7), and CAZ-DP7-ODEX hydrogel (3 wt% DP7). No alterations in morphology were observed when bacteria were mixed with CAZ or LB agar, whereas the mixing with the DP7-ODEX hydrogel and CAZ-DP7-ODEX hydrogel resulted in wrinkling and disruption of the bacterial outer membrane.152 Copyright 2021 Elsevier Ltd. (g) Representative live/dead staining images of methicillin resistant Staphylococcus aureus (MRSA) and E. coli cultured on MeTro/GelMA-AMP hydrogels with varied AMP content (0%, 0.1%, and 0.3% w/v) in combination with 3% (w/v) ZnO nanoparticles.153 Copyright 2017 Elsevier Ltd. (h) Schematic illustration of TCS-GA-GFFY hydrogel nano-antibiotics formation and in vivo bacterial infection treatment.159 Copyright 2020 Published by Elsevier B.V. | ||
Additionally, stimulus-responsive peptide hydrogels serving as wound dressings can also be applied for wound hemostasis, thereby promoting wound healing. Chen et al. engineered an amphiphilic short peptide, I3QGK, which, upon the incorporation of transglutaminase (TGase), assembles into a non-covalently crosslinked hydrogel with remarkable shear-thinning behavior and rapid self-recovery properties.145 This peptide hydrogel exhibits swifter and more effective hemostatic efficacy when compared to alternative hemostasis methods or materials due to its ability to promote blood clot formation and enhance platelet adhesion (Fig. 10c). In a separate investigation, Wu et al. employed solid-phase synthesis to fabricate a dual-function, degradable, non-covalently crosslinked self-assembling nonapeptide (SAP) hydrogel that is regulated by metal ions such as Na+ or K+, designated as RADA16-I.146 This hydrogel not only exhibited outstanding hemostatic properties but also demonstrated efficacy in promoting bone regeneration.
During the wound healing process, peptide hydrogels can also stimulate angiogenesis within the wound, thereby accelerating wound healing. Huang et al. co-assembled salvianolic acid B (SAB) and a soluble phosphopeptide to design a robust and adhesive non-covalently crosslinked hydrogel called 1&SAB.147 This hydrogel possesses antioxidant properties, enhances cell migration, accelerates angiogenesis, and prolongs the residence time of therapeutic agents on skin wound surfaces, thereby promoting wound healing. Xing et al. designed a chiral non-covalently crosslinked hydrogel dressing (HA-LM2-RMR) composed of l-phenylalanine and cationic hexapeptide co-assembled helical nanofibers cross-linked with hyaluronic acid via hydrogen bonding.148 This hydrogel dressing contains abundant chiral and cationic sites that can selectively interact with the negatively charged microbial membrane. Notably, HA-LM2-RMR promotes angiogenesis of human umbilical vein endothelial cells by upregulating the expression of vascular endothelial growth factor (VEGF) and optic atrophy 1 (OPA1). Moreover, Shang et al. synthesized a covalently crosslinked dual-network hydrogel loaded with chlorhexidine (CHX) through free radical polymerization of sulfobetaine and oxidative cross-linking of reduced keratin.149 The introduced keratin and CHX imparted cell compatibility and antioxidant properties to the hydrogel. Interestingly, the freeze-dried hydrogel could be milled into a powder, sprinkled onto irregular wound beds, and subsequently rehydrated by wound fluid to reform the gel in situ. This dry hydrogel powder dressing facilitated collagen deposition and alleviated inflammatory responses, thus expediting skin wound closure and regeneration (Fig. 10d).
Peptide hydrogels can be used as wound dressings by incorporating hemostatic peptide sequences, antimicrobial peptide sequences, and bioactive factors, exerting multifunctional effects such as antimicrobial, hemostatic, and proangiogenic properties, thereby promoting wound healing. The basic strategy for using stimulus-responsive peptide hydrogels to promote wound healing is to design environment-sensitive peptide sequences and construct a hydrogel network to achieve controlled release of incorporated antimicrobial factors, pro-angiogenic factors, and other bioactive molecules, thus exerting multi-directional biological regulatory functions. However, most of these designs are preliminary laboratory explorations, and there are still many challenges to achieve clinical translation. The wound microenvironment is complex and dynamic, and in vitro experiments have limited capability of mimicking in vivo situations. In the design of hydrogel materials, the characteristics of different wound types and healing stages should be considered to enable customized modulation of the treatment process, which may improve their clinical applicability. In addition, some studies have mentioned approaches to alleviate the pain caused by wound dressing changes, which is also a direction that can be explored regarding the application of peptide hydrogels in wound healing. At the technical level, the introduction of advanced nanotechnology and chemical biology strategies is expected to overcome the limitations of existing peptide sequence functions and achieve multi-target regulation of the wound healing process.
3.4. Anti-bacterial
Bacterial infection remains a ubiquitous threat across biomedical applications, necessitating anti-bacterial interventions. Ideal anti-bacterial hydrogels should exhibit direct bactericidal activity and/or the capability of delivering anti-bacterial agents. Therefore, peptides that possess both anti-bacterial activities (such as AMPs) and the ability to conjugate with anti-bacterial drugs are an outstanding choice for constructing anti-bacterial stimulus-responsive hydrogels. This dual anti-bacterial mechanism highlights peptide hydrogels as advantageous biomaterials to combat bacterial infections for diverse therapeutic needs.AMPs constitute essential elements of the innate immune system, exhibiting a ubiquitous presence throughout the animal and plant kingdoms. Their primary function is to safeguard organisms against bacterial infections.150,151 In recent years, as bacterial resistance to antibiotics and related agents has steadily escalated, AMP hydrogels have emerged as important biomaterials in the realm of bacterial infection treatment.
Currently, most AMP hydrogels exhibit broad-spectrum antibacterial activity inhibiting both Gram-positive and Gram-negative bacteria. For instance, as previously mentioned, stimulus-responsive peptide hydrogels that leverage the abilities of AMPs have been utilized to promote wound healing and prevent Gram-positive and Gram-negative bacterial infections.143,144 Additionally, Wu et al. designed a dual-function, pH-sensitive covalently crosslinked hydrogel based on the antimicrobial peptide DP7 (VQWRIRVAVIRK) and oxidized dextran (ODEX).152 This hydrogel demonstrated potent anti-bacterial activity against common pathogens involved in traumatic infections, including Staphylococcus aureus, Escherichia coli, and Pseudomonas aeruginosa (Fig. 10e). Moreover, the combination of DP7 with ceftazidime (CAZ) in the hydrogel relatively enhanced its anti-bacterial efficacy compared to CAZ alone (Fig. 10f). Annabi et al. also prepared a covalently crosslinked composite hydrogel with anti-bacterial capabilities by visible light-induced cross-linking of gelatin methacryloyl and methacryloyl-substituted recombinant human tropoelastin (MeTro/GelMA) in conjunction with the AMP Tet213 (Fig. 10g).153 Adak et al. designed and synthesized a biocompatible non-covalently crosslinked hydrogel featuring a hexapeptide as the key component, with hydrophobic long-chain and hydrophilic tri-lysine units attached at the N- and C-termini, respectively.154 This hydrogel possesses the capability of disrupting bacterial cell membranes, making it a material suitable for inhibiting bacterial growth. The amphiphilic peptide I4K2 belongs to a class of peptides capable of self-assembling into nanofibers and forming hydrogels in response to plasma amine oxidase (PAO). The resulting non-covalently crosslinked hydrogel demonstrates anti-bacterial activity against both Gram-positive and Gram-negative bacteria.155 In conclusion, an increasing number of broad-spectrum antibacterial AMP hydrogels have become attractive materials to combat bacterial infections.156–158
Moreover, in addition to the utilization of AMPs, hydrogels created via cross linking peptides with anti-bacterial agents can also exhibit broad-spectrum bioactivity against pathogenic bacteria. Pham et al. developed a covalently crosslinked GPLD peptide hydrogel, which not only demonstrates potential for effective DOX delivery to effectively eliminate cancer cells but also showcases remarkable anti-bacterial activity, owing to the incorporation of metal nanoparticles (NPs).71 Yang et al. crafted a non-covalently crosslinked supramolecular hydrogel nano-antibiotic by utilizing triclosan (TCS) conjugated with self-assembling peptides (Fig. 10h).159 This supramolecular hydrogel demonstrates significantly increased anti-bacterial efficacy compared to conventional antibiotics, as substantiated by both in vitro and in vivo assessments.
Additionally, some peptide hydrogels exhibit selective antibacterial activity against either Gram-positive or Gram-negative bacteria. For example, Durão et al. synthesized an AMP with the ability to self-assemble into a non-covalently crosslinked hydrogel in response to pH changes and exhibit anti-bacterial activity against Escherichia coli (Gram-negative bacteria).160 On the other hand, in another study, Zhu et al. utilized TCS and preloaded it onto a non-covalently crosslinked hybrid hydrogel composed of peptide-based bis-acrylate and acrylic acid (AAc), and this hydrogel demonstrated anti-bacterial properties against Staphylococcus aureus (Gram-positive bacteria).161
In summary, most current peptide hydrogels that are used for antimicrobial treatment have broad-spectrum antibacterial activity, being able to inhibit both Gram-positive and Gram-negative bacteria. Some hydrogels have demonstrated selective antibacterial activity against either Gram-positive or Gram-negative bacteria. However, the antibacterial properties of peptide hydrogels mainly originate from the embedded antimicrobial peptides, which are susceptible to environmental pH, enzymatic degradation, and inactivation. Therefore, antimicrobial peptides with higher stability should be selected or modified to improve their stability in vivo. One current cutting-edge technique is synergistic antibacterial hydrogels that integrate multiple antibacterial mechanisms into one entity, such as the combination of antimicrobial peptides and antibiotics. This increases the antibacterial pathways and reduces the risk of bacterial drug resistance. Moreover, current research is limited to preliminary in vitro antibacterial experiments without an in-depth evaluation of the dose–effect relationship of the hydrogels. Animal model validations are still lacking. Further systematic and comprehensive assessments of their antibacterial capabilities are warranted.
3.5. 3D cell culture
3D cell culture is indispensable in regenerative medicine and preclinical evaluation,162 with hydrogels having become an ideal culture platform due to their capacity to mimic the extracellular matrix.163 Among the array of hydrogels, peptide hydrogels distinguish themselves owing to their inherent attributes that promote enhanced cell adhesion and compatibility with cells, and interconnected porous structures that facilitate cell spreading. Furthermore, peptide hydrogels offer the potential for further customization, responding to specific cues to fine-tune their interactions with cells and their microenvironment. Therefore, stimuli-responsive peptide hydrogels, endowed with these advantageous features, represent versatile and excellent biomaterials to support a multitude of 3D cell cultures.Peptide hydrogel culture of normal cells can provide materials for tissue engineering and cell functional studies. The construction of 3D biomimetic scaffolds using peptide hydrogels is a critical strategy in 3D cell culture, as it enables efficient cell adhesion, proliferation, and differentiation on the scaffold, thereby achieving the goal of 3D cell culture.107,164 Zanna et al. crafted a series of thixotropic peptide-based non-covalently crosslinked physical hydrogels, incorporating D-Oxd or DpGlu [Oxd = (4R,5S)-4-methyl-5-carboxyl-oxazolidin-2-one, pGlu = pyroglutamic acid], for 3D cell culture applications.165 Additionally, the properties of peptide hydrogels can be controlled by adjusting the type of peptides, their concentration, and the degree of cross-linking, enabling a bespoke tailoring of hydrogel properties to meet the demands of diverse cell types and applications. Gould et al. synthesized bioactive covalently crosslinked peptide-functionalized PEG hydrogels by introducing three small adhesive peptide sequences, RGDS, VGVAPG, and GTPGPQGIAGQRGVV (P15), onto a thiol–ene polymerization platform.166 These hydrogels were subsequently employed for the cultivation and observation of aortic valve interstitial cells (VICs), demonstrating their practicality in addressing specific biological hypotheses. Likewise, Yamaguchi et al. developed a covalently crosslinked photo-responsive hydrogel composed of avidin and biotinylated PEG, serving as a multifunctional substrate for 3D cell culture and sorting.167 They gained control of the adhesion and viability of fibroblast cells through the incorporation of the biotin-RGDS peptide into the hydrogel. Additionally, cells embedded within it could recover without any cellular damage due to the photo-induced degradation capability of this hydrogel (Fig. 11a). In addition to conventional approaches focused on the design of various peptide hydrogels, Song et al. embarked on a strategy involving the utilization of mechanical motion to accelerate cell proliferation.168 They reported a mechanically dynamic covalently crosslinked hydrogel characterized by oriented domain sliding motion. In contrast to thermodynamically static hydrogels, the mechanical cues produced by the sliding motion of these oriented structural domains within the mechanically dynamic hydrogel effectively accelerated the proliferation of human hepatocyte LO2 cells and rat aortic vascular smooth muscle A10 cells when cells were cultured on it (Fig. 11b).
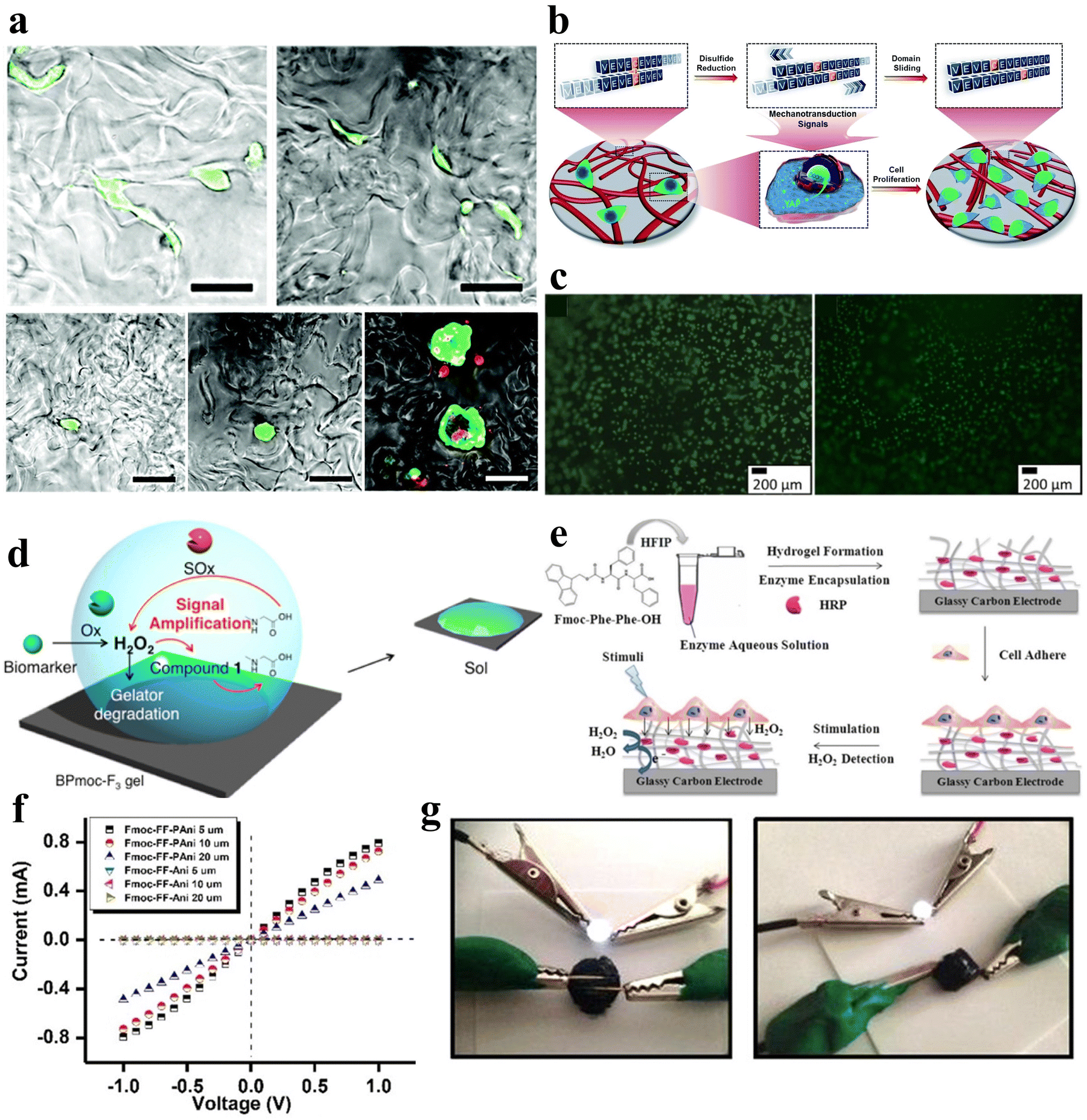 | ||
| Fig. 11 Stimuli-responsive peptide hydrogels for 3D cell culture and biosensors. (a) Confocal microscopy images of adherent cells after 2 days of incubation in hydrogels functionalized with RGDS peptides at varying concentrations (from top to bottom, left to right: 1000, 500, 250, 125, 0 μM).167 Copyright 2021 Royal Society of Chemistry. (b) Schematic illustration of a mechanically dynamic hydrogel, demonstrating well-defined mechanical movements. The enhanced cell proliferation induced by directed mechanical motion on hydrogels is attributed to a mechanotransduction mechanism involving the translocation of yes-associated protein (YAP) into the nucleus.168 Copyright 2020 Royal Society of Chemistry. (c) Fluorescence staining images of HeLa cells on the D-P1 hydrogel (left) and L-P1 hydrogel (right) (fetal bovine serum (FBS)-free) following 72 hours of incubation. Green fluorescence signifies sustained cell viability after 72 hours of incubation.170 Copyright 2020 Elsevier B.V. (d) Schematic illustration of biomarker detection within a supramolecular hydrogel (BPmoc-F3) incorporating a signal amplification system (amplifier 1 + SOx).175 Copyright 2015 American Chemical Society. (e) Schematic representation of the Fmoc-FF hydrogel designed as a biointerface for enzyme-based electrochemical biosensing and cellular monitoring. HRP was encapsulated within the hydrogel matrix during the self-assembly process of Fmoc-FF to measure H2O2 released by living cells.176 Copyright 2016 American Chemical Society. (f) Current–voltage (I–V) characteristic curves for dried Fmoc-FF-PAni and Fmoc-FF-Ani hydrogels. The I–V curves of dried Fmoc-FF-PAni hydrogels exhibit nearly ohmic behavior within the measured voltage range. (g) Illustration of the conductivity of Fmoc-FF-PAni hydrogels, serving as a bridge to establish an electrical circuit with LEDs connected in series (Fmoc-FF concentration = 2% (w/v)).108 Copyright 2018 American Chemical Society. | ||
Apart from supporting normal cell growth, peptide hydrogels can also be used to culture cancer cells for cancer basic research, drug evaluation, and clinical detection. For instance, Xu et al. also utilizing a non-covalently crosslinked 3D hydrogel system developed a novel 3D h9e hydrogel concurrently employed for the cultivation of liver cancer HepG2 and colorectal adenocarcinoma SW480 cells.169 Restu et al. reported a study involving a short pentapeptide, D-P1 (Ac-D-Phe–D-Phe–D-Phe-Gly–D-Lys), composed of only five D-amino acids.170 This peptide possesses the ability to self-assemble into helical nanofibers, forming a non-covalently crosslinked hydrogel that serves as a scaffold for 3D cell culture. Notably, this oligopeptide hydrogel exhibits non-toxic properties not only towards human induced pluripotent stem cell-derived embryoid bodies (EBs) but also towards human cancer cells, as exemplified by the successful formation of HeLa cell spheroids on the D-P1 hydrogel (Fig. 11c).
Stimuli-responsive peptide hydrogels have become an ideal platform for three-dimensional cell culture to support the cultivation of diverse cell types owing to their capacity to mimic the extracellular matrix. Although peptide hydrogels possess advantages in tunable structures, the mechanisms underlying how different peptide sequences impact cell adhesion and proliferation remain elusive. To enable custom-tailored construction of ideal cell microenvironments, systematic assessments of bioactivities across various peptide sequences coupled with detailed structure–function relationship studies are still necessitated to provide a guiding principle for further optimization of peptide hydrogels with superior and predictable performance. If precise control over cell microenvironments could be achieved via stimulus-responsive mechanisms, such technology would facilitate further advances in cell culture and in vitro construction of multicellular tissues and organs.
3.6. Biosensors
Sensing is a highly critical field within applied science, and especially in the context of the rapid advancements in the field of medicine, there is a growing demand for fast, cost-effective, and reliable sensing tools, particularly in crucial domains such as diagnosis and treatment.171,172 Peptide hydrogels have garnered significant interest as materials for biosensing due to their exceptional biocompatibility, tunability, water-absorbing capacity, and stability.173 Particularly, peptide hydrogels endowed with various stimulus-responsive properties and sensitive responses offer versatile options for a wide range of biosensor design applications.A diverse array of biomolecules within the biological system play a significant role as essential markers for health assessment and diagnosis. Consequently, the detection of these biomolecules constitutes a significant facet of biosensing applications. For instance, King et al. designed a covalently crosslinked peptide-based hydrogel (Val–Lys–Val–Lys–Val–Glu–Val–Lys) serving as a 3D solid carrier for a DNA biosensor.174 This hydrogel, when interacting with oligonucleotides ranging from 22 pM, enables the detection of repeatable fluorescence signals for biomolecule sensing. Additionally, certain stimulus-responsive peptide hydrogels exhibit phase transition properties. The interaction of these hydrogels with detectable molecules induces volume-phase transition, leading to modifications in hydrogel properties. These alterations can be effectively harnessed in the production of sensor probes for detecting biomolecules. Continuing to harness the phase transition properties of hydrogels, Yoshii et al. proposed a multi-component strategy for the fabrication of non-covalently crosslinked hydrogel-based sensors.175 They integrated dendritic-triggered self-degrading molecules and their corresponding enzymes to establish a signal amplification system, stably embedding them alongside other enzymes within a supramolecular hydrogel (BPmoc-F3) with a specific responsiveness to hydrogen peroxide (H2O2). Utilizing the gel–sol transition detection methodology, they achieved highly sensitive and visually discernible detection of biomarkers, including H2O2, glucose, and uric acid (Fig. 11d).
H2O2, NO, H2S, and superoxide anions (O2˙) play significant roles in regulating the redox balance in organisms. Monitoring these biological signaling molecules helps to understand and track the dynamic changes in redox balance within living systems. For example, Lian et al. utilized non-covalently crosslinked hydrogels based on Fmoc-FF self-assembled peptide nanofibers to construct intelligent biointerfaces for enzyme-based electrochemical biosensing and cell monitoring.176 Fmoc-FF peptides can embed HRP, which exhibits biological activity towards H2O2 during the self-assembly process, thereby forming an electrochemical H2O2 biosensor that demonstrated good analytical performance, stability, and selectivity, achieving a low detection limit of 18 nM (Fig. 11e). Furthermore, they conducted another study in which they also employed HRP.177 They immobilized cells, HRP, and superoxide dismutase as sensing elements within a non-covalently crosslinked peptide-derived hydrogel matrix to create an electrochemical biosensor for in situ detection of released superoxide anions (O2˙). The proximity of the enzyme and O2-producing cells enabled the sensor device to attain a high level of sensitivity, yielding precise responses to minuscule quantities of O2˙ molecules. Inspired by this approach, Zheng et al. similarly adopted a strategy and developed a series of non-covalently crosslinked peptide-based supramolecular hydrogels, where they achieved the detection of biologically important signaling molecules such as GSH, NO, and H2S by inducing a “triggered self-degradation” process.96
Monitoring physiological indices is significant for understanding the body's state and predicting disease trends. The use of polypeptide hydrogels offers a new avenue for real-time monitoring, providing fresh opportunities in medical research and clinical practice. Conductive polymer hydrogels (CPHs) constitute a unique class of materials that combine the advantages of hydrogels and organic conductors, finding applications in various fields.178 Nevertheless, the applications of CPHs as functional materials in biomedical contexts are currently restricted by the requirement for biocompatibility, self-repair capability, and mechanical performance. Chakraborty et al. developed an approach involving the use of biocompatible and highly gelation-capable protective dipeptides as non-covalently crosslinked supramolecular gelators.108 These dipeptides were integrated with an in situ polymerized conductive polymer, polyaniline (PAni), to create a dual-component, entirely organic conductive hydrogel. This hydrogel demonstrates pressure sensitivity and is suitable for a range of pressure sensing applications, serving as a conductive interface for electrogenic cardiac cells (Fig. 11f and g). This sensitive monitoring system is significant for early disease diagnosis and treatment.
Stimuli-responsive peptide hydrogels, as an emerging biosensor carrier material, possess good biocompatibility, tunability, and multifunctionality. They can detect biomolecules, biological signaling molecules, and physiological indices by external environmental stimuli or specific interactions with target molecules. Such sensors can visually detect low concentrations of biomolecules through signal amplification mechanisms. Although signal amplification helps improve the sensitivity of biosensors, it also leads to more complex result interpretation. Ensuring detection sensitivity while making the results more intuitive still needs further research. In addition, selectively detecting and identifying various biomolecules in in vivo complex biological environments remains a pressing technical barrier to overcome. Furthermore, the development of miniaturized and implantable devices is expected to facilitate the clinical translation of stimuli-responsive peptide hydrogel-based biosensors.
4. Conclusions
The escalating demand for “smart” materials in biomedicine has intensified the focus on stimulus-responsive hydrogels. Peptides, with their unique ability for molecular self-assembly and ease of functionalization and responsiveness, epitomize an ideal choice for the construction of stimuli-responsive hydrogels. Peptide hydrogels, as a distinct class of materials, find widespread use in biomedical applications.In this review, we center on stimuli-responsive peptide hydrogels, highlighting the most recent progress in their design and biomedical applications. Peptide hydrogels can be rationally engineered to respond to a diverse array of stimuli, including temperature, pH, light, enzymes, ions, and more, which are intricately linked with biological processes. Extensive research efforts have been devoted to designing peptide hydrogels that exhibit responsiveness to these environmental cues. Various strategies for designing stimulus-responsive peptide hydrogels can be categorized generally into two overarching themes: firstly, harnessing the intrinsic stimulus-responsiveness of peptides themselves, either directly or by cross-linking with other materials to form hydrogels; secondly, incorporating substances that respond to specific stimuli into peptides to confer desired responsiveness. It is noteworthy that the stimulus responsiveness of peptide hydrogels is not confined to a singular factor. When hydrogels acquire the capability of responding to stimuli, they provide even more possibilities in the biomedical field. They can be used in various biomedical applications, including tissue repair and regeneration, precise control of drug release, expedited wound healing, and the treatment of bacterial infections, among others.
Despite the promising progress in the design and biomedical utilization of stimuli-responsive peptide hydrogels, further efforts are required to fully realize advanced biomedical applications. Currently, several key aspects warrant more research attention. Firstly, the selectivity and precision of stimulus-responsive peptide hydrogels need to be further improved. The complex physiological milieu contains identical stimuli at multiple sites, which can elicit unintended responses from the hydrogels in off-target areas. A key challenge is improving the accuracy of peptide hydrogels to selectively reach only intended sites without off-target triggering. Secondly, peptides typically exhibit weak intermolecular cross-linking and unstable supramolecular interactions, which can lead to insufficient mechanical strength and stability. It is imperative to enhance the stability of peptide hydrogels while retaining their bioactivity. This can be achieved by optimizing peptide structures to increase cross-linking density or incorporating materials with robust mechanical properties to form double networks. Thirdly, research on hydrogels has focused on dermal applications, with minimal focus on implantable systems for in vivo use. Further developing injectable peptide hydrogels that gel in situ is necessary to expand their utility in internal biomedical applications. These challenges require focused research efforts to unlock the full potential of stimulus-responsive peptide hydrogels in advanced biomedical applications.
In summary, stimuli-responsive peptide hydrogels are extremely promising biomaterials for biomedical applications. Over the past few years, significant progress has been achieved in the design of these hydrogels and their biomedical applications. Currently, stimuli-responsive peptide hydrogels demonstrate significant potential, with ample opportunities for further advancement in this domain. This review provides a comprehensive overview of the latest advancements, remaining challenges and future perspectives regarding stimuli-responsive peptide hydrogels. We believe that this review will inspire the rational design of more innovative stimuli-responsive peptide hydrogels for biomedical applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 52103148), the Start-up Funding from Chongqing University. The authors thank Mr Jiawei Sun for helpful discussions.References
- J. D. Hartgerink, E. Beniash and S. I. Stupp, Science, 2001, 294, 1684–1688 CrossRef CAS PubMed.
- Y. S. Zhang and A. Khademhosseini., Science, 2017, 356, eaaf3627 CrossRef PubMed.
- D. Seliktar, Science, 2012, 336, 1124–1128 CrossRef CAS PubMed.
- E. R. Draper and D. J. Adams, Chem. Soc. Rev., 2018, 47, 3395–3405 RSC.
- Y. Y. Yin, S. H. Yang, D. Q. Ai, H. Qin, Y. Sun, X. Y. Xia, X. H. Xu, W. Ji and J. L. Song, Adv. Funct. Mater., 2023, 33, 2301062 CrossRef CAS.
- Y. H. Li, G. Y. Huang, X. H. Zhang, B. Q. Li, Y. M. Chen, T. L. Lu, T. J. Lu and F. Xu, Adv. Funct. Mater., 2013, 23, 660–672 CrossRef CAS.
- Y. L. Li, J. Rodrigues and H. Tomas, Chem. Soc. Rev., 2012, 41, 2193–2221 RSC.
- A. S. Hoffman, Adv. Drug Delivery Rev., 2012, 64, 18–23 CrossRef.
- X. Q. Dou, N. Mehwish, C. L. Zhao, J. Y. Liu, C. Xing and C. L. Feng, Accounts Chem Res, 2020, 53, 852–862 CrossRef CAS PubMed.
- H. L. Qin, T. Zhang, N. Li, H. P. Cong and S. H. Yu, Nat. Commun., 2019, 10, 2202 CrossRef PubMed.
- W. Lu, X. X. Le, J. W. Zhang, Y. J. Huang and T. Chen, Chem. Soc. Rev., 2017, 46, 1284–1294 RSC.
- V. Yesilyurt, M. J. Webber, E. A. Appel, C. Godwin, R. Langer and D. G. Anderson, Adv. Mater., 2016, 28, 86–91 CrossRef CAS PubMed.
- J. M. Hu, G. Q. Zhang and S. Y. Liu, Chem. Soc. Rev., 2012, 41, 5933–5949 RSC.
- Y. Yang, C. Li, L. C. Palmer and S. I. Stupp, Sci. Adv., 2023, 9, eadi4566 CrossRef CAS PubMed.
- P. Gupta, K. Vermani and S. Garg, Drug Discovery Today, 2002, 7, 569–579 CrossRef CAS PubMed.
- K. Nagahama, T. Ouchi and Y. Ohya, Adv. Funct. Mater., 2008, 18, 1220–1231 CrossRef CAS.
- M. R. Dethe, A. Prabakaran, H. Ahmed, M. Agrawal, U. Roy and A. Alexander, J. Controlled Release, 2022, 343, 217–236 CrossRef CAS PubMed.
- H. J. Dai, H. Zhang, L. Ma, H. Y. Zhou, Y. Yu, T. Guo, Y. H. Zhang and H. H. Huang, Carbohydr. Polym., 2019, 209, 51–61 CrossRef CAS PubMed.
- B. L. Lu, F. C. Lin, X. Jiang, J. J. Cheng, Q. L. Lu, J. B. Song, C. Chen and B. Huang, ACS Sustainable Chem. Eng., 2017, 5, 948–956 CrossRef CAS.
- S. N. Dobic, J. M. Filipovic and S. L. Tomic, Chem. Eng. J., 2012, 179, 372–380 CrossRef CAS.
- H. Mackova, Z. Plichta, H. Hlidkova, O. Sedlacek, R. Konefal, Z. Sadakbayeva, M. Duskova-Smrckova, D. Horak and S. Kubinova, ACS Appl. Mater. Interfaces, 2017, 9, 10544–10553 CrossRef CAS PubMed.
- N. Samiraninezhad, K. Asadi, H. Rezazadeh and A. Gholami, Int. J. Biol. Macromol., 2023, 252, 126573 CrossRef CAS PubMed.
- N. Ninan, A. Forget, V. P. Shastri, N. H. Voelcker and A. Blencowe., ACS Appl. Mater. Interfaces, 2016, 8, 28511–28521 CrossRef CAS PubMed.
- N. Bhattarai, J. Gunn and M. Q. Zhang, Adv. Drug Delivery Rev., 2010, 62, 83–99 CrossRef CAS PubMed.
- K. Yue, G. Trujillo-de Santiago, M. M. Alvarez, A. Tamayol, N. Annabi and A. Khademhosseini., Biomaterials, 2015, 73, 254–271 CrossRef CAS PubMed.
- J. Wang, K. Liu, R. R. Xing and X. H. Yan, Chem. Soc. Rev., 2016, 45, 5589–5604 RSC.
- W. Ji, S. J. Zhang, S. Yukawa, S. Onomura, T. Sasaki, K. Miyazawa and Y. Zhang, Angew. Chem., Int. Ed., 2018, 57, 3636–3640 CrossRef CAS PubMed.
- S. A. Fisher, A. E. G. Baker and M. S. Shoichet, J. Am. Chem. Soc., 2017, 139, 7416–7427 CrossRef CAS PubMed.
- J. Chen and X. N. Zou, Bioact Mater, 2019, 4, 120–131 Search PubMed.
- I. W. Hamley, Chem. Rev., 2017, 117, 14015–14041 CrossRef CAS PubMed.
- A. Levin, T. A. Hakala, L. Schnaider, G. J. L. Bernardes, E. Gazit and T. P. J. Knowles, Nat. Rev. Chem., 2020, 4, 615–634 CrossRef CAS.
- W. Ji, C. Q. Yuan, P. Chakraborty, P. Makam, S. Bera, S. Rencus-Lazar, J. B. Li, X. H. Yan and E. Gazit, ACS Nano, 2020, 14, 7181–7190 CrossRef CAS PubMed.
- W. Ji, Y. M. Tang, P. Makam, Y. F. Yao, R. R. Jiao, K. Y. Cai, G. H. Wei and E. Gazit, J. Am. Chem. Soc., 2021, 143, 17633–17645 CrossRef CAS PubMed.
- K. Tao, P. Makam, R. Aizen and E. Gazit, Science, 2017, 358, eaam9756 CrossRef PubMed.
- F. Sheehan, D. Sementa, A. Jain, M. Kumar, M. Tayarani-Najjaran, D. Kroiss and R. V. Ulijn, Chem. Rev., 2021, 121, 13869–13914 CrossRef CAS PubMed.
- Y. W. Hao, J. X. Meng and S. T. Wang, Chin. Chem. Lett., 2017, 28, 2085–2091 CrossRef CAS.
- L. Li, J. M. Scheiger and P. A. Levkin, Adv. Mater., 2019, 31, 1807333 CrossRef PubMed.
- H. Shigemitsu, R. Kubota, K. Nakamura, T. Matsuzaki, S. Minami, T. Aoyama, K. Urayama and I. Hamachi, Nat. Commun., 2020, 11, 3859 CrossRef PubMed.
- M. Fu, C. Y. Zhang, Y. X. Dai, X. Li, M. B. Pan, W. L. Huang, H. Qian and L. Ge, Biomater. Sci., 2018, 6, 1480–1491 RSC.
- Y. P. Liang, X. Zhao, T. L. Hu, B. J. Chen, Z. H. Yin, P. X. Ma and B. L. Guo, Small, 2019, 15, 1900046 CrossRef PubMed.
- R. Binaymotlagh, L. Chronopoulou and C. Palocci, J. Funct. Biomater., 2023, 14, 233 CrossRef CAS PubMed.
- Y. Q. Hou, T. Y. Tan, Z. Guo, Y. W. Ji, J. Hu and Y. Zhang, Biomater. Sci., 2022, 10, 3831–3844 RSC.
- A. M. Hilderbrand, E. M. Ford, C. Guo, J. D. Sloppy and A. M. Kloxin, Biomater. Sci., 2020, 8, 1256–1269 RSC.
- J. Qu, X. Zhao, Y. P. Liang, Y. M. Xu, P. X. Ma and B. L. Guo, Chem. Eng. J., 2019, 362, 548–560 CrossRef CAS.
- J. Kim, I. Jeerapan, S. Imani, T. N. Cho, A. Bandodkar, S. Cinti, P. P. Mercier and J. Wang, ACS Sens., 2016, 1, 1011–1019 CrossRef CAS.
- S. Panja and D. J. Adams, Chem. Soc. Rev., 2021, 50, 5165–5200 RSC.
- M. C. Koetting, J. T. Peters, S. D. Steichen and N. A. Peppas, Mater. Sci. Eng., R, 2015, 93, 1–49 CrossRef PubMed.
- M. Dzuricky, S. Roberts and A. Chilkoti., Biochemistry, 2018, 57, 2405–2414 CrossRef CAS PubMed.
- K. M. Ruff, S. Roberts, A. Chilkoti and R. V. Pappu, J. Mol. Biol., 2018, 430, 4619–4635 CrossRef CAS PubMed.
- J. S. Lee, M. J. Kang, J. H. Lee and D. W. Linl, Biomacromolecules, 2022, 23, 2051–2063 CrossRef CAS PubMed.
- Y. Mizuguchi, Y. Mashimo, M. Mie and E. Kobatake, Biomacromolecules, 2020, 21, 1126–1135 CrossRef CAS PubMed.
- R. C. Op′t Veld, O. I. van den Boomen, D. M. S. Lundvig, E. M. Bronkhorst, P. H. J. Kouwer, J. A. Jansen, E. Middelkoop, J. W. Von den Hoff, A. E. Rowan and F. Wagener., Biomaterials, 2018, 181, 392–401 CrossRef PubMed.
- Z. Q. Lin, J. F. Ding, X. S. Chen and C. L. He, Chem. – Asian J., 2023, 18, e202300021 CrossRef CAS PubMed.
- Y. C. Zhang, M. S. Desai, T. Wang and S. W. Lee, Biomacromolecules, 2020, 21, 1149–1156 CrossRef CAS PubMed.
- S. Schmidt, M. Zeiser, T. Hellweg, C. Duschl, A. Fery and H. Mohwald, Adv. Funct. Mater., 2010, 20, 3235–3243 CrossRef CAS.
- Y. Cheng, C. Huang, D. Yang, K. Ren and J. Wei, J. Mater. Chem. B, 2018, 6, 8170–8179 RSC.
- Y. Fang, J. J. Tan, S. Lim and S. L. Soh, NPG Asia Mater., 2018, 10, e465 CrossRef CAS.
- T. Guan, J. Y. Li, C. Y. Chen and Y. Liu, Adv. Sci., 2022, 9, 2104165 CrossRef CAS PubMed.
- Y. Zhu, L. Y. Wang, Y. P. Li, Z. W. Huang, S. Y. Luo, Y. He, H. Han, A. S. Raza, J. Wu and L. Ge, Biomater. Sci., 2020, 8, 5415–5426 RSC.
- J. Gačanin, J. Hedrich, S. Sieste, G. Glasser, I. Lieberwirth, C. Schilling, S. Fischer, H. Barth, B. Knoll, C. V. Synatschke and T. Weil., Adv. Mater., 2019, 31, 1805044 CrossRef PubMed.
- P. Frederix, G. G. Scott, Y. M. Abul-Haija, D. Kalafatovic, C. G. Pappas, N. Javid, N. T. Hunt, R. V. Ulijn and T. Tuttle, Nat. Chem., 2015, 7, 30–37 CrossRef CAS PubMed.
- S. Fleming and R. V. Ulijn, Chem. Soc. Rev., 2014, 43, 8150–8177 RSC.
- G. Fichman and E. Gazit, Acta Biomater., 2014, 10, 1671–1682 CrossRef CAS PubMed.
- D. E. Clarke, C. D. J. Parmenter and O. A. Scherman, Angew. Chem., Int. Ed., 2018, 57, 7709–7713 CrossRef CAS PubMed.
- H. Kaur, P. Sharma, N. Patel, V. K. Pal and S. Roy, Langmuir, 2020, 36, 12107–12120 CrossRef CAS PubMed.
- Y. P. Tong, Z. C. Wang, Y. Xiao, W. Liu, J. H. Pan, Y. Zhou and M. D. Lang, ACS Appl. Bio Mater., 2019, 2, 4545–4556 CrossRef CAS PubMed.
- Y. X. Xiang, C. Liu, S. A. Ma, X. T. Wang, L. Y. Zhu and C. Y. Bao, Adv. Funct. Mater., 2023, 33, 2300416 CrossRef CAS.
- T. Koga, Y. Oatari, H. Motoda, S. N. Nishimura, Y. Sasaki, Y. Okamoto, D. Yamamoto, A. Shioi and N. Higashi, Biomacromolecules, 2022, 23, 2941–2950 CrossRef CAS PubMed.
- A. Reinecke, G. Brezesinski and M. J. Harrington, Adv. Mater. Interfaces, 2017, 4, 1600416 CrossRef.
- M. Rammal, C. Li, J. Reeves, C. Moraes and M. J. Harrington, ACS Appl. Mater. Interfaces, 2023, 15, 29004–29011 CrossRef CAS PubMed.
- T. N. Pham, C. F. Su, C. C. Huang and J. S. Jan, Colloids Surf., B, 2020, 196, 111316 CrossRef CAS PubMed.
- L. C. Liu, H. Wang, Y. Y. Han, S. S. Lv and J. F. Chen, J. Mater. Chem. B, 2018, 6, 5303–5312 RSC.
- H. Tsutsumi, M. Kawamura and H. Mihara, Bioorg. Med. Chem., 2018, 26, 3126–3132 CrossRef CAS PubMed.
- M. K. Pradhan, D. Gupta, K. R. Namdev, A. S. Anjali, C. Miglani, A. Pal and A. Srivastava, Nanoscale, 2022, 14, 15079–15090 RSC.
- C. S. Fox, H. A. Berry and S. Pedigo, Biomacromolecules, 2020, 21, 2073–2086 CrossRef CAS PubMed.
- J. R. Luo and F. Sun, Polym. Chem., 2020, 11, 4973–4977 RSC.
- B. B. Sun, K. Tao, Y. Jia, X. H. Yan, Q. L. Zou, E. Gazit and J. B. Li, Chem. Soc. Rev., 2019, 48, 4387–4400 RSC.
- X. J. Liu, M. Gao, J. Y. Chen, S. Guo, W. Zhu, L. C. Bai, W. Z. Zhai, H. J. Du, H. Wu, C. Z. Yan, Y. S. Shi, J. W. Gu, H. J. Qi and K. Zhou, Adv. Funct. Mater., 2022, 32, 2203323 CrossRef CAS.
- D. D. Wu, X. Xie, A. A. Kadi and Y. Zhang, Chin. Chem. Lett., 2018, 29, 1098–1104 CrossRef CAS.
- B. P. Nowak and B. J. Ravoo, Faraday Discuss., 2019, 219, 220–228 RSC.
- B. Li, A. Adam, M. Criado-Gonzalez, L. Jierry, J. Bizeau, A. Chaumont, S. Harlepp, C. Mélart, S. Begin-Colin, D. Begin and D. Mertz, Materialia, 2022, 22, 101414 CrossRef CAS.
- T. O. Omosun, M. C. Hsieh, W. S. Childers, D. Das, A. K. Mehta, N. R. Anthony, T. Pan, M. A. Grover, K. M. Berland and D. G. Lynn, Nat. Chem., 2017, 9, 805–809 CrossRef CAS PubMed.
- I. W. Hamley, Angew. Chem., Int. Ed., 2007, 46, 8128–8147 CrossRef CAS PubMed.
- Y. Inoue, P. Kuad, Y. Okumura, Y. Takashima, H. Yamaguchi and A. Harada., J. Am. Chem. Soc., 2007, 129, 6396–6397 CrossRef CAS PubMed.
- T. Sakai, H. Murayama, S. Nagano, Y. Takeoka, M. Kidowaki, K. Ito and T. Seki, Adv. Mater., 2007, 19, 2023–2025 CrossRef CAS.
- Y. C. Huang, Z. J. Qiu, Y. M. Xu, J. F. Shi, H. K. Lin and Y. Zhang, Org. Biomol. Chem., 2011, 9, 2149–2155 RSC.
- J. F. Shi, G. Fichman and J. P. Schneider, Angew. Chem., Int. Ed., 2018, 57, 11188–11192 CrossRef CAS PubMed.
- C. Wu, C. L. Wang, Y. Y. Zheng, Y. X. Zheng, Z. Q. Liu, K. M. Xu and W. Y. Zhong, Adv. Funct. Mater., 2021, 31, 2104418 CrossRef CAS.
- J. F. Shi and J. P. Schneider, Angew. Chem., Int. Ed., 2019, 58, 13706–13710 CrossRef CAS PubMed.
- L. P. da Silva, A. K. Jha, V. M. Correlo, A. P. Marques, R. L. Reis and K. E. Healy, Adv. Healthcare Mater., 2018, 7, 1700686 CrossRef PubMed.
- G. J. Feng, Z. S. Zha, Y. Huang, J. J. Li, Y. H. Wang, W. D. Ke, H. Y. Chen, L. M. Liu, Y. M. Song and Z. S. Ge, Adv. Healthcare Mater., 2018, 7, 1800623 CrossRef PubMed.
- C. X. Fan, J. J. Shi, Y. Zhuang, L. L. Zhang, L. Huang, W. Yang, B. Chen, Y. Y. Chen, Z. F. Xiao, H. Shen, Y. N. Zhao and J. W. Dai, Adv. Mater., 2019, 31, 1902900 CrossRef CAS PubMed.
- B. B. Hu, N. Song, Y. W. Cao, M. M. Li, X. Liu, Z. F. Zhou, L. Q. Shi and Z. L. Yu, J. Am. Chem. Soc., 2021, 143, 13854–13864 CrossRef CAS PubMed.
- M. J. Zhou, Y. Qian, Y. M. Zhu and J. Matson, Chem. Commun., 2020, 56, 1085–1088 RSC.
- R. Liu, Z. K. Shi, J. Sun and Z. B. Li, Sci. China: Chem., 2018, 61, 1314–1319 CrossRef CAS.
- D. B. Zheng, Z. F. Gao, T. Y. Xu, C. H. Liang, Y. Shi, L. Wang and Z. M. Yang, Nanoscale, 2018, 10, 21459–21465 RSC.
- R. Otter, C. M. Berac, S. Seiffert and P. Besenius, Eur. Polym. J., 2019, 110, 90–96 CrossRef CAS.
- A. Rani, L. M. De Leon-Rodriguez, I. Kavianinia, D. J. McGillivray, D. E. Williams and M. A. Brimble, Org. Biomol. Chem., 2021, 19, 3665–3677 RSC.
- A. R. Mohanty, A. Ravikumar and N. A. Peppas, Regener. Biomater., 2022, 9, rbac056 CrossRef CAS PubMed.
- M. C. Manas-Torres, C. Gila-Vilchez, F. J. Vazquez-Perez, P. Kuzhir, D. Momier, J. C. Scimeca, A. Borderie, M. Goracci, F. Burel-Vandenbos, C. Blanco-Elices, I. A. Rodriguez, M. Alaminos, L. A. de Cienfuegos and M. T. Lopez-Lopez, ACS Appl. Mater. Interfaces, 2021, 13, 49692–49704 CrossRef CAS PubMed.
- B. P. Nowak, M. Niehues and B. J. Ravoo, Soft Matter, 2021, 17, 2857–2864 RSC.
- S. Z. Kou, X. Yang, Z. G. Yang, X. T. Liu, S. V. Wegner and F. Sun, ACS Macro Lett., 2019, 8, 773–778 CrossRef CAS PubMed.
- Y. Zhang, B. Zhang, Y. Kuang, Y. Gao, J. F. Shi, X. X. Zhang and B. Xu, J. Am. Chem. Soc., 2013, 135, 5008–5011 CrossRef CAS PubMed.
- Y. H. Huo, J. Hu, Y. Y. Yin, P. Liu, K. Y. Cai and W. Ji, ChemBioChem, 2023, 24, e202200582 CrossRef CAS PubMed.
- S. Bera, S. Mondal, B. Xue, L. J. W. Shimon, Y. Cao and E. Gazit, Nat. Mater., 2019, 18, 503–509 CrossRef CAS PubMed.
- S. Bera, S. Guerin, H. Yuan, J. O'Donnell, N. P. Reynolds, O. Maraba, W. Ji, L. J. W. Shimon, P. A. Cazade, S. A. M. Tofail, D. Thompson, R. S. Yang and E. Gazit, Nat. Commun., 2021, 12, 2634 CrossRef CAS PubMed.
- X. Q. Dou and C. L. Feng, Adv. Mater., 2017, 29, 1604062 CrossRef PubMed.
- P. Chakraborty, T. Guterman, N. Adadi, M. Yadid, T. Brosh, L. Adler-Abramovich, T. Dvir and E. Gazit, ACS Nano, 2019, 13, 163–175 CrossRef CAS PubMed.
- R. Chang, L. Y. Zhao, R. R. Xing, J. B. Li and X. H. Yan, Chem. Soc. Rev., 2023, 52, 2688–2712 RSC.
- M. Vazquez-Gonzalez and I. Willner, Angew. Chem., Int. Ed., 2020, 59, 15342–15377 CrossRef CAS PubMed.
- T. Li, X. M. Lu, M. R. Zhang, K. Hu and Z. Li, Bioact. Mater., 2022, 11, 268–282 CAS.
- A. S. Mao and D. J. Mooney, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 14452–14459 CrossRef CAS PubMed.
- T. Hoffman, P. Antovski, P. Tebon, C. Xu, N. Ashammakhi, S. Ahadian, L. Morsut and A. Khademhosseini, Adv. Funct. Mater., 2020, 30, 1909882 CrossRef CAS.
- K. Y. Lee and D. J. Mooney, Chem. Rev., 2001, 101, 1869–1879 CrossRef CAS PubMed.
- S. Bera, P. A. Cazade, S. Bhattacharya, S. Guerin, M. Ghosh, F. Netti, D. Thompson and L. Adler-Abramovich, ACS Appl. Mater. Interfaces, 2022, 14, 46827–46840 CrossRef CAS PubMed.
- J. Qu, X. Zhao, Y. P. Liang, T. L. Zhang, P. X. Ma and B. L. Guo, Biomaterials, 2018, 183, 185–199 CrossRef CAS PubMed.
- X. Liu, X. M. Wang, X. J. Wang, H. Ren, J. He, L. Qiao and F. Z. Cui, Acta Biomater., 2013, 9, 6798–6805 CrossRef CAS PubMed.
- A. Pal, C. I. Smith, J. Palade, S. Nagarajua, B. A. Alarcon-Benedetto, J. Kilbourne, A. Rawls, J. Wilson-Rawls, B. L. Vernon and M. Nikkhah, Acta Biomater., 2020, 107, 138–151 CrossRef CAS PubMed.
- X. Li, S. Q. Bian, M. D. Zhao, X. W. Han, J. Liang, K. F. Wang, Q. Jiang, Y. Sun, Y. J. Fan and X. D. Zhang, Acta Biomater., 2021, 131, 128–137 CrossRef CAS PubMed.
- Z. H. Yang, H. Y. Xu and X. J. Zhao, Adv. Sci., 2020, 7, 1903718 CrossRef CAS PubMed.
- E. F. Banwell, E. S. Abelardo, D. J. Adams, M. A. Birchall, A. Corrigan, A. M. Donald, M. Kirkland, L. C. Serpell, M. F. Butler and D. N. Woolfson, Nat. Mater., 2009, 8, 596–600 CrossRef CAS PubMed.
- N. Mehrban, E. Abelardo, A. Wasmuth, K. L. Hudson, L. M. Mullen, A. R. Thomson, M. A. Birchall and D. N. Woolfson, Adv. Healthcare Mater., 2014, 3, 1387–1391 CrossRef CAS PubMed.
- S. Das and D. Das, Front. Chem., 2021, 9, 770102 CrossRef CAS PubMed.
- M. J. Webber and E. T. Pashuck, Adv. Drug Delivery Rev., 2021, 172, 275–295 CrossRef CAS PubMed.
- M. W. Cao, Y. Wang, X. Z. Hu, H. N. Gong, R. H. Li, H. Cox, J. Zhang, T. A. Waigh, H. Xu and J. R. Lu, Biomacromolecules, 2019, 20, 3601–3610 CrossRef CAS PubMed.
- M. K. Monroe, H. Wang, C. F. Anderson, M. Qin, C. L. Thio, C. Flexner and H. G. Cui, Biomater. Sci., 2023, 11, 489–498 RSC.
- H. K. Noddeland, M. Lind, L. B. Jensen, K. Petersson, T. Skak-Nielsen, F. H. Larsen, M. Malmsten and A. Heinz, Acta Biomater., 2023, 157, 149–161 CrossRef CAS PubMed.
- Y. Q. Zhang, M. G. Qin, C. Xing, C. L. Zhao, X. Q. Dou and C. L. Feng, ACS Nano, 2020, 14, 17151–17162 CrossRef CAS PubMed.
- S. Mansoor, P. P. D. Kondiah and Y. E. Choonara, Pharmaceutics, 2021, 13, 2113 CrossRef CAS PubMed.
- X. Bao, X. H. Si, X. Y. Ding, L. J. Duan and C. S. Xiao, J. Polym. Res., 2019, 26, 278 CrossRef CAS.
- M. E. Roth-Konforti, M. Comune, M. Halperin-Sternfeld, I. Grigoriants, D. Shabat and L. Adler-Abramovich, Macromol. Rapid Commun., 2018, 39, 1800588 CrossRef PubMed.
- M. H. Turabee, T. Thambi, J. S. Lym and D. S. Lee, Biomater. Sci., 2017, 5, 837–848 RSC.
- G. Cirillo, U. G. Spizzirri, M. Curcio, F. P. Nicoletta and F. Iemma, Pharmaceutics, 2019, 11, 486 CrossRef CAS PubMed.
- R. R. Mehra, P. Tiwari, A. Basu and A. DuttKonar, New J. Chem., 2019, 43, 11666–11678 RSC.
- T. Mateti, K. Likhith, A. Laha and G. Thakur, Curr. Opin. Biomed. Eng., 2023, 25, 100424 CrossRef CAS.
- Y. F. Liu, Y. C. Ran, Y. Ge, F. Raza, S. S. Li, H. Zafar, Y. Q. Wu, A. C. Paiva-Santos, C. Y. Yu, M. Sun, Y. Zhu and F. Li, Pharmaceutics, 2022, 14, 652 CrossRef CAS PubMed.
- X. A. Yu, H. Wang, X. Liu, L. Huang, N. Song, Y. Q. Song, X. W. Mo, S. F. Lou, L. Q. Shi and Z. L. Yu, Nano Today, 2022, 46, 101594 CrossRef CAS.
- M. T. Tao, S. Y. He, J. Liu, H. M. Li, L. X. Mei, C. Wu, K. M. Xu and W. Y. Zhong, J. Mater. Chem. B, 2019, 7, 469–476 RSC.
- L. X. Mei, K. M. Xu, Z. R. Zhai, S. Y. He, T. T. Zhu and W. Y. Zhong, Org. Biomol. Chem., 2019, 17, 3853–3860 RSC.
- J. Zhu, R. Gao, Z. S. Wang, Z. M. Cheng, Z. H. Xu, Z. Y. Liu, Y. Q. Wu, M. Wang and Y. Zhang, Pharmaceutics, 2023, 15, 668 CrossRef CAS PubMed.
- P. H. Li, X. Y. Dai, L. J. Qu, Y. L. Sui and C. L. Zhang, RSC Adv., 2020, 10, 24507–24514 RSC.
- M. Farahani and A. Shafiee, Adv. Healthcare Mater., 2021, 10, 2100477 CrossRef CAS PubMed.
- S. H. Jeong, S. A. Cheong, T. Y. Kim, H. Choi and S. K. Hahn, ACS Appl. Mater. Interfaces, 2023, 15, 16471–16481 CrossRef CAS PubMed.
- H. N. Suo, M. Hussain, H. Wang, N. Y. Zhou, J. Tao, H. Jiang and J. T. Zhu, Biomacromolecules, 2021, 22, 3049–3059 CrossRef CAS PubMed.
- C. X. Chen, Y. Zhang, R. Fei, C. H. Cao, M. Wang, J. X. Wang, J. K. Bai, H. Cox, T. Waigh, J. R. Lu and H. Xu, ACS Appl. Mater. Interfaces, 2016, 8, 17833–17841 CrossRef CAS PubMed.
- M. Wu, Z. Y. Ye, H. Y. Zhu and X. J. Zhao, Biomacromolecules, 2015, 16, 3112–3118 CrossRef CAS PubMed.
- X. J. Huang, T. T. Li, X. Y. Jiang, Z. L. Wang, M. S. Wang, X. Wu, J. Li and J. F. Shi, ACS Appl. Mater. Interfaces, 2023, 15, 45606–45615 CrossRef CAS PubMed.
- C. Xing, H. T. Zhu, X. Q. Dou, L. B. Gao, S. Baddi, Y. Q. Zou, C. L. Zhao, Y. B. Peng, Y. Fang and C. L. Feng, ACS Nano, 2023, 17, 6275–6291 CrossRef CAS PubMed.
- Y. S. Shang, P. H. Wang, X. Z. Wan, L. J. Wang, X. Liu, J. Yuan, B. Chi and J. Shen, Int. J. Biol. Macromol., 2023, 242, 124754 CrossRef CAS PubMed.
- G. Y. Li, Z. H. Lai and A. S. Shan, Adv. Sci., 2023, 10, e2206602 CrossRef PubMed.
- J. J. Wang, X. J. Dou, J. Song, Y. F. Lyu, X. Zhu, L. Xu, W. Z. Li and A. S. Shan, Med. Res. Rev., 2019, 39, 831–859 CrossRef CAS PubMed.
- S. W. Wu, Y. L. Yang, S. A. Wang, C. Y. Dong, X. Y. Zhang, R. Zhang and L. Yang, Carbohydr. Polym., 2022, 278, 118994 CrossRef CAS PubMed.
- N. Annabi, D. Rana, E. S. Sani, R. Portillo-Lara, J. L. Gifford, M. M. Fares, S. M. Mithieux and A. S. Weiss, Biomaterials, 2017, 139, 229–243 CrossRef CAS PubMed.
- A. Adak, S. Ghosh, V. Gupta and S. Ghosh, Biomacromolecules, 2019, 20, 1889–1898 CrossRef CAS PubMed.
- J. K. Bai, Z. Y. Gong, J. X. Wang and C. D. Wang, RSC Adv., 2017, 7, 48631–48638 RSC.
- Z. Guo, Y. J. Wang, T. Y. Tan, Y. W. Ji, J. Hu and Y. Zhang, ACS Biomater. Sci. Eng., 2021, 7, 1703–1712 CrossRef CAS PubMed.
- S. Atefyekta, E. Blomstrand, A. K. Rajasekharan, S. Svensson, M. Trobos, J. Hong, T. J. Webster, P. Thomsen and M. Andersson, ACS Biomater. Sci. Eng., 2021, 7, 1693–1702 CrossRef CAS PubMed.
- S. X. Liu, H. L. Cong, B. Yu and Y. Q. Shen, Int. J. Biol. Macromol., 2023, 238, 124056 CrossRef CAS PubMed.
- L. J. Yang, C. R. Zhang, F. Huang, J. J. Liu, Y. M. Zhang, C. H. Yang, C. H. Ren, L. P. Chu, B. Liu and J. F. Liu, J. Controlled Release, 2020, 324, 354–365 CrossRef CAS PubMed.
- J. Durao, N. Vale, S. Gomes, P. Gomes, C. C. Barrias and L. Gales, Biomolecules, 2019, 9, 4 CrossRef PubMed.
- J. Zhu, H. Han, T. T. Ye, F. X. Li, X. L. Wang, J. Y. Yu and D. Q. Wu, Molecules, 2018, 23, 3383 CrossRef PubMed.
- N. Annabi, A. Tamayol, J. A. Uquillas, M. Akbari, L. E. Bertassoni, C. Cha, G. Camci-Unal, M. R. Dokmeci, N. A. Peppas and A. Khademhosseini, Adv. Mater., 2014, 26, 85–124 CrossRef CAS PubMed.
- F. Z. Volpato, T. Führmann, C. Migliaresi, D. W. Hutmacher and P. D. Dalton, Biomaterials, 2013, 34, 4945–4955 CrossRef CAS PubMed.
- A. S. Weingarten, R. V. Kazantsev, L. C. Palmer, M. McClendon, A. R. Koltonow, A. P. S. Samuel, D. J. Kiebala, M. R. Wasielewski and S. I. Stupp, Nat. Chem., 2014, 6, 964–970 CrossRef CAS PubMed.
- N. Zanna, S. Focaroli, A. Merlettini, L. Gentilucci, G. Teti, M. Falconi and C. Tomasini, ACS Omega, 2017, 2, 2374–2381 CrossRef CAS PubMed.
- S. T. Gould, N. J. Darling and K. S. Anseth, Acta Biomater., 2012, 8, 3201–3209 CrossRef CAS PubMed.
- S. Yamaguchi, N. Ohashi, K. Minamihata and T. Nagamune, Biomater. Sci., 2021, 9, 6416–6424 RSC.
- S. X. Song, J. Y. Wang, Z. F. Cheng, Z. M. Yang, L. Q. Shi and Z. L. Yu, Chem. Sci., 2020, 11, 1383–1393 RSC.
- J. W. Xu, G. Y. Qi, C. X. Sui, W. Q. Wang and X. Z. Sun, Toxicol. Vitro, 2019, 61, 104599 CrossRef CAS PubMed.
- W. K. Restu, S. Yamamoto, Y. Nishida, H. Ienaga, T. Aoi and T. Maruyama, Mater. Sci. Eng., C, 2020, 111, 110746 CrossRef CAS PubMed.
- A. C. Chen and S. Chatterjee, Chem. Soc. Rev., 2013, 42, 5425–5438 RSC.
- J. N. Tiwari, V. Vij, K. C. Kemp and K. S. Kim, ACS Nano, 2016, 10, 46–80 CrossRef CAS PubMed.
- Q. Li, M. J. Xian, A. H. Wang, Y. Jia, S. Bai, X. H. Yen and J. B. Li, Matter, 2022, 5, 3643–3658 CrossRef CAS.
- P. J. S. King, A. Saiani, E. V. Bichenkova and A. F. Miller, Chem. Commun., 2016, 52, 6697–6700 RSC.
- T. Yoshii, S. Onogi, H. Shigemitsu and I. Hamachi, J. Am. Chem. Soc., 2015, 137, 3360–3365 CrossRef CAS PubMed.
- M. L. Lian, X. Chen, Y. L. Lu and W. S. Yang, ACS Appl. Mater. Interfaces, 2016, 8, 25036–25042 CrossRef CAS PubMed.
- M. L. Lian, L. Xu, X. W. Zhu, X. Chen, W. S. Yang and T. Wang, Anal. Chem., 2017, 89, 12843–12849 CrossRef CAS PubMed.
- L. J. Pan, G. H. Yu, D. Y. Zhai, H. R. Lee, W. T. Zhao, N. Liu, H. L. Wang, B. C. K. Tee, Y. Shi, Y. Cui and Z. N. Bao, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 9287–9292 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |